

Abstract
MET exon 14 alterations are oncogenic drivers of non-small cell lung cancers (NSCLCs).1 These alterations are associated with increased MET activity and preclinical sensitivity to MET inhibition.2 Crizotinib is a multikinase inhibitor with potent activity against MET.3 The antitumor activity and safety of crizotinib were assessed in 69 patients with advanced NSCLCs harboring MET exon 14 alterations in an expansion cohort of an open-label phase 1 study of crizotinib (NCT00585195). The confirmed objective response rate was 32% (95% confidence interval [CI], 21–45) among 65 response-evaluable patients. Objective responses were observed independent of the molecular heterogeneity that characterizes these cancers and did not vary by MET exon 14 alteration splice site region and mutation type, concurrent increased MET copy number, or the detection of a MET exon 14 alteration in ctDNA. The median duration of response was 9.1 months (95% CI, 6.4–12.7). The median progression-free survival was 7.3 months (95% CI, 5.4–9.1). MET exon 14 alteration defines a molecular subgroup of NSCLCs for which MET inhibition with crizotinib is active. These results address an unmet need for targeted therapy in patients with MET exon 14-altered lung cancers and adds to an expanding list of genomically-driven therapies for oncogenic subsets of NSCLC.
Introduction
MET exon 14 alterations are identified in 3 to 4% of non-small cell lung cancers (NSCLCs)4–6 and less frequently in other cancers.7 These drivers are notable in that an extremely diverse set of mutations (point mutations, indels, or large deletions) can result in a convergent biology that is reliant on increased MET activity.8 Base substitutions and indels of varying lengths disrupt spatially distinct splicing sites that flank MET exon 14 and can individually lead to the exclusion of MET exon 14 at the RNA level.7,9 MET exon 14 encodes a juxtamembrane region of the MET receptor that contains the CBL E3 ubiquitin ligase binding site, Y1003.10 Large deletions encompassing MET exon 14 and mutations involving Y1003 similarly interfere with CBL binding.11 In the absence of Y1003, CBL is unable to regulate ubiquitin-mediated degradation of the MET receptor.12 This leads to decreased turnover of MET and increased MET signaling that drives oncogenesis, recognizing that additional mechanisms contributing to altered MET kinase activity cannot be excluded.
Crizotinib is a multi-tyrosine kinase inhibitor that is approved for the treatment of ALK- or ROS1-rearranged advanced NSCLCs. Apart from its activity against ALK and ROS1, it has potent activity against MET and low nanomolar potency in cell lines that harbor MET exon 14 alterations.3,13 In 2015, we reported the first cases of response to crizotinib in patients with MET exon 14-altered lung cancers in a small retrospective series.14 This established early proof of principle of the activity of a MET inhibitor in these cancers and was followed by additional reports.4,7,13,15–20 In this paper, we evaluated the antitumor activity of crizotinib in MET exon 14-altered advanced NSCLCs in an expansion cohort of the PROFILE 1001 trial. In contrast to prior reports, this was the first prospective study to characterize the activity of a MET inhibitor in a dedicated cohort of patients with MET exon 14-altered NSCLCs.
Results
Patient Characteristics
From September 11, 2014 through January 26, 2018, 69 patients with MET exon 14-altered NSCLCs were enrolled and treated with crizotinib (Table 1, Extended Data Fig. 1). The median age was 72 years. The majority (61%) were former smokers. Most patients (84%) had adenocarcinoma; 9% had sarcomatoid carcinoma. The majority of patients (62%) had received ≥1 previous lines of treatment for advanced disease. MET exon 14 alterations were identified by local testing, primarily using DNA- or RNA-based next-generation sequencing (NGS) in 96% of patients (Table 1, Supplementary Table 1). The median duration of treatment was 7.4 months (95% confidence interval [CI], 5.5–9.1); 20 patients (29%) continued to receive crizotinib after the data cutoff date.
Table 1.
Patient Characteristics at Baseline.
| Patients with MET exon 14-altered NSCLC | N = 69 |
|---|---|
|
| |
| Age — yr | |
| Median | 72 |
| Range | 34–91 |
|
| |
| Sex — no. (%) | |
| Female | 40 (58) |
| Male | 29 (42) |
|
| |
| Race — no. (%) | |
| White | 50 (73) |
| Asian | 11 (16) |
| Black/African American | 2 (3) |
| Other | 6 (9) |
|
| |
| Smoking history — no. (%) | |
| Former | 42 (61) |
| Never | 26 (38) |
| Current | 1 (1) |
|
| |
| ECOG performance status — no. (%) | |
| 0 | 19 (28) |
| 1 | 49 (71) |
| 2 | 1 (1) |
|
| |
| Tumor histology — no. (%) | |
| Adenocarcinoma | 58 (84) |
| Sarcomatoid carcinoma | 6 (9) |
| Squamous cell carcinoma | 3 (4) |
| Adenosquamous carcinoma | 2 (3) |
|
| |
| Prior treatments for advanced disease — no. (%)* | |
| 0 | 26 (38) |
| 1 | 29 (42) |
| ˃1 | 14 (20) |
|
| |
| Local assay — no. (%) | |
| Next-generation sequencing (NGS) | 66 (96) |
| RT-PCR | 3 (4) |
Of the 43 patients who received ≥1 prior systemic therapy for advanced disease, 20 (47%) received immunotherapy.
Data cutoff was January 31, 2018
ECOG, Eastern Cooperative Oncology Group; NSCLC, non-small cell lung cancer; RT-PCR, reverse transcription polymerase chain reaction.
Anti-tumor Activity
Sixty-five patients were tumor response-evaluable. The objective response rate (ORR) was 32% (95% CI, 21–45). Among the 65 patients, 3 patients (5%) had a confirmed complete response, 18 patients (28%) had a confirmed partial response, 29 patients (45%) had stable disease, and 4 (6%) had disease progression as their best overall response. Best overall response could not be determined for 11 patients (17%). These included 11 patients whose target lesions could not be measured at each follow-up timepoint or who did not undergo ≥1 post-baseline disease assessment secondary to toxicity, withdrawal of consent, or death (Supplementary Table 2). Objective response by age, prior line of therapy, smoking history, and histology is described in Supplementary Table 3. A decrease in target lesion size was observed in most patients (Fig. 1). The median time to tumor response was 7.6 weeks (range, 3.7 to 16.3), corresponding approximately to the first protocol-mandated tumor assessment. The median duration of response was 9.1 months (95% CI, 6.4–12.7). Of 21 patients with an objective response, 12 (57%) had a duration of response of ≥6 months.
Figure 1. Best Percent Change in Target Lesions from Baseline in MET Exon 14-Altered NSCLCs.

A plot of the best response to crizotinib in 52 patients with MET exon 14-altered NSCLCs is shown. Cases not evaluable for response, with early death, and without measurable disease in the response-evaluable population of 65 patients were excluded from this plot. The bars indicate the best percentage change in the sum of target tumor measurements from baseline. As indicated by the asterisk, one patient was determined to have MET exon 14-altered NSCLC by local testing and ROS1-rearranged (and MET wild type) NSCLC by central testing. Below the plot, local testing results for the MET exon 14 alteration splice site region (A) mutation type (B), and the presence of concurrent increased (↑) MET copy number (C), as well as the presence of a MET exon 14 alteration in ctDNA, by central testing (D) are depicted in relation to best response. Note that while the two cases with increased MET copy number were called MET-amplified by local testing, none of these tumors had high-level amplification. In row A, the splice acceptor region includes alterations in the splice acceptor region, polypyrimidine tract, and branch point. Cases classified as unknown include MET exon 14 alterations for which DNA coding region information was not available, such as alterations detected using an RNA-based assay. In rows B and C, a white space indicates that no results were reported by the local assay, or that the reported results could not be analyzed for the biomarker of interest. In row D, a white space indicates that a plasma sample was not available for ctDNA testing, such as in patients who were treated prior to an amendment that included plasma ctDNA collections.
A swimmer plot of the duration of crizotinib therapy is shown in Fig. 2. The median progression-free survival (PFS) was 7.3 months (95% CI, 5.4–9.1) (Fig. 3). The probability of being event free at 6 months was 54% (95% CI, 39.2–66.9). The median overall survival (OS) was 20.5 months (95% CI, 14.3–21.8); OS data were not mature, with 24 patients (35%) having died and 28 (41%) still in follow-up. The probability of survival at 6 and 12 months was 87% (95% CI, 74.7–93.1) and 70% (95% CI, 54.7–81.1), respectively. The estimated median duration of follow-up for OS was 11.5 months (95% CI, 7.9–16.7).
Figure 2. Duration of Crizotinib Therapy in MET Exon 14-Altered NSCLCs.

A plot of the duration of crizotinib therapy in 65 response-evaluable patients with MET exon 14-altered NSCLCs is shown. To the left of the plot, local testing results for the MET exon 14 alteration splice site region (A) mutation type (B) and the presence of concurrent increased (↑) MET copy number (C), as well as the presence of a MET exon 14 alteration in ctDNA by central testing (D) are depicted in relation to the duration of therapy and best response. While the cases with increased MET copy number were called MET-amplified by local testing, none of these tumors had high-level amplification. Arrows indicate patients for whom crizotinib therapy was ongoing at the time of data cutoff. As indicated by the asterisk, one patient was determined to have MET exon 14-altered NSCLC by local testing and ROS1-rearranged NSCLC by central testing. In column A, the splice acceptor region includes alterations in the splice acceptor region, polypyrimidine tract, and branching point. Cases classified as unknown include MET exon 14 alterations for which DNA coding region information was not available, such as alterations detected using an RNA-based assay. In columns B and C, a white space indicates that no results were reported by the local assay, or that the reported results could not be analyzed for the biomarker of interest. In column D, a white space indicates that a plasma sample was not available for ctDNA testing, such as in patients who were treated prior to an amendment that included plasma ctDNA collections.
Figure 3. Progression-Free Survival.

The Kaplan-Meier curve for progression-free survival in patients with MET exon 14-altered NSCLCs treated with crizotinib is shown. Progression-free survival was defined as the time from the date of the first dose of crizotinib to objective disease progression or death from any cause. The shaded area represents the 95% Hall–Wellner confidence bands. Hash marks on the survival curve indicate censoring of data. PFS, progression-free survival.
Biomarker Analysis
MET exon 14 alterations were genomically diverse by local testing (Supplementary Table 4A). In terms of splice site, these alterations most commonly involved the splice donor site (57%), followed by the splice acceptor site (25%). The most frequent mutation type (regardless of splice site) was base substitution (51%) followed by the occurrence of an indel (31%); a whole exon deletion occurred in 1 (2%) patient. The genomic sequence of the MET exon 14 alteration (region/type) was unknown in 11 (17%) patients (included a MET rearrangement resulting in MET exon 14 exclusion; Supplementary Table 4A). Objective responses to crizotinib were observed independent of the MET exon 14 alteration splice site (ORR 12/37 [32%] for splice donor site and 5/16 [31%] for splice acceptor site) or mutation type (ORR 12/33 [36%] for base substitution and 5/20 [25%] for indel). The overall association of tumor response with MET exon 14 alteration (splice site region/mutation type, including patients with unknown MET exon 14 alteration status) was not significant (P=0.65, Supplementary Table 4A; Fig. 1). Similar results were observed with central testing (P=0.53, Supplementary Table 4B, Extended Data Fig. 2). Durable disease control was observed across different MET exon 14 alterations (Fig. 2).
Concurrent increased MET copy number was observed via local testing in 2 of 54 (4%) tumors in which testing was performed (Supplementary Table 5). While local testing reported these 2 cases as MET-amplified, the FISH-positive case (MET/CEP7 ratio 3.6) did not have high-level amplification (MET/CEP7 ratio of >4 to 5),21 and the fold change in the NGS-positive case was low (2.3-fold change). While the true impact of these concurrent copy number changes remains unclear, both patients had durable disease control that exceeded 6 months (Fig. 2). Central testing of analyzable tumor tissue revealed an increase in MET copy number in 2 of 40 (5%) patients (Supplementary Table 5).
The most common (≥20%) concurrent non-MET genomic alterations in tumor by central testing (Extended Data Fig. 3) involved TP53 (38%), MDM2 (20%), and CDKN2A (20%). ALK or ROS1 rearrangements were not observed by initial local molecular testing. However, central NGS testing showed that 1 patient was MET exon 14 alteration-negative and ROS1-rearranged (Fig. 1; Fig. 2).
Among 37 patients with MET exon 14-altered tumors and analyzable plasma samples, 18 (49%) had a MET exon 14 alteration detected in circulating tumor DNA (ctDNA-positive). Mean allele frequencies ranged from 0.23% to 54%. There was no statistically significant difference in ORR between ctDNA-positive versus ctDNA-negative cases (ORR 19% versus 24%; P=1.00). The duration of treatment was shorter in ctDNA-positive versus ctDNA-negative cases with a median of 3.6 months (95% CI, 1.2–7.3) versus 7.8 months (95% CI, 5.5–20.9; HR=2.27, 95% CI, 0.94–5.48; P=0.06), respectively. PFS was significantly shorter in ctDNA-positive versus ctDNA-negative cases with a median of 3.7 months (95% CI, 1.6–7.2) versus 8.1 months (95% CI, 7.3–not estimable; HR=3.85, 95% CI, 1.27–11.66; P=0.01), respectively (Extended Data Fig. 4). The most common concurrent alterations in ctDNA (≥10%; Extended Data Fig. 5) involved TP53 (28%), and JAK2 (11%), recognizing that these alterations can occasionally be attributed to clonal hematopoiesis.22
Safety
The overall safety profile of crizotinib in all 69 patients was similar to that reported previously for patients with ALK- and ROS1-rearranged NSCLCs.23–25 Treatment-related adverse events (TRAEs) seen in ≥10% of patients are listed in Supplementary Table 6. The most common TRAEs were edema (51%), vision disorder (45%), nausea (41%), diarrhea (39%), and vomiting (29%). Most TRAEs were grade 1/2. The most common (≥3%) grade 3 TRAEs were elevated transaminases (4%), and dyspnea (4%). There were three grade 4 TRAEs: hypophosphatemia, lymphopenia, and pulmonary embolism. One patient had grade 5 treatment-related interstitial lung disease (Supplementary Table 2). TRAEs associated with a dose reduction or permanent treatment discontinuation occurred in 38% and 7% of patients, respectively.
Discussion
In this study, we showed that crizotinib is active in patients with MET exon 14-altered NSCLCs. This underscores the importance of testing for MET exon 14 alterations in the clinic. NGS using DNA-based hybrid capture is an ideal assay to use as it can detect a wide variety of mutations that result in MET exon 14 skipping or a comparable biology. It is important to note, however, that tumor RNA sequencing can detect MET exon 14 alterations in cancers where these were missed by DNA-based sequencing.26 Beyond tumor-based testing, plasma-derived ctDNA assays can also be used to detect these alterations, as was demonstrated in this paper.
PROFILE 1001 was the first prospective trial to demonstrate that MET inhibition is active in MET exon 14-altered NSCLCs.27 In this seminal data set, crizotinib achieved an ORR of 32% (95% CI, 21–45), a median duration of response of 9.1 months, and a median PFS of 7.3 months. These outcomes clearly exceed those observed with second-line chemotherapy (ORR 7–23%, median PFS 2.4–4.5 months)28,29 and are comparable to that of first-line platinum doublet-chemotherapy (ORR 31–35%, median time to progression 4.8–6.2 months).30,31 While these do not surpass the outcomes observed with select first-line chemoimmunotherapy combinations in unselected NSCLCs,32 MET inhibition with crizotinib remains an important treatment option for MET exon 14-altered NSCLCs, as is currently supported by National Comprehensive Cancer Center Guidelines.33
While robust, the ORR of crizotinib in MET exon 14-altered NSCLCs was lower compared to the ORRs of ~60–80% are achieved with targeted therapy for other NSCLC drivers.34,35 As MET exon 14-altered NSCLCs are heterogeneous, we posited that distinct subpopulations may be less likely to respond to crizotinib, contributing to a lower response rate. An exploratory analysis of this series showed, however, that this was not the case. First, mutation type and splice site region did not affect outcomes and MET copy number was not a major differentiating factor (recognizing that few tumors had concurrent MET amplification, and none had high-level MET amplification). Second, a strong association between concurrent genomic alterations and outcomes was not found, despite preclinical studies suggesting that select co-occurring alterations can confer resistance.13,36 Finally, ctDNA status was not significantly associated with ORR, acknowledging that the median PFS was significantly shorter in ctDNA-positive versus negative cases. The latter should be interpreted with caution as the sample size was small and few events were observed. Other factors that potentially affect crizotinib outcomes should continue to be explored. It was recently shown, for example, that no responses were observed with crizotinib in MET exon 14-altered NSCLCs without MET protein expression.37
Ultimately, beyond these analyses, the type of MET inhibitor used to treat MET exon 14-altered cancers may affect clinical outcomes. While crizotinib, a type Ia inhibitor, was the first MET TKI to be explored in MET exon 14-altered NSCLCs, preliminary reports of prospective trials of type Ib TKIs such as capmatinib,38 tepotinib,39 and savolitinib40 have subsequently emerged. These agents are more selective compared to crizotinib and are potentially more potent in preclinical models. The ORRs ranged from 40–55% in the post-platinum doublet or all-comers setting (for all 3 drugs) and ranged from 44–68% in treatment-naïve patients (for tepotinib and capmatinib). Additional data and longer follow up will determine if these agents are markedly different in activity compared to crizotinib. Furthermore, a careful analysis of relative toxicity will be important given that increased anti-MET activity could result in higher rates of on-target adverse events (e.g. peripheral edema) in patients.
In summary, crizotinib is active in patients with advanced MET exon 14-altered NSCLC. The results of PROFILE 1001 established a clinical paradigm for the treatment of MET exon 14-altered lung cancers with MET-directed targeted therapy, paving the way for additional research to move forward in this space with the goal of regulatory approval of one or more MET inhibitors for this genomic indication.
Methods
Patients
Eligible patients had histologically confirmed NSCLC that harbored a MET exon 14-alteration (local molecular testing; Supplementary Table 1). Increased MET copy number was determined by assay-defined thresholds for both local and central molecular testing (FoundationOne CDx).41,42
Other eligibility criteria included an age of ≥18 years (≥20 years for Japanese patients), an Eastern Cooperative Oncology Group performance status43 of 0 to 1 (2 was allowed with sponsor approval), adequate organ function, and measurable disease (non-measurable disease was allowed with sponsor approval) per Response Evaluation Criteria in Solid Tumors (RECIST), version 1.0.44 For a full list of inclusion/exclusion criteria, see Supplementary Information.
The protocol (see Supplementary Information) was approved by the institutional review board or independent ethics committee at each site and complied with the International Ethical Guidelines for Biomedical Research Involving Human Subjects, Good Clinical Practice guidelines, the Declaration of Helsinki, and local laws. All patients provided written informed consent.
Study Design and Treatment
PROFILE 1001 is an ongoing open-label phase 1 study with a dose-escalation phase followed by a Recommended Phase 2 Dose (RP2D) expansion phase.45 Details on dose escalation and results from the expansion phase in ALK-rearranged and ROS1-rearranged NSCLCs have been published.46,47,48 The study protocol was amended (amendment no. 21, April 7, 2015) to allow the retrospective and prospective enrollment of patients with MET exon 14-altered advanced NSCLCs in a separate expansion cohort, including sample collection for circulating nucleic acid analysis and documenting survival information every 3 months after discontinuation of study treatment up to at least 1 year after the first crizotinib dose for the last patient enrolled into the MET exon 14 cohort. Subsequent protocol amendments (nos. 22 and 23) are described in the statistical analysis section. Objective response was the primary end point. Duration of response (the time from the first documentation of objective tumor response [complete response or partial response] that was subsequently confirmed, to the documentation of objective tumor progression or death on study due to any cause, whichever occurred first), time to tumor response (time from first dose to first documentation of objective tumor response [complete response or partial response] that was subsequently confirmed), PFS (time from first dose to first documentation of tumor progression or death on study due to any cause, whichever occurred first), OS (time from first dose to death due to any cause), and safety were additional end points.
Crizotinib was administered orally at the standard starting dose of 250 mg twice daily in continuous 28-day cycles. Disease progression or clinical deterioration, unacceptable toxicity, consent withdrawal, or death were reasons for treatment discontinuation. Treatment could be continued beyond progression at the investigator’s discretion and with sponsor approval.
Study Assessments
Patients underwent baseline tumor imaging with computed tomography or magnetic resonance imaging (chest, abdomen, and pelvis). Brain/bone scans were obtained if disease at these sites was suspected. Tumor assessments were performed every 8 weeks and could be performed every 16 and 24 weeks starting with cycles 15 and 24, respectively. Response was confirmed ≥4 weeks after initial response documentation.
Adverse events were assessed from the first day of study treatment until ≥28 days after the last dose or until all serious/drug-related toxicities resolved or were determined to be stable/chronic, whichever was later. The type, incidence, and severity of adverse events were determined by the Common Terminology Criteria for Adverse Events, version 3.0 (http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf).
Central Biomarker Analyses
MET exon 14 alterations were retrospectively evaluated by central testing on archival tumor tissue using a validated DNA-based next-generation sequencing (NGS; FoundationOne CDx™, Foundation Medicine, Cambridge, MA) assay. MET exon 14 alteration status was retrospectively evaluated by ctDNA analyses using a validated assay (PlasmaSELECT™-R64, Personal Genome Diagnostics, Boston, MA) in baseline or end-of-treatment plasma samples. The same assays identified concurrent genomic alterations in tumor tissue and ctDNA.
Study Oversight
The study was designed jointly by the investigators and representatives of the sponsor, Pfizer Incorporated. All authors were involved in data analysis and manuscript preparation and attest to the completeness and accuracy of the data, analyses, and study protocol adherence.
Statistical Analysis
It was originally anticipated that 33 patients would need to be enrolled in the MET exon 14-altered NSCLC group. This would achieve a power of ≥90% to test the null hypothesis that the ORR of crizotinib would be ≤10%, versus the alternative hypothesis that the ORR would be >10% (one-sided alpha level of 0.05, single-stage design). For the alternative hypothesis, the target ORR was 30%. On this basis, the null hypothesis would be rejected if ≥7 objective responses were observed among the first 33 evaluable patients (protocol amendment no. 22, April 27, 2016). As of August 01, 2016, there were 11 objective responses (among 28 evaluable patients); thus, the null hypothesis was rejected. To report antitumor activity and safety with greater precision, an increase in the sample size beyond the original 33 required for rejection of the null hypothesis was planned (protocol amendment no. 23, February 21, 2017). Thus, the sample size was increased to 68 patients as of May 15, 2017. Because the null hypothesis was already rejected with the first 33 patients enrolled, adjustment of type I error rate was not considered necessary for subsequent analyses. This dataset met the planned cutoff of an expanded sample size of at least 68 patients for formal reporting. As of December 07, 2017, the sample size was augmented to 81 patients; however, because enrollment is still ongoing (at the data cutoff date of January 31, 2018), a final analysis will be conducted after the last patient accrued completes the last survival follow-up visit. Patients who received ≥1 dose of crizotinib were included in the safety population and analyses of PFS and OS. The response-evaluable population was defined as all patients in the safety population who had an adequate baseline and ≥1 post-baseline disease assessment (≥6 weeks from the first dose), or who withdrew from the study, progressed, or died. CIs for the ORR were estimated using an exact binomial method based on the F-distribution. A Kaplan–Meier analysis of time-to event data to estimate medians and the Brookmeyer–Crowley method to calculate two-sided 95% CIs for the median were used. Only descriptive statistics were produced for time to tumor response. The associations of tumor response with splice site region/mutation type or with ctDNA results were evaluated using 2-sided Fisher exact tests. Hazard ratios and associated 95% CIs were estimated by Cox regression models and 2-sided log-rank tests were performed to compare the duration of treatment and PFS between patients ctDNA-positive versus ctDNA-negative. P-values were not adjusted for multiple comparisons. Study database data were analyzed with the use of SAS statistical software, version 9.2 or later (SAS Institute, Cary, NC, USA). The data cutoff date was January 31, 2018.
Data Availability
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1. Patient Evaluation Groups.
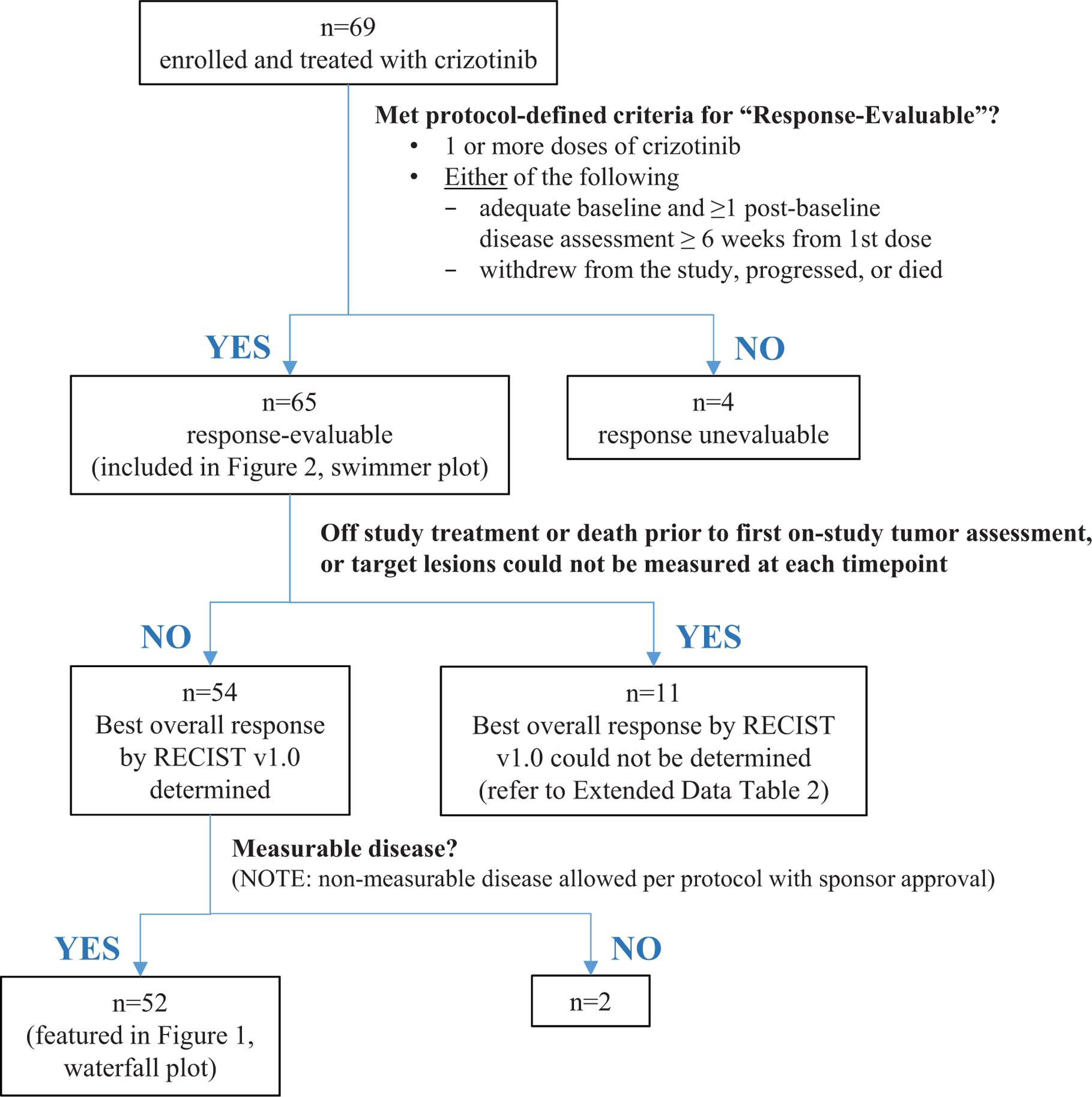
Shown is a flow diagram summarizing study enrollment and patient evaluation groups in the MET exon 14-altered advanced NSCLC expansion cohort of the ongoing PROFILE 1001 study as of January 31, 2018. Patients who received ≥1 dose of crizotinib were included in the safety population and analyses of PFS and OS. OS, overall survival; PFS, progression-free survival.
Extended Data Fig. 2. Best Percent Change in Target Lesions from Baseline in MET Exon 14-Altered NSCLCs and MET Exon 14 Alterations (Splice Site Region and Type) by Central and Local Testing.
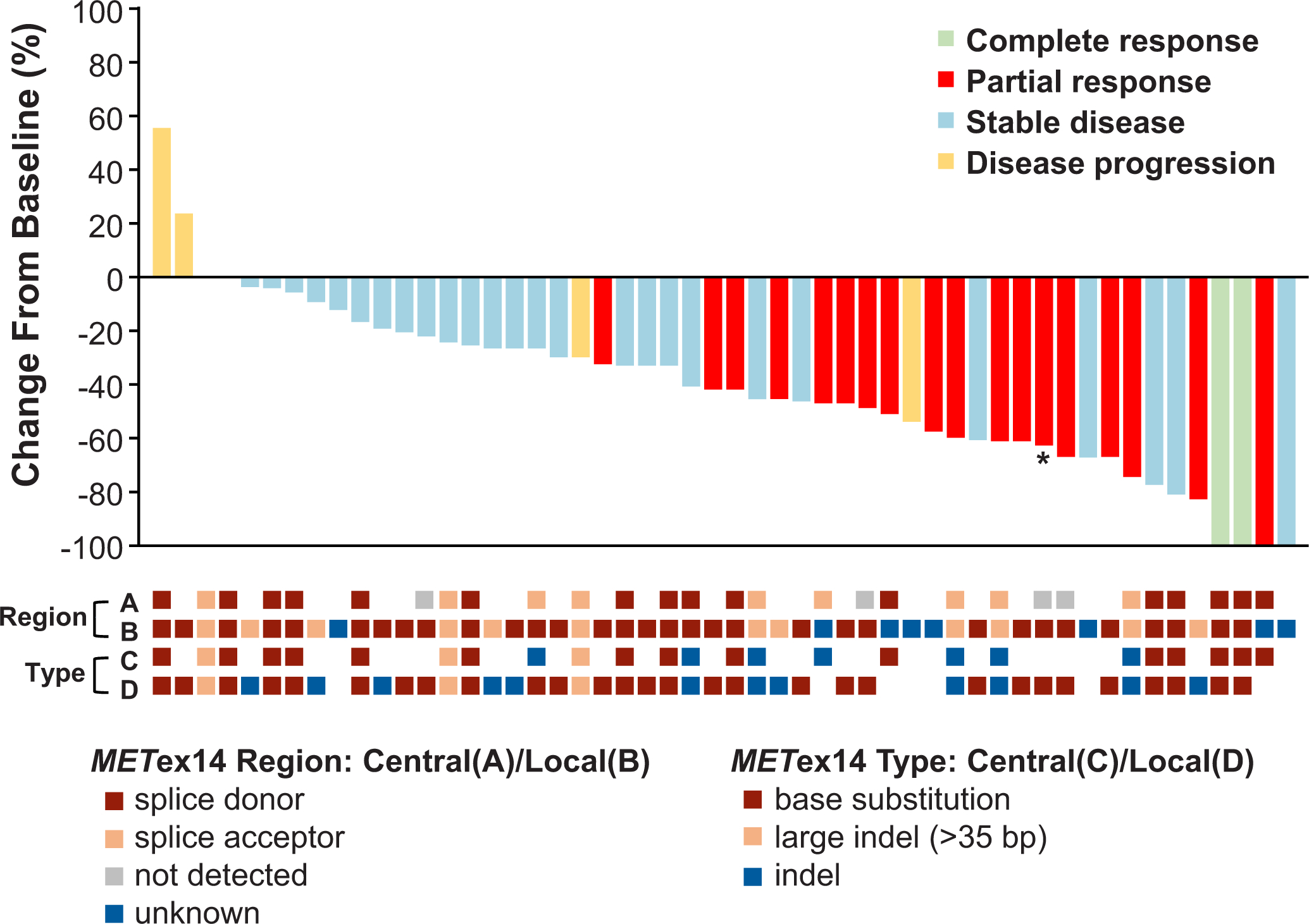
A plot of the best response to crizotinib in 52 patients with MET exon 14-altered NSCLCs is shown. The bars indicate the best percentage change in the sum of target tumor measurements from baseline. Below the plot, retrospective central testing results for the MET exon 14 alteration splice site region (A) and mutation type (C) are depicted in relation to best response. For comparison, prospective local testing results for region (B) and type (D) are also included. In rows A and B, the splice acceptor region includes alterations in the splice acceptor region, polypyrimidine tract, and branching point. Cases classified as unknown include MET exon 14 alterations for which DNA coding region information was not available, such as alterations detected using an RNA-based assay. White space indicates that no results were reported by the central (rows A, C) or local (rows B, D) assay, or that the reported results could not be analyzed for the biomarker of interest. Retrospective central testing confirmed the presence of a MET exon 14 alteration in 88% of the 40 patients with tumor tissue analyzed. Central testing confirmation was not obtained for five patients with MET exon-14-altered NSCLC as determined by local testing methods. Of these five central testing-negative patients, four did not pass full quality control metrics (mostly attributed to low tumor purity or tumor input issues) but were reportable. One patient was determined to have MET exon 14-altered NSCLC by local testing and ROS1 rearranged NSCLC by central testing (as indicated by an asterisk).
Extended Data Fig. 3. Concurrent Alterations in Tumor.

Shown are concurrent alterations observed by retrospective molecular profiling of archival tumor tissue (FoundationOne CDx). Each patient with available data is represented by a column. The colored rectangles above each column represent the best objective response to crizotinib. Within a column, each gene of interest for which a concurrent alteration is present is represented by a colored rectangle corresponding to the alteration type. Concurrent genomic alterations (average number of alterations per patient was 4.3 [range: 0 to 12]) were identified in tumor tissue from 35 of 40 (88%) patients with analyzable samples. MDM2 amplification was detected in non-responders, but not observed in responders. No notable response differences were seen in relation to absence or presence of TP53 mutation.
Extended Data Fig. 4. Progression-Free Survival by MET Exon 14 Alteration Detection in ctDNA.

The Kaplan-Meier curves for progression-free survival in patients treated with crizotinib are shown according to detection of MET Exon 14 alterations in ctDNA by plasma profiling. Progression-free survival was defined as the time from the date of the first dose of crizotinib to objective disease progression or death from any cause. Hash marks on the survival curve indicate censoring of data. *P-value from 2-sided log-rank test comparing survival distributions among ctDNA-positive versus ctDNA-negative patients. †HR from Cox proportional hazards regression – assuming proportional hazards, an HR >1 indicates a greater risk of disease progression or death among ctDNA-positive versus ctDNA-negative patients. ctDNA, circulating tumor DNA; HR, hazard ratio; NE, not estimable; PFS, progression-free survival.
Extended Data Fig. 5. Concurrent Alterations in ctDNA.
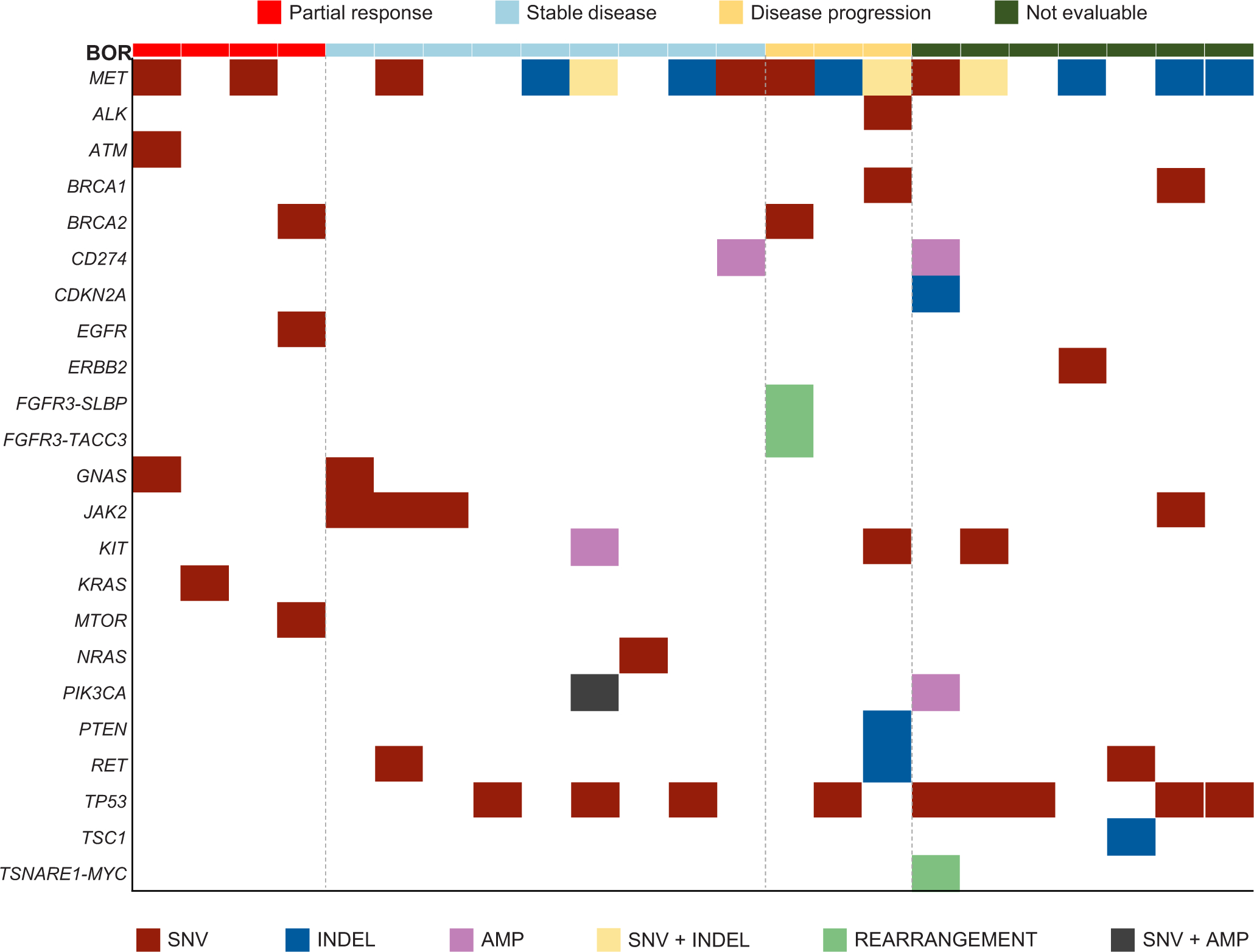
Shown are concurrent alterations observed by retrospective molecular profiling of baseline plasma samples (PlasmaSELECTR64). Each patient with available data is represented by a column. The colored rectangles above each column represent the best objective response to crizotinib. Within a column, each gene of interest for which a concurrent alteration is present is represented by a colored rectangle corresponding to the alteration type. Concurrent genomic alterations (average number of alterations per patient was 2.36 [range: 0 to 8]) were identified in ctDNA from 25 of 36 (69%) patients with analyzable samples. ctDNA, circulating tumor DNA.
Supplementary Material
Acknowledgments
We would like to thank our patients, their families and caregivers, and participating clinical sites and teams. For biomarker analyses, we acknowledge Amber Donahue and Chetan Deshpande for their support. Jonathan N. Raksin, Ph.D. and Julian Martins, M.A., inScience Communications, Springer Healthcare (Philadelphia, PA, USA) provided medical writing support, funded by Pfizer Inc.
Funding
This study was supported by Pfizer. AD, GJR, and PKP are supported by the National Institutes of Health award P30 CA008748.
Disclosure
The authors disclose the following: Dr. Drilon reports receiving fees for consulting/advisory board roles from AstraZeneca, Bayer, BeiGene, Blueprint Medicines, Genentech/Roche, Helsinn, Ignyta, Loxo/Lilly, Pfizer, TP Therapeutics, BergenBio, Exelixis, Tyra, Verastem, MORE Health, Abbvie, GlaxoSmithKlein, Teva, PharmaMar, Taiho, Merck, Puma, Merus and Takeda/Ariad/Millenium. Dr. Clark has nothing to disclose. Dr. Weiss reports receiving fees for speaker or advisory board roles from AstraZeneca, Biodesix, BioMarck Pharmaceuticals, Clovis Oncology, Eli Lilly, EMD Serono, Genentech, and Inivata, and that his institution has received grant/research support from AstraZeneca, Astellas, Celgene, Merck, Novartis and Pfizer. Dr. Ou reports receiving fees for serving on speaker bureaus for AstraZeneca, Genentech, and Takeda, fees for speaker or advisory board roles from AstraZeneca, Foundation Medicine, Novartis, Pfizer, Roche/Genentech, and Takeda, honoraria from ARIAD/Takeda, AstraZeneca, Foundation Medicine, Genentech/Roche, Novartis, Pfizer, and Roche Pharma AG, and that his institution has received grant/research support from ARIAD, Astellas Pharma, AstraZeneca, AstraZeneca/MedImmune, Chugai Pharma, Clovis Oncology, GlaxoSmithKline, Ignyta, Peregrine Pharmaceuticals, Pfizer and Roche Pharma AG. Dr. Camidge reports that his institution has received grant/research support from Pfizer. Dr. Solomon reports receiving fees for speaker or advisory board roles for AstraZeneca, Bristol-Myers Squibb, Merck Sharp & Dohme, Novartis, Pfizer, and Roche/Genentech, honoraria from AstraZeneca and Bristol-Myers Squibb, that his institution has received grant/research support from Pfizer, that he has collected royalties and/or is an IP rights/patent holder with Veristrat (Biodesix), and that he has received other payment (travel, accommodations, expenses) from AstraZeneca, Bristol-Myers Squibb, Merck, Novartis, and Roche. Dr. Otterson reports receiving fees for advisory board roles with Amgen, Genentech, Novartis, Pfizer, and Takeda, and that his institution has received grant/research support from Astra Zeneca, BMS, Clovis, Genentech, Ignyta, Merck, Novartis, and Pfizer. Dr. Villaruz reports receiving fees for speaker or advisory board roles for Pfizer. Dr. Riely reports receiving fees for speaker or advisory board roles from Genentech, and that his institution has received grant/research support from Ariad, GlaxoSmithKline, Infinity Pharmaceuticals, Millenium, Novartis, Pfizer, and Roche/Genetech. Dr. Heist reports receiving honoraria from Boehringer Ingelheim, and that her institution has received grant/research support from Abbvie, Agios, Celgene, Corvus, Daichii, Debiopharm, Genentech/Roche, Incyte, Millenium, Novartis and Peregrine. Dr. Awad reports receiving fees for consulting/advisory board roles with Abbvie, ARIAD, AstraZeneca/MedImmune, Boehringer Ingelheim, Bristol-Myers Squibb, Clovis Oncology, Foundation Medicine, Genentech, Merck, Nektar, Novartis, Pfizer, and Syndax, and that his institution has received grant/research support from Bristol-Myers Squibb. Dr. Shapiro reports receiving fees for speaker or advisory board roles from G1 Therapeutics, Lilly, Pfizer, Roche, and Vertex Pharmaceuticals, and that his institution has received grant/research support from Pfizer and Lilly. Dr. Satouchi reports receiving honoraria from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Chugai Pharmaceutical, Eli Lilly Japan, Merck, Novartis, Ono Pharmaceutical, Pfizer Japan, and Taiho Pharmaceutical, and that his institution has received grant/research support from AstraZeneca, Boehringer Ingelheim, Bristol-Myers Squibb, Chugai Pharmaceutical, Eli Lilly Japan, Merck, Novartis, Ono Pharmaceutical, Pfizer Japan, and Taiho Pharmaceutical. Dr. Hida reports receiving fees for consulting/advisory board roles from Novartis, and receiving honoraria from AstraZeneca, Boehringer Ingelheim, Bristol-Meyers Squibb, Chugai Pharmaceutical, Clovis Oncology, Eli Lilly, Nippon, Novartis, Ono Pharmaceutical, Pfizer, and Taiho Pharmaceutical. Dr. Hayashi reports receiving fees for consulting/advisory board roles from AstraZeneca, Boehringer Ingelheim, Chugai Pharma, and Lilly. Dr. Murphy reports being an employee of, and owning stock in, Pfizer. Dr. Wang reports being an employee of, and owning stock in, Pfizer. Ms. Li reports being an employee of, and owning stock in, Pfizer. Ms. Usari reports being an employee of, and owning stock in, Pfizer. Dr. Wilner reports being an employee of, and owning stock in, Pfizer. Dr. Paik has nothing to disclose. No other potential conflict of interest relevant to this article was reported.
References
- 1.Kong-Beltran M et al. Somatic mutations lead to an oncogenic deletion of met in lung cancer. Cancer Res. 66, 283–289 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Ma PC et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 65, 1479–1488 (2005). [DOI] [PubMed] [Google Scholar]
- 3.Cui JJ et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK). J. Med. Chem. 54, 6342–6363 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Awad MM et al. MET Exon 14 Mutations in Non-Small-Cell Lung Cancer Are Associated With Advanced Age and Stage-Dependent MET Genomic Amplification and c-Met Overexpression. J. Clin. Oncol. 34, 721–730 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Tong JH et al. MET amplification and exon 14 splice site mutation define unique molecular subgroups of Non-small Cell Lung Carcinoma with poor prognosis. Clin. Cancer Res. 22, 3048–3056 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Schrock AB et al. Characterization of 298 patients with lung cancer harboring MET exon 14 skipping alterations. J. Thorac. Oncol. 11, 1493–1502 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Frampton GM et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 5, 850–859 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Drilon A, Cappuzzo F, Ou SI & Camidge DR Targeting MET in Lung Cancer: Will Expectations Finally Be MET? J. Thorac. Oncol. 12, 15–26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onozato R et al. Activation of MET by gene amplification or by splice mutations deleting the juxtamembrane domain in primary resected lung cancers. J. Thorac. Oncol. 4, 5–11 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Drilon A MET Exon 14 Alterations in Lung Cancer: Exon Skipping Extends Half-Life. Clin. Cancer Res. 22, 2832–2834 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peschard P et al. Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 8, 995–1004 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Awad MM Impaired c-Met receptor degradation mediated by MET exon 14 mutations in non-small-cell lung cancer. J. Clin. Oncol. 34, 879–881 (2016). [DOI] [PubMed] [Google Scholar]
- 13.Liu X et al. Next-generation sequencing of pulmonary sarcomatoid carcinoma reveals high frequency of actionable MET gene mutations. J. Clin. Oncol. 34, 794–802 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Paik PK et al. Response to MET inhibitors in patients with stage IV lung adenocarcinomas harboring MET mutations causing exon 14 skipping. Cancer Discov. 5, 842–849 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heist RS et al. MET Exon 14 Skipping in Non-Small Cell Lung Cancer. Oncologist 21, 481–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jenkins RW et al. Response to Crizotinib in a Patient With Lung Adenocarcinoma Harboring a MET Splice Site Mutation. Clin. Lung Cancer 16, e101–104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee C et al. MET 14 Deletion in Sarcomatoid Non-Small-Cell Lung Cancer Detected by Next-Generation Sequencing and Successfully Treated with a MET Inhibitor. J. Thorac. Oncol. 10, e113–114 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Mendenhall MA & Goldman JW MET-mutated NSCLC with major response to crizotinib. J. Thorac. Oncol. 10, e33–34 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Awad MM et al. Impact of MET inhibitors on survival among patients with non-small cell lung cancer harboring MET exon 14 mutations: a retrospective analysis. Lung Cancer 133, 96–102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Landi L et al. Crizotinib in MET deregulated or ROS1 rearranged pretreated non-small-cell lung cancer (METROS): a phase II, prospective, multicentre, two-arms trial. Clin. Cancer Res, doi: 10.1158/1078-0432.CCR-19-0994 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Noonan SA et al. Identifying the appropriate FISH criteria for defining MET copy number-driven lung adenocarcinoma through oncogene overlap analysis. J. Thorac. Oncol. 11, 1293–1304 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Genovese G et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 371, 2477–2487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Camidge DR et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 13, 1011–1019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw AT et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 371, 1963–1971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw AT et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 368, 2385–2394 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Benayed R et al. High Yield of RNA Sequencing for Targetable Kinase Fusions in Lung Adenocarcinomas with No Mitogenic Driver Alteration Detected by DNA Sequencing and Low Tumor Mutation Burden. Clin. Cancer Res. 25, 4712–4722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Drilon A et al. Efficacy and safety of crizotinib in patients with advanced MET exon 14-altered non-small cell lung cancer (NSCLC). J. Clin. Oncol. 34, doi: 10.1200/JCO.2016.34.15_suppl.108 (2016). [DOI] [Google Scholar]
- 28.Shepherd FA et al. Prospective randomized trial of docetaxel versus best supportive care in patients with non-small-cell lung cancer previously treated with platinum-based chemotherapy. J. Clin. Oncol. 18, 2095–2103 (2000). [DOI] [PubMed] [Google Scholar]
- 29.Garon EB et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet 384, 665–673 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Scagliotti GV et al. Phase III study comparing cisplatin plus gemcitabine with cisplatin plus pemetrexed in chemotherapy-naive patients with advanced-stage non-small-cell lung cancer. J. Clin. Oncol. 26, 3543–3551 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Sandler A et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N. Engl. J. Med. 355, 2542–2550 (2006). [DOI] [PubMed] [Google Scholar]
- 32.Socinski MA et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. N. Engl. J. Med. 378, 2288–2301 (2018). [DOI] [PubMed] [Google Scholar]
- 33.National Comprehensive Cancer Network. NCCN clinical practice guidelines: Non-Small Cell Lung Cancer (Version 6.2019), (2019). [Google Scholar]
- 34.Soria JC et al. Osimertinib in Untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N. Engl. J. Med. 378, 113–125 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Camidge DR et al. Brigatinib versus Crizotinib in ALK-Positive Non-Small-Cell Lung Cancer. N. Engl. J. Med. 379, 2027–2039 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Lee GD et al. MET Exon 14 Skipping Mutations in Lung Adenocarcinoma: Clinicopathologic Implications and Prognostic Values. J. Thorac. Oncol. 12, 1233–1246 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Guo R et al. MET inhibitor resistance in patients with MET exon 14-altered lung cancers. J. Clin. Oncol. 37, doi: 10.1200/JCO.2019.37.15_suppl.9006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf J et al. Capmatinib (INC280) in METΔex14-mutated advanced non-small cell lung cancer (NSCLC): Efficacy data from the phase II GEOMETRY mono-1 study. J. Clin. Oncol. 37, doi: 10.1200/JCO.2019.37.15_suppl.9004 (2019). [DOI] [Google Scholar]
- 39.Paik PK et al. Phase II study of tepotinib in NSCLC patients with METex14 mutations. J. Clin. Oncol. 37, doi: 10.1200/JCO.2019.37.15_suppl.9005 (2019). [DOI] [Google Scholar]
- 40.Lu S et al. Preliminary efficacy and safety results of savolitinib treating patients with pulmonary sarcomatoid carcinoma (PSC) and other types of non-small cell lung cancer (NSCLC) harboring MET exon 14 skipping mutations. Cancer Res. 79, doi: 10.1158/1538-7445.AM2019-CT031 (2019). [DOI] [Google Scholar]
Methods References
- 41.Frampton GM et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 5, 850–859 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Palma NA et al. Frequency of MET amplification determined by comprehensive next-generation sequencing (NGS) in multiple solid tumors and implications for use of MET inhibitors. J. Clin. Oncol. 31, doi: 10.1200/jco.2013.31.15_suppl.11068 (2013). [DOI] [Google Scholar]
- 43.Oken MM et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am. J. Clin. Oncol. 5, 649–655 (1982). [PubMed] [Google Scholar]
- 44.Therasse P et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J. Natl. Cancer Inst. 92, 205–216 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Kwak EL et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N. Engl. J. Med. 363, 1693–1703 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Camidge DR et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 13, 1011–1019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shaw AT et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N. Engl. J. Med. 371, 1963–1971 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kwak EL et al. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066. J. Clin. Oncol. 27, doi: 10.1200/jco.2009.27.15s.3509 (2009). [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Upon request, and subject to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.