

SUMMARY
Citrate lies at a critical node of metabolism, linking tricarboxylic acid metabolism and lipogenesis via acetyl-coenzyme A. Recent studies have observed that deficiency of the sodium-dependent citrate transporter (NaCT), encoded by SLC13A5, dysregulates hepatic metabolism and drives pediatric epilepsy. To examine how NaCT contributes to citrate metabolism in cells relevant to the pathophysiology of these diseases, we apply 13C isotope tracing to SLC13A5-deficient hepatocellular carcinoma (HCC) cells and primary rat cortical neurons. Exogenous citrate appreciably contributes to intermediary metabolism only under hypoxic conditions. In the absence of glutamine, citrate supplementation increases de novo lipogenesis and growth of HCC cells. Knockout of SLC13A5 in Huh7 cells compromises citrate uptake and catabolism. Citrate supplementation rescues Huh7 cell viability in response to glutamine deprivation or Zn2+ treatment, and NaCT deficiency mitigates these effects. Collectively, these findings demonstrate that NaCT-mediated citrate uptake is metabolically important under nutrient-limited conditions and may facilitate resistance to metal toxicity.
Graphical abstract
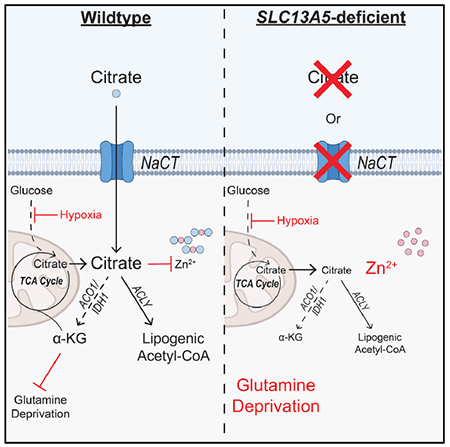
In brief
Using isotope tracing, 13C MFA, and proliferation assays, Kumar et al. show that NaCT-mediated extracellular citrate is cytosolically catabolized, contributing to de novo lipogenesis (DNL) in HCC cells. Furthermore, extracellular citrate import by NaCT promotes growth in oxygen- and glutamine-limited conditions and protects against zinc-induced toxicity.
INTRODUCTION
Citrate serves as a critical substrate for biosynthesis, acetylation, and the regeneration of NAD(P)H. Within mitochondria, citrate is synthesized by citrate synthase and metabolized in the tricarboxylic acid (TCA) cycle to support bioenergetics. Citrate is also exported to the cytosol via the mitochondrial citrate transporter (SLC25A1/CTP) and metabolized by ATP-citrate lyase (ACLY) to generate acetyl-coenzyme A (acetyl-CoA) for downstream metabolic processes, including lipid biosynthesis and acetylation (Wakil and Abu-Elheiga, 2009). Although mitochondrial production of citrate is the primary source for most cells, plasma concentrations are relatively high (Costello and Franklin, 2016). In addition, dysregulation of plasma citrate homeostasis has pathophysiological consequences including impaired blood clotting and bone disorders (Costello and Franklin, 2016; Zuckerman and Assimos, 2009). The functional importance of exogenous citrate transport by cells has therefore garnered increasing interest (Bhutia et al., 2017; Costello and Franklin, 1991, 2016).
A plasma membrane-specific variant of SLC25A1, pmCIC, allows for import and catabolism of extracellular citrate in prostate cancer cells (Mycielska et al., 2018). Furthermore, several members of the SLC13 sodium sulfate/carboxylate symporter gene family are capable of importing citrate into cells in a sodium-coupled mechanism (Pajor, 2006, 2014). Both SLC13A2, primarily expressed in the kidney and small intestine, and SLC13A3, widely expressed across tissues, are capable of importing citrate, although with significantly lower affinity than dicarboxylic TCA intermediates (Pajor, 2006, 2014). Liver and brain tissue expresses the sodium-dependent citrate transporter (NaCT; also known as mINDY), encoded by SLC13A5, which preferentially transports citrate across the plasma membrane (Inoue et al., 2002; Pajor, 1996).
SLC13A5 function has been linked to hepatic glucose and fatty acid metabolism, and development of NaCT inhibitors has garnered therapeutic interest (Huard et al., 2015; Sauer et al., 2021). Deletion of Slc13a5 is protective against high-fat diet-induced insulin resistance and attenuates hepatic gluconeogenesis and lipogenesis (Birkenfeld et al., 2011). Furthermore, pharmacological inhibition of NaCT reduced hepatic lipid accumulation in mice fed a high-fat diet (Huard et al., 2015). Additionally, hepatic SLC13A5 expression correlated positively with both body and liver fat in a cohort of non-alcoholic fatty liver disease (NAFLD) patients (von Loeffelholz et al., 2017). Functional impacts of NaCT inhibition or knockdown have been proposed in cells, although findings are not necessarily dependent on the presence of extracellular citrate (Li et al., 2017; Phokrai et al., 2018; Poolsri et al., 2018). Citrate transport may also influence acetyl-CoA metabolism and ionic homeostasis in the nervous system, as loss-of-function mutations in SLC13A5 have been linked to pediatric epilepsy, Kohlschütter-Tönz syndrome, and other brain disorders (Hardies et al., 2015; Klotz et al., 2016; Matricardi et al., 2020; Schossig et al., 2017; Thevenon et al., 2014). Notably, citrate levels were significantly increased in plasma and cerebrospinal fluid (CSF) in such epilepsy patients (Bainbridge et al., 2017). Mice deficient in this transporter accumulated citrate in CSF, while plasma abundances were not affected; moreover, Slc13a5−/− mice exhibited an increased propensity for seizures as well as impaired neuronal function (Henke et al., 2020). While citrate’s role in chelating metal cations has been hypothesized to drive this pathophysiology (Bhutia et al., 2017; Glusker, 1980), the mechanism(s) through which SLC13A5 deficiency drives pathogenesis in mammalian cells warrants further study.
Here, we have applied mass spectrometry and metabolic flux approaches to genetically engineered hepatocellular carcinoma (HCC) cell lines and primary cortical neurons to decipher the impact of extracellular citrate import on metabolism and cell viability. SLC13A5 was expressed in both cell types, and exogenous citrate was imported and metabolized to fatty acids and TCA cycle intermediates. However, citrate only contributed appreciably to these pathways under hypoxic conditions where pyruvate dehydrogenase (PDH) flux is reduced. Under these conditions, citrate was predominantly catabolized in the cytosol to support acetyl-CoA generation. We also used CRISPR-Cas9 to generate SLC13A5-deficient HCC cell lines, which lacked the ability to transport and metabolize exogenous citrate. In addition, we observed that SLC13A5 expression was required to increase the growth of hypoxic HCC cells under glutamine-deprived conditions. Finally, NaCT function was also necessary to allow for citrate-mediated protection against Zn2+ toxicity. Collectively, our study highlights the biological roles of NaCT and citrate metabolism in mammalian cells, emphasizing the importance of metabolic stress in observing significant phenotypes.
RESULTS
Extracellular citrate uptake is tissue specific
Recent findings have highlighted the importance of circulating TCA intermediates as metabolic substrates or regulators of tissue function (Mills et al., 2018). We quantified levels of TCA intermediates in human plasma and observed that the concentration of citrate was 108 ± 23 μM, while other TCA cycle intermediates were 10-fold less abundant (all <10 μM) (Figure 1A). Although citrate is not present in typical culture media such as DMEM, RPMI, or OptiMEM, we found that complete medium including 10% fetal bovine serum (FBS) contained 16 ± 5 μM citrate, significantly lower than that observed in human plasma (Figure 1B).
Figure 1. Extracellular citrate uptake is tissue specific.
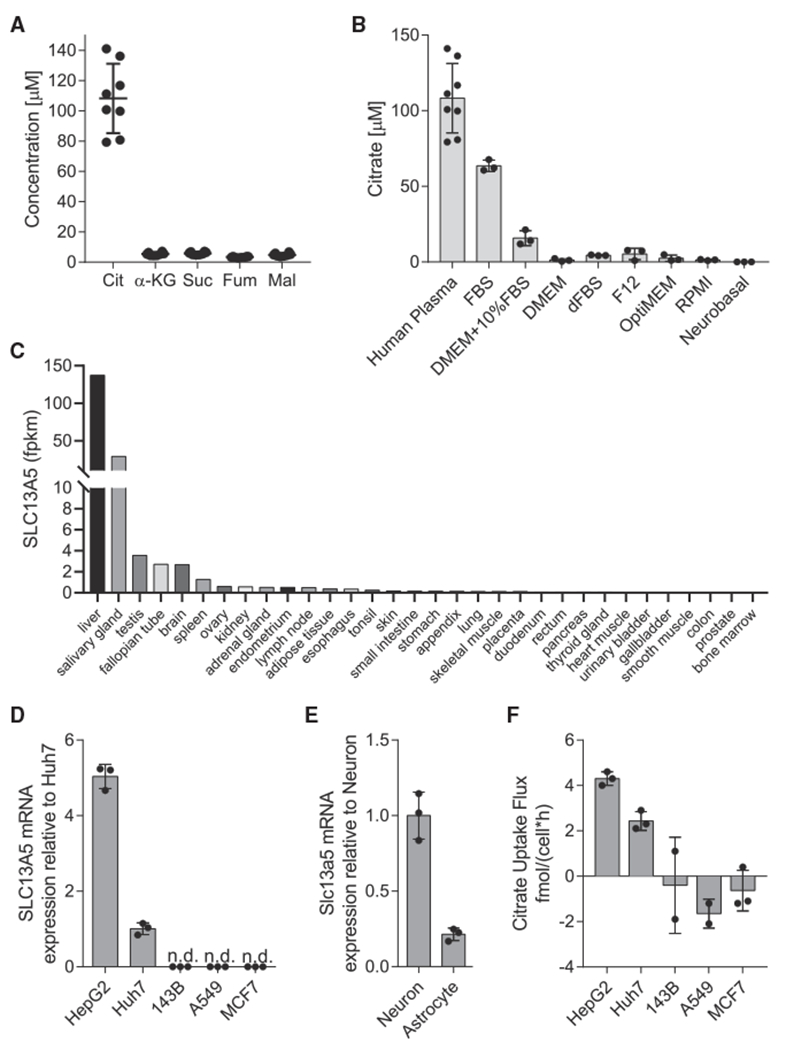
(A) Plasma concentration of TCA intermediates in humans (n = 8).
(B) Citrate concentration in cell culture medias (n = 3) and human plasma (n = 8) same as in (A).
(C) SLC13A5 mRNA expression in human tissues, from (Uhlén et al., 2015).
(D) SLC13A5 mRNA expression in cancer cells grown in normoxia relative to Huh7s (n = 3).
(E) Slc13a5 mRNA expression in primary rat neuron and astrocyte cells in normoxia relative to neurons (n = 3).
(F) Net citrate uptake flux in cancer cell lines over 48 h (n = 3).
Cit, citrate; α-KG, α-ketoglutarate; Suc, succinate; Fum, fumarate; Mal, malate. All data are presented as means ± SD. All data are representative of three sample replicates.
To identify cell types that might readily consume extracellular citrate, we first used a publicly available transcriptomics dataset from Uhlén et al. and compared SLC13A5 expression across 32 human tissues (Uhlén et al., 2015). SLC13A5 transcripts were highest in liver, brain, reproductive tissues, and salivary gland (Figure 1C). Next, we quantified the expression of SLC13A5 in a panel of cells of different tissue origin. Consistent with published data (Uhlén et al., 2015), we found that the HCC cell lines HepG2 and Huh7 as well as primary cortical neurons express detectable SLC13A5 mRNA, while little was detected in other cell lines (Figures 1D and 1E). We next cultured cells in DMEM supplemented with 500 μM extracellular citrate for 48 h and quantified uptake flux. Only the HCC cell lines exhibited net uptake of citrate, which correlated with SLC13A5 transcription (Figure 1F).
Citrate dilutes central carbon metabolism in HCC cells and neurons in hypoxia
The aforementioned results indicate that HCC cells import exogenous citrate. To quantify the contribution of citrate to central carbon metabolism in more detail, we cultured HepG2 and Huh7 cells in medium containing uniformly 13C-labeled ([U-13C5]) glutamine and quantified metabolite abundance and isotope enrichment (Figure 2A). Studies were performed under normoxic (21% oxygen) or hypoxic (1% oxygen) conditions for 48 h, as hypoxia is known to potently reduce citrate abundances due to decreased PDH flux (Metallo et al., 2011; Mullen et al., 2011; Wise et al., 2011). Indeed, TCA intermediate abundances were significantly decreased in hypoxia, with citrate being the most decreased of those measured (Figure S1A). As expected, labeling from [U-13C5]glutamine indicated that cells increased the contribution of reductive carboxylation to synthesis of aspartate and fatty acids as well as glutamine anaplerosis under hypoxia (Figures S1B–S1D). To indirectly determine whether exogenous citrate was metabolized in cells, we cultured Huh7 and HepG2 cells with and without 500 μM unlabeled (12C) citrate in medium containing [U-13C5]glutamine or [U-13C6]glucose, respectively. Citrate addition resulted in a dilution of glutamine’s contribution to TCA cycle anaplerosis (Figures 2B and S1E) and de novo lipogenesis through reductive carboxylation in HCC cell lines (Figures 2C and 2D). We performed similar studies using primary rat cortical neurons, although [U-13C6]glucose was used given the higher enrichment obtained in differentiated neurons (Divakaruni et al., 2017). Although labeling of the intracellular citrate pool was significantly diluted, glucose-derived TCA labeling was not impacted (Figure 2E). On the other hand, we observed a significant impact on the contribution of glucose to lipogenic acetyl-CoA using isotopomer spectral analysis (ISA) modeling (Figure 2F). Our findings suggest that extracellular citrate metabolically contributes as an anaplerotic and/or lipogenic substrate in low oxygen conditions and further highlight the need for some metabolic stress to observe a significant contribution.
Figure 2. Citrate dilutes central carbon pathway labeling in hepatocellular carcinoma and neuronal cells in hypoxia.
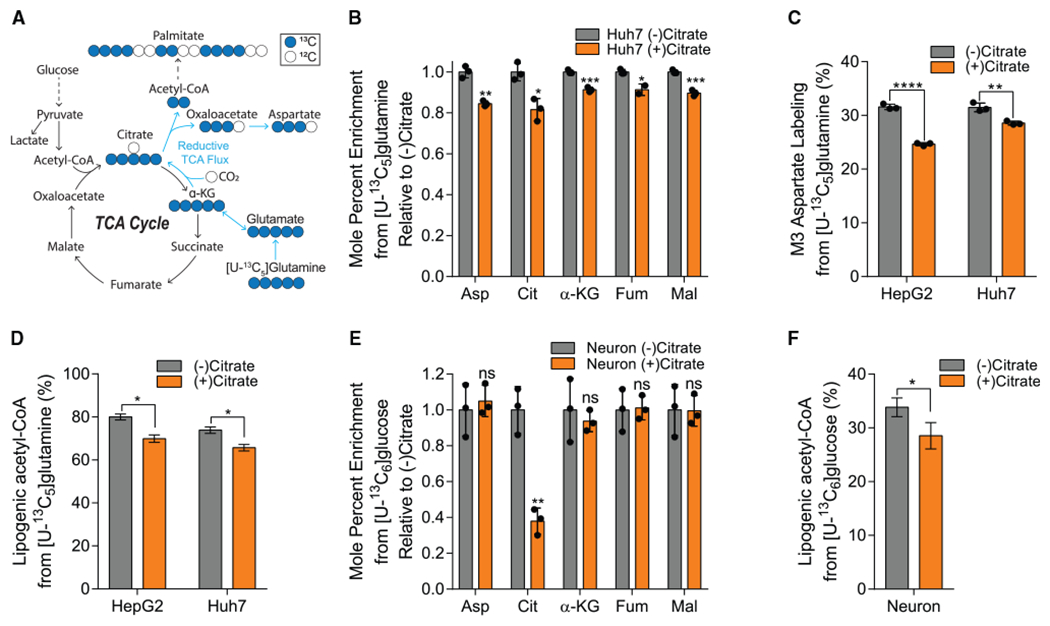
(A) Atom transition map depicting catabolism of [U-13C5]glutamine. Closed circles indicate 13C carbon, and open circles indicate 12C carbon.
(B) Mole percent enrichment of TCA intermediates from [U-13C5]glutamine in Huh7 cells grown in hypoxia ± 500 μM citrate for 48 h, relative to (−) citrate (n = 3).
(C) Percent labeling of M3 aspartate from [U-13C5]glutamine in HepG2 and Huh7 cells grown in hypoxia ± 500 μM citrate for 48 h (n = 3).
(D) Percentage of lipogenic acetyl-CoA contributed by [U-13C5]glutamine in HepG2 and Huh7 cells grown in hypoxia ± 500 μM citrate for 48 h (n = 3).
(E) Mole percent enrichment of TCA intermediates from [U-13C6]glucose in primary rat neuron cells grown in 3% oxygen ± 500 μM citrate for 48 h, relative to (−) citrate (n = 3).
(F) Percent of lipogenic acetyl-CoA contributed by [U-13C6]glucose in primary rat neuron cells grown in 3% oxygen ± 500 μM citrate for 48 h (n = 3).
Asp, aspartate; Cit, citrate; α-KG, α-ketoglutarate; Fum, fumarate; Mal, malate. In (B), (C), and (E), data are plotted as mean ± SD. Statistical significance is relative to (−) citrate as determined by two-sided Student’s t test with *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. In (D) and (F), data are plotted as mean ± 95% confidence interval (CI). Asterisk (*) indicates statistical significance by non-overlapping CIs. Unless indicated, all data represent biological triplicates. Data shown are from one of at least two separate experiments. See also Figure S1.
To more directly quantify how extracellular citrate is metabolized in SLC13A5-expressing cells, we cultured the aforementioned cell types in growth medium supplemented with 500 μM [2,4-13C2]citrate. Cells were rinsed two times in NaCl prior to extraction to ensure accurate analysis of intracellular pools. In HepG2 and Huh7 cells, extracellular citrate contributed significantly to TCA labeling, with ~4% enrichment of citrate and ~1% enrichment of downstream intermediates in normoxic cells (Figure 3A). Under hypoxia, enrichment of intracellular citrate and other TCA intermediates was significantly elevated (Figure 3A). In Huh7 cells, net uptake of extracellular citrate was significantly lower than that of glucose or glutamine in both normoxia and hypoxia (Figure S2A). However, although citrate uptake was not significantly increased in hypoxia, the ratio of net citrate uptake to anaplerotic glutamine usage (glutamine uptake versus glutamate efflux) flux was increased in hypoxia (3.0) compared with normoxia (1.3) (Figure S2B), reflecting the increased dependence on exogenous citrate driven by reduced PDH flux in hypoxia. Intracellular citrate was also highly enriched by [2,4-13C2]citrate in neonatal rat cortical neurons, although detectable labeling was only observed on α-ketoglutarate (Figure 3B). Furthermore, no changes in enrichment were observed when comparing normoxic to 3% oxygen. These differences are likely due to the reduced anaplerotic/biosynthetic needs of post-mitotic neurons. Our results confirm that extracellular citrate is transported into these SLC13A5-expressing cell types. Importantly, we were unable to detect significant isotope enrichment on TCA intermediates in other cell types tested that lack SLC13A5 expression, suggesting this transporter is important for citrate import and metabolism (Figure S2C).
Figure 3. Exogenous citrate is metabolized for TCA anaplerosis and fatty acid synthesis.

(A) Mole percent enrichment of TCA intermediates from [2,4-13C2]citrate in Huh7 (left) and HepG2 (right) cells grown in normoxia or hypoxia for 48 h (n = 3).
(B) Mole percent enrichment of TCA intermediates from [2,4-13C2]citrate in primary rat neuron cells grown in normoxia or 3% oxygen for 48 h (n = 3).
(C) Mole percent enrichment of fatty acids from [2,4-13C2]citrate in Huh7 (left) and HepG2 (right) cells grown in normoxia or hypoxia for 48 h (n = 3).
(D) Mole percent enrichment of fatty acids from [2,4-13C2]citrate in primary rat neuron cells grown in normoxia or 3% oxygen for 48 h (n = 3).
(E) Percent of lipogenic acetyl-CoA contributed by [U-13C6]glucose, [U-13C5]glutamine, and [2,4-13C2]citrate, in the presence or absence of 500 μM unlabeled citrate in Huh7 (left) and HepG2 (right) cells grown in hypoxia for 48 h (n = 3).
Cit, citrate; α-KG, α-ketoglutarate; Suc, succinate; Fum, fumarate; Mal, malate; Asp, aspartate; Glu, glutamate; Gln, glutamine; Glc, glucose. In (A)–(D), data are plotted as means ± SD. Statistical significance is relative to normoxia as determined by two-sided Student’s t test with *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Unless indicated, all data represent biological triplicates. In (E), data are plotted as mean ± 95% CI. Asterisk (*) indicates statistical significance by non-overlapping CIs. Data shown are from one of at least two separate experiments. See also Figure S2.
Next, we examined whether extracellular citrate was metabolized to acetyl-CoA by quantifying enrichment of fatty acids in cells cultured with the isotope tracers noted above. We detected significant enrichment of palmitate and stearate from [2,4-13C2]citrate, demonstrating that HCC cell lines and neurons generate acetyl-CoA from exogenous citrate (Figures 3C and 3D). No enrichment of palmitate was observed in other cell types that lack SLC13A5 expression (Figure S2D). We then compared the relative contributions of [U-13C5]glucose, [U-13C5]glutamine, and [2,4-13C2]citrate with the lipogenic acetyl-CoA fueling palmitate synthesis under hypoxia (Figure 3E). As expected, glutamine is the primary source of lipogenic carbon under these conditions. However, when citrate was present in the medium, HCC cells actively metabolized this substrate and the contribution of exogenous citrate to palmitate was greater than that of glucose, although glutamine remained the major lipogenic substrate in these hypoxic conditions. Notably, addition of extracellular citrate did not alter the rate of de novo lipogenesis (Figure S2E). Thus, exogenous citrate is taken up by SLC13A5-expressing cell types and is metabolized to fuel TCA cycle metabolism and lipogenesis, thereby diluting the corresponding glutamine contribution in low oxygen conditions. These findings are reminiscent of the usage of acetate under hypoxia in cells expressing cytosolic acetyl-CoA synthetase (ACSS2) (Kamphorst et al., 2014).
Extracellular citrate is primarily catabolized in the cytosol
The [2,4-13C2]citrate tracer enables more detailed insights into the compartment-specific catabolism of extracellular citrate when considering isotopolog distributions in more detail. In hypoxic, proliferating cancer cells, TCA intermediates are predominantly obtained from glutaminolysis (M4 labeling from [U-13C5] glutamine) or reductive carboxylation (M3 labeling from [U-13C5]glutamine), with TCA (re)cycling contributing to a lesser degree (Fan et al., 2013; Metallo et al., 2011). Therefore, TCA substrates in such cells are mostly present for one pass of the cycle. When [2,4-13C2]citrate is catabolized directly in the cytosol by ACLY, M1 acetyl-CoA and M1 oxaloacetate are formed, which further yielded M1 TCA intermediates via malate-aspartate shuttling and malic enzyme activity (Figure 4A). Alternatively, if citrate is imported into the mitochondria directly by SLC25A1, as has been shown to occur in anchorage-independent cultures (Jiang et al., 2016, 2017), [2,4-13C2]citrate will generate M2 isotopologues of TCA intermediates (Figure 4B). When culturing HCC cell lines in the presence of 500 μM [2,4-13C2]citrate for 48 h in either normoxia or hypoxia, we found that the M1 isotopolog of aspartate, malate, and fumarate was more abundant than the M2 isotopolog, and the ratio of M1/M2 labeling increased for these metabolites in hypoxic conditions (Figure 4C). On the other hand, relative abundance of M2 α-ketoglutarate was similar to or greater than M1 labeling, which indicates that a high exchange flux exists for the cytosolic aconitase (ACO1) and isocitrate dehydrogenase (IDH1) reactions (Figures 4A and 4D). To resolve these intracellular fluxes in a more unbiased manner that allows for multiple turns of the TCA cycle, we generated a 13C metabolic flux analysis (MFA) model encompassing glycolysis, compartmentalized TCA and amino acid metabolism, pyruvate cycling, de novo lipogenesis, and citrate import/exchange (Data S1) using the INCA software suite (Young, 2014). This model incorporated steady-state labeling of Huh7 cells grown in hypoxia with citrate across three independent experiments using either [U-13C6]glucose, [U-13C5]glutamine, or [2,4-13C2]citrate. We also included direct flux measurements of glucose, lactate, glutamine, glutamate, and citrate uptake and/or secretion. The model output confirmed that extracellular citrate (cit.x) contributes appreciably to lipogenesis and TCA metabolism and is significantly catabolized in the cytosol by ACLY and ACO1/IDH1 (Figure 4E). Importantly, the model estimated that reductive carboxylation exchange flux occurs primarily in the mitochondrial matrix via IDH2/ACO2 under these citrate-replete conditions, although cytosolic exchange cannot be entirely ruled out from this model (Tables S2 and S3). Glutamine-to-fatty acid flux was reconciled by high rates of pyruvate cycling and malic enzyme flux, consistent with the maintained activity of glutaminolysis that occurs in hypoxia (Fan et al., 2013; Vacanti et al., 2014). Collectively, these results suggest that exogenous citrate is primarily metabolized in the cytosol by ACLY and ACO1/IDH1 and predominantly enters TCA metabolism via α-ketoglutarate under the conditions tested (Figures 4C and 4D).
Figure 4. Extracellular citrate is primarily catabolized in the cytosol.
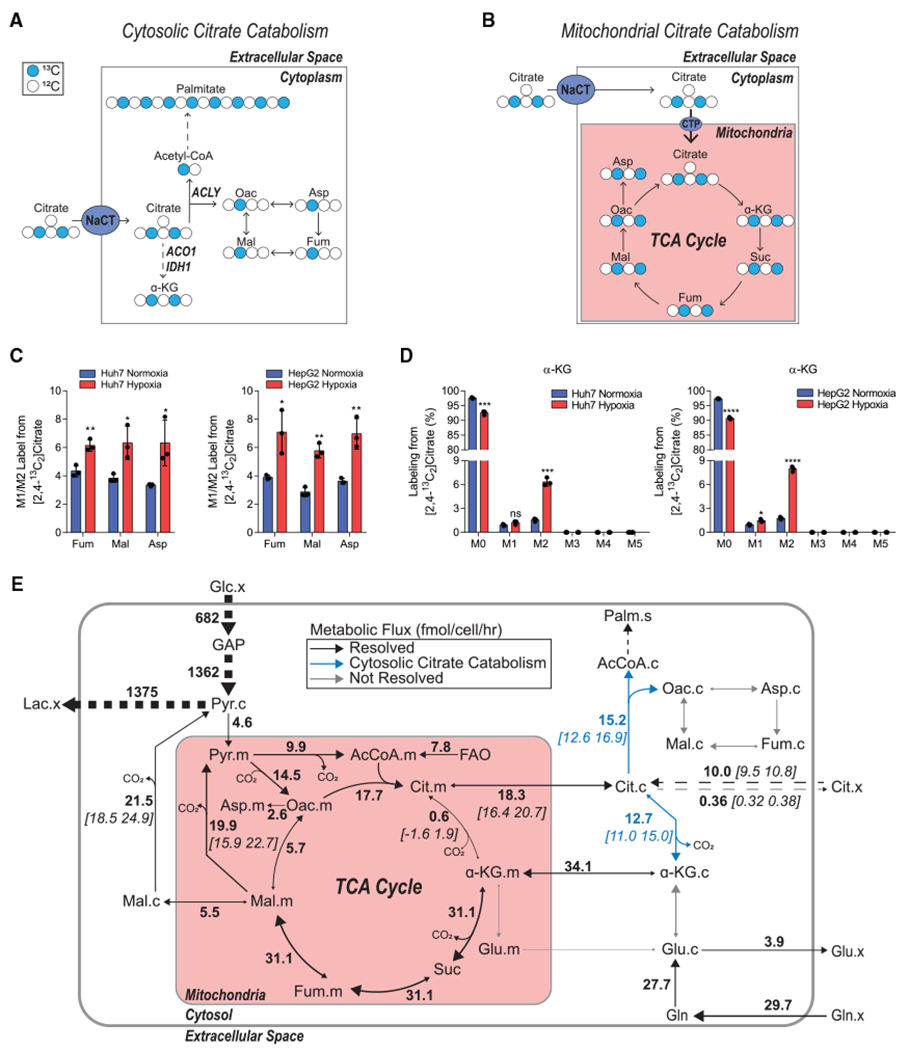
(A) Schematic of extracellular [2,4-13C2]citrate catabolism in the cytosol. Closed circles indicate 13C carbon, and open circles indicate 12C carbon.
(B) Schematic of extracellular [2,4-13C2]citrate catabolism in the mitochondria. Closed circles indicate 13C carbon, and open circles indicate 12C carbon.
(C) Ratio of the relative abundance of M1/M2 TCA intermediates from 500 μM [2,4-13C2]citrate in Huh7 (left) and HepG2 (right) cells grown in normoxia or hypoxia for 48 h (n = 3).
(D) Mass isotopomer distribution of α-KG from 500 μM [2,4-13C2]citrate in Huh7 (left) and HepG2 (right) cells grown in hypoxia for 48 h (n = 3).
(E) Schematic of central carbon metabolism with net fluxes and selected CIs estimated by 13C MFA for Huh7 cells grown in DMEM with 500 μM citrate in hypoxia for 48 h. Net flux values are listed adjacent to reaction arrows, with 95% CIs in square brackets. Dashed arrows represent grouped reactions. Some net or exchange fluxes for compartmentalized, parallel reactions were not resolvable.
Cit, citrate; Oac, oxaloacetate; Fum, fumarate; Mal, malate; Asp, aspartate; α-KG, α-ketoglutarate; Glc, glucose; Gln, glutamine; Glu, glutamate; Lac, lactate; GAP, glyceraldehyde 3-phosphate; Palm, palmitate; FAO, fatty acid oxidation. In all graphs, data are plotted as means ± SD. Statistical significance is relative to normoxia as determined by two-sided Student’s t test with *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Unless indicated, all data represent biological triplicates. Data shown are from one of at least two separate experiments.
NaCT supports extracellular citrate import and metabolism in HCC cells
To directly interrogate the function of NaCT with respect to citrate metabolism, we engineered SLC13A5-deficient knockout (KO) HepG2 and Huh7 cells using CRISPR-Cas9 and compared their metabolic phenotype with cells expressing a non-targeting control (NTC) single guide RNA (sgRNA). Two clones were tested for each KO cell line. SLC13A5 KO clones were validated by sequencing the region of interest (Figure S3). In both cell lines, KO of SLC13A5 resulted in reduced citrate uptake flux when the cells were cultured in growth medium supplemented with 500 μM exogenous citrate (Figures 5A and S4A), suggesting these cells lacked NaCT activity. To further verify the functional impact of NaCT deficiency, we quantified citrate transport using [1,5-14C] citrate (Pajor et al., 2016). Sodium-induced citrate uptake was significantly reduced in the SLC13A5 KO cells (Figure 5B). Notably, NaCT deficiency induced no significant metabolic phenotype in cells grown in the absence of extracellular citrate, as fatty acid synthesis rates were unchanged and alterations to metabolite abundances were either moderate or inconsistent in the KO clones (Figures S4B and S4C). Additionally, although previous studies have found that SLC13A5 knockdown alone induced a reduction in the expression of fatty acid synthesis gene expression in HCC cells (Li et al., 2017), we observed no impact on ACLY expression in HepG2 or Huh7 SLC13A5 KO cells (Figure S4D). These data indicate that the citrate transporter induces a metabolic change that is selective to citrate uptake.
Figure 5. NaCT supports extracellular citrate import and metabolism in hepatocellular carcinoma cells.
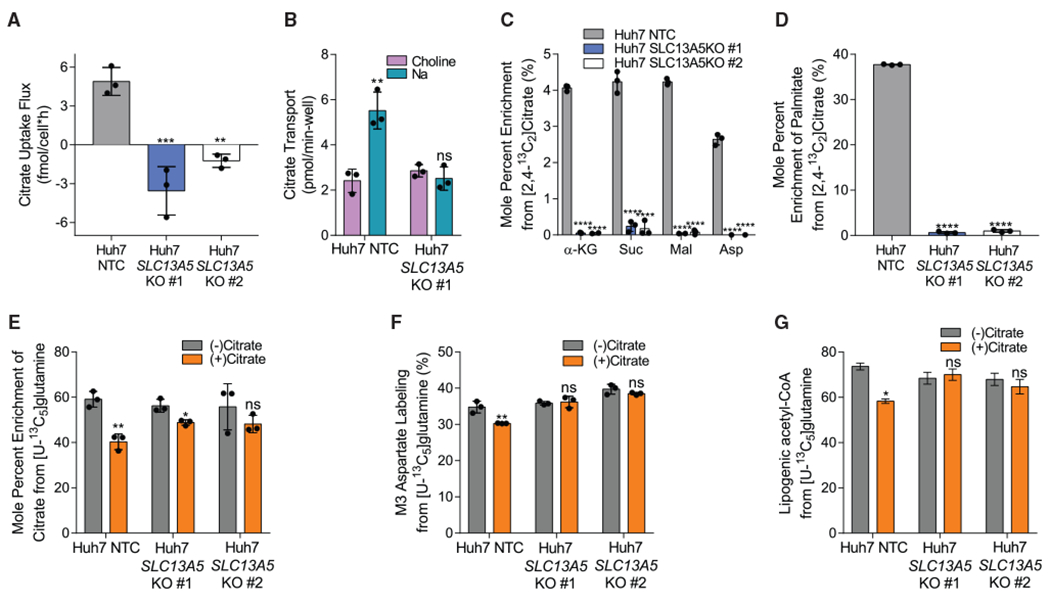
(A) Citrate uptake flux in Huh7 NTC and SLC13A5 KO cells grown in hypoxia for 48 h (n = 3).
(B) Citrate transport in Huh7 NTC and SLC13A5 KO #1 cells (n = 3).
(C) Mole percent enrichment of TCA intermediates from [2,4-13C2]citrate in Huh7 NTC and SLC13A5 KO cells grown in hypoxia for 48 h (n = 3).
(D) Mole percent enrichment of palmitate from [2,4-13C2]citrate in Huh7 NTC and SLC13A5 KO cells grown in hypoxia for 48 h (n = 3).
(E) Mole percent enrichment of citrate from [U-13C5]glutamine in Huh7 NTC and SLC13A5 KO cells ± 500 μM citrate grown in hypoxia for 48 h (n = 3).
(F) Percent labeling of M3 aspartate from [U-13C5]glutamine in Huh7 NTC and SLC13A5 KO cells ± 500 μM citrate grown in hypoxia for 48 h (n = 3).
(G) Percentage of lipogenic acetyl-CoA contributed by [U-13C5]glutamine in Huh7 NTC and SLC13A5 KO cells ± 500 μM citrate grown in hypoxia for 48 h (n = 3).
α-KG, α-ketoglutarate; Suc, succinate; Mal, malate; Asp, aspartate. In all graphs data are plotted as means ± SD. Statistical significance is relative to NTC as determined by one-way ANOVA with Dunnett’s method for multiple comparisons (A, C, and D) or relative to (−) citrate as determined by two-sided Student’s t test (B, E, and F) with *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. In (G), data are plotted as mean ± 95% CI. Asterisk (*) indicates statistical significance by non-overlapping CIs. Unless indicated, all data represent biological triplicates. See also Figures S3 and S4. (C–G) Data shown are from one of at least two separate experiments.
To elucidate how impaired NaCT activity influences citrate catabolism, we cultured each cell line for 48 h in the presence of 500 μM [2,4-13C2]citrate under hypoxic conditions. Huh7 SLC13A5 KO cells had no measurable enrichment from [2,4-13C2]citrate on TCA cycle intermediates or fatty acids (Figures 5C and 5D). A reduction in enrichment was also observed with HepG2 SLC13A5 KO cells (Figures S4E and S4F). Our data confirm that NaCT is the primary importer of extracellular citrate in the tested HCC cell lines. Next, we cultured NaCT-deficient cells in the presence or absence of exogenous 12C citrate in [U-13C5]glutamine tracer medium and quantified enrichment on TCA cycle intermediates and fatty acids compared with control cells. When unlabeled citrate was present in the media, we only observed dilution of [U-13C5]glutamine on TCA cycle intermediates in SLC13A5-expressing cells (Figures 5E and S4G). Furthermore, we observed no dilution in isotopologues downstream of reductive carboxylation (i.e., M3 aspartate) or glutamine-derived lipogenic acetyl-CoA with extracellular unlabeled citrate addition in the Huh7 SLC13A5 KO cells (Figures 5F and 5G). Thus, our results indicate that NaCT is required for the import and metabolism of extracellular citrate, in HCC cells.
NaCT facilitates growth under nutrient stress and resistance to Zn2+ toxicity
Next, we hypothesized that citrate import and catabolism could fulfill certain metabolic roles of glutamine, which serves as a key substrate for anaplerosis and acetyl-CoA synthesis under hypoxia. We cultured the aforementioned HCC cell lines in glutamine-deficient media containing [U-13C6]glucose supplemented with exogenous citrate. Notably, addition of citrate in the absence of glutamine significantly increased fatty acid synthesis in control cells (Figure 6A). Furthermore, citrate supplementation generally decreased enrichment of TCA intermediates and associated amino acids from [U-13C6]glucose, but increased the abundance of α-ketoglutarate in NTC cells (Figures 6B and S5A), no change was observed in NaCT-deficient cell lines. Next, we cultured SLC13A5-expressing Huh7 cells with [2,4-13C2]citrate and found that in the absence of glutamine, enrichment from 13C citrate on metabolites associated with glutamine synthesis increased significantly (Figure 6C). These data indicate that NaCT facilitates citrate-mediated anaplerosis under nutrient-limited conditions.
Figure 6. NaCT facilitates growth under nutrient stress and resistance to Zn2+ toxicity.
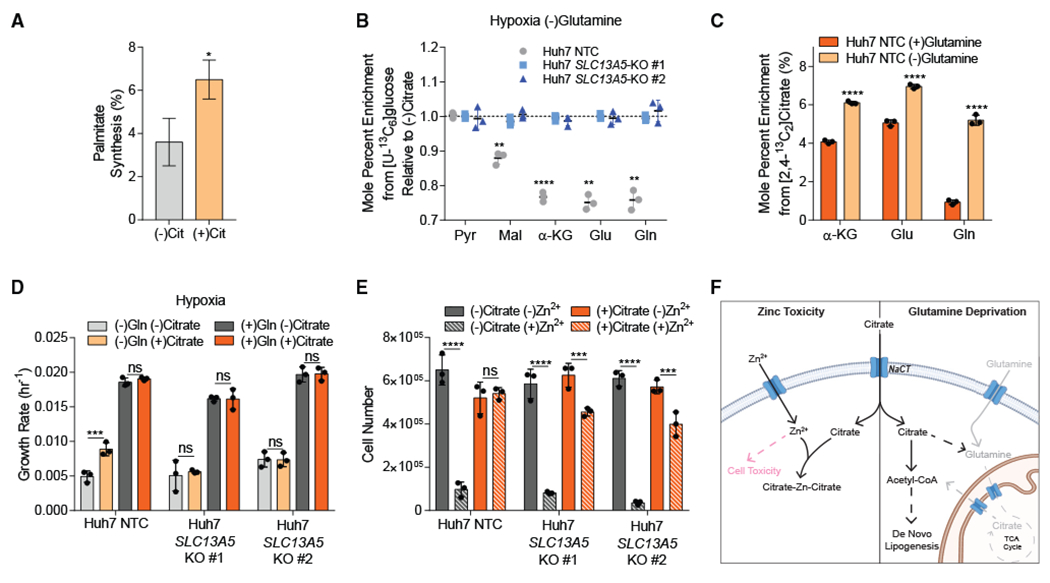
(A) Palmitate synthesis in Huh7 NTC cells grown in high glucose DMEM without glutamine ± 500 μM citrate in hypoxia for 48 h (n = 3).
(B) Mole percent enrichment of metabolites from [U-13C6]glucose in Huh7 SLC13A5 KO cells grown without glutamine in hypoxia ± 500 μM citrate for 48 h, relative to (−) citrate (n = 3).
(C) Mole percent enrichment of TCA intermediates from [2,4-13C2]citrate in Huh7 NTC cells grown in hypoxia ± 4 mM glutamine for 48 h (n = 3).
(D) Growth rates of Huh7 NTC and SLC13A5-KO cells grown in high glucose DMEM ± 4 mM glutamine ± 500 μM citrate in hypoxia for 4 days.
(E) Cell numbers of Huh7 NTC and SLC13A5-KO cells grown in high glucose DMEM ± 5 mM citrate ± 300 μM ZnCl2 24 h after Zn2+ treatment in hypoxia.
(F) Schematic depicting extracellular citrate utilization during cellular stress. Gray indicates removed metabolites, and pink indicates cytotoxic. Created with Biorender.com.
Cit, citrate; Pyr, pyruvate; Mal, malate; α-KG, α-ketoglutarate; Glu, glutamate; Gln, glutamine. In (A), data are plotted as mean ± 95% CI. Asterisk (*) indicates statistical significance by non-overlapping CIs. In (B)–(E), data are plotted as means ± SD. Statistical significance is determined by two-sided Student’s t test relative to (−) citrate (B), (+) glutamine (C), or determined by two-way ANOVA with Tukey’s method for multiple comparisons relative to (−) citrate (D) or (−) Zn2+ (E) conditions with *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. Unless indicated, all data represent biological triplicates. See also Figure S5. Data shown are from one of at least two separate experiments.
Since glutamine is critical for numerous biological processes, including anaplerosis and lipogenesis, we next tested whether NaCT-mediated citrate uptake impacted cell proliferation upon glutamine deprivation. We observed that exogenous citrate addition increased the growth rate of the Huh7 NTC cells by 80% in glutamine-depleted media under hypoxic conditions (Figure 6D). Notably, no growth alteration was observed with citrate addition in glutamine-rich media (Figure 6D) or normoxia (Figure S5B). Importantly, citrate supplementation did not increase proliferation of NaCT-deficient cell lines (Figures 6D and S5B). These data indicate that NaCT-mediated citrate transport supports metabolism under nutrient-limited conditions. We next tested whether NaCT-mediated citrate transport influences resistance to metal toxicity against ions such as zinc (Zn2+). Regions of the brain including the hippocampus and cerebral cortex are Zn2+ enriched (Assaf and Chung, 1984). Under pathological conditions including epilepsy, ischemia, and traumatic brain injury, cerebral Zn2+ levels increase and may cause neuronal cell death (Assaf and Chung, 1984; Frederickson et al., 1983; Weiss et al., 2000). Intracellular citrate has been suggested to be a protective chelator against Zn2+ in rat cortical neurons (Sul et al., 2016); however, the role of NaCT in this process remains unclear. Given the established link between pediatric epilepsy and SLC13A5 mutations, we hypothesized that NaCT-mediated citrate uptake could generally facilitate protection against Zn2+ toxicity. Hepatocytes can also exhibit sensitivity to Zn2+ (Lemire et al., 2008; Steinebach and Wolterbeek, 1993), so we used the Huh7 NaCT-deficient cells described above to determine whether NaCT function mediates protection against Zn2+ toxicity. We cultured hypoxic Huh7 NTC and NaCT-deficient cells in the presence or absence of citrate and ZnCl2 for 24 h and quantified cell viability. While citrate supplementation protected Huh7 NTC against Zn2+ toxicity, cell lines lacking NaCT showed reduced viability (Figure 6E). Thus, our data highlight that extracellular citrate import via NaCT facilitates protection against nutrient deprivation as well as Zn2+ toxicity in human cells (Figure 6F).
DISCUSSION
Cells take up diverse nutrients from the extracellular microenvironment, and each metabolite may serve a distinct purpose or function. Nutrient transport is selective and regulated, in part, through cell-specific expression of transporters such as NaCT, which is restricted to liver and neural tissue. Here, we investigated how NaCT influences citrate metabolism in HCC cell lines and primary rat cortical neurons. We observed that NaCT-mediated citrate import contributes to cytosolic acetyl-CoA pools in HCC cells and neurons under distinct conditions. Hypoxic HCC cells metabolized extracellular citrate to fatty acids and TCA intermediates in a NaCT-dependent manner. When glutamine (and other nutrients) became limited, citrate supported the growth and lipid synthesis of HCC cell lines expressing NaCT. Furthermore, citrate supplementation enhanced the viability of HCC cell lines exposed to Zn2+ toxicity, but this protective effect was mitigated in SLC13A5 KO cells that lacked NaCT expression.
Many cancer cell types increase glutamine utilization to support the TCA cycle and fatty acid biosynthesis in vitro (Metallo et al., 2011; Mullen et al., 2011); however, reproducing this phenotype in vivo has generated mixed results (Davidson et al., 2016; Le et al., 2012; Son et al., 2013; Tardito et al., 2015). One potential explanation for this discrepancy is the difference in nutrient profile of cell culture media compared with in vivo microenvironments (Ackermann and Tardito, 2019; Rossiter et al., 2021; Vande Voorde et al., 2019). Plasma and interstitial fluid contain metabolites that impact cellular metabolism, but are routinely excluded from most cell culture medias. For instance, uric acid, a metabolite abundant in human plasma but absent in many cell culture medias, inhibited de novo pyrimidine synthesis in cancer cells in vitro (Cantor et al., 2017). Previous in vitro studies have observed that upregulated reductive glutamine catabolism supports fatty acid biosynthesis and defuses mitochondrial redox stress in hypoxia (Jiang et al., 2016; Metallo et al., 2011). In our study, we found that reductive carboxylation was elevated in hypoxia in HepG2 and Huh7 cells, but observed that extracellular citrate addition reduced flux through this pathway. Thus, uptake of extracellular citrate may provide an additional carbon source which is accessible to ACLY and downstream pathways, curtailing the need for other nutrients to support these processes. Similar observations of citrate import and metabolism were made in prostate cancer cells, which express pmCIC (Mycielska et al., 2018). On the other hand, we only observed appreciable citrate contributions to biosynthesis under selected conditions (hypoxia and glutamine restriction). These findings collectively suggest that citrate is not a major biosynthetic substrate, but serves as a resource to cells when they experience distinct stresses (e.g., ischemia, hypoxia, metal toxicity).
We also demonstrated that SLC13A5-encoded NaCT is the primary mechanism for citrate import in HCC cells. No major changes in metabolism were observed beyond acetyl-CoA and TCA metabolism, and phenotypic responses were dependent upon expression of functional NaCT as well as citrate supplementation. Importantly, some prior studies observed signaling and growth phenotypes in cells upon knockdown or inhibition of NaCT in the absence of extracellular citrate (Li et al., 2017; Phokrai et al., 2018; Poolsri et al., 2018), but our results indicate that citrate transport is a key function of the SLC13A5 gene product. Indeed, we also found that citrate uptake by NaCT was protective against Zn2+ cytotoxicity in Huh7 cells. This finding mirrors those of previous studies that demonstrated that citrate or pyruvate administration were protective against Zn2+ cytotoxicity in neurons (Sul et al., 2016; Yoo et al., 2004). Loss-of-function mutations in SLC13A5 have been associated with early-onset epilepsy, but the role of citrate metabolism in the pathophysiology of these patients is not well understood (Hardies et al., 2015; Klotz et al., 2016; Thevenon et al., 2014). Various mechanisms have been proposed for this phenotype, including the susceptibility of NaCT-deficient neurons to increased synaptic Zn2+ concentrations induced upon neuronal activation (Sul et al., 2016). Alternatively, others have hypothesized that improper NaCT function leads to increased synaptic citrate concentrations and excessive Zn2+ chelation, which impairs NMDA receptor function and drives neuronal dysfunction (Bhutia et al., 2017). Notably, we performed our study in HCC cells using supraphysiological concentrations of citrate and Zn2+ that might only occur transiently in the body. Furthermore, application of relatively severe metabolic stress (i.e., 1% oxygen and glutamine deprivation) to limit endogenous citrate was required to observe a growth phenotype in HCC cells. These findings suggest that biosynthetic defects may not be as relevant to the neurological phenotype of NaCT-deficient patients. As such, our results should motivate further investigation into the impact of SLC13A5 mutations on homeostasis of potentially toxic metal cations such as Zn2+ and its relation to epilepsy. Ideally, such studies would be performed in a more physiological model system such as NaCT KO in brain organoids or animals, which recapitulate the physiology of citrate better than 2D cell culture. Collectively, our results demonstrate that NaCT mediates citrate transport and metabolism under conditions of metabolic stress. Our approach also highlights the utility of coordinated studies that involve targeting of disease-related metabolic genes in relevant cell types and deep metabolic profiling using flux analysis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christian M. Metallo (cmetallo@ucsd.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
All original code is available in this paper’s supplemental information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Samples
Plasma samples from healthy fasted adults (29-42 years, 5 male, 3 female) were obtained from a clinical cohort recruited from Tompkins County, New York area as described previously (Gheller et al., 2021). Participants were excluded if they had a history of negative or allergic reactions to local anesthetic, used immunosuppressive medications, were prescribed anti-coagulation therapy, were pregnant, had a musculoskeletal disorder, suffered from alcoholism (> 11 drinks per week for women and > 14 drinks per week for men) or other drug addictions or were acutely ill at the time of participation (Gheller et al., 2019; Riddle et al., 2018). The Cornell University Institutional Review Board approved the protocol and all the subjects provided written informed consent in accordance with the Declaration of Helsinki.
Cell Lines
HepG2, Huh7, A549, MCF7, and 143B cells were obtained from ATCC and were incubated at 37C with 5% CO2 and cultured using Dulbecco’s Modified Eagle Media (DMEM) with 10% Fetal Bovine Serum and 1% Penicillin-Streptomycin. Cells tested negative for mycoplasma contamination. For hypoxia experiments, cells were maintained in a humidified glove box (Coy) at 5% CO2 and either 1% O2 (cancer cell lines) or 3% O2 (primary cortical neurons). Primary cortical neurons and astrocytes were isolated and cultured in Neurobasal medium (neuron) or DMEM medium supplemented with 10 mM glucose, 0 mM glutamine, and 10% FBS (astrocytes) as described in detail by Cordes et al. (2020). All media were adjusted to pH = 7.3.
METHOD DETAILS
Cell Proliferation and 13C Tracing
Proliferation studies were performed on 12 well plates with an initial cell number of 50,000/well for Huh7s and 100,000/well for HepG2s. Cells were plated in growth media and allowed to adhere for 24 hours before changing to the specified growth media. Cell counts were performed at days 0 and 4 using a hemocytometer.
For Zn2+ tolerance studies, cells were plated at 50,000/well and allowed to adhere for 24 hours. They were then placed in the hypoxia chamber for 24 hours before swapping to ± 5 mM citrate media. After changing media, cells were allowed to acclimate for 24 hours before 300 μM ZnCl2 in water was spiked into the designated wells. Cells were counted 24 hours after Zn2+ treatment using a hemocytometer.
13C isotope tracing media was formulated using glucose and glutamine free DMEM 5030 supplemented with either 20 mM [U-13C6] glucose, 4 mM [U-13C5]glutamine, or 500 μM [2,4-13C2]citrate (Cambridge Isotopes) and 10% dialyzed FBS. All studies were performed with a final concentration of 20 mM glucose and 4 mM glutamine. Cultured cells were washed with 1 mL PBS prior to applying tracing media for 24-72 hours as indicated in figure legends. For tracing experiments performed in hypoxia, cancer cells were acclimated to the hypoxia chamber in basal media for 24 hours prior to applying pre-hypoxified tracing media. Mole percent enrichment (MPE) of isotopes were calculated as the percent of all atoms within the metabolite pool that are labeled, using Escher-Trace (Kumar et al., 2020):
where n is the number of carbon atoms in the metabolite and Mi is the relative abundance of the ith mass isotopomer. The MPE of [2,4-13C2]citrate is 33%, while MPE for [U-13C6]glucose and [U-13C5]glutamine is 100%.
Isotopomer Spectral Analysis (ISA)
Isotopomer spectral analysis (ISA) was performed to estimate the percent of newly synthesized palmitate as well as the contribution of a tracer of interest to the lipogenic acetyl-CoA pool (Cordes and Metallo, 2019; Young, 2014). Parameters for contribution of 13C tracers to lipogenic acetyl-CoA (D value) and percentage of newly synthesized fatty acid (g(t) value) and their 95% confidence intervals are then calculated using a best-fit model from INCA MFA software. Experimental fatty acid labeling from [U-13C6]glucose, [U-13C5]glutamine, or [2,4-13C2]citrate after 48 hour trace was compared to simulated labeling using a reaction network where C16:0 is condensation of 8 AcCoA. ISA data plotted as mean ± 95% CI. * indicates statistical significance by non-overlapping confidence intervals.
CRISPR/Cas9 engineered knockout cell lines
SLC13A5 knockout clones were generated using the strategy described previously (Ran et al., 2013). Briefly, a guide RNA (gRNA) was designed to target the human SLC13A5 gene using the CRISPOR webtool (gRNA sequence: AGGCACAATGAATAGCAGGG) (Concordet and Haeussler, 2018). The gRNA duplex was cloned into lentiCRISPRv2 (Addgene #52961) (Sanjana et al., 2014). HepG2 and Huh7 cells were transfected with the SLC13A5 specific gRNA to generate pooled knockouts. After puromycin selection, single-cell clones were isolated by diluting the pooled knockout lines at 1 cell/100 μL and plating 100 μL into each well of a 96 well plate. Clones were maintained by exchanging media every 3-5 days. HepG2 clones were cultured in 50% conditioned media to enhance proliferation at early stages of cloning. Conditioned media was generated by collecting the spent media that had been culturing HepG2s after 2 days, spinning down at 300g for 5 min, and filtering using a 0.2 micron filter to clear cellular debris. This media was diluted 1:1 with DMEM with 10% FBS 1% penstrep to generate the 50% conditioned media. Clones were validated via Sanger sequencing. Sequences were aligned using the NCBI BLASTN suite (Agarwala et al., 2018).
Lentivirus Production
One 10cm dish of HEK293FT cells at 60% confluency were transfected with 1.3 μg VSV.G/pMD2.G, 5.3 μg lenti-gag/pol/pCMVR8.2, and 4 μg of the gRNA duplexed lentiCRISPRv2 using 16 μL Lipofectamine 3000 diluted in 0.66 mL of OPTI-MEM. Medium containing viral particles was harvested 48 and 72 hours after transfection, then concentrated by Centricon Plus-20 100,000 NMWL centrifugal ultrafilters, divided into aliquots and frozen at −80°C.
Determination of Extracellular Fluxes
Metabolic fluxes for citrate were calculated by collecting media at time 0 and spent media after 48 hours. Spent media was centrifuged at 300 g for 5 min, to remove cell debris. Cell counts were performed at time 0 and after 48 hours as well. To calculate citrate uptake fluxes, citrate was quantified by GC-MS via standard curve. To calculate glucose, lactate, glutamine, and glutamate uptake fluxes media metabolites were quantified using a Yellow Springs Instruments (YSI) Biochemistry Analyzer 2950.
Metabolite Extraction and GC-MS Analysis
At the conclusion of the tracer experiment, media was aspirated. Then, cells were rinsed twice with 0.9% saline solution and lysed with 250 μL ice-cold methanol. After 1 minute, 100 μL water containing 1 μg/ml norvaline was added to each sample and vortexed for one minute. 250 μL chloroform was added to each sample, and all were vortexed again for 1 minute. After centrifugation at 21,130 g for 10 minutes at 4°C, 250 μL of the upper aqueous layer was collected and evaporated under vacuum at 4°C. The lower organic layer was evaporated under air at room temperature.
Plasma metabolites were extracted and quantified as follows. For metabolite extraction, 10 μL of each plasma sample was utilized. First, 90 μL of a 9:1 methanol:water mix was added to each sample and the samples were vortexed for 1 minute. After centrifugation at 16,000 g for 10 minutes, 90 μL of supernatant was collected, evaporated under vacuum at −4°C and analyzed using GC/MS. Metabolite levels of TCA intermediates were quantified using external standard curves.
Dried polar and nonpolar metabolites were processed for gas chromatography (GC) mass spectrometry (MS) as described previously in Cordes and Metallo (2019). Briefly, polar metabolites were derivatized using a Gerstel MultiPurpose Sampler (MPS 2XL). Methoxime-tBDMS derivatives were formed by addition of 15 μL 2% (w/v) methoxylamine hydrochloride (MP Biomedicals, Solon, OH) in pyridine and incubated at 45°C for 60 minutes. Samples were then silylated by addition of 15 μL of N-tert-butyldimethylsily-N-methyltrifluoroacetamide (MTBSTFA) with 1% tert-butyldimethylchlorosilane (tBDMS) (Regis Technologies, Morton Grove, IL) and incubated at 45°C for 30 minutes. Nonpolar metabolites were saponified and transesterified to fatty acid methyl esters (FAMEs) by adding 500 μL of 2% H2SO4 in methanol to the dried nonpolar layer and heating at 50°C for 1 hour. FAMEs were then extracted by adding 100 μL of a saturated salt solution and 500 μL hexane and vortexing for 1 minute. The hexane layer was removed, evaporated, and resuspended with 60μL hexane for injection.
Derivatized polar samples were injected into a GC-MS using a DB-35MSUI column (30 m x 0.25mm i.d. x 0.25 μm, Agilent J&W Scientific, Santa Clara, CA) installed in an Agilent 7890B GC system integrated with an Agilent 5977a MS. Samples were injected at a GC oven temperature of 100°C which was held for 1 minute before ramping to 255°C at 3.5°C/min then to 320°C at 15°C/min and held for 3 minutes. Electron impact ionization was performed with the MS scanning over the range of 100-650 m/z for polar metabolites.
Derivatized nonpolar samples were injected into a GC-MS using a Fame Select column (100 m x 0.25mm i.d. x 0.25 μm, Agilent J&W Scientific, Santa Clara, CA) installed in an Agilent 7890A GC system integrated with an Agilent 5977A MS. Samples were injected at a GC oven temperature of 80°C which was held for 1 minute before ramping to 170°C at 20°C/min then to 188°C at 1°C/min then to 250°C at 20°C/min and held for 10 minutes. Electron impact ionization was performed with the MS scanning over the range of 54-400 m/z for nonpolar metabolites.
Metabolite levels and mass isotopomer distributions were analyzed with an in-house MATLAB script which integrated the metabolite fragment ions and corrected for natural isotope abundances.
13C Metabolic Flux Analysis
13C Metabolic Flux Analysis (MFA) was performed using INCA (Young, 2014). The model was comprised of the chemical reactions and atom transitions of central carbon metabolism, as well as extracellular fluxes of glucose, lactate, glutamine, glutamate, and citrate and the isotopic labeling distributions of intracellular metabolites from Huh7 cells traced with DMEM and 500μM citrate containing [U-13C6]glucose, [U-13C5]glutamine, or [2,4-13C2]citrate under hypoxia.
The 13C MFA was performed with the following assumptions:
Cellular metabolism and isotopic labeling were assumed to be at steady state.
Cells were assumed to grow exponentially.
Labeled CO2 formed from decarboxylation reactions is dilute and assumed to never reincorporate in carboxylation reactions.
Separate mitochondrial and cytosolic pools of aspartate, oxaloacetate, malate, fumarate, α-KG, citrate, glutamate, pyruvate, and acetyl-CoA were modeled with exchange fluxes for malate, aspartate, glutamate, α-KG, and citrate.
The gross, per cell biomass requirements of proliferating Huh7 hepatoma cells were similar to those reported previously for mammalian cells (Grassian et al., 2014).
Additional information regarding the estimated fluxes generated by the model, as well as the data incorporated into the model can be found in the supplementary material (Tables S2 and S3; Data S1).
RNA isolation and quantitative RT-PCR
Total RNA was purified from cultured cells using Trizol Reagent (Life Technologies) per manufacturer’s instructions. First-strand cDNA was synthesized from 1 μg of total RNA using iScript Reverse Transcription Supermix for RT-PCR (Bio-Rad Laboratories) according to the manufacturer’s instructions. Individual 20 μL SYBR Green real-time PCR reactions consisted of 1 μL of diluted cDNA, 10 μL of SYBR Green Supermix (Bio-Rad), and 1 μL of each 5 μM forward and reverse primers. For standardization of quantification, 18S was amplified simultaneously. The PCR was carried out on 96-well plates on a CFX Connect Real time System (Bio-Rad), using a three-stage program provided by the manufacturer: 95°C for 3 min, 40 cycles of 95°C for 10 s and 60°C for 30 s. Gene-specific primers are tabulated in Table S1.
14C citrate uptake assay
Uptake of 14C citrate was quantified as described previously (Pajor and Sun, 2010). Briefly, cells were seeded at 3 × 105 cells/well on 6 well collagen treated plates. After 24 hours, cells were acclimated to serum free media for 24 hours. To carry out the assay, each well was washed twice with choline buffer (mM: 140 Choline, 2 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, pH adjusted to 7.4 with 1M Tris), then incubated with 1 mL sodium buffer (mM: 140 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, pH adjusted to 7.4 with 1M Tris) or choline buffer containing 100 μM [14C]-citrate for 30 minutes at 37°C with rocking. The uptake assays were stopped, and the cells were washed four times with 2.5 mL choline buffer. Cells were dissolved in 0.4 ml/well 1% SDS, transferred to scintillation vials with Econoscint Scintillation cocktail and then the radioactivity in the plates was counted using a scintillation counter.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using Graphpad PRISM software. Unless indicated, all results shown as mean ± SD of cellular triplicates obtained from one representative experiment as specified in each figure legend. P values were calculated using a Student’s two-tailed t test, One-way ANOVA w/ Dunnet’s method for multiple comparisons, or Two-way ANOVA w/ Tukey’s method for multiple comparisons; *, P value < 0.05; **, P value < 0.01; ***, P value < 0.001, ****, P value < 0.0001. Unless indicated, all normalization and statistical tests compared to NTC cells, normoxia, or (−)citrate conditions.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| One Shot Stbl3 E. Coli | ThermoFisher Scientific | Cat#C7373 |
| Library Efficiency DH5α Competent Cells | ThermoFisher Scientific | Cat#18263012 |
| Biological samples | ||
| Healthy human plasma | Thalacker-Mercer Lab; Gheller et al., 2021 | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| [U-13C6] Glucose | Cambridge Isotope Labs | Cat#CLM-1396 |
| [U-13C5] Glutamine | Cambridge Isotope Labs | Cat#CLM-1822 |
| [2,4-13C2] Citrate | Cambridge Isotope Labs | Cat#CLM-148 |
| [1,5-14C2] Citrate | Morovak Inc. | N/A |
| Zinc Chloride | Sigma-Aldrich | CAS# 7646-85-7 |
| Sodium Citrate | Sigma-Aldrich | CAS# 6132-04-3 |
| Methoxylamine Hydrochloride | MP Biomedicals | Cat#02155405 |
| MTBSTFA + 1% TBDMSCl | Regis Technologies | Cat#1-270143-200 |
| BsmBI | New England Biolabs | Cat#R0580L |
| Critical commercial assays | ||
| Lipofectamine 3000 | ThermoFisher Scientific | Cat#L3000008 |
| KAPA HiFi HotStart Ready Mix | Kapa Biosystems | Cat#KK2602 |
| iTaq Universal SYBR Green | Biorad | Cat#1725121 |
| iScript Reverse Transcription Supermix | Biorad | Cat#1708840 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| Zero Blunt TOPO PCR Cloning Kit | ThermoFisher Scientific | Cat#450245 |
| prepGEM gold buffer | Zygem | N/A |
| Experimental models: Cell lines | ||
| HepG2 | ATCC | Cat#HB-8065 |
| Huh7 | Provided by M. Hermann, MIT, Cambridge, MA, USA | N/A |
| A549 | ATCC | Cat#CCL-185 |
| 143B | ATCC | Cat#CRL-8303 |
| MCF7 | ATCC | Cat#HTB-22 |
| HEK293FT | ThermoFisher Scientific | Cat#R70007 |
| Primary rat cortical neurons | This paper | N/A |
| Primary rat astrocytes | This paper | N/A |
| Oligonucleotides | ||
| See Table S1 | N/A | N/A |
| Recombinant DNA | ||
| LentiCRISPR v2 plasmid | Addgene | Cat#52961 |
| pCMVR8.2 | Addgene | Cat#12263 |
| pMD2.G | Addgene | Cat#12259 |
| Software and algorithms | ||
| MATLAB 2014b and 2016b | MathWorks | https://www.mathworks.com/products/matlab.html |
| Escher-Trace | Kumar et al., 2020 | https://escher-trace.github.io/ |
| GraphPad Prism 7.00 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| INCA 1.5 | Young, 2014 | https://mfa.vueinnovations.com/ |
| CRISPOR | Concordet and Haeussler, 2018 | http://crispor.tefor.net/ |
| NCBI BLAST | Agarwala et al., 2018 | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
Highlights.
HCC cells utilize NaCT-mediated citrate uptake for lipogenesis
Citrate import supports lipogenesis and anaplerosis upon glutamine deprivation
NaCT-mediated citrate uptake facilitates protection against zinc toxicity
ACKNOWLEDGMENTS
We thank all members of the Metallo laboratory for support and helpful discussions. This study was supported, in part, by National Institutes of Health (NIH) grants R01CA234245 (C.M.M.) and R21NS104513 (A.N.M., A.M.P.) and a TESS Research Foundation grant (A.N.M., A.M.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109701.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ackermann T, and Tardito S (2019). Cell Culture Medium Formulation and Its Implications in Cancer Metabolism. Trends Cancer 5, 329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwala R, Barrett T, Beck J, Benson DA, Bollin C, Bolton E, Bourexis D, Brister JR, Bryant SH, Canese K, et al. (2018). Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 46 (D1), D8–D13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaf SY, and Chung SH (1984). Release of endogenous Zn2+ from brain tissue during activity. Nature 308, 734–736. [DOI] [PubMed] [Google Scholar]
- Bainbridge MN, Cooney E, Miller M, Kennedy AD, Wulff JE, Donti T, Jhangiani SN, Gibbs RA, Elsea SH, Porter BE, and Graham BH (2017). Analyses of SLC13A5-epilepsy patients reveal perturbations of TCA cycle. Mol. Genet. Metab 121, 314–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutia YD, Kopel JJ, Lawrence JJ, Neugebauer V, and Ganapathy V (2017). Plasma membrane Na+-coupled citrate transporter (SLC13A5) and neonatal epileptic encephalopathy. Molecules 22, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkenfeld AL, Lee HY, Guebre-Egziabher F, Alves TC, Jurczak MJ, Jornayvaz FR, Zhang D, Hsiao JJ, Martin-Montalvo A, Fischer-Rosinsky A, et al. (2011). Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 14, 184–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A Jr., Lewis CA, and Sabatini DM (2017). Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concordet JP, and Haeussler M (2018). CRISPOR: intuitive guide selection for CRISPR/Cas9 genome editing experiments and screens. Nucleic Acids Res. 46 (W1), W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes T, and Metallo CM (2019). Quantifying Intermediary Metabolism and Lipogenesis in Cultured Mammalian Cells Using Stable Isotope Tracing and Mass Spectrometry. In High-Throughput Metabolomics: Methods and Protocols, D’Alessandro A, ed. (Springer; New York: ), pp. 219–241. [DOI] [PubMed] [Google Scholar]
- Cordes T, Lucas A, Divakaruni AS, Murphy AN, Cabrales P, and Metallo CM (2020). Itaconate modulates tricarboxylic acid and redox metabolism to mitigate reperfusion injury. Mol. Metab 32, 122–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello LC, and Franklin RB (1991). Concepts of citrate production and secretion by prostate. 1. Metabolic relationships. Prostate 18, 25–46. [DOI] [PubMed] [Google Scholar]
- Costello LC, and Franklin RB (2016). Plasma Citrate Homeostasis: How It Is Regulated; And Its Physiological and Clinical Implications. An Important, But Neglected, Relationship in Medicine. HSOA J. Hum. Endocrinol 1, 1–8. [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Wallace M, Buren C, Martyniuk K, Andreyev AY, Li E, Fields JA, Cordes T, Reynolds IJ, Bloodgood BL, et al. (2017). Inhibition of the mitochondrial pyruvate carrier protects from excitotoxic neuronal death. J. Cell Biol 216, 1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, and Rabinowitz JD (2013). Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol 9, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frederickson CJ, Klitenick MA, Manton WI, and Kirkpatrick JB (1983). Cytoarchitectonic distribution of zinc in the hippocampus of man and the rat. Brain Res. 273, 335–339. [DOI] [PubMed] [Google Scholar]
- Gheller BJ, Blum JE, Merritt EK, Cummings BP, and Thalacker-Mercer AE (2019). Peptide YY (PYY) is expressed in human skeletal muscle tissue and expanding human muscle progenitor cells. Front. Physiol 10, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheller BJ, Blum JE, Lim EW, Handzlik MK, Hannah Fong EH, Ko AC, Khanna S, Gheller ME, Bender EL, Alexander MS, et al. (2021). Extracellular serine and glycine are required for mouse and human skeletal muscle stem and progenitor cell function. Mol. Metab 43, 101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glusker JP (1980). Citrate Conformation and Chelation: Enzymatic Implications. Acc. Chem. Res 13, 345–352. [Google Scholar]
- Grassian AR, Parker SJ, Davidson SM, Divakaruni AS, Green CR, Zhang X, Slocum KL, Pu M, Lin F, Vickers C, et al. (2014). IDH1 mutations alter citric acid cycle metabolism and increase dependence on oxidative mitochondrial metabolism. Cancer Res. 74, 3317–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardies K, de Kovel CGF, Weckhuysen S, Asselbergh B, Geuens T, Deconinck T,Azmi A, May P, Brilstra E, Becker F, et al. (2015). Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 138, 3238–3250. [DOI] [PubMed] [Google Scholar]
- Henke C, Töllner K, van Dijk RM, Miljanovic N, Cordes T, Twele F, Bröer S, Ziesak V, Rohde M, Hauck SM, et al. (2020). Disruption of the sodium-dependent citrate transporter SLC13A5 in mice causes alterations in brain citrate levels and neuronal network excitability in the hippocampus. Neurobiol. Dis 143, 105018. [DOI] [PubMed] [Google Scholar]
- Huard K, Brown J, Jones JC, Cabral S, Futatsugi K, Gorgoglione M, Lanba A, Vera NB, Zhu Y, Yan Q, et al. (2015). Discovery and characterization of novel inhibitors of the sodium-coupled citrate transporter (NaCT or SLC13A5). Sci. Rep 5, 17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Zhuang L, Maddox DM, Smith SB, and Ganapathy V (2002). Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem 277, 39469–39476. [DOI] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. (2016). Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Boufersaoui A, Yang C, Ko B, Rakheja D, Guevara G, Hu Z, and DeBerardinis RJ (2017). Quantitative metabolic flux analysis reveals an unconventional pathway of fatty acid synthesis in cancer cells deficient for the mitochondrial citrate transport protein. Metab. Eng 43 (Pt B), 198–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Chung MK, Fan J, and Rabinowitz JD (2014). Quantitative analysis of acetyl-CoA production in hypoxic cancer cells reveals substantial contribution from acetate. Cancer Metab. 2, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klotz J, Porter BE, Colas C, Schlessinger A, and Pajor AM (2016). Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med 22, 310–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Mitchener J, King ZA, and Metallo CM (2020). Escher-Trace: a web application for pathway-based visualization of stable isotope tracing data. BMC Bioinformatics 21, 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire J, Mailloux R, and Appanna VD (2008). Zinc toxicity alters mitochondrial metabolism and leads to decreased ATP production in hepatocytes. J. Appl. Toxicol 28, 175–182. [DOI] [PubMed] [Google Scholar]
- Li Z, Li D, Choi EY, Lapidus R, Zhang L, Huang SM, Shapiro P, and Wang H (2017). Silencing of solute carrier family13 member 5 disrupts energy homeostasis and inhibits proliferation of human hepatocarcinoma cells. J. Biol. Chem 292, 13890–13901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matricardi S, De Liso P, Freri E, Costa P, Castellotti B, Magri S, Gellera C, Granata T, Musante L, Lesca G, et al. (2020). Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia 61, 2474–2485. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481,380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Pierce KA, Jedrychowski MP, Garrity R, Winther S, Vidoni S, Yoneshiro T, Spinelli JB, Lu GZ, Kazak L, et al. (2018). Accumulation of succinate controls activation of adipose tissue thermogenesis. Nature 560, 102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, and DeBerardinis RJ (2011). Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mycielska ME, Dettmer K, Rümmele P, Schmidt K, Prehn C, Milenkovic VM, Jagla W, Madej GM, Lantow M, Schladt M, et al. (2018). Extracellular citrate affects critical elements of cancer cell metabolism and supports cancer development in vivo. Cancer Res. 78, 2513–2523. [DOI] [PubMed] [Google Scholar]
- Pajor AM (1996). Molecular cloning and functional expression of a sodium-dicarboxylate cotransporter from human kidney. Am. J. Physiol 270, F642–F648. [DOI] [PubMed] [Google Scholar]
- Pajor AM (2006). Molecular properties of the SLC13 family of dicarboxylate and sulfate transporters. Pflugers Arch. 451, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajor AM (2014). Sodium-coupled dicarboxylate and citrate transporters from the SLC13 family. Pflugers Arch. 466, 119–130. [DOI] [PubMed] [Google Scholar]
- Pajor AM, and Sun NN (2010). Single nucleotide polymorphisms in the human Na+-dicarboxylate cotransporter affect transport activity and protein expression. Am. J. Physiol. Ren. Physiol 299, 704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajor AM, de Oliveira CA, Song K, Huard K, Shanmugasundaram V, and Erion DM (2016). Molecular Basis for Inhibition of the Na+/Citrate Transporter NaCT (SLC13A5) by Dicarboxylate Inhibitors. Mol. Pharmacol 90, 755–765. [DOI] [PubMed] [Google Scholar]
- Phokrai P, Poolsri WA, Suwankulanan S, Phakdeeto N, Kaewkong W, Pekthong D, Richert L, and Srisawang P (2018). Suppressed de novo lipogenesis by plasma membrane citrate transporter inhibitor promotes apoptosis in HepG2 cells. FEBS Open Bio 8, 986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolsri W-A, Phokrai P, Suwankulanan S, Phakdeeto N, Phunsomboon P, Pekthong D, Richert L, Pongcharoen S, and Srisawang P (2018). Combination of Mitochondrial and Plasma Membrane Citrate Transporter Inhibitors Inhibits De Novo Lipogenesis Pathway and Triggers Apoptosis in Hepatocellular Carcinoma Cells. BioMed Res. Int 2018, 3683026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J,Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddle ES, Bender EL, and Thalacker-Mercer AE (2018). Expansion capacity of human muscle progenitor cells differs by age, sex, and metabolic fuel preference. Am. J. Physiol. Cell Physiol 315, C643–C652. [DOI] [PubMed] [Google Scholar]
- Rossiter NJ, Huggler KS, Adelmann CH, Keys HR, Soens RW, Sabatini DM, and Cantor JR (2021). CRISPR screens in physiologic medium reveal conditionally essential genes in human cells. Cell Metab. 33, 1248–1263.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer DB, Song J, Wang B, Hilton JK, Karpowich NK, Mindell JA, Rice WJ, and Wang D-N (2021). Structure and inhibition mechanism of the human citrate transporter NaCT. Nature 591, 157–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schossig A, Bloch-Zupan A, Lussi A,Wolf NI, Raskin S, Cohen M, Giuliano F, Jurgens J, Krabichler B, Koolen DA, et al. (2017). SLC13A5 is the second gene associated with Kohlschütter-Tönz syndrome. J. Med. Genet 54, 54–62. [DOI] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. (2013). Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinebach OM, and Wolterbeek HT (1993). Effects of zinc on rat hepatoma HTC cells and primary cultured rat hepatocytes. Toxicol. Appl. Pharmacol 118, 245–254. [DOI] [PubMed] [Google Scholar]
- Sul JW, Kim TY, Yoo HJ, Kim J, Suh YA, Hwang JJ, and Koh JY (2016). A novel mechanism for the pyruvate protection against zinc-induced cytotoxicity: mediation by the chelating effect of citrate and isocitrate. Arch. Pharm. Res 39, 1151–1159. [DOI] [PubMed] [Google Scholar]
- Tardito S, Oudin A,Ahmed SU, Fack F, Keunen O, Zheng L, Miletic H, Sakariassen P∅, Weinstock A, Wagner A, et al. (2015). Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol 17, 1556–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenon J, Milh M, Feillet F, St-Onge J, Duffourd Y, Jugé C, Roubertie A, Héron D, Mignot C, Raffo E, et al. (2014). Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet 95, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Tissue-based map of the human proteome. Science 347, 1260419. 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Vacanti NM, Divakaruni AS, Green CR, Parker SJ, Henry RR, Ciaraldi TP, Murphy AN, and Metallo CM (2014). Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol. Cell 56, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Voorde J, Ackermann T, Pfetzer N, Sumpton D, Mackay G, Kalna G, Nixon C, Blyth K, Gottlieb E, and Tardito S (2019). Improving the metabolic fidelity of cancer models with a physiological cell culture medium. Sci. Adv 5, eaau7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Loeffelholz C, Lieske S, Neuschäfer-Rube F, Willmes DM, Raschzok N, Sauer IM, König J, Fromm MF, Horn P, Chatzigeorgiou A, et al. (2017). The human longevity gene homolog INDY and interleukin-6 interact in hepatic lipid metabolism. Hepatology 66, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakil SJ, and Abu-Elheiga LA (2009). Fatty acid metabolism: target for metabolic syndrome. J. Lipid Res 50 (Suppl), S138–S143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL, and Koh JY (2000). Zn(2+): a novel ionic mediator of neural injury in brain disease. Trends Pharmacol. Sci 21, 395–401. [DOI] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JES, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, and Thompson CB (2011). Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. USA 108, 19611–19616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo MH, Lee JY, Lee SE, Koh JY, and Yoon YH (2004). Protection by pyruvate of rat retinal cells against zinc toxicity in vitro, and pressure-induced ischemia in vivo. Invest. Ophthalmol. Vis. Sci 45, 1523–1530. [DOI] [PubMed] [Google Scholar]
- Young JD (2014). INCA: a computational platform for isotopically non-stationary metabolic flux analysis. Bioinformatics 30, 1333–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuckerman JM, and Assimos DG (2009). Hypocitraturia: pathophysiology and medical management. Rev. Urol 11, 134–144. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
All original code is available in this paper’s supplemental information.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.