

Abstract
Purpose:
Immune checkpoint blockade has shown remarkable efficacy, but in only a minority of patients with cancer, suggesting the need to develop additional treatment strategies. Aberrant glycosylation in tumors, resulting from the dysregulated expression of key enzymes in glycan biosynthesis, modulates the immune response. However, the role of glycan biosynthesis enzymes in antitumor immunity is poorly understood. We aimed to study the immunomodulatory effects of these enzymes.
Experimental Design:
We integrated transcriptional profiles of treatment-naïve human tumors and functional CRISPR screens to identify glycometabolism genes with immunomodulatory effects. We further validated our findings using in vitro coculture and in vivo syngeneic tumor growth assays.
Results:
We identified MAN2A1, encoding an enzyme in N-glycan maturation, as a key immunomodulatory gene. Analyses of public immune checkpoint blockade trial data also suggested a synergy between MAN2A1 inhibition and anti–PD-L1 treatment. Loss of Man2a1 in cancer cells increased their sensitivity to T-cell–mediated killing. Man2a1 knockout enhanced response to anti–PD-L1 treatment and facilitated higher cytotoxic T-cell infiltration in tumors under anti–PD-L1 treatment. Furthermore, a pharmacologic inhibitor of MAN2A1, swainsonine, synergized with anti–PD-L1 in syngeneic melanoma and lung cancer models, whereas each treatment alone had little effect.
Conclusions:
Man2a1 loss renders cancer cells more susceptible to T-cell–mediated killing. Swainsonine synergizes with anti–PD-L1 in suppressing tumor growth. In light of the limited efficacy of anti–PD-L1 and failed phase II clinical trial on swainsonine, our study reveals a potential therapy combining the two to overcome tumor immune evasion.
Introduction
Immune checkpoint blockade (ICB), which aims to boost the host immune system to attack cancer cells, could induce durable responses in multiple cancers (1, 2). However, most patients develop primary or acquired resistance to immunotherapy (3, 4), highlighting the importance of identifying biomarkers and regulators of tumor immune response (5). Genomic and transcriptomic profiling of tumors has facilitated the identification of factors that may affect tumor response to ICB treatment (6). Multiple factors have been reported to affect ICB response and resistance, including cytotoxic T-cell infiltration (7), mutation load (8–10), antigen presentation (11–14), PD-L1 expression (15, 16), gut microbiome (17), interferon signaling (14, 18, 19), PI3K pathway (20, 21), and beta-catenin signaling (22). Elucidation of these mechanisms provides important insights into the discovery of predictive biomarkers of response (23, 24). However, the identification of novel therapeutic strategies that improve ICB response is still a critical challenge in this field.
Protein glycosylation is a ubiquitous posttranslational modification and is essential in the development and physiology of living organisms (25). Glycans have been found to regulate many key biological processes, including protein folding, cellular adhesion, extracellular matrix interactions, molecular trafficking and clearance, signal transduction, and endocytosis (25–28). Compared with the normal tissues, tumors often show altered glycosylation patterns (29). Aberrant glycosylation is often observed as a driver of the malignant phenotype, directly affecting key processes in tumor progression and metastasis (29–31). In addition, altered glycosylation in cancer cells allows new interactions with immune cells to suppress antitumor immunity (32, 33). Immune cells can sense and respond to changes in the glycans of their microenvironment via glycan-binding proteins called lectins, which often leads to the attenuation of immune activation and the induction of inhibitory immune processes (32). Altogether, the abnormal glycosylation in cancer cells is associated with tumor development and progression and the ability of cancer cells to evade immune surveillance (34, 35).
Protein glycosylation consists of two major types, namely N-linked and O-linked (36). N-linked glycoproteins are frequently found on secretory and membrane-bound glycoproteins, thus are accessible for therapeutic purposes (37, 38). A large number of cell surface molecules involved in the innate and adaptive immune responses are N-linked glycoproteins, such as T-cell coreceptors, Toll-like receptors, cytokines, and cytokine receptors (39). The expression and function of these glycoproteins are dependent on normal glycosylation (39). Changes in their glycosylation patterns may lead to impaired protein expression and function, and significant alterations in immune pathway signaling (33, 39). In addition, N-glycosylation is also required for some immunosuppressive receptor-ligand engagement, such as PD-1/PD-L1, B7-1/PD-L1, and PD-1/PD-L2 (40). Among these, the study of the N-glycosylation of PD-L1 is the most detailed and systematic (41). PD-L1 is highly glycosylated in the majority of cells in which it is expressed, and N-glycosylation has been shown to stabilize PD-L1 (42). Thus, N-linked glycosylation is a critical regulator in the tumor microenvironment and could be manipulated to enhance antitumor immunity.
Despite the knowledge of the regulatory effect of glycosylation in tumor progression and the interplay of glycosylation with the immune response, how glycosylation modulates biological processes is still an open question. Thus, the development of strategies targeting key glycan modulators to improve antitumor immunity remains a relatively unexploited field. Given that the dysregulated gene expression of key enzymes in the glycosylation pathway is a major contributor to alterations in glycosylation patterns (35, 36). Glycan biosynthesis enzymes are poised to be a major source of druggable targets that can be manipulated to enhance immune response. Because N-linked glycosylation has been reported to regulate the expression and function of many cell surface glycoproteins involved in immune responses and affect the immune receptor/ligand binding (39, 40), we focused on genes participating in N-glycan biosynthesis. Using a large-scale data integration approach, we evaluated the association between N-glycan biosynthesis genes and resistance to T-cell cytotoxicity. This effort identified a novel N-glycan biosynthesis gene as a regulator of antitumor immune response, which we experimentally validated as a potential target to improve ICB response.
Materials and Methods
Cell culture and compounds
B16F10, A375, HEK293FT, and LLC cells were maintained in complete DMEM media (10% FBS, 1% l-glutamine, and 1% penicillin/streptomycin). B16F10-Cas9 cells were maintained in complete DMEM media with 2.5 to 5 μg/mL of blasticidin. CD8 T cells isolated from mice were cultured in complete RPMI 1640 media (10% FBS, 20 mmol/L HEPES, 1 mmol/L sodium pyruvate, 0.05 mmol/L 2-mercaptoethanol, 2 mmol/L l-glutamine, and 50 U/mL streptomycin and penicillin). All cell lines are tested for mycoplasma contamination.
Generation of Man2a1-knockout GFP+ cells
To knockout Man2a1, CRISPR gRNA sequences targeting Man2a1 or nontargeting control were cloned into a PLKO3G-GFP vector and confirmed by sequencing. Knockout constructs were cotransfected with pMD2.G and psPAX2 into HEK293FT cells to generate lentivirus. Transfection was performed using X-tremeGENE HP DNA Transfection Reagent (Roche, #6366546001) following the manufacturer’s protocol. Lentivirus was collected at 48 and 72 hours. Then B16F10-Cas9 cells were infected with a lentivirus driving expression of a single gRNA for 48 hours to inactivate Man2a1 genes individually. Infected cells were sorted on the basis of GFP expression by BD FACS Aria II.
Western blotting
For Western blotting, cells were lysed in RIPA buffer [50 mmol/L Tris-HCl (pH 7.5), 150 mmol/L NaCl, 1% NP-40, 2 mmol/L EDTA, 0.5% Na Deoxycholate, and 0.1% SDS] supplemented with protease and phosphatase inhibitor cocktail (Cell Signaling Technology, #5872s). Protein concentrations were measured with BCA Protein Assay Kit (Sangon Biotech, #C503021). For Western blotting, equal amounts of protein were heat denatured in the presence of a reducing agent and separated on 4%–12% or 10% Bis-Tris SurePAGE gels (GeneScript, #M00652, #M00665), and transferred to PVDF membranes. Antibodies used for Western blotting were as follows: anti-mouse MAN2A1 Antibody (Santa Cruz Biotechnology, #sc-376909), anti-human MAN2A1 antibody (Santa Cruz Biotechnology, #sc-377204), pSTAT1 (Cell Signaling Technology, #9167), STAT1 (Cell Signaling Technology, #9172), ERK2 (Santa Cruz Biotechnology, #sc-1647), GAPDH (Sigma, #G9545), PD-L1 (E1L3N) XP Rabbit mAb (Cell Signaling Technology, #13684T), vinculin (Santa Cruz Biotechnology, #sc-73614), Goat anti-rabbit secondary antibody (Cell Signaling Technology, #7074s), and Goat anti-mouse secondary antibody (Cell Signaling Technology, #7076s). The blots were imaged using CLINX imaging system according to the manufacturer’s instruction.
In vitro T-cell coculture assay
Pmel-1 TCR transgenic mice were purchased from The Jackson Laboratory (stock no. 005023). CD8 T cells were isolated from spleen and lymph nodes from Pmel-1 TCR transgenic mice using the EasySep mouse CD8+ T-cell isolation kit (STEMCELL #19753) according to the manufacturer’s protocol. Freshly isolated CD8 T cells were stimulated with anti-CD3/CD28 beads (Thermo Fisher Scientific, #11452D) at a bead to cell ratio of 1:2. On day 3, Recombinant mouse IL2 (Biolegend, #575406) was added to the culture at 20 ng/mL. The animal experiments have complied with all relevant ethical regulations. The study protocol was approved by the Institutional Care and Use Committee at Dana-Farber Cancer Institute.
For optimal killing by Pmel-1 T cells, B16F10 cells were pretreated with 10 ng/mL IFNγ for 24 hours prior to coculture to enhance MHC-I expression. In a competition assay, Man2a1-knockout cells or nontargeting control B16F10-Cas9 cells (GFP positive) were mixed with parental B16F10-Cas9 cells (GFP negative) at a 1:1 ratio. Mixed cells were then cocultured with in vitro–activated Pmel-1 T cells at effector-to-target ratios of 1:0, 1:1, 1:2, or 1:3. There were three cell-culture replicates for each condition. After a 3-day coculture with T cells, the percentage of GFP-positive cells was determined by FACS. T cells present in these cultures were gated out on the basis of antibodies specific for CD45 (APC–Cy7; Biolegend, #103115).
N-glycan profiling
Note that 5 × 106 cells were used as starting material and are lyophilized. The lyophilized proteins were digested into peptides by trypsin (Sigma-Aldrich) and then treated with PNGase F (New England Biolabs) to release N-Glycans. The released N-glycans were collected, pooled, and lyophilized. The lyophilized N-glycan samples were incubated with 1 mL of a NaOH/DMSO (Sigma-Aldrich)–NaOH (Sigma-Aldrich) slurry solution and 500 μL of methyl iodide (Sigma-Aldrich) for 30 minutes to permethylate the N-glycans. The permethylated N-glycans were then loaded into a conditioned [1 CV methanol, 1 CV MilliQ water, 1 CV acetonitrile (Sigma-Aldrich), and 1 CV MilliQ Water] C18 50 mg Sep-Pak column. The C18 column was washed with 3 mL of 15% acetonitrile and then eluted with 3 mL of 50% acetonitrile. The eluted fraction was lyophilized and then redissolved in 15 μL of 75% methanol from which 1 μL was mixed with 1 μL 2,5-dihydroxybenzoic acid (Sigma-Aldrich) and spotted on a MALDI-polished steel target plate (Bruker). Mass spectrum (MS) data were acquired on a Bruker UltraFlex II MALDI-TOF Mass Spectrometer instrument. Reflective positive mode was used and data recorded between 500 m/z and 6,000 m/z. MS profiles represent the aggregation of at least 20,000 laser shots. Mass peaks were annotated and assigned to N-glycans when a match was found. MS data were further analyzed and processed with mMass (43).
In vivo experiments
Note that 1 × 106 B16F10 or LLC cells were resuspended in Hank’s Balanced Salt Solution (Gibco, #14170112) and subcutaneously injected into the right flank of 6- to 8-week-old female C57BL/6 mice. Anti–PD-L1 (Bio X Cell, clone 10F.9G2, #BE0101, 100 μg/mouse) mAbs and IgG isotype control antibodies (BioX Cell, clone LTF-2, #BE0090, 100 μg/mouse) were administered on days 6, 9, and 12. For the drug combination experiment, mice were treated with swainsonine (APExBIO, #B7316) by oral gavage daily in 1 mg/kg/day from day 6 for 2 weeks. For CD8 depletion studies, anti-CD8 (Bio X Cell, clone YTS 169.4, #BE0117,150 μg/mouse) was administered 1 day prior to treatment and then every 3 days for a total of 3 doses. Tumors were measured every 3 days beginning on day 6 after tumor challenge until the survival endpoint was reached. Measurements were assessed manually by assessing the longest dimension (length) and the longest perpendicular dimension (width). Tumor volume was calculated using the formula 1/2 × length × width2. All mouse experiments were carried out at the Shanghai Model Organisms Center. The mouse experiments were approved by the Institutional Animal Care and Use Committee of the Institute of Biochemistry and Cell Biology and performed in accordance with this committee’s guidelines.
FACS analysis with tumor-infiltrating lymphocyte cells
B16F10 cells were subcutaneously injected into 6- to 8-week-old female C57BL/6 mice. Mice were administrated with anti–PD-L1 or IgG Isotype Control starting from day 6 and then every third day. For the drug combination experiment, mice were treated with swainsonine (APExBIO, #B7316) by oral gavage daily in 1 mg/kg/day from day 6. Tumors were harvested on day 15, weighed, dissected into small pieces, and incubated with 0.1% Collagenase Type I (Invitrogen, #17100-017) supplemented HBSS medium (Gibco, #2005368) for 20 minutes at 37°C. After incubation, tumor cells were passed through 70-μm filters to remove the undigested tumors. Single-cell suspensions were stained with the following antibodies: BD Horizon Fixable viability stain 450 (BD Pharmingen, #562247), Ms CD45 Percy-cy5.5 30-F11 (BD Pharmingen, #550994), APC anti-mouse CD3 (Biolegend, #100236), FITC anti-mouse CD4 (Biolegend, #100405), FITC anti-mouse CD8a (Biolegend, #100705), APC anti-mouse NK1.1 (Biolegend, #108709), FITC anti-mouse F4/80 (Biolegend, #123107), Ms CD86 PE GL1 (BD Pharmingen, #561963), and CD206(MMR) Monoclonal antibody (MR6F3) PE (eBioscience, #12-2061-80). BD FACSVerse was used for data acquisition, and FlowJo was used for data analysis.
Cytokine analysis from tumor lysate
Tumors were isolated from mice on day 15 after inoculation. One hundred milligrams of tissues were collected in a tube with prechilled 500 μL of cell lysis buffer [50 mmol/L Tris-HCl (pH 8.0), 150 mmol/L NaCl, 0.5% NP40, 10% Glycerol] supplemented with 1 mmol/L PMSF and cOmplete Protease Inhibitor Cocktail tablets (Roche, #11836153001). Tissues were first homogenized by tissue tearor. Then ultrasonic homogenization was performed by ultrasonic homogenizer with sonication duration for 60 seconds, at an ultrasonic cycle mode of 3 seconds sonication, and 3 seconds resting time. Then the samples were centrifuged at 12,000 × g for 20 minutes at 4°C. Protein concentration was assessed by BCA assay (Sangon Biotech, C503021). Lysates were then probed for IFNγ (ExCell Bio, #EM007-96, sensitivity: 4 pg/mL) and TNFα (ExCell Bio, #EM008-96, sensitivity: 7 pg/mL) protein levels by ELISA.
RNA sequencing data analysis
RNA was extracted using TRIzol reagent (Invitrogen Life Technologies) according to the manufacturer’s instructions. Quality and quantity of RNA were analyzed using NanoDrop, agarose gel electrophoresis, and Agilent 2100 (Thermo Fisher Scientific). Oligo(dT)-attached magnetic beads were used to purify mRNA. Purified mRNA was fragmented into short fragments with fragment buffer, and cDNAs were synthesized using the RNA fragments as templates for random primer, followed by end reparation and ligation to adapters. The cDNA libraries for B16F10 cancer cell line were sequenced using Illumina Nova 6000 by Berry Genomics Co. Ltd. For B16F10 tumors, the libraries were sequenced on BGIseq500 platform (BGI-Shenzhen). Data were aligned to mouse reference genome mm10 using STAR. RNA-Seq by Expectation-Maximization (RSEM) was used to map aligned reads and to generate a gene count matrix. Differential expression was performed using Deseq2. We performed gene set enrichment analysis (GSEA) using “MAGeCKFlute” R package (44) and GSEA preranked mode (45).
Histopathology analysis of internal organs
Mouse tissues were perfusion fixed in 4% paraformaldehyde solution overnight, and then embedded in paraffin. Four-micrometer-thick sections were cut and stained with hematoxylin and eosin (H&E). H&E-stained tissue sections were imaged using NIKON ECLIPSE CI. The pathology of mouse colon and liver sections was scored by referring to McGuckin’s (46) and Ishak’s (47) standards respectively, and was scored double-blind. The toxicity score for each organ is the sum of individual scores.
Data processing for publicly available datasets
The method to calculate the T-cell dysfunction score was described in the Tumor Immune Dysfunction and Exclusion (TIDE) algorithm, which we published before (48). Briefly, the interaction test in multivariate Cox proportional hazards (Cox-PH) regression was applied to identify gene association with the T-cell dysfunction phenotype. The T-cell dysfunction score for each gene is defined as the Wald test z score. For visualization purposes, samples were split to optimize the expression coefficients of interested genes in the Cox-PH regression model. However, the P-value significance computation was continuous without any cutoffs. The cytotoxic T-lymphocyte (CTL) level was estimated through bulk-tumor expression average of CD8A, CD8B, GZMA, GZMB, and PRF1. For public T-cell coculture screen analysis, the raw count was downloaded, and MAGeCK (49) was used to compute the log2 fold change for all gRNAs by comparing the experimental and control conditions.
Statistical analysis
Statistical tests employed with the number of replicates and independent experiments are listed in the text and figure legends. Statistical analyses were performed either with the R software (http://www.R-project.org/) or Prism 6 (GraphPad). Statistical analyses gathering more than two groups were performed using ANOVA. Otherwise, for two groups, statistical analyses were performed using the unpaired t-test. Log-rank test was applied when assessing the impact of the treatment on mice survival. Multiple hypothesis testing corrections were applied where multiple hypotheses were tested and were indicated by the use of FDR.
Results
MAN2A1 is associated with T-cell dysfunction
Previously our group developed a computational model TIDE (48), which integrated the expression signatures of T-cell dysfunction and T-cell exclusion to evaluate tumor immune evasion and predict response to ICB. TIDE examined 188 clinical study cohorts with expression profiles and clinical outcome information of over 33K cancer samples from The Cancer Genome Atlas (TCGA; ref. 50), PRECOG (51), and METABRIC (52) databases. To model T-cell dysfunction, TIDE applied a Cox-PH regression to compute how the expression of each gene interacts with CTL infiltration to influence patient survival. To model T-cell exclusion, TIDE calculated the gene expression correlation with that of three immunosuppressive cell types reported to restrict T-cell infiltration into tumors. Recently, we significantly expanded the scope of TIDE by incorporating many new datasets and functional modules (53). We integrated nearly 1,000 omics data from 12 published ICB clinical studies and 8 published CRISPR screens that identified genes that regulate lymphocyte-driven cancer killing and/or immunotherapy response. These efforts in the reuse of public data significantly improved the power of TIDE in hypothesis generation and enabled us to identify novel genes with immunomodulatory effects.
Because N-linked glycoproteins are essential to the antigen presentation and subsequent triggering of T-cell–mediated immune responses (54), we evaluated all the N-glycan biosynthesis genes through TIDE to identify their association with T-cell dysfunction. Several genes in this pathway (Supplementary Table S1), such as MGAT5, MAN1C1, and MAN2A1, showed high T-cell dysfunction scores across multiple cohorts (Fig. 1A). High expression of these genes is negatively associated with cytotoxic T-cell–mediated survival benefit (Fig. 1B; Supplementary Fig. S1), suggesting that T cells might be dysfunctional when these genes are highly expressed in the tumor microenvironment. To distinguish whether these genes are regulators or merely markers of T-cell dysfunction, we examined public CRISPR screens in mouse cancer cells cocultured with mouse primary T cells that specifically target the cancer cell antigen. Based on five such screens from two independent studies (55, 56), we found that gRNAs targeting Man2a1 were consis tently negatively selected (Fig. 1C). These results implicated MAN2A1 as a regulator of T-cell dysfunction, whose inactivation may sensitize cancer cells to T-cell–mediated lysis.
Figure 1.
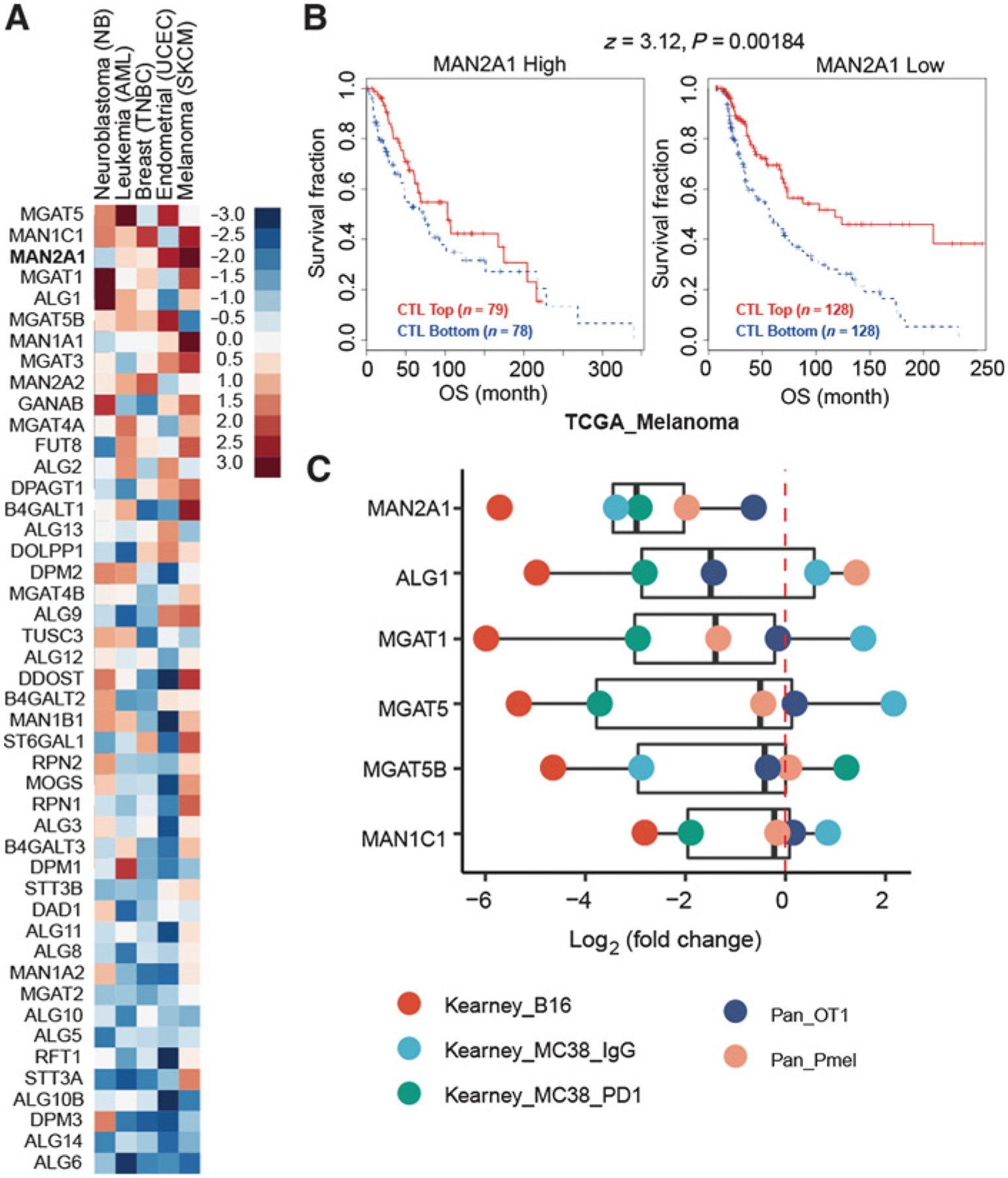
MAN2A1 is associated with T-cell dysfunction. A, T-cell dysfunction scores of N-glycan biosynthesis genes in the five TIDE core datasets. N-glycan biosynthesis genes were collected from KEGG_N_GLYCAN_ BIOSYNTHESIS (hsa00510). Five data sets, representing five cancer types, had more than 1% of genes with FDR > 0.1. The gene list was ranked by the average T-cell dysfunction score of the five cancer types. B, The association between the CTL level and overall survival for patients with different MAN2A1 levels in TCGA melanoma cohort. The CTL infiltration level was estimated as the average expression level of CD8A, CD8B, GZMA, GZMB, and PRF1. The association between the CTL level and overall survival was computed through the two-sided Wald test in the Cox-PH regression. “CTL Top” means samples have above-average CTL values among all samples, whereas “CTL Bottom” means below average. Samples were split by the best separation strategy according to the MAN2A1 expression coefficients in the Cox-PH regression model. C, Log2 fold change of genes with high dysfunction score in the public T-cell coculture screen data. MAGeCK was used to compute the log2 fold change for all gRNAs by comparing the experimental and control conditions.
MAN2A1 encodes a glycosyl hydrolase that is normally expressed in Golgi and catalyzes the final step in the N-linked glycan maturation pathway (57). To evaluate in which cancer type(s) MAN2A1 regulates the response to T-cell–driven cytotoxicity, we examined the expression of MAN2A1 in patient samples using publicly available databases, such as TCGA and NCBI Gene Expression Omnibus (GEO). We found MAN2A1 is expressed at a higher level in tumor tissues of melanoma and lung cancer as compared with their adjacent normal tissues (Supplementary Fig. S2A–S2D). In addition, MAN2A1 showed high T-cell dysfunction scores in most of the melanoma and lung cancer cohorts (Supplementary Fig. S2E and S2F). These data suggest that MAN2A1 may promote the resistance to T-cell–mediated killing in melanoma and lung cancer.
Man2a1-null cancer cells are more sensitive to T-cell–mediated killing
To validate the function of Man2a1 in T-cell–mediated tumor killing, we transduced B16F10-Cas9 cells with lentivirus coexpressing GFP and CRISPR guide RNAs (gRNA) targeting Man2a1. Western blot experiment confirmed the diminished level of MAN2A1 in the knockout cell line (Fig. 2A). Next, Man2a1-null or control sgRosa26 B16F10 cells (GFP positive) were mixed with equal number of parental B16F10 cells (GFP negative), and cocultured with in vitro–activated Pmel-1 T cells at different ratios. After 3 days of coculture, the percentage of Man2a1-null B16F10 cells in the presence or absence of T cells was determined by FACS (Fig. 2B; Supplementary Fig. S3). Consistent with the public coculture CRISPR screen result, Man2a1-null B16F10 cells were significantly more sensitive to T-cell–mediated cytotoxicity compared with sgRosa26 control cells (Fig. 2C).
Figure 2.
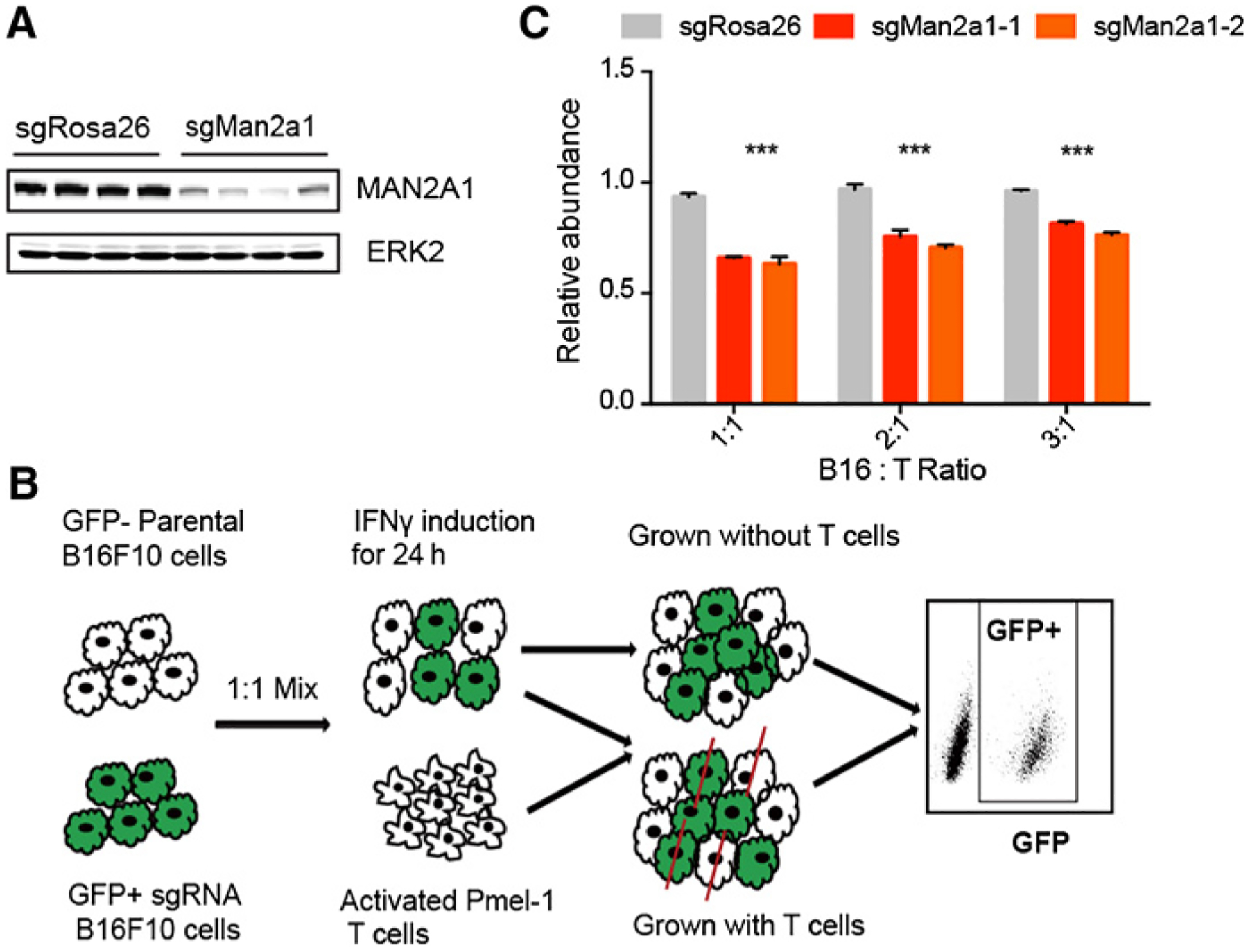
Man2a1-null cancer cells are more sensitive to T-cell–mediated killing. A, Western blot of MAN2A1 following genetic knockout. There are four independent CRISPR guides targeting Man2a1 and control nontargeting sgRosa26. We selected the two guides with best knockout efficiency (the middle two lanes of sgMan2a1 group) for further functional study. B, Experimental schematic of T-cell coculture assay. Man2a1-null (GFP+) B16F10 cells and parental (GFP−) B16F10 were mixed and cocultured with activated Pmel-1 T cells under IFNγ induction for 24 hours. After coculture, the ratio of GFP+ cells to parental cells (GFP−) was determined by flow cytometry. C, The effect of Man2a1 knockout on T-cell–mediated tumor killing. The y axis is the ratio of GFP+ cells to GFP− cells normalized to the untreated condition. Two-way ANOVA with Benjamini–Hochberg post-test comparison was used to determine statistical significance (***, P < 0.001). Values represent mean ± SD.
CD8+ T-cell–mediated tumor attack involves the secretion of key cytokines, such as IFNγ and TNFα, to induce proliferative arrest or apoptosis signaling (58). Suppression of cytokine signaling is one of the key mechanisms by which tumors evade attack by cytotoxic T cells (56, 59). To investigate whether Man2a1 depletion sensitized B16F10 cells to T-cell–mediated killing via the IFNγ signaling, we examined the responsiveness of cancer cells to IFNγ treatment. The biological effects of IFNγ are elicited mainly through the activation of the JAK/STAT pathway (60). Man2a1-knockout cells exhibited no significant difference from the control cells in the induction of STAT1 signaling upon IFNγ treatment, as measured by the phosphorylation status of STAT1 (Supplementary Fig. S4A). In addition, mouse Man2a1-null B16F10 cells and human MAN2A1-null A375 cells showed no significant proliferative difference compared with their respective control cells in response to IFNγ (Supplementary Fig. S4B and S4C). Similarly, loss of MAN2A1 in these cells had no significant impact on the growth inhibition by either TNFα alone or combined treatment of IFNγ and TNFα (Supplementary Fig. S4D and S4E). Collectively, these data suggest that the enhanced sensitivity to T-cell–mediated killing through Man2a1 knockout is not mediated by enhanced sensitivity to IFNγ or TNFα by cytokine-intrinsic mechanisms, implying instead potentially novel mechanisms of this phenomenon.
Man2a1 knockout changes glycometabolism
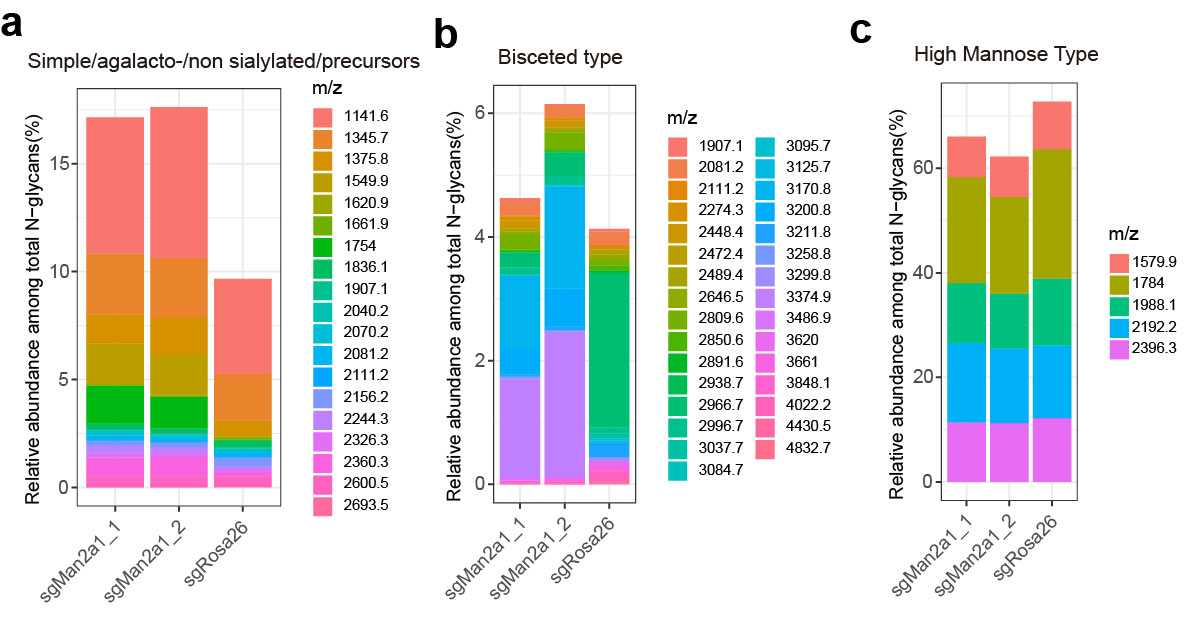
To investigate how Man2a1 knockout regulates cancer cell sensitivity to T-cell–driven cytotoxicity, we examined the transcriptome of Man2a1-null B16F10 cells by RNA sequencing (RNA-seq). Man2a1 knockout in B16F10 cells did not induce any marked gene expression change in the immune-related pathways (Supplementary Table S2). Interestingly, genes involved in the glyco-metabolic pathways, such as glycolysis, starch and sucrose metabolism, and galactose metabolism, were concordantly downregulated in Man2a1-knockout cells (Fig. 3A and B), indicating that Man2a1 loss may affect cancer cell glycometabolism. MAN2A1 encodes α-mannosidase II, a key enzyme in the N-glycan biosynthesis that converts precursor high mannose type N-glycans to matured complex-type structures (61). Glucose is necessary for glycosylation through its utilization in the hexosamine biosynthetic pathway, which produces UDP-N-acetylglucosamine (UDP-GlcNAc), the substrate for glycosylation (62). Thus, Man2a1 knockout might change glucose metabolism through the utilization of glucose in the N-glycan biosynthesis process.
Figure 3.
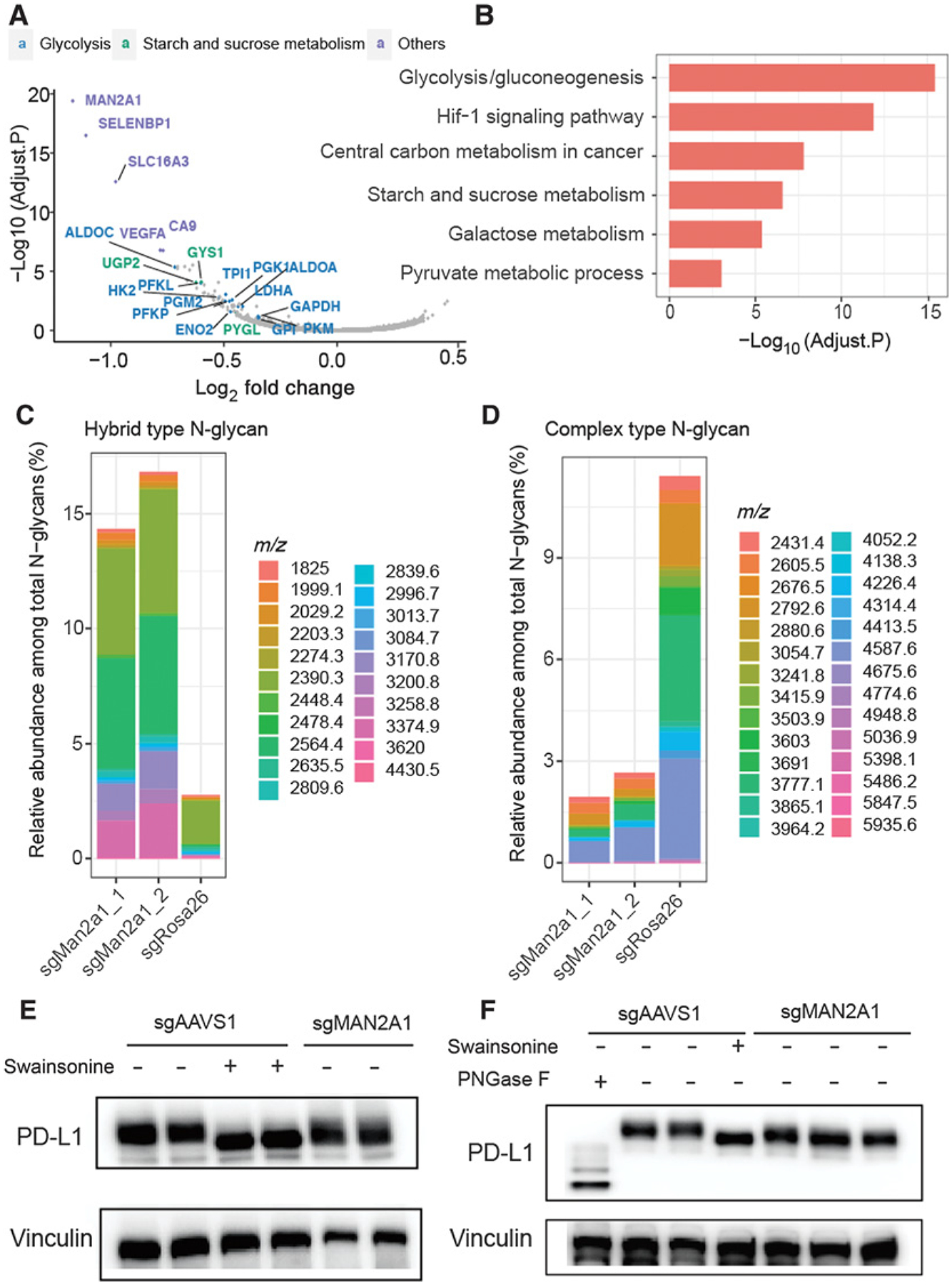
Man2a1 knockout changes glycol-metabolism and glycosylation of PD-L1. A, Volcano plot for differentially expressed genes in Man2a1-null B16F10 cells compared with control cells. Log2 fold change and adjusted P values were computed by DESeq2. Annotated genes represent downregulated genes (fold change < 1.2 & adjusted P value < 0.1) which are enriched in glycolysis, starch, and sucrose metabolism pathways or the others (top 5 hits with the most significant adjust P value). B, Gene ontology analysis for downregulated genes between Man2a1-null and control B16F10 cells. “MAGeCKFlute” R package was used to perform the enrichment analysis. C and D, Bar plot of the relative abundance of representative N-glycans among total N-glycans. (C) Hybrid type and (D), Complex type. E, Western blot analysis of PD-L1 expression in MAN2A1-knockout and control A375 cells treated with/without swainsonine. F, Western blot analysis of PD-L1 expression in MAN2A-1-knockout and control Colo205 cells treated with/without swainsonine and PNGase F.
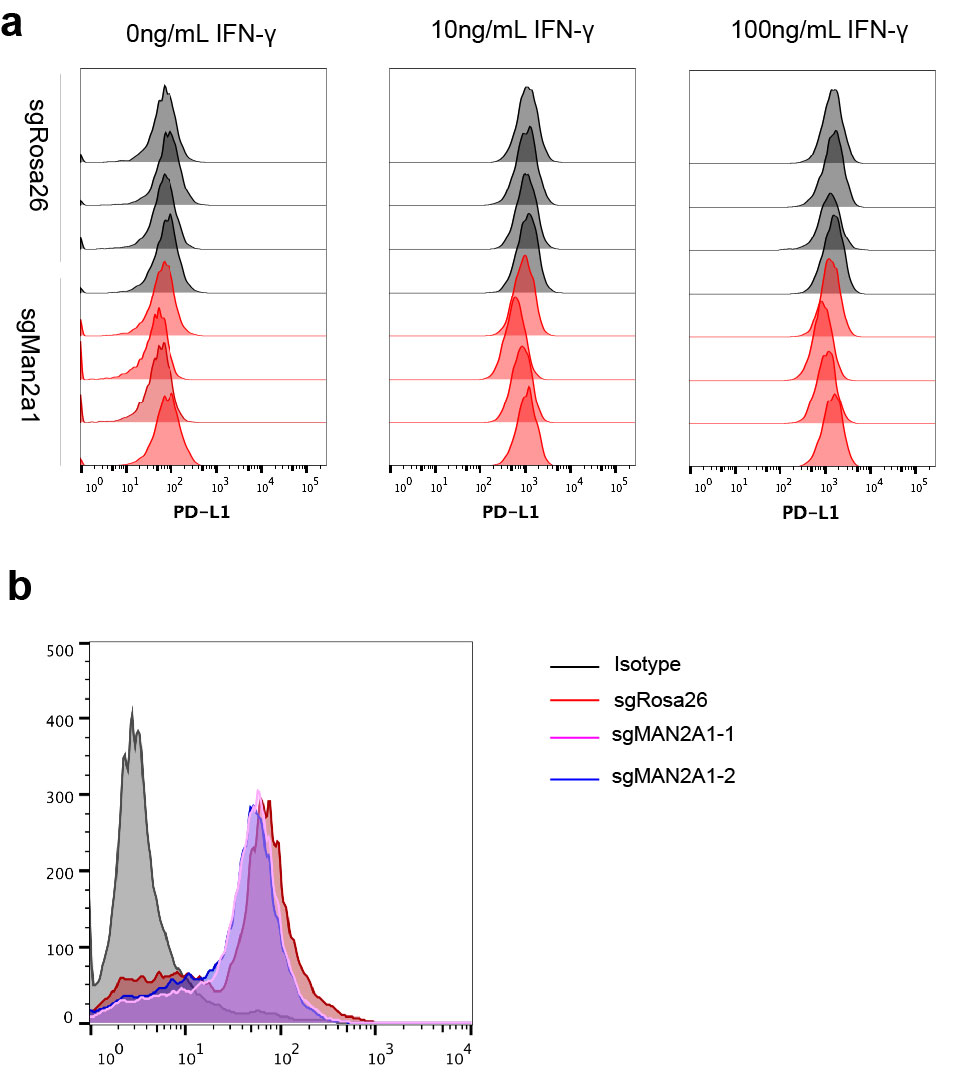
Of note, we did not observe significant expression changes in the N-glycan biosynthesis pathway or protein N-glycosylation pathway upon Man2a1 knockout (Supplementary Table S2), which is expected because N-glycosylation is a posttranslational modification. To examine the effect of Man2a1 knockout on N-glycan changes, we used mass spectrometry to evaluate the relative abundance of N-glycan component in the B16F10 cells. Consistent with MAN2A1’s known function, upon Man2a1 knockout, simple/precursor type (Supplementary Fig. S5A) and hybrid type (Fig. 3C) N-glycans increased, whereas complex type N-glycans decreased (Fig. 3D). In contrast, bisected (Supplementary Fig. S5B) and high mannose type (Supplementary Fig. S5C) N-glycans showed no significant change. Given that MAN2A1 knockout changes the N-glycan composition, we hypothesized that loss of MAN2A1 might alter the glycosylation pattern of some cell surface glycoproteins, thus affecting the interaction with T cells. Li and colleagues reported that inhibitors blocking N-linked glycosylation, such as swainsonine, altered the glycosylation of PD-L1 and reduced the PD-1/PD-L1 interaction (40). Glycosylation of PD-L1 is essential to the immune inhibitory effect of PD-L1, and nonglycosylated PD-L1 is unable to bind to PD-1 (40). Cells expressing nonglycosylated PD-L1 are more sensitive to T-cell–mediated killing (40). Indeed, we found that the molecular weight of PD-L1 in MAN2A1-knockout or swainsonine-treated cells was lower than in control cells, whereas expression remained similar (Fig. 3E and F; Supplementary Fig. S6), consistent with its altered glycosylation. These data demonstrate that the immunomodulatory effect of MAN2A1 is associated with global glycosylation changes, consistent with reported effects of altered PD-L1 glycosylation.
Man2a1 loss in cancer cells enhances anti–PD-L1 response
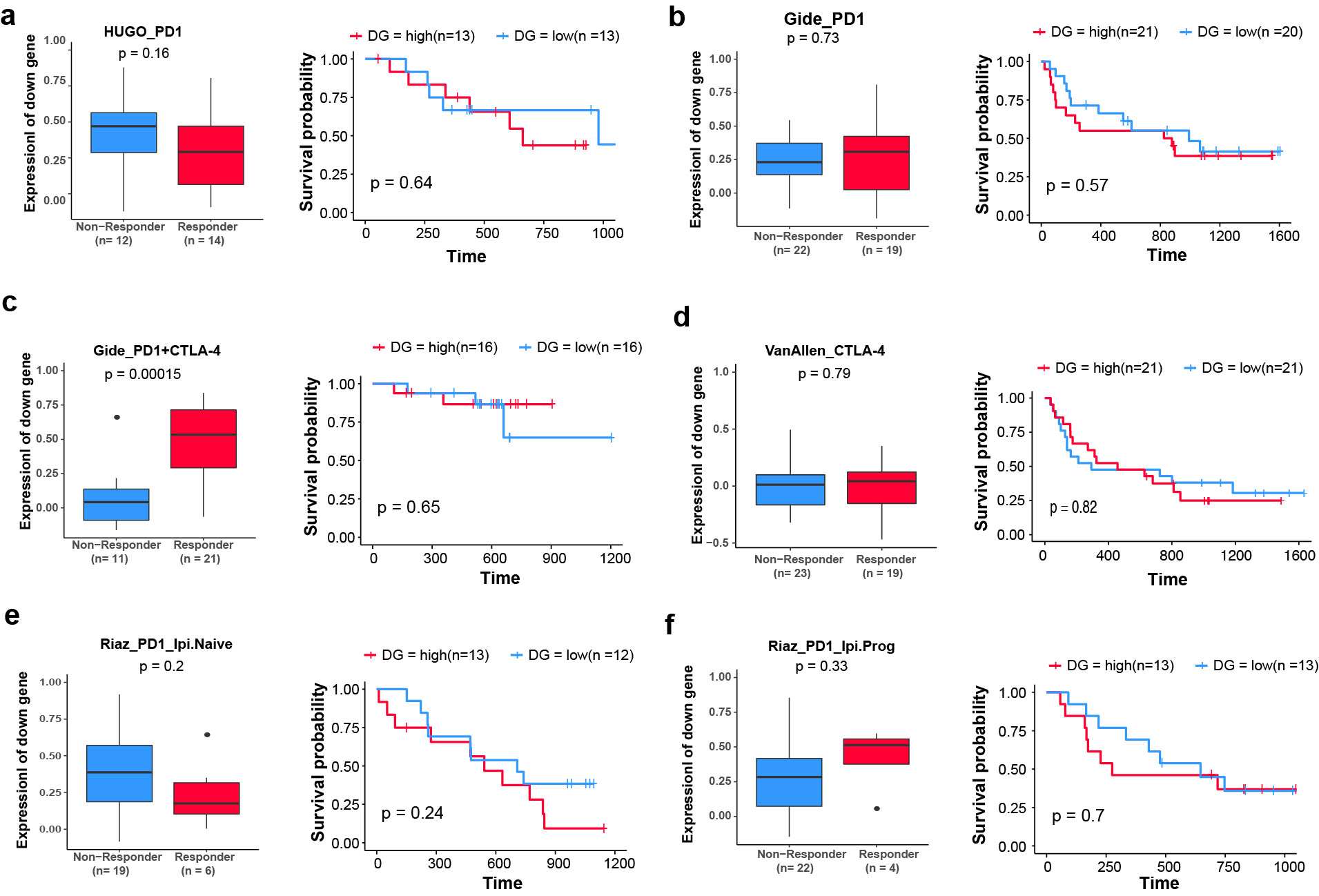
Lee and colleagues have demonstrated that N-linked glycosylation of PD-L1 hinders its binding with PD-L1 antibodies, and the removal of N-linked glycosylation enhances its predictive value for anti–PD-1/PD-L1 efficacy (63). We therefore asked whether MAN2A1 knockout would influence the efficacy of ICB treatment. To study the association between MAN2A1 and tumor ICB response, we used the downregulated genes in Man2a1-knockout cells to generate a signature. Then we used this signature as a proxy for MAN2A1 activity to test its association with response to ICB. Out of the five clinical cohorts we collected, we found lower MAN2A1 activity to be associated with better response and better overall survival in a large anti–PD-L1 trial (ref. 64; Fig. 4A and B), but not consistently in the anti–PD-1 (65–67) or anti–CTLA-4 trials (ref. 7; Supplementary Fig. S7). These data suggest that inhibition of MAN2A1 might improve tumor response to anti–PD-L1.
Figure 4.

Man2a1 loss in cancer cells enhances anti–PD-L1 response. A, mRNA levels of Man2a1-knockout gene signature (downregulated genes in Man2a1 knockout cells) between the responding versus nonresponding pretreatment tumors. ssGSEA was used to calculate the single sample signature score. Two-sided Student t test was used to compute the P value. B, Overall survival of anti–PD-L1–treated patients with urothelial cancer harboring high (above the group median) or low (below the group median) expression levels of Man2a1-knockout gene signature. Log-rank test was used to compute the P value in survival analysis. C, Tumor volume of control and Man2a1-null B16F10 tumors in C57BL/6 mice treated with IgG isotype control or anti–PD-L1. N = 9 mice for each group. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01, and *, P < 0.05 by two-way ANOVA. Data are mean ± SEM. D, Survival analysis of control and Man2a1-null B16F10 tumors treated with IgG isotype control or anti–PD-L1. N = 9 mice for each group. *, P < 0.05 by log-rank test.
To test whether Man2a1 knockout synergizes with anti–PD-L1 treatment in vivo, we used the B16F10 syngeneic tumor model that is resistant to current ICB (67–69). Man2a1-null B16F10 tumors showed similar growth with nontargeting control B16F10 tumors when treated with IgG isotype control antibody (Fig. 4C). However, PD-L1 blocking antibody conferred therapeutic benefit in mice bearing Man2a1-null B16F10 tumors, but this treatment was ineffective against control B16F10 tumors (Fig. 4C and D). Altogether, these data indicate that inactivation of Man2a1 in cancer cells could sensitize tumors to anti–PD-L1 immunotherapy.
Man2a1 knockout increases the cytotoxic immune cell infiltration and activities
To better understand the effect of Man2a1 depletion on the tumor microenvironment, we performed RNA-seq and flow cytometry on the syngeneic tumors in each of the above four conditions. RNA-seq analysis found immune-related pathways, such as lymphocyte migration and activation, adaptive immune response, cytokine and chemokine signaling, and IFNγ production, to be significantly enriched in the Man2a1-null tumors treated with anti–PD-L1 (Fig. 5A; Supplementary Table S3). In addition, upregulation of genes involved in antigen presentation, inflammatory response, and T-cell activation was consistently observed in Man2a1-null tumors with anti–PD-L1 treatment (Fig. 5B). The immune-stimulatory phenomena were not observed in other conditions (Supplementary Tables S4 and S5), except that anti–PD-L1 treatment of the control tumors increased CD45+ lymphocyte infiltration (Fig. 5C). Furthermore, Man2a1-null tumors had significantly increased proportions of CD3+ T cells and CD8+ T cells in response to anti–PD-L1 (Fig. 5D and E), whereas CD4+ T cells, natural killer (NK) cells, and M1 or M2 macrophages had no significant change (Supplementary Fig. S8). Finally, a significant increase of IFNγ and TNFα protein secretion was observed (Fig. 5F and G), indicating the activation of cytotoxic lymphocytes and effective tumor killing. Taken together, our data suggest that inactivation of Man2a1 in tumors treated with anti–PD-L1 results in not only increased immune cell infiltration but also enhanced cytotoxic tumor killing.
Figure 5.
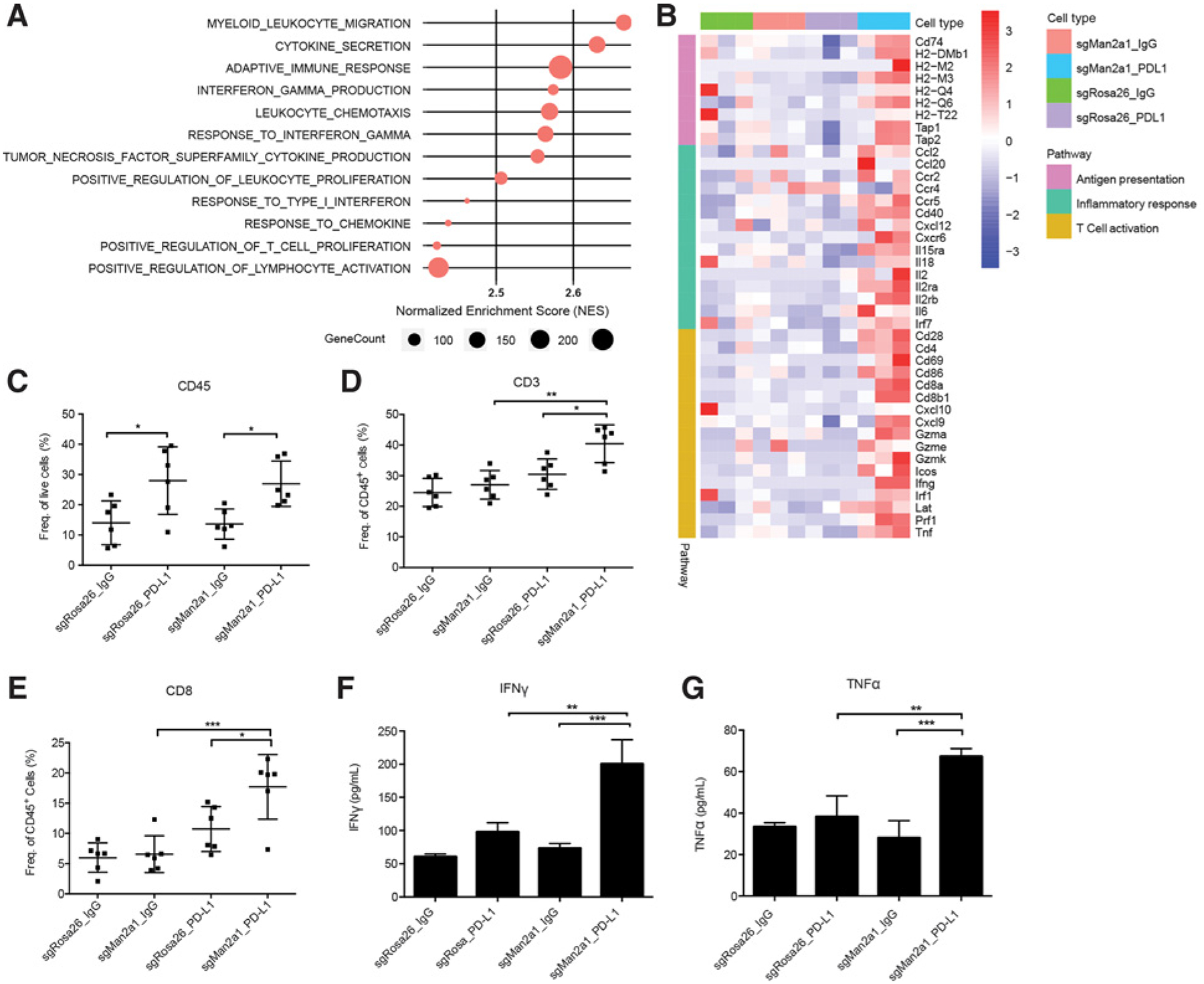
Man2a1 knockout increases the cytotoxic immune cell infiltration and activities. A, Gene sets enriched in Man2a1-knockout tumors treated with PD-L1 blockade compared with control tumors with anti–PD-L1 treatment. Preranked GSEA was used to calculate the enrichment. B, Heat map showing expression value (z-score based on TPM) of antigen presentation, inflammatory response, and T-cell activation genes in Man2a1-null or control B16F10 tumors treated with IgG isotype control or anti–PD-L1. C–E, Flow cytometry of immune populations from control and Man2a1-null B16F10 tumors treated with IgG or anti–PD-L1. (C) CD45; (D) CD3; and (E) CD8. Data are mean ± SD, representative of at least two independent experiments. N = 6 mice for each group. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; and *, P < 0.05 by one-way ANOVA with Tukey multiple comparisons test. F and G, IFNγ and TNFα protein levels in tumor lysates of Man2a1-null and control B16F10 tumors. (F) IFNγ protein and (G) TNFα protein. N = 3 mice for each group. Data are mean ± SD, representative of at least two independent experiments. ****, P < 0.0001; ***, P < 0.001; **, P < 0.01; and *, P < 0.05 by one-way ANOVA with Tukey multiple comparisons test.
Pharmacologic inhibition of Man2a1 sensitizes tumors to anti–PD-L1
To further evaluate the efficacy of targeting MAN2A1, we tested the in vivo effect of combined treatment of a MAN2A1 inhibitor swainsonine and anti–PD-L1 antibody. Swainsonine is a pharmacologic inhibitor of α-mannosidase, and it inhibits both Golgi alpha-mannosidase and lysosomal alpha mannosidase (70). It has been shown to stimulate lymphocyte proliferation, enhance T-cell stimulation by antigen, and activate natural antitumor immunity (71, 72). Furthermore, it has been shown to inhibit tumor growth and metastasis in hepatocellular carcinoma (73), melanoma (74, 75), and lymphoma (76) mouse models. Although swainsonine had promising results in initial clinical settings (77, 78), it failed a phase II clinical trial on advanced or metastatic renal cell carcinoma due to limited efficacy (79). In B16F10 tumors, coadministration of swainsonine and anti–PD-L1 significantly enhanced the antitumor effects and survival benefits, whereas anti–PD-L1 or swainsonine treatment alone had little impact on tumor growth or mouse survival (Fig. 6A and B). We also tested this synergistic effect in the LLC lung cancer model and observed similar effects (Fig. 6C and D). Notably, depletion of CD8+ T cells by anti-CD8 treatment significantly abrogated the antitumor effects of anti–PD-L1 and swainsonine combination therapy (Fig. 6C and D), suggesting that CD8+ T cells play important roles in the tumor rejection effect of the combination therapy.
Figure 6.
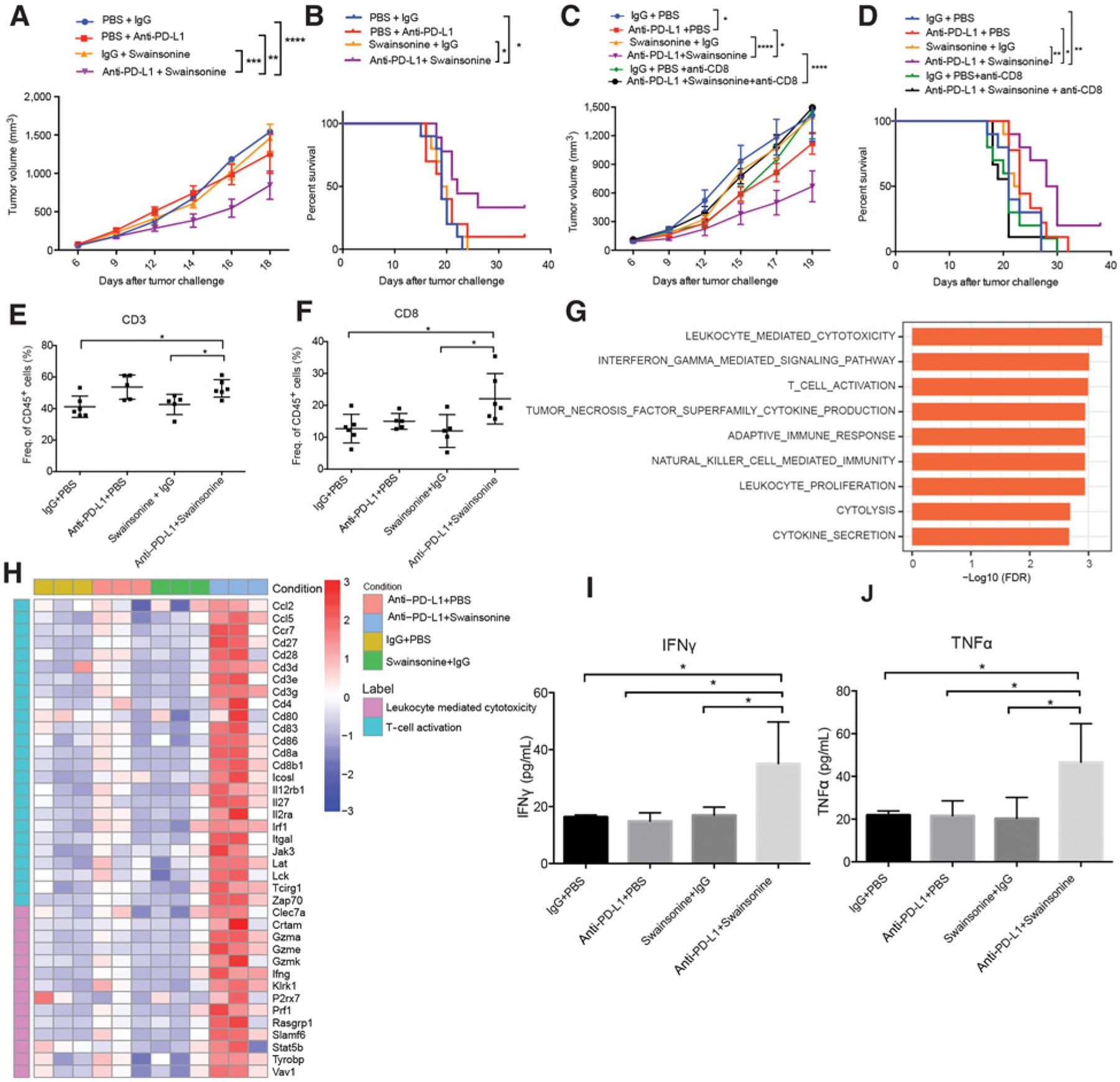
Pharmacologic inhibition of Man2a1 sensitizes tumors to anti–PD-L1. A, B16F10 tumor volume in response to swainsonine and anti–PD-L1. N = 10 mice per group. Data are shown as the mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 by two-way ANOVA. B, Survival curve of B16F10 tumors in response to swainsonine and anti–PD-L1. N = 10 mice per group. Log-rank test, *, P < 0.05. C, LLC tumor volume in response to swainsonine and anti–PD-L1. N = 10 mice per group. Data are shown as the mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 by two-way ANOVA. D, Survival curve of LLC tumors in response to swainsonine and anti–PD-L1. N = 10 mice per group. Log-rank test, *, P < 0.05. E and F, Flow cytometry of immune populations from B16F10 tumors treated with different drug combinations. (E) CD3 and (F) CD8. Data are mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 by one-way ANOVA with Tukey multiple comparisons test. G, Gene sets enriched in B16F10 tumors treated with combined anti–PD-L1 and swainsonine compared with tumors treated with anti–PD-L1. Preranked GSEA was used to calculate the enrichment. H, Heat map showing expression value (z-score based on TPM) of T-cell activation and leukocyte-mediated cytolysis genes in B16F10 tumors treated with different drug combinations. I and J, IFNγ and TNFα protein levels in tumor lysates of B16F10 tumors treated with different drug combinations. (I) IFNγ protein and (J) TNFα protein. N = 4 mice for each group. Data are mean ± SD, representative of at least two independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001 by one-way ANOVA with Tukey multiple comparisons test.
To further characterize the enhanced immune response to combination therapy, we performed flow cytometry and RNA-seq of B16F10 tumors treated with either monotherapy or combination therapy. Flow cytometry analysis showed a significant increase of tumor-infiltrating CD3+ and CD8+ T cells in tumors receiving anti–PD-L1 plus swainsonine (Fig. 6E and F), whereas CD4+ T cells, NK cells, and M1 or M2 macrophages had no significant change (Supplementary Fig. S9). RNA-seq analysis revealed upregulation of immune-related pathways, such as adaptive immune response, IFNγ production, T-cell activation, and cytokine secretion, in tumors treated with combined anti–PD-L1 and swainsonine relative to tumors treated with either monotherapy or control antibody (Fig. 6G, Supplementary Tables S6 and S7). Upregulation of gene signatures associated with T-cell activation and leukocyte-mediated cytolysis was also observed in tumors treated with the combination therapy (Fig. 6H). In addition, both IFNγ and TNFα protein levels were significantly higher in tumors treated with the combination therapy, consistent with enhanced T-cell function (Fig. 6I and J). Altogether, our findings suggest that swainsonine may be combined with anti–PD-L1 immunotherapy to enhance antitumor immunity.
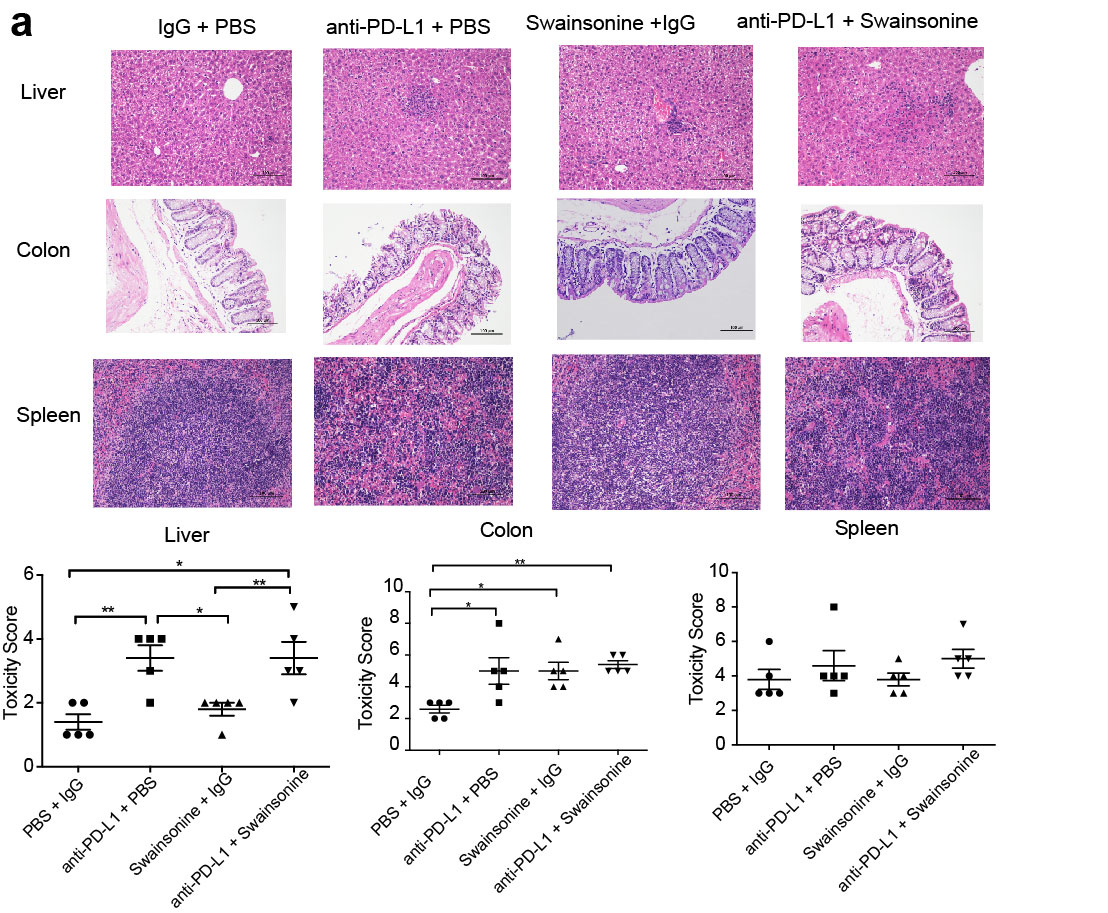
Patients receiving combination immunotherapy are at an increased risk of immune-related adverse events (irAE; ref. 80). Although fatal high-grade irAEs are rare, adverse events remain a major limitation of checkpoint blockade and frequently result in treatment termination. To evaluate whether the combination of anti–PD-L1 and swainsonine would aggravate irAEs, we harvested organs from B16F10 tumor–bearing mice that had received either monotherapy or combination therapy. Examination of tissue sections revealed a pathology of low/moderate portal and lobular inflammation in the liver as well as moderate colon inflammation with anti–PD-L1 monotherapy (Supplementary Fig. S10). Colon and liver inflammation was not worsened by combination therapy with anti–PD-L1 and swainsonine compared with anti–PD-L1 monotherapy (Supplementary Fig. S10). No significant histologic changes were observed in the spleen in any of the treatment groups (Supplementary Fig. S10). These preclinical safety data demonstrate that the addition of swainsonine did not increase inflammatory lesions associated with anti–PD-L1 therapy.
Discussion
Cancer treatment by ICB achieved striking successes in the last decade, but the majority of patients do not respond (81). Although N-glycosylation has been known to be important for cancer immunology, previous studies have not identified the key regulators that can be targeted to improve immunotherapy efficacy. In this study, we used a comprehensive data integration approach to identify and in vitro experiments to characterize MAN2A1, encoding an enzyme involved in N-glycan maturation, as a novel target to sensitize tumors to T-cell–mediated cytotoxicity. Man2a1 loss changed the overall glycosylation pattern in cancer cells, and Man2a1 knockout in cancer cells rendered B16F10 tumors sensitive to PD-L1 blockade therapy and activated the tumor immune microenvironment. We also identified that swainsonine, an inhibitor of alpha-mannosidase II, might be an ideal drug that can be combined with anti–PD-L1 immunotherapy to enhance immune response. The synergy we observed between anti–PD-L1 and swainsonine in vivo, whereas each treatment alone had little effect, is an important finding. This is especially of potential clinical significance, in light of the limited efficacy of anti–PD-L1 and failed clinical trial on swainsonine, and underlies the importance of data integration on cancer immunology studies.
Previous studies reported that Man2a1-null mice exhibited a 3-fold increase in the abundance of hybrid type N-glycans and approximately half the level of complex type N-glycans (61). Aged mice with constitutive Man2a1 deficiency show systemic autoimmune disease similar to human systemic lupus erythematosus (82), although lymphoid lineage development and activation responses were unaffected (83). The autoimmune syndrome of Man2a1-deficient mice can be explained by a pathogenic proinflammatory condition in the absence of infection due to the aberrant synthesis of endogenous glycans (37, 83). These symptoms indicated a prominent role of endogenous α-mannosidase II in the regulation of inflammatory immune responses. We also observed the immune-stimulatory signaling in mice injected with Man2a1-null B16F10 tumors treated with anti–PD-L1 but not in those treated with IgG isotype control antibody. The lack of immune stimulation with Man2a1 inhibition alone in the B16F10 model and maybe other tumors might explain the reason why swainsonine failed the previous clinical trial. Our data suggest that anti–PD-L1 treatment might enhance the proinflammatory benefit of MAN2A1 inhibition.
There are several limitations of our study. First, in the B16F10 and LLC model we tested, Man2a1 inhibition alone does not affect tumor growth and tumor microenvironment. However, other studies reported tumor growth rejection and immune stimulation with swainsonine treatment alone (84), suggesting that Man2a1 may have strong effect in other tumor models. Second, we observed reduced molecular weight of PD-L1 in MAN2A1-knockout and swainsonine-treated cells, suggesting the impaired glycosylation of PD-L1. Previous studies demonstrated that glycosylation of PD-L1 is necessary for PD-1/PD-L1 interaction (40) and impedes the binding with anti–PD-L1 antibodies (63). Thus, we hypothesized that PD-L1 expressed in MAN2A1-null cells might have lower binding affinity with PD-1, thus reducing the suppression of T-cell activity by PD-1/PD-L1 interaction and enhancing the sensitivity to T-cell killing. Moreover, MAN2A1 knockout may also affect the glycosylation of other immunoregulatory proteins and influence the function of these glycoproteins and also the interaction of important immune receptor/ligand. Changes of the glycosylation pattern of immunomodulatory glycoproteins might affect the activity of multiple immune cells and immune responses, such as antigen presentation and T-cell priming (32). However, additional detailed experiments are needed to fully elucidate the full mechanism. Third, single-cell RNA-seq (scRNA-seq) provides an unbiased profiling of immune cells and enables us to study the clonal expansion, migration, and functional state transition of T cells or other immune cells in the tumor microenvironment (85, 86). Although tumor bulk RNA-seq revealed the upregulation of proinflammatory pathways in Man2a1-null tumors in response to anti–PD-L1 and B16F10 tumors treated with the combined anti–PD-L1 and swainsonine, scRNA-seq will allow us to systematically study the tumor microenvironment and gene expression changes induced by Man2a1 knockout at finer resolution. Although there have been numerous scRNA-seq studies published on tumors, some treated with ICB, none were on tumors treated with anti–PD-L1. Finally, swainsonine treatment could sensitize tumors to anti–PD-L1 treatment. However, swainsonine might affect the glycosylation pattern of other cells in the tumor microenvironment or in normal tissues besides cancer cells, and these effects deserve more detailed characterization.
Despite these limitations, our discovery that inhibition of MAN2A1 sensitizes tumors to PD-L1 blockade therapy represents the first work identifying a regulator in the N-glycan biosynthesis pathway that can be targeted to improve ICB response. Our work not only lays the foundation for the combination of MAN2A1 inhibitor and anti–PD-L1 antibody to improve response in future cancer therapeutics, but also sets the bridge for immunology and glycobiology. Manipulation of glycosylation might be an effective strategy to improve ICB response and overcome current limitations in the treatment of cancer.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Translational Relevance.
Immune checkpoint blockade has shown remarkable efficacy, but in only a minority of patients with cancer, suggesting the need to develop additional treatment strategies. In this study, we integrated analyses of clinical transcription profiles of human tumors and functional CRISPR screens to discover combination therapy candidates. We identified a novel target involved in glycometabolism, MAN2A1, which suppressed the sensitivity of cancer cells to T-cell–mediated killing. Pharmacologic inhibition of MAN2A1 by swainsonine, an indolizidine alkaloid that can be tolerated by patients with cancer, synergized with anti–PD-L1 in the treatment of melanoma and lung cancer. In light of the limited efficacy of anti–PD-L1 and failed phase II clinical trial of swainsonine on advanced or metastatic renal cell carcinoma, our study reveals a potential therapy combining the two to overcome tumor immune evasion.
Acknowledgments
This work was partially supported by NIH R01CA234018 and the National Natural Science Foundation of China (81872290).
Disclosure of Potential Conflicts of Interest
M. Brown reports grants from Novartis (sponsored research), other from Novartis (unpaid consultant), personal fees and other from Kronos Bio (consultant and SAB member), personal fees and other from H3 Biomedicine (consultant and SAB member), and personal fees and other from GV20 Oncotherapeutics (consultant and SAB member) outside the submitted work. K.W. Wucherpfennig reports grants from NIH during the conduct of the study; personal fees from TCR2 Therapeutics, personal fees from TScan Therapeutics, personal fees from Nextech Invest, personal fees from Immunitas Therapeutics, and personal fees from SQZ Biotech outside the submitted work. X.S. Liu reports grants from National Institute of Health (R01CA234018) and grants from National Natural Science Foundation of China (81872290) during the conduct of the study; and co-founder, board member, and SAB of GV20 Oncotherapy; SAB of 3DMedCare; received research funding from Takeda and Sanofi; consultant for Genentech; and stockholder in BMY, CL, TMO, WBA, ABT, ABBV, and JNJ. No potential conflicts of interest were disclosed by the other authors.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Data Availability
All data presented in this article are available from the corresponding author upon reasonable request. RNA-seq data have been deposited at the GEO under accession number GSE152925.
References
- 1.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest 2015;125:3384–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory pathways in immunotherapy for cancer. Annu Rev Immunol 2016;34:539–73. [DOI] [PubMed] [Google Scholar]
- 3.Kalbasi A, Ribas A. Tumour-intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 2020;20:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017;168:707–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature 2017;541:321–30. [DOI] [PubMed] [Google Scholar]
- 6.van’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AAM, Mao M, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 2002;415: 530–6. [DOI] [PubMed] [Google Scholar]
- 7.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51:202–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; 348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane J-P, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 2017;8:1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marincola FM, Jaffee EM, Hicklin DJ, Ferrone S. Escape of human solid tumors from T-cell recognition: molecular mechanisms and functional significance. Adv Immunol 2000;74:181–273. [DOI] [PubMed] [Google Scholar]
- 13.Sucker A, Zhao F, Real B, Heeke C, Bielefeld N, Massen S, et al. Genetic evolution of T-cell resistance in the course of melanoma progression. Clin Cancer Res 2014; 20:6593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med 2016;375:819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nishino M, Ramaiya NH, Hatabu H, Hodi FS. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol 2017;14:655–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shien K, Papadimitrakopoulou VA, Wistuba II. Predictive biomarkers of response to PD-1/PD-L1 immune checkpoint inhibitors in non-small cell lung cancer. Lung Cancer 2016;99:79–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science 2018;359:91–7. [DOI] [PubMed] [Google Scholar]
- 18.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell 2016;167:397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jacquelot N, Yamazaki T, Roberti MP, Duong CPM, Andrews MC, Verlingue L, et al. Sustained type I interferon signaling as a mechanism of resistance to PD-1 blockade. Cell Res 2019;29:846–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng W, Pollard JW. Inhibiting macrophage PI3Kgamma to enhance immunotherapy. Cell Res 2016;26:1267–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kgamma in myeloid cells. Nature 2016;539:443–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature 2015;523:231–5. [DOI] [PubMed] [Google Scholar]
- 23.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19: 133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 2017;551:517–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohtsubo K, Marth JD. Glycosylation in cellular mechanisms of health and disease. Cell 2006;126:855–67. [DOI] [PubMed] [Google Scholar]
- 26.Lau KS, Partridge EA, Grigorian A, Silvescu CI, Reinhold VN, Demetriou M, et al. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell 2007;129:123–34. [DOI] [PubMed] [Google Scholar]
- 27.Pinho SS, Reis CA. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 2015;15:540–55. [DOI] [PubMed] [Google Scholar]
- 28.Mereiter S, Balmana M, Campos D, Gomes J, Reis CA. Glycosylation in the era of cancer-targeted therapy: where are we heading? Cancer Cell 2019;36:6–16. [DOI] [PubMed] [Google Scholar]
- 29.Cheng WK, Oon CE. How glycosylation aids tumor angiogenesis: An updated review. Biomed Pharmacother 2018;103:1246–52. [DOI] [PubMed] [Google Scholar]
- 30.Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Cell surface protein glycosylation in cancer. Proteomics 2014;14:525–46. [DOI] [PubMed] [Google Scholar]
- 31.Fuster MM, Esko JD. The sweet and sour of cancer: glycans as novel therapeutic targets. Nat Rev Cancer 2005;5:526–42. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez E, Schetters STT, van Kooyk Y. The tumour glyco-code as a novel immune checkpoint for immunotherapy. Nat Rev Immunol 2018;18:204–11. [DOI] [PubMed] [Google Scholar]
- 33.van Kooyk Y, Rabinovich GA. Protein-glycan interactions in the control of innate and adaptive immune responses. Nat Immunol 2008;9:593–601. [DOI] [PubMed] [Google Scholar]
- 34.Vajaria BN, Patel PS. Glycosylation: a hallmark of cancer? Glycoconj J 2017;34: 147–56. [DOI] [PubMed] [Google Scholar]
- 35.van den Elsen JM, Kuntz DA, Rose DR. Structure of Golgi alpha-mannosidase II: a target for inhibition of growth and metastasis of cancer cells. EMBO J 2001;20: 3008–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Silsirivanit A. Glycosylation markers in cancer. Adv Clin Chem 2019;89: 189–213. [DOI] [PubMed] [Google Scholar]
- 37.Marth JD, Grewal PK. Mammalian glycosylation in immunity. Nat Rev Immunol 2008;8:874–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tian Y, Zhang H. Characterization of disease-associated N-linked glycoproteins. Proteomics 2013;13:504–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyons JJ, Milner JD, Rosenzweig SD. Glycans instructing immunity: the emerging role of altered glycosylation in clinical immunology. Front Pediatr 2015;3:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li C-W, Lim S-O, Chung EM, Kim Y-S, Park AH, Yao J, et al. Eradication of triple-negative breast cancer cells by targeting glycosylated PD-L1. Cancer Cell 2018;33:187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hsu JM, Li CW, Lai YJ, Hung MC. Posttranslational modifications of PD-L1 and their applications in cancer therapy. Cancer Res 2018;78:6349–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C-W, Lim S-O, Xia W, Lee H-H, Chan Li-C, Kuo C-W, et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun 2016;7:12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strohalm M, Hassman M, Kosata B, Kodicek M. mMass data miner: an open source alternative for mass spectrometric data analysis. Rapid Commun Mass Spectrom 2008;22:905–8. [DOI] [PubMed] [Google Scholar]
- 44.Wang B, Wang M, Zhang W, Xiao T, Chen C-H, Wu A, et al. Integrative analysis of pooled CRISPR genetic screens using MAGeCKFlute. Nat Protoc 2019;14: 756–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102: 15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heazlewood CK, Cook MC, Eri R, Price GR, Tauro SB, Taupin D, et al. Aberrant mucin assembly in mice causes endoplasmic reticulum stress and spontaneous inflammation resembling ulcerative colitis. PLoS Med 2008;5:e54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22:696–9. [DOI] [PubMed] [Google Scholar]
- 48.Jiang P, Gu S, Pan D, Fu J, Sahu A, Hu X, et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat Med 2018;24:1550–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li W, Xu H, Xiao T, Cong Le, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 2014;15:554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KR, Ozenberger BA, et al. The cancer genome atlas pan-cancer analysis project. Nat Genet 2013;45:1113–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015;21:938–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012;486:346–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fu J, Li K, Zhang W, Wan C, Zhang J, Jiang P, et al. Large-scale public data reuse to model immunotherapy response and resistance. Genome Med 2020;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wall KA, Pierce JD, Elbein AD. Inhibitors of glycoprotein processing alter T-cell proliferative responses to antigen and to interleukin 2. Proc Natl Acad Sci U S A 1988;85:5644–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay REn, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science 2018;359:770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kearney CJ, Vervoort SJ, Hogg SJ, Ramsbottom KM, Freeman AJ, Lalaoui N, et al. Tumor immune evasion arises through loss of TNF sensitivity. Sci Immunol 2018;3:eaar3451. [DOI] [PubMed] [Google Scholar]
- 57.Moremen KW, Robbins PW. Isolation, characterization, and expression of cDNAs encoding murine alpha-mannosidase II, a Golgi enzyme that controls conversion of high mannose to complex N-glycans. J Cell Biol 1991;115: 1521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Turner MD, Nedjai B, Hurst T, Pennington DJ. Cytokines and chemokines: at the crossroads of cell signalling and inflammatory disease. Biochim Biophys Acta 2014;1843:2563–82. [DOI] [PubMed] [Google Scholar]
- 59.Vredevoogd DW, Kuilman T, Ligtenberg MA, Boshuizen J, Stecker KE, de Bruijn B, et al. Augmenting immunotherapy impact by lowering tumor TNF cytotoxicity threshold. Cell 2019;178:585–99. [DOI] [PubMed] [Google Scholar]
- 60.Castro F, Cardoso AP, Goncalves RM, Serre K, Oliveira MJ. Interferon-gamma at the crossroads of tumor immune surveillance or evasion. Front Immunol 2018; 9:847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chui D, Oh-Eda M, Liao Y-F, Panneerselvam K, Lal A, Marek KW, et al. Alpha-mannosidase-II deficiency results in dyserythropoiesis and unveils an alternate pathway in oligosaccharide biosynthesis. Cell 1997;90:157–67. [DOI] [PubMed] [Google Scholar]
- 62.Wellen KE, Thompson CB. A two-way street: reciprocal regulation of metabolism and signalling. Nat Rev Mol Cell Biol 2012;13:270–6. [DOI] [PubMed] [Google Scholar]
- 63.Lee H-H, Wang Y-N, Xia W, Chen C-H, Rau K-M, Ye L, et al. Removal of N-linked glycosylation enhances PD-L1 detection and predicts anti-PD-1/PD-L1 therapeutic efficacy. Cancer Cell 2019;36:168–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171:934–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gide TN, Quek C, Menzies AM, Tasker AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti-PD-1 monotherapy and anti-PD-1/anti-CTLA-4 combined therapy. Cancer Cell 2019;35:238–55. [DOI] [PubMed] [Google Scholar]
- 67.Hugo W, Zaretsky JM, Sun Lu, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016;165:35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chen S, Lee Li-F, Fisher TS, Jessen B, Elliott M, Evering W, et al. Combination of 4–1BB agonist and PD-1 antagonist promotes antitumor effector/memory CD8 T cells in a poorly immunogenic tumor model. Cancer Immunol Res 2015;3:149–60. [DOI] [PubMed] [Google Scholar]
- 69.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 1999;190:355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rose DR. Structure, mechanism and inhibition of Golgi alpha-mannosidase II. Curr Opin Struct Biol 2012;22:558–62. [DOI] [PubMed] [Google Scholar]
- 71.White SL, Schweitzer K, Humphries MJ, Olden K. Stimulation of DNA synthesis in murine lymphocytes by the drug swainsonine: immunomodulatory properties. Biochem Biophys Res Commun 1988;150:615–25. [DOI] [PubMed] [Google Scholar]
- 72.Humphries MJ, Matsumoto K, White SL, Molyneux RJ, Olden K. Augmentation of murine natural killer cell activity by swainsonine, a new antimetastatic immunomodulator. Cancer Res 1988;48:1410–5. [PubMed] [Google Scholar]
- 73.You N, Liu W, Wang T, Ji Ru, Wang X, Gong Z, et al. Swainsonine inhibits growth and potentiates the cytotoxic effect of paclitaxel in hepatocellular carcinoma in vitro and in vivo. Oncol Rep 2012;28:2091–100. [DOI] [PubMed] [Google Scholar]
- 74.Humphries MJ, Matsumoto K, White SL, Olden K. Oligosaccharide modification by swainsonine treatment inhibits pulmonary colonization by B16-F10 murine melanoma cells. Proc Natl Acad Sci U S A 1986;83:1752–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dennis JW. Effects of swainsonine and polyinosinic:polycytidylic acid on murine tumor cell growth and metastasis. Cancer Res 1986;46:5131–6. [PubMed] [Google Scholar]
- 76.Cornil I, Kerbel RS, Dennis JW. Tumor cell surface beta 1–4-linked galactose binds to lectin(s) on microvascular endothelial cells and contributes to organ colonization. J Cell Biol 1990;111:773–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goss PE, Baptiste J, Fernandes B, Baker M, Dennis JW. A phase I study of swainsonine in patients with advanced malignancies. Cancer Res 1994;54: 1450–7. [PubMed] [Google Scholar]
- 78.Goss PE, Reid CL, Bailey D, Dennis JW. Phase IB clinical trial of the oligosaccharide processing inhibitor swainsonine in patients with advanced malignancies. Clin Cancer Res 1997;3:1077–86. [PubMed] [Google Scholar]
- 79.Shaheen PE, Stadler W, Elson P, Knox J, Winquist E, Bukowski RM. Phase II study of the efficacy and safety of oral GD0039 in patients with locally advanced or metastatic renal cell carcinoma. Invest New Drugs 2005;23: 577–81. [DOI] [PubMed] [Google Scholar]
- 80.Almutairi AR, McBride A, Slack M, Erstad BL, Abraham I. Potential immune-related adverse events associated with monotherapy and combination therapy of ipilimumab, nivolumab, and pembrolizumab for advanced melanoma: a systematic review and meta-analysis. Front Oncol 2020;10:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang RF. A special issue on cancer immunotherapy. Cell Res 2017;27:1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Akama TO, Nakagawa H, Wong NK, Sutton-Smith M, Dell A, Morris HR, et al. Essential and mutually compensatory roles of {alpha}-mannosidase II and {alpha}-mannosidase IIx in N-glycan processing in vivo in mice. Proc Natl Acad Sci U S A 2006;103:8983–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Green RS, Stone EL, Tenno M, Lehtonen E, Farquhar MG, Marth JD. Mammalian N-glycan branching protects against innate immune self-recognition and inflammation in autoimmune disease pathogenesis. Immunity 2007;27:308–20. [DOI] [PubMed] [Google Scholar]
- 84.Lowe JB. Glycosylation, immunity, and autoimmunity. Cell 2001;104:809–12. [DOI] [PubMed] [Google Scholar]
- 85.Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature 2018;564:268–72. [DOI] [PubMed] [Google Scholar]
- 86.Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med 2018;24:978–85. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data presented in this article are available from the corresponding author upon reasonable request. RNA-seq data have been deposited at the GEO under accession number GSE152925.