

Abstract
The human leukocyte antigen (HLA) system is the most polymorphic in the human genome that has been associated with protection and predisposition to a broad array of infectious, autoimmune, and malignant diseases. More recently over the last two decades, HLA class I alleles have been strongly associated with T-cell mediated drug hypersensitivity reactions. In the case of abacavir hypersensitivity and HLA-B*57:01, the 100% negative predictive value and low number needed to test to prevent a single case has led to a durable and effective global pre-prescription screening strategy. However, HLA associations are still undefined for most drugs clinically-associated with different delayed drug hypersensitivity phenotypes, and an HLA association relevant to one population is not generalizable across ethnicities. Furthermore, while a specific risk HLA allele is necessary for drug-induced T-cell activation, it is not sufficient. The low and incomplete positive predictive value has hindered efforts at clinical implementation for many drugs, however, has provided the impetus to understand the mechanisms of HLA class I restricted T-cell mediated drug hypersensitivity reactions. Current research has focused on defining the contribution of additional elements of the adaptive immune response and other genetic and ecologic risk factors that contribute to drug hypersensitivity risk. In this review we focus on new insights into immunological, pharmacological, and genetic mechanisms underpinning HLA associated drug reactions and the implications for future translation into clinical care.
Keywords: human leukocyte antigen, T-cell receptor, endoplasmic reticulum aminopeptidase, adverse drug reactions, killer-cell immunoglobulin-receptor
Introduction to adverse drug reactions.
Adverse drug reactions (ADRs) are leading causes of morbidity and mortality that are classified based on whether they result from pharmacologically predictable (“on-target”, type A) effects or are otherwise unpredictable (“off-target”, type B). Type B reactions comprise <20% of ADRs and include those that are immunologically mediated, themselves subcategorized into (i) immediate onset antibody-mediated reactions, including anaphylaxis, but also (ii) delayed hypersensitivity reactions (DHR), occurring ≥6 hours post drug administration and associated with cytotoxic T-cell activation towards self-tissue, most often skin but also other organs such as the liver, lung, kidney, or heart. Cutaneous DHR are diverse in clinical presentation, most commonly manifesting as non-lethal maculopapular exanthemas (MPE). However, patients can also develop life-threatening phenotypes including drug reaction with eosinophilia and systemic symptoms (DRESS), or Stevens-Johnsons syndrome and toxic epidermal necrolysis (SJS/TEN), associated with overall mortality of 20% (1). Despite clinical distinction, less common diseases such as SJS/TEN, estimated to affect 1–5/1,000,000 patients annually in the United States, are usually detected in the post-marketing phase of drug development. Significant immunogenic discoveries in the last twenty years have driven understanding of ‘at-risk’ patients towards preventative screening, initiated by seminal discovery of strong association between the reverse-transcriptase inhibitor abacavir, development of abacavir hypersensitivity syndrome (AHS), and carriage of human leukocyte antigen (HLA)-B*57:01. The PREDICT-1 double-blind clinical trial (2), which randomized individuals to receive real time HLA-B*57:01 screening versus the standard of care, demonstrated that HLA-B*57:01 had 100% negative predictive value (NPV) and a positive predictive value (PPV) of 55%. In defining HLA-B*57:01 as necessary, high confidence for prescribing abacavir to HLA-B*57:01 negative individuals has eliminated false positive clinical diagnosis and near eliminated AHS. Many other HLA associations have since been described, however many show weaker NPV without applicability across race and all have incomplete PPV (1), limiting cost-benefit and highlighting the need for continued vigilance. This also suggests that other genetic and ecological risk factors are important in patients carrying a known HLA risk factor. In this review we focus on new insights into immunological, pharmacological, and genetic mechanisms underpinning HLA associated drug reactions and the implications for increased predictivity for future translation into clinical care.
HLA and risk of Drug Hypersensitivity.
HLA plays a fundamental role in the presentation of self-antigen to T-cells for tolerance, but also foreign antigens for cytotoxic destruction. Highly specific interaction between HLA, antigen, and T-cell, through specific T-cell receptors (TCR), is defined signal 1 and a requirement for T-cell activation. Non-classical HLA loci (-E, -F, -G, -H) have few polymorphisms and typically inhibitory roles spanning innate and adaptive immune response. However, classical HLA class I (-A, -B, -C) expressed on all nucleated cell and class II (-DP, -DQ, -DR) expressed only on antigen presenting cells are highly polymorphic with >25,000 alleles annotated and HLA-B the most polymorphic gene in the human genome. Importantly, single HLA allele associations described since the 1970’s, with protection from or susceptibility to a broad array of infectious, autoimmune, and malignant disease (Figure 1A), led understanding for a critical role in T-cell function and implicated similar role in inter-individual susceptibility to CD8+ T-cell mediated DHR. Indeed, HLA risk alleles for different DHR have been described since 2002 driven by the advent of high-resolution HLA typing (Figure 1A, Table S1). Importantly this allele-dependant specificity is a product of structure, with HLA class I heterodimers composed of a heavy α chain forming a highly-specific peptide binding groove, as well as a conserved, constant immunoglobulin-like domain (β2-microglobulin) (Figure 1B). The HLA class I-binding groove, characterized by a highly conserved structure of α-helices on both sides and a β-pleated sheet on the bottom/floor (Figure 1C), is divided into 6 pockets (A-F) with closed conformation in which antigen of typically 8–11 amino acids may bind (1). Although the polymorphic residues and pockets impart differential binding specificities, three overarching but non-mutually exclusive mechanisms proposed by which small molecular weight drugs can stimulate T-cells (hapten hypothesis, pharmacological interaction (PI) concept, and altered peptide repertoire (1)), understanding of which is critical to determine HLA-restricted structural risk.
Figure 1. Association and structural role of HLA in drug-induced T-cell activation.
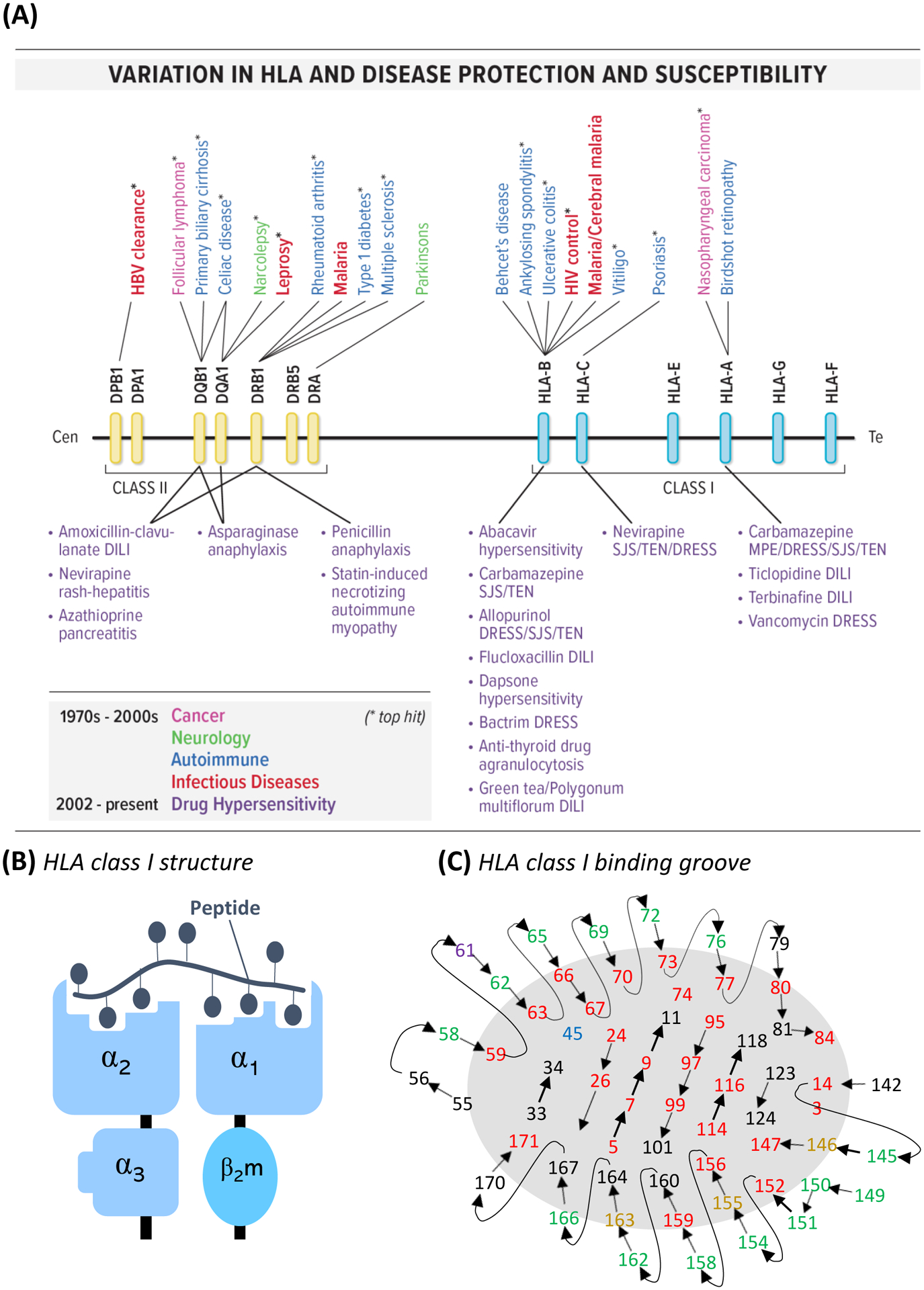
(A) HLA class I and II disease susceptibility and protection highlighting the discovery of numerous associations with drug hypersensitivity over the last 2 decades. *indicates top hit for disease. (B) Schematic of HLA class I molecule structure and (C) HLA class I peptide-binding groove with residues labelled with potential to influence peptide/T-cell binding. Central connected residues in lines form those of the beta strand platform floor, with connected residues 55–84 and 142–171 form those of the respective α1 and α2 helices. Green residues interact with the T-cell receptor. Red residues interact with peptide. Yellow residues point up and into the binding site, while the purple residue points away, and the blue residue points towards. Adapted from Garrett et al, 1989 (24).
Current models of HLA-restricted T-cell activation by drug-derived antigens.
Landsteiner and Jacobs proposed the hapten hypothesis; a process by which chemically-reactive compounds covalently bind self-proteins to form hapten-carrier complexes and undergo intracellular processing to drug hapten-modified peptide(s) that subsequently elicit T-cell response (Figure 2A, i). The beta-lactam antibiotic flucloxacillin is a key example of a hapten-forming drug, described to bind protein via lysine residues with risk HLA-B*57:01-presented flucloxacillin-modified peptides recently annotated as derived from self-protein (3). However, the hapten hypothesis is not suitable for chemically inert drugs such as sulfamethoxazole, which may similarly stimulate patient T-cells in cell cultures without capacity for antigen uptake and processing. Thus, the pharmacological interaction (PI) concept was established with direct, labile, and non-covalent interaction between drug antigen and HLA or TCR driving rapid T-cell activation (Figure 2A, ii, iii). However, models are not mutually exclusive, with both parent drug- and reactive metabolite-responsive T-cells detected from patients with sulfamethoxazole-induced reactions, stimulated via direct PI or after initial hapten formation, respectively. Finally, in the altered peptide model, the drug modifies the risk HLA itself, changing the conformational specificity of the binding groove and the self-peptides presented, subsequently recognized as foreign (Figure 2A, iv). Crystal structure of abacavir bound to HLA-B*57:01 and synthetic or natural peptide was solved by two independent groups (4). To date, abacavir remains the only drug specifically associated with the altered peptide repertoire model, with altered immunopeptidome loading of HLA-B*57:01 described in the endoplasmic reticulum (5). Recently this hypothesis has been expanded to an altered TCR model, postulating that drug may bind TCR and alter the T-cell-recognized repertoire (Figure 2A, v). Future strategies to characterize risk HLA-presented immunogenic peptides are now required to define relevant structural risk signatures and screen out cross-reactivities, critical for safety of ongoing therapeutic treatment and future drug development.
Figure 2. Models of drug-induced HLA-restricted T-cell activation and heterologous immunity.
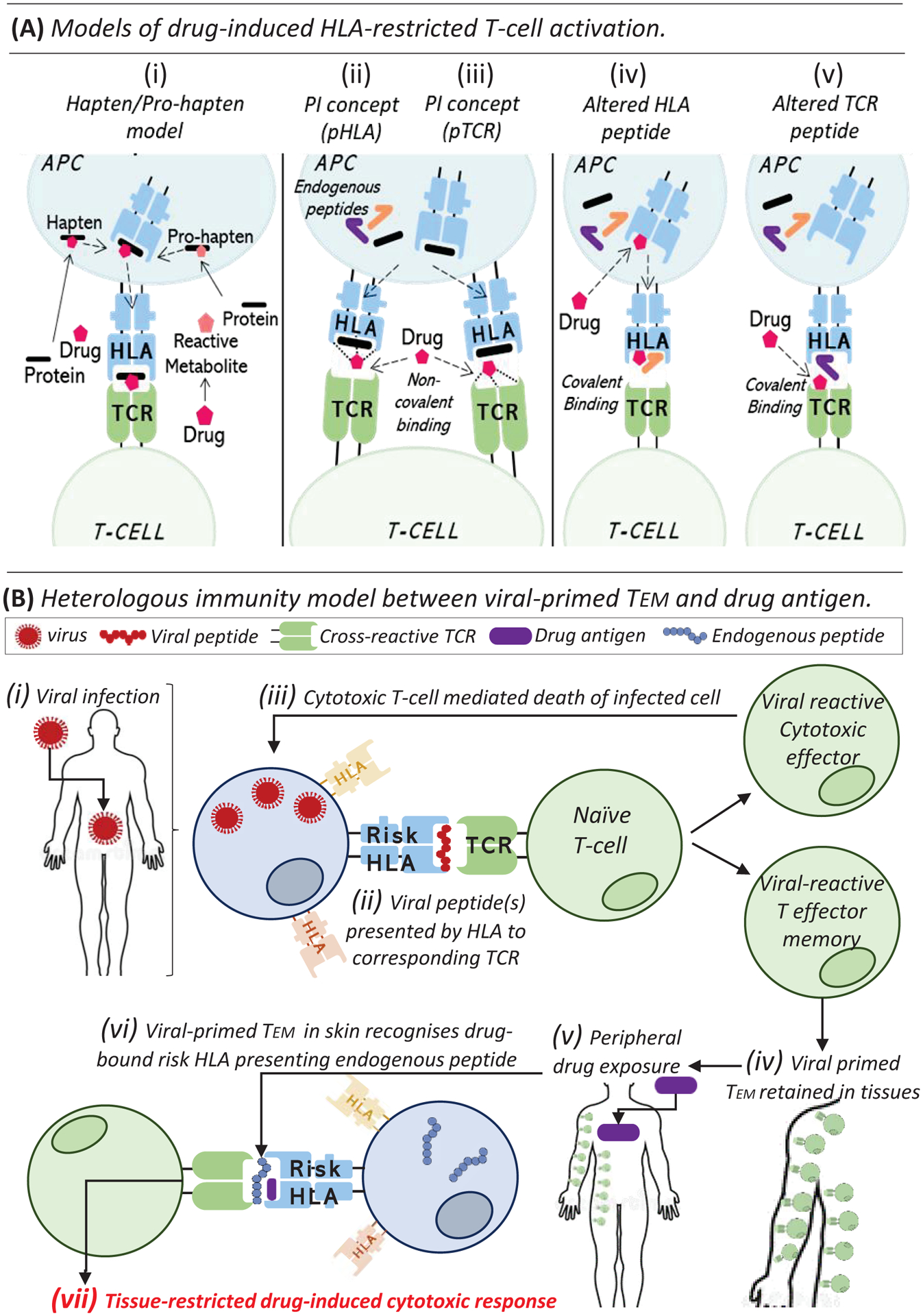
(A) Models of drug-induced HLA-restricted T-cell activation. (i) Hapten/pro-hapten model whereby drug-antigen binds to self-protein prior to intracellular processing to form immunogenic drug-modified peptide, presented on risk HLA to corresponding TCR. (ii, iii) The pharmacological interaction of drugs with immune receptors (PI) concept postulates that drug-antigen directly and non-covalently binds to risk HLA (ii) or TCR (iii) to stimulate T-cells, bypassing requirement for antigen-processing. (iv) The altered peptide repertoire model shows drug-antigens can bind to HLA altering the repertoire of self-peptides that bind and thus seen as foreign and immunogenic. (v) Similarly, an altered TCR repertoire model proposed similar drug binding to the TCR skews HLA-presented peptides recognized as immunogenic by the T-cell. (B) Heterologous immunity model. (i) Patient infected with virus earlier in life, with viral peptides presented on risk HLA to prime naïve T-cells for clonal expansion and differentiation into (ii) viral-reactive T-cell cytotoxic effectors to destroy virus-infected cells and (iii) T effector memory (TEM) for long-term immunological memory and protection upon re-exposure and (iv) positioned in tissues such as skin. (v) Drug antigen exposure later in life leads to (vi) cross-reactive response with viral-reactive TEM resulting in (vii) drug-induced tissue-restricted T-cell activation.
HLA alleles are risk factors for the specific drug and phenotype of drug hypersensitivity.
Although a number of HLA class II associations have been identified (Table S1), evidence of preferential drug allele binding remains limited and risk associations for different DHR remain statistically weighted towards HLA class I and the pure T-cell mediated reactions such as AHS. Additional strong associations include allopurinol-DRESS and SJS/TEN with HLA-B*58:01, carbamazepine (CBZ)-SJS/TEN with HLA-B*15:02 in Southeast Asians, and, in 2019, HLA-A*32:01 with vancomycin-DRESS in Europeans (6). In vitro cross-reactivity with HLA-A*32:01 for other glyco/lipoglycopeptides highlighted that the minority of patients displayed cross-reactivity and those that did shared a HLA class II haplotype (DQA1*01:01, DQB1*05:03) (7), highlighting how understanding of extended HLA-type may be of utility to select safe drugs for ongoing treatment and the potential for class II-restricted CD8+ T-cell cross-recognition, warranting further study (7). While robust, inexpensive, and rapid single-allele screening assays now exist for HLA-B*57:01, -B*15:02, -B*58:01, -B*13:01, and - A*32:01, global utility is hindered by lack of generalized risk across diverse populations with divergent rates of allelic carriage. Indeed, while HLA-B*58:01 risk is replicated for allopurinol-SCAR in Koreans, Southern Chinese, and Southeast Asian populations, Europeans have far lower prevalence of this allele and both the low PPV and the incomplete NPV impede screening efforts. Further example is strong association of CBZ-SCAR with HLA-B*15:02 in Taiwanese, where benefit of preventative HLA-screening is highlighted by a report where 0/4120 HLA-B*15:02 negative Taiwanese patients developed CBZ-SJS/TEN (8). However, with <1% carriage in Africans and Europeans, alternate risk alleles for CBZ hypersensitivities are recently identified, including HLA-A*31:01 with CBZ-SCAR and drug induced liver injury (DILI) in Europeans. Importantly, similar associations have yet to be defined across drugs for minority populations without ethnicity-matched tolerant cohorts required for population-wide study. An intriguing prospect to define population-shared risk is the potential for shared binding specificities of different HLA alleles, with nevirapine-induced rash associated with HLA-C*04:01 in Malawian HIV patients (9), but also across HLA-C*04 alleles with similar F pockets, including HLA-C*05:01 and HLA-C*18:01 (10). Alternatively, an additional report highlighted HLA-B*56:02 carriage among Indigenous Australian patients with phenytoin-DRESS through simple observation of shared allelic presence in carefully curated ethnicity-matched samples (11). While this imparts a need for ethnic awareness during (i) clinical treatment and (ii) reporting of drug-specific reactions, crucially all HLA associations described to date maintain an incomplete, often low PPV, meaning that while HLA is necessary it is not the sole risk factor, and other parameters drive risk of disease in HLA-predisposed patients.
Antigen presentation and peptide processing.
Carr confirmed HLA-C*04:01 predisposition for nevirapine-rash in sub-Saharan Africans, but also observed a protective effect from an allotype of endoplasmic reticulum aminopeptidase (ERAP), a polymorphic enzyme involved in N-terminal peptide processing immediately prior to HLA loading (9). Importantly, inter-individual ERAP polymorphisms are previously described which dictate trimming capacity with hypo- or hyper-active variants either over- or under-trimming peptides. Crucially, longer less immunogenic peptides impair HLA-class I-restricted CD8+ T-cell response and clearance of infectious disease. In 2020, hypoactive allotypes of ERAP1 were similarly reported as protective from AHS in risk HLA-B*57:01+ patients (12). Intriguingly, abacavir-treatment of cells alters the amino acid preference of HLA-B*57:01-bound peptides to those with hydrophobic or aromatic C-terminal residues, the same residues favoured for efficient trimming by ERAP1. Similar investigation of ERAP allotypes across different drug-induced hypersensitivities is now warranted with clinical implication of protective alleles to de-risk drug use in risk-HLA presenting patients, of particular utility for personalised treatment of front-line drugs that remain without safe and effective alternatives.
Drug Metabolism.
Inter-individual differences in drug metabolism rate also have propensity to alter immunogenic exposures but to upstream parent drug, metabolite(s), or hapten(s). A key example is association between poor renal function, delayed clearance of oxypurinol, and increased risk of and mortality associated with allopurinol-SCAR (13). Yet despite metabolism representing an extensive, multi-enzyme step process for many drugs involving highly polymorphic enzymes, few specific variants have been associated with risk of DHR. The main exception is phenytoin hypersensitivity associated with CYP2C9 (14). Here, the CYP2C9*3 variant reduces the metabolic capacity by 90%, slowing clearance of immunogenic phenytoin. In updated review of the only current clinical recommendation of dose adjustment to prevent HLA-restricted DHR, CYP2C9 intermediate or poor metabolising patients with risk HLA-B genotypes should have their starting phenytoin dose reduced by 25–50%, respectively (14).
Proposed influence of HLA ligation by killer-cell immunoglobulin like-receptors.
During characterisation of flucloxacillin-modified peptides from the HLA-B*57:01-presented ligandome, Puig reported modification of a lysine residue on the HLA molecule itself, questioning how this affects binding of peptides, corresponding TCR, but also Killer-cell immunoglobulin like receptors (KIR) (3). Indeed, polymorphic inhibitory or activating KIR are epistatically associated with and distinct ligands for HLA, presented on both natural killer (NK) and T-cells to control cytotoxic degranulation. KIR selectivity is based on three distinct HLA epitopes; C1 and C2 found on HLA-C alleles, and Bw4, a motif at positions 77–83 of select HLA-A and B alleles. Bw4 itself encodes three separate isoforms impacting KIR3DL1 recognition: one relevant to HLA-A, and two split for HLA-B around position dimorphism for threonine (80T) or isoleucine (80I). Bw4-80I is previously associated with more inhibitory allotypes with higher expression driving weaker cell activation and cytotoxicity. As such, Bw4-80I has been associated with non-progression of HIV (15). Thus, as KIR alleles influence HLA-restricted adaptive response against virus, they are proposed to similarly alter response to drug antigen. Although it has been shown that T-cells in the peripheral blood of patients with SJS/TEN overexpress KIR2DL2 and KIR2DL3 (16), implying a role in HLA-regulation during DHR, genetic association with DHR remains to be elucidated.
Distribution of T-cells with HLA-antigen corresponding TCR.
For host defence, a substantial repertoire of unique TCR clonotypes is required to recognise diversity in possible foreign antigen but with specificity such to avoid self. To accommodate, the TCR α and β loci, consisting of variable (V), joining (J) and constant (C) regions, undergo rearrangement during T-cell development resulting in an extensive repertoire of TCR with unique complimentary determining region (CDR)3 sequence for antigen interaction. Intriguingly, TCR repertoires may be unevenly distributed in the human body with tissue-resident memory (Trm) CD8+ T-cell populations of the mucous membranes and skin retaining TCR specificities for previously encountered viral antigen. These tissues, selected for immunological memory as likely re-exposure sites, are also the specific sites of drug-induced damage during SJS/TEN. Importantly, cross-reactive and HLA-restricted drug-induced activation of viral peptide-primed but otherwise quiescent Trm T-cells is proposed as a heterologous immunity model of why HLA-restricted DHR, despite peripheral distribution of drug, often distinctly targets the skin (Figure 2B). Importantly, response during DHR is independent of active pathogenic replication or reactivation, with supporting evidence by Lucas demonstrating pre-existence of abacavir-responsive CD8+ T-cells in all risk HLA-B*57:01+ drug-naïve healthy individuals (17). Stable interaction of HLA-antigen and TCR initiates a complex intracellular signalling cascade resulting in T-cell activation. Briefly, lymphocyte-specific protein tyrosine kinase (LCK) is recruited and phosphorylates an immunoreceptor tyrosine-based activation motif (ITAM) and Zap70. Zap70 is then able to phosphorylate the linker for activation of T-cells (LAT) that recruits and activates various signalling pathway components that comprise the LAT signalosome. While this cascade is well-documented, how TCR binding translates signal across the cell membrane remains ill-defined, with multiple models proposed (Figure 3). As a requirement to complete signal 1, TCRs are undeniably involved in HLA-restricted DHR, however unique specificity of an individual’s repertoire is now considered a risk factor in itself, with identification of a dominant TCR in the blister of patients with HLA-B*15:02-restricted CBZ-SJS/TEN, with specific “VFDNTDKLI” TCRα CDR3 and “ASSLAGELFF” TCRβ CDR3 (18). Importantly, this single, drug-reactive TCRαβ clonotype was not dominantly expanded in paired peripheral blood and was absent from CBZ-tolerant controls who expressed the risk HLA, directly distinguishing hypersensitive from tolerant patients and directing need to explore cells from affected tissue to delineate disease. Villani has also since reported highly restricted preferential TCRβ V-chain usage on cytotoxic CD8+ blister T-cells from patients with different culprit drug-induced TEN (19). While single, TCRβ CDR3 are similarly dominantly expanded in the blister of patients with HLA-B*58:01-restricted allopurinol-SJS/TEN (20), defining the complete TCRαβ sequence is crucial to determine antigen-reactivity, possible by single-cell sequencing, and now warranted across drugs, disease phenotypes and populations.
Figure 3. Models of HLA-associated drug-induced TCR triggering.
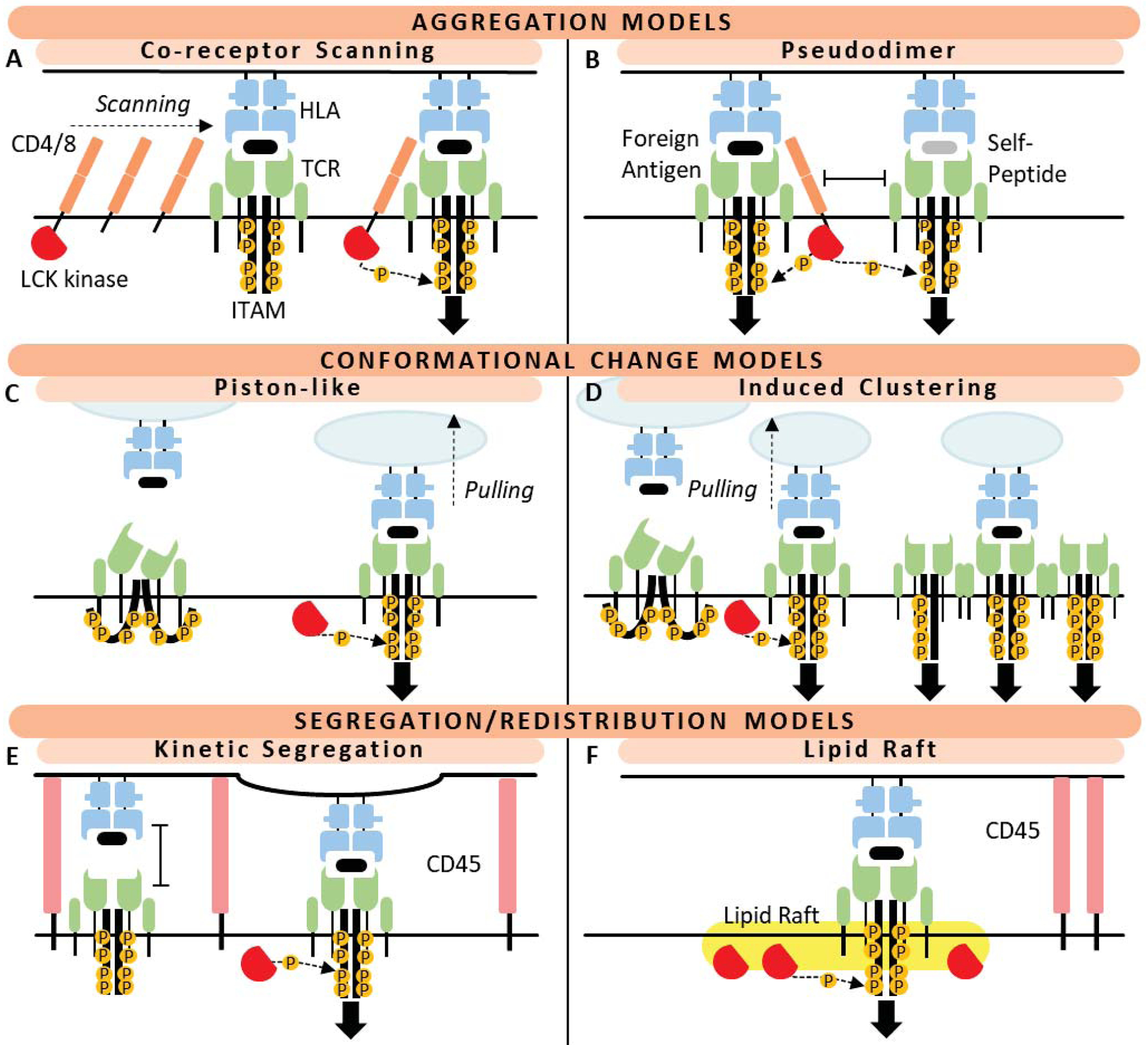
Co-receptor aggregation models: (A) The co-receptor heterodimerisation model states HLA-TCR interaction is accompanied by binding of coreceptor (CD4/8). Lck, a tyrosine kinase, is bound to the coreceptor and thus brought into close proximity with immunoreceptor tyrosine-based activation motif (ITAM) on the TCR CD3 chain to begin phosphorylation cascade for intracellular signalling. (B) The pseudodimer model requires additional presence of a second HLA-TCR interaction presenting self-peptide to boost downstream signalling. Conformational change models: (C) The ‘piston-like’ model states interaction of TCR with HLA-peptide initiates an extracellular conformational change that translates to the intracellular portion of the TCR to ‘open’ the folded conformation CD3 chain to expose ITAMs for phosphorylation. (D) The induced clustering model is an adaption of piston-like movement that allows TCR clustering required for sufficient amplification of signal for downstream signalling. Segregation and redistribution models: (E) The kinetic model identifies that large fluid cell surface proteins such as CD45 prevent T-cell activation. Exclusion of these longer protruding inhibitory molecules from activation sites allow close contact between APCs and T-cells for stable HLA-TCR interaction. (F) The lipid-raft model states that lipid rafts containing T-cell activation molecules such as Lck, but excluding inhibitory proteins (CD45), may associate with TCR to provide sufficient phosphorylation activity. Mechanisms and figure adapted from Van der Merwe et al (25).
Dynamic co-signalling proposed to regulate HLA-restricted tolerance or cytotoxic response.
Following engagement of TCR, co-signalling receptor-ligand interactions determine whether tolerogenic or cytotoxic response ensues. These checkpoints combine to fine-tune immune regulation as signal 2, manipulating the T-cell activation threshold with co-stimulation pathways driving cytotoxic activation, and those of co-inhibition towards tolerance. Importantly, co-inhibitory checkpoint receptors have polymorphic variants described that associate with onset of HLA-restricted autoimmune disease. Although there is yet no direct association with DHR, blockade of co-inhibitory programmed death-1 (PD-1) and cytotoxic lymphocyte antigen-4 (CTLA-4) is shown to increase the proliferative response of drug-reactive T-cells in vitro (21). Interestingly, immune checkpoint inhibitors (ICI) are now commonplace in cancer immunotherapy, with off-target ADR commonly reported including increased incidence of DHR. In 2018, Cardone (22) reported that transgenic HLA-B*57:01+ mice remained abacavir tolerant, yet prior CD4+ T-cell depletion led to greater maturation of dendritic cells and abacavir-induced CD8+ T-cell inflammatory response in skin. Importantly, blockade of co-stimulatory receptors prevented CD8+ T-cell activation suggesting that CD4+ T-cells enabled tolerance. While therapeutic dysregulation could impart risk, co-signalling may also be altered by other environment exposures. Thus, it may be that T-cells exhibit dynamic loss of tolerance, suggesting not just inter- but intra-patient susceptibility. Importantly, other pathways may also regulate T-cell response, with a missense variant of protein tyrosine phosphatase PTPN22 associated with and proposed to reduce tolerance in patients with DILI (23).
Conclusions and future needs.
The guideline-based pre-prescription screening strategy implemented for HLA-B*57:01 and AHS provided a clear translational roadmap for HLA-B*15:02 pre-prescription to prevent carbamazepine SJS/TEN in Southeast Asia (8), with other emerging examples including HLA-B*58:01 with allopurinol (Southeast Asians) (20). However, HLA associations remain undefined for most drugs and clinical phenotypes, and an association relevant to one population may not be relevant across ethnicities (8). Furthermore, while a specific risk HLA allele is necessary for drug-induced T-cell activation, it is not sufficient. Low PPV has stalled clinical implementation efforts for many drugs but has provided impetus to understand the mechanisms of HLA class I-restricted T-cell mediated DHR with several other risk factors now described albeit largely yet to translate clinical guidelines and summarised in Figure 4. Steps in antigen processing may be important and genetic variation in ERAP impacts availability of risk HLA-presented immunodominant moieties (9, 12). Additionally, at site of tissue-specific damage there must be availability of co-stimulated T-cells expressing corresponding TCR specificities to surpass tolerance and drive cytotoxic activation, with implication of heterologous response from a “double HLA hit” model between remote viral infection and the drug altered endotype many years later. CD4+ T cells may play an important role in immune tolerance through suppression of dendritic cell maturation (22), and altered rates of drug metabolism, particularly in the setting of a risk HLA allele, may modify risk of DHR (14). Consequently, studies examining multiple genetic and ecological effects are now warranted across drugs and reactions to increase PPV of relevant HLA risk alleles. However, where HLA risk remains undefined, strategies are also needed to propose and validate similar risk, including for minority populations who remain understudied. Understanding of shared HLA drug/peptide-binding specificities for prevalent alleles specific to populations represents one option (10). Another is careful curation of ethnicity, reaction, and drug-matched clinical samples (11). This will require continued development of internationally collaborative biorepositories, led by specialist clinicians to collect cells, DNA, RNA and tissue samples to look at genetic and functional correlates from well-phenotyped patients. We anticipate that multi-omic studies across diverse populations will be necessary to define the contribution of genetic, epigenetic, cellular risk factors and mechanisms that will help define strategies to improve targeted preventive, diagnostic and treatment efforts.
Figure 4. Cellular risk factors associated with or proposed to influence HLA-restricted drug-induced T-cell activation.
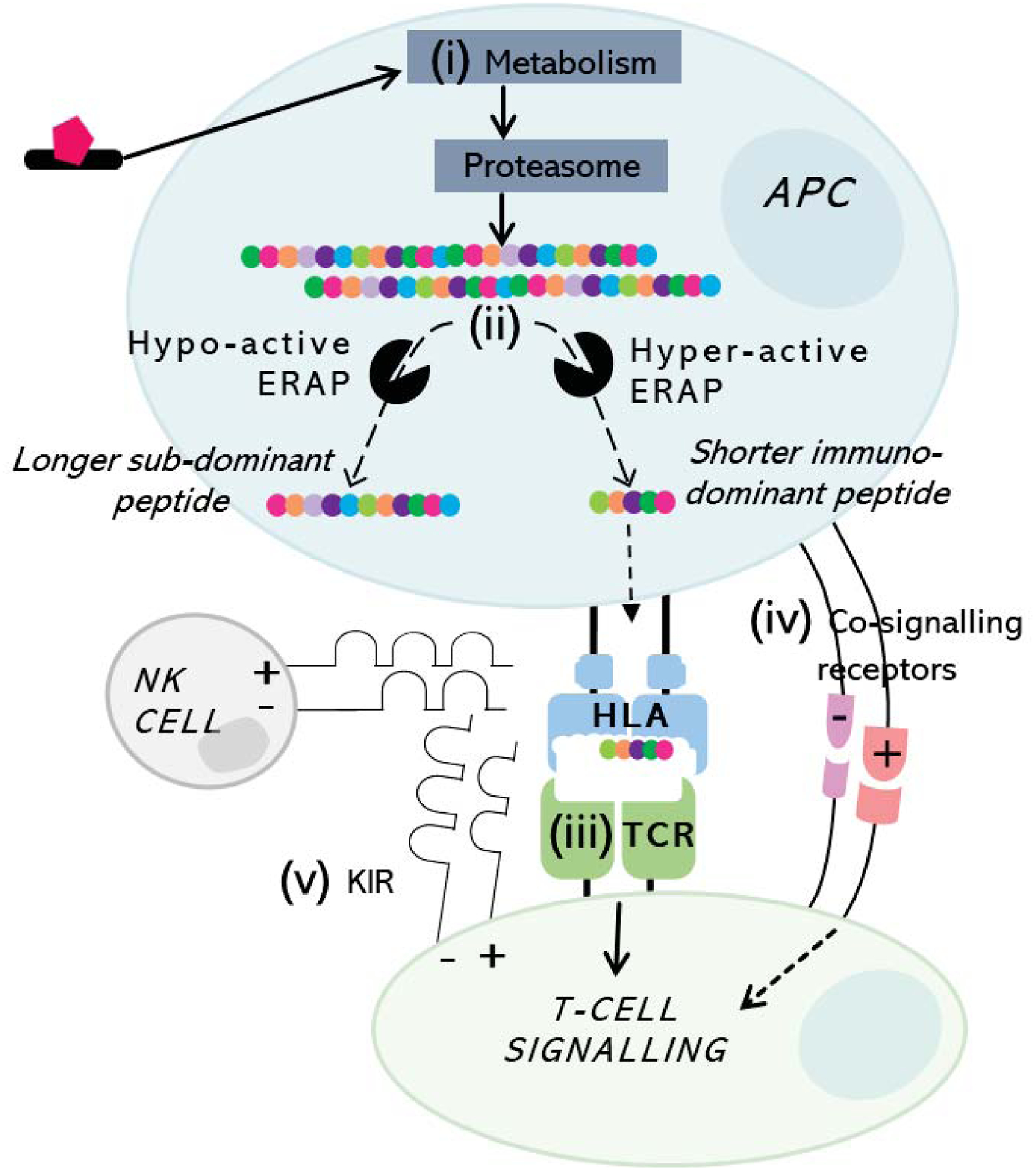
Antigen may undergo (i) metabolism and proteasomal processing to peptides which finally undergo trimming via (ii) endoplasmic reticulum aminopeptidases (ERAP) to an optimal length before loading into the HLA class I-binding groove. Risk HLA-presented peptide is then presented to T-cells with corresponding (iii) TCR specificity, forming signal 1 (stable HLA-antigen-TCR) in the presence of (iv) conversely co-signalling pathways skewing resultant response as signal 2 towards cytotoxicity (co-stimulation) or tolerance (co-inhibition). (v) KIR expressed on both NK- and T-cells bind to distinct HLA epitopes with specificity for presented peptide to regulate degranulation and cytotoxic response.
Supplementary Material
Acknowledgements.
The authors would like to thank Karen Adamson for illustration work on Figure 1A.
Funding.
E.J.P. reports grants from National Institutes of Health (P50GM115305, R01HG010863, R01AI152183, R21AI139021, U01AI154659) and from the National Health and Medical Research Council of Australia.
Footnotes
Conflicts of Interest. EJP is codirector of IIID Pty Ltd that holds a patent for HLA-B*57:01 testing for abacavir hypersensitivity; she holds a provisional patent for HLA-A*32:01 testing for vancomycin hypersensitivity. All other authors declared no competing interests for this work.
References.
- (1).Redwood AJ, Pavlos RK, White KD & Phillips EJ HLAs: Key regulators of T-cell-mediated drug hypersensitivity. Hla 91, 3–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mallal S et al. HLA-B*5701 screening for hypersensitivity to abacavir. The New England journal of medicine 358, 568–79 (2008). [DOI] [PubMed] [Google Scholar]
- (3).Puig M et al. Alterations in the HLA-B*57:01 Immunopeptidome by Flucloxacillin and Immunogenicity of Drug-Haptenated Peptides. Front Immunol 11, 629399 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Illing PT et al. Immune self-reactivity triggered by drug-modified HLA-peptide repertoire. Nature 486, 554–8 (2012). [DOI] [PubMed] [Google Scholar]
- (5).Illing PT et al. Kinetics of Abacavir-Induced Remodelling of the Major Histocompatibility Complex Class I Peptide Repertoire. Frontiers in Immunology 12, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Konvinse KC et al. HLA-A*32:01 is strongly associated with vancomycin-induced drug reaction with eosinophilia and systemic symptoms. The Journal of allergy and clinical immunology 144, 183–92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Nakkam N et al. Cross-reactivity between vancomycin, teicoplanin, and telavancin in patients with HLA-A*32:01-positive vancomycin-induced DRESS sharing an HLA class II haplotype. The Journal of allergy and clinical immunology 147, 403–5 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Chen P et al. Carbamazepine-Induced Toxic Effects and HLA-B*1502 Screening in Taiwan. New England Journal of Medicine 364, 1126–33 (2011). [DOI] [PubMed] [Google Scholar]
- (9).Carr DF et al. Genome-wide association study of nevirapine hypersensitivity in a sub-Saharan African HIV-infected population. Journal of Antimicrobial Chemotherapy 72, 1152–62 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pavlos R et al. Shared peptide binding of HLA Class I and II alleles associate with cutaneous nevirapine hypersensitivity and identify novel risk alleles. Sci Rep 7, 8653 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Somogyi AA, Barratt DT, Phillips EJ, Moore K, Ilyas F & Gabb GM High and variable population prevalence of HLA-B* 56: 02 in indigenous Australians and relation to phenytoin-associated drug reaction with eosinophilia and systemic symptoms. British journal of clinical pharmacology 85, 2163–9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Pavlos R et al. New genetic predictors for abacavir tolerance in HLA-B* 57: 01 positive individuals. Human immunology, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chung W-H et al. Insights into the poor prognosis of allopurinol-induced severe cutaneous adverse reactions: the impact of renal insufficiency, high plasma levels of oxypurinol and granulysin. Annals of the Rheumatic Diseases 74, 2157 (2015). [DOI] [PubMed] [Google Scholar]
- (14).Karnes JH et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2C9 and HLA-B Genotypes and Phenytoin Dosing: 2020 Update. Clinical Pharmacology & Therapeutics 109, 302–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jiang Y et al. KIR3DS1/L1 and HLA-Bw4-80I are associated with HIV disease progression among HIV typical progressors and long-term nonprogressors. BMC Infectious Diseases 13, 405 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Morel E, Escamochero S, Cabañas R, Díaz R, Fiandor A & Bellón T CD94/NKG2C is a killer effector molecule in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. Journal of Allergy and Clinical Immunology 125, 703–10.e8 (2010). [DOI] [PubMed] [Google Scholar]
- (17).Lucas A et al. Abacavir-reactive memory T cells are present in drug naïve individuals. PLoS One 10, e0117160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Pan R-Y et al. Identification of drug-specific public TCR driving severe cutaneous adverse reactions. Nature communications 10, 1–13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Villani AP et al. Massive clonal expansion of polycytotoxic skin and blood CD8(+) T cells in patients with toxic epidermal necrolysis. Sci Adv 7, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Chung W-H et al. Oxypurinol-Specific T Cells Possess Preferential TCR Clonotypes and Express Granulysin in Allopurinol-Induced Severe Cutaneous Adverse Reactions. Journal of Investigative Dermatology 135, 2237–48 (2015). [DOI] [PubMed] [Google Scholar]
- (21).Gibson A et al. The Effect of Inhibitory Signals on the Priming of Drug Hapten-Specific T Cells That Express Distinct Vβ Receptors. Journal of immunology (Baltimore, Md : 1950) 199, 1223–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Cardone M et al. A transgenic mouse model for HLA-B*57:01-linked abacavir drug tolerance and reactivity. J Clin Invest 128, 2819–32 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Cirulli ET et al. A Missense Variant in PTPN22 is a Risk Factor for Drug-induced Liver Injury. Gastroenterology 156, 1707–16.e2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Garrett TP, Saper MA, Bjorkman PJ, Strominger JL & Wiley DC Specificity pockets for the side chains of peptide antigens in HLA-Aw68. Nature 342, 692–6 (1989). [DOI] [PubMed] [Google Scholar]
- (25).van der Merwe PA & Dushek O Mechanisms for T cell receptor triggering. Nature Reviews Immunology 11, 47–55 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.