

Abstract
The COVID-19 pandemic has highlighted the need to come out with quick interventional solutions that can now be obtained through the application of different bioinformatics software to actively improve the success rate. Technological advances in fields such as computer modeling and simulation are enriching the discovery, development, assessment and monitoring for better prevention, diagnosis, treatment and scientific evidence generation of specific therapeutic strategies. The combined use of both molecular prediction tools and computer simulation in the development or regulatory evaluation of a medical intervention, are making the difference to better predict the efficacy and safety of new vaccines. An integrated bioinformatics pipeline that merges the prediction power of different software that act at different scales for evaluating the elicited response of human immune system against every pathogen is proposed. As a working example, we applied this problem solving protocol to predict the cross-reactivity of pre-existing vaccination interventions against SARS-CoV-2.
Keywords: SARS-CoV-2, in silico trial, multi-step algorithm, multi-scale approach, vaccine
Introduction
It was just over one year ago that the COVID-19 pandemic officially began. Since then, SARS-CoV-2 has claimed more than 2.6 million lives and upended millions more [1]. COVID-19 symptoms involve generally the respiratory tract, ranging from mild/moderate to severe manifestations. SARS-CoV-2 infection can also affect other organs such as the gastrointestinal compartment, liver and pancreas, cardiovascular system and in some cases can promote renal dysfunction and neurological disorders [2–4].
COVID-19 vaccines, developed with unprecedented speed, are now rolling out worldwide to stop the outbreak. However, the onset of a wide variety of SARS-CoV-2 variants urge to investigate if COVID-19 vaccines will also provide protection against these variants [5, 6]. The EMA/FDA has currently authorized four vaccines for emergency but researchers are currently testing about 90 vaccines in clinical trials on humans, and 27 have reached the final stages of testing [7].
Along with these scenarios, scientists all over the world are also exploring alternative strategies for the protection and treatment of COVID-19. One of the most interesting hypotheses for a potential COVID-19 vaccine repositioning is the under investigated cross-reactive immunity acquired from pediatric vaccinations or other already existing vaccine formulations [8]. This strategy is supported by the fact that both antibody and T cell responses has been detected in unexposed subjects, although they have been linked to previous exposure to circulating common cold coronaviruses [9–11].
Technological advances in fields such as computer modeling and simulation are enriching the discovery, development, assessment and monitoring for better prevention, diagnosis, treatment and scientific evidence generation of specific therapeutic strategies. In this perspective, in silico platforms, are making the difference to better predict the efficacy and safety of new pharmacological tools. The COVID-19 pandemic has, and continues to have, a very serious impact on the challenging development of interventional solutions for SARS-CoV-2 and never as now, the application of in silico technology can actively improve success rates of these trials [12–14].
Here we describe a multi-step and multi-scale bioinformatic problem solving protocol to discover and test potential components that could be implemented in potential effective vaccines against SARS-CoV-2 and potentially to every disease. In particular, as a working example, we investigated potential sources of cross-reactive immunity to SARS-CoV-2 to identify cross-reactive epitopes between SARS-CoV-2 and antigens included in Bacillus Calmette–Guérin (BCG) vaccine and within other common antigenic subunits commonly used for vaccine formulations against tuberculosis (TB) [15]. We then compared these identified cross-reactive epitopes with the ones previously detected between antigens in tetanus, diphtheria and pertussis (DTP) vaccines and SARS-CoV-2 [8, 16] to obtain a scale of cross-reactivity through such different antigens.
Potential immune response able to elicit a degree of protection against SARS-CoV-2 infection should then be tested at a cellular and organ level. This is the reason why we selected, as a final step in the problem-solving protocol we are describing, an immune system simulator to predict the induced immune response and its degree of efficacy. The Universal Immune System Simulator for SARS-CoV-2 (UISS-SARS-CoV-2 for short) is an in silico trial platform based on agent-based methodology, which is able to simulate the intricate human immune system dynamics in the response to SARS-CoV-2 insult [17].
The obtained framework, used as a combination of protocols for predicting the potential cross immunity induced by different existing vaccines, is applicable as a comprehensive computational pipeline to envisage the efficacy of new developed vaccines.
Methods and materials
Workflow of the multi-step and multi-scale bioinformatic approach
The problem-solving protocol consists of four distinct phases. In the first processing phase, a search for similarity between the sequences generated by the genome of SARS-COV-2 and the antigens of our interest was performed, through a series of queries on BLASTP database (https://blast.ncbi.nlm.nih.gov/Blast.cgi). BLASTP is one of the most popular BLAST variations and it is used for aligning protein query sequences against protein DB sequences. BLAST queries were performed with default parameters and only hit sequences consisting of eight or more residues with an identity ≥70% with SARS-CoV-2 were selected as potential cross-reactivity sources. BLASTP queries were executed through a local Python script which takes SARS-CoV-2 sequences as ‘query’ input and the antigens under examination as ‘subject’ input. The output of this script is a table containing all the information about hit sequences found, i.e. the matching sequences, the sequence interval where they are located and the identity percentage value. The amino-acid coding sequences (CDS) encoded by SARS-CoV-2 reference genome have been fragmented into overlapping 15 mer peptides with 10 residues overlaps by using an ad hoc PERL script before being submitted to BLAST searches in order to enhance the epitopes mapping resolution.
In the second phase, we tested T cell reactivity of hit sequences identified through BLASTP queries, by predicting their binding to class I and II human leukocyte antigen (HLA I and HLA II) molecules. Peptide binding was predicted to the following HLA I molecules: HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*11:01, HLA-A*23:01, HLA-A*24:02, HLA-B*07:02, HLA-B*08:01, HLA-B*35:01, HLA-B*40:01, HLA-B*44:02, HLA-B*44:03 using IEDB MHC I binding tool (http://tools.iedb.org/mhci/) with default recommended method through the RESTful interface. To assert that the binding had happened a percentile rank of 2% cut-off was used. Regarding HLA II, we tested the following molecules: HLA-DRB1*01:01, HLA-DRB1*03:01, HLA-DRB1*04:01, HLA-DRB1*04:05, HLA-DRB1*07:01, HLA-DRB1*08:02, HLA-DRB1*09:01, HLA-DRB1*11:01, HLA-DRB1*12:01, HLA-DRB1*13:02, HLA-DRB1*15:01 HLA-DRB3*01:01 HLA-DRB3*02:02 HLA-DRB4*01:01, HLA-DRB5*01:01 using the IEDB MHC II binding tool (http://tools.iedb.org/mhcii/) with default recommended method through the RESTful interface. To assert that the binding had happened a percentile rank of 10% cut-off was used.
In the third phase, we tested B cell reactivity by using BediPred software at the IEDB Analysis Resource (http://http://tools.iedb.org/bcell/). BediPred calculates an antigenicity value for each residue (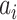 ) and then, a global value of antigenicity for the entire peptide (l is the total number of residues of the peptide) is calculated using the following formula:
) and then, a global value of antigenicity for the entire peptide (l is the total number of residues of the peptide) is calculated using the following formula:
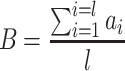 |
Peptides showing B values ≥0.4 were considered antigenic.
The fourth phase consists in simulating the induced immune response at cellular and organ level using UISS-SARS-CoV-2. It uses bit-string model (0 and 1 s) to represent specific elements or binding properties in the same way that Farmer, Packard and Perelson [18] did. In particular, a string of NBIT bits is used to define the immune system repertoire. Using binary strings of length NBIT mimics a 2NBIT repertoire. Considering that the immune system repertoire diversity could be quantified in about 1015 for B cells [19] and 1020 for T cells [20], binary strings of about NBIT = 60 should be used to represent the diversity of the immune system repertoire at natural scale. Each different bit-string defines an element of the repertoire. An m-bit match is obtained when exactly m bits complement each other and the others NBIT—m are equal.
The function match(a, b) = hamming(a, b) is defined to give us the number of matching bits between two strings a and b and is computed as the Hamming distance in the space of the bit- strings. Another function, affinity(m), is defined by a vector of length bits, called bit match vector, with each component of the vector giving the affinity of an m-bit match. To specify the vector affinity, we use the additional parameters m0, A0, ∂A to calculate the vector in the following way: (i) first, set affinity(m) = 0 for m < m0; this provides a threshold level below which binding cannot occur; (ii) set affinity(m0) to the parameter A0; (iii) set the increase of strength on increasing a match by one bit to be the inverse of the ratio of number of clones with match m + 1 and m multiplied by ∂A. In formulas:
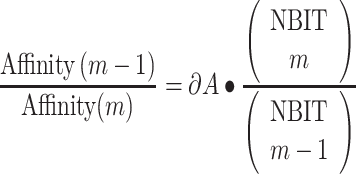 |
As we are now calculating the affinity scores using BediPred for B cells reactivity and IEDB tools coupled with python scripts for T cells epitope prediction, we imported such scores directly in the Affinity vector for the epitopes and peptides match the residues we would like to test. Figure 1 summarizes the multi-step and multi-scale bioinformatic approach used.
Figure 1.
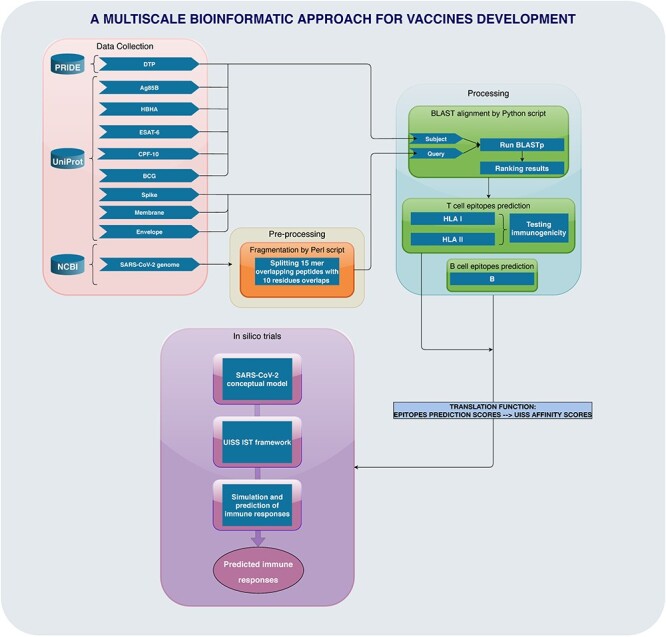
Workflow of the multi-scale bioinformatic approach.
SARS-CoV-2 genome
SARS-CoV-2 reference sequence deals with the RNA genome isolated from one of the first cases in Wuhan, China, and is known as ‘severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome’. This sequence is broadly used as a standard reference and, because of its early identification, is used as the origin genome in phylogenetic trees produced by Nextstrain, COVID-19 Genomics UK (COG-UK) and the China National Center for Bioinformation project.
SARS-CoV-2 reference genome was retrieved by National Center for Biotechnology Information (NCBI), through NCBI Reference Sequence: NC_045512 within GenBank. Its Spike (S), Membrane (M) and Envelope (E) proteins were retrieved from UniProt database annotation records (P0DTC2, P0DTC5 and P0DTC4, respectively).
Existing vaccinations tested
BCG is the only vaccine against TB available today, even if its efficacy still remains controversial [21]. It shows good results against the severe forms of TB and its use prevents a large number of deaths every year especially in children, but it fails to confer protection against adult TB. The choice of the BCG strain to be used for vaccination remains crucial for the efficacy of the vaccination itself. By the end of 1940, several studies provided evidence for the utility of BCG in protection against tuberculosis, so that the majority of the world, including east European countries, introduced routine BCG vaccination according to various schedules (e.g. at birth, school entry, school leaving).
The amino-acid sequence encoded by the genome of BCG vaccine has been downloaded from UniProt database (https://www.uniprot.org/proteomes/) after its GenBank accession (NC_008769).
By the end of 1990, whole-cell pertussis vaccines combined with diphtheria and tetanus toxoids (diphtheria-tetanus toxoids-pertussis (DTP)) were used to vaccinate children against pertussis, diphtheria, and tetanus with a 5-dose series administered at 2 months, 4 months, 6 months, 12–18 months and 4–6 years of age. At the beginning of 1990s, two acellular pertussis vaccines (containing purified components of B pertussis) combined with diphtheria and tetanus toxoids (diphtheria-tetanus toxoids-acellular pertussis (DTaP)) were licensed for use as the fourth and fifth doses of the vaccination series among children who had received three doses of whole-cell DTP [22].
The sequences of the antigens of D (diphtheria), T (tetanus) and P (pertussis) vaccines have been downloaded from the Proteomics Identification Database (PRIDE, https://www.ebi.ac.uk/pride/) upon the proteomics projects PXD009289 and PXD013804.
Antigens tested
Ag85B, a fibronectin-binding protein with mycolyltransferase activity, is the main secretory protein in actively replicating M. tuberculosis (MTB). Because of its high immunogenicity, as it can easily detect specific humoral and cell-mediated immune responses both in latently and actively infected TB patients, Ag85B has been investigated as a potential candidate for subunit TB vaccines [23]. Ag85B sequence has been downloaded from UniProt (Q847N4 (Q847N4_MYCTX)).
The heparin-binding hemagglutinin adhesin (HBHA) is an important surface-displayed protein that serves as an adhesin for non-phagocytic cells and is involved in extra-pulmonary dissemination of the tubercle bacillus [24]. HBHA is present at the outermost layer of the bacterial cell, mediates the attachment of the bacilli to non-phagocytic cells, induces mycobacterial aggregation and is involved in extrapulmonary dissemination of MTB. For these reasons, is also considered as an important marker of latency, inducing a strong interferon gamma response in latently infected subjects. HBHA sequence has been downloaded from UniProt (P9WIP9 (HBHA_MYCTU)).
The early secretory antigenic target (ESAT-6), which is secreted along with its chaperone culture filtrate protein (CFP-10), is one of the most important virulent factors for MTB [25]. ESAT-6 and CFP-10 have been implicated in several virulence mechanisms of mycobacteria, even if the exact mechanism of virulence of ESAT-6 is not totally clear yet. They are capable of modulating both innate and adaptive immune responses and inactivation of ESAT-6 results in dramatical reduction of the MTB virulence. ESAT-6 and CFP-10 sequences have been downloaded from UniProt, respectively, B5TV89 (B5TV89_MYCTU) and B5TV88 (B5TV88_MYCTU).
MTB32A and MTB39A are two antigens expressed in M. tuberculosis (MTB) and in BCG and comprised in the formulation of candidate MTB vaccine Mtb72F/AS02A, which has been developed to boost specific, pre-existing immunity induced by BCG and MTB. [26] MTB32A and MTB39A have been selected by T cell antigen screening because of their ability to restimulate, in vitro, peripheral blood mononuclear cells (PBMCs) from healthy PPD (Purified Protein Derivative) -positive individuals, to induce Th1 responses in mice and to induce protection in animal models of TB. MTB32A and MTB39A sequences have been downloaded from UniProt database (O07175_MYCTU and A0A7U4J0T5_MYCTU respectively).
Results and discussion
Several studies suggest that vaccines commonly used during infancy may be associated with effects on child morbidity and mortality that are unrelated to protection against the diseases they were designed for, including BCG and DTP vaccine [27]. For example, it has been found that BCG promotes a Th1 response in new-borns (Th1-type cytokines, such as interferon gamma, tend to produce the proinflammatory reaction responsible for killing intracellular parasites and for perpetuating autoimmune responses) and is associated with less atopy, better response to other vaccines and lower mortality, particularly for children with a positive tuberculin response [28].
Here, we propose a vaccine discovery in silico trial pipeline that using different sets of programs (e.g. databases, software and tools) aims to predict the elicited immune response against a particular pathogen. As a working example, we report the application of the computational framework to predict the potential cross-reactive immunity induced by existing vaccinations against SARS-CoV-2. For this purpose, we searched for peptide matches to SARS-CoV-2 in the proteomes of BCG and DTP vaccine and of some of the most frequently used antigenic subunits in vaccines, and subsequently tested their T and B cell reactivity in order to identify potential cross-reactive epitopes.
BLASTP similarity search phase results
Since a structural resemblance often corresponds to a functional resemblance, this first phase focuses on the assessment of the level of similarity between the sequences resulting from the fragmentation of the SARS-CoV-2 CDS and the BCG, DTP and all other antigens covered by this study. We obtained a total of 759 peptides that mapped to BCG sequence and 748 to DTP with a similarity level equal or greater than 70%. The results obtained for the other antigens were considerably less significant: 2 sequences for Ag85B, 2 for CFP-10, 1 for ESAT-6, 3 for HBHA, 1 for MTB32A and 3 for MTB39A. The complete data can be found in Supplementary Material available online at http://bib.oxfordjournals.org/.
All the subject hits with eight or more residues and ≥70% identity to SARS-CoV-2 have been selected as potential cross-reactivity sources and then tested for the prediction of T cell and B cell reactivity in next phases of the protocol.
T cell epitopes prediction results
For assessing that a binding between peptides and HLA I/II alleles has occurred we consider all values below 2% percentile rank for MHC I and below 10% for MHC II molecules. We selected as potential sources of cross-reactivity only those query peptides that bind to the same alleles to which the corresponding subject peptides bind. Furthermore, the selection ends successfully if and only if the percentage rank is lower than the allowed thresholds. With the sequences that mapped to BCG, we obtained 127 query hits and 127 subject hits satisfying the acceptance criteria for MHC I, 37 query hits and 27 for MHC II. For DTP, we obtained 225 subject hits and 188 query hits for MHC I, 54 subject hits and 53 query hits for MHC II.
The T cell response prediction for the other antigens under examination did not produce any significant results. The complete data can be found in Supplementary Material available online at http://bib.oxfordjournals.org/. Figure 2 shows the correlation between percentile rank and score, both for BCG and DTP, in MHC-I and MHC-II on four different clustered heatmaps. Each row on the heatmap represents one of the alleles we tested for cross-reactivity prediction, and the columns represent, respectively, the scores and the corresponding percentile ranks obtained as results from the prediction analysis. The color range on the top left side of each panel represents the legend for how each color maps specific numeric value. These heatmaps, built on non-normalized data, show the inverse correlation existing between percentile ranks and corresponding scores: the lower the rank, the higher the score. This kind of observation is not so intuitive in the heatmaps related to MHC-II as the fragmentary and less homogeneous results we obtained. There are ‘jumps’ (white spaces) that make it almost impossible to interpret the existing relationship between the variables under examination, even if Tables 1 and 2 highlight these correlations for some of them.
Figure 2.
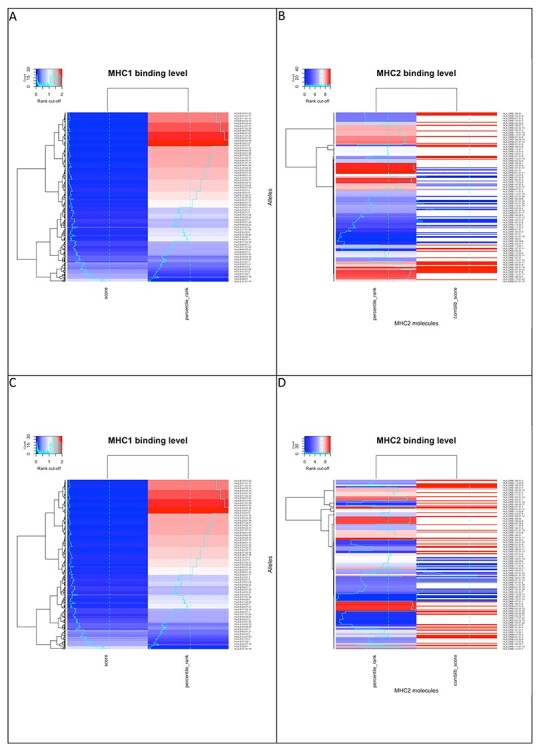
Inverse correlation heatmap for BCG (panel A, MHC-I and panel B, MHC-II) and DTP (panel C, MHC-I and panel D, MHC-II).
Table 1.
BCG B cell or antibody reactivity (presence of B cell epitopes) of peptides
| ALLELE;QUERY_SEQUENCE (SARS-CoV-2);QUERY_SCORE_MHC1;PERCENTILE_RANK;SUBJECT_SEQUENCE (BCG);SUBJECT_SCORE_MHC1;PERCENTILE_RANK | ||||||||||
| HLA-B*35:01;SPDAVTAY;0.8;0.08;SPDVLTTY;814;0.07 | ||||||||||
| HLA-B*08:01;NITRFQTL;0.59;0.1;NISRFRTL;508;0.15 | ||||||||||
| HLA-B*07:02;PGLPGTIL;0.0971;1.2;APGHPGSI;378;0.36 | ||||||||||
| Allele;QUERY_SEQUENCE (SARS-CoV-2);PERCENTILE_RANK;QUERY_SCORE_MHC2;SUBJECT_SEQUENCE (BCG);PERCENTILE_RANK;SUBJECT_SCORE_MHC2 | ||||||||||
| HLA-DRB1*01:01;KHVYQLRARSV;0.59;0.42;NP;NP;NP | ||||||||||
| HLA-DRB1*01:01;DLFMRIFTIGT;2.50;0.71;NP;NP;NP | ||||||||||
| HLA-DRB1*01:01;HTVLQAVGACV;3.50;0.41;NP;NP;NP | ||||||||||
| HLA-DRB1*01:01;FACVVADAVIK;7.70;4.09;APLLSAGATAA;0.71;0.01 | ||||||||||
| HLA-DRB1*07:01;GKSHFAIGLAL;4.80;2.08;GKTHLAVGLAI;4.70;5.99 | ||||||||||
| HLA-DRB1*01:01;NP;NP;NP;DPFMMIFTSGT;0.72;18.00 | ||||||||||
| QUERY_SEQUENCE (SARS-CoV-2);B SCORE_QUERY;SUBJECT_SEQUENCE (BCG);B SCORE_SUBJECT | ||||||||||
| DLGDELGTDP;1.1655;DAGETLPTMP;1.1065 | ||||||||||
| GPSDSTGSNQ;2.2545;GPSDATGIPQ;1.7147999999999999 | ||||||||||
| DTANPKTPKY;1.9848;DTADPKGAKY;1.8296 | ||||||||||
Table 2.
DTP B cell or antibody reactivity (presence of B cell epitopes) of peptides
| ALLELE;QUERY_SEQUENCE (DTP);QUERY_MHC1_score;percentile_rank;SUBJECT_SEQUENCE (DTP);SUBJECT_MHC1_score;percentile_rank;;; | ||||||||||
| HLA-A*01:01;ITQMNLKY;294;0.38;ITDLSLKY;0.96;0.01;;; | ||||||||||
| HLA-B*40:01;LEDEFTPF;0.0822;1.1;LEFDFTSF;0.19;0.66;;; | ||||||||||
| HLA-A*01:01;AVFDKNLY;0.0436;1.7;AVFDPELY;0.0939;0.98;;; | ||||||||||
| ALLELE;QUERY_SEQUENCE (DTP);QUERY_MHC2_score;percentile_rank;SUBJECT_SEQUENCE (DTP);SUBJECT_MHC2_score;percentile_rank | ||||||||||
| HLA-DRB1*09:01;AQFAPSASAFF;0.01;0.01;AVFTPSALAFF;0.01;0.27 | ||||||||||
| HLA-DRB3*01:01;TGLFKDCSKVI;32.93;0.77;TKLFKNTSKVI;4548.53;7.30 | ||||||||||
| HLA-DRB1*01:01;KLKTLVATAEA;0.01;5.70;KLKTLEASAQA;0.26;5.90 | ||||||||||
| QUERY_SEQUENCE;B_SCORE;SUBJECT_SEQUENCE;B_SCORE | ||||||||||
| SAKPPPGD;2.9286250000000003;TAAPPPGD;2.8165 | ||||||||||
| GPPGTGKS;2.648625;GPPGSGKT;2.6715 | ||||||||||
| LQGPPGTGKS;2.2448;LVGPPGSGK;1.9208888888888889 | ||||||||||
B cell reactivity prediction
B cells are the core of the adaptive humoral immune system and are primarily involved in the production of antigen-specific immunoglobulins against invasive pathogens. Using BediPred software, we predicted the presence of B cell epitopes by calculating the antigenicity value for each residue in the peptides under examination. Then, we obtained the global antigenicity value (B) for the entire peptide, representing its ability to be specifically recognized by the antibodies generated due to immune response [29].
Only peptides showing B ≥0.4 have been considered to be antigenic. We obtained a total of 288 antigenic peptides from BCG query sequences and 356 from BCG subject sequences. For DTP, we obtained 354 antigenic peptides from its query sequences and 314 from DTP subject sequences. The other antigens under study did not produce any relevant results. The complete results dataset can be accessible through the Supplementary Material section available online at http://bib.oxfordjournals.org/.
Tables 1 and 2 depict the best peptides found for both BCG and DTP that we used as input for the prediction of antibody reactivity at cellular level.
Prediction of the induced immune system response at cellular and organ level
As final step, we run the UISS-SARS-CoV-2 simulations at cellular and organ level importing all the predictions made at molecular scale in the way we described above. We generated 1000 digital twins cohort using a sequential approach. This allows the sampling from the joint features population distribution to create a cohort of virtual patients with Gaussian distributions of immune system profiles, resembling the recruitment process for the target clinical trial [30]. Then we challenged each digital twin with a 1.5 × 102 plaque forming units (PFU). This is a good estimate (we used the in vivo study of [31] to have an estimate from ferrets models and, moreover, we used the same PFU dosage previous modeling experiences [17]) that eventually leads to the COVID-19 disease development. To guarantee a sufficient statistical diversity in terms of immunological repertoire, we randomly generated 1000 digital twins cohort. We then extracted 300 digital twins subdividing them in three different in silico cohorts, i.e. light, mild/moderate and severe disease outcome scenario. We measured the cytopathic effects (CPE) on the lung compartment to establish the severity of the disease. Light and mild/moderate scenarios are available as Supplementary Material available online at http://bib.oxfordjournals.org/. Figure 3 shows the simulation results (mean values over a 100 digital twins) for the severe scenario. Panel A depicts the dynamics of the cytopathic effects induced by SARS-CoV-2 on the lung epithelial cells (LPE). In the untreated case, an important damage is predicted (about 53% of LPE eventually die). For BCG and DTP treated virtual patients one can see how the CPE is limited (both cases) and the recovery is faster for DTP than BCG treated individuals. Panel B shows the predicted humoral immune response in terms of immunoglobulins class G anti SARS-CoV-2. Comparing to the untreated case, both BCG and DTP show a non-negligible increase in terms of geometric mean titers (GMT). Panel C shows that IL-6 levels are considerably reduced for both BCG and DTP treated digital twins, along with the levels of CD8+ T cells. Hence the predictions clearly demonstrate that a considerable protective effect from cytokines storm induced inflammation (and the consequent cellular damage) is elicited by both BCG and DTP vaccinations.
Figure 3.
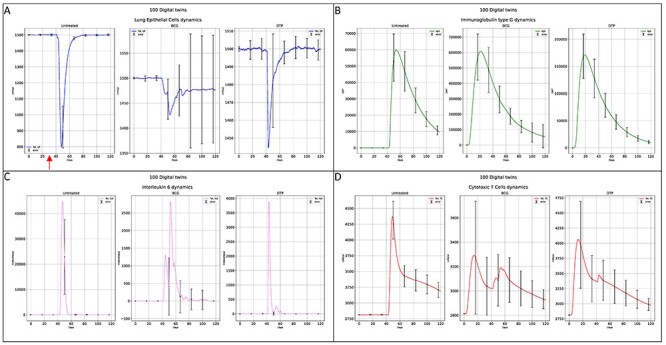
Immune response elicited by BCG and DTP vaccine interventions compared to the untreated case. Panel A shows the cytopathic effects on lung epithelial cells to evaluate the severity of the SARS-CoV-2 infection. Panel B depicts the humoral response measured through the immunoglobulins class G (geometric mean titers, GMT). Panel C reports IL-6 as a predictor of the severity of inflammation and the potentiality to develop a cytokines storm that can eventually lead to severe damage and fatal prognosis. Panel D highlights cellular response through the dynamics of specific CD8+ T cells.
Conclusions
The opportunity to echo and resonate the importance of the combination of bioinformatics software solutions in healthcare, especially in this pandemic situation, sheds the light on the fact that they can support the ‘3 Rs’ principles (replace, reduce, refine) in drug development and better predict the efficacy of new medicines such as vaccines.
Here, we presented a problem solving protocol that makes use of different software acting at different scales (molecular to organ) that can be potentially applied to predict the elicited human immune response to pathogens. As a specific application, we used this computational pipeline to evaluate the cross-reactivity potentiality of pre-existing vaccine formulations that can lead to an immune response specifically targeted against SARS-CoV-2. One of the limitations of this approach is that it cannot provide, at this stage, advices and/or suggestions on the best vaccine formulation ready to be used at the production stage. There are, however, different solutions that can be embedded to the proposed pipeline to achieve this target.
Moreover, in the context of the ‘variants of concern’, the proposed in silico pipeline can be applied to predict the potential cross-reactive immunity induced by existing vaccinations against SARS-CoV-2 new emerging variants (e.g. B.1.1.7, B.1.351 and other lineages). Together with the possibility to use this problem solving protocol to estimate the degree of the induced immune response by completely new developed vaccines, it can indeed speed up the development of vaccines tailored to the emerging antigenic variants.
Key Points
A multi-step and multi-scale protocol based on a combination of different programs could be effectively used for accelerating and optimizing vaccine discovery, especially in emergency scenarios.
The protocol was applied, as a working example, to predict potential cross-reactivity immunity to SARS-CoV-2 and, potentially, its variants.
Accurate prediction of cross-reactivity immunity antigens provides important clues to consider alternative protective vaccination strategies against COVID-19.
Supplementary Material
Acknowledgments
Authors of this paper acknowledge support from the STriTuVaD project. The information and views set out in this article are those of the authors and do not necessarily reflect the official opinion of the European Commission. Neither the European Commission institutions and bodies nor any person acting on their behalf may be held responsible for the use which may be made of the information contained therein. Publication costs are covered by STriTuVaD project.
Funding
Horizon 2020 Framework Programme, H2020-EU-3.1.5, In Silico Trial for Vaccine Development, acronym ``STriTuVaD'' project, grant id number: 777123.
Giulia Russo is an assistant professor at the University of Catania. Her research activity is focused on computational modeling in biomedical sciences with a particular interest in modeling methodologies at the molecular level.
Valentina Di Salvatore is a post-doc researcher at the University of Catania. Her research activity is focused on the usage of specific bioinformatic tools in the field of computational biology.
Giuseppe Sgroi is a PhD candidate at the University of Catania. His research activity is focused on data science with a particular interest in modeling methodologies for systems biomedicine.
Giuseppe Alessandro Parasiliti Palumbo is a PhD candidate at the University of Catania. His research activity is focused on the development of biomedical applications and data privacy.
Pedro A. Reche is an associate professor in immunology at the Complutense University of Madrid. His research focuses on a wide range of topics ranging from deciphering the factors that determine immunogenicity and epitope-vaccine design to the study of immunomodulation by epithelial cells.
Francesco Pappalardo is an associate professor at the University of Catania. He was a visiting research scientist at the Dana-Farber Cancer Institute in Boston and at the Molecular Immunogenetics Labs, IMGT in Montpellier. His major research area is computational biomedicine.
Contributor Information
Giulia Russo, Department of Drug and Health Sciences, University of Catania, Catania, Italy.
Valentina Di Salvatore, Department of Drug and Health Sciences, University of Catania, Catania, Italy.
Giuseppe Sgroi, Department of Drug and Health Sciences, University of Catania, Catania, Italy.
Giuseppe Alessandro Parasiliti Palumbo, Department of Drug and Health Sciences, University of Catania, Catania, Italy.
Pedro A Reche, Department of Drug and Health Sciences, University of Catania, Catania, Italy.
Francesco Pappalardo, Department of Drug and Health Sciences, University of Catania, Catania, Italy.
References
- 1. Carvalho T, Krammer F, Iwasaki A. The first 12 months of COVID-19: a timeline of immunological insights. Nat Rev Immunol 2021;21:245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Tsatsakis A, Calina D, Falzone L, et al. SARS-CoV-2 pathophysiology and its clinical implications: an integrative overview of the pharmacotherapeutic management of COVID-19. Food Chem Toxicol 2020;146:111769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kordzadeh-Kermani E, Khalili H, Karimzadeh I. Pathogenesis, clinical manifestations and complications of coronavirus disease 2019 (COVID-19). Future Microbiol 2020;15:1287–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pennisi M, Lanza G, Falzone L, et al. SARS-CoV-2 and the nervous system: from clinical features to molecular mechanisms. Int J Mol Sci 2020;21:5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Davies NG, Abbott S, Barnard RC, et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021;372:eabg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Collier DA, De Marco A, Ferreira IATM, et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature 2021;593:136–41. [DOI] [PubMed] [Google Scholar]
- 7. Forni G, Mantovani A. COVID-19 Commission of Accademia Nazionale dei Lincei R. COVID-19 vaccines: where we stand and challenges ahead. Cell Death Differ 2021;28:626–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Reche PA. Potential cross-reactive immunity to SARS-CoV-2 from common human pathogens and vaccines. Front Immunol 2020;11:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mateus J, Grifoni A, Tarke A, et al. Selective and cross-reactive SARS-CoV-2 T cell epitopes in unexposed humans. Science 2020;370:89–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lipsitch M, Grad YH, Sette A, et al. Cross-reactive memory T cells and herd immunity to SARS-CoV-2. Nat Rev Immunol 2020;20:709–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sharwani K, Sharma R, Krishnan M, et al. Detection of serum cross-reactive antibodies and memory response to SARS-CoV-2 in pre-pandemic and post-COVID-19 convalescent samples. J Infect Dis 2021;23:jiab333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Falzone L, Gattuso G, Tsatsakis A, et al. Current and innovative methods for the diagnosis of COVID-19 infection (review). Int J Mol Med 2021;47:100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Li X, Yu J, Zhang Z, et al. Network bioinformatics analysis provides insight into drug repurposing for COVID-19. Med drug Discov 2021;10:100090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hufsky F, Lamkiewicz K, Almeida A, et al. Computational strategies to combat COVID-19: useful tools to accelerate SARS-CoV-2 and coronavirus research. Brief Bioinform 2021;22:642–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bettencourt P, Müller J, Nicastri A, et al. Identification of antigens presented by MHC for vaccines against tuberculosis. NPJ Vaccines. 2020;5(1):2. doi: 10.1038/s41541-019-0148-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reche PA. Cross-reactive immunity from combination DTP vaccines could protect against COVID-19, 2020. doi: 10.31219/osf.io/sbgy3. [DOI]
- 17. Russo G, Pennisi M, Fichera E, et al. In silico trial to test COVID-19 candidate vaccines: a case study with UISS platform. BMC Bioinformatics 2020;21:527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Farmer JD, Packard NH, Perelson AS. The immune system, adaptation, and machine learning. Phys D Nonlinear Phenom 1986;22:187–204. [Google Scholar]
- 19. Briney B, Inderbitzin A, Joyce C, et al. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 2019;566:393–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mora T, Walczak AM. Quantifying lymphocyte receptor diversity. Syst Immunol 2018;183–98. doi: 10.1201/9781315119847-11. [DOI] [Google Scholar]
- 21. Luca S, Mihaescu T. History of BCG vaccine. Maedica (Buchar) 2013;8:53–8. [PMC free article] [PubMed] [Google Scholar]
- 22. Bisgard KM, Rhodes P, Connelly BL, et al. Pertussis vaccine effectiveness among children 6 to 59 months of age in the United States, 1998-2001. Pediatrics 2005;116:e285–94. [DOI] [PubMed] [Google Scholar]
- 23. Palma C, Iona E, Giannoni F, et al. The Ag85B protein of Mycobacterium tuberculosis may turn a protective immune response induced by Ag85B-DNA vaccine into a potent but non-protective Th1 immune response in mice. Cell Microbiol 2007;9:1455–65. [DOI] [PubMed] [Google Scholar]
- 24. Raze D, Verwaerde C, Deloison G, et al. Heparin-binding hemagglutinin adhesin (HBHA) is involved in intracytosolic lipid inclusions formation in mycobacteria. Front Microbiol 2018;9:2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sreejit G, Ahmed A, Parveen N, et al. The ESAT-6 protein of Mycobacterium tuberculosis interacts with Beta-2-microglobulin (β2M) affecting antigen presentation function of macrophage. PLoS Pathog 2014;10(10):e1004446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Von Eschen K, Morrison R, Braun M, et al. The candidate tuberculosis vaccine Mtb72F/AS02A: tolerability and immunogenicity in humans. Hum Vaccin 2009;5:475–82. [DOI] [PubMed] [Google Scholar]
- 27. Messina NL, Zimmermann P, Curtis N. The impact of vaccines on heterologous adaptive immunity. Clin Microbiol Infect 2019;25:1484–93. [DOI] [PubMed] [Google Scholar]
- 28. Aaby P, Jensen H, Gomes J, et al. The introduction of diphtheria-tetanus-pertussis vaccine and child mortality in rural Guinea-Bissau: an observational study. Int J Epidemiol 2004;33:374–80. [DOI] [PubMed] [Google Scholar]
- 29. Ilinskaya AN, Dobrovolskaia MA. Understanding the immunogenicity and antigenicity of nanomaterials: past, present and future. Toxicol Appl Pharmacol 2016;299:70–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Juárez MA, Pennisi M, Russo G, et al. Generation of digital patients for the simulation of tuberculosis with UISS-TB. BMC Bioinformatics 2020;21:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. An D, Li K, Rowe DK, et al. Protection of K18-hACE2 mice and ferrets against SARS-CoV-2 challenge by a single-dose mucosal immunization with a parainfluenza virus 5–based COVID-19 vaccine. Sci Adv 2021;7:eabi5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.