

Abstract
Background & Aims:
Despite surgical and chemotherapeutic advances, the five-year survival rate for Stage IV Hepatoblastoma (HB), the predominant pediatric liver tumor, remains at 27%. YAP1 and β-Catenin co-activation occurs in 80% of children’s HB; however, a lack of conditional genetic models precludes tumor maintenance exploration. Thus, the need for a targeted therapy remains unmet. Given the predominance of YAP1 and β-Catenin activation in HB, we sought to evaluate YAP1 as a therapeutic target in HB.
Approach & Results:
We engineered the first conditional HB murine model using hydrodynamic injection to deliver transposon plasmids encoding inducible YAP1S127A, constitutive β-CateninDelN90, and a luciferase reporter to murine liver. Tumor regression was evaluated using bioluminescent imaging, and tumor landscape characterized using RNA and ATAC sequencing, and DNA foot-printing. Here we show that YAP1S127A withdrawal mediates >90% tumor regression with survival for 230+ days in mice. YAP1 S127A withdrawal promotes apoptosis in a subset of tumor cells and in remaining cells induces a cell fate switch driving therapeutic differentiation of HB tumors into Ki-67 negative “hbHep cells” with hepatocyte-like morphology and mature hepatocyte gene expression. YAP1 S127A withdrawal drives formation of hbHeps by modulating liver differentiation transcription factor (TF) occupancy. Indeed, tumor-derived hbHeps, consistent with their reprogrammed transcriptional landscape, regain partial hepatocyte function and rescue liver damage in mice.
Conclusions:
YAP1S127A withdrawal, without silencing oncogenic β-Catenin, significantly regresses hepatoblastoma, providing the first in vivo data to support YAP1 as a therapeutic target for HB. YAP1S127A withdrawal alone sufficiently drives long-term regression in hepatoblastoma because it promotes cell death in a subset of tumor cells and modulates transcription factor occupancy to reverse the fate of residual tumor cells to mimic functional hepatocytes.
Keywords: Therapeutic Differentiation, Targeted Therapy, Pediatric Cancer, Oncogene, Liver cancer
Introduction
Hepatoblastoma (HB) is the predominant primary pediatric liver cancer and most often diagnosed in children under five years of age. Advances in surgical resection and chemotherapy regimens have saved many young lives; however, the five-year survival rate remains at ~75% overall and ~27% for Stage IV tumors (1–3). To date, the clinical need for a HB-targeted therapeutic remains unmet.
Yes-Associated Protein 1 (YAP1) and β-Catenin are co-activated in nearly 80% of children’s HB tumors (4). In mice, hepatoblastoma initiation requires oncogenic cooperation between β-Catenin and YAP1 (4, 5). However, it remains unclear if YAP1 is essential for HB tumor maintenance and if withdrawing oncogenic YAP1expression would be clinically beneficial for pediatric patients. To our knowledge, there are limited in vivo models to study tumor maintenance in childhood cancers, and to date, no conditional mouse models for HB to facilitate genetic evaluation of oncogenic YAP1 withdrawal.
Understanding the fate of liver cancer cells after oncogene inhibition is critical when evaluating targeted therapies. Previously, oncogene inhibition in cancer cells has been shown to induce markers of differentiation; however, these cells are thought to be dormant tumor cells, non-functional and prone to relapse (6–11). The reversibility of liver cancer cells remains incompletely understood, specifically, whether oncogene inactivation can therapeutically differentiate liver cancer cells into functional somatic cells that contribute to normal tissue architecture and participate in tissue homeostasis.
In this study, we evaluate the requirement of oncogenic YAP1 for hepatoblastoma tumor maintenance. We find that in established hepatoblastoma tumors withdrawal of YAP1 overexpression without modulation of oncogenic β-Catenin activates a cell fate switch promoting a subset of cells to undergo apoptosis leading to regression, and the remaining tumor cells to re-differentiate towards hepatocyte-like cells. We termed these reprogrammed cells, hepatoblastoma-derived hepatocyte-like cells, “hbHeps.” Consistent with their hepatocyte-like transcriptional profiles, hbHeps participate in tissue homeostasis and rescue liver cell damage.
Methods
Experimental Procedures Data Accession
RNA sequencing and ATAC sequencing data herein are available for download at NCBI GEO GSE146548, or by reasonable request from the authors.
ATAC Sequencing Library Preparation and Analysis
Library Preparation
ATAC-seq was performed according to Corces et al. (2017; Supplementary Protocol 2) (12). Briefly, mCherry-positive hbHep cell nodules were extracted from fresh liver under a Leica MZ FL II stereo-dissection fluorescent scope. hbHep libraries were prepared from tissue at D33 and D64. YAP1 ON Tumor, YAP1 OFF D14 tumor and WT liver libraries were prepared in biologic replicate (n=2 mice). Representative replicate shown in main figures. Freshly extracted liver tissue was flash-frozen, transported and resuspended in homogenization buffer prior to douncing. 50,000 nuclei isolated by gradient centrifugation were used for 30min transposition reaction. Tagmented DNA was purified using Zymo Clean and Concentrator-5 (Zymo D4013) and amplified with custom oligonucleotides listed in Buenrostro et al. 2013 Supplementary Table 1 (13). Barcoded libraries were pooled for paired-end sequencing on two Illumina NextSeq High-Output 150-cycle cartridges.
ATAC-seq Pre-Analysis Processing
Demultiplexed FASTQs were trimmed by Cutadapt (13) and aligned to the mm10 reference genome with BWA-MEM (14). Alignments underwent PCR duplicate removal (https://github.com/broadinstitute/picard) and compression (15). Peaks/pileup-containing files (bigWig, bedGraph, bed) were generated by ZPeaks (https://github.com/krews-community/zpeaks) for visualization.
ATAC-seq Analysis
To make direct comparisons between ATAC-seq samples, each sorted, de-duplicated BAM was normalized using deepTools bamCoverage with 1nt bin size and CPM normalization. Coverage matrices depicting occupancy around enhancers and cCREs (screen.umassmed.edu) were built using deepTools computeMatrix (“--reference-point” mode). BigWig correlation matrices were built by deepTools multiBigWigSummary with 1kb bin size and visualized by plotCorrelation, discarding outliers (“--removeOutliers” mode) (Fig. 6D) (16). Pairwise comparison of open enhancer regions (Fig. 6A–C) was conducted using Bedtools intersect (17). To minimize false-positive intersects, only peaks scoring >1.64 standard deviations above the corresponding sample mean (Z>1.64) were retained.
Figure 6. Induction of mature hepatocyte gene expression in hbHep cells results from YAP1 modulation of liver differentiation transcription factor occupancy.
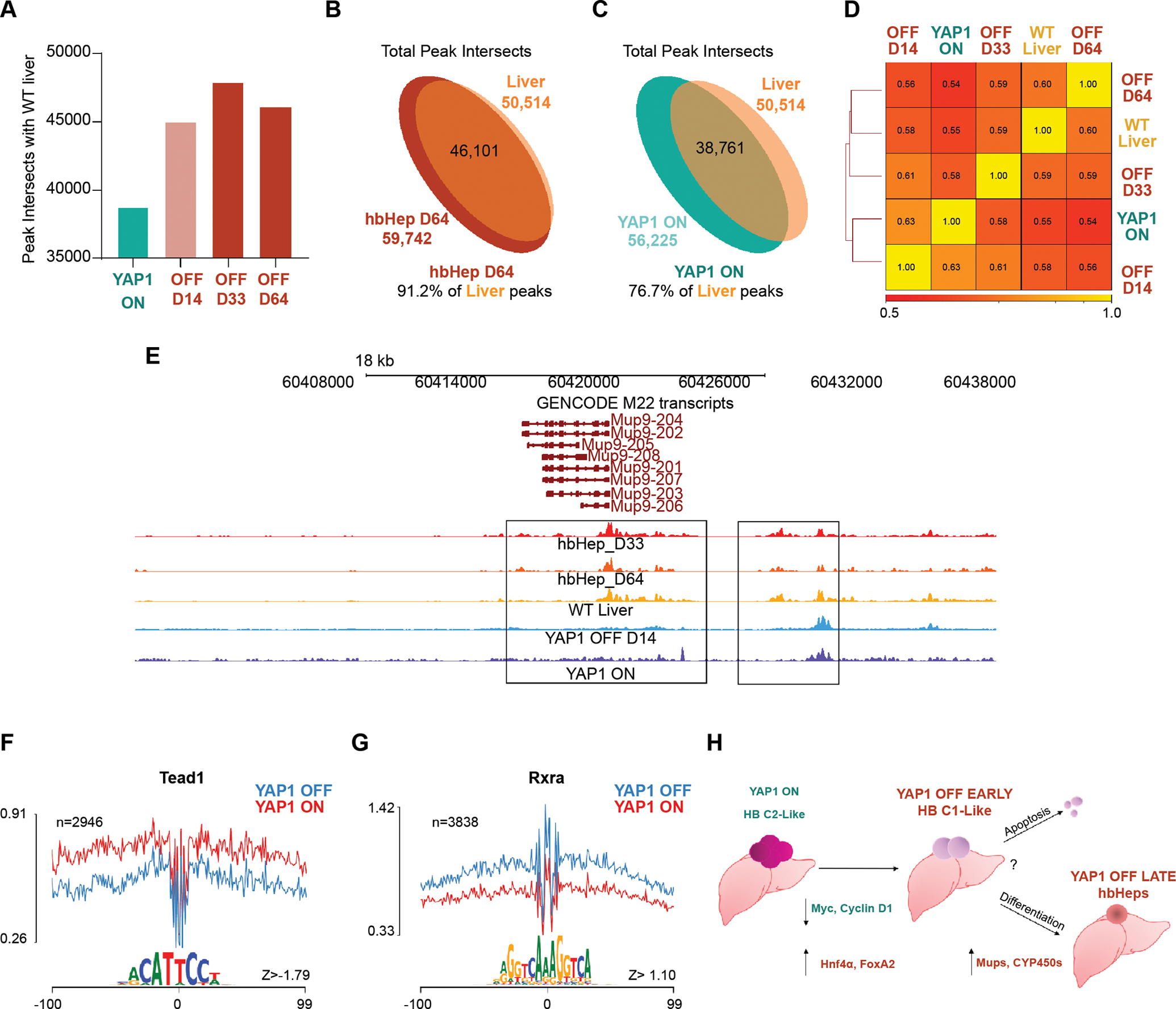
(A) ATAC peak intersects shared with WT liver in YAP1 ON, YAP1 OFF D14 regressing tumor, and hbHeps YAP1 OFF D33 and D64. Representative ATAC peaks from one representative sequencing replicate for YAP1 ON, YAP1 OFF D14 and WT liver sequencing experiment shown for Figure 6. (B-C) Total ATAC peak intersects between hbHeps D64 and WT liver, WT liver and YAP1 ON tumor. Not to scale. (D) Spearman correlation values with hierarchical clustering YAP1 ON, YAP1 OFF D14, hbHeps D33 and D64 (E) Representative genome browser ATAC tracks for Mup9, gene found to be highly upregulated in RNA-seq YAP1 OFF D6/YAP1 ON, (F-G) DNA Footprinting analysis of ATAC Seq YAP1 OFF hbHeps D33 shows decreased occupancy at Tead1 motif (F) and increased occupancy at Rxra motif compared to YAP1 ON tumors (H) Model: YAP1 ON mouse tumors resemble aggressive C2 human tumors. YAP1 withdrawal promotes downregulation of Myc, Cyclin D1, and HB prognostic markers promoting a C1-like favorable prognosis tumor. Long-term YAP1 withdrawal promotes durable tumor regression by promoting apoptosis in a subset of tumor cells, and differentiation towards a hepatocyte-like state in other tumor cells, i.e. “hbHeps.”
Transcription Factor Footprinting
ATAC-seq output was further processed using software and instructions from the Regulatory Genomics Toolbox (RGT) HMM-based footprinting pipeline, HINT, available at http://regulatory-genomics.org/ (14). Preceding analysis, we dockerized RGT-HINT to overcome dependency conflicts. Transcription factor occupancy in regulatory regions was determined by matching motifs flanked by ATAC signal to known binding sequences in the JASPAR database (Khan et al. 2018) (15). Occupancy was scored and compared across samples in a pairwise manner. Line graphs for each motif are the default output of RGT-HINT. Z-score ranked scatterplots (Z=1.64 cutoff) were made by the R plot function. RGT motif “match” BED files were reformatted for use with bedtools intersect (steps available upon request) to isolate factor-specific binding sites. BED files were converted to BigBed format with bedToBigBed (Kent et al. 2010) (16) and displayed on the UMMS Genome Browser.
Animal Studies
All studies were performed with approval and in accordance with the University of Massachusetts Medical School Institutional Animal Care and Use Committee (IACUC). Mice were housed at University of Massachusetts Medical School Animal Facilities Core. Healthy male and female mice were used for studies and hydrodynamically injected at >8 weeks of age. Mouse strains purchased from Jax laboratories include: B6.Cg-Gt(ROSA)26Sortm1.1(CAG-rtTA3)Slowe/LdowJ (Strain#029627), B6N.FVB(Cg)-Tg(CAG-rtTA3)4288Slowe/J (Strain #016532) and FVB/NJ (Strain #001800). FAH−/− (deltaExon5) mice were a gift from Dr. Markus Grompe (Oregon Health & Science University).
Results
Conditional YAP1 cooperates with constitutive β-Catenin to drive formation of hepatoblastoma in vivo
To engineer a conditional model of hepatoblastoma (HB), we used hydrodynamic injection to co-deliver Sleeping Beauty transposon plasmids encoding human oncogenes including a TET-ON inducible YAP1S127A, a constitutive β-CateninDelN90 oncogene, and a luciferase reporter to the livers of mice transgenic for reverse tetracycline transactivator (Tg-rtTA) (Figure 1A) (17–21). The luciferase reporter and Sleeping Beauty transposase are encoded on the same plasmid (Figure 1A). As an alternative approach for tumor initiation in vivo, we co-delivered a sleeping beauty rtTA (SB-rtTA) plasmid with the oncogenes and luciferase plasmids into wildtype FVB mice (Figure 1A). Previous work showed that co-expression of constitutive oncogenic YAP1S127A and β-CateninDelN90 in mouse liver induces hepatoblastoma (4). YAP1S127A and β-CateninDelN90 protein remain constitutively active in the nucleus by evading cytoplasmic retention and degradation (19, 20). Importantly, previous work demonstrated that hydrodynamic transposon-based expression of either YAP1S127A or β-CateninDelN90 alone does not induce hepatoblastoma (4).Using either transgenic Tg-rtTA or transposon SB-rtTA, human YAP1 oncogene overexpression can be reversibly regulated by feeding mice a diet with doxycycline (YAP1 ON) or without doxycycline (YAP1 OFF), allowing us to study the relevance of withdrawing YAP1 expression in vivo in HB tumors (Figure 1A).
Figure 1. Conditional murine hepatoblastoma gene expression mimics the poor prognosis C2 subtype of human hepatoblastoma (HB).
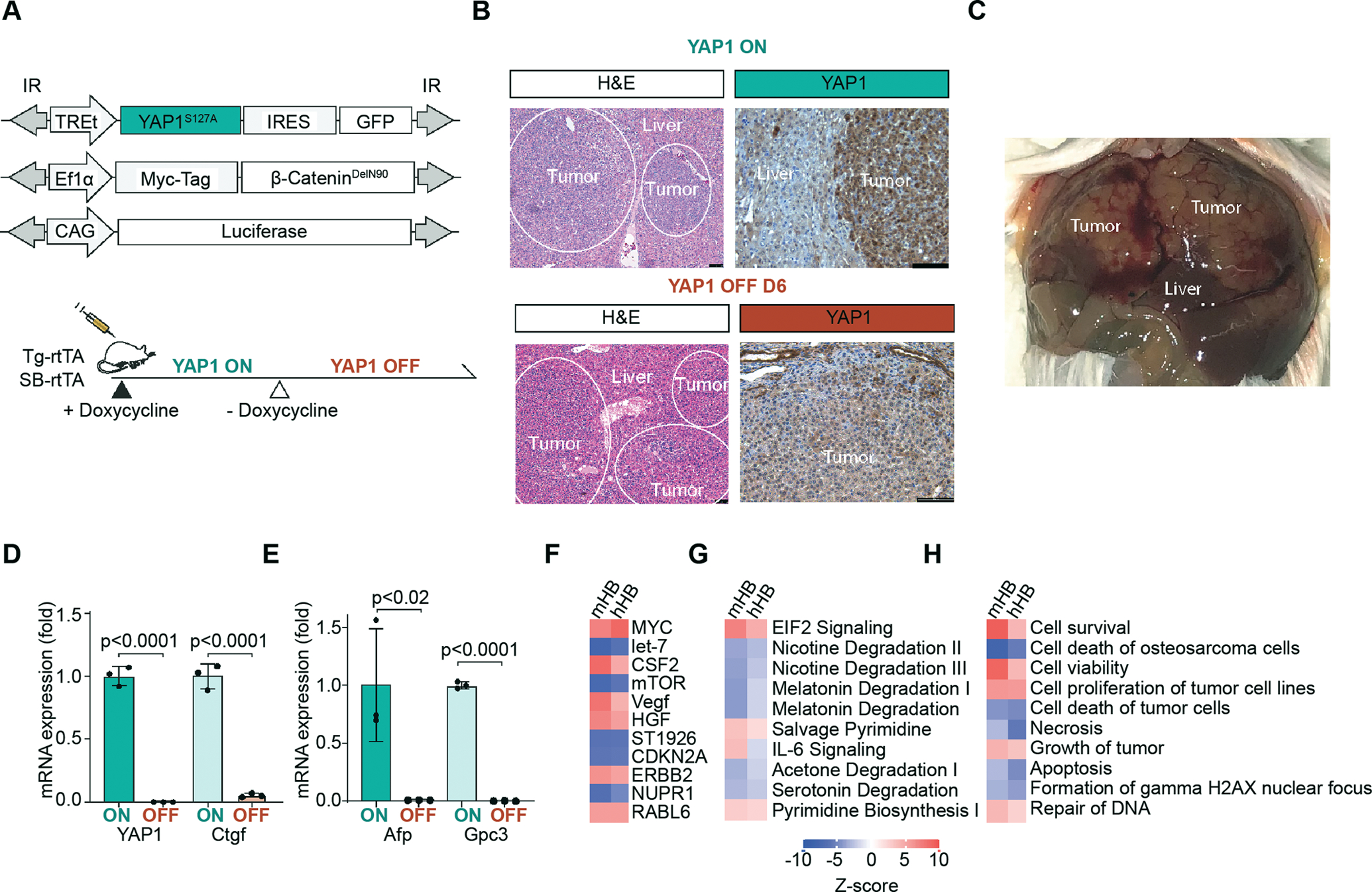
(A) Sleeping Beauty transposon plasmids are hydrodynamically injected into the liver of transgenic rtTA mice (Tg-rtTA). TREt encodes a TET-ON promoter, IR marks inverted repeats. Alternatively, rtTA was co-delivered with sleeping beauty plasmids to FVB mice by hydrodynamic injection (SB-rtTA). (B) Top: Representative H&E (10X lens), scale bars 75 μm and IHC in YAP1 ON tumors (20X lens), n=3 mice, scale bars 100 μm. Bottom: Representative H&E (10X lens), scale bars 75 μm and IHC in YAP1 OFF D6 tumors (20X lens), n=3 mice, scale bars 100 μm) (C) Representative gross live tumor image in YAP1 ON SB-rtTA mouse (D) qPCR YAP1, Ctgf in YAP1 ON and YAP1 OFF D14–21, n=3 tumors per mouse, p <0.0001 unpaired t-test, n=3 mice. (E) qPCR of clinical HB marker Afp and Gpc3 in YAP1 ON and YAP1 OFF D14–21, n=3 tumors per mouse, p <0.02, p<0.0001, respectively, unpaired t-test, n=3 mice. (F-H) Mouse HB (mHB) gene signatures resemble human HB (hHB). IPA ingenuity analysis comparing YAP1 ON/OFF mouse HB to C2-poor/C1-favorable prognosis human HB microarray data from Cairo et al. Top 10 IPA Upstream Gene pathways I, Canonical Pathways, Disease/function signature pathways respectively
Consistent with previous work using constitutive YAP1 to establish HB in mice, in vivo cooperation between inducible YAP1 and constitutive β-Catenin drives the formation of large multi-focal liver tumors with hepatoblastoma features (Figure 1B–C, 3D). Similar to the constitutive HB model (4), the earliest detectable tumor by histology appeared at 4 weeks post-injection. At ~6–8 weeks of doxycycline treatment, mice had extensive (>30) gross surface tumors, including punctate and confluent lesions (Figure 1C, 3D).
Figure 3. YAP1 withdrawal results in >90% tumor regression in a conditional mouse model of hepatoblastoma.
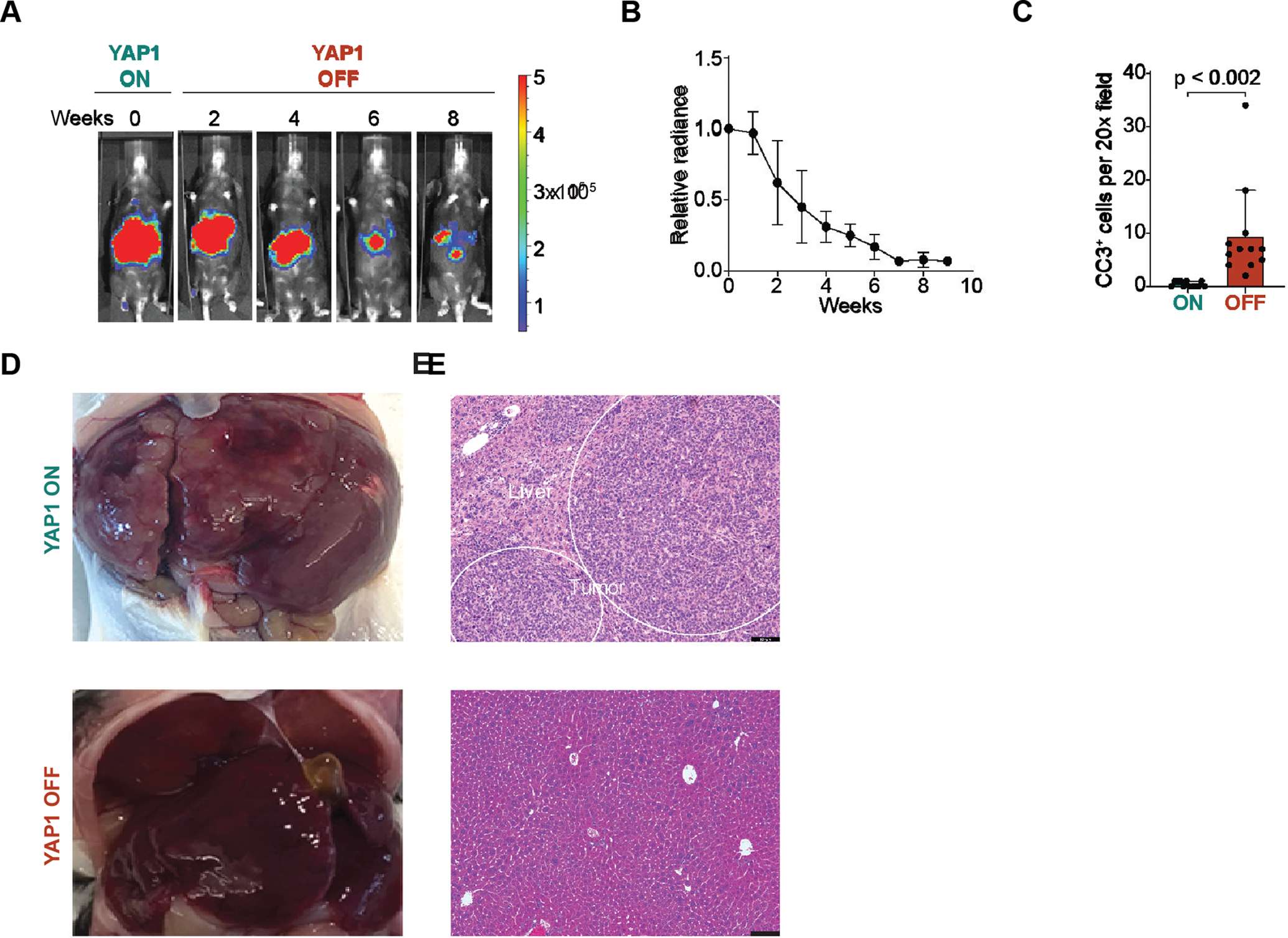
(A) Representative Luciferase imaging shows YAP1 OFF tumor regression, n=3 mice (B) Quantification of a. Mean relative luciferase; error bars are SEM, n=3 mice. (C) Cleaved caspase 3 (CC3) positive cells in YAP1 ON and YAP1 OFF D3–14 tumors, n=4 20X sections per mouse, n=3 mice. (D) 10 weeks YAP1 OFF livers do not harbor macroscopic surface tumors compared to YAP1 ON. Representative liver images, YAP1 ON SB-rtTA mouse and YAP1 OFF Tg-rtTA mice. (E) Paired histology from Panel D, YAP1 ON SB-rtTA mouse and YAP1 OFF Tg-rtTA mice are shown, scale bars 100 μm.
By immunohistochemistry, YAP1 ON tumors were GFP-positive and express both nuclear β-Catenin/Myc-Tag and nuclear YAP1 (Figure 1B, Supplemental Figure 1B). To functionally validate the conditional mouse model, we removed doxycycline to withdraw oncogenic YAP1 (i.e. YAP1 OFF, Figure 1A). After 3 days of (D3) doxycycline withdrawal, nuclear YAP1S127A was dramatically reduced in histologic sections, and virtually absent at D6 and D14 (Figure 1B, Supplemental Figure 1A–D). The TET-ON YAP1S127A plasmid encodes a GFP reporter by an internal ribosomal entry site (IRES). Additionally, we found that doxycycline withdrawal also silenced expression of GFP downstream of YAP1 by D14 (Figure 1A, Supplemental Figure 1A, 1E).
The expression of YAP1 and its target gene, Ctgf, were significantly reduced in day 14 to 21 (D14–21) YAP1 OFF tumors demonstrating that the inducible YAP1 oncogene is rapidly inactivated (Figure 1B,D). Concordantly, α-fetoprotein (Afp) and Glypican C3 (Gpc3), stem-like clinical and prognostic markers of HB, are also reduced significantly in YAP1 OFF D14–21 tumors (Figure 1E) (22).
YAP1 withdrawal reprograms the transcriptional landscape in murine hepatoblastoma
To characterize the transcriptional landscape associated with YAP1 withdrawal, we compared the mRNA profiles of YAP1 ON and YAP1 OFF D6 early regressing tumors (Figure 2A–B, Supplemental Table 1). Consistent with the rapid inactivation of YAP1, many canonical YAP1 target genes, including Birc5 (Survivin), Amotl2, and Foxm1, were reduced in YAP1 OFF D6 tumors (Figure 2C–E). In addition, six days of YAP1 withdrawal suffices to downregulate many tumor-associated signatures, including proliferation signatures (Hallmark G2M checkpoint, Mitotic Spindle, E2F targets) (Figure 2F, J–L).
Figure 2. YAP1 withdrawal reprograms gene expression in murine hepatoblastoma.
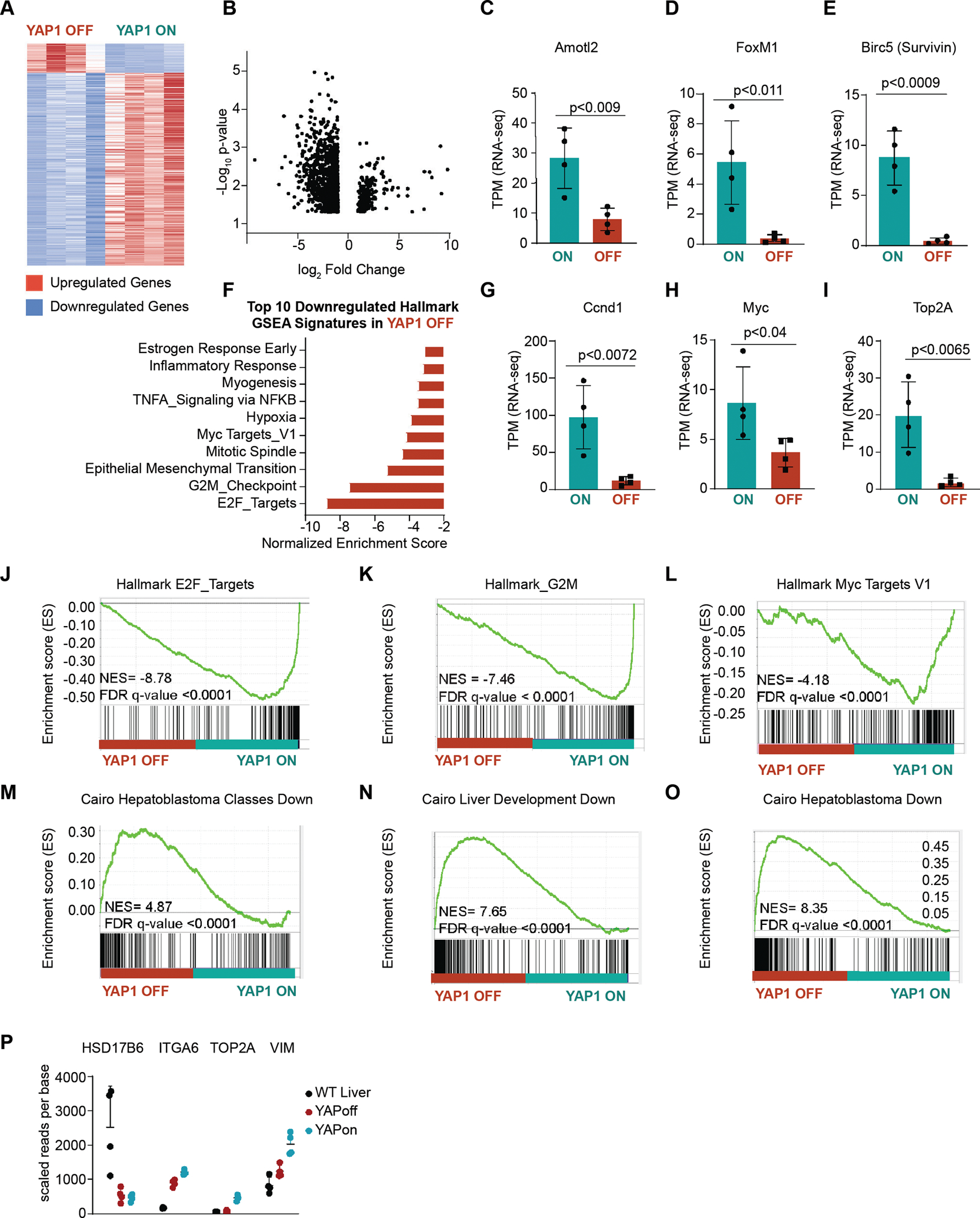
(A) Heatmap of differentially expressed transcripts log2fold change in YAP1 OFF D6 tumors compared to YAP1 ON tumors, n=4 tumors per group, genes shown in heatmap >2 upregulated or <0.5 down-regulated fold change, Student’s t-test, p<0.01, Full fold change gene list shown in Supplemental Table 1. (B) Volcano plot of differentially expressed genes in YAP1 OFF D6/YAP1 ON tumors, genes with log2fold >1 and <−1 shown, p<0.05. (C-H) TPM RNA-seq counts in YAP1 ON and YAP1 OFF D6 tumors, n=4 tumors per group, unpaired t-test. (I) Top 10 most downregulated Gene Set Enrichment Analysis (GSEA) Hallmark pathways in YAP1 OFF D6/ YAP1 ON tumors. (J-O) GSEA analysis in YAP1 OFF D6/ YAP1 ON tumors, FDR q-value <0.0001 (P) Hooks Four gene signature for C2 poor prognosis and C1 favorable prognosis tumors in YAP1 ON and YAP1 OFF D6 tumors gene expression, n=4, control FVB liver shown for WT
Previous studies showed that YAP1 can co-regulate β-Catenin target gene expression in vitro (4). We observed that YAP1 withdrawal alone, without modulation of oncogenic β-Catenin, downregulates two canonical β-Catenin target genes, Myc and CyclinD1 (Figure 2F–G). Gene set enrichment analysis (GSEA) further suggests downregulation of Myc targets (Hallmark_Myc_TargetsV1) in YAP1 OFF D6 early regressing tumors (Figure 2L). Thus, YAP1 withdrawal may partially abrogate the oncogenicity of β-Catenin through downregulation of its target genes.
Conditional murine hepatoblastoma gene expression mimics the poor prognosis C2 HB pediatric subtype
Differentiation status impacts HB prognosis. Indeed, Cairo et al. first characterized children’s hepatoblastoma tumors as either well-differentiated human pediatric hepatoblastoma Class 1 (C1) tumors with favorable outcomes, or as undifferentiated highly proliferative Class 2 (C2) tumors with poor prognosis (23). C1 and C2 tumors have comparable rates of Wnt pathway mutations, suggesting β-Catenin alone does not determine the aggressiveness of HB tumors. Indeed, unlike C1 tumors, poor prognosis C2 tumors differentially activate YAP1, express YAP1 target genes, and have high levels of proliferation and stem markers (23, 24).
To characterize the subtype of hepatoblastoma recapitulated in our conditional YAP1 ON model, we returned to the RNA-sequencing of YAP1 ON tumors and YAP1 OFF D6 regressing tumors (Figure 2A–B). Using GSEA, we found that six days of YAP1 withdrawal was sufficient to downregulate aggressive “C2” tumor associated genes. Specifically, we find that YAP1 OFF D6 tumors are enriched for GSEA signatures Cairo Hepatoblastoma Classes Down, Cairo Hepatoblastoma Down, and Cairo Liver Development Down (Figure 2M–O). The Cairo Hepatoblastoma Classes Down predefined gene signature uses genes downregulated in invasive C2 tumors compared to favorable C1 tumors. Similarly, the Cairo Hepatoblastoma Down signature uses genes downregulated in all HB tumors compared to normal liver. Thus, YAP1 OFF D6 tumors downregulate C2 associated genes, and upregulate genes associated with C1 tumors and normal liver.
To further characterize our conditional murine HB as congruent with a well-differentiated favorable C1 or a poor prognosis C2 tumor subtypes, we performed Ingenuity Pathway Analysis (IPA) comparing YAP1 ON/YAP1 OFF D6 gene expression to C2/C1 human hepatoblastoma microarray from Cairo et al. (23). Similarly, to our YAP1 ON and YAP1 OFF mouse tumors, C1 and C2 human tumors have comparable Wnt activation, but C2 tumors have differential YAP1 activation (23–25). Like GSEA, IPA analyses allows for comparison analyses of curated gene signatures. IPA revealed that top Upstream Gene Pathways I, Canonical Pathways, and Disease/function IPA signatures were congruent between YAP1 ON/OFF D6 mouse HB tumors and C2/C1 human HB tumors (26) (Figure 1F–H). Indeed, we observe upregulation of proliferation and cell survival, and downregulation of liver metabolism and cell death signatures in C2 human and YAP1 ON mouse tumors.
C2 tumors can be further stratified into the Hooks highly aggressive C2A and intermediate C2B subtypes (24, 25). Analyzing the Hooks signature (25), we found that YAP1 ON tumors closely mimic the aggressive C2A tumors that express high levels of Top2A, and reduced levels of differentiation maker HSD17B6 (compared to WT FVB mice; Figure 2I) and YAPOFF D6 tumors more closely resemble C1 tumors (Figure 2P), suggesting that YAP1 status determines tumor aggressiveness. YAP1 ON tumors also display some features of moderately aggressive C2B tumors, i.e. their increased levels of vimentin.
Taken together, these analyses suggest that YAP1 ON in conditional murine hepatoblastoma drives an aggressive phenotype consistent with the “C2” subgroup of pediatric tumors. Moreover, withdrawal of YAP1 alone—i.e. without silencing oncogenic β-Catenin—suffices to modulate the hepatoblastoma subtype from poor prognosis to favorable.
Withdrawal of oncogenic YAP1 alone mediates >90% tumor regression
Using the luciferase reporter, we then serially monitored tumor progression over time in live mice using in vivo bioluminescent imaging. We found that YAP1 OFF tumors gradually regressed, and by 9 weeks luminescent signal was reduced 93% ± 6% (n=3; Figure 3A–B). Moreover, histological examination revealed ~10-fold fewer Ki67-positive proliferating cells in regressing YAP1 OFF D6–14 tumors than in YAP1 ON tumors (YAP1 ON, 242.5 ± 53.9 vs. YAP1 OFF, 26.8 ± 8.7 positive cells per 20× field; n=3) and a slight increase in cleaved caspase 3-positive apoptotic cells (YAP1 ON 0.416 ± 0.51 vs. YAP1 OFF 9.33 ± 8.75 positive cells per 20× field; n=3; Figure 3C, 4B, and Supplemental Figure 2A–B). These results indicate that genetic withdrawal of YAP1 can regress hepatoblastoma driven by oncogenic YAP1 and β-Catenin.
Figure 4. Sustained YAP1 withdrawal results in durable tumor regression and generates non-proliferative tumor lineage cells with hepatocyte morphology.
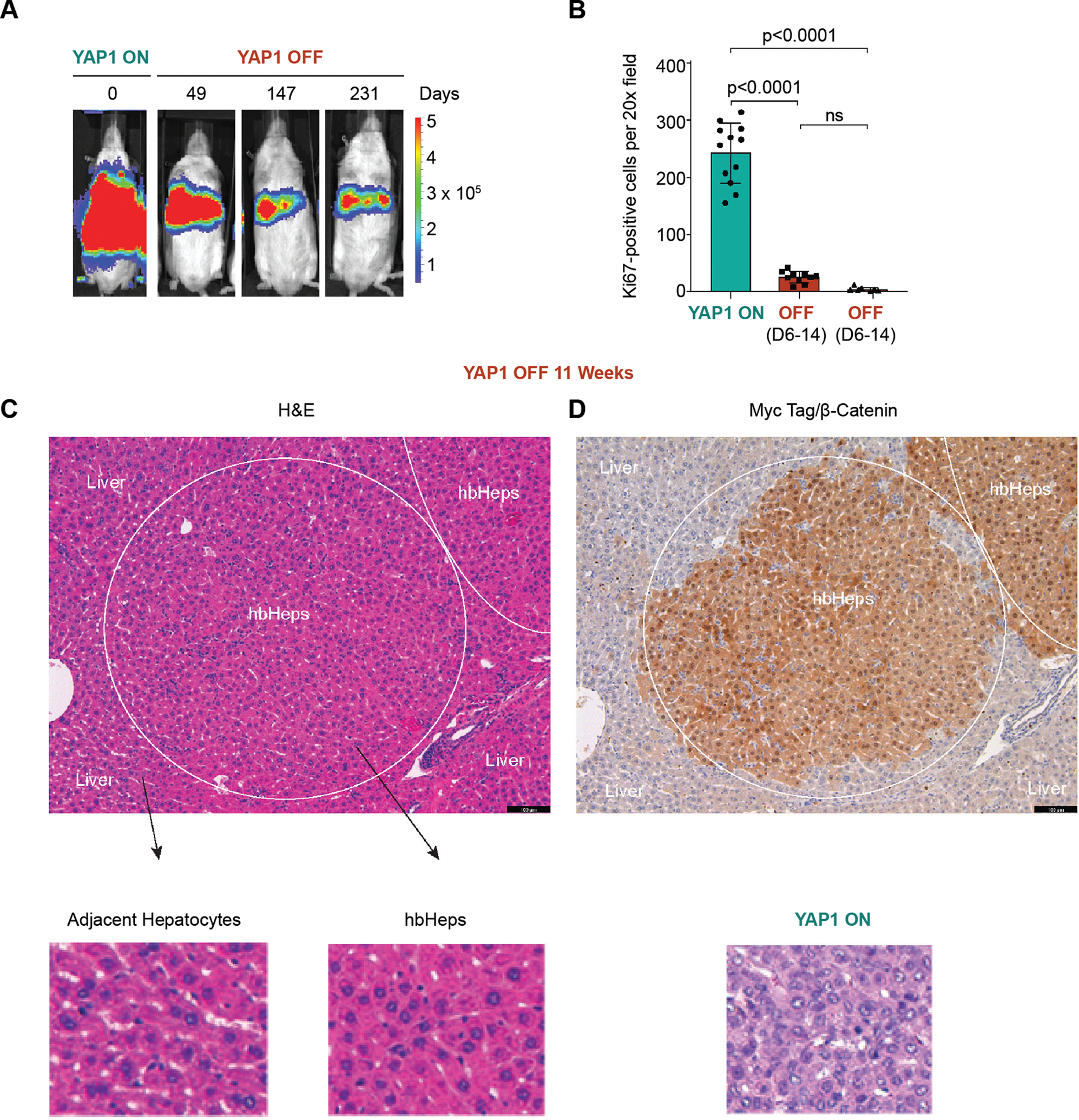
(A) Long-term tumor regression in YAP1 OFF mice. Residual luminescent signal persists in YAP1 OFF mice (SB-rtTA plasmid) 231 days post-doxycycline withdrawal, n=2 mice. (B) Ki67+ cells per 20X field, n=4 20X sections per mouse, n=3 mice. One-Way Anova with Tukey multiple comparisons test. (C-D) Myc-tag IHC detects residual cells of tumor lineage (circled area) with hepatocyte morphology in YAP1 OFF mice (n=4). We termed these cells hepatoblastoma-derived hepatocyte-like cells (hbHep). Scale bars are 100μm, YAP1 ON tumor H&E serves as a control. IHC and H&E shown are serial sections.
YAP1 withdrawal results in durable tumor regression for >230 days
We observed ~93% regression of HB tumors in YAP1 OFF livers by week 7, and no evidence of further regression or tumor relapse in weeks 8 or 9 (Figure 3A–B), suggesting that tumor regression might be stable. After 10 weeks of YAP1 withdrawal (YAP1 OFF Day 70 (D70+), macroscopic examination of livers revealed no evidence of surface tumors (n=3; Figure 3D). Moreover, liver histology revealed lack of obvious tumor nodules (Figure 3E), and proliferating Ki67-positive cells were virtually absent from YAP1 OFF D70+ livers (3.75 ± 2.83 positive cells per 20X field, n=3; Figure 4B and Supplemental Figure 3A) compared to YAP1 ON tumors.
To evaluate long-term survival of YAP1 OFF mice, we allowed a cohort of mice to proceed with sustained YAP1 withdrawal (Figure 4A). Whereas YAP1 ON mice reached a lethal burden of tumors by 65.6 days on average post tumor initiation (n=8), YAP1 OFF mice were viable 230 days after YAP1 OFF (n=2), and low levels of luciferase signal persisted in YAP1 OFF livers as mice aged (Figure 4A and Supplemental Figure 3B). Thus, sustained YAP1 withdrawal alone, without inactivation of oncogenic β-Catenin, in this HB mouse model sufficiently drives durable tumor regression, providing long-term survival benefit.
Long-term YAP1 OFF livers harbor non-proliferative tumor lineage cells with hepatocyte morphology
The persistent luminescence in YAP1 OFF D230+ livers suggested that, although tumors were significantly regressed, YAP1 OFF livers retain residual cells of tumor lineage. Indeed, staining YAP1 OFF D70+ livers for the Myc-tag fused to oncogenic β-Catenin (Figure 1A) revealed regions of Myc-tag-positive cells with hepatocyte morphology (13 ± 9.9 residual nodules per liver section, n=4 mice; Figure 4C–D). Further, reintroducing doxycycline to the diet after tumor regression induced tumor growth (Supplemental Figure 4A–C). Nevertheless, with sustained YAP1 withdrawal, residual tumor lineage cells remain camouflaged within the liver (Figure 4C–D). Because they are morphologically similar to normal hepatocytes, we have termed these residual tumor lineage cells “hepatoblastoma-derived hepatocyte-like cells” (hbHep cells).
YAP1 withdrawal induces therapeutic differentiation of HB tumor cells
In our RNA-Seq analysis of early regressing YAP1 OFF D6 tumors (Figure 2), the top 10 upregulated GSEA hallmark pathways included multiple liver metabolic pathways associated with normal hepatocyte function, such as xenobiotic and fatty acid metabolism pathways suggesting that YAP1 withdrawal induces mature hepatocyte gene expression in HB tumors (Figure 5A–C and Supplemental Figure 5A,D, Supplementary Table 1). Consistent with the hepatocyte-like morphology of hbHeps, we hypothesized that YAP1 withdrawal activated a cell fate switch driving differentiation of HB tumor cells towards mature hepatocytes.
Figure 5. YAP1 withdrawal promotes hepatocyte differentiation in mouse hepatoblastoma.
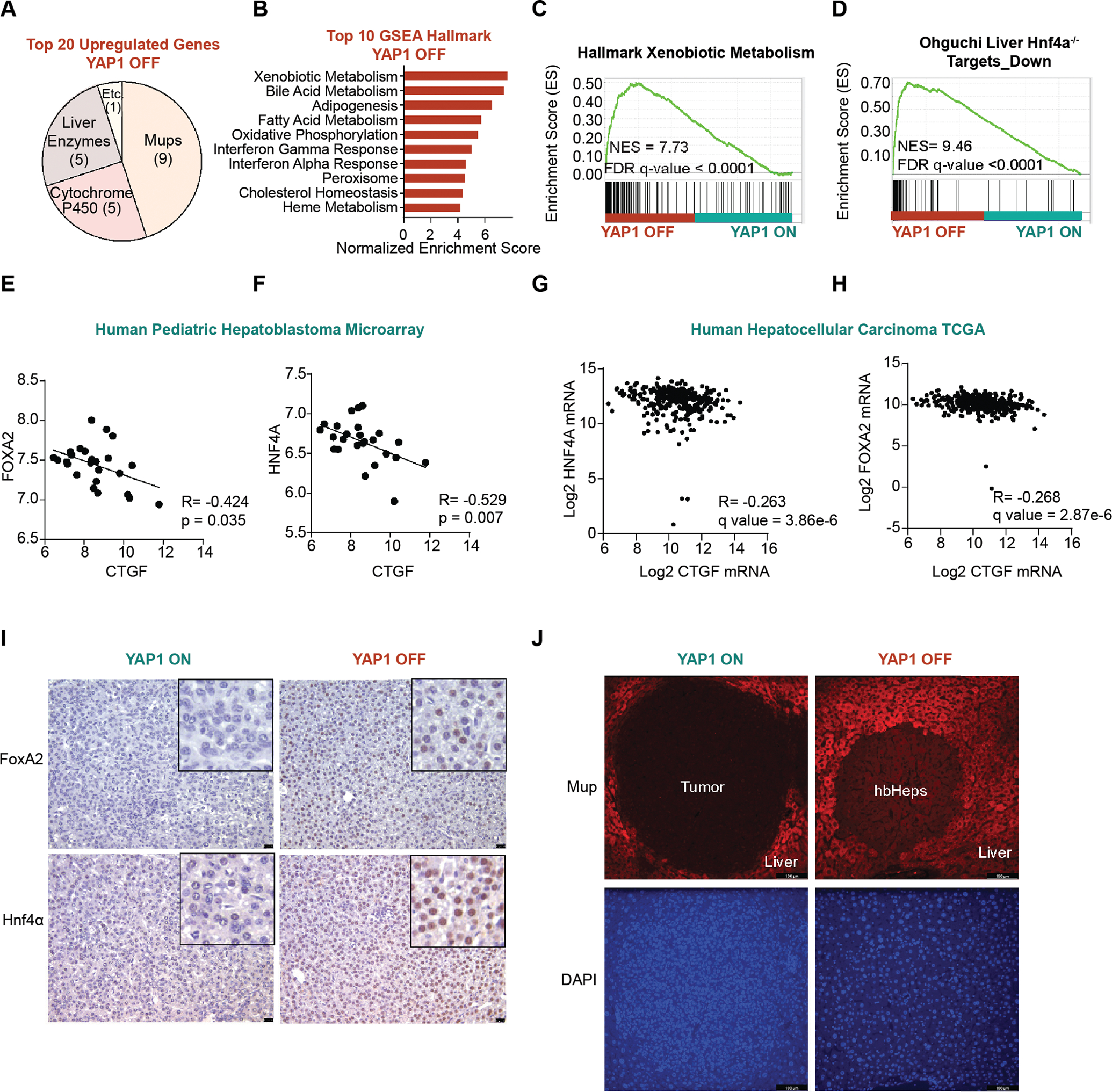
(A) 20 most significant upregulated genes in YAP1 OFF tumor RNA-Seq include major urinary proteins (Mups), cytochrome p450 enzymes, and other metabolic liver enzymes, p<0.05 (B) Top GSEA signatures. (C-D) Representative GSEA Signatures. (E-F) Inverse correlation between YAP1 target gene CTGF and differentiation markers in human hepatoblastoma microarray data (n=25) from Cairo et al, Spearman’s Correlation analysis. (G-H) Inverse correlation between YAP1 target gene CTGF and differentiation markers in human hepatocellular carcinoma TCGA data (n=348) from Uhlen at al, Spearman’s Correlation analysis. (I) YAP1 OFF induces hepatocyte differentiation factors FoxA2 and Hnf4α. Representative immunohistochemistry in YAP1 OFF D6 compared to YAP1 ON, scale bars 25 μm. (J) Hepatocyte marker MUP is partially restored in long-term YAP1 OFF D70 hbHep cells compared to YAP1 ON tumors, scale bars 100 μm. Representative immunofluorescence shown.
It has been previously shown that YAP1 and differentiation signaling are often polarized in the liver (27–32). Indeed, GSEA analysis shows target genes of Hnf4α and Foxa2—transcription factors that promote hepatocyte differentiation (28, 33, 34)—were both upregulated in YAP1 OFF tumors (Fig 5D and Supplemental Figure 5B). Further, immunohistochemistry revealed the presence of nuclear HNF4α and FoxA2 in YAP1 OFF D6 tumors but not in YAP1 ON tumors (Figure 5I). Concordantly, in both human pediatric HB microarray (23) and human hepatocellular carcinoma TCGA datasets (35), the YAP1 target gene CTGF and HNF4α/FOXA2 are inversely correlated, indicating the general role of YAP1 in suppressing differentiation in human liver cancer (Figure 5E–H).
We further explored this phenomenon in human hepatoblastoma cells. We used CRISPR/Cas9 to knock out YAP1 in Huh6 hepatoblastoma cells, which were derived from a 1-year old male patient, and which possess a mutation in CTNNB1 and express nuclear YAP1 (4, 36). Following YAP1 knockout, we observed an increase in canonical liver markers, including ApoA2, Ces3, Cyp2A6, Cyp2e1, Cyp3A5, and OAT (Supplemental Figure 5E–F), suggesting a more differentiated state.
Further, the top 5 genes upregulated in YAP1 OFF D6 tumors belong to the family of major urinary proteins (Mups) (Figure 5A and Supplemental Figure 5C, Supplemental Table 1). Mup proteins serve as a marker of mature hepatocytes (37, 38). Immunofluorescence detected virtually no Mup expression in YAP1 ON tumors, and partially restored levels of Mup in hbHep cells compared to adjacent normal hepatocytes in YAP1 OFF D70+ livers (Figure 5J). These findings further support YAP1 withdrawal promoting induction of mature hepatocyte gene expression in HB tumor cells.
YAP1 withdrawal promotes remodeling of the chromatin landscape in HB tumor cells
We hypothesized that YAP1 withdrawal drives transcriptional reprogramming for upregulation of mature hepatocyte gene expression, and the subsequent generation of hbHep cells, in part through modulation of liver chromatin accessibility. It has been previously demonstrated that YAP1 can exert transcriptional control through regulation of chromatin structure, specifically enhancer regions (39, 40). To assess open chromatin structure, we performed Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) (12) in YAP1 ON, YAP1 OFF D14 tumors, hbHep cells (D33 and D64) and normal liver. We isolated hbHep cells by fluorescently labeling them with mCherry upstream of TET-ON YAP1S127A (Supplemental Figure 6A). Using a Leica MZ FL II stereo-dissection fluorescent scope, mCherry-positive hbHep cell nodules were extracted using fluorescence. Further, using mCherry as a lineage tracing marker, we confirmed that the mCherry+ area in the YAP1 OFF livers were of the tumor lineage (Supplemental Figure 6B–C). Grossly and histologically, mCherry+ hbHep cells were similar in appearance to WT liver.
By analyzing intersected ATAC-seq peaks between YAP1 ON, YAP1 OFF D14 regressing tumors, hbHep cells, and WT liver, we observed that hbHep cells share a higher percent of intersected ATAC peaks and enhancers with WT liver than with YAP1 ON (Figure 6A–C and Supplemental Figure 6D–G). Next, we performed hierarchical clustering for total peaks. Two hbHep samples and WT Liver clustered together whereas YAP1 ON and YAP1 OFF D14 regressing tumor formed another group (Figure 6D).
When we examined enhancer occupancy in biological replicates of YAP1 ON tumor, YAP1 OFF D14, and WT liver, we found decreasing occupancy in YAP1 OFF D14 livers at adult and embryonic enhancers (Supplemental Figure 7). Interestingly, when we examined embryonic enhancer occupancy for hbHeps, we observe a transient increase in enhancer occupancy at the D33 timepoint compared to the later D64 hbHeps. We suspect this transient increase reflects the dynamic chromatin remodeling and unique transcription profile that we observe in early hbHeps; however, further detailed investigation will be required to define the genomic transitions that accompany the adoption of the hbHep cell fate (Supplemental Figure 8).
Finally, if we look at individual ATAC-seq tracks, we observe the YAP1 OFF D6 RNA-seq upregulated gene and mature hepatocyte marker, Mup9, has differential peaks in hbHeps and WT liver compared to YAP1 ON tumor (Figure 6E).
YAP1 withdrawal modulates liver transcription factor occupancy promoting the generation of hbHep cells
Finally, to understand more clearly how YAP1 withdrawal promoted differentiation of HB tumor cells to hbHeps, we performed computational DNA footprinting in regulatory regions in our ATAC-seq dataset. As expected, we observe decreased occupancy at Tead and Fos-Jun motifs in YAP1 OFF D33 hbHeps compared to YAP1 ON tumors (Fig 6F and Supplemental Figure 9A–D, Supplemental Table 2). Further in YAP1 OFF D33 hbHeps, we observe increased occupancy at the motif of Retinoid X receptor alpha (Rxra), known to mediate xenobiotic CYP450 expression, and multiple canonical liver differentiation transcription factors belonging to the Hnf and FoxA families, including Hnf4α, Hnf1α, Hnf1B, Onecut1, Onecut3, FoxA1, and FoxA2 (Figure 6G and Supplemental Figure 10A–D, Supplemental Table 3). These data indicate that YAP1 withdrawal drives transcriptional reprogramming and differentiation of HB tumor cells into hbHeps by modulating occupancy of canonical liver differentiation transcription factors to promote mature hepatocyte gene expression (Figure 6H).
hbHep cells rescue liver damage phenotype in Fah−/− mice
Oncogene withdrawal in mouse models of adult liver cancer has been shown to induce features of hepatocyte differentiation, but it remains unknown if the differentiated tumor cells are functional in a tissue context in vivo (8, 41). To determine if hbHep cells have somatic hepatocyte function in vivo, we assayed their ability to rescue liver damage in a mouse model of tyrosinemia. Fah–/– mice (deficient for the fumarylacetoacetate enzyme) fail to fully metabolize tyrosine and accumulate toxic metabolites that cause liver damage, weight loss, and death. Fah deficiency can be rescued by NTBC (supplied in water), which blocks the buildup of toxic metabolites (42–44), or by repopulating the liver with Fah-positive hepatocytes following NTBC withdrawal (42, 44–47). The proliferation of Fah-positive hepatocytes restores the functional/metabolic capacity of the liver rescuing the weight loss phenotype. If the liver is repopulated by Fah-positive cells that lack expression of critical liver metabolic enzymes, the weight loss phenotype will not be rescued.
We engineered a dual-function transposon plasmid that encodes a constitutively expressed Fah gene and TET-ON inducible YAP1S127A (YAP1-Fah; Figure 7A,B). We delivered YAP1-Fah, β-CateninDelN90, and SB-rtTA plasmids to Fah–/– mice by hydrodynamic injection and fed mice with doxycycline to induce YAP1 expression and HB tumor formation. Once tumors were established, we removed doxycycline and allowed tumors to regress (Figure 7A). We then withdrew NTBC from the water supply to induce liver damage and monitored mice for 10% total body weight loss.
Figure 7. Hepatoblastoma-derived hepatocyte-like cells (hbHep cells) rescue liver damage in Fah mutant mice.
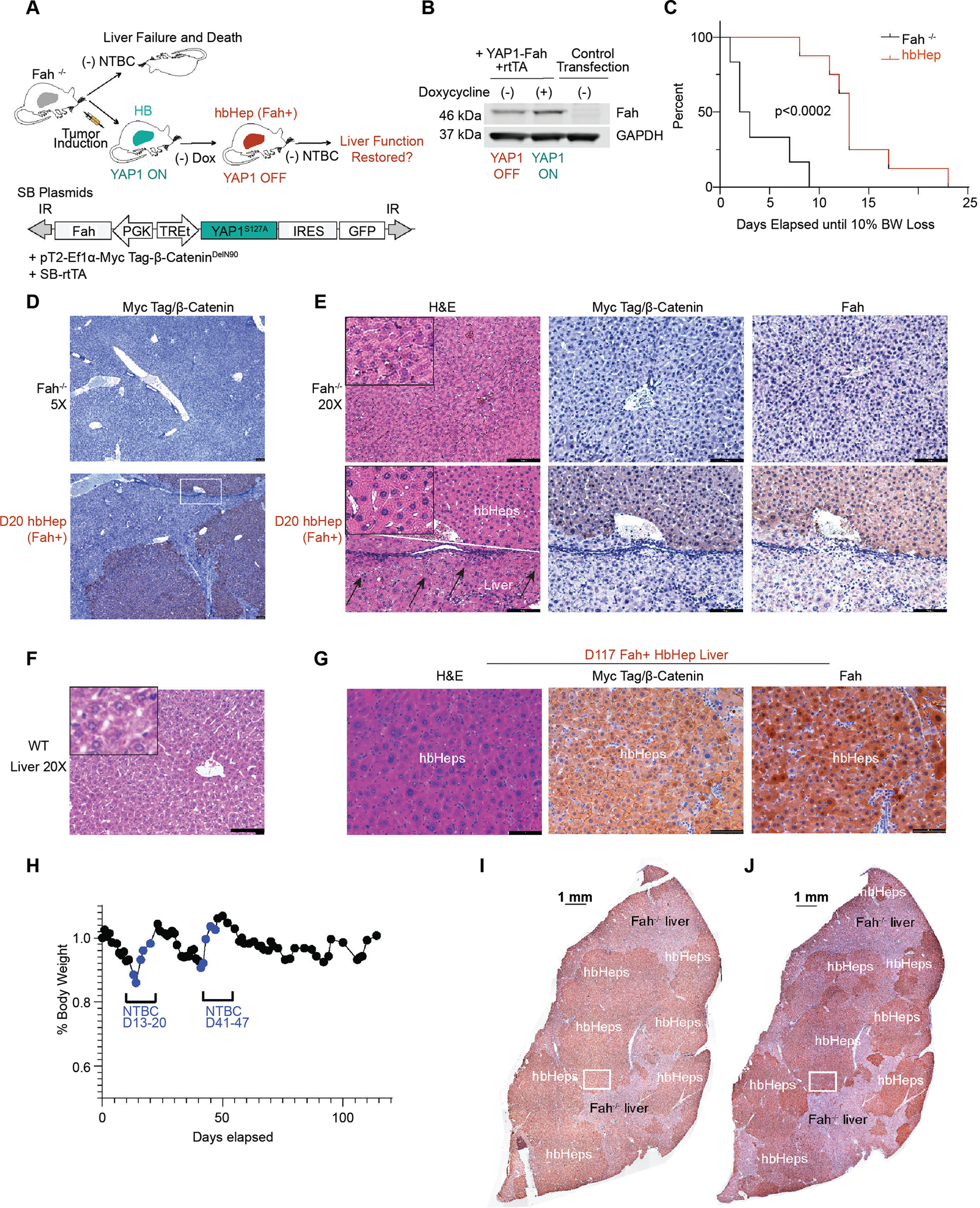
(A) Experimental Design. A dual function transposon plasmid encodes a constitutive Fah gene and TET-ON inducible YAP1 (termed YAP1-Fah plasmid). The plasmid combination was injected in Fah−/− mice to model HB tumor induction and regression. Only hbHep cells, but not host liver hepatocytes, express the Fah enzyme. (B) Validation of YAP1-Fah plasmid. Western blot in HCT116 cells transfected with indicated plasmids. (C) Kaplan Meier curve of hbHep expressing Fah (n=8) compared to untreated control Fah−/− mice (n=6) following NTBC withdrawal. Number of days until 10% body weight loss is shown, p<0.002, Log-rank (Mantel-Cox) test. (D) hbHep mice post NTBC withdrawal harbor Myc-tag positive hbHep cells. Representative immunohistochemistry in control Fah−/− mice and hbHep mice, n=3, scale bars 100 μm (5X lens). (E) The Myc tag positive hbHep cells are also positive for Fah. Representative immunohistochemistry (20X lens) in Fah−/− mice and hbHep mice (n=3). WT liver H&E is for comparison, scale bars 100 μm. The hbHep group shows serial liver sections of the white square region from panel d. White line denotes the border between hbHep area and host liver. Arrows denote dead hepatocytes in the Fah−/− liver area. (F) Representative WT liver (G) Representative immunohistochemistry (20X) lens in hbHep mice D117 (n=3 mice), white box from Panel I and J (H) Representative weight trace in hbHep mouse, blue marks NTBC cycling, (n=3 mice) (I-J) Scan of liver slice, scale bar 1mm, Myc-tag and Fah
Untreated Fah–/– control mice rapidly lost 10% of their total body weight and needed to be euthanized within 10 days, with a median of 2.5 days. Unlike Fah–/– control mice, Fah-positive hbHeps maintained weight out to 23 days, with median weight maintenance of 13 days (Figure 7C). Immunohistochemistry revealed that livers harbor hbHep cells positive for Myc-tagged β-Catenin and the Fah enzyme (Figure 7D–E). Whereas Fah-negative liver (in the control mice and in tissue adjacent to hbHep regions) showed abundant cell death, the Fah-positive hbHep areas of the liver appeared healthy (Figure 7E).
To promote expansion and recovery of hbHep cells into the damaged Fah–/– liver, Fah-positive hbHep mice were fed NTBC until normal weight was restored. NTBC was then removed, and mice were monitored for weight loss. When mice lost 10% of their body weight, NTBC was provided at least 2X to promote hbHep expansion (n=3) (Figure 7H). Fah-positive hbHep mice maintained weight (>90%) for a maximum of 39.6 days (SD 23.7 days; range 24 to 67 days; n=3) before NTBC needed to be provided (Figure 7H). By contrast, untreated Fah–/– control mice rapidly lost 10% of their total body weight required euthanasia between 2.5 and 10 days (Figure 7C).
On immunohistochemical examination, D114+ liver sections were grossly positive for Fah and the Myc epitope (n=3 mice, D114+) (Figure 7G). Myc-positive and Fah-positive nodular areas overlap in serial sections of liver, allowing hbHep cells to be tracked easily (Figure 7I–J). Gross examination of the liver under a dissection scope revealed 2 to 3 GFP-positive lesions per mouse (data not shown). GFP-positive tumors are presumed to be TET-ON escaper nodules. No other gross or histologic abnormalities were noted in the D114+ hbHep mice.
To further investigate the gene expression profile of hbHeps, we performed RNA-sequencing analysis in the three D114+ hbHep mice compared to Fah–/– mice on NTBC. Control Fah–/– mice on NTBC do not accumulate toxic metabolites, and therefore maintain healthy hepatocytes and normal hepatocyte gene expression. Gene expression comparisons between YAP1 ON, YAP1 OFF D6 regressing tumors, Fah-positive D114+ hbHeps, and NTBC-fed Fah–/– mice suggest that YAP1 withdrawal results in a gradual progression over time toward normal hepatocyte function (Supplemental Figure 11, Supplemental Table 6–8).
Specifically, Ingenuity Pathway Analysis (IPA) revealed a loss of Disease and Function liver cancer signatures (Liver Cancer, Liver Tumor, Liver Carcinoma) in hbHeps compared to YAP1 ON tumors (Supplemental Figure 11A, Supplemental Table 7). Further, hbHeps and WT liver also trend together on Hnf4α and Myc IPA Upstream Regulator signatures (Supplemental Table 6). Indeed, examining scaled reads per base (i.e., normalized transcript count), we observed comparable levels of Myc expression between YAP1 OFF D6 tumors, D114+ hbHeps, and WT liver (Supplemental Figure 11G). This finding suggests that YAP1 withdrawal sufficiently dampens the hepatoblastoma MYC signature observed in children’s aggressive hepatoblastoma tumors.
Besides loss of tumorigenic signaling in hbHeps, we also observed restoration of some hepatocyte metabolic function. We observe comparable scores for hbHeps and WT liver in liver-specific IPA Canonical Pathways, including PXR/RXR Activation, Xenobiotic Metabolism, Oxidative Phosphorylation, and Super-pathway of Cholesterol Biosynthesis (Supplemental Tables 6–8). Indeed, hbHeps do not fully recapitulate WT liver gene expression, but do partially recover expression of Mups, the mature hepatocyte marker (Supplemental Figure 11B,D–F). This finding is consistent with the immunofluorescence in hbHeps of Mup family proteins (Figure 5J). We observe increased levels of Mups in hbHeps compared to tumor, but the level is not fully comparable to WT liver. Finally, using the Hooks Four-Gene Signature, whereas YAP1 ON tumors resemble C2 tumors and YAP1 OFF D6 tumors resemble C1 tumors, hbHeps more closely resemble WT liver (Supplemental Figure 11C).
Thus, hbHep cells engineered to express Fah can rescue liver function in Fah-deficient mice, demonstrating that the transcriptionally reprogrammed hbHeps resemble functional hepatocytes that can repair liver damage.
Discussion
Using a conditional mouse model of the aggressive C2 subtype of pediatric HB, we have shown that sustained withdrawal of YAP1 results in >90% tumor regression, demonstrating that HB tumor maintenance requires YAP1. Though driven by both β-Catenin and YAP1, HB is sensitive to the inactivation of YAP1 alone, perhaps because YAP1 inactivation also dampens β-Catenin signaling in this model (4). Despite the persistence of residual tumor-lineage cells, sustained YAP withdrawal mediates durable tumor regression and long-term survival. These results provide preclinical evidence in favor of evaluating YAP1-targeted therapies, either alone or in combination with other agents, in the treatment of HB. Moreover, the >90% tumor regression suggests that YAP1 inhibition may benefit children as a neoadjuvant therapy to improve the resection of HB tumors, or as a post-resection adjuvant therapy to promote regression of residual disease. Further investigation is needed to understand how human hepatoblastoma will respond to pharmacologic inhibition of oncogenic YAP1. Nevertheless, YAP1 inhibition presents a potentially tractable therapeutic strategy in children with HB.
We propose that withdrawal of YAP1 overexpression in hepatoblastoma tumor cells activates a cell fate switch that promotes apoptosis and, in a subset of tumor cells, differentiation (Figure 6H). In our system, sustained YAP1 rehabilitates a subset of tumor cells into a hepatocyte-like state, hbHep cells, that can rescue liver damage. While oncogene inactivation has been shown to induce features of differentiation, if the differentiated tumor cells are functional in a tissue context in vivo remained unclear (8, 41). Our data show that YAP1withdrawal in mouse hepatoblastoma activates a cell fate switch allowing HB tumor cells to reprogram and differentiate toward a functional hepatocyte-like state that can rescue liver damage.
This work provides clear functional evidence that hepatoblastoma cells can be therapeutically differentiated to mimic a normal somatic cell state, offering a new outcome in the traditional paradigm of oncogene addiction and inactivation. Future work will determine how HB cells choose the fate between cell death and differentiation and which signals maintain the survival of hbHep cells. Our work supports YAP1 inhibition as a rational targeted therapeutic for the treatment of hepatoblastoma and provides a new in vivo model to explore cell fate decisions as tumor cells are therapeutically differentiated into a functional somatic-like state.
Supplementary Material
Acknowledgements
We thank P. Zamore, C. Mello, A. Mercurio, J. Shohet, P. Newburger, J. Aidlen, S. Corvera, L. Shaw and H. Yin for discussions. We thank Drs. Scott Lowe and Holger Willenbring for sharing plasmids and reagents and Dr. Markus Grompe (Oregon Health & Science University) for providing the Fah mice. We thank Y. Liu and E. Kittler in the UMass Morphology and Deep Sequencing Cores for support. We thank D. Conte and E. Mohn for critical manuscript feedback. We thank members of the Xue lab for constructive suggestions. We thank the UMassMed Animal Core, specifically Greg Cottle, for assistance with mouse injections.
Grant Support:
W.X. was supported by grants from the National Institutes of Health (DP2HL137167, P01HL131471 and UG3HL147367), American Cancer Society (129056-RSG-16-093), and the Cystic Fibrosis Foundation. Z.W. was supported by U24HG009446. T.Y. was supported by a student fellowship by the China Scholarship Council. J.L.S. was supported by the National Cancer Institute F30CA239483, the University of Massachusetts Medical School Cancer Biology T32CA130807, and the University of Massachusetts Medical School MSTP T32GM107000.
Abbreviations
- HB
Hepatoblastoma
- ATAC-seq
Assay for Transposase Accessible Chromatin Sequencing
- hbHeps
Hepatocyte-Like Hepatoblastoma Cells
- YAP1
Yes-Associated Protein 1
- TET-ON
Tetracycline inducible-ON
- rtTA
Reverse tetracycline transactivator
- Tg-rtTA
Transgenic-rtTA
- SB-rtTA
Sleeping Beauty Transposase rtTA
- IRES
Internal ribosomal entry site
- GFP
Green fluorescent protein
- CTGF
Connective tissue growth factor
- Afp
α-fetoprotein
- IPA
Ingenuity pathway analysis
- GSEA
Gene set enrichment analysis
- Birc5/Survivin
Baculoviral inhibitor of apoptosis repeat-containing
- Amotl2
Angiomotin Like 2
- FoxM1
Forkhead box protein M1
- Hnf4a
Hepatocyte nuclear factor 4
- FoxA2
Forkhead box A2
- MUP
Major urinary protein
- FAH
Fumarylacetoacetate
- Rxra
Retinoid X receptor alpha
- Hnf1a
Hepatocyte nuclear factor 1 1
- Hnf1b
Hepatocyte nuclear factor 1 b
- FoxA1
Forkhead box A1
- Onecut1
One cut homeobox 1
- Onecut3
One cut homeobox 3
- NTBC
Nitisione
Footnotes
Disclosures: Authors do not disclose any potential conflicts of interest
References
Author names in bold designate shared co-first authorship
- 1.Trobaugh-Lotrario AD, Meyers RL, O’Neill AF, Feusner JH. Unresectable hepatoblastoma: current perspectives. Hepat Med 2017;9:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ortega JA, Douglass EC, Feusner JH, Reynolds M, Quinn JJ, Finegold MJ, Haas JE, et al. Randomized comparison of cisplatin/vincristine/fluorouracil and cisplatin/continuous infusion doxorubicin for treatment of pediatric hepatoblastoma: A report from the Children’s Cancer Group and the Pediatric Oncology Group. J Clin Oncol 2000;18:2665–2675. [DOI] [PubMed] [Google Scholar]
- 3.Lopez-Terrada D, Alaggio R, de Davila MT, Czauderna P, Hiyama E, Katzenstein H, Leuschner I, et al. Towards an international pediatric liver tumor consensus classification: proceedings of the Los Angeles COG liver tumors symposium. Mod Pathol 2014;27:472–491. [DOI] [PubMed] [Google Scholar]
- 4.Tao J, Calvisi DF, Ranganathan S, Cigliano A, Zhou L, Singh S, Jiang L, et al. Activation of beta-catenin and Yap1 in human hepatoblastoma and induction of hepatocarcinogenesis in mice. Gastroenterology 2014;147:690–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Min Q, Molina L, Li J, Adebayo Michael AO, Russell JO, Preziosi ME, Singh S, et al. beta-Catenin and Yes-Associated Protein 1 Cooperate in Hepatoblastoma Pathogenesis. Am J Pathol 2019;189:1091–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Felsher DW, Bishop JM. Reversible tumorigenesis by MYC in hematopoietic lineages. Mol Cell 1999;4:199–207. [DOI] [PubMed] [Google Scholar]
- 7.Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, et al. Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 2002;297:102–104. [DOI] [PubMed] [Google Scholar]
- 8.Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature 2004;431:1112–1117. [DOI] [PubMed] [Google Scholar]
- 9.Wu CH, van Riggelen J, Yetil A, Fan AC, Bachireddy P, Felsher DW. Cellular senescence is an important mechanism of tumor regression upon c-Myc inactivation. Proc Natl Acad Sci U S A 2007;104:13028–13033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, Shen Q, et al. Essential role for oncogenic Ras in tumour maintenance. Nature 1999;400:468–472. [DOI] [PubMed] [Google Scholar]
- 11.Fitamant J, Kottakis F, Benhamouche S, Tian Helen S, Chuvin N, Parachoniak Christine A, Nagle Julia M, et al. YAP Inhibition Restores Hepatocyte Differentiation in Advanced HCC, Leading to Tumor Regression. Cell Reports 2015;10:1692–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat Methods 2017;14:959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 2013;10:1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Z, Schulz MH, Look T, Begemann M, Zenke M, Costa IG. Identification of transcription factor binding sites using ATAC-seq. Genome Biology 2019;20:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, et al. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res 2018;46:D260–d266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kent WJ, Zweig AS, Barber G, Hinrichs AS, Karolchik D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics (Oxford, England) 2010;26:2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014;184:912–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci U S A 1996;93:10933–10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, Kay MA, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A 2007;104:14771–14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tschaharganeh DF, Xue W, Calvisi DF, Evert M, Michurina TV, Dow LE, Banito A, et al. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014;158:579–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wiesner SM, Decker SA, Larson JD, Ericson K, Forster C, Gallardo JL, Long C, et al. De novo induction of genetically engineered brain tumors in mice using plasmid DNA. Cancer Res 2009;69:431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koh KN, Park M, Kim BE, Bae KW, Kim KM, Im HJ, Seo JJ. Prognostic implications of serum alpha-fetoprotein response during treatment of hepatoblastoma. Pediatr Blood Cancer 2011;57:554–560. [DOI] [PubMed] [Google Scholar]
- 23.Cairo S, Armengol C, De Reynies A, Wei Y, Thomas E, Renard CA, Goga A, et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008;14:471–484. [DOI] [PubMed] [Google Scholar]
- 24.Sumazin P, Chen Y, Trevino LR, Sarabia SF, Hampton OA, Patel K, Mistretta TA, et al. Genomic analysis of hepatoblastoma identifies distinct molecular and prognostic subgroups. Hepatology 2017;65:104–121. [DOI] [PubMed] [Google Scholar]
- 25.Hooks KB, Audoux J, Fazli H, Lesjean S, Ernault T, Dugot-Senant N, Leste-Lasserre T, et al. New insights into diagnosis and therapeutic options for proliferative hepatoblastoma. Hepatology 2018;68:89–102. [DOI] [PubMed] [Google Scholar]
- 26.Kramer A, Green J, Pollard J Jr., Tugendreich S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014;30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alder O, Cullum R, Lee S, Kan AC, Wei W, Yi Y, Garside VC, et al. Hippo signaling influences HNF4A and FOXA2 enhancer switching during hepatocyte differentiation. Cell Rep 2014;9:261–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costa RH, Kalinichenko VV, Holterman AX, Wang X. Transcription factors in liver development, differentiation, and regeneration. Hepatology 2003;38:1331–1347. [DOI] [PubMed] [Google Scholar]
- 29.Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, et al. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell 2007;130:1120–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A 2010;107:1437–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 2008;22:1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 2009;16:425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J, Ning G, Duncan SA. Mammalian hepatocyte differentiation requires the transcription factor HNF-4alpha. Genes Dev 2000;14:464–474. [PMC free article] [PubMed] [Google Scholar]
- 34.Spath GF, Weiss MC. Hepatocyte nuclear factor 4 provokes expression of epithelial marker genes, acting as a morphogen in dedifferentiated hepatoma cells. J Cell Biol 1998;140:935–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, et al. A pathology atlas of the human cancer transcriptome. Science 2017;357. [DOI] [PubMed] [Google Scholar]
- 36.Matsumoto S, Yamamichi T, Shinzawa K, Kasahara Y, Nojima S, Kodama T, Obika S, et al. GREB1 induced by Wnt signaling promotes development of hepatoblastoma by suppressing TGFbeta signaling. Nat Commun 2019;10:3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fan B, Malato Y, Calvisi DF, Naqvi S, Razumilava N, Ribback S, Gores GJ, et al. Cholangiocarcinomas can originate from hepatocytes in mice. J Clin Invest 2012;122:2911–2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malato Y, Naqvi S, Schurmann N, Ng R, Wang B, Zape J, Kay MA, et al. Fate tracing of mature hepatocytes in mouse liver homeostasis and regeneration. J Clin Invest 2011;121:4850–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, et al. YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS Genet 2015;11:e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol 2015;17:1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fitamant J, Kottakis F, Benhamouche S, Tian HS, Chuvin N, Parachoniak CA, Nagle JM, et al. YAP Inhibition Restores Hepatocyte Differentiation in Advanced HCC, Leading to Tumor Regression. Cell Rep 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grompe M, al-Dhalimy M, Finegold M, Ou CN, Burlingame T, Kennaway NG, Soriano P. Loss of fumarylacetoacetate hydrolase is responsible for the neonatal hepatic dysfunction phenotype of lethal albino mice. Genes Dev 1993;7:2298–2307. [DOI] [PubMed] [Google Scholar]
- 43.Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, Hu Y, et al. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature 2011;475:386–389. [DOI] [PubMed] [Google Scholar]
- 44.Huch M, Dorrell C, Boj SF, van Es JH, Li VS, van de Wetering M, Sato T, et al. In vitro expansion of single Lgr5+ liver stem cells induced by Wnt-driven regeneration. Nature 2013;494:247–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yao X, Wang X, Liu J, Hu X, Shi L, Shen X, Ying W, et al. CRISPR/Cas9 - Mediated Precise Targeted Integration In Vivo Using a Double Cut Donor with Short Homology Arms. EBioMedicine 2017;20:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wangensteen KJ, Wilber A, Keng VW, He Z, Matise I, Wangensteen L, Carson CM, et al. A facile method for somatic, lifelong manipulation of multiple genes in the mouse liver. Hepatology 2008;47:1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yin H, Song CQ, Dorkin JR, Zhu LJ, Li Y, Wu Q, Park A, et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat Biotechnol 2016;34:328–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.