

Summary
We describe PROPER-seq (protein-protein interaction sequencing) to map protein-protein interactions (PPI) en masse. PROPER-seq first converts transcriptomes of input cells into RNA-barcoded protein libraries, in which all interacting protein pairs are captured through nucleotide barcode ligation, recorded as chimeric DNA sequences, and decoded at once by sequencing and mapping. We applied PROPER-seq to human embryonic kidney cells, T lymphocytes, and endothelial cells and identified 210,518 human PPIs (collected in PROPER v.1.0 database). Among these, 1,365 and 2,480 PPIs are supported by published co-IP and AP-MS data, 17,638 predicted by prePPI algorithm but without previous experimental validation, and 100 overlapping with human synthetic lethal gene pairs. Additionally, four previously uncharacterized interaction partners with PARP1 (a critical protein in DNA repair), including XPO1, MATR3, IPO5, and LEO1 are validated in vivo. PROPER-seq presents a time-effective technology to map PPIs at the transcriptome scale and PROPER v.1.0 provides a rich resource for studying PPI.
eTOC blurb:
Johnson et al. introduce PROPER-seq for large-scale and time-effective mapping of protein-protein interactions (PPI) in various cell types based on DNA sequencing. Collected in PROPER v.1.0, PROPER-seq adds more than 200,000 previously uncharacterized PPIs to the reference human protein interactome and provide experimental support to more than 17,000 computationally predicted human PPIs.
Graphical Abstract
Introduction
Our ability to interpret the human genome function is greatly improved by our understanding of the interaction networks formed by the genome products. Recent technological breakthroughs enabled genome-wide mapping of DNA-DNA (Dekker et al., 2017), protein-DNA (Consortium, 2004, Consortium, 2012), RNA-DNA (Sridhar et al., 2017, Yan et al., 2019, Li et al., 2017), and RNA-RNA (Lu et al., 2018, Sharma et al., 2016, Aw et al., 2017, Nguyen et al., 2016) interactions. However, genome-wide mapping of human protein-protein interactions (PPI) remains a resource-intensive task.
Large-scale PPI mapping methods can be grouped into 3 classes, that are “parallelized one-to-one”, “one-to-many”, and “many-to-many” approaches. The “parallelized one-to-one” methods leverage automation and parallelization to enhance throughput of yeast two-hybrid (Y2H) assays(Rual et al., 2005, Luck et al., 2020, Rolland et al., 2014). These include High-Throughput Y2H (Walhout and Vidal, 2001), mammalian protein-protein interaction trap (MAPPIT) (Lievens et al., 2009), quantitative interactor screening with next-generation sequencing (QIS-seq) (Lewis et al., 2012), which massively parallelized the binary interactions, recombination-based library versus library high-throughput Y2H (RLL-Y2H) (Yang et al., 2018), Stitch-seq (Kawalia et al., 2015), Cre recombinase yeast two-hybrid (CrY2H-seq) (Trigg et al., 2017), and barcode fusion genetics-yeast two-Hybrid (BFG-Y2H) (Yachie et al., 2016), in which gene sequences of interacting PPI pairs were fused and sequenced. The “one-to-many” methods start with purifying or tagging a target (or “bait”) protein to identify the co-purified proteins in spatial proximity using affinity purification(Vermeulen et al., 2008), proximity biotinylation (BioID) (Touchette et al., 2017), green fluorescent protein (GFP) fusion (Zhang et al., 2017), or protein microarray (Kukar et al., 2002). The “many-to-many” approach, aiming to read out all the pairwise PPIs from a single experiment, has been applied to resolve ligand-target pairs (McGregor et al., 2014) and antibody-antigen pairs (Gu et al., 2014).
The aforementioned methods can be also grouped into protein interaction assays and spatial proximity assays, depending upon the property of protein pair unraveled. The protein interaction assays can be further divided into binary and non-binary assays (Yu et al., 2008). Whereas binary assays such as Y2H yield direct pairwise protein interactions, non-binary assays such as affinity purification-mass spectrometry (AP-MS) and co-immunoprecipitation (coIP) yield physical associations, where each protein identified in a pair may not directly interact with each other, as in a multi-protein complex. Finally, spatial proximity assays including BioID (Touchette et al., 2017) reveals proteins that may not form physical interactions or associations, other than being spatially proximal.
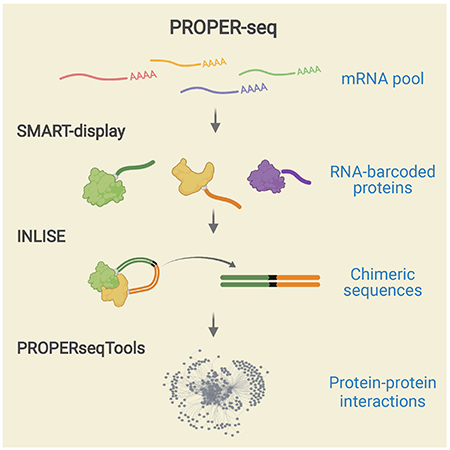
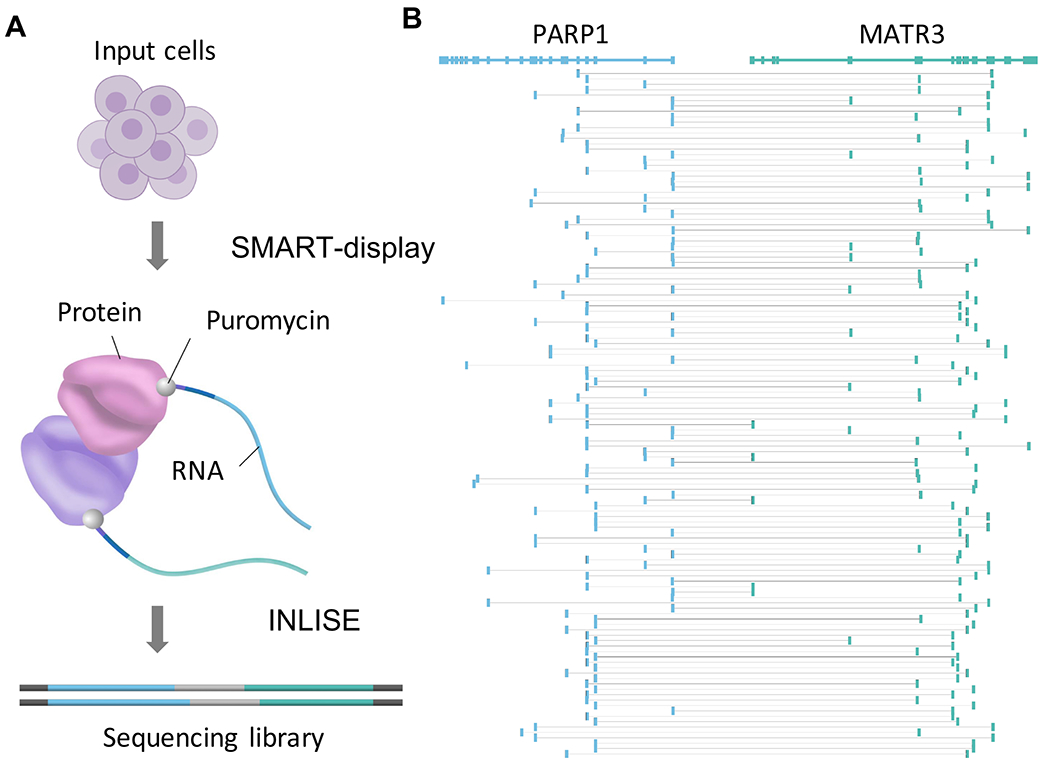
In this work, we introduce PROPER-seq, a resource-efficient “many-to-many” non-binary assay for PPI mapping. The central idea of PROPER-seq is to convert each PPI into a unique sequence of DNA, and then to leverage the extremely high throughput of DNA sequencing to decode these PPIs. To implement this idea, we developed a technique called SMART-display to attach a unique RNA barcode to every protein (Figure 1A) and a method called “Incubation, ligation and sequencing” (INLISE) to sequence the pair of DNA barcodes that are attached to two interacting proteins (Figure 1B). We named the overall technology combining SMART-diplay and INLISE as PROPER-seq (Figure 1A). The input to PROPER-seq is a group of cells and the PROPER-seq’s output is a list of identified PPIs and their associated read counts and test statistics. We demonstrate that PROPER-seq is capable of scanning the order of 10,000×10,000 protein pairs in one experiment and identify both binary and multiway protein interactions. Applying PROPER-seq on human embryonic kidney cells, T lymphocytes, and endothelial cells, we constructed a reference map of human PPIs (PROPER v.1.0) that include 210,518 PPIs involving 8,635 proteins.
Figure 1.
Overview of PROPER-seq experimental pipeline. (A) PROPER-seq starts with SMART-display, that transforms the input cells into a library of RNA-barcoded proteins (the first arrow), followed by INLISE, that transforms the barcoded proteins in a sequencing library, such that the barcodes of interacting protein pairs form a chimeric sequence (the second arrow). (B) Alignment of the barcodes to reveal the identities of the two genes (top track) between which the chimeric sequences (rows) were formed.
Design
SMART-display: efficient labeling of proteins of RNA barcodes
We developed a modified mRNA-display method, called SMART-display, to efficiently generate a protein library where the proteins are conjugated with their mRNA (Figure 1A and Figure 2). Thus, the mRNA serves as the unique nucleic acid barcode for each protein. Similar to mRNA-display(Roberts and Szostak, 1997, Barendt et al., 2013), SMART-display is designed to create mRNA-protein fusions, specifically by adding an amino acid analog puromycin (“P” in purple circle, Figure S1A) near the 3’ end of the mRNA. The translated protein from this mRNA is then covalently linked with its mRNA when puromycin enters the A site of the ribosome and is joined to the amino acid chain. This generates an mRNA-protein fusion, which is then released from the ribosome (Figure S1).
Figure 2.
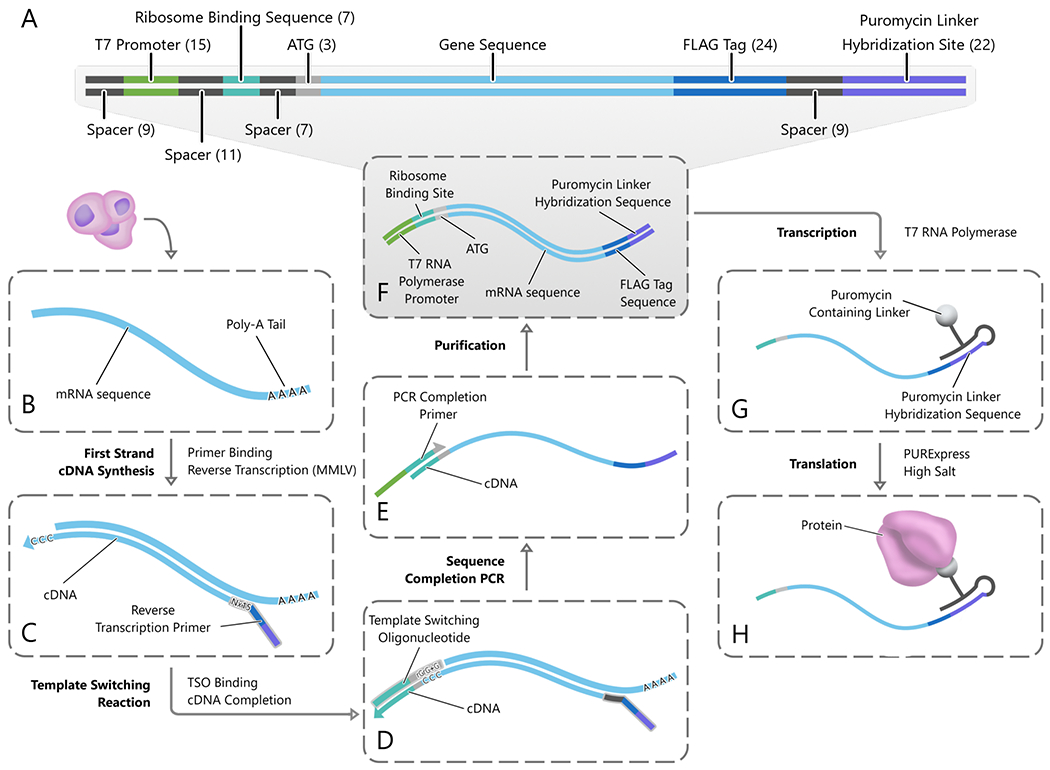
SMART-display. (A) The structure of gene templates produced by SMART-display (the product of step (F)). (B) Poly-A selected and rRNA depleted mRNA is collected from the input cells. (C) A reverse transcription primer containing a random sixteen base-pair region followed by the sequences for a FLAG tag and a GC-rich puromycin linker hybridization site is annealed to the mRNA. (D) Reverse transcription and incorporation of the template switching oligo (TSO). (E) PCR is performed with a primer that partially overlaps the TSO sequences to introduce the T7 promoter and complete the ribosome binding site. (F) Double-stranded DNA is purified. (G-H) The transcribed RNA is ligated to a puromycin-containing linker sequence (G) and subsequently translated to form mRNA-protein fusion products (H).
In SMART-display, we replaced the gene-by-gene cloning (or gene-by-gene PCR) step in mRNA-display by reactions that can be carried out with a mixture of genes (or mRNAs) without the need for independent purification of each gene. This was achieved by replacing the gene-specific primers in mRNA-display with template switching oligos (TSO) (Petalidis et al., 2003, Zhu et al., 2001) that are universal for all genes. The input to SMART-display is a user selected cell population. An important intermediate product of SMART-display is a gene library suitable for mRNA display, where the sequences for transcription initiation, translation initiation, and puromycin attachment have been incorporated in the appropriate places for every gene (Figure 2A). The output of SMART-display is a library of display complexes in the form of mRNA–linker–protein (Figure 2H and Figure S1D).
Incubation, ligation and sequencing (INLISE)
As the second key step of PROPER-seq, INLISE is to convert PPIs into chimeric sequences with the structure: cDNA1-linker-cDNA2 (Figure 3). The input of INLISE are two display libraries generated by SMART-display. Each display library contains approximately 15,000 mRNA-protein fusions. One library, called the “bait” library, is immobilized on streptavidin beads through the biotin on the puromycin linker sequence (“B” in blue circle, Figure S1A). The other library, called the “prey” library, is not immobilized, as the biotin is cleaved from the puromycin linker, and is mixed with the bait library to allow for interactions. After removal of spurious interactions, the mRNA barcodes of interacting proteins are ligated to create a chimeric sequence in the form of cDNA1-linker-cDNA2, where cDNA1 and cDNA2 represent the two interacting proteins. These chimeric sequences are subsequently selected for and subjected to paired-end sequencing (INLISE, STAR Methods).
Figure 3.
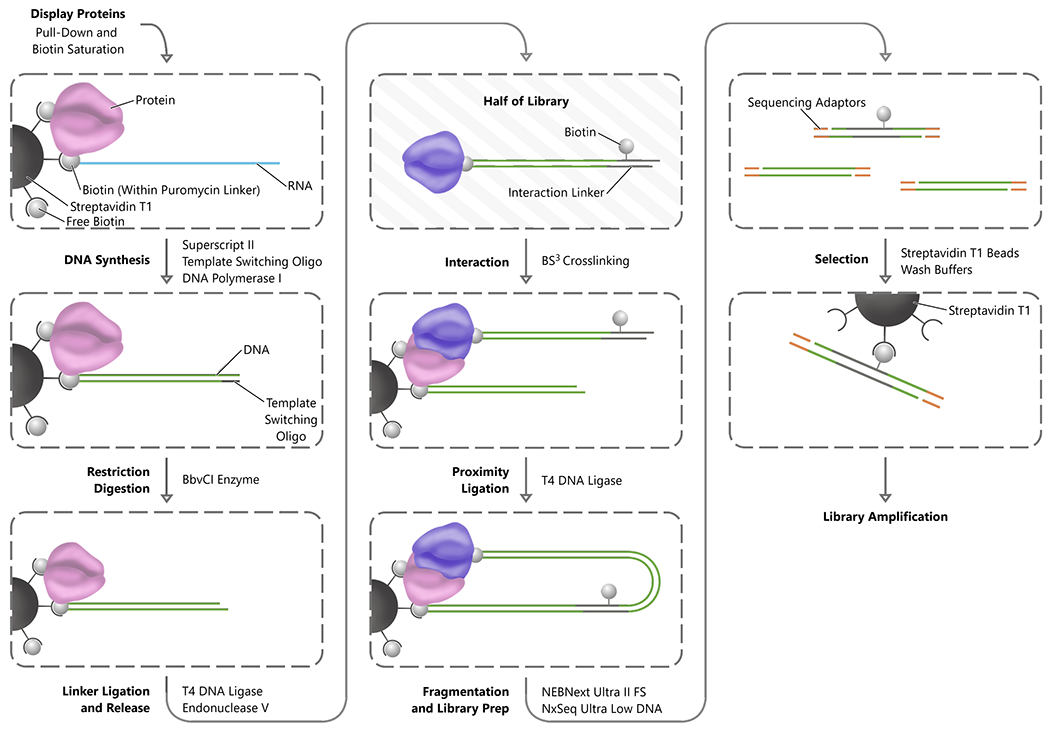
INLISE. Steps are indicated in bold font to the left of each process arrow, and the primary enzymes or reagents used to accomplish each step are indicated to the right of the process arrow. The process begins with the stabilization of the display complexes on streptavidin magnetic beads. Subsequently, the RNA component of each display complex is converted to double-stranded DNA and digested with a non-palindromic restriction enzyme. The library of display proteins is then split into two populations. One half of the display protein complex is ligated to the biotinylated interaction linker and then digested to remove the complexes from the streptavidin beads. The free half of the display protein library is combined with the half still on the beads to perform the interaction step and the interacting proteins crosslinked. The beads are washed to remove nonspecific interactions and then proximity ligation between the display nucleic acids is performed. The DNA is then fragmented and adaptor ligation for sequencing is performed before a final streptavidin selection for the biotin containing interaction linker and library amplification.
Identification of PPIs by statistical tests
Our overarching goal is to examine as many protein pairs as possible and assign a binary indicator (interacting or not) to every protein pair. Toward this goal, we subjected the mapped read pairs on each gene pair to an association test. The null hypothesis is that the mapping of a read pair to one gene is independent of the mapping of this read pair to the other gene (Figure S2A). We used Bonferroni-Hochberg (BH) correction to account for multiple hypothesis tests (Benjamini and Hochberg, 1995). To facilitate reproducible analysis, we have implemented all data processing and statistical test steps into an open-source software package called PROPERseqTools (Figure S2B) (https://doi.org/10.5281/zenodo.50091711.
Results
Assessments of SMART-display and INLISE
We assessed SMART-display in two aspects. First, we asked if the display products exhibit any specificity in antibody-protein interactions. To test whether a specific PPI can be detected by using the mRNA “barcode” on the display protein, we used the GFP antibody and GFP protein as the testbed PPI. We constructed a small SMART-display library as follows. We started from four full-length mRNAs, GFP, creatine kinase, mitochondrial 2 (CKMT2), MAPK activated protein kinase 2 (MAPKAPK2), and dihydrofolate reductase (DHFR). After the display process (Figure S3 A and B), we mixed the resulting mRNA-protein fusions equimolarly to create a small SMART-display library. We used qPCR to quantify each mRNA in this mixture (pre-selection value), used GFP antibody for pulldown on magnetic beads, and applied stringent washes to remove non-specific attached RNA-bead attachments. qPCR was then used to quantify each mRNA in the mixture (post-selection value). A greater ratio of post- to pre-selection values suggests a higher anti-GFP antibody interaction with the protein. As expected, the ratios of the other three mRNAs (CKMT2, MAPKAPK2, and DHFR) were much lower than that of the GFP mRNA (Figure S3C). This test suggests that the display protein can be specifically recognized by its antibody and that the mRNA “barcodes” could provide quantitative readout of the PPIs.
Second, we evaluated the proportion of mRNAs from the original sample that were converted to display complexes by SMART-display. To this end, we split a population of HEK293T cells equally into two, one for RNA sequencing (RNA-seq), and the other for SMART-display, where we purified the display complexes by their protein moiety and sequenced the co-purified RNA moiety. While the RNA-seq reads were mapped to 15,191 protein-coding genes (Transcripts per million [TPM]>0.1), the sequencing reads from SMART-display were mapped to 14,805 protein-coding genes (Transcripts per million [TPM]>0.1) (displayed genes), 14,658 of which overlapped with those revealed by the RNA-seq (Figure S3D). This level of overlap in the detected mRNAs is comparable to that between two RNA-seq experiments carried out with the same cell type (Li et al., 2014, Su et al., 2014). Thus, SMART-display-generated product library recapitulated to a large extent the diversity of mRNAs from input cells. We subjected two HEK293T samples to the SMART-display. The samples yielded 14,805 and 14,104 displayed genes (Figure S3E), with 13,835 overlapping (odds ratio = 274.8, Chi-square p-value < 10−32), suggesting a limited variation between two SMART-display repeats.
Several experimental steps in INLISE were designed to promote the formation chimeric sequences. To test if this design goal was achieved, we carried out the INLISE procedure with two variations, one with interaction linker excluded (no-linker column, Figure S4A), and the other with the bait library pre-incubated with proteinase (Proteinase column, Figure S4A). Compared to the standard INLISE procedure, both variations yielded less DNA in the second last step (Streptavidin T1 Selection) (Figure S4 A and B) and final sequencing libraries with lower concentrations (Figure S4 C and D). These results suggest that INLISE’s experimental steps improved the efficiency of forming chimeric sequences, in line with our design goal.
Validations of PROPER-seq identified PPIs
We evaluated PROPER-seq based on its reproducibility, precision, and recall. To test these properties, we generated six PROPER-seq libraries from HEK293T, Jurkat, and HUVEC cells. Two biological replicates from each cell type were used to generate two libraries of that cell type. These libraries are named HEK1, HEK2, JKT1, JKT2, HUVEC1, HUVEC2 (Table S1). Sequencing of these libraries yielded approximately 350 million read pairs per library. Among these, approximately 8 million are non-duplicate chimeric read pairs, each mapped to two different coding genes (# chimeric reads, Table S1). These chimeric read paris were then used as the input for association tests (Figure S2A). A pair of proteins was identified as interacting (i.e., a PPI) by two criteria. First, the BH-corrected p value derived from an association (Chi-square) test is smaller than 0.05 (Figure S2A). Second, the number of the chimeric read pairs mapped to this gene pair is no less than 4 times the average number of chimeric reads mapped to any gene pair (4 × # all mapped chimeric read pairs / # all mapped gene pairs). Hereafter, we call these the default threshold, denoted as BH-corrected p-value < 0.05 and # read pairs > 4X, where X is the expected number of read pairs mapped on a randomly chosen gene pair. Unless otherwise specified, all PPIs presented in the rest of this manuscript were identified based on this default threshold.
Reproducibility between biological replicates
To test reproducibility, we identified PPIs from each library separately. HEK1 and HEK2 libraries identified 62,637 and 51,611 PPIs, respectively. A total of 34,244 PPIs was shared between the two biological replicates (odds ratio = 14,242, p-value < 2.2×10−16, Chi-square test) (Figure S5A), suggesting a significant overlap between experimental repeats. We also tested how sensitive the reproducibility is to the threshold applied for PPI calling. We started from the default threshold and then varied the threshold (BH-corrected p-value < 0.05, # read pairs > nX) by changing n from 4 (default) to 40 (Figure S5 B and C). As the criterion (n) increased, the number of identified PPIs decreased as expected. However, the relative size of the overlap exhibited monotonic increase (Figure S5C). These data suggest that the reproducibility of PROPER-seq increases as the threshold increases. We repeated these analyses with the two Jurkat libraries and the two HUVEC libraries and detected a similar increase in reproducibility, evident by the monotonic increase of the proportions of the overlaps, as the threshold increases (Figure S5 D–I). These results indicate that among the statistically significant PPIs, the more read pairs supporting a PPI, the more likely this PPI is reproducible by another repeat experiment.
Precision and recall of PROPER-seq identified PPIs
Next, we evaluated the precision and recall (Saito and Rehmsmeier, 2015) of the PROPER-seq identified PPIs (PROPER) with reference to known PPIs. We obtained reference datasets from the Agile Protein Interactomes DataServer (APID) (Alonso-Lopez et al., 2019, Alonso-Lopez et al., 2016), which has integrated experimentally reported PPIs from more than 6,689 curated articles and the Biomolecular Interaction Network Database (BIND) (Bader et al., 2003), BioGRID (Stark et al., 2006), the database of interacting proteins (DIP) (Xenarios et al., 2000), Human Protein Reference Database (HPRD) (Peri et al., 2003), the IntAct database (Hermjakob et al., 2004), and the molecular interaction (MINT) databases. Based on this most up-to-date archive of PPIs (Alonso-Lopez et al., 2019), three types of non-binary assays yielded more than 10,000 PPIs per experimental type. These are affinity purification-mass spectrometry (AP-MS), co-immunoprecipitation (co-IP), and liquid chromatography-mass spectrometry (LC-MS), which have reported 131,224, 50,290, and 33,195 human PPIs, respectively (Table S2). These were then compared with 109,539 PPIs identified in two merged PROPER-seq libraries from HEK (Figure S6, A-C). We plotted the precision and recall using the collection of all human coding genes as the search space (Venkatesan et al., 2009) and generated a dataset by permutating the assignment of chimeric read pairs to gene pairs. The precision-recall curve of this permutated dataset (grey dots, Figure S6A) is far beneath that of the actual data (black dots, Figure S6A), confirming that PROPER-seq’s read pairs were distinguished from the background of randomly sampled gene pairs. We repeated these analyses with PROPER-seq data from Jurkat and HUVEC, using the merged data of two replicates (Figure S6) or each replicate separately (Figure S7). In all analyses, increases of thresholds resulted in larger precisions and smaller recalls (Figure S6 and Figure S7). Furthermore, PROPER-seq identified PPIs exhibited better precisions and recalls than the permutation data (Figure S6 and Figure S7). Taken together, PROPER-seq identified PPIs are supported by the PPIs identified by previous literature.
PROPER v.1.0: a reference human PPI network
To generate a reference human PPI network, we combined all six PROPER-seq libraries (HEK1, HEK2, JKT1, JKT2, HUVEC1, and HUVEC2) into one dataset, composed of approximately 1.4 billion read pairs. This combined dataset revealed 210,518 pairwise PPIs involving 8,635 proteins, which are collectively termed the PROPER v.1.0 network (Figure 4A). We have developed a web interface to download, search, and visualize PROPER v.1.0 https://genemo.ucsd.edu/proper).
Figure 4.
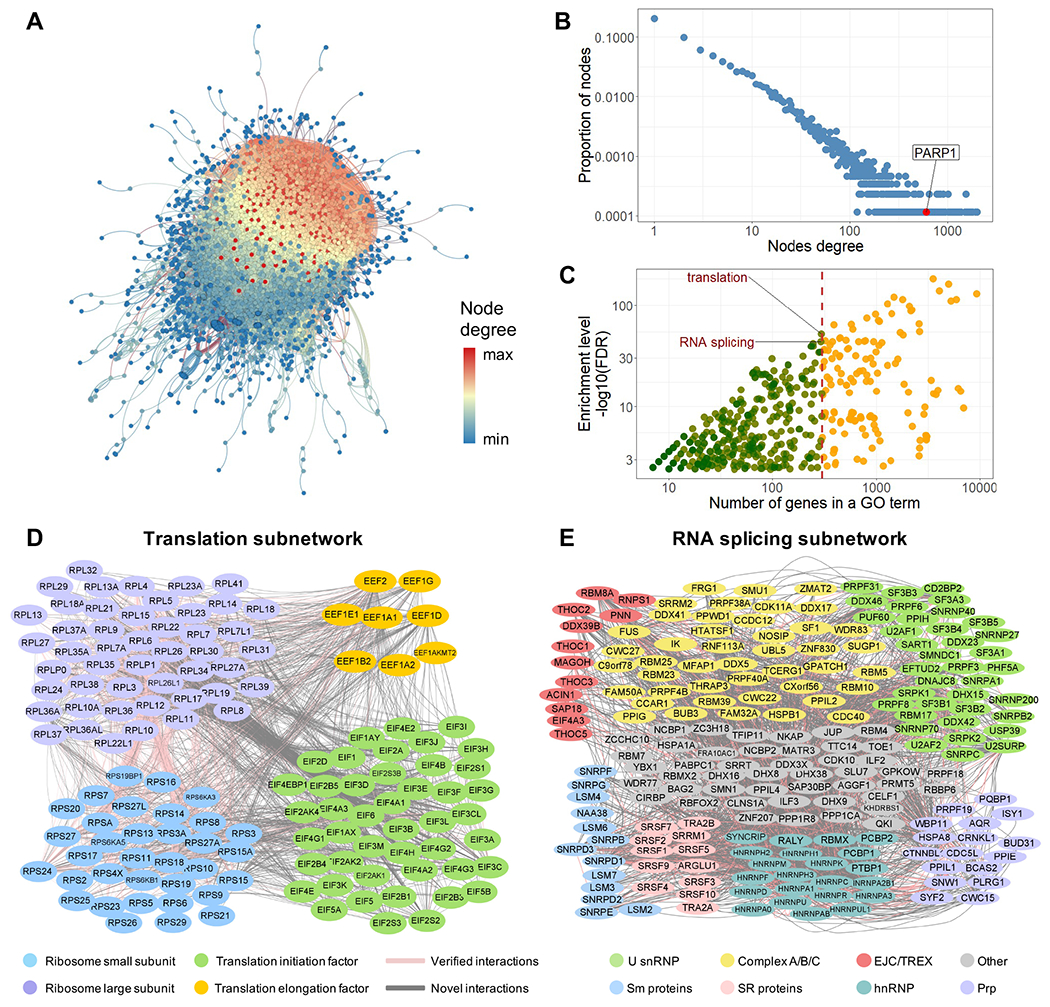
PROPER v.1.0. (A) The entire PROPER v.1.0 network with proteins as nodes and PPIs as edges. The degree of nodes is color-coded from high (red) to low (blue). (B) PROPER’S degree distribution, with the degree (number of connections of a node) (x axis) plotted against the proportion of nodes in that degree (y axis). Arrow: the PARP1 node. The fitted probability density function of the degree distribution is proportional to k−1076, where k is the degree. (C) The number of genes (x axis) of each GO term (dot) vs. the enrichment level of this GO term in PROPER v.1.0 (y axis). Color of the dots: the GO terms with less (green) and more (yellow) than 300 genes. (D) The translation subnetwork. (E) The RNA splicing subnetwork, including the core components of human spliceosomes (U snRNP), components of the pre-spliceosome complex, the precatalytic spliceosome and catalytic step 1 spliceosome (Complex A/B/C), the exon junction complex (EJC), and the transcription and export complex (TREX), as well as SR proteins, Sm proteins, heterogeneous nuclear ribonucleoproteins (hnRNP) and pre-mRNA processing factors (Prp). Pink edges: known PPIs (as documented in APID database). Grey edges: previously uncharacterized PPIs.
To evaluate the topology of the network, we examined the degree distribution of PROPER v.1.0 (Barabasi, 2009, Barabasi and Bonabeau, 2003, Navlakha et al., 2014). The proportion of proteins (nodes) is inversely correlated with the number of interactions (edges) (Figure 4B), suggesting that PROPER v.1.0 is a scale-free network (Barabasi, 2009, Barabasi and Bonabeau, 2003). A major characteristic of scale-free networks is that they contain a small proportion of highly connected nodes, called hubs (Barabasi, 2009, Barabasi and Bonabeau, 2003). For example, Poly(ADP-Ribose) Polymerase 1 (PARP1), a key regulator of a variety of biological processes, emerged as a hub of PROPER v.1.0 by participating in 605 PPIs (edges) (Figure 4B and Figure 5A). PROPER v.1.0’s clustering coefficient (C(k)) exhibits a reverse correlation to the degree (k) (Figure S8I), which is in line with hierarchical networks’ C(k) distributions (Barabasi and Oltvai, 2004). PROPER’S C(k) approaches 1 when k becomes small, suggesting that the nodes with small degrees are embedded in highly connected neighborhoods.
Figure 5.
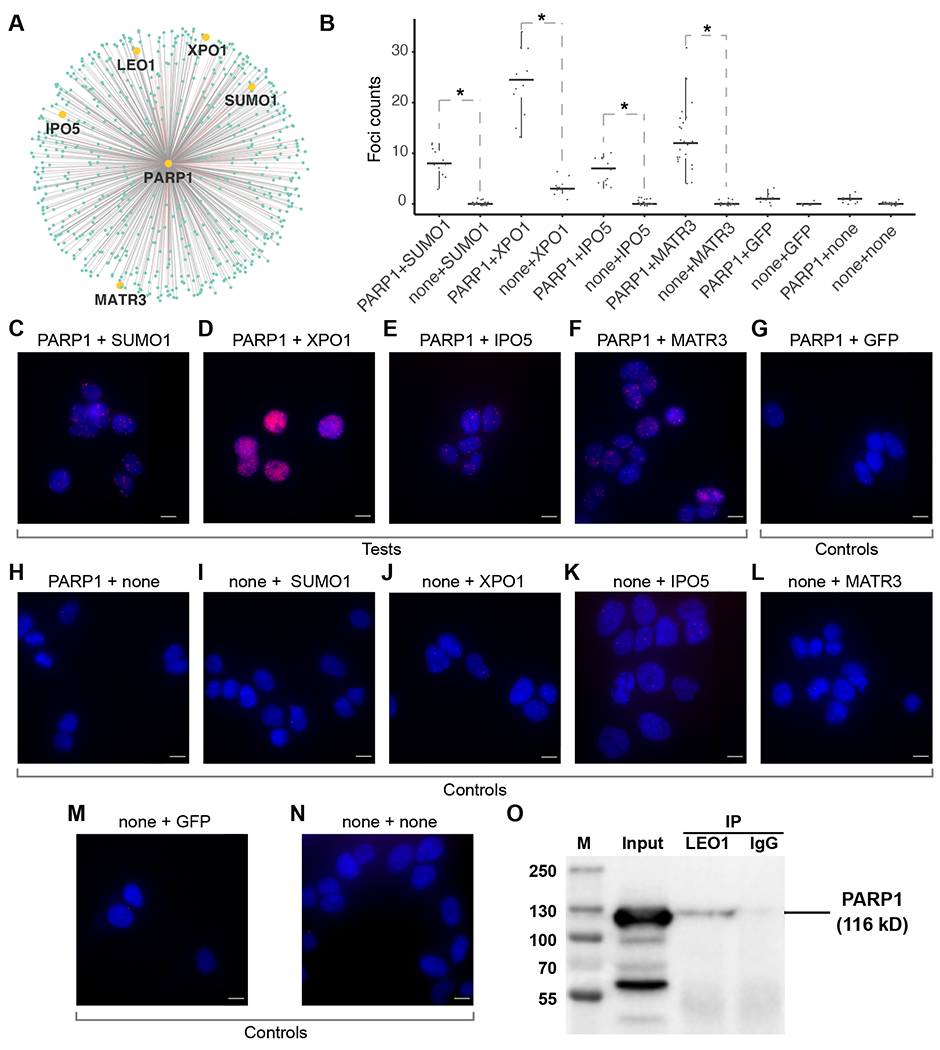
Experimental validations of previously uncharacterized PPIs. (A) The 605 PPIs involving PARP1. Pink edges: known PPIs. Grey edges: previously uncharacterized PPIs. The 5 PPIs tested are labeled. (B) Box plots of the number of PLA foci. Columns: experimental conditions, including 4 test conditions (PARP1+SUMO1, PARP1+XPO1, PARP1+IPO5, PARP1+MATR3) and 8 control conditions (the other columns). *: p-value < 0.05, Wilcoxon test. (C-N) Representative microscopic images in each experimental condition corresponding to columns C-N in panel B, with DAPI staining (blue) and PLA signals (red). Scale bar: 10 μm. (O) co-IP analysis of PARP1 and LEO1. PARP1 immunoblots in LEO1 antibody (IP/LEO1) and IgG antibody immunoprecipitated materials (IP/IgG). M: Marker lane from a pre-stained protein ladder. Input: 5% of precleared cell lysates.
We asked if any functional groups are enriched in PROPER v.1.0. We plotted the enrichment level of every biological process GO term in PROPER v.1.0 against the total number of human genes of that GO term (Figure 4C). To avoid the generic GO terms that involve too many genes, we focused our analysis on GO terms that contained no more than 300 genes (green dots, Figure 4C). The most enriched GO terms were “Translation” (Bonferroni corrected p-value < 9.4×10−51) and “RNA splicing” (Bonferroni corrected p-value < 8.9×10−41, Figure 4C). By intersecting PROPER v.1.0 with each GO term, we obtained a subnetwork associated with each GO term, including a translation subnetwork and an RNA splicing subnetwork. Considering the successes of previous research in elucidating the central dogma, we expected large fractions of the PPIs in the translation and the RNA splicing subnetworks to be known PPIs. Indeed, the Translation subnetwork included 2,520 PPIs, in which 1,185 PPIs (47%) overlapped with APID documented PPIs (Figure 4D). The RNA splicing subnetwork included 2,081 PPIs, where 468 PPIs (23%) overlapped with APID documented PPIs (Figure 4E).
Following Yu et al. and Venkatesan et al. (Yu et al., 2008, Venkatesan et al., 2009, Cusick et al., 2009), we calculated the screening completeness, sampling sensitivity, assay sensitivity, overall sensitivity, precision of PROPER v.1.0 (Venkatesan et al., 2009) (Table S3). PROPER v.1.0’s sequencing reads covered 16,305 human protein coding genes, in which 8,635 protein coding genes were involved in PROPER v.1.0’s PPIs (Table S2). We further tested if PROPER v.1.0 is enriched with either binary or non-binary PPIs by comparing with three pairs of binary and non-binary PPIs, namely APID-binary vs. APID-non-binary, Lit-BM-13 vs. Lit-NB-13 (Kovacs et al., 2019), and L3-BM vs. L3-NB (Kovacs et al., 2019) (Table S2). Association tests suggested enrichments of non-binary PPIs in PROPER v.1.0 (p-value < 2.2×10−16, p-value = 0.081, p-value = 9.8×10−9, Chi-square tests with the three pairs of binary and non-binary datasets). These results are consistent with our expectation that PROPER v.1.0 includes both binary and non-binary PPIs, because both binary and multiway interactions are allowed when the two display libraries are incubated at the INLISE step. Taken together, PROPER v.1.0 expands the reference map of human protein interactome with more than 200,000 previously uncharacterized PPIs.
Support of 17,638 computationally predicted PPIs by PROPER v.1.0
A genome-wide structure-based prediction of human PPIs was accomplished based on the prePPI (Predicting Protein-Protein Interactions) algorithm (Zhang et al., 2012, Zhang et al., 2013). Among the 1,273,679 computationally predicted and previously uncharacterized human PPIs (previously uncharacterized prePPIs) that currently do not have experimental support (not recorded in the APID database), 17,638 previously uncharacterized prePPIs appeared in PROPER v.1.0 (1.38% of the previously uncharacterized prePPIs, 8.38% of PROPER v.1.0, odds ratio = 14.83, p-value < 2.2×10−16, Chi-square test). We also examined whether the PROPER-seq-supported prePPIs were enriched with predicted domain-domain or domain-peptide interactions (Zhang et al., 2012, Chen et al., 2015, Garzon et al., 2016). As expected, PROPER-seq-supported prePPIs exhibited smaller structure scores that reflect a direct interaction between two protein domains (Zhang et al., 2012, Chen et al., 2015, Garzon et al., 2016) as compared to the entire prePPI (Figure S9D). This is because the prePPI algorithm used the structure score as an important component to predict what protein pairs can interact (Zhang et al., 2012). However, the PROPER-seq-supported prePPIs exhibited a similar distribution of domain-peptide scores (Zhang et al., 2012, Chen et al., 2015, Garzon et al., 2016) as that of the entire prePPI (Figure S9H), suggesting little difference in domain-peptide interactions between computationally-derived and PROPER-seq-supported PPIs.
Experimental validation of previously uncharacterized PPIs with proximity ligation assay (PLA) and co-IP
We subjected select previously uncharacterized PPIs to experimental validation. We first investigated whether any previously uncharacterized PPIs in PROPER v.1.0 exhibit spatial proximity in situ by PLA (Gullberg et al., 2004, Soderberg et al., 2006), which enables direct observation of protein interactions by generating fluorescence signals specifically from interacting protein pairs in unmodified cells (Gullberg et al., 2004, Soderberg et al., 2006). We decided to choose a hub in PROPER v.1.0 and selectively test a few previously uncharacterized PPIs involving this hub. We elected several previously uncharacterized PARP1-participating PPIs, i.e., PARP1-exportin 1 (XPO1), PARP1-matrin 3 (MATR3), and PARP1-importin 5 (IPO5) to PLA tests. XP01 (Exportin 1) and IP05 (Importin 5) regulate export and import through nuclear pores (Fornerod et al., 1997, Jäkel and Görlich, 1998). MATR3 is a nuclear matrix protein.
As a positive control, we assayed for PARP1-small ubiquitin-like modifier 1 (SUMO1), a known PPI (Messner et al., 2009). The HEK293 cells co-incubated with PARP1 and SUM01 antibodies exhibited 3 to 12 PLA foci per cell, as compared to 0 to 2 foci per cell in the control cells (p-value = 1.1×10−4 for PARP1+none control, p-value = 1.2×10−6 for none+SUM01 control, Wilcoxon test, Figure 5 B, C, H and I). In parallel, cells co-incubated with PARP1 and XP01 antibodies exhibited 13 to 34 PLA foci per cell, as compared to 0 to 6 foci per cell in the cells incubated with PARP1 or XP01 antibody alone (p-value = 7.4×10−5, for PARP1+none control, p-value = 7.7×10−5 for none+XP01 control, Wilcoxon test, Figure 5 B, D, H and J). Similarly, tests for PARP1-IP05 and PARP1-MATR3 also yielded more PLA foci per cell than their respective controls (the largest p-value = 1×10−4, Wilcoxon test, Figure 5 B, E, F, H, K, L). Furthermore, all the additional controls including co-incubation of PARP1 and GFP antibodies, GFP antibody alone, and a no antibody control, yielded fewer foci as compared to the experimental groups (the largest p-value = 4.5×10−4, Wilcoxon test, Figure 5 B, G, M, and N).
We selected another previously uncharacterized PPI, PARP1-LEO1, for a co-IP test. LEO1 is a component of the PAF1 complex that associates with the RNA polymerase II (Pol II) (Yu et al., 2015). In HEK293, IP with LEO1 antibody (Figure S10) resulted in co-IP of PARP1 (IP/LEO1 lane and Input lane, Figure 5O), whereas the lysates immunoprecipitated with IgG antibody did not exhibit any signal when immunoblotted with PARP1 antibody (IP/IgG lane, Figure 5O). Taken together, 4 out the 4 previously uncharacterized PPIs have been confirmed by PLA or co-IP.
Correlation between human synthetic lethal (SL) gene pairs and human PPIs
We asked whether human genetic interactions exhibit any correlation with physical interactions. To this end, we compared DAISY (data mining synthetic lethality identification pipeline) (Jerby-Arnon et al., 2014, Lee et al.,2018 identified human SL gene pairs with three sets of human PPIs, namely PROPER v.1.0, APID, and HuRI (Luck et al., 2020). DAISY included 2,816 SL pairs (Jerby-Arnon et al., 2014), whereas PROPER v.1.0, APID, and HuRI contained 210,518, 322,260, and 52,544 human PPIs, respectively. DAISY and PROPER v.1.0 shared 100 gene pairs (odds ratio = 27.6, p-value < 2.2×10−16, hypergeometric test) (Figure S11A); DAISY and APID shared 74 gene pairs (odds ratio = 13.2, p-value < 2.2×10−16, hypergeometric test); and DAISY and HuRI shared 4 gene pairs (odds ratio = 4.2, p-value = 0.015, hypergeometric test). Although the association between DAISY and HuRI was weaker than DAISY’S associations with PROPER v.1.0 and APID, all three comparisons revealed positive associations. These data suggest a positive correlation between human SL gene pairs and human PPIs.
Next, we tested whether the hubs (proteins with many interactions) and the other nodes of PROPER v.1.0 are equally likely to participate in synthetic lethality. To this end, we identified the 121 nodes in PROPER v.1.0 that are involved in the human SL pairs (SL nodes) (Figure S11A). The SL nodes exhibited an average degree of 538 in PROPER v.1.0, far above the average degree of the entire PROPER v.1.0 (p-value < 2.2×10−16, Kolmogorov–Smirnov test) (Figure S11B). These data suggest that the human genes involved in SL tend to be the hubs of the human PPI network, in line with the notion that the hubs of a scale-free network are more important than the other nodes for maintaining the integrity of the network (Buldyrev et al., 2010).
Cell type-associated subnetworks
When we designed PROPER-seq, we did not anticipate it to be sensitive enough to reveal cell type differences. After evaluating PROPER v.1.0 (the integrated result from three input cell lines), we tested if the cell type-specific gene expression could lead to differential contribution of PROPER-seq data from each cell type to the identified PPIs in PROPER v.1.0. We tested this possibility at two levels, namely for every PPI and for every subnetwork (as defined by GO terms). At the level of individual PPIs, approximately 33% of PROPER v.1.0’s PPIs were identified primarily due to the read pairs from a specific cell type, including approximately 14,000 (6.8%), 25,000 (12%), and 29,000 (14.1%) PPIs attributable to HEK, Jurkat, and HUVEC data, respectively (Figure 6A).
Figure 6.
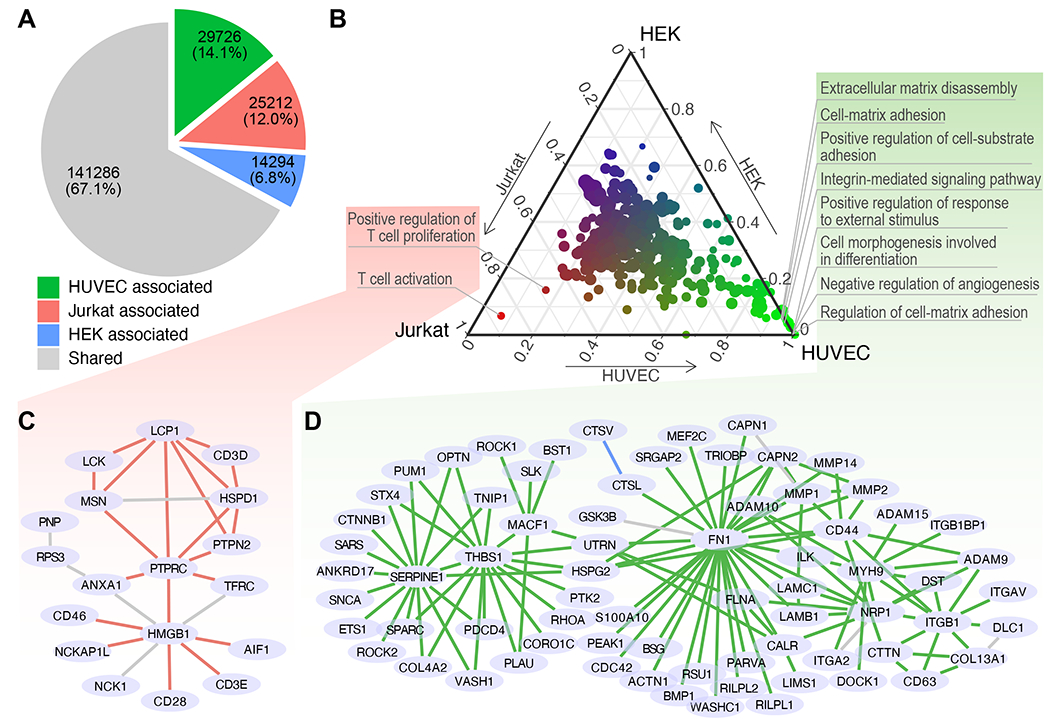
Cell type-associated subnetworks. (A) Numbers of PPIs associated with HEK, Jurkat, HUVEC and those that did not associate with any cell type (shared). (B) Associations of subnetworks and cell types. The proportion of PPIs that are associated with each cell type (each axis on the edge of the triangle) in every GO term-defined subnetwork (dot). The relative associations to the three cell types are also represented in a color gradient from red (Jurkat) to green (HUVEC) and to blue (HEK). Dot size: the number of genes in a GO term. (C-D) Expanded view of the combined subnetwork of the subnetworks associated with Jurkat (C) and those associated with HUVEC (D). Edge colors denote shared PPIs (grey) as well as the PPIs associated with Jurkat (red) or HUVEC (green).
At the subnetwork level, we obtained 431 subnetworks by extracting the nodes in PROPER v.1.0 associated with each GO term and the edges connecting the extracted nodes. We quantified the association of each subnetwork to each cell type by the proportions of PPIs (edges) attributable to that cell type. Most subnetworks (402 out of 431) did not preferentially associate with any one of the three cell types (clustered at the center, Figure 6B), consistent with the idea that most biological processes as defined by GO terms are shared across these cell types. Specifically, no subnetwork exhibited preferential association with HEK (top corner, Figure 6B). The “T cell activation” and “positive regulation of T cell proliferation” subnetworks emerged as the top 2 subnetworks with the strongest associations with Jurkat cells, consistent with the T lymphocyte origin of Jurkat cells (lower left corner, Figure 6 B and C). Several subnetworks were associated with vascular endothelial cells, including “regulation of extracellular matrix”, “cell mobility”, “cell-matrix and cell-substrate adhesion”, and “integrin-mediated signaling pathway” (lower right corner, Figure 6 B and D), reflecting the crucial functional properties of endothelial cells (Deanfield et al., 2007). These data suggested a strong potential of applying PROPER-seq to reveal cell type-specific PPIs.
Discussion
PROPER-seq provides a time-effective approach to mapping PPIs at the transcriptome scale in a single experiment. It does not require specialized resource or reagents such as antibodies, and can be applied to a variety of input cells. Thus, PROPER-seq may be a useful profiling tool to assist users in a broad scientific community to discover PPIs relevant to many cells or tissue of interest.
The PROPER v.1.0 database expands the human reference protein interactome by contributing approximately 200,000 previously uncharacterized PPIs. For example, PROPER v.1.0 adds several hundred interaction partners to PARP1. Markedly, PROPER v.1.0 lends experimental supports to more than 17,000 computationally predicted PPIs that have not been experimentally validated, suggesting the strong predictive ability of structure-based computational models. Furthermore, the hub proteins of PROPER v.1.0 are more likely to overlap with the genes in SL gene pairs than the non-hub proteins, suggesting a connection between the human protein interactome’s connectivity and human genes’ sythetical lethality.
Limitations
This study has several limitations. First, PROPER-seq is an in vitro assay and it may miss PPIs that rely on posttranslational modifications or in vivo protein localizations. Second, we have only validated a very small number of previously uncharacterized PPIs and future studies are warranted to interrogate many other previously uncharacterized PPIs. Third, we have not tested whether the DNA tags of proteins can interfere with protein-protein interactions. Fourth, we cannot rule out all possible false positive interactions, e.g. those due to high-abundance proteins (Mellacheruvu et al., 2013) and protein-DNA interactions. To control for high-abundance proteins, we accounted for unligated reads belonging to each protein in the Chi-square test; we also marked 13 PPIs in PROPER v.1.0 as potential background contaminations, because they include proteins that appear at high frequencies in negative control AP-MS experiments (Mellacheruvu et al., 2013). To minimize protein-DNA binding, PROPER-seq uses a protein specific cross-linker, BS3, which only crosslinks amines to other amines. After cross-linking by BS3, we included multiple rounds of washes in PROPER-seq to minimize spurious binding.
This study is not designed to identify cell-type-specific interactions with statistical rigor. To identify cell-type-specific PPIs, we anticipate that future work is required to characterize the within-cell-type variation and dissect the with-cell-type variation into biological variation (amongst different cell sources, batches, culture conditions, cell cycle phases) and technical variation (amongst sufficient replicate experiments on the cells with the biological variation controlled for). With within-cell-type variation fully characterized and accounted for, we anticipate that a comparison among different cell types can identify cell-type-specific PPIs.
STAR Methods
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sheng Zhong (szhonq@ucsd.edu).
Materials availability
This study did not generate any unique reagents.
Data and code availability
All sequencing data have been uploaded to GEO with accession number: GSE150818.
PROPERseqTools is available at DOI: https://doi.org/10.5281/zenodo.5009171. PROPER v.1.0 database is at https://genemo.ucsd.edu/proper.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
Experimental model and subject details
All cell lines were obtained from ATCC. Female human embryonic kidney cells (HEK 293T) were cultured in Dulbecco’s modified Eagle medium (DMEM; GIBCO, 11960044) supplemented with 10% FBS (Gemini, 100-500), 2 mM Glutamax (GIBCO, 35050061), and 5,000 U/ml penicillin/streptomycin (GIBCO, 15070063), at 37°C with 5 % CO2.
Female primary umbilical vein endothelial cells (HUVEC) and male human T-lymphocyte (Jurkat) cells were cultured in RPMI-1640 Medium (ATCC, 30-2001) supplemented with 10% FBS (Gemini, 100-500), 10 mM HEPES (Sigma-Aldrich, H0887-100ML), and 5,000 U/ml penicillin/streptomycin (GIBCO, 15070063), at 37°C with 5 % CO2.
Method details
SMART-display
Overview of SMART-display
Because mRNA-display has been thoroughly tested (Barendt et al., 2013, Seelig, 2011), our goal is to simplify the mRNA-display process, so that mRNA-display can be performed at genome scale. Our simplification was achieved by replacing the most time-consuming experimental step in mRNA-display (Cotten et al., 2011). The most time-consuming step in mRNA-display is creating a gene library for in vitro transcription and translation. For each gene in this library, specific sequences for transcription initiation (T7 RNA polymerase promoter), translation initiation (Ribosome binding site), and puromycin attachment (Puromycin linker hybridization site) must be incorporated into appropriated places (Figure 2A). Generating such a library for numerous genes requires laborious cloning. Alternatively, starting from commercial collections, called ORFeomes (Matsuyama et al., 2006), of protein coding genes contained within expression plasmids, would save some time. However, these plasmids still lack the sequences required to hybridize to the puromycin linker. In order to introduce the puromycin linker hybridization sequence to every gene, the appropriate bacterial strain must be grown, the plasmid purified, and PCR performed with primers containing the desired sequences. This process must be performed for each strain to be used in the assay. Thus, even if one starts with an ORFome, it still requires a large effort to create a gene library suitable for mRNA-display is not trivial. In addition, one of the largest human ORFemones, the Human ORFeome V8.1 (Yang et al., 2011), contains approximately 12,000 open reading frames; this is less than half of the known human coding genes.
The SMART-display procedure starts by extracting the transcriptome from the input cells. Next, leveraging the property of template switching oligo (TSO) mediated cDNA synthesis that can extend the cDNA with help of a TSO-containing primer, SMART-display uses a 5’ primer that contains the translation initiation site and the bases required for template switching (Figure 2C) and a 3’ primer that contains a random priming sequence followed by the linker hybridization sequence. Using SMART-RACE based cDNA synthesis (Figure 2 C and D) and PCR (adding the transcription initiation sequence, Figure 2E), SMART-display generates an entire gene library suitable for mRNA-display. Next, this gene library is transcribed. The transcripts are ligated with a universal puromycin-containing linker oligonucleotide, facilitated by hybridization of the puromycin-containing linker oligo to the linker hybridization sequence at the 3’ end of the transcript (Figure 2G). Finally, the puromycin ligated transcripts are translated into display complexes in the form of mRNA-linker-protein (Figure 2H, Figure S1).
mRNA Purification
Total RNA was isolated from HEK with TRIzol™ Reagent (Invitrogen, 15596026) according to the manufacturer’s recommendations. Subsequently, poly-A RNAs were enriched with the Dynabeads™ mRNA Purification Kit (Invitrogen, 61006). The reduction of rRNA was evaluated against the total RNA using Agilent’s Bioanalyzer RNA 6000 Pico Kit (Agilent Technologies, 5067-1513). The remaining rRNA was depleted with the Ribo-Zero H/M/R Kit (illumina, MRZH116) or the RiboMinus Transcriptome Isolation Kit (Invitrogen, K155002) adjusting the input amount based on the estimated rRNA removed by the oligo-dT selection (For example, if rRNA was 50% depleted, input was twice as much RNA as recommended). The final quality of the RNA as assessed with Agilent’s Bioanalyzer RNA 6000 Pico Kit.
Generation of DNA Library
To hybridize the Right/Random primer (5′ TTT CCC CGC CGC CCC CCG TCC TGC TGC CGC CCT TGT CGT CAT CGT CTT TGT AGT C(Nx15) 3′), 0.5 pmols of mRNA, 2.33 uM primer, and 2.33 mM dNTPs were mixed in a total volume of 10.75 uLs. This reaction was brought to 72 °C for 3 minutes and then cooled to 25 °C for 10 minutes. The template switching reaction was performed by adding 250 U Superscript II Reverse Transcriptase (Thermo Scientific, 18064014), Superscript II First Strand Buffer (to 1X), 5 mM DTT, 20 U SUPERase•In™ RNase Inhibitor (Thermo Scientific, AM2694), 1 M Betaine (Sigma-Aldrich, 61962), 6 mM MgCl2 (Invitrogen, AM9530G), and 1 uM Library TSO (5′ /5Biosg/GGC TCA CGA GTA AGG AGG ATC CAA CAT rGrGrG 3′) to a total volume of 25 uLs. The reaction was incubated at 25 °C for 2 minutes, 42 °C for 50 minutes, 10 cycles of 50 °C for 2 minutes and 42 °C for 2 minutes, and 70 °C for 15 minutes. Purification was performed with 1.8x Agencourt RNAClean XP Beads (Beckman Coulter, A63987) and the product was quantified with the Qubit™ dsDNA BR Assay Kit (Invitrogen, Q32853).
Amplification of 1 ng of cDNA/RNA product was performed per 25 uL NEBNext High-Fidelity 2X PCR Master Mix (NEB, M0541L) reaction, containing 0.5 uM Left PCR primer (5′ GCG AAT TAA TAC GAC TCA CTA TAG GGC TCA CGA GTA AGG AGG 3′) and 0.3 uM Right PCR primer (5′ TTT CCC CGC CGC CCC CCG TC 3′). Reactions were cycled twice with a 65 °C annealing step and a 3 minute 72 °C extension step, and 13 cycles with a single 3 minute 72 °C combined annealing and extension step. Approximately 24 reactions were performed simultaneously to generate enough material for in vitro transcription; the products were co-purified with 1.8x Agencourt AMPure XP Beads (Beckman Coulter, A63881) and quantified with the Qubit™ dsDNA BR Assay Kit.
Synthesis of Puromycin containing linker
All oligo components of the puromycin containing linker were reconstituted to 1 mM with 1x PBS pH 7.2 (Thermo Scientific, 20012027). To generate the dl containing puromycin containing linker, the Biotin Arm (w/dl) (5’ /5Phos/CC/ideoxyl/ C/iBiodT/C /ideoxyl/AC CCC CCG CCC CCC CCG /iAzideN/CCT 3’) was mixed in a 1:1 ratio with the Puromycin Arm (5’ /5DBCON/TCT /iSp18/iSp18/iSp18/iSp18/CC/3Puro/ 3’). To generate puromycin containing linker without dl bases, the Biotin Arm (w/o dl) (5’ /5Phos/CCG C/iBiodT/C GAC CCC CCG CCC CCC CCG /iAzideN/CCT 3’) was mixed in a 1:1 ratio with the Puromycin Arm (5’ /5DBCON/TCT /iSp18/iSp18/iSp18/iSp18/CC/3Puro/ 3’). The mixtures were incubated at 40 °C overnight with agitation.
The mixtures were run on a 15% TBE-UREA Gel (Invitrogen, EC6885BOX) prepared in a 1:1 ratio with Formamide Running Buffer (1 part 10x TBE Buffer Running Buffer (Invitrogen, LC6675), 9 parts Deionized Formamide (EMD Millipore, 4610-100ML)) at 200V for 1 hour. The gel was removed from the cassette and a exposed to UV while on a TLC Silica gel 60 F254 Plate (EMD Millipore, 1.05715.0001) to visualize the DNA bands. Two bright bands appeared, the largest was removed with a clean scalpel and transferred to a clean 2 mL tube. The gel fragment was crushed with the plunger from a 1 mL syringe and suspended in 500 uLs Elution Buffer (0.5M Ammonium Acetate (Invitrogen, AM9070G), 10 mM Magnesium Acetate (Sigma-Aldrich, 63052-100ML)). The gel fragment was incubated at room temperature with rotation overnight. The gel and buffer mixture was transferred to a 0.45 uM Nanosep® MF spin filter (Pall Corporation, ODM45C33), and the liquid collected by spinning at 5,000 xg for 10 minutes. The flow through was precipitated with 0.5x volume LiCl Precipitation Solution (Invitrogen, AM9480), 6 uLs Co-Precipitant Pink (Bioline, BIO-37075), and 3x volume of 100% Ethyl Alcohol (Sigma-Aldrich, 493546) and incubated overnight at −80 °C. The linker was then pelleted by centrifugation at 22,000 xg for 20 minutes, washed with 70% Ethyl Alcohol, and air dried. The pelleted linker was suspended in nuclease-free water (Thermo Scientific, 10977023).
Generation of Puromycin Ligated RNA Library
RNA libraries were generated with 500 ngs of DNA Library using the HiScribe™ T7 High Yield RNA Synthesis Kit (NEB, E2040S). After synthesis, DNA was removed with TURBO™ DNase (Invitrogen, AM2238). The RNA was precipitated with 2.5 M LiCl Precipitation Solution, quantified with the Qubit™ dsDNA BR Assay Kit (Invitrogen, Q32853), and the distribution checked with the Agilent RNA 6000 Pico Kit.
RNA libraries were annealed to the appropriate puromycin containing linker in a 1:1.25 molar ratio in Annealing Buffer (10x: 100 mM Tris-HCl Buffer, pH 7.5 (Invitrogen, 15567027), 500 mM NaCl (Thermo Fisher Scientific, AM9759), 10 mM EDTA (Research Products International, E14100-50.0)), incubating at 75 °C for 5 minutes and cooling slowly to 25 °C. Ligation was performed with 0.4 U/uL of T4 RNA Ligase 1 (NEB, M0204S), 1 mM ATP, and 1.6 U/uL of SUPERase• In™ RNase Inhibitor for 30 minutes at 25 °C. NEBuffer 4 was added to 1x, and unligated linker was digested with 0.2 U/uL of T5 Exonuclease (NEB, M0363S) at 37 °C for 30 minutes. The ligated RNA was purified with an RNeasy Mini Column (Qiagen, 74104).
Translation and Display
Protein products were generated using 25 pmols of ligated RNA product per 25 uL reaction of the PURExpress® In Vitro Protein Synthesis Kit (NEB, E6800S). Translation reactions were performed in an air incubator for 90 minutes at 37 °C. After translation, KCl (Invitrogen, AM9640G) and MgCl2 (Invitrogen, AM9530G) were added to a final concentration of 800 mM and 80 mM respectively. The reaction was incubated at room temperature for 30 minutes and then stored at −20 °C for a minimum of 12 hours.
VALIDATION by anti-GFP Selection
Preparation of SMART-display Library
Templates for the target genes were ordered from IDT with all display sequences already incorporated on the 5’ and 3’ ends of the template. From these templates, RNA was generated and SMART-display proceeded as described above.
Pull-Down with anti-GFP antibody
The products of the SMART-display process for each of the target genes were mixed in a 1:1 ratio. The mixture was precleared with 50 μL of Streptavidin T1 magnetic beads. The mixture was incubated at 4°C with gentle rotation for 1 hour. The Streptavidin T1 beads were separated with a magnetic rack for 1 minute and the supernatant was transferred into a new microcentrifuge tube placed on ice.
To the precleared solution, Normal Goat Serum (NGS) (Thermo Fisher Scientific, 31873) in PBS was added to 5% for blocking. Primary anti-GFP antibody (Thermo Fisher Scientific, A10259) diluted in PBS was added to a final concentration of 0.2 μg/mL. The sample was incubated at 4°C overnight with gentle rotation.
50 uLs Streptavidin T1 magnetic beads were added to the samples and incubated at room temperature for 1 hour with gentle rotation. The tubes were placed on a magnetic rack for 1 minute and the supernatant discarded. The beads were suspended in wash buffer (5% NGS in PBS, 1% Triton® X-100, 3% BSA (NEB, B9000S) by pipetting gently up and down. The tubes were rotated gently for 10 minutes. The wash process was repeated two more times.
cDNA Synthesis
A reverse transcription reaction solution was prepared for the selected sample (immobilized on the Streptavidin T1 beads) and for the pre-selection samples. The 100 uL reactions contained 800 U Superscript II, 1x First Strand buffer, 10 mM DTT, and 0.5 mM dNTPs. The same volume of pre-selection sample was used for each of the genes; the entire bead volume was use in the post-selection reactions. The reactions were incubated at 42°C for 90 min with agitation.
Protein Removal
1.6 units of Proteinase K was added to each sample and incubated for 15 minutes at 65°C. Samples were purified with 1.2x Ampure beads and eluted in 30 uL of water.
Gene Identification using qPCR
Three 25 uL qPCR reaction containing 1x Power SYBR® Green PCR Master Mix (Thermo Fisher Scientific, 4367659) and 10 mM of each of the gene specific primers was prepared for each sample and for the no template controls. Three 25 uL reactions were also prepared for each sample without primers as a no primer control. 1 uL of sample was used in each reaction. The qPCR assay was run on a QuantStudio 3 Real-Time PCR System with an initial denaturation of 95 °C for 2 minutes, 30 cycles of 95 °C for 30 seconds, 55 °C for 15 seconds, and 72 °C for 30 seconds, and a final extension of 72 °C for 5 minutes. A melt curve was run to assess the purity of the qPCR products.
Comparison of SMART-display product library and control libraries
SMART-display libraries were prepared as described above up to the puromycin containing linker ligation.
Generation of Puromycin Ligated RNA Library
RNA libraries were annealed to a puromycin containing linker with no biotin (5’ /5Phos/CC/ideoxyl/CTC/ideoxyl/ACCCCCCGCCGCCCCCCGTCCT/iSp18/iSp18/iSp18/iSp18/CC/3Puro/ 3’) in a 1:1.25 molar ratio in Annealing Buffer (10x: 100 mM Tris-HCl Buffer, pH 7.5, 500 mM NaCl, 10 mM EDTA). The “no puromycin” control was subject to the same reaction with the omission of the puromycin containing linker. The reactions were incubated at 75 °C for 5 minutes and cooled slowly to 25 °C. Ligation was performed with 0.4 U/uL of T4 RNA Ligase 1, 1 mM ATP, and 1.6 U/uL of SUPERase• In™ RNase Inhibitor for 30 minutes at 25 °C. NEBuffer 4 was added to 1x, and unligated linker was digested with 0.2 U/uL of T5 Exonuclease at 37 °C for 30 minutes. The ligated RNA was purified with an RNeasy Mini Column.
Translation and Display
Protein products were generated using 25 pmols of RNA product and 2 uLs Transcend™ tRNA (Promega, L5061) per 25 uL reaction of the NEB PURExpress IVT kit. 2 uLs of Proteinase K was added to the “protein digested control”. Translation reactions were performed in an air incubator for 90 minutes at 37 °C. After translation, KCl and MgCl2 were added to a final concentration of 800 mM and 80 mM respectively. The reaction was incubated at room temperature for 30 minutes and then stored at −20 °C for a minimum of 12 hours.
Protein Selection and Pull-Down
75 uLs of Dynabeads MyOne Streptavidin T1 Beads were prepared per IVT reaction according to the manufacturer’s directions. The IVT reaction was added to the suspended beads and incubated for 1 hour with rotation at room temperature. The beads were washed 3 times with 8M Urea wash buffer (8M Urea, 50 mM Tris, 5 mM EDTA, 0.1% NP40, 500 mM LiCl, 2% SDS), and 3 times with 1x B&W buffer (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1M NaCl).
Library Preparation and Sequencing
The beads were subject to a Superscript III One-Step RT-PCR (Invitrogen, 12574018) reaction at 5x the original volume of streptavidin beads, with 0.5 uM of each a universal forward primer (5’ AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCGAGTAAGGAGGATCCAACATG 3’) and an indexed reverse primer (5’ CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTTGTCGTCATCGTCTTTGTAGTC 3’, where X represents the index bases). The cycle number was optimized for each sample, using the minimum number of cycles to generate a library. Samples were mixed 3:2 with PhiX and sequenced 150 base pairs from each end on an illumina MiniSeq.
INLISE
Overview of INLISE
INLISE includes 3 steps. The first step is to create the bait and the prey libraries. In this step, The SMART-display gene library is split and processed in two independent display reactions. One library is ligated to a puromycin containing linker sequence with a non-cleavable biotin and will become the ‘bait’ library, permanently immobilized on a magnetic bead (pink fusion, Figure 3). The other library is ligated with a puromycin containing linker sequence containing a cleavable biotin, and becomes the free, unbound, ‘prey’ library (blue fusion, Figure 3). Next, both libraries are pulled down onto streptavidin magnetic beads, and their mRNA tags are stabilized by reverse transcription. Next, free biotin is added to the bait population to block remaining binding sites on the streptavidin beads and prevent non-specific binding. Meanwhile, the prey population is released from the streptavidin beads via digestion of the inosine nucleotides found in the loop region of the puromycin containing linker (“I” in pink, Figure S1). Next, the double-stranded cDNA barcode is treated with a non-palindromic restriction enzyme (BbvCI) to generate sticky ends. A biotin-labeled linker sequence (grey bars, Figure 3), designed with complementary sticky ends to the BbvCI restriction site, is introduced and ligated to the BbvCI trimmed cDNA of the prey library only.
The second step is to ligate the barcodes from interacting proteins. In this step, the free and bound libraries are mixed and incubated in buffer. The interacting proteins will be cross-linked. Stringent washes will be applied to remove non-specifically bound proteins or RNAs. The linker containing ends of the prey library are then ligated to the sticky end of the interacting bait protein. The use of a non-palindromic restriction enzyme prevents self-ligation of the cDNA or self-ligation of the linker sequence. At this point, PPIs have been recorded by chimeric sequences of the form cDNA1-linker-cDNA2.
The third step is to construct the sequencing library. The cDNAs, including those that have been ligated (cDNA1-linker-cDNA2), are subjected by to enzymatic fragmentation and addition of sequencing adapters (NEBNext Ultra II FS). The chimeric sequences (cDNA1-linker-cDNA2) are selected for using the biotin on the linker sequence. The resulting library is amplified and subjected to paired-end sequencing. Taken together, SMART-display and INLISE constitute the entire PROPER-seq experimental pipeline.
Purification and Immobilization of Display Products
75 uLs of Dynabeads™ MyOne™ Streptavidin T1 (Thermo Fisher Scientific, 65601) were prepared by washing twice in an equivalent volume of 1x PBS pH 7.4 (Thermo Fisher Scientific, 70011044). The IVT reaction was added to the suspended beads in 1.8 mLs of 1x PBS pH 7.4 (Thermo Fisher Scientific, 70011044) with 0.1% Triton™ X-100 (Sigma-Aldrich, T8787-50ML) and incubated for 1 hour with rotation at room temperature. D-Biotin (Ivitrogen, B20656) was added to 2.25 uM and incubated at room temperature for 10 minutes with rotation. The beads were washed 2 times for 5 minutes with 500 uLs 1x PBS pH 7.4 with 0.1% Triton™ X-100 (Sigma-Aldrich, T8787-50ML).
DNA Synthesis
50 uLs of first strand reaction was mixed per sample containing 500 U of Superscript II Reverse Transcriptase (Thermo Scientific, 18064014), 1x Superscript II FS Buffer, 5 mM DTT, 1 uM dNTP mix (NEB, N0447S), 1 M Betaine (Sigma-Aldrich, 61962), 6 mM MgCl2, 500 pmol of End Capture TSO (5’ /5dSp/AGT AAA GGA GAC CTC AGC TTC ACT GGA rGrGrG 3’), and 40 U of SUPERase· In™ RNase Inhibitor. The mix was added to the beads and incubated at 42°C for 50 minutes with agitation, and then cycled 10 times at 50°C for 2 minutes followed by 42°C for 2 minutes. The beads were washed 2 times for 5 minutes with 500 uLs 1x PBS pH 7.4 with 0.1% Triton™ X-100. 100 uLs of first strand reaction was mixed per sample containing 20 U DNA Polymerase I (NEB, M0209S), 1x NEBuffer 2, 2.4 mM DTT, and 0.25 mM dNTP mix. The mix was added to the beads and incubated at 37°C for 30 minutes with agitation. The beads were washed 2 times for 5 minutes with 500 uLs 1x PBS pH 7.4 with 0.1% Triton™ X-100.
Restriction Digestion and Control Digestion
All samples were digested with 10 U of BbvCI (NEB, R0601S) in 1x CutSmart Buffer at 500 uLs. The digestion was incubated at 37°C for 1 hour with agitation. After the restriction enzyme digestion, but without washing the beads, the bait population used in the Proteinase control was generated by the addition of 5 uLs of Proteinase K (NEB, P8107S) to the sample. The sample was incubated an additional 30 minutes at 37°C with agitation. All samples were then washed 2 times for 5 minutes with 500 uLs 1x PBS pH 7.4 with 0.1% Triton™ X-100.
Synthesis of Interaction Linker
The top and bottom strands of the interaction linker were reconstituted to 200 uM with Annealing Buffer. The two strands were mixed in a 1:1 molar ratio, incubated at 75 °C for 5 minutes and cooled slowly to 25 °C.
Interaction Linker Ligation and Release of Prey
Samples with a dl containing puromycin containing linker were ligated to the Interaction Linker and subsequently released from the Dynabeads™ MyOne™ Streptavidin T1 beads to generate the prey population. Ligation was performed at 37°C with agitation for 30 minutes, with 200 pmol Interaction Linker, 4000 U T4 DNA Ligase (NEB, M0202M), and 1x T4 DNA Ligase Buffer in 500 uLs. The interaction linker was omitted in the prey reaction used in the No-linker control. The beads were washed 2 times for 5 minutes with 500 uLs 1x PBS pH 7.4 with 0.1% Triton™ X-100. The release of the complexes from the beads was performed at 37°C with agitation for 30 minutes, with 40 U of Endonuclease V (NEB, M0305S) in 50 uLs of 1x NEBuffer™ 3 (NEB, B7003S).
Interaction
The sample without deoxyinosine (dl) bases in the puromycin containing linker were retained on the Dynabeads™ MyOne™ Streptavidin T1 beads to become the bait libraries. These samples were suspended in 150 uLs Binding Buffer (10 mM HEPES (Fisher Scientific, BP299100), 50 mM KCl, 4 mM MgCl2, 2mM DTT, 0.2 mM EDTA, 0.1% Tween® 20 (Sigma-Aldrich, P9416-100ML)). The 50 uL of supernatant from the Endonuclease V digestion (the prey library), was added to the bait samples with the following conventions. PROPER-seq reaction: bait and prey libraries with the full PROPER-seq protocol; No-linker control: bait library with the full PROPER-seq proctol, prey library created without the interaction linker ligated; and Proteinase control: bait library treated with Proteinase K and the prey library created with the full PROPER-seq protocol. The mixtures were incubated at room temperature with rotation for 1 hour. 800 uLs of Binding Buffer was added to each reaction to bring the volume to 1 mL, and they were rotated an additional 10 minutes at room temperature.
Crosslinking and Proximity Ligation
Crosslinking was performed at room temperature for 30 minutes with 0.5 mM BS3 (Thermo Fisher Scientific, A39266). The reaction was quenched with 50 mM Tris-HCl Buffer, pH 7.5 with rotation for 15 minutes. The beads were washed 3 times for 5 minutes with 500 uLs 1x PBS pH 7.4 with 0.1% Triton™ X-100.
Proximity ligation was performed with 20,000 U of T4 DNA Ligase in 1 mL of 1x T4 DNA Ligase Buffer. The reaction was incubated with constant rotation for 30 minutes at room temperature. The enzyme was inactivated before the beads were gathered by heating to 65°C for 10 minutes. The beads were washed 2 times for 5 minutes with 500 uLs 1x PBS pH 7.4 with 0.1% Triton™ X-100.
Sequencing Library Generation and Sequencing
The DNA was released from the beads with the NEBNext® Ultra™ II FS DNA Module (NEB, E7810S) using twice the reaction volume and a fragmentation time of 5 minutes. The end repair step was not performed. Libraries were then generated with the NxSeq® UltraLow DNA Library Kit (Lucigen, 15012-1) up to the final AMPure XP Bead purification before amplification. Each sample was eluted in 50 uLs Nuclease-free water, and added to 10 uLs of Dynabeads™ MyOne™ Streptavidin T1 beads suspended in 50 uLs 1x PBS pH 7.4 with 0.1% Triton X-100. The selection was performed at room temperature for 1 hour. Beads were washed 2 times with 500 uLs Low Salt buffer [0.1% SDS (Invitrogen, AM9820), 0.1% Triton™ X-100, 2 mM EDTA, 20 mM Tris-HCl buffer, pH 8 (Invitrogen, 15568025), 150 mM NaCl], 2 times with 500 uLs 1x B&W buffer (5 mM Tris-HCl pH 7.5, 0.5 mM EDTA, 1M NaCl), and 2 times with 500 uLs 1x PBS pH 7.4 with 0.1% Triton™ X-100. Library amplification was then performed with the NxSeq® UltraLow DNA Library Kit as directed.
Each library was paired end sequenced for 100 cycles on each end on an lllumina HiSeq 4000 or NovaSeq 6000.
Validation by proximity ligation assay (PLA)
Cell Culture
HEK 293T cells were cultured in Dulbecco’s modified Eagle medium (DMEM; GIBCO, 11960044) supplemented with 10% FBS (Gemini, 100-500), 2 mM Glutamax (GIBCO, 35050061), and 5,000 U/ml penicillin/streptomycin (GIBCO, 15070063), at 37°C with 5 % CO2.
Fixation and Permeabilization
Approximately 0.5 million HEK cells per well were fixed with 4% formaldehyde (Thermo Fisher Scientific, 28906) in PBS pH 7.2 (Life Technologies, 20012027) at room temperature for 30 minutes on a Lab-Tek 8-well Chamber Slide (Thermo Fisher Scientific, 154534). Cells were washed once with PBS pH 7.2, then permeablized with 200 uLs of 0.1% Triton X-100 (Sigma-Aldrich, T8787-50ML) in PBS for 15 minutes at room temperature with rocking.
Blocking
Cells were blocked by adding 40 uLs Duolink Blocking Solution (Sigma-Aldrich, DUO92101-1KT) and incubating in a humidity chamber for 1 hour at 37°C.
Staining with Primary Antibody
Primary antibodies were added to the cells at the dilutions listed below in a total of 40 uLs. The slides were incubating in a humidity chamber for 1 hour at 37°C.
| Target | Manufacturer | Catalog Number | Dilution |
|---|---|---|---|
| PARP1 | Abeam | Ab227244 | 1:250 |
| PARP1 | Atlas Antibodies | AMAb90959 | 1:200 |
| SUMO1 | Abeam | Ab32058 | 1:250 |
| XP01 | Atlas Antibodies | HPA042933 | 1:500 |
| MATR3 | Atlas Antibodies | HPA036565 | 1:250 |
| IP05 | Santa Cruz Biotechnology | Sc-55527 | 1:1000 |
| GFP | Thermo Fisher Scientific | A10259 | 1:250 |
Staining with PLA Probes, Ligation, and Amplification
Slides were wash 2x with 70 mL of wash buffer A, and stained with PLA probes according to the Duolink Assay instructions. Slides were wash 2x with 70 mL of wash buffer A, and ligation performed according to the Duolink Assay instructions. Slides were wash 2x with 70 mL of wash buffer A, and amplification performed according to the Duolink Assay instructions. Slides were then wash 2x with wash buffer B and 1x with 1:100 wash buffer B.
Imaging
Coverslips were mounted with 12 uLs Duolink PLA mounting medium with DAPI per well and sealed with clear nail polish. Images were acquired on Olympus Inverted Microscope using a 60X/1.518 oil objective (GE Healthcare Life Sciences) (pixel size = 0.1075 μm). A series of z-stack images across the cells were acquired with 0.3 μm sample thickness (3 sections).
Validation by co-IP
Five million HEK293T cells were lysed in RIPA buffer [150 mM NaCl, 5 mM EDTA, 50 mM Tris pH 7.5, 1% NP-40, 0.5% sodium deoxycholate (Sigma-Aldrich, 30970-25G), 0.1% SDS, and a protease inhibitor cocktail (Sigma Aldrich, P8340)] for 30 minutes on ice and subsequently centrifuged at 10,000 xg for 10 minutes. The supernatants were precleared by incubation with Protein-G Dynabeads (Thermo Fisher Scientific, 10003D) for 30 minutes at 4°C. Antibody-coated beads were prepared by incubating rabbit anti-human Leo1 antibody (5 μg per sample, Bethyl Laboratories, A300-175A) or control rabbit IgG (5 μg per sample; Abeam, AB37415) with pre-washed Protein-G Dynabeads for 2-3 hours at room temperature. 5% of the precleared lysate (input) was saved for later analysis, and the remaining lysate was split equally among the Leo1- or IgG-coated beads for immunoprecipitation (IP). IP was carried out overnight at 4°C. 10% of the flow through (FT) was retained for analysis. The Dynabeads were washed 3 times for 5 mins each with RIPA buffer. The washed beads were eluted in reducing sample buffer (Thermo Fisher Scientific, 39000) before resolving on an 8% SDS-PAGE and immunoblotting (IB) with indicated antibodies.
| Target | Manufacturer | Catalog Number | Dilution |
|---|---|---|---|
| Leo1 | Bethyl Laboratories | A300-175A | 1:1000 |
| PARP1 | Thermo Fisher | MA3950 | 1:500 |
Quantification and Statistical Analysis
Processing proper-seq read pairs
The following data processing steps are implemented in the PROPERseqTools pipeline: https://qithub.com/Zhong-Lab-UCSD/PROPERseqTools. The sequencing reads were subjected to Cutadapt 2.5(Martin, 2011) to remove the 3′ linker sequence and the 5′ adapter sequence. The remaining read pairs were subsequently subjected to Fastp 0.20.0(Chen et al., 2018) to remove low-quality reads (average quality per base < Q20) and short reads (<20 bp). The remaining read pairs were subsequently mapped to RefSeq transcripts (O’Leary et al., 2016) (based on GRCh38.p13, NCBI Homo sapiens Annotation Release 109.20190607) using BWA-MEM 0.7.12-r1039 (Li, 2013) with the default parameters. A read was regarded as mapped to a gene if this read was mapped to any of the Refseq transcripts of this gene. The read pairs where the two ends were mapped to two different protein coding genes were identified. Any duplicated chimeric read pairs were subsequently removed to obtain non-duplicate chimeric read pairs.
Test of association between a gene pair and the chimeric read pairs
A Chi-square test was carried out on every gene pair. The null hypothesis is that the mapping of one end of a chimeric read pair to a gene is independent of the mapping of the other end of this chimeric read pair to the other gene. The contingency table of this association test is given in Figure S4A. FDR computed from the Benjamini-Hochberg procedure was used to control for family-wise errors.
Downloading APID data and its subsets
PPIs were downloaded as a MITAB file from the Agile Protein Interactomes DataServer (APID) at http://cicblade.dep.usal.es:8080/APID/init.action. The AP-MS and co-IP derived PPIs were identified by the corresponding labels in the ‘Interaction detection method’ column of the downloaded MITAB file. The LC-MS derived PPIs identified by the label of “biochemistry” in the ‘Interaction detection method’ column and specifying “Publication first author” as “Wan, C. et al. (2015)” (Wan et al., 2015), “Havugimana, PC. et al. (2012)” (Havugimana et al., 2012) and “Kristensen, AR. et al. (2012)” (Kristensen et al., 2012).
Quantifying reproducibility by odds ratio
The odds ratio was used to quantify the degree of overlap between two sets of PPIs. The odds ratio (OR) of the following contingency table is calculated as OR=(A×D)/(C×B), where A, B, C, D are numbers of PPIs in the corresponding cell in the contingency table.
| Within set II | Outside set II | |
| Within set I | A | B |
| Outside set I | C | D |
Comparison to structurally predicted PPIs
The human prePPIs were downloaded from the prePPI database (https://honiglab.c2b2.columbia.edu/prePPI/ref/preppi%20final600.txt.tar.gz). The Uniprot protein IDs used in prePPI were converted to gene symbols using the org.Hs.eg.db Bioconductor package in R.
GO term defined subnetworks
The subnetwork associated with a GO term (Ashburner et al., 2000) was retrieved by the PROPER v.1.0 nodes that were annotated by this GO term and all the edges connecting these nodes. GO term enrichment analysis was based on hypergeometric tests between the genes annotated by every GO term and the PROPER v.1.0 nodes. FDR computed from the Benjamini-Hochberg procedure was used to control for family-wise errors. The entire PROPER v.1.0 was plotted with Gephi (0.9.2, https://gephi.org/) (Bastian et al., 2009). All other network figures were plotted with Cytoscape (Shannon et al., 2003).
Test of cell type association
A Chi-square test was applied to every PPI to test the association of this PPI with a cell type. The null hypothesis is that whether a chimeric read pair is mapped to this gene pair is independent to whether this chimeric read pair was generated from this cell type. A PPI was regarded as attributable to a cell type if Chi-square test FDR < 0.05 and odds ratio > 2, where the odds ratio for the following contingency table is calculated as OR=(A×D)/(C×B).
| The read pairs is generated from this cell type | |||
| Yes | No | ||
| Mapped to this gene pair | Yes | A | B |
| No | C | D | |
A GO term defined subnetwork was included in the analysis of cell type association when this GO term contained at least 50 genes (regardless of whether these genes were included in PROPER v.1.0) and this GO term defined subnetwork contained at least 10 edges. The association of a subnetwork to a cell type was quantified by the proportions of PPIs (edges) associated with that cell type among all the PPIs of this subnetwork.
Calculating screening completeness, sampling sensitivity, assay sensitivity, precision, and protein interactome size for PROPER v.1.0
Screening completeness, sampling sensitivity, assay sensitivity, precision, and protein interactome size were defined by Yu et al. (Yu et al., 2008) and Venkatesan et al. (Venkatesan et al., 2009). We calculated these metrics for PROPER v.1.0 based on the methods described by Venkatesan et al. (Venkatesan et al., 2009) and the following positive reference set (PRS), random reference set (RRS) and orthogonal validation sets.
Positive reference set (PRS).
The CORUM database (Giurgiu et al., 2019) contains 2417 human protein complexes, corresponding to 3433 proteins and 39,103 protein pairs. These 39,103 protein pairs are used as our PRS.
Random reference set (RRS). Following Venkatesan et al. (Venkatesan et al., 2009), RRS was randomly sampled from PROPER-seq’s search space outside the PRS to contain the same number of gene pairs as PROPER v.1.0.
Orthogonal validation assay.
Targeted co-IP is used as the orthogonal validation assay. The targeted co-IP data were retrieved from APID based on two MI Ontology terms: Anti bait coimmunoprecipitation (MI:0006) and Anti tag coimmunoprecipitation (MI:0007)).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal Biotinylated Anti-GFP | Thermo Fisher Scientific | Cat#A10259; RRID: AB_2534021 |
| Rabbit Polyclonal Anti-PARP1 | Abcam | Cat#Ab227244 |
| Mouse Monoclonal Anti-PARP1 | Atlas Antibodies | Cat#AMAb90959; RRID: AB_2665732 |
| Rabbit Monoclonal Anti-SUMO1 | Abcam | Cat#Ab32058; RRID: AB_778173 |
| Rabbit Polyclonal Anti-XPO1 | Atlas Antibodies | Cat#HPA042933; RRID: AB_2678229 |
| Rabbit Polyclonal Anti-MATR3 | Atlas Antibodies | Cat#HPA036565; RRID: AB_10673623 |
| Mouse Monoclonal Anti-IPO5 | Santa Cruz Biotechnologies | Cat#sc-55527; RRID: AB_2127684 |
| Rabbit Polyclonal Anti-LEO1 | Thermo Fisher Scientific | Cat#A300-175A; RRID: AB_2135932 |
| Mouse Monoclonal Anti-PARP1 | Thermo Fisher Scientific | Cat#MA3-950; RRID: AB_325523 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Transcend™ tRNA | Promega | Cat#L5061 |
| BS3 | Thermo Fisher Scientific | Cat#A39266 |
| Critical Commercial Assays | ||
| PURExpress® In Vitro Protein Synthesis Kit | NEB | Cat#E6800S |
| SuperScript™ III One-Step RT-PCR System | Invitrogen | Cat#12574018 |
| NEBNext® Ultra™ II FS DNA Module | NEB | Cat#E7810S |
| NxSeq® UltraLow DNA Library Kit | Lucigen | Cat#15012-1 |
| Duolink™ In Situ Red Starter Kit Mouse/Rabbit | Millipore Sigma | Cat#DUO92101-1KT |
| Deposited Data | ||
| All sequencing data have been uploaded to GEO. | This paper | GSE150818 |
| Experimental Models: Cell Lines | ||
| Human T-Cells (Jurkat) | ATCC | Cat#TIB-152; RRID: CVCL 0255 |
| Human Embryonic Kidney Cells (HEK 293T) | ATCC | Cat#CRL-3216; RRID: CVCL_0063 |
| Human Umbilical Vein Cells (HUVEC) | ATCC | Cat#CRL-1730; RRID: CVCL_2959 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Right/Random primer: 5′ TTT CCC CGC CGC CCC CCG TCC TGC TGC CGC CCT TGT CGT CAT CGT CTT TGT AGT C(Nx15) 3′ | This paper | N/A |
| Library TSO: 5′ /5Biosg/GGC TCA CGA GTA AGG AGG ATC CAA CAT rGrGrG 3′ | This paper | N/A |
| Left PCR primer: 5′ GCG AAT TAA TAC GAC TCA CTA TAG GGC TCA CGA GTA AGG AGG 3′ | This paper | N/A |
| Right PCR primer: 5′ TTT CCC CGC CGC CCC CCG TC 3′ | This paper | N/A |
| Biotin Arm (w/dI): 5’ /5Phos/CC/ideoxyI/ C/iBiodT/C /ideoxyI/AC CCC CCG CCC CCC CCG /iAzideN/CCT 3’ | This paper | N/A |
| Biotin Arm (w/o dI): 5’ /5Phos/CCG C/iBiodT/C GAC CCC CCG CCC CCC CCG /iAzideN/CCT 3’ | This paper | N/A |
| Puromycin Arm: 5’ /5DBCON/TCT /iSp18/iSp18/iSp18/iSp18/CC/3Puro/ 3’ | This paper | N/A |
| Puromycin containing linker with no biotin: 5’ /5Phos/CC/ideoxyI/CTC/ideoxyI/ACCCCCCGCCGCCCCCCGTCCT/iSp18/iSp18/iSp18/iSp18/CC/3Puro/ 3’ | This paper | N/A |
| Universal forward primer: 5’ AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTCGAGTAAGGAGGATCCAACATG 3’ | This paper | N/A |
| Indexed reverse primer: 5’ CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTTGTCGTCATCGTCTTTGTAGTC 3’, where X represents the index bases | This paper | N/A |
| End Capture TSO: 5’ /5dSp/AGT AAA GGA GAC CTC AGC TTC ACT GGA rGrGrG 3’ | This paper | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| PROPERseqTools is available at https://github.com/Zhong-Lab-UCSD/PROPERseqTools. | This paper | DOI: 10.5281/zenodo.5009171 |
| other | ||
Please upload a single Word document that includes your Highlights and eTOC Blurb. Highlights are 3–4 bullet points of no more than 85 characters in length, including spaces, and they summarize the core results of the paper in order to allow readers to quickly gain an understanding of the main take-home messages. An eTOC blurb should also be included that is no longer than 50 words describing the context and significance of the findings for the broader journal readership. When writing this paragraph, please target it to non-specialists by highlighting the major conceptual point of the paper in plain language, without extensive experimental detail. The blurb must be written in the third person and refer to “First Author et al.”
Highlights:
PROPER-seq maps protein-protein interactions (PPI) en masse through DNA sequencing.
PROPER-seq reveals over 200,000 previously uncharacterized human PPIs.
PROPER-seq validates over 17,000 computationally predicted human PPIs.
The hubs of the human protein interactome tend to be synthetic lethal genes.
Acknowledgements
This work is supported by NIH grants R01GM138852, DP1DK126138, R01HL145170, and Ella Fitzgerald Charitable Foundation. The graphical abstract is created with BioRender.com.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
S.Z. is a founder, a board member and shareholder of Genemo Inc.
Reference
- ALONSO-LOPEZ D, CAMPOS-LABORIE FJ, GUTIERREZ MA, LAMBOURNE L, CALDERWOOD MA, VIDAL M & DE LAS RIVAS J 2019. APID database: redefining protein-protein interaction experimental evidences and binary interactomes. Database (Oxford), 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALONSO-LOPEZ D, GUTIERREZ MA, LOPES KP, PRIETO C, SANTAMARIA R & DE LAS RIVAS J 2016. APID interactomes: providing proteome-based interactomes with controlled quality for multiple species and derived networks. Nucleic Acids Res, 44, W529–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ASHBURNER M, BALL CA, BLAKE JA, BOTSTEIN D, BUTLER H, CHERRY JM, DAVIS AP, DOLINSKI K, DWIGHT SS, EPPIG JT, HARRIS MA, HILL DP, ISSEL-TARVER L, KASARSKIS A, LEWIS S, MATESE JC, RICHARDSON JE, RINGWALD M, RUBIN GM & SHERLOCK G 2000. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature genetics, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AW JGA, SHEN Y, NAGARAJAN N & WAN Y 2017. Mapping RNA-RNA Interactions Globally Using Biotinylated Psoralen. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BADER GD, BETEL D & HOGUE CW 2003. BIND: the Biomolecular Interaction Network Database. Nucleic Acids Res, 31, 248–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARABASI AL 2009. Scale-free networks: a decade and beyond. Science, 325, 412–3. [DOI] [PubMed] [Google Scholar]
- BARABASI AL & BONABEAU E 2003. Scale-free networks. Sci Am, 288, 60–9. [DOI] [PubMed] [Google Scholar]
- BARABASI AL & OLTVAI ZN 2004. Network biology: understanding the cell’s functional organization. Nat Rev Genet, 5, 101–13. [DOI] [PubMed] [Google Scholar]
- BARENDT PA, NG DT, MCQUADE CN & SARKAR CA 2013. Streamlined protocol for mRNA display. ACS Comb Sci, 15, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BASTIAN M, HEYMANN S & JACOMY M Gephi: an open source software for exploring and manipulating networks. Third international AAAI conference on weblogs and social media, 2009. [Google Scholar]
- BENJAMINI Y & HOCHBERG Y 1995. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological), 57, 289–300. [Google Scholar]
- BULDYREV SV, PARSHANI R, PAUL G, STANLEY HE & HAVLIN S 2010. Catastrophic cascade of failures in interdependent networks. Nature, 464, 1025–8. [DOI] [PubMed] [Google Scholar]
- CHEN S, ZHOU Y, CHEN Y & GU J 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics, 34, i884–i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN TS, PETREY D, GARZON JI & HONIG B 2015. Predicting peptide-mediated interactions on a genome-wide scale. PLoS Comput Biol, 11, e1004248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CONSORTIUM, E. P. 2004. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science, 306, 636–40. [DOI] [PubMed] [Google Scholar]
- CONSORTIUM, E. P. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COTTEN SW, ZOU J, VALENCIA CA & LIU R 2011. Selection of proteins with desired properties from natural proteome libraries using mRNA display. Nat Protoc, 6, 1163–82. [DOI] [PubMed] [Google Scholar]
- CUSICK ME, YU H, SMOLYAR A, VENKATESAN K, CARVUNIS AR, SIMONIS N, RUAL JF, BORICK H, BRAUN P, DREZE M, VANDENHAUTE J, GALLI M, YAZAKI J, HILL DE, ECKER JR, ROTH FP & VIDAL M 2009. Literature-curated protein interaction datasets. Nat Methods, 6, 39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEANFIELD JE, HALCOX JP & RABELINK TJ 2007. Endothelial function and dysfunction: testing and clinical relevance. Circulation, 115, 1285–95. [DOI] [PubMed] [Google Scholar]
- DEKKER J, BELMONT AS, GUTTMAN M, LESHYK VO, LIS JT, LOMVARDAS S, MIRNY LA, O’SHEA CC, PARK PJ, REN B, POLITZ JCR, SHENDURE J, ZHONG S & NETWORK DN 2017. The 4D nucleome project. Nature, 549, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FORNEROD M, OHNO M, YOSHIDA M & MATTAJ IW 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell, 90, 1051–60. [DOI] [PubMed] [Google Scholar]
- GARZON JI, DENG L, MURRAY D, SHAPIRA S, PETREY D & HONIG B 2016. A computational interactome and functional annotation for the human proteome. Elife, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GIURGIU M, REINHARD J, BRAUNER B, DUNGER-KALTENBACH I, FOBO G, FRISHMAN G, MONTRONE C & RUEPP A 2019. CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res, 47, D559–D563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GU L, LI C, AACH J, HILL DE, VIDAL M & CHURCH GM 2014. Multiplex single-molecule interaction profiling of DNA-barcoded proteins. Nature, 515, 554–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GULLBERG M, GUSTAFSDOTTIR SM, SCHALLMEINER E, JARVIUS J, BJARNEGARD M, BETSHOLTZ C, LANDEGREN U & FREDRIKSSON S 2004. Cytokine detection by antibody-based proximity ligation. Proc Natl Acad Sci U S A, 101, 8420–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAVUGIMANA PC, HART GT, NEPUSZ T, YANG H, TURINSKY AL, LI Z, WANG PI, BOUTZ DR, FONG V, PHANSE S, BABU M, CRAIG SA, HU P, WAN C, VLASBLOM J, DAR VU, BEZGINOV A, CLARK GW, WU GC, WODAK SJ, TILLIER ER, PACCANARO A, MARCOTTE EM & EMILI A 2012. A census of human soluble protein complexes. Cell, 150, 1068–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERMJAKOB H, MONTECCHI-PALAZZI L, LEWINGTON C, MUDALI S, KERRIEN S, ORCHARD S, VINGRON M, ROECHERT B, ROEPSTORFF P, VALENCIA A, MARGALIT H, ARMSTRONG J, BAIROCH A, CESARENI G, SHERMAN D & APWEILER R 2004. IntAct: an open source molecular interaction database. Nucleic Acids Res, 32, D452–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JÄKEL S & GÖRLICH D 1998. Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. Embo j, 17, 4491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JERBY-ARNON L, PFETZER N, WALDMAN YY, MCGARRY L, JAMES D, SHANKS E, SEASHORE-LUDLOW B, WEINSTOCK A, GEIGER T, CLEMONS PA, GOTTLIEB E & RUPPIN E 2014. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell, 158, 1199–1209. [DOI] [PubMed] [Google Scholar]
- KAWALIA A, MOTAMENY S, WONCZAK S, THIELE H, NIERODA L, JABBARI K, BOROWSKI S, SINHA V, GUNIA W, LANG U, ACHTER V & NURNBERG P 2015. Leveraging the power of high performance computing for next generation sequencing data analysis: tricks and twists from a high throughput exome workflow. PLoS One, 10, e0126321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOVACS IA, LUCK K, SPIROHN K, WANG Y, POLLIS C, SCHLABACH S, BIAN W, KIM DK, KISHORE N, HAO T, CALDERWOOD MA, VIDAL M & BARABASI AL 2019. Network-based prediction of protein interactions. Nat Commun, 10, 1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRISTENSEN AR, GSPONER J & FOSTER LJ 2012. A high-throughput approach for measuring temporal changes in the interactome. Nat Methods, 9, 907–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUKAR T, ECKENRODE S, GU Y, LIAN W, MEGGINSON M, SHE JX & WU D 2002. Protein microarrays to detect protein-protein interactions using red and green fluorescent proteins. Anal Biochem, 306, 50–4. [DOI] [PubMed] [Google Scholar]
- LEE JS, DAS A, JERBY-ARNON L, ARAFEH R, AUSLANDER N, DAVIDSON M, MCGARRY L, JAMES D, AMZALLAG A, PARK SG, CHENG K, ROBINSON W, ATIAS D, STOSSEL C, BUZHOR E, STEIN G, WATERFALL JJ, MELTZER PS, GOLAN T, HANNENHALLI S, GOTTLIEB E, BENES CH, SAMUELS Y, SHANKS E & RUPPIN E 2018. Harnessing synthetic lethality to predict the response to cancer treatment. Nat Commun, 9, 2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEWIS JD, WAN J, FORD R, GONG Y, FUNG P, NAHAL H, WANG PW, DESVEAUX D & GUTTMAN DS 2012. Quantitative Interactor Screening with next-generation Sequencing (QIS-Seq) identifies Arabidopsis thaliana MLO2 as a target of the Pseudomonas syringae type III effector HopZ2. BMC Genomics, 13, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI H 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv preprint arXiv:1303.3997 [Google Scholar]
- LI S, TIGHE SW, NICOLET CM, GROVE D, LEVY S, FARMERIE W, VIALE A, WRIGHT C, SCHWEITZER PA, GAO Y, KIM D, BOLAND J, HICKS B, KIM R, CHHANGAWALA S, JAFARI N, RAGHAVACHARI N, GANDARA J, GARCIA-REYERO N, HENDRICKSON C, ROBERSON D, ROSENFELD J, SMITH T, UNDERWOOD JG, WANG M, ZUMBO P, BALDWIN DA, GRILLS GS & MASON CE 2014. Multi-platform assessment of transcriptome profiling using RNA-seq in the ABRF next-generation sequencing study. Nat Biotechnol, 32, 915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI X, ZHOU B, CHEN L, GOU LT, LI H & FU XD 2017. GRID-seq reveals the global RNA-chromatin interactome. Nat Biotechnol, 35, 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LICATA L, BRIGANTI L, PELUSO D, PERFETTO L, IANNUCCELLI M, GALEOTA E, SACCO F, PALMA A, NARDOZZA AP, SANTONICO E, CASTAGNOLI L & CESARENI G 2012. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res, 40, D857–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIEVENS S, VANDERROOST N, VAN DER HEYDEN J, GESELLCHEN V, VIDAL M & TAVERNIER J 2009. Array MAPPIT: high-throughput interactome analysis in mammalian cells. J Proteome Res, 8, 877–86. [DOI] [PubMed] [Google Scholar]
- LU Z, GONG J & ZHANG QC 2018. PARIS: Psoralen Analysis of RNA Interactions and Structures with High Throughput and Resolution. Methods Mol Biol, 1649, 59–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUCK K, KIM DK, LAMBOURNE L, SPIROHN K, BEGG BE, BIAN W, BRIGNALL R, CAFARELLI T, CAMPOS-LABORIE FJ, CHARLOTEAUX B, CHOI D, COTE AG, DALEY M, DEIMLING S, DESBULEUX A, DRICOT A, GEBBIA M, HARDY MF, KISHORE N, KNAPP JJ, KOVACS IA, LEMMENS I, MEE MW, MELLOR JC, POLLIS C, PONS C, RICHARDSON AD, SCHLABACH S, TEEKING B, YADAV A, BABOR M, BALCHA D, BASHA O, BOWMAN-COLIN C, CHIN SF, CHOI SG, COLABELLA C, COPPIN G, D’AMATA C, DE RIDDER D, DE ROUCK S, DURAN-FRIGOLA M, ENNAJDAOUI H, GOEBELS F, GOEHRING L, GOPAL A, HADDAD G, HATCHI E, HELMY M, JACOB Y, KASSA Y, LANDINI S, LI R, VAN LIESHOUT N, MACWILLIAMS A, MARKEY D, PAULSON JN, RANGARAJAN S, RASLA J, RAYHAN A, ROLLAND T, SAN-MIGUEL A, SHEN Y, SHEYKHKARIMLI D, SHEYNKMAN GM, SIMONOVSKY E, TASAN M, TEJEDA A, TROPEPE V, TWIZERE JC, WANG Y, WEATHERITT RJ, WEILE J, XIA Y, YANG X, YEGER-LOTEM E, ZHONG Q, ALOY P, BADER GD, DE LAS RIVAS J, GAUDET S, HAO T, RAK J, TAVERNIER J, HILL DE, VIDAL M, ROTH FP & CALDERWOOD MA 2020. A reference map of the human binary protein interactome. Nature, 580, 402–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN M 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011, 17, 3. [Google Scholar]
- MATSUYAMA A, ARAI R, YASHIRODA Y, SHIRAI A, KAMATA A, SEKIDO S, KOBAYASHI Y, HASHIMOTO A, HAMAMOTO M, HIRAOKA Y, HORINOUCHI S & YOSHIDA M 2006. ORFeome cloning and global analysis of protein localization in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol, 24, 841–7. [DOI] [PubMed] [Google Scholar]
- MCGREGOR LM, JAIN T & LIU DR 2014. Identification of Ligand-Target Pairs from Combined Libraries of Small Molecules and Unpurified Protein Targets in Cell Lysates. 136, 3264–3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MELLACHERUVU D, WRIGHT Z, COUZENS AL, LAMBERT JP, ST-DENIS NA, LI T, MITEVA YV, HAURI S, SARDIU ME, LOW TY, HALIM VA, BAGSHAW RD, HUBNER NC, AL-HAKIM A, BOUCHARD A, FAUBERT D, FERMIN D, DUNHAM WH, GOUDREAULT M, LIN ZY, BADILLO BG, PAWSON T, DUROCHER D, COULOMBE B, AEBERSOLD R, SUPERTI-FURGA G, COLINGE J, HECK AJ, CHOI H, GSTAIGER M, MOHAMMED S, CRISTEA IM, BENNETT KL, WASHBURN MP, RAUGHT B, EWING RM, GINGRAS AC & NESVIZHSKII AI 2013. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat Methods, 10, 730–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MESSNER S, SCHUERMANN D, ALTMEYER M, KASSNER I, SCHMIDT D, SCHÄR P, MÜLLER S & HOTTIGER MO 2009. Sumoylation of poly(ADP-ribose) polymerase 1 inhibits its acetylation and restrains transcriptional coactivator function. Faseb j, 23, 3978–89. [DOI] [PubMed] [Google Scholar]
- NAVLAKHA S, HE X, FALOUTSOS C & BAR-JOSEPH Z 2014. Topological properties of robust biological and computational networks. J R Soc Interface, 11, 20140283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NGUYEN TC, CAO X, YU P, XIAO S, LU J, BIASE FH, SRIDHAR B, HUANG N, ZHANG K & ZHONG S 2016. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat Commun, 7, 12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’EARY NA, WRIGHT MW, BRISTER JR, CIUFO S, HADDAD D, MCVEIGH R, RAJPUT B, ROBBERTSE B, SMITHWHITE B, AKO-ADJEI D, ASTASHYN A, BADRETDIN A, BAO Y, BLINKOVA O, BROVER V, CHETVERNIN V, CHOI J, COX E, ERMOLAEVA O, FARRELL CM, GOLDFARB T, GUPTA T, HAFT D, HATCHER E, HLAVINA W, JOARDAR VS, KODALI VK, LI W, MAGLOTT D, MASTERSON P, MCGARVEY KM, MURPHY MR, O'NEILL K, PUJAR S, RANGWALA SH, RAUSCH D, RIDDICK LD, SCHOCH C, SHKEDA A, STORZ SS, SUN H, THIBAUD-NISSEN F, TOLSTOY I, TULLY RE, VATSAN AR, WALLIN C, WEBB D, WU W, LANDRUM MJ, KIMCHI A, TATUSOVA T, DICUCCIO M, KITTS P, MURPHY TD & PRUITT KD 2016. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res, 44, D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERI S, NAVARRO JD, AMANCHY R, KRISTIANSEN TZ, JONNALAGADDA CK, SURENDRANATH V, NIRANJAN V, MUTHUSAMY B, GANDHI TK, GRONBORG M, IBARROLA N, DESHPANDE N, SHANKER K, SHIVASHANKAR HN, RASHMI BP, RAMYA MA, ZHAO Z, CHANDRIKA KN, PADMA N, HARSHA HC, YATISH AJ, KAVITHA MP, MENEZES M, CHOUDHURY DR, SURESH S, GHOSH N, SARAVANA R, CHANDRAN S, KRISHNA S, JOY M, ANAND SK, MADAVAN V, JOSEPH A, WONG GW, SCHIEMANN WP, CONSTANTINESCU SN, HUANG L, KHOSRAVI-FAR R, STEEN H, TEWARI M, GHAFFARI S, BLOBE GC, DANG CV, GARCIA JG, PEVSNER J, JENSEN ON, ROEPSTORFF P, DESHPANDE KS, CHINNAIYAN AM, HAMOSH A, CHAKRAVARTI A & PANDEY A 2003. Development of human protein reference database as an initial platform for approaching systems biology in humans. Genome Res, 13, 2363–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PETALIDIS L, BHATTACHARYYA S, MORRIS GA, COLLINS VP, FREEMAN TC & LYONS PA 2003. Global amplification of mRNA by template-switching PCR: linearity and application to microarray analysis. Nucleic Acids Res, 31, e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBERTS RW & SZOSTAK JW 1997. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl AcadSci U S A, 94, 12297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROLLAND T, TASAN M, CHARLOTEAUX B, PEVZNER SJ, ZHONG Q, SAHNI N, Yl S, LEMMENS I, FONTANILLO C, MOSCA R, KAMBUROV A, GHIASSIAN SD, YANG X, GHAMSARI L, BALCHA D, BEGG BE, BRAUN P, BREHME M, BROLY MP, CARVUNIS AR, CONVERY-ZUPAN D, COROMINAS R, COULOMBE-HUNTINGTON J, DANN E, DREZE M, DRICOT A, FAN C, FRANZOSA E, GEBREAB F, GUTIERREZ BJ, HARDY MF, JIN M, KANG S, KIROS R, LIN GN, LUCK K, MACWILLIAMS A, MENCHE J, MURRAY RR, PALAGI A, POULIN MM, RAMBOUT X, RASLA J, REICHERT P, ROMERO V, RUYSSINCK E, SAHALIE JM, SCHOLZ A, SHAH AA, SHARMA A, SHEN Y, SPIROHN K, TAM S, TEJEDA AO, TRIGG SA, TWIZERE JC, VEGA K, WALSH J, CUSICK ME, XIA Y, BARABASI AL, IAKOUCHEVA LM, ALOY P, DE LAS RIVAS J, TAVERNIER J, CALDERWOOD MA, HILL DE, HAO T, ROTH FP & VIDAL M 2014. A proteome-scale map of the human interactome network. Cell, 159, 1212–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUAL JF, VENKATESAN K, HAO T, HIROZANE-KISHIKAWA T, DRICOT A, LI N, BERRIZ GF, GIBBONS FD, DREZE M, AYIVI-GUEDEHOUSSOU N, KLITGORD N, SIMON C, BOXEM M, MILSTEIN S, ROSENBERG J, GOLDBERG DS, ZHANG LV, WONG SL, FRANKLIN G, LI S, ALBALA JS, LIM J, FRAUGHTON C, LLAMOSAS E, CEVIK S, BEX C, LAMESCH P, SIKORSKI RS, VANDENHAUTE J, ZOGHBI HY, SMOLYAR A, BOSAK S, SEQUERRA R, DOUCETTE-STAMM L, CUSICK ME, HILL DE, ROTH FP & VIDAL M 2005. Towards a proteome-scale map of the human protein-protein interaction network. Nature, 437, 1173–8. [DOI] [PubMed] [Google Scholar]
- SAITO T & REHMSMEIER M 2015. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One, 10, e0118432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEELIG B 2011. mRNA display for the selection and evolution of enzymes from in vitro-translated protein libraries. Nat Protoc, 6, 540–52. [DOI] [PubMed] [Google Scholar]
- SHANNON P, MARKIEL A, OZIER O, BALIGA NS, WANG JT, RAMAGE D, AMIN N, SCHWIKOWSKI B & IDEKER T 2003. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res, 13, 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARMA E, STERNE-WEILER T, O’HANLON D & BLENCOWE BJ 2016. Global Mapping of Human RNA-RNA Interactions. Mol Cell, 62, 618–26. [DOI] [PubMed] [Google Scholar]
- SODERBERG O, GULLBERG M, JARVIUS M, RIDDERSTRALE K, LEUCHOWIUS KJ, JARVIUS J, WESTER K, HYDBRING P, BAHRAM F, LARSSON LG & LANDEGREN U 2006. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat Methods, 3, 995–1000. [DOI] [PubMed] [Google Scholar]
- SRIDHAR B, RIVAS-ASTROZA M, NGUYEN TC, CHEN W, YAN Z, CAO X, HEBERT L & ZHONG S 2017. Systematic Mapping of RNA-Chromatin Interactions In Vivo. Curr Biol, 27, 610–612. [DOI] [PubMed] [Google Scholar]
- STARK C, BREITKREUTZ BJ, REGULY T, BOUCHER L, BREITKREUTZ A & TYERS M 2006. BioGRID: a general repository for interaction datasets. Nucleic Acids Res, 34, D535–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SU Z, ŁABAJ PP, LI S, THIERRY-MIEG J, THIERRY-MIEG D, SHI W, WANG C, SCHROTH GP, SETTERQUIST RA, THOMPSON JF, JONES WD, XIAO W, XU W, JENSEN RV, KELLY R, XU J, CONESA A, FURLANELLO C, GAO H, HONG H, JAFARI N, LETOVSKY S, LIAO Y, LU F, OAKELEY EJ, PENG Z, PRAUL CA, SANTOYO-LOPEZ J, SCHERER A, SHI T, SMYTH GK, STAEDTLER F, SYKACEK P, TAN X-X, THOMPSON EA, VANDESOMPELE J, WANG MD, WANG J, WOLFINGER RD, ZAVADIL J, AUERBACH SS, BAO W, BINDER H, BLOMQUIST T, BRILLIANT MH, BUSHEL PR, CAI W, CATALANO JG, CHANG C-W, CHEN T, CHEN G, CHEN R, CHIERICI M, CHU T-M, CLEVERT D-A, DENG Y, DERTI A, DEVANARAYAN V, DONG Z, DOPAZO J, DU T, FANG H, FANG Y, FASOLD M, FERNANDEZ A, FISCHER M, FURIÓ-TARI P, FUSCOE JC, CAI MET F, GAJ S, GANDARA J, GAO H, GE W, GONDO Y, GONG B, GONG M, GONG Z, GREEN B, GUO C, GUO L, GUO L-W, HADFIELD J, HELLEMANS J, HOCHREITER S, JIA M, JIAN M, JOHNSON CD, KAY S, KLEINJANS J, LABABIDI S, LEVY S, LI Q-Z, LI L, LI L, LI P, LI Y, LI H, LI J, LI S, LIN SM., et al. 2014. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium. Nature Biotechnology, 32, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOUCHETTE ΜH, VAN VLACK ER, BAI L, KIM J, COGNETTA AB 3RD, PREVITI ML, BACKUS KM, MARTIN DW, CRAVATT BF & SEELIGER JC. 2017. A Screen for Protein-Protein Interactions in Live Mycobacteria Reveals a Functional Link between the Virulence-Associated Lipid Transporter LprG and the Mycolyltransferase Antigen 85A. ACS Infect Dis, 3, 336–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TRIGG SA, GARZA RM, MACWILLIAMS A, NERY JR, BARTLETT A, CASTANON R, GOUBIL A, FEENEY J, O’MALLEY R, HUANG S-SC, ZHANG ZZ, GALLI M & ECKER JR 2017. CrY2H-seq: a massively multiplexed assay for deep-coverage interactome mapping. Nature Methods, 14, 819–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VENKATESAN K, RUAL JF, VAZQUEZ A, STELZL U, LEMMENS I, HIROZANE-KISHIKAWA T, HAO T, ZENKNER M, XIN X, GOH KI, YILDIRIM MA, SIMONIS N, HEINZMANN K, GEBREAB F, SAHALIE JM, CEVIK S, SIMON C, DE SMET AS, DANN E, SMOLYAR A, VINAYAGAM A, YU H, SZETO D, BORICK H, DRICOT A, KLITGORD N, MURRAY RR, LIN C, LALOWSKI M, TIMM J, RAU K, BOONE C, BRAUN P, CUSICK ME, ROTH FP, HILL DE, TAVERNIER J, WANKER EE, BARABASI AL & VIDAL M 2009. An empirical framework for binary interactome mapping. Nat Methods, 6, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VERMEULEN M, HUBNER NC & MANN M 2008. High confidence determination of specific protein-protein interactions using quantitative mass spectrometry. Curr Opin Biotechnol, 19, 331–7. [DOI] [PubMed] [Google Scholar]
- WALHOUT AJ & VIDAL M 2001. High-throughput yeast two-hybrid assays for large-scale protein interaction mapping. Methods, 24, 297–306. [DOI] [PubMed] [Google Scholar]
- WAN C, BORGESON B, PHANSE S, TU F, DREW K, CLARK G, XIONG X, KAGAN O, KWAN J, BEZGINOV A, CHESSMAN K, PAL S, CROMAR G, PAPOULAS O, NI Z, BOUTZ DR, STOILOVA S, HAVUGIMANA PC, GUO X, MALTY RH, SAROV M, GREENBLATT J, BABU M, DERRY WB, TILLIER ER, WALLINGFORD JB, PARKINSON J, MARCOTTE EM & EMILI A 2015. Panorama of ancient metazoan macromolecular complexes. Nature, 525, 339–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XENARIOS I, RICE DW, SALWINSKI L, BARON MK, MARCOTTE EM & EISENBERG D 2000. DIP: the database of interacting proteins. Nucleic Acids Res, 28, 289–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YACHIE N, PETSALAKI E, MELLOR JC, WEILE J, JACOB Y, VERBY M, OZTURK SB, LI S, COTE AG, MOSCA R, KNAPP JJ, KO M, YU A, GEBBIA M, SAHNI N, YI S, TYAGI T, SHEYKHKARIMLI D, ROTH JF, WONG C, MUSA L, SNIDER J, LIU YC, YU H, BRAUN P, STAGLJAR I, HAO T, CALDERWOOD MA, PELLETIER L, ALOY P, HILL DE, VIDAL M & ROTH FP 2016. Pooled-matrix protein interaction screens using Barcode Fusion Genetics. 12, 863–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAN Z, HUANG N, WU W, CHEN W, JIANG Y, CHEN J, HUANG X, WEN X, XU J, JIN Q, ZHANG K, CHEN Z, CHIEN S & ZHONG S 2019. Genome-wide colocalization of RNA-DNA interactions and fusion RNA pairs. Proc Natl Acad Sci U S A, 116, 3328–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG F, LEI Y, ZHOU M, YAO Q, HAN Y, WU X, ZHONG W, ZHU C, XU W, TAO R, CHEN X, LIN D, RAHMAN K, TYAGI R, HABIB Z, XIAO S, WANG D, YU Y, CHEN H, FU Z & CAO G 2018. Development and application of a recombination-based library versus library high- throughput yeast two-hybrid (RLL-Y2H) screening system. Nucleic Acids Res, 46, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG X, BOEHM JS, YANG X, SALEHI-ASHTIANI K, HAO T, SHEN Y, LUBONJA R, THOMAS SR, ALKAN O, BHIMDI T, GREEN TM, JOHANNESSEN CM, SILVER SJ, NGUYEN C, MURRAY RR, HIERONYMUS H, BALCHA D, FAN C, LIN C, GHAMSARI L, VIDAL M, HAHN WC, HILL DE & ROOT DE 2011. A public genome-scale lentiviral expression library of human ORFs. Nat Methods, 8, 659–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU H, BRAUN P, YILDIRIM MA, LEMMENS I, VENKATESAN K, SAHALIE J, HIROZANE-KISHIKAWA T, GEBREAB F, LI N, SIMONIS N, HAO T, RUAL JF, DRICOT A, VAZQUEZ A, MURRAY RR, SIMON C, TARDIVO L, TAM S, SVRZIKAPA N, FAN C, DE SMET AS, MOTYL A, HUDSON ME, PARK J, XIN X, CUSICK ME, MOORE T, BOONE C, SNYDER M, ROTH FP, BARABASI AL, TAVERNIER J, HILL DE & VIDAL M 2008. High-quality binary protein interaction map of the yeast interactome network. Science, 322, 104–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU M, YANG W, NI T, TANG Z, NAKADAI T, ZHU J & ROEDER RG 2015. RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science, 350, 1383–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG QC, PETREY D, DENG L, QIANG L, SHI Y, THU CA, BISIKIRSKA B, LEFEBVRE C, ACCILI D, HUNTER T, MANIATIS T, CALIFANO A & HONIG B 2012. Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature, 490, 556–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG QC, PETREY D, GARZON JI, DENG L & HONIG B 2013. prePPI: a structure-informed database of protein-protein interactions. Nucleic Acids Res, 41, D828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Y, KU WL, LIU S, CUI K, JIN W, TANG Q, LU W, NI B & ZHAO K 2017. Genome-wide identification of histone H2A and histone variant H2A.Z-interacting proteins by bPPI-seq. Cell Res, 27, 1258–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHU YY, MACHLEDER EM, CHENCHIK A, LI R & SIEBERT PD 2001. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques, 30, 892–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been uploaded to GEO with accession number: GSE150818.
PROPERseqTools is available at DOI: https://doi.org/10.5281/zenodo.5009171. PROPER v.1.0 database is at https://genemo.ucsd.edu/proper.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.