

Abstract
Artemisinin-based combination therapies (ACTs), the World Health Organization-recommended first-line therapy for uncomplicated falciparum malaria, has led to significant decreases in malaria-associated morbidity and mortality in the past two decades. Decreased therapeutic efficacy of artemisinins, the cornerstone of ACTs, is threatening the gains made against this disease. As such, novel therapeutics with uncompromised mechanisms of action are needed to combat parasite-mediated antimalarial resistance. We have previously reported the antimalarial activity of Plasmodium falciparum-specific proteasome inhibitors in conjunction with a variety of antimalarials in clinical use or in preclinical investigations and of proteasome mutants generated in response to these inhibitors. Here, we discover that despite harboring K13C580Y, which has conventionally mediated artemisinin resistance in vitro as measured by increased survival in ring-stage survival assays (RSA), the Cam3.II strain parasites of Cambodian origin that have acquired an additional mutation in the proteasome display increased susceptibility to DHA and OZ439. This discovery implicates the proteasome in peroxide susceptibilities and has favorable implications on the use of peroxide and proteasome inhibitor combination therapy for the treatment of artemisinin-resistant malaria.
Keywords: Plasmodium falciparum, proteasome inhibitor, DHA, OZ439, artemisinin resistance
Graphical Abstract
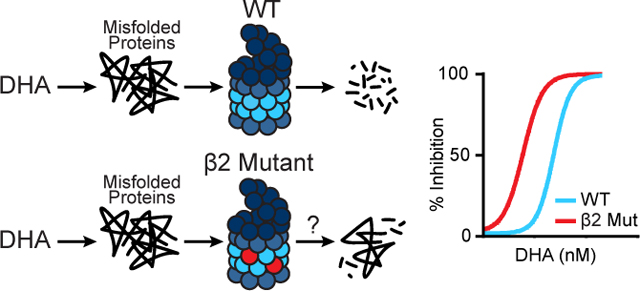
Dihydroartemisinin (DHA) and OZ439 cause an accumulation of misfolded proteins in P. falciparum. In order to survive these antimalarials, parasites rely on the proteasome to degrade damaged proteins (left, top). Cam3.II K13C580Y parasites with a mutation in the β2 proteasome subunit, indicated by red circles (left, bottom), display increased sensitivity to DHA and OZ439 as assayed by growth inhibition studies (right). Our data directly implicate the proteasome in peroxide susceptibilities. For simplicity, the graphic is shown with DHA as a representative of peroxides.
Malaria remains a significant global health concern, responsible for a recorded 229 million cases and 409,000 deaths in 2019 1. There are five species of the eukaryotic protozoan parasite Plasmodium that cause malaria in humans, of which P. falciparum is the deadliest. Artemisinin-based combination therapies (ACTs), comprised of a fast-acting artemisinin derivative and a long-lasting partner drug, are largely responsible for the substantial (44%) decline in malaria-related deaths over the past two decades 1. Alarmingly, resistance to artemisinins has developed in Cambodia, Thailand, Laos, Myanmar, and Vietnam, countries that comprise the Greater Mekong Subregion (GMS) 2–4. Outside of Southeast Asia, several cases of artemisinin resistance have been reported in Equatorial Guinea 5, Uganda 6, Rwanda 7, Senegal 8, India 9, and the Guiana Shield 10, 11. In vivo, artemisinin resistance is defined as parasite clearance half-life greater than 5 h following treatment with an artemisinin or an ACT 3 and can result in treatment failure if WHO-recommended ACT administration schedules of multiple dosing are not followed. Artemisinin resistance is strongly associated with point mutations in the propeller domain of kelch 13 (K13, PF3D7_1343700) 3, 12, and a handful of K13 mutations including M476I, Y493H, R539T, I543T, and C580Y have been experimentally confirmed to confer resistance 13, 14. The most prevalent clinical mutation in the GMS is K13 C580Y 15, 16. A predominant theory of K13-mediated artemisinin resistance is that parasite hemoglobin import and digestion is reduced in K13 propeller domain mutants (K13PDmut) 17–19, thus reducing the amount of free heme available to activate artemisinins.
Upon activation by heme, artemisinins nonspecifically alkylate adjacent proteins 20, 21, causing widespread protein damage. This suggests that parasites that survive artemisinin treatment possess increased capacity to restore proteostasis. In eukaryotes, the ubiquitin proteasome pathway serves as the primary mechanism to degrade and recycle misfolded and damaged proteins 22. Substrates tagged with K48- or K11-linked ubiquitin moieties are targeted for proteasome-mediated degradation 23. The 26S proteasome has two components: the 19S regulatory particle (RP) and the 20S core particle (CP). The RP is responsible for substrate recognition, deubiquitination, unfolding, and insertion into the CP 22. The CP, composed of two heptameric rings of β subunits sandwiched by heptameric rings of α subunits, carries out the catalytic function 22. β1 has caspase-like activity, cleaving after acidic residues; β2 has trypsin-like activity, cleaving after basic residues; and β5 has chymotrypsin-like activity, cleaving after non-polar residues 24. In P. falciparum, the CP has been visualized by cryoelectron microscopy 25, 26 and all components of the CP and RP have been biochemically identified as an interacting complex 27.
Chemical inhibitors developed against human proteasomes have antimalarial activity across the lifecycle of Plasmodium parasites in the liver stages, blood stages, and mosquito stages 28, 29. Specifically, lactacystin prevents intraerythrocytic growth as well as the establishment of liver stages 28. Epoxomicin and ALLN prevent intraerythrocytic development and maturation to stage V gametocytes, and incubation of these inhibitors with stage V gametocytes prior to mosquito feeding inhibited oocyst production within the mosquito midgut 29. Dihydroartemisinin (DHA), the active metabolite of all clinical artemisinins, synergizes with epoxomicin in vitro and with carfilzomib in a mouse model 30.
In the last five years, parasite-specific proteasome inhibitors of various chemotypes have been developed 25, 31–35. We have recently reported antimalarial activity of the covalent vinyl sulfone inhibitors WLW-vs (WLW), which inhibits the P. falciparum β2 subunit, and WLL-vs (WLL), which inhibits both the β2 and β5 subunits of the P. falciparum proteasome 25, 36. These inhibitors demonstrate favorable characteristics of an antimalarial candidate, including a high therapeutic index, high potency against artemisinin-resistant parasites with IC50 values in the low nanomolar range, a low propensity of parasite resistance acquisition, and potent synergy with the clinical drug DHA and OZ439, which is in clinical trials 25, 36. Although it was difficult to generate parasite resistance to these proteasome inhibitors, we nonetheless did manage to isolate parasites with mutations in the 26S proteasome that could survive WLL or WLW 36. 78% of WLW-selected parasites were found to have a mutation in the β2 subunit, while 50% of WLL-selected parasites were found to have a mutation in the β5 subunit 36. In this study, we investigate the peroxide susceptibility of parasites of Cambodian origin (Cam3.II strain) harboring mutations at the K13, β2, and β5 loci.
Results
A Mutation in the β2 Proteasome Subunit Sensitizes Cam3.II K13C580Y Parasites to DHA
Susceptibility to DHA was measured using the ring stage survival assay (RSA0–3h, also referred to as RSA) 37 and determination of ring-stage half-maximal inhibitory concentrations (ring IC50) using dose-response inhibition assays, as described in the literature 38. A 1% survival threshold in the RSA is used to designate artemisinin sensitivity vs resistance in vitro and correlates with fast-clearing and slow-clearing parasites, respectively 12, 14, 37. RSA and IC50 values measure related but distinct features of artemisinin susceptibility, and genome-wide association studies (GWAS) using IC50 values or RSA values identified different genes with strong associations to that particular measurement 39, indicating that there is a qualitative difference between these two measurements. To examine the susceptibility of proteasome mutants to DHA, RSAs were conducted on tightly synchronized 0–3hpi ring stage parasites isolated from the following parasites: Cam3.II K13WT, Cam3.II K13C580Y, Cam3.II K13C580Y β2 C31Y, Cam3.II K13C580Y β2 C31F, and Cam3.II K13C580Y β5 A20S (Figures 1, S1, and S2). Cam3.II K13WT and Cam3.II K13R539T strains were included as internal experimental controls for artemisinin susceptibility and resistance (Figure S1A–C) as they display known low and high RSA values, respectively. Cam3.II K13WT, Cam3.II K13R539T, and Cam3.II K13C580Y parasites displayed survival rates of 0.7%, 34.3%, and 11.4%, respectively (Figures S1 A, B), which is consistent with previously reported values 14, 38. Cam3.II K13C580Y β2 C31F and Cam3.II K13C580Y β5 A20S parasites displayed survival rates similar to the parental Cam3.II K13C580Y of 11.4% and 15.7%, respectively (Figure 1A). In contrast, Cam3.II K13C580Y β2 C31Y parasites displayed a 2-fold decrease in survival rate (6.1%) relative to its parent (Figure 1A), although this was not statistically significant (p = 0.056). In dose-response inhibition assays, the Cam3.II K13WT, Cam3.II K13R539T, and Cam3.II K13C580Y parasites had similar ring-stage IC50 values (Figure S1C), as previously reported 38. The β5 A20S mutant displayed similar IC50 values to its parent (15.2 nM vs. 19.5 nM) (Figure 1B). In contrast, both β2 C31Y and β2 C31F mutants were sensitized to DHA and had IC50 values of 7.3 nM (p = 0.016) and 10.4 nM (p = 0.19), respectively (Figure 1B). Note that DHA IC50 values in the β2 mutants, which harbor K13C580Y, are even lower than the 18.1 nM IC50 value for the artemisinin-sensitive Cam3.II K13WT parasite (Figure 1B). Dose-response curves are shown in Figure S2A.
Figure 1. Cam3.II K13C580Y β2 proteasome mutants display increased sensitivity to DHA.

(A and B) 0–3hpi ring stages and (C) 26–30hpi trophozoite stages isolated from Cam3.II K13WT, Cam3.II K13C580Y, Cam3.II K13C580Y β2 C31Y, Cam3.II K13C580Y β2 C31F, and Cam3.II K13C580Y β5 A20S parasites were exposed to a range of DHA concentrations for 3 h, the drug was washed out, and then parasitemia was assessed approximately 66 h later. (D) Asynchronous cultures were exposed to a range of DHA for 72 h. Bar graphs depict (A) quantification of percent survival at 700 nM DHA, and (B-D) mean IC50 values ± SEM. Values were calculated from at least three biological replicates. Statistical significance was examined between proteasome mutants and Cam3.II K13WT or Cam3.II K13C580Y parasites using a Mann-Whitney U test. * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001; n.s. = not significant.
Since the proteasome is upregulated during the trophozoite stages 40, 41, we repeated the experiments outlined above but used tightly synchronized 26–30hpi trophozoites (Figure 1C). Cam3.II K13WT, Cam3.II K13R539T, and Cam3.II K13C580Y parasites demonstrated similar IC50 values (Figures S1D, E), as expected. Cam3.II K13C580Y and Cam3.II K13C580Y β5 A20S parasites had similar DHA trophozoite IC50 values of 12.4 nM and 13.7 nM, respectively (Figure 1C). In contrast, the β2 C31Y and β2 C31F mutants had a statistically significant 2-fold decrease in DHA IC50 compared to the parental strain (C580Y: 12.4 nM vs. β2 C31Y: 5.2 nM and β2 C31F: 6.2 nM, p < 0.0001) (Figure 1C), mirroring the increase in sensitivity that we observed at the 0–3hpi ring stages. This shift in IC50 prompted us to investigate the sensitivity of these proteasome mutants in asynchronous cultures. Compared to Cam3.II K13WT, Cam3.II K13R539T, Cam3.II K13C580Y, and Cam3.II K13C580Y β5 A20S, which all displayed similar dose-response curves and IC50 values of approximately 5 nM (Figures S1F, G), the β2 C31Y and β2 C31F mutants displayed a significant (p = 0.002) 2-fold increased sensitivity to DHA with IC50 values of 2.5 nM and 2.6 nM, respectively (Figure 1D). These data show that mutations in the β2 proteasome catalytic subunit sensitizes parasites to DHA not only at ring stages, but throughout the asexual life cycle. As assessed by the RSA, β2 C31Y and β2 C31F parasites would be classified as artemisinin-resistant, since they displayed survival rates > 1%. However, relative to their Cam3.II K13C580Y parent, these proteasome mutants display 2–3 fold lower DHA IC50 values throughout their asexual lifecycles and that were below the IC50 values displayed by Cam3.II K13WT parasites, which are classified to be artemisinin-sensitive by RSA. This discrepancy highlights the subtle differences these two tests measure. The dose-response curves for trophozoite stage parasites and asynchronous parasites are shown in Figures S2B, C.
A Mutation in the β2 Proteasome Subunit Sensitizes Cam3.II K13C580Y Parasites to OZ439
Similar to DHA, OZ439 (artefenomel) contains a peroxide bond that reacts with heme, which produces carbon-centered radicals that mediate widespread protein damage 42. No cross-resistance between DHA and OZ439 has been observed in parasites harboring K13R539T or K13C580Y in the Cam3.I and Cam3.II strains 38, 43. Dose-response inhibition assays with OZ439 were conducted on 0–3hpi ring stages, 26–30hpi trophozoite stages, or with asynchronous parasites (Figures 2, S3, and S4). As expected, Cam3.II K13WT, Cam3.II K13R539T and Cam3.II K13C580Y parasites behaved similarly in response to OZ439 at all stages tested (Figure S3) and as previously shown 38. However, Cam3.II K13C580Y β2 C31Y and β2 C31F mutants were sensitized to OZ439 compared to parental strains when tested against rings, trophozoites, or asynchronous cultures (Figure 2A–C). 0–3hpi ring stages of the Cam3.II K13C580Y β2 mutants were 2–4-fold more susceptible to OZ439 than the parental Cam3.II K13C580Y parasites (C580Y: 62.7 nM vs β2 C31F: 14.8 nM (p = 0.008) and β2 C31Y: 27.1 nM (p = 0.032)) (Figure 2A). At the trophozoite stages, both the β2 C31Y and β2 C31F mutants displayed 2-fold increased sensitivity to OZ439 compared to the parental line (C580Y: 27.9 nM vs β2 C31Y: 10.8 nM and β2 C31F: 12.7 nM, p < 0.0001) (Figure 2B). This result was recapitulated when tested in asynchronous cultures (C580Y: 10.8 nM vs β2 C31Y: 4.8 nM and β2 C31F: 4.8 nM, p = 0.004) (Figure 2C). Cam3.II K13C580Y β5 A20S displayed similar OZ439 IC50 values compared to Cam3.II K13C580Y when assayed in ring, trophozoite, or asynchronous cultures (Figure 2). Dose-response curves for parasites assayed against OZ439 are depicted in Figure S4. DHA and OZ439 IC50 values determined in ring-stage, trophozoite-stage, and asynchronous parasites are summarized in Table S1. Note that K13 variants conferring DHA resistance do not display cross-resistance to OZ439 but the β2 C31Y and C31F mutations on a K13C580Y background confer sensitivity to both peroxides.
Figure 2. Cam3.II K13C580Y β2 proteasome mutants display increased susceptibility to OZ439.

(A) 0–3hpi ring stages and (B) 26–30hpi trophozoite stages isolated from Cam3.II K13WT, Cam3.II K13C580Y, Cam3.II K13C580Y β2 C31Y, Cam3.II K13C580Y β2 C31F, and Cam3.II K13C580Y β5 A20S parasites were exposed to a range of OZ439 concentrations for 3 h, the drug was washed out, and then parasitemia was assessed approximately 66 h later. (C) Asynchronous cultures were exposed to a range of OZ439 for 72 h. Parasitemia was assessed and mean IC50 values ± SEM were calculated from at least three biological replicates. Statistical significance was examined between proteasome mutants and Cam3.II K13WT or Cam3.II K13C580Y parasites using a Mann-Whitney U test. * p < 0.05; ** p < 0.01; **** p < 0.0001; n.s. = not significant.
Peroxides and Proteasome Inhibitors Strongly Synergize in Cam3.II K13C580Y β5 A20S Parasites
Previously, it was shown that WLW and WLL synergize with DHA and OZ439 in Cam3.II K13WT and Cam3.II K13C580Y parasites at ring, trophozoite, and asynchronous stages 36. To determine if the changes in susceptibility to DHA and OZ439 observed in the β2 proteasome mutants would alter the drug-drug interactions of peroxides and proteasome inhibitors, we conducted isobolograms on 26–30hpi trophozoites isolated from proteasome mutant parasites as well as their parental strain. Parasites were exposed to fixed ratios (1:0, 4:1, 2:1, 1:1, 1:2, 1:4, 0:1) of WLW or WLL in combination with DHA or OZ439. Fractional IC50 (FIC50) values were calculated from these data as described 36 and plotted on a Cartesian graph. Data points lying along the dotted line indicate additivity, data points below the dotted line forming a convex curve indicate synergy, and data points above the dotted line forming a concave curve indicate antagonism. As previously shown, there is synergy in Cam3.II K13C580Y for all tested drug combinations (WLL and DHA, WLL and OZ439, WLW and DHA, and WLW and OZ439) (Figure 3, blue dots) 36. Interestingly, the β5 A20S mutant displayed even greater synergy with WLL and peroxides compared to Cam3.II K13C580Y (Figure 3A, B panels 1 vs. 4, and Figure 4), despite demonstrating increased resistance to WLL 36, confirmed in (Figure S5A). The susceptibility of Cam3.II K13C580Y β2 C31F to WLL and WLW was previously unknown. Here, we determined that the β2 C31F mutant, like the previously tested β2 C31Y mutant, exhibited increased sensitivity to WLL (Figure S5A). Cam3.II K13C580Y β2 C31F was more resistant to WLW than its parent Cam3.II K13C580Y but not as resistant as Cam3.II K13C580Y β2 C31Y (Figure S5B). Both β2 mutants demonstrated additive effects when exposed to WLL and DHA, WLL and OZ439, or WLW and DHA (Figure 3A–C). However, WLW and OZ439 retained synergistic activities in the β2 mutants (Figure 3D). A global overview of these drug-drug interactions is shown in Figure 4. Together, these data demonstrate that in the unlikely event that parasite resistance arises to WLL or WLW, the most common mutations will not compromise susceptibility to DHA or drug-drug interactions. In some instances, proteasome subunit mutations will confer increased susceptibility to DHA and OZ439, or maintain synergistic interactions between peroxides and proteasome inhibitors.
Figure 3. Peroxides and proteasome inhibitors are strongly synergistic in Cam3.II K13C580Y β5 A20S parasites.

Isobolograms were conducted on 26–30hpi trophozoites isolated from Cam3.II K13C580Y, Cam3.II K13C580Y β2 C31Y, Cam3.II K13C580Y β2 C31F, and Cam3.II K13C580Y β5 A20S strains. Parasites were exposed to a range of (A) WLL and DHA, (B) WLL and OZ439, (C) WLW and DHA, or (D) WLW and OZ439 at fixed ratios (1:0, 4:1, 2:1, 1:1, 1:2, 1:4, 0:1) for 3 h, and then the drugs were washed out and parasitemia was assessed in the following replication cycle. Fractional IC50 (FIC50) values from 2 independent experiments (indicated by different shading) were calculated and plotted on a Cartesian graph. The diagonal dotted black line represents the isobole line which represents perfect additivity. Data points along this line indicate additivity and data points that fall below the isobole line along a convex curve indicate synergy. Cam3.II K13C580Y, Cam3.II K13C580Y β2 C31Y, Cam3.II K13C580Y β2 C31F, and Cam3.II K13C580Y β5 A20S are indicated in blue, pink, orange, and green, respectively.
Figure 4. Proteasome inhibitors and peroxides are synergistic or additive in Cam3.II K13C580Y β2 and β5 proteasome mutants.

The heatmap represents mean ∑FIC50 values derived from isobologram analyses performed on 26–30hpi trophozoite stages isolated from the indicated parasites. Values obtained range from 0.57, indicating synergy in blue, to values of 1.01, indicating additivity in white. The following drug combinations were tested: WLL and DHA, WLL and OZ439, WLW and DHA, and WLW and OZ439. Mean ∑FIC50 values close to 0 indicates synergy, values close to 1 indicates additivity, while values close to 2 indicate antagonism.
Discussion
The Plasmodium-specific vinyl sulfone proteasome inhibitors WLL and WLW potently inhibit both artemisinin-sensitive and artemisinin-resistant parasites and demonstrate strong synergy with DHA and OZ439 36. Thus, proteasome inhibitors are attractive partner drugs to overcome current problems with artemisinin resistance. In vitro, resistance to WLL and WLW was difficult to generate, with the majority of resistance-associated mutations observed in the β2 and β5 catalytic subunits 36. Here, we show that the most frequently recovered WLW-selected mutations, β2 C31Y and β2 C31F, sensitize Cam3.II K13C580Y parasites to DHA and OZ439 when exposed to these drugs at the early ring stages, mid-trophozoite stages, and in asynchronous cultures. In fact, these parasites displayed approximately 2-fold higher sensitivity to DHA and OZ439 than artemisinin-sensitive Cam3.II K13WT parasites. The singular change of K13WT to K13C580Y confers artemisinin resistance in the Cam3.II strain 14 but does not confer resistance to the related peroxide OZ439 38, 43. Yet we observe that an additional mutation at the β2 locus 36 sensitizes Cam3.II K13C580Y parasites to both DHA and OZ439 at early ring stages, trophozoite stages and in asynchronous parasites. This demonstrates an uncoupling between the contribution of K13 and proteasomes to differential peroxide susceptibility and underscores the developmental stage specificity of each peroxide. Our data also show that it is difficult to predict drug-drug interactions. For example, only the WLW and OZ439 drug combination displayed synergy in Cam3.II K13C580Y β2 mutants, which had increased susceptibility to both DHA and OZ439. In contrast, all tested combinations of peroxides and proteasome inhibitors demonstrated synergy in Cam3.II K13C580Y β5 A20S parasites, which have a 4.5-fold increased resistance to WLL, a 2-fold decreased resistance to WLW, and no difference in susceptibilities to DHA and OZ439 when compared to its parent Cam3.II K13C580Y. This demonstrates the unpredictability of drug-drug interactions and necessity for empirical determination of emergent combinatorial drug effects.
K13PDmut are believed to mediate resistance by decreasing hemoglobin uptake in early ring stages, thus reducing peroxide activation 17–19. In contrast, proteasomes are important throughout the parasite lifecycle 28, 29 and are upregulated during the mature stages 40, 41. Recently, proteasomes have been shown to be secreted in extracellular vesicles delivered to naive RBC, where this proteolytic complex degrades cytoskeleton proteins, thus modifying the host RBC to promote parasite invasion 44. While differential K13-mediated artemisinin drug responses are not observed outside of early ring stages, β2 proteasome mutants show a marked decrease in sensitivity to DHA and OZ439 compared to the parental Cam3.II K13C580Y throughout the asexual life cycle. There are a number of reasons that can explain these phenomena. After heme-mediated activation, DHA and OZ439 are known to nonspecifically alkylate heme and parasite proteins in the immediate vicinity 20, 21, 42, 45. Presumably, alkylation of parasite proteins impairs protein function, thus impairing parasite viability. These nonfunctional proteins are recognized by chaperones as misfolded or damaged proteins which will then be ubiquitinated and targeted for proteasome-mediated degradation. Whether K13 acts as an E3 ubiquitin ligase substrate adaptor in this cellular quality control process remains to be seen. DHA has been shown to inhibit proteolytic activity of the β5 subunit, although only at about 50% of the β5-targeting proteasome inhibitor epoxomicin 46. Whether DHA inhibits the other catalytic subunits is unknown. Our data implicate the importance of the proteasome in artemisinin and OZ439 response. Whether the β2 C31Y and C31F mutations directly impair proteolytic activity or if these mutations allosterically regulate proteasome activity, and if these changes in activity impact proteasome-mediated RBC remodelling remains to be explored. Although the proteasome possesses three catalytic subunits, most of the proteolytic activity is attributed to the β5 subunit. One would surmise that it would be difficult to obtain a viable parasite with a mutation in the main catalytic subunit that compromised its ability to degrade proteins, given the necessity of the proteasome throughout the parasite lifecycle 28, 29, 44. Thus, it is perhaps unsurprising that the β5 mutant obtained in our in vitro drug selections does not sensitize Cam3.II K13C580Y to peroxides, if this sensitization is due to loss of proteolytic activity.
The data presented here provide compelling evidence toward the future of proteasome inhibitor-peroxide combinations to combat artemisinin resistance. Should these combination therapies advance to clinical use, we will likely not see resistant parasites arise because: (1) WLL and WLW are equally potent on artemisinin-sensitive and artemisinin-resistant parasites, (2) it was difficult to generate resistance against WLL and WLW in vitro, (3) the majority of the mutations which mediate resistance to WLW (β2 mutants) will be selected against due to their increased sensitivity to peroxides, (4) the WLL-selected β5 mutation did not compromise DHA and OZ439 potency, and (5) peroxides and proteasome inhibitors were highly synergistic in the β5 mutant. Mutations in other proteasome subunits and on different parasite backgrounds have yet to be investigated, and it remains to be seen if these results will be recapitulated in vivo.
Methods
Parasite Culturing
Cam3.II K13WT, Cam3.II K13R539T (RF967), and Cam3.II K13C580Y are laboratory-adapted Cambodian isolates that were genetically engineered at the K13 locus as described in the literature 14. Cam3.II K13C580Y parasites expressing β2 C31Y, β2 C31F, or β5 A20S were obtained from in vitro selection studies with WLL and WLW as described in the literature 36. Parasite cultures were propagated in O+ red blood cells (RBCs) (anonymous donors, purchased from Interstate Blood Bank, Memphis, Tennessee). RBCs were stored at 50% hematocrit in ADSOL (2 mM adenine (Alfa Aesar, Haverhill, Massachusetts), 111 mM dextrose (Fisher BioReagents, Pittsburgh, Pennsylvania), 41.2 mM mannitol (Acros Organics, Fair Lawn, New Jersey), and 154 mM sodium chloride (Fisher BioReagents) for prolonged RBC vitality as described in ref 47 at 4oC. Parasites were cultured in complete media (RPMI 1640 media supplemented with 0.01 mg/mL gentamicin (Gibco, Dun Laoghaire, Co Dublin, Ireland), 50 mg/mL hypoxanthine (Acros Organics), and 0.5% Albumax II (Invitrogen, Carlsbad, California)) at 5% hematocrit, and were maintained at 37oC in a Heracell™ VIOS 160i CO2 Incubator (Thermo Fisher Scientific, Waltham, Massachusetts) under hypoxic conditions (5% O2, 5% CO2, 90% N2). Gas was purchased from Matheson Gas (Irving, Texas).
Stage Synchronization
0–3hpi ring stages were obtained as previously described 37 with minor modifications. Briefly, cultures were treated with 5% sorbitol (Acros Organics) at 37 oC for 15 min and then cultured for 33 h. Schizont-enriched cultures were resuspended in RPMI 1640 supplemented with 14.3 U/mL sodium heparin (Merck, Darmstadt, Germany) and incubated at 37 oC for 30 minutes with intermittent vortexing. Schizont stages were purified using a 75% Percoll (GE Healthcare) density gradient followed by centrifuging at 2200g for 15 min. Purified schizont stages were washed once with RPMI 1640 supplemented with 14.3 U/mL sodium heparin, then propagated in fresh RBCs and complete media at 2% hematocrit. Parasite cultures were incubated for 3 h, and any remaining late-stage parasites were removed with 5% sorbitol, leaving 0–3hpi rings. Trophozoite stage parasites (26–30hpi) were obtained by exposing cultures to two consecutive treatments of 5% sorbitol, 12 h apart. Parasites were then cultured for an additional 12 h. Stage isolation was verified using Giemsa-stained thin blood smears and imaged via light microscopy using a 100× oil immersion objective.
Growth Inhibition and Survival Assays
Parasites were synchronized as described above, and then seeded in 96-well plates (Thermo Fisher Scientific) at 1% hematocrit and 0.7% parasitemia for ring stage survival assays (RSAs) and ring-stage growth inhibition assays, or 0.2% parasitemia for trophozoite stage and asynchronous growth inhibition assays in a total of 200 μL per well. Custom drug concentrations were dispensed with a MANTIS® microfluidic liquid handler (FORMULATRIX®, Bedford, Massachusetts) or were dispensed by hand in 2-fold serial dilutions. Ring and trophozoite stage parasites were treated for 3 h in U-bottom plates, then drug was washed out by centrifuging 96-well plates at 1500 rpm for 1 min, removing media, and resuspending in 190 μL of complete media, which removed >95% of medium per wash 38, 48. Following four washes, cultures were transferred to a new flat-bottom 96-well plate (Thermo Fisher Scientific) 38, 48 and cultured for an additional 66 h. Asynchronous parasites were treated for 72 h in flat-bottom plates.
Parasites were measured by staining parasite nuclei with 2 μg/mL Hoechst 33342 (Thermo Fisher Scientific) and respirating mitochondria with 100 nM MitoTracker Deep Red FM (Thermo Fisher Scientific). CellCarrier-96 Ultra Microplates (PerkinElmer, Waltham, MA) were coated with 0.1 mg/mL poly-L-lysine (MP Biomedicals, Irvine, CA). 40 μL of stain was added per well. Then, 10 μL of parasite culture at 1% hematocrit, approximately 10 million RBC, was added per well. Cells were allowed to settle for 1 h at 37 oC under normal culturing conditions as described above. Plates were washed twice with 1 × PBS, taking care to not disturb RBCs. Cells were fixed by adding 50 μL of 2% paraformaldehyde (Alfa Aesar) and 2% glutaraldehyde (Fisher Scientific) to each well and incubating at room temperature for 30 min. Following fixation, the fixative was removed and 150 μL 1 × PBS was added to each well. Plates were imaged with an Operetta CLS High-Content Imaging System (PerkinElmer) using the following settings: 20× air objective, nonconfocal, binning 2. Channels were captured in the following order: Hoechst (excitation/emission (ex/em) 355–385/430–500), Mitotracker Deep Red (ex/em 615–645/655–760), and brightfield. For each channel, focus height was determined, and exposure time and percent power were adjusted so that the intensity for all channels was approximately 4000–5000 counts/pixel. At least 10,000 cells were imaged per sample. Images were analyzed using Harmony 4.9 software with PhenoLOGIC. GraphPad Prism software version 9 was used to calculate IC50 values by nonlinear regression analysis. Survival in RSAs were calculated by dividing the parasitemia of cultures treated at 700nM by the parasitemia of DMSO-treated cultures. Statistical analyses were performed using Mann-Whitney U tests. At least three biological replicates were performed for each assay.
Isobolograms
Trophozoite stages were isolated as described above and then seeded in round-bottom 96-well plates at 1% hematocrit and 0.2% parasitemia. Starting concentrations of drug were prepared in 1:0, 4:1, 2:1, 1:1, 1:2, 1:4 ratios and 2-fold serial dilutions were dispensed by hand or using a MANTIS microfluidic liquid handler. Drug incubation, washout, and analysis were performed as described above. FIC50 values were calculated by dividing the IC50 value obtained when exposed to both compounds by the IC50 obtained when exposed to one compound alone.
FIC50 values are plotted on a Cartesian graph in Figure 3. Mean ∑FIC50 was calculated by averaging the sums of FIC50 values at each combination and plotted as a heat map in Figure 4. Isobolograms and heat maps were plotted using GraphPad Prism 9 software.
Drug Compounds
DHA was purchased from Sigma-Aldrich (St. Louis, MO). OZ439 was kindly provided by Dr. J. L. Vennerstrom (University of Nebraska Medical Center). WLL and WLW were kindly provided by Dr. M. Bogyo (Stanford School of Medicine). All compounds were solubilized by dissolving in DMSO (Fisher Scientific), and maximum DMSO concentrations in all assays were less than 0.2%.
Supplementary Material
Figure S1. DHA dose-response inhibition assays conducted on K13 isogenic Cam3.II parasites.
Figure S2. Dose-response curves show increased susceptibility of Cam3.II K13C580Y β2 proteasome mutants to DHA.
Figure S3. OZ439 dose-response inhibition assays conducted on K13 isogenic Cam3.II parasites.
Figure S4. Dose-response curves show increased susceptibility of Cam3.II K13C580Y β2 proteasome mutants to OZ439.
Figure S5. Susceptibility of Cam3.II K13C580Y parent and the β2 and β5 proteasome mutants to WLL and WLW.
Table S1. Summary of DHA and OZ439 IC50 values.
Acknowledgments
Funding for this work was provided by NIH R21 AI137900 to C.L.N. as well as a pilot grant to C.L.N. under NIH P20 GM121316 (overall grant awarded to R.E.L). C.L.N. also gratefully acknowledges her UNMC Start-up Funds and a UNMC Diversity Fund Grant. We would like to thank Dr. S.P. Reid for use of the Operetta CLS High-Content Imaging System and Dr. R.E. Lewis for use of the MANTIS microfluidic liquid handler.
Abbreviations
- DHA
dihydroartemisinin
- FIC50
Fractional IC50
- hpi
hours post invasion
- RSA
ring-stage survival assay
Footnotes
Conflict of Interest
The authors declare no competing financial interest.
Supporting Information
Supporting information is available free of charge on the ACS publication website.
References
- 1.WHO, World Malaria Report. 2020.
- 2.Imwong M; Dhorda M; Myo Tun K; Thu AM; Phyo AP; Proux S; Suwannasin K; Kunasol C; Srisutham S; Duanguppama J; Vongpromek R; Promnarate C; Saejeng A; Khantikul N; Sugaram R; Thanapongpichat S; Sawangjaroen N; Sutawong K; Han KT; Htut Y; Linn K; Win AA; Hlaing TM; van der Pluijm RW; Mayxay M; Pongvongsa T; Phommasone K; Tripura R; Peto TJ; von Seidlein L; Nguon C; Lek D; Chan XHS; Rekol H; Leang R; Huch C; Kwiatkowski DP; Miotto O; Ashley EA; Kyaw MP; Pukrittayakamee S; Day NPJ; Dondorp AM; Smithuis FM; Nosten FH; White NJ, Molecular epidemiology of resistance to antimalarial drugs in the Greater Mekong subregion: an observational study. Lancet Infect Dis 2020, 20, 1470–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashley EA; Dhorda M; Fairhurst RM; Amaratunga C; Lim P; Suon S; Sreng S; Anderson JM; Mao S; Sam B; Sopha C; Chuor CM; Nguon C; Sovannaroth S; Pukrittayakamee S; Jittamala P; Chotivanich K; Chutasmit K; Suchatsoonthorn C; Runcharoen R; Hien TT; Thuy-Nhien NT; Thanh NV; Phu NH; Htut Y; Han KT; Aye KH; Mokuolu OA; Olaosebikan RR; Folaranmi OO; Mayxay M; Khanthavong M; Hongvanthong B; Newton PN; Onyamboko MA; Fanello CI; Tshefu AK; Mishra N; Valecha N; Phyo AP; Nosten F; Yi P; Tripura R; Borrmann S; Bashraheil M; Peshu J; Faiz MA; Ghose A; Hossain MA; Samad R; Rahman MR; Hasan MM; Islam A; Miotto O; Amato R; MacInnis B; Stalker J; Kwiatkowski DP; Bozdech Z; Jeeyapant A; Cheah PY; Sakulthaew T; Chalk J; Intharabut B; Silamut K; Lee SJ; Vihokhern B; Kunasol C; Imwong M; Tarning J; Taylor WJ; Yeung S; Woodrow CJ; Flegg JA; Das D; Smith J; Venkatesan M; Plowe CV; Stepniewska K; Guerin PJ; Dondorp AM; Day NP; White NJ; Tracking Resistance to Artemisinin, C., Spread of artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 2014, 371, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thanh NV; Thuy-Nhien N; Tuyen NT; Tong NT; Nha-Ca NT; Dong LT; Quang HH; Farrar J; Thwaites G; White NJ; Wolbers M; Hien TT, Rapid decline in the susceptibility of Plasmodium falciparum to dihydroartemisinin-piperaquine in the south of Vietnam. Malar J 2017, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu F; Culleton R; Zhang M; Ramaprasad A; von Seidlein L; Zhou H; Zhu G; Tang J; Liu Y; Wang W; Cao Y; Xu S; Gu Y; Li J; Zhang C; Gao Q; Menard D; Pain A; Yang H; Zhang Q; Cao J, Emergence of indigenous artemisinin-resistant Plasmodium falciparum in Africa. N Engl J Med 2017, 376, 991–993. [DOI] [PubMed] [Google Scholar]
- 6.Hawkes M; Conroy AL; Opoka RO; Namasopo S; Zhong K; Liles WC; John CC; Kain KC, Slow clearance of Plasmodium falciparum in severe pediatric malaria, Uganda, 2011–2013. Emerging infectious diseases 2015, 21, 1237–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uwimana A; Legrand E; Stokes BH; Ndikumana JM; Warsame M; Umulisa N; Ngamije D; Munyaneza T; Mazarati JB; Munguti K; Campagne P; Criscuolo A; Ariey F; Murindahabi M; Ringwald P; Fidock DA; Mbituyumuremyi A; Menard D, Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat Med 2020, 26, 1602–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Madamet M; Kounta MB; Wade KA; Lo G; Diawara S; Fall M; Bercion R; Nakoulima A; Fall KB; Benoit N; Gueye MW; Fall B; Diatta B; Pradines B, Absence of association between polymorphisms in the K13 gene and the presence of Plasmodium falciparum parasites at day 3 after treatment with artemisinin derivatives in Senegal. Int J Antimicrob Agents 2017, 49, 754–756. [DOI] [PubMed] [Google Scholar]
- 9.Das S; Saha B; Hati AK; Roy S, Evidence of artemisinin-resistant Plasmodium falciparum malaria in eastern India. N Engl J Med 2018, 379, 1962–1964. [DOI] [PubMed] [Google Scholar]
- 10.Vreden SG; Jitan JK; Bansie RD; Adhin MR, Evidence of an increased incidence of day 3 parasitaemia in Suriname: an indicator of the emerging resistance of Plasmodium falciparum to artemether. Mem Inst Oswaldo Cruz 2013, 108, 968–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Douine M; Lazrek Y; Blanchet D; Pelleau S; Chanlin R; Corlin F; Hureau L; Volney B; Hiwat H; Vreden S; Djossou F; Demar M; Nacher M; Musset L, Predictors of antimalarial self-medication in illegal gold miners in French Guiana: a pathway towards artemisinin resistance. J Antimicrob Chemother 2018, 73, 231–239. [DOI] [PubMed] [Google Scholar]
- 12.Ariey F; Witkowski B; Amaratunga C; Beghain J; Langlois AC; Khim N; Kim S; Duru V; Bouchier C; Ma L; Lim P; Leang R; Duong S; Sreng S; Suon S; Chuor CM; Bout DM; Menard S; Rogers WO; Genton B; Fandeur T; Miotto O; Ringwald P; Le Bras J; Berry A; Barale JC; Fairhurst RM; Benoit-Vical F; Mercereau-Puijalon O; Menard D, A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 2014, 505, 50–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghorbal M; Gorman M; Macpherson CR; Martins RM; Scherf A; Lopez-Rubio JJ, Genome editing in the human malaria parasite Plasmodium falciparum using the CRISPR-Cas9 system. Nat Biotechnol 2014, 32, 819–821. [DOI] [PubMed] [Google Scholar]
- 14.Straimer J; Gnadig NF; Witkowski B; Amaratunga C; Duru V; Ramadani AP; Dacheux M; Khim N; Zhang L; Lam S; Gregory PD; Urnov FD; Mercereau-Puijalon O; Benoit-Vical F; Fairhurst RM; Menard D; Fidock DA, K13-propeller mutations confer artemisinin resistance in Plasmodium falciparum clinical isolates. Science 2015, 347, 428–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson TJ; Nair S; McDew-White M; Cheeseman IH; Nkhoma S; Bilgic F; McGready R; Ashley E; Pyae Phyo A; White NJ; Nosten F, Population parameters underlying an ongoing soft sweep in Southeast Asian malaria parasites. Mol Biol Evol 2017, 34, 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zaw MT; Lin Z; Emran NA, Importance of kelch 13 C580Y mutation in the studies of artemisinin resistance in Plasmodium falciparum in Greater Mekong Subregion. J Microbiol Immunol Infect 2020, 53, 676–681. [DOI] [PubMed] [Google Scholar]
- 17.Siddiqui G; Srivastava A; Russell AS; Creek DJ, Multi-omics based identification of specific biochemical changes associated with PfKelch13-mutant artemisinin-resistant Plasmodium falciparum. J Infect Dis 2017, 215, 1435–1444. [DOI] [PubMed] [Google Scholar]
- 18.Yang T; Yeoh LM; Tutor MV; Dixon MW; McMillan PJ; Xie SC; Bridgford JL; Gillett DL; Duffy MF; Ralph SA; McConville MJ; Tilley L; Cobbold SA, Decreased K13 abundance reduces hemoglobin catabolism and proteotoxic stress, underpinning artemisinin resistance. Cell Rep 2019, 29, 2917–2928 e2915. [DOI] [PubMed] [Google Scholar]
- 19.Birnbaum J; Scharf S; Schmidt S; Jonscher E; Hoeijmakers WAM; Flemming S; Toenhake CG; Schmitt M; Sabitzki R; Bergmann B; Frohlke U; Mesen-Ramirez P; Blancke Soares A; Herrmann H; Bartfai R; Spielmann T, A Kelch13-defined endocytosis pathway mediates artemisinin resistance in malaria parasites. Science 2020, 367, 51–59. [DOI] [PubMed] [Google Scholar]
- 20.Wang J; Zhang CJ; Chia WN; Loh CC; Li Z; Lee YM; He Y; Yuan LX; Lim TK; Liu M; Liew CX; Lee YQ; Zhang J; Lu N; Lim CT; Hua ZC; Liu B; Shen HM; Tan KS; Lin Q, Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat Commun 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ismail HM; Barton V; Phanchana M; Charoensutthivarakul S; Wong MH; Hemingway J; Biagini GA; O’Neill PM; Ward SA, Artemisinin activity-based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proceedings of the National Academy of Sciences of the United States of America 2016, 113, 2080–2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Collins GA; Goldberg AL, The logic of the 26S proteasome. Cell 2017, 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yau R; Rape M, The increasing complexity of the ubiquitin code. Nature cell biology 2016, 18, 579–586. [DOI] [PubMed] [Google Scholar]
- 24.Kisselev AF; van der Linden WA; Overkleeft HS, Proteasome inhibitors: an expanding army attacking a unique target. Chem Biol 2012, 19, 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li H; O’Donoghue AJ; van der Linden WA; Xie SC; Yoo E; Foe IT; Tilley L; Craik CS; da Fonseca PC; Bogyo M, Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 2016, 530, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xie SC; Metcalfe RD; Hanssen E; Yang T; Gillett DL; Leis AP; Morton CJ; Kuiper MJ; Parker MW; Spillman NJ; Wong W; Tsu C; Dick LR; Griffin MDW; Tilley L, The structure of the PA28–20S proteasome complex from Plasmodium falciparum and implications for proteostasis. Nat Microbiol 2019, 4, 1990–2000. [DOI] [PubMed] [Google Scholar]
- 27.Wang L; Delahunty C; Fritz-Wolf K; Rahlfs S; Helena Prieto J; Yates JR; Becker K, Characterization of the 26S proteasome network in Plasmodium falciparum. Sci Rep 2015, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gantt SM; Myung JM; Briones MR; Li WD; Corey EJ; Omura S; Nussenzweig V; Sinnis P, Proteasome inhibitors block development of Plasmodium spp. Antimicrob Agents Chemother 1998, 42, 2731–2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Czesny B; Goshu S; Cook JL; Williamson KC, The proteasome inhibitor epoxomicin has potent Plasmodium falciparum gametocytocidal activity. Antimicrob Agents Chemother 2009, 53, 4080–4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dogovski C; Xie SC; Burgio G; Bridgford J; Mok S; McCaw JM; Chotivanich K; Kenny S; Gnadig N; Straimer J; Bozdech Z; Fidock DA; Simpson JA; Dondorp AM; Foote S; Klonis N; Tilley L, Targeting the cell stress response of Plasmodium falciparum to overcome artemisinin resistance. PLoS biology 2015, 13, e1002132–e1002157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaMonte GM; Almaliti J; Bibo-Verdugo B; Keller L; Zou BY; Yang J; Antonova-Koch Y; Orjuela-Sanchez P; Boyle CA; Vigil E; Wang L; Goldgof GM; Gerwick L; O’Donoghue AJ; Winzeler EA; Gerwick WH; Ottilie S, Development of a potent inhibitor of the Plasmodium proteasome with reduced mammalian toxicity. J Med Chem 2017, 60, 6721–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yoo E; Stokes BH; de Jong H; Vanaerschot M; Kumar T; Lawrence N; Njoroge M; Garcia A; Van der Westhuyzen R; Momper JD; Ng CL; Fidock DA; Bogyo M, Defining the determinants of specificity of Plasmodium proteasome inhibitors. J Am Chem Soc 2018, 140, 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kirkman LA; Zhan W; Visone J; Dziedziech A; Singh PK; Fan H; Tong X; Bruzual I; Hara R; Kawasaki M; Imaeda T; Okamoto R; Sato K; Michino M; Alvaro EF; Guiang LF; Sanz L; Mota DJ; Govindasamy K; Wang R; Ling Y; Tumwebaze PK; Sukenick G; Shi L; Vendome J; Bhanot P; Rosenthal PJ; Aso K; Foley MA; Cooper RA; Kafsack B; Doggett JS; Nathan CF; Lin G, Antimalarial proteasome inhibitor reveals collateral sensitivity from intersubunit interactions and fitness cost of resistance. Proceedings of the National Academy of Sciences of the United States of America 2018, 115, e6863–e6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xie SC; Gillett DL; Spillman NJ; Tsu C; Luth MR; Ottilie S; Duffy S; Gould AE; Hales P; Seager BA; Charron CL; Bruzzese F; Yang X; Zhao X; Huang SC; Hutton CA; Burrows JN; Winzeler EA; Avery VM; Dick LR; Tilley L, Target validation and identification of novel boronate inhibitors of the Plasmodium falciparum proteasome. J Med Chem 2018, 61, 10053–10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhan W; Visone J; Ouellette T; Harris JC; Wang R; Zhang H; Singh PK; Ginn J; Sukenick G; Wong TT; Okoro JI; Scales RM; Tumwebaze PK; Rosenthal PJ; Kafsack BFC; Cooper RA; Meinke PT; Kirkman LA; Lin G, Improvement of asparagine ethylenediamines as anti-malarial Plasmodium-selective proteasome inhibitors. J Med Chem 2019, 62, 6137–6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stokes BH; Yoo E; Murithi JM; Luth MR; Afanasyev P; da Fonseca PCA; Winzeler EA; Ng CL; Bogyo M; Fidock DA, Covalent Plasmodium falciparum-selective proteasome inhibitors exhibit a low propensity for generating resistance in vitro and synergize with multiple antimalarial agents. PLoS Pathog 2019, 15, e1007722–e1107750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Witkowski B; Amaratunga C; Khim N; Sreng S; Chim P; Kim S; Lim P; Mao S; Sopha C; Sam B; Anderson JM; Duong S; Chuor CM; Taylor WR; Suon S; Mercereau-Puijalon O; Fairhurst RM; Menard D, Novel phenotypic assays for the detection of artemisinin-resistant Plasmodium falciparum malaria in Cambodia: in-vitro and ex-vivo drug-response studies. Lancet Infect Dis 2013, 13, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Straimer J; Gnadig NF; Stokes BH; Ehrenberger M; Crane AA; Fidock DA, Plasmodium falciparum K13 mutations differentially impact ozonide susceptibility and parasite fitness in vitro. MBio 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z; Cabrera M; Yang J; Yuan L; Gupta B; Liang X; Kemirembe K; Shrestha S; Brashear A; Li X; Porcella SF; Miao J; Yang Z; Su XZ; Cui L, Genome-wide association analysis identifies genetic loci associated with resistance to multiple antimalarials in Plasmodium falciparum from China-Myanmar border. Sci Rep 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ponts N; Saraf A; Chung DW; Harris A; Prudhomme J; Washburn MP; Florens L; Le Roch KG, Unraveling the ubiquitome of the human malaria parasite. J Biol Chem 2011, 286, 40320–40330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bozdech Z; Llinas M; Pulliam BL; Wong ED; Zhu J; DeRisi JL, The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS biology 2003, 1, 85–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ismail HM; Barton VE; Panchana M; Charoensutthivarakul S; Biagini GA; Ward SA; O’Neill PM, A click chemistry-based proteomic approach reveals that 1,2,4-trioxolane and artemisinin antimalarials share a common protein alkylation profile. Angewandte Chemie 2016, 55, 6401–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walz A; Leroy D; Andenmatten N; Maser P; Wittlin S, Anti-malarial ozonides OZ439 and OZ609 tested at clinically relevant compound exposure parameters in a novel ring-stage survival assay. Malar J 2019, 18, 427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dekel E; Yaffe D; Rosenhek-Goldian I; Ben-Nissan G; Ofir-Birin Y; Morandi MI; Ziv T; Sisquella X; Pimentel MA; Nebl T; Kapp E; Ohana Daniel Y; Karam PA; Alfandari D; Rotkopf R; Malihi S; Temin TB; Mullick D; Revach OY; Rudik A; Gov NS; Azuri I; Porat Z; Bergamaschi G; Sorkin R; Wuite GJL; Avinoam O; Carvalho TG; Cohen SR; Sharon M; Regev-Rudzki N, 20S proteasomes secreted by the malaria parasite promote its growth. Nat Commun 2021, 12, 1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jourdan J; Matile H; Reift E; Biehlmaier O; Dong Y; Wang X; Maser P; Vennerstrom JL; Wittlin S, Monoclonal antibodies that recognize the alkylation signature of antimalarial ozonides OZ277 (arterolane) and OZ439 (artefenomel). ACS Infect Dis 2016, 2, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bridgford JL; Xie SC; Cobbold SA; Pasaje CFA; Herrmann S; Yang T; Gillett DL; Dick LR; Ralph SA; Dogovski C; Spillman NJ; Tilley L, Artemisinin kills malaria parasites by damaging proteins and inhibiting the proteasome. Nat Commun 2018, 9, 3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heaton A; Miripol J; Aster R; Hartman P; Dehart D; Rzad L; Grapka B; Davisson W; Buchholz DH, Use of Adsol preservation solution for prolonged storage of low viscosity AS-1 red blood cells. Br J Haematol 1984, 57, 467–478. [DOI] [PubMed] [Google Scholar]
- 48.Yang T; Xie SC; Cao P; Giannangelo C; McCaw J; Creek DJ; Charman SA; Klonis N; Tilley L, Comparison of the exposure time dependence of the activities of synthetic ozonide antimalarials and dihydroartemisinin against K13 wild-type and mutant Plasmodium falciparum strains. Antimicrob Agents Chemother 2016, 60, 4501–4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. DHA dose-response inhibition assays conducted on K13 isogenic Cam3.II parasites.
Figure S2. Dose-response curves show increased susceptibility of Cam3.II K13C580Y β2 proteasome mutants to DHA.
Figure S3. OZ439 dose-response inhibition assays conducted on K13 isogenic Cam3.II parasites.
Figure S4. Dose-response curves show increased susceptibility of Cam3.II K13C580Y β2 proteasome mutants to OZ439.
Figure S5. Susceptibility of Cam3.II K13C580Y parent and the β2 and β5 proteasome mutants to WLL and WLW.
Table S1. Summary of DHA and OZ439 IC50 values.