

Abstract
Although the current coronavirus disease 2019 (COVID-19) vaccines have been used worldwide to halt spread of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the emergence of new SARS-CoV-2 variants with E484K mutation shows significant resistance to the neutralization of vaccine sera. To better understand the resistant mechanism, we calculated the binding affinities of 26 antibodies to wild-type (WT) spike protein and to the protein harboring E484K mutation, respectively. The results showed that most antibodies (~85%) have weaker binding affinities to the E484K mutated spike protein than to the WT, indicating the high risk of immune evasion of the mutated virus from most of current antibodies. Binding free energy decomposition revealed that the residue E484 forms attraction with most antibodies, while the K484 has repulsion from most antibodies, which should be the main reason of the weaker binding affinities of E484K mutant to most antibodies. Impressively, a monoclonal antibody (mAb) combination was found to have much stronger binding affinity with E484K mutant than WT, which may work well against the mutated virus. Based on binding free energy decomposition, we predicted that the mutation of four more residues on receptor-binding domain (RBD) of spike protein, viz., F490, V483, G485 and S494, may have high risk of immune evasion, which we should pay close attention on during the development of new mAb therapeutics.
Keywords: COVID-19, spike, E484K, antibody, MM/GBSA
Introduction
The SARS-CoV-2 has already caused over 168 million confirmed cases and over 3 million deaths worldwide by the end of May 2021 [1]. Although there are no specifically effective drugs on market to cure the COVID-19, exceeding 80 vaccines are in clinical development and exceeding 180 vaccines are in pre-clinical development [2]. In fact, vaccines have been widely used to curtail the disease, and mAb therapeutic has attracted great concerns [3, 4].
Current mAbs and vaccines were designed based on the initial SARS-CoV-2 identified at the end of 2019. However, SARS-CoV-2, as an RNA virus, was expected to have a high mutation rate [3]. Since identified in the early phases of the COVID-19 pandemic, considerable variants have been reported, including fast-spreading variants appeared in the UK (B.1.1.7), South Africa (B.1.351) and Brazil (B.1.1.248). Several studies have shown that neutralization by some mAbs and immune sera was diminished against the SARS-CoV-2 variants, particularly for the B.1.351 harboring E484K [5]. For instance, Novavax vaccine (NVX-CoV2373), which shows high effective rates of 96 and 86% for the common strains of SARS-CoV-2 and the B.1.1.7, respectively, has greatly reduced effective rate to 51.0% for the B.1.351 in the HIV-negative population and to <50% in entire population, respectively [6].
Recently, experiments in vitro have shown that E484K mutation occurs when SARS-CoV-2 was co-incubated in a highly neutralizing COVID-19 convalescent plasma [7]. The single mutation (E484K) residing in RBD makes the SARS-CoV-2 variant strongly resist to plasma neutralization, indicating that the survivors may be reinfected by the variants carrying with E484K mutation. Recently emerged SARS-CoV-2 variants B.1.1.33, P1 and P2 are also identified as the E484K mutant in the Brazilian territory, which are of serious concern due to the possibility of escaping from neutralizing antibodies [8]. Additionally, a recent experimental study suggested that the E484K mutation impairs the efficacy of current mAbs targeting the angiotensin-converting enzyme 2 (ACE2) binding site, such as REGN10933 [9]. Therefore, the E484K mutation has received particular attention among the SARS-CoV-2 variants due to its worrisome implications in vaccination and passive immune therapies.
Although the E484 resides at the interface of RBD and ACE2 [10], there is no obvious interactions formed by E484 with ACE2 or with other two monomers of spike protein trimer (Figure S1). A number of biophysical and simulation studies also demonstrated the little role of the E484 for ACE2-RBD interactions [11–14]. A deep mutational scanning study revealed that the E484K mutant only leads to slightly increased ACE2 binding affinity of the mutated RBD [15]. Hence, the strong immune evasion of the E484K mutant is unlikely caused by its ACE2 binding affinity but by its mAb interaction.
Considering that the RBD is a dominant target of neutralizing antibodies and its E484K mutation has led to unsettling immune evasion, it is of great importance to understand the underlying mechanism for better coping with the E484K mutation. In this paper, we calculated the binding affinities of current 26 mAbs to RBDWT and to RBDE484K, respectively, alarming that 22 mAbs have decreased binding affinity to the E484K mutant. Impressively, a mAb combination (52 & 298) was predicted to have enhanced binding affinity. In addition, we predicted other four detrimental mutations that may cause potential immune evasion from most of the currently available mAbs.
Methods
Preparation of mAb-spike complexes
Twenty-five mAb-spike complex structures including 26 mAbs that bind to the RBM of spike protein were downloaded directly from Protein Data Bank (PDB). The missing residues were built by using SWISS-MODEL webserver [16, 17]. The E484K mutation was built by PyMOL based on initial wild-type (WT) structures. Two glycosylated spike protein models (PDB ID: 7K43 and 7K4N) were built as representatives of RBD ‘up’ and ‘down’ states, respectively, by using GLYCAM-Web (www.glycam.org).
A cubic box of TIP3P water was used to solvate the complex system, which was extended by 12 Å from the solute. Each system was neutralized by a number of Na+ or Cl−. The protein complexes were parameterized by Amber ff14SB force field [18], and the glycans were parameterized by GLYCAM_06j-1 force field [19]. About, 10 000 steps of minimization, including 5000 steps of steepest descent minimization and 5000 steps of conjugate gradient minimization, were performed to remove bad contacts formed during the system preparation. Each system was heated to 300 K within 0.2 ns, and then 0.1 ns of equilibration was performed in NPT ensemble. Sander program in Amber 18 was used to run the minimization, heating and equilibrium simulations with constraints on heavy atoms [20].
Molecular dynamics simulations
To assess the dynamic interactions of mAb-RBD, pmemd.cuda in Amber 18 was used to perform MD simulations for mAb-RBD complexes at 300 K. Temperature was controlled by Langevin dynamics, and bonds involving hydrogen atoms were fixed by the SHAKE algorithm [21]. The cutoff distance applied for van der Waals interactions was 8.0 Å. And long-range electrostatic interactions were addressed by the particle mesh Ewald method [22]. About, 20 ns of production MD simulations of 25 mAb-spike complexes were run for both the WT and E484K mutant. To explore the binding free energy variance with simulation time, additional 500 ns MD simulations were performed for REGN10933-RBD complexes (PDB ID: 6XDG) [23] (Figure S2 and Table S1).
Binding free energy calculation
Binding free energy (ΔG) of mAb-RBD complexes was calculated by end-point molecular mechanics generalized Born surface area (MM/GBSA) [24]. In this study, the dielectric constants of solute and solvent were set to 1.0 and 80.0, respectively. The OBC solvation model (igb = 5) was used. And the binding free energy was further decomposed into energy contribution of each residue in Amber18 (idecomp = 1).
Results and discussion
Mapping the binding sites of current mAbs on spike protein
Figure 1 shows that the binding sites of current mAbs are largely focused on the S1 fragment of the spike protein, including the N-terminal domain (NTD), the base and the receptor-binding motif (RBM) of RBD, respectively. There are more than ten mAbs targeting NTD that have available crystal structures in PDB, for instance, 4A8 (PDB ID: 7C2L) [25], FC05 (PDB ID: 7CWU) [26] and 2–17 (PDB ID: 7LQW) [27]. Besides, Acharya P. et al. presented a cryo-EM structure of a glycan-dependent mAb (PDB ID: 2G12) targeting N709, N717 and N801 [28], revealing a new S2 epitope on SARS-CoV-2 spike. Although the RBD ‘up’ state is required for the ACE2 binding of spike protein [29], RBD could bind to mAbs in its ‘down’ state. For example, S2E12 (PDB ID: 7K4N) binds to RBD that is in ‘up’ state, while S2M11 (PDB ID: 7K43) binds to RBD that is in ‘down’ state [30]. By structural analysis, we found 26 mAbs bind to RBM and even near the position of E484, indicating that most of the current mAbs are likely influenced by the E484K mutation.
Figure 1.
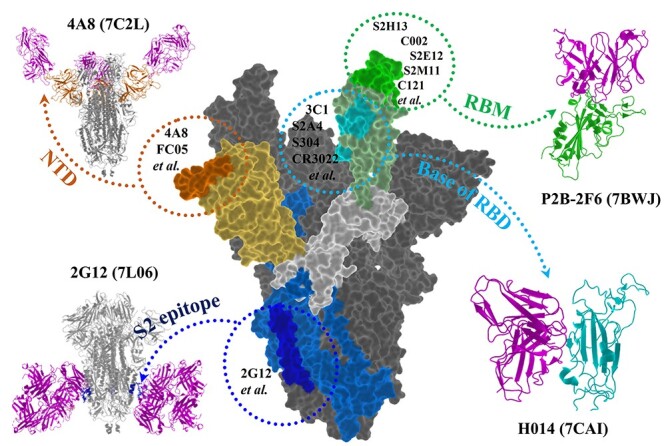
The binding sites of current mAbs on spike protein. The spike protein is represented as surface. The RBM (green), base of RBD (cyan), NTD (orange) and S2 epitope (blue) bound with their representative mAbs (magenta) are shown as cartoon.
The difference of binding free energy between WT and E484K mutant RBDs to the mAb REGN10933
Recently, Hansen et al. found that the neutralization IC50 of REGN10933 on the E484K mutant and SA∆9 (B.1.351) variant was 10.5 and 58.8 times lower than that of the WT, respectively [9]. We calculated the binding free energy (∆G) of REGN10933 to the WT and E484K mutant, respectively. As shown in Figure 1 (Table S2 for details), the ∆G to the mutant RBD is −24.71 ± 0.67, −21.13 ± 0.67, −29.61 ± 0.77 kcal/mol, respectively, in triple runs, while that to the WT is −36.19 ± 0.91, −36.12 ± 0.79, −41.61 ± 0.69 kcal/mol, respectively, in triple runs. The significantly reduced ∆G of the SARS-CoV-2 harboring E484K mutation to REGN10933 suggests its weakened neutralization efficacy, which is consistent with the experimentally determined neutralization IC50 values (resistance 10.5-fold) [9].
To evaluate whether the E484K mutation affects the ACE2 binding affinity, we calculated the ∆G of ACE2 to the WT and the E484K mutant, which are −36.43 ± 0.77 (∆GWT) and − 41.52 ± 0.69 kcal/mol (∆GE484K), respectively. The slightly increased binding free energies demonstrated that E484K mutation has a little beneficial effect on the binding strength of ACE2 to the RBD, which is consistent with biophysical studies [15]. We decomposed the binding free energy and found that the residue E484 contributed 0.82 kcal/mol to the overall binding strength, suggesting that E484 is not a key residue for the RBD-ACE2 interactions. Therefore, the special role of E484K mutation in immune evasion could be attributed to its significantly reduced binding affinity to mAbs.
The diverse impacts of E484K mutation on its binding affinities to 26 mAbs
There are 25 mAb-RBD complex structures found in PDB, including 26 mAbs. To explore the potential effect of the E484K mutation on its binding to those mAbs, we constructed 25 complex structures of the E484K mutant RBD and the 26 mAbs. Then, the binding free energy calculation was performed for the 50 complexes. After analyzing the ∆G of seven different lengths of MD simulation time (Table S3), viz. 4–8, 4–12, 4–16, 4–20, 8–20, 12–20 and 16–20 ns, we found that the average ∆G fluctuation between 4–16 and 4–20 ns is only 1.25 kcal/mol (P = 0.99), showed that 20 ns MD simulation is long enough to obtain reliable ∆G, which is consistent with our previous study [13, 31] and other literature reports [11, 32, 33]. To further explore the impact of simulation time on binding free energy, two REGN10933-RBD complexes for WT and E484K mutant, respectively, were run for 500 ns. As shown in Figure S2 and Table S1, the same variation trend of binding free energy change was observed by the two different simulation time.
With 2.00 kcal/mol as criteria, 22 systems show weaker binding affinity of mAb-RBDE484K than mAb-RBDWT (Table S2), indicating that 85% of the RBD-targeting mAbs might exhibit weaker neutralization ability to the E484K mutated virus. Tortorici M. A. et al. showed that S2M11 (PDB ID: 7K43) possessed lower neutralization ability in the E484K mutant [30], and Chen R. E. et al. showed that the neutralization ability of S2E12 (PDB ID: 7K4N) was reduced about 5 folds against the E484K virus [34]. Therefore, our predictions (Figure 2) are in good agreement with these experimental results, indicating the high reliability of the simulation results.
Figure 2.
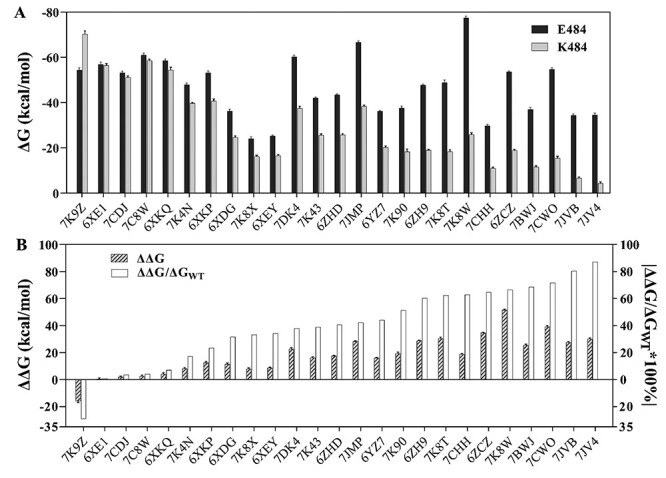
The binding free energy of mAb to RBDWT or RBDE484K. (A) The binding free energy of RBDWT to mAb (black), and of RBDE484K to mAb (gray). (B) The relative binding free energy (ΔΔG = ΔGE484K-ΔGWT) between mAb-RBDWT and mAb-RBDE484K (twill), and the percentage of |ΔΔG/ΔGWT| (white).
Impressively, one mAb combination system (PDB ID: 7K9Z) with two mAbs (52 & 298) has stronger binding affinity to E484K mutant than to WT (ΔΔG = −15.87 ± 1.13 kcal/mol), which may work well to fight against the mutated virus. Two systems, viz., 6XE1 (CV30) [35] and 7CDJ (P2C-1A3) [36], have similar binding free energies between WT and E484K mutant (ΔΔG < 2.00 kcal/mol, Table S2), indicating that the neutralization of these mAbs is insensitive to the E484K mutation. In summary, the E484K mutation might cause an obviously weakened binding affinity of about 85% mAbs to the spike protein of SARS-CoV-2.
Effect of protein glycosylation on the calculated binding free energy
Protein glycosylation plays a crucial role in modulating the conformational dynamics of spike’s RBD. Specifically, N-glycans at N165 and N234 potentially affect RBD binding to ACE2 [37]. As representatives of RBD-up and RBD-down states, we built two glycosylated SARS-CoV-2 spike models at GLYCAM-Web (www.glycam.org) based on the structures of 7K43 (RBD-down, Figure S3) and 7K4N (RBD-up, Figure S4). In total, 51 N-linked glycans (Table S4) were added to the spike protein, and the calculated binding free energy of mAb-spike (glycan) was summarized in Table 1.
Table 1.
The predicted mAb-spike binding free energies of the systems 7K43 and 7K4N (kcal/mol)
| PDB ID | System-Type | ΔGWT | ΔGE484K | ΔΔG | |ΔΔG/ΔGWT| |
|---|---|---|---|---|---|
| 7K43 (S2M11) | Glyco-trimer | −60.41 ± 0.96 | −37.90 ± 1.10 | 22.51 ± 1.03 | 37.26% |
| Trimer | −52.39 ± 0.64 | −34.86 ± 0.92 | 17.53 ± 0.78 | 33.46% | |
| RBD | −42.02 ± 0.53 | −25.67 ± 0.56 | 16.34 ± 0.55 | 38.90% | |
| 7K4N (S2E12) | Glyco-trimer | −36.96 ± 0.48 | −28.22 ± 0.62 | 8.74 ± 0.55 | 23.65% |
| Trimer | −47.13 ± 0.60 | −24.79 ± 0.48 | 22.34 ± 0.54 | 47.40% | |
| RBD | −47.93 ± 0.73 | −39.70 ± 0.49 | 8.23 ± 0.61 | 17.17% |
For the RBD-down state (7K43), the sum of N-glycan contribution to the overall binding free energy is −1.06 ± 0.16 kcal/mol (WT) and 2.20 ± 0.06 kcal/mol (E484K), respectively. The ΔΔG between the WT and E484K mutant are 22.51 ± 1.03, 17.53 ± 0.78 and 16.34 ± 0.55 kcal/mol for glycosylated spike trimer, the spike trimer and the RBD, respectively (Table 1). The results showed that the glycosylation of spike protein may change the specific values of the calculated free energy, but does not alter the variation tendency of the E484K mutation effect.
For the RBD-up state (7K4N), similar results were observed, the sum of N-glycan contribution in the predicted binding free energy is also very small, which is 0.06 ± 0.00 kcal/mol (WT) and 0.97 ± 0.04 kcal/mol (E484K), respectively. Additionally, we compared seven pairs of mAb-spike (trimer) and mAb-RBD (Table 2), the same effect tendency of E484K mutation on binding free energy was observed by using trimer structure or RBD only. Hence, we postulated that the binding free energy of mAb-RBD can reflect the overall change tendency of mAb-spike binding free energy.
Table 2.
The predicted binding free energies of 7 mAbs to spike trimer or to RBD (kcal/mol)
| PDB ID | System type | ΔGWT | ΔGE484K | ΔΔG | |ΔΔG/ΔGWT| |
|---|---|---|---|---|---|
| 7K43 (S2M11) | Trimer | −52.39 ± 0.64 | −34.86 ± 0.92 | 17.53 | 33.46% |
| RBD | −42.02 ± 0.53 | −25.67 ± 0.56 | 16.34 | 38.90% | |
| 7K4N (S2E12) | Trimer | −47.13 ± 0.60 | −24.79 ± 0.48 | 22.34 | 47.40% |
| RBD | −47.93 ± 0.73 | −39.70 ± 0.49 | 8.23 | 17.17% | |
| 7K8T (C002) | Trimer | −45.25 ± 1.06 | −13.37 ± 0.85 | 31.89 | 70.46% |
| RBD | −48.90 ± 1.11 | −18.39 ± 0.78 | 30.51 | 62.39% | |
| 7DK4 (2H2) | Trimer | −60.54 ± 1.16 | −29.52 ± 1.17 | 31.01 | 51.23% |
| RBD | −60.29 ± 0.72 | −37.51 ± 0.97 | 22.78 | 37.79% | |
| 7K8X (C121) | Trimer | −19.77 ± 0.72 | −16.97 ± 0.66 | 2.79 | 14.13% |
| RBD | −24.16 ± 0.77 | −16.15 ± 0.66 | 8.00 | 33.14% | |
| 7K8W (C119) | Trimer | −49.03 ± 1.08 | −14.98 ± 1.22 | 34.05 | 69.45% |
| RBD | −77.42 ± 0.78 | −25.92 ± 0.74 | 51.50 | 66.52% | |
| 7 K90 (C144) | Trimer | −61.03 ± 0.81 | −47.90 ± 1.03 | 13.14 | 21.52% |
| RBD | −37.67 ± 0.86 | −18.34 ± 1.14 | 19.33 | 51.32% |
The role of E484 and K484
To further explore the mechanism of the E484K mutation affecting the neutralization ability of mAbs, we compared energy contributions of E484 and K484 to the overall binding free energy (Figure 3, Table S2). In most of mAb-RBD systems (72%), E484 have obvious greater contribution (> 1.00 kcal/mol) to the binding free energy than that of K484. To further look into different binding modes, 3 mAb-RBD complexes are taken as representative examples, whose neutralization ability is decreased (PDB ID: 7CWO), unchanged (PDB ID: 6XE1) and enhanced (PDB ID: 7K9Z) after the E484K mutation, respectively.
Figure 3.
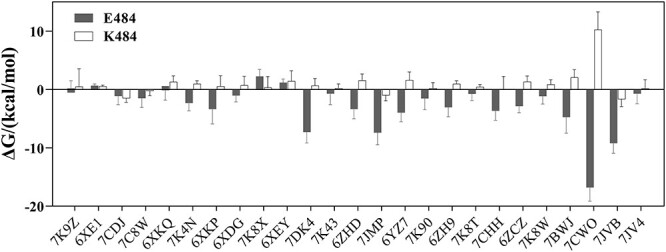
The contribution of E484 or K484 to overall binding free energy. Per residue energy decomposition is based on 4–20 ns MD trajectories of mAb-RBD systems.
The energy contribution of E484 in 7CWO (P17) is −16.80 ± 2.35 kcal/mol (a strong attraction), while the energy contribution of K484 is 10.24 ± 3.08 kcal/mol (a strong repulsion). By structural analyses, we found that the negatively charged E484 could form a strong electrostatic interaction with mAb, while the positively charged K484 should be repulsive to the positively charged binding site of the mAb (Figure 4A and B). For the two mAb-RBD systems (6XE1 and 7CDJ) that share similar binding affinities between K484 mutant and WT, the difference in the residue contribution to overall binding free energy is also slight. By analyzing the crystal structure, e.g., 6XE1, we found that the position of E484 is 7.7 Å away from the mAb-RBD interface (Figure 4C and D); therefore, the E484K mutation should have little effect on the overall binding free energy. However, the binding site for E484 in 7K9Z is negatively charged (Figure 4E); therefore, the E484K mutation should enhance the binding of RBD to mAbs (Table S2). By decomposing the binding free energy of 7K9Z complexes, four residues in the K484 mutant RBD (Q474, T478, E465 and R466) have stronger binding strength with 7K9Z than WT (≤ − 1.00 kcal/mol for each of the residues, Figure 4F). Furthermore, by structural analyses, we found that the E484K mutation causes the four residues closer to the mAb, enhancing their interactions with the mAb.
Figure 4.
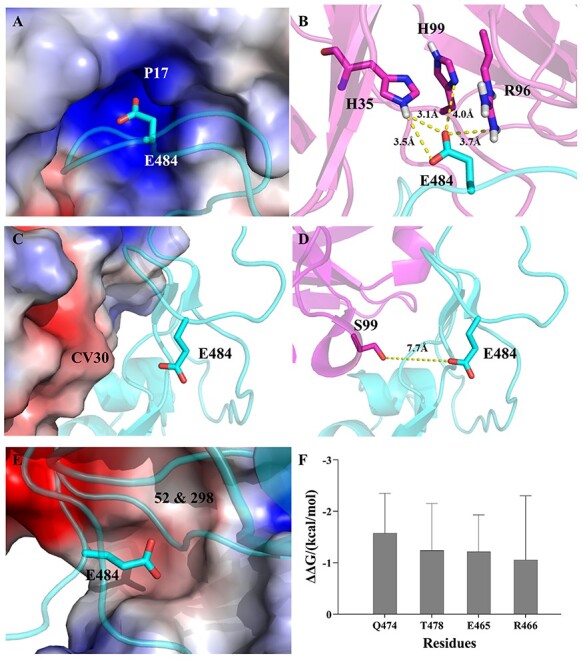
Three types of mAb-spike binding modes. (A) The mAb P17 (PDB ID: 7CWO) is showed by vacuum electrostatic potential, blue (positive charge), red (negative charge). The RBD is showed by cartoon (cyan). (B) Interactions of E484 (cyan) and mAb P17 (magenta). The distances are shown with yellow dotted line. (C) The mAb CV30 (PDB ID: 6XE1) is showed by vacuum electrostatic potential. The RBD is showed by cartoon (cyan). (D) The distance between E484 and CV30. (E) The mAbs 52 & 98 (PDB ID: 7K9Z) are showed by vacuum electrostatic potential. The RBD is showed by cartoon (cyan). (F) Key residues of RBD interacting with mAbs 52 & 98. The residues with binding free energy change ≤ −1.00 kcal/mol.
Other potential mutations with high risk of immune evasion
By binding free energy decomposition (Table S5), we systematically investigated the detailed roles of the key residues playing in the RBD-mAb interaction with ΔG ≤ −1.00 kcal/mol (Figure 5). Among the 25 complexes, E484 was found to be the key interaction residue of 17 systems, accounting for 68% of the mAbs. In addition to E484, we found 10 residues (Figure 5A) that are mostly involved in the mAb binding with occupancy >30%, which are Y489 (84%), Y449 (80%), F490 (80%), F486 (68%), Q493 (56%), V483 (52%), G485 (44%), S494 (40%), F456 (36%) and N487 (32%). Recently, Starr et al. found that a lysine mutation at residue 486 (F486K) of the RBD helps SARS-CoV-2 escape neutralization by REGN10933 with slightly decreased binding affinity to ACE2. However, the F486K was not accessible via a single-nucleotide change [38].
Figure 5.
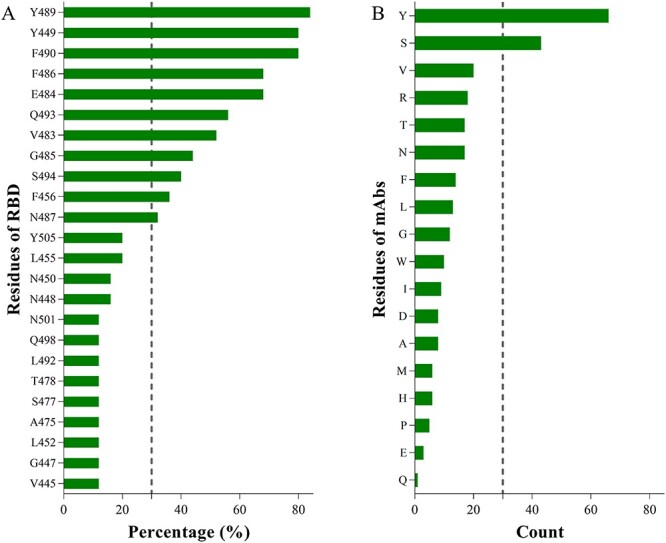
Residues on the mAb-RBD interface. (A) Occupancy (%) of spike residues found in 26 mAb-RBD interfaces. (B) Occurrence number of residues on mAbs counted in 26 mAb-RBD interfaces. Residues with energy contribution≤ − 1.00 kcal/mol are counted.
The major difference of the interaction between the RBD-ACE2 and RBD-mAb is the key binding residues (Table S6). In details, Y489, Y449, F486, Q493, F456 and N487 can form stable interactions with ACE2 [13, 39], their mutations may obviously affect the infection efficacy of the SARS-CoV-2, which may lead to less risk. Differently, the rest of four residues, viz., F490, V483, G485 and S494, without strong binding to ACE2, could form stable interaction with most mAbs. Therefore, the mutations in F490, V483, G485 and S494 may impair the binding of the new RBD mutants to mAbs, causing immune evasion. Although there is no evidence showing the immune evasion caused by F490 mutation [38], the high occupancy (80%) of F490 among 26 mAbs indicates its dangerous potential to escape vaccine-induced immunity. Therefore, close attention should be paid to the four residues in developing new mAbs against the RBM of spike protein. In addition, it is not surprising that the residues of mAb (Figure 5B) that are high frequently observed on the RBD-mAb interface could match well the residues of RBD, to form strong binding interaction. For instance, tyrosine and serine on mAb appear most frequently on the interface of RBD-mAb, which can form hydrogen bonds with residues on RBD (Y489, Y449, E484, Q493, etc.), and the four residues on RBD (Y489, Y449, F490 and F486) with the highest occupancy can form π-π stacking with tyrosine on mAb.
Conclusion
The rapid development and use of vaccines and mAbs have made people see the dawn of success in the fight against the COVID-19 pandemic. However, the immune evasion caused by the mutation of SARS-CoV-2 spike protein, such as the B.1.351 harboring E484K, may reinfect the survivors, weaken or even invalidate the efficacy of mAbs and vaccines. To understand the underlined mechanism, we calculated and analyzed the binding affinities of 26 mAbs to WT spike protein and to the E484K mutant, respectively. The results reveal that the E484K mutation impairs most of mAbs (~85%) to bind to the spike RBD. Further calculations on the glycosylated and trimer spike proteins show the same change tendency as that based on the mAb-RBD. In addition, we predicted that the mutation of the four residues in the RBD, viz., F490, V483, G485 and S494, are also likely to cause immune evasion. Overall, this work sheds light on the impact of the E484K mutation on mAb efficacy and predicts other potential residue mutations with risk of immune evasion, which may serve as an alarm for future mAb and vaccine development.
Key Points
The recently identified SARS-CoV-2 variants harboring E484K mutation was found to have great resistance to numerous mAbs.
We calculated the binding free energy of current 26 mAbs to WT spike protein and its E484K mutant, respectively. Twenty-two mAbs (85% of all the studied mAbs) have significantly decreased binding affinity to E484K mutant, indicating that E484K mutant is likely resistant to most mAbs.
In most of mAb-RBD complexes (72%), E484 has obvious greater contribution to the binding strength of the RBD to the mAb (>1.00 kcal/mol) than K484 does.
The mutations of F490, V483, G485 and S494 are predicted to have immune evasion risk, as these residues are involved in the binding of most mAbs.
Supplementary Material
Leyun Wu is a postgraduate at Shanghai Institute of Materia Medica. Her research interests are computer-aided drug design and molecular dynamics. Her affiliation is Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Cheng Peng is a Ph.D. student at Shanghai Institute of Materia Medica. His research interest is molecular dynamics. His affiliation is Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Yanqing Yang is a postgraduate at Shanghai Institute of Materia Medica. His research interests are molecular docking, virtual screening and molecular dynamics. His affiliation is Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Yulong Shi is a Ph.D. student at Shanghai Institute of Materia Medica. His research interest is molecular docking method development. His affiliation is Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Liping Zhou is a Ph.D. student at Shanghai Institute of Materia Medica. Her research interests are molecular dynamics simulations. Her affiliation is Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No.19A Yuquan Road, Beijing, 100049, China.
Zhijian Xu got his Ph.D. degree and is an associate professor at Shanghai Institute of Materia Medica in 2012. His research interests include drug-target non-covalent interaction, virtual screening and target prediction.
Weiliang Zhu is a professor who received his Ph.D. degree from Shanghai Institute of Materia Medica in 1998. His main research fields are computer-aided drug design, computational biology, computational chemistry and pharmaceutical chemistry, with a special focus on the theoretical research and method development of drug design.
Contributor Information
Leyun Wu, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China.
Cheng Peng, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China.
Yanqing Yang, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China.
Yulong Shi, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China.
Liping Zhou, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China.
Zhijian Xu, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China.
Weiliang Zhu, Drug Discovery and Design Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, 201203, China; School of Pharmacy, University of Chinese Academy of Sciences, No. 19A Yuquan Road, Beijing, 100049, China.
Data Availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Funding
The National Key Research and Development Program of China (2016YFA0502301 & 2017YFB0202601) and Natural Science Foundation of Shanghai (21ZR1475600). The simulations were partially run at the TianHe 1 supercomputer in Tianjin and TianHe 2 supercomputer in Guangzhou.
References
- 1. WHO . Coronavirus disease (COVID-2019) situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports/ (28 May 2021, date last accessed). 2021.
- 2. WHO . Draft landscape and tracker of COVID-19 candidate vaccines. https://www.who.int/publications/m/item/draft-landscape-of-COVID-19-candidate-vaccines 2020.
- 3. Forni G, Mantovani A. COVID-19 vaccines: where we stand and challenges ahead. Cell Death Differ 2021;28:626–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sempowski GD, Saunders KO, Acharya P, et al. Pandemic preparedness: developing vaccines and therapeutic antibodies for COVID-19. Cell 2020;181:1458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mahase E. Covid-19: Novavax vaccine efficacy is 86% against UK variant and 60% against south African variant. Br Med J 2021;372:n296. [DOI] [PubMed] [Google Scholar]
- 6. Shinde V, Bhikha S, Hoosain Z, et al. Efficacy of NVX-CoV2373 Covid-19 Vaccine against the B.1.351 Variant. N Engl J Med 2021;384:1899–1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andreano E, Piccini G, Licastro D, et al. SARS-CoV-2 escape from a highly neutralizing COVID-19 convalescent plasma. Proc Natl Acad Sci U S A 2021;118(36):e2103154118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ferrareze PAG, Franceschi VB, Mayer AM, et al. E484K as an innovative phylogenetic event for viral evolution: Genomic analysis of the E484K spike mutation in SARS-CoV-2 lineages from Brazil. Infect Genet Evol 2021;93:104941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang P, Nair MS, Liu L, et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021;593(7857):130–135. [DOI] [PubMed] [Google Scholar]
- 10. Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581:215–20. [DOI] [PubMed] [Google Scholar]
- 11. Adhikari P, Li N, Shin M, et al. Intra- and intermolecular atomic-scale interactions in the receptor binding domain of SARS-CoV-2 spike protein: implication for ACE2 receptor binding. Phys Chem Chem Phys 2020;22:18272–83. [DOI] [PubMed] [Google Scholar]
- 12. Amin M, Sorour MK, Kasry A. Comparing the binding interactions in the receptor binding domains of SARS-CoV-2 and SARS-CoV. J Phys Chem Lett 2020;11:4897–900. [DOI] [PubMed] [Google Scholar]
- 13. Peng C, Zhu Z, Shi Y, et al. Computational insights into the conformational accessibility and binding strength of SARS-CoV-2 spike protein to human angiotensin-converting enzyme 2. J Phys Chem Lett 2020;11:10482–8. [DOI] [PubMed] [Google Scholar]
- 14. Wang Y, Liu M, Gao J. Enhanced receptor binding of SARS-CoV-2 through networks of hydrogen-bonding and hydrophobic interactions. Proc Natl Acad Sci U S A 2020a;117:13967–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Starr TN, Greaney AJ, Hilton SK, et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020;182:1295–310 e1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biasini M, Bienert S, Waterhouse A, et al. SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 2014;42:W252–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Waterhouse A, Bertoni M, Bienert S, et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 2018;46:W296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Duan Y, Wu C, Chowdhury S, et al. A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J Comput Chem 2003;24:1999–2012. [DOI] [PubMed] [Google Scholar]
- 19. Kirschner KN, Yongye AB, Tschampel SM, et al. GLYCAM06: a generalizable biomolecular force field. Carbohydrates, J Computational Chemistry 2008;29:622–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Case DA, Ben-Shalom IY, Brozell SR, et al. AMBER 2018. San Francisco: University of California, 2018. [Google Scholar]
- 21. Ryckaert JPC, G.; Berendsen, H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 1997;23:327–41. [Google Scholar]
- 22. Essmann U, Perera L, Berkowitz ML, et al. A smooth particle mesh Ewald method. J Chem Phys 1995;103:8577–93. [Google Scholar]
- 23. Hansen J, Baum A, Pascal KE, et al. Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail. Science 2020;369(6506):1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kollman PA, Massova I, Reyes C, et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc Chem Res 2000;33:889–97. [DOI] [PubMed] [Google Scholar]
- 25. Chi X, Yan R, Zhang J, et al. A neutralizing human antibody binds to the N-terminal domain of the spike protein of SARS-CoV-2. Science 2020;369:650–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang N, Sun Y, Feng R, et al. Structure-based development of human antibody cocktails against SARS-CoV-2. Cell Res 2021b;31:101–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cerutti G, Guo Y, Zhou T, et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe 2021;29(5):819–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Acharya P, Williams W, Henderson R, et al. A glycan cluster on the SARS-CoV-2 spike ectodomain is recognized by Fab-dimerized glycan-reactive antibodies. bioRxiv 2020:doi: 10.1101/2020.1106.1130.178897. preprint: not peer reviewed [DOI] [Google Scholar]
- 29. Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020;367:1260–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tortorici MA, Beltramello M, Lempp FA, et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 2020;370:950–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peng C, Wang J, Yu Y, et al. Improving the accuracy of predicting protein–ligand binding-free energy with semiempirical quantum chemistry charge. Future Med Chem 2019;11:303–21. [DOI] [PubMed] [Google Scholar]
- 32. Hou T, Wang J, Li Y, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J Chem Inf Model 2011;51:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Xu L, Sun H, Li Y, et al. Assessing the performance of MM/PBSA and MM/GBSA methods. 3. The impact of force fields and ligand charge models. J Phys Chem B 2013;117:8408–21. [DOI] [PubMed] [Google Scholar]
- 34. Chen RE, Zhang X, Case JB, et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med 2021;27:717–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hurlburt NK, Seydoux E, Wan YH, et al. Structural basis for potent neutralization of SARS-CoV-2 and role of antibody affinity maturation. Nat Commun 2020;11(1):5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ge J, Wang R, Ju B, et al. Antibody neutralization of SARS-CoV-2 through ACE2 receptor mimicry. Nat Commun 2021;12(1):250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Casalino L, Gaieb Z, Goldsmith JA, et al. Beyond shielding: the roles of Glycans in the SARS-CoV-2 spike protein. ACS Central Science 2020;6:1722–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Starr TN, Greaney AJ, Addetia A, et al. Prospective mapping of viral mutations that escape antibodies used to treat COVID-19. Science 2021;371:850–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shang E, Axelsen PH. The potential for SARS-CoV-2 to evade both natural and vaccine-induced immunity. bioRxiv 2020. 13 December 2020, doi 10.1101/2020.12.13.422567, Arxiv biorxiv;2020.12.13.422567v1, preprint: not peer reviewed [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.