

SUMMARY
BRCA1/2-mutant tumor cells display an elevated mutation burden, the etiology of which remains unclear. Here, we report that these cells accumulate ssDNA gaps and spontaneous mutations during unperturbed DNA replication due to repriming by the DNA primase-polymerase PRIMPOL. Gap accumulation requires the DNA glycosylase SMUG1 and is exacerbated by depletion of the translesion synthesis (TLS) factor RAD18 or inhibition of the error-prone TLS polymerase complex REV1-Polζ by the small molecule JH-RE-06. JH-RE-06 treatment of BRCA1/2-deficient cells results in reduced mutation rates and PRIMPOL- and SMUG1-dependent loss of viability. Through cellular and animal studies, we demonstrate that JH-RE-06 is preferentially toxic towards HR-deficient cancer cells. Furthermore, JH-RE-06 remains effective towards PARP inhibitor (PARPi)-resistant BRCA1-mutant cells and displays additive toxicity with crosslinking agents or PARPi. Collectively, these studies identify a protective and mutagenic role for REV1-Polζ in BRCA1/2-mutant cells and provide the rationale for using REV1-Polζ inhibitors to treat BRCA1/2-mutant tumors.
Keywords: Homologous recombination, ssDNA gaps, BRCA1 and BRCA2, translesion synthesis, RAD18, REV1 and Polζ, DNA repriming, PRIMPOL, breast and ovarian cancer, synthetic lethality
Graphical Abstract
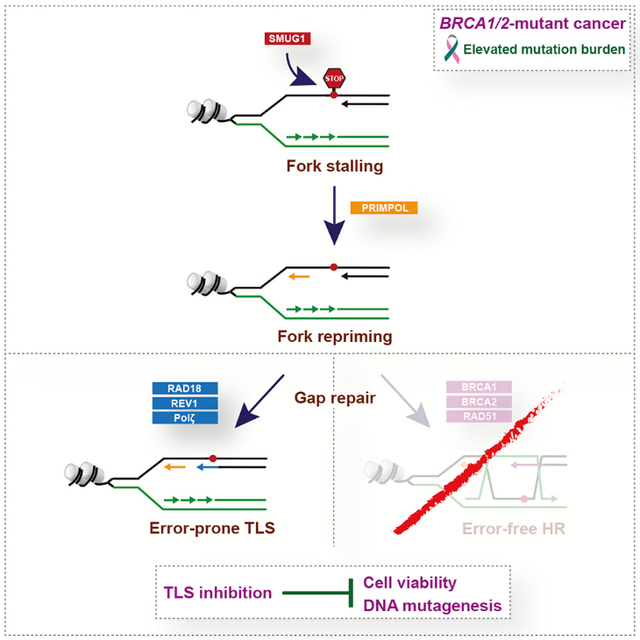
eTOC
Taglialatela et al. report that homologous recombination (HR)-deficient cancer cells, like BRCA1/2-mutant cells, display increased reliance on error-prone translesion synthesis (TLS) for the repair of ssDNA gaps arising spontaneously during DNA replication. TLS inhibition shows exquisite toxicity in BRCA1/2-deficient cancer cells, providing the basis for alternative therapies against BRCA1/2-mutant tumors.
INTRODUCTION
Germline mutations of BRCA1 and BRCA2 (BRCA1/2) are the most common genetic lesions associated with familial breast and ovarian cancer (Apostolou and Fostira, 2013). BRCA1/2 act to preserve genome integrity through multiple functions, including the repair of DNA double-strand breaks (DSBs) by homologous recombination (HR) (Moynahan et al., 1999; Moynahan et al., 2001) and the protection of stalled replication forks (Schlacher et al., 2011; Schlacher et al., 2012). Given their defects in preserving genome stability, BRCA1/2-deficient cancer cells are exquisitely sensitive to DNA damaging agents, such as DNA crosslinking agents (e.g., cisplatin) and PARP inhibitors (Bryant et al., 2005; Farmer et al., 2005; Lord and Ashworth, 2016). However, with time BRCA1/2-mutant cancers acquire resistance to these treatments (Gogola et al., 2019). Developing alternative targeted therapies for BRCA1/2-mutant cancer patients is therefore of paramount importance.
DNA replication stress is one of the main sources of DNA damage in BRCA1/2-mutant cells (Feng and Jasin, 2017; Pathania et al., 2014). Replication-associated DNA damage induces the monoubiquitination of PCNA on lysine 164 (K164) by RAD18/RAD6, resulting in the binding of translesion synthesis (TLS) DNA polymerases to PCNA (Ulrich and Walden, 2010). TLS polymerases enable the bypass of replication fork-stalling lesions, albeit in a potentially error-prone manner, given their low fidelity (Yang and Gao, 2018). TLS-mediated lesion bypass is thought to occur in two steps involving the insertion of a DNA base opposite the lesion by Y-family DNA polymerases (Polη, Polι, Polκ and REV1), followed by the extension of DNA synthesis by the B-family Polζ complex (REV3L/REV7/POLD2/POLD3) (Vaisman and Woodgate, 2017; Yang and Gao, 2018). Polζ can synthesize up to 1,000 nucleotides (Kochenova et al., 2015) with ~100-fold less accuracy than the replicative polymerases Polδ and Polɛ (Zhong et al., 2006). Coordination of TLS activities is mediated by REV1, which acts as a TLS scaffold protein that binds PCNA through its N-terminal BRCT domain (de Groote et al., 2011; Guo et al., 2006) and Polη, Polι and Polκ or the REV7 subunit of the Polζ complex through distinct C-terminal interaction surfaces (Liu et al., 2013; Pozhidaeva et al., 2012; Pustovalova et al., 2016). REV1 can also incorporate dCMP opposite to damaged bases and abasic sites (Kim et al., 2011; Nelson et al., 1996; Weerasooriya et al., 2014).
In addition to restarting DNA synthesis at stalled forks, TLS polymerases also fill in single-stranded DNA (ssDNA) gaps remaining after DNA replication (Gallo et al., 2019; Hashimoto et al., 2010; Lopes et al., 2006). Post-replicative repair of ssDNA gaps can also occur through HR-mediated processes, such as template switching, a pathway dependent on PCNA polyubiquitination that utilizes the sister chromatid as a template for DNA synthesis (Branzei and Szakal, 2017; Garcia-Rodriguez et al., 2016; Hoege et al., 2002). Growing evidence indicates that TLS and HR can act as alternative compensatory processes for ssDNA gap repair. In bacteria, the RecA recombinase promotes ssDNA gap repair (Rupp et al., 1971; West et al., 1982) and defective HR leads to increased TLS (Fujii et al., 2018; Naiman et al., 2016). Furthermore, yeast TLS mutants accumulate ssDNA gaps upon UV radiation that require HR for repair (Lopes et al., 2006; Ma et al., 2013), and TLS impairment in RAD51-depleted Xenopus egg extracts leads to enhanced accumulation of ssDNA gaps (Hashimoto et al., 2010). Additionally, combinatorial inactivation of RAD18 and the HR factor RAD54 induces synthetic lethality in chicken B cells (Yamashita et al., 2002), while simultaneous deficiency of TLS and HR factors causes additive sensitivity to replication stress-inducing agents in human cancer cells (Thakar et al., 2020; Villafanez et al., 2019; Yang et al., 2017). Interestingly, BRCA1-deficient human cells exhibit elevated PCNA ubiquitination upon DNA damage (Pathania et al., 2011) and BRCA1/2-deficient tumors display increased rates of base substitutions (Alexandrov et al., 2013; Lal et al., 2019; Nik-Zainal et al., 2012; Nik-Zainal et al., 2016). Nonetheless, it remains to be determined whether BRCA1/2-deficient cancer cells are inherently addicted to TLS-mediated ssDNA gap repair for their normal growth and whether TLS inhibition could serve as a targeted therapy for BRCA1/2-mutant cancer patients.
In this study, we report that BRCA1-deficient cancer cells accumulate spontaneous ssDNA gaps that are repaired by the TLS factors RAD18 and REV1. We show that loss of RAD18 or REV1 results in synthetic lethality with BRCA1/2 deficiency and that inhibition of REV1-Polζ using the small-molecule JH-RE-06 (Wojtaszek et al., 2019) reduces the rate of spontaneous mutagenesis in BRCA1/2-deficient cells and induces cytotoxicity in BRCA1-mutant cancer cells in vitro and in vivo. Finally, we demonstrate that the ssDNA gap accumulation and cell lethality observed in BRCA1/2-deficient cells upon RAD18 loss or JH-RE-06 treatment require both the DNA glycosylase SMUG1 and DNA repriming by the DNA primase-polymerase PRIMPOL. These studies uncover a dependency on REV1-Polζ in BRCA1/2-mutant cancer cells and establish REV1-Polζ inhibition as a potential treatment for BRCA1/2-mutant tumors.
RESULTS
BRCA1/2-deficient cells depend on RAD18 for their viability
To investigate the nature of the spontaneous DNA damage induced by BRCA1 deficiency, we performed electron microscopy (EM) studies on DNA replication structures in BRCA1-mutant HCC1937 breast cancer cells. This analysis revealed the presence of ssDNA at replication fork junctions and internal ssDNA gaps behind the fork junctions (Figure 1A–B), which were suppressed upon expression of BRCA1 cDNA (Figures 1A–B and S1A), in line with the role of HR proteins in promoting post-replicative repair of ssDNA gaps (Hashimoto et al., 2010).
FIGURE 1. Replication fork gaps and cellular viability in BRCA1/2-deficient cells following RAD18 loss.

A) Representative image of a replication fork detected by EM in BRCA1-mutant HCC1937 cells. ssDNA gaps at the fork junction and internal gaps are indicated by brown and green arrows, respectively.
B) Fold change in the percentage of replication forks with the indicated number of internal ssDNA gaps in HCC1937 cells relative to BRCA1-reconstituted HCC1937 cells. Values of individual experiments are indicated as dots. Columns represent the mean ± SEM of independent biological replicates (n = 3). Statistical analysis was conducted by unpaired t-test (*p < 0.05).
C) BRCA1, RAD18, PCNA and ubiquitinated PCNA (PCNA-Ub) levels in WT and RAD18 KO (#1, #2) U2OS cells transfected with the indicated siRNAs, as detected by western blot. Vinculin is shown as control.
D) Viability of WT and RAD18 KO (#1, #2) U2OS cells at day 5 and 10 after treatment with control or BRCA1 siRNA. Cell viability is represented as a percentage of viable BRCA1-depleted cells relative to control siRNA-treated cells. Values of individual experiments are indicated as dots. Columns represent the mean ± SEM of independent biological replicates (n ≥ 3). Statistical analysis was conducted by unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.001).
E) BRCA2 and RAD18 levels in WT and RAD18 KO (#1, #2) U2OS cells transfected with the indicated siRNAs, as detected by western blot. Vinculin is shown as control.
F and H) Viability of WT and RAD18 KO (#1, #2) U2OS cells after treatment with the indicated siRNAs. Graphical representation and statistical analysis were conducted as in (D) (n = 3).
G) Percentage of apoptotic cells determined by propidium iodide/Annexin V staining and flow cytometry after transfection of the indicated siRNAs. Graphical representation and statistical analysis were conducted as in (D) (n = 3).
To examine whether HR-deficient cells are specifically dependent on alternative pathways for post-replicative repair of ssDNA gaps, U2OS cells were subjected to siRNA-mediated depletion of BRCA1 and/or RAD18, a ubiquitin ligase that promotes post-replication repair by TLS and template switching (Garcia-Rodriguez et al., 2016) (Figure S1B). Notably, combined depletion of BRCA1 and RAD18 synergistically reduced the viability of U2OS cells under untreated conditions (Figure S1B–C). To further investigate this phenotype, we used CRISPR-Cas9 to generate RAD18 knockout U2OS cells (RAD18 KO #1 and #2) (Figure 1C). In line with the above observations, siRNA-mediated BRCA1 or BRCA2 depletion led to a 2–3 fold reduction in the viability of RAD18 KO cells relative to WT cells (Figure 1C–F). Likewise, CRISPR-mediated disruption of BRCA1 or BRCA2 decreased the viability of RAD18 KO cells 2-fold relative to WT cells (Figure S1D–E). Importantly, reconstitution with RAD18 cDNA suppressed the sensitivity of RAD18 KO cells to BRCA1 depletion, excluding the possibility of clonal effects and confirming the specificity of our findings (Figure S1F–G). Propidium iodide and Annexin V staining revealed a 2–3-fold increase in the percentage of apoptotic cells in BRCA1/2-depleted RAD18 KO, but not WT, cells (Figures 1G and S1H), indicating that the simultaneous loss of BRCA1/2 and RAD18 induces cell death by apoptosis. Notably, 53BP1 depletion rescued the viability defect of RAD18 KO cells caused by depletion of BRCA1 but not BRCA2 (Figures 1H and S1I), in line with the known role of 53BP1 loss in restoring HR in BRCA1-deficient, but not BRCA2-deficient, cells (Bouwman et al., 2010; Bunting et al., 2010). Together, these findings identify a synthetic lethal interaction between BRCA1/2 deficiency and RAD18 loss in cancer cells under unperturbed conditions.
RAD18 loss exacerbates the formation of DNA damage and accumulation of unrepaired ssDNA in BRCA1-deficient cells
To elucidate the mechanisms responsible for the aforementioned synthetic lethal interaction, we measured the levels of spontaneous DNA damage that occur during unperturbed DNA replication in WT and RAD18 KO cells following BRCA1 depletion. As shown in Figures 2A–B and S2A, BRCA1 depletion caused a 2-fold increase in the percentage of γH2AX foci-positive cells, which was further enhanced by ~2-fold following RAD18 loss. In addition, BRCA1 depletion led to a 2-fold increase in the formation of 53BP1 nuclear bodies (NBs), a marker of G1-phase DNA lesions resulting from under-replicated DNA (Lukas et al., 2011; Spies et al., 2019), in RAD18-depleted or RAD18 KO cells relative to control cells (Figures 2C–D and S2B). These studies indicate that RAD18 loss induces DNA damage in BRCA1-deficient cells.
FIGURE 2. Analysis of DNA damage induced by RAD18 loss in BRCA1/2-deficient cells.

A) Representative images of γH2AX foci (green) with and without DAPI staining (blue) in WT and RAD18 KO U2OS cells after treatment with the indicated siRNAs. Scale bar = 10 μm.
B) Percentage of cyclin A-positive cells with >5 γH2AX foci per nucleus in WT and RAD18 KO U2OS cells treated with the indicated siRNAs. Values of individual experiments are indicated as dots. Columns represent the mean ± SEM of independent biological replicates (n = 3). Statistical analysis was conducted by unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.001).
C) Representative images of cyclin A (red) and 53BP1 (green) staining with and without merge with DAPI staining (blue) in WT and RAD18 KO U2OS cells after treatment with the indicated siRNAs. Scale bar = 10 μm.
D) Percentage of cyclin A-negative cells with >3 53BP1 nuclear bodies (NBs) per nucleus in WT and RAD18 KO U2OS cells treated with the indicated siRNAs. Graphical representation and statistical analysis were conducted as in (B).
E) Representative images of CldU staining under non-denaturing conditions (red) with DAPI staining (blue) in WT and RAD18 KO U2OS cells after treatment with the indicated siRNAs. Scale bar = 10 μm.
F) Percentage of cyclin A-positive cells with >3 CldU foci per nucleus in WT and RAD18 KO U2OS cells treated with the indicated siRNAs. Graphical representation and statistical analysis were conducted as in (B).
G) Schematic of the IdU/CldU pulse-labeling protocol followed by S1 nuclease treatment (top). Dot plot and median of CldU tract lengths (μm) in RAD18 KO cells per indicated experimental condition (bottom). P-values were calculated by Mann-Whitney test (*p < 0.05, ***p < 0.001, ****p < 0.0001).
Based on the above observations, we reasoned that RAD18 may exert its protective role in HR-deficient cells by promoting post-replicative repair of ssDNA gaps formed during unperturbed DNA replication. In support of this hypothesis, BRCA1 or BRCA2 depletion induced a significant increase in the number of RAD18 foci in S/G2-phase cells (Figure S2C–D). To assess the presence of ssDNA in cells deficient for BRCA1 and RAD18, WT and RAD18 KO U2OS cells were incubated with 5-chloro-2′-deoxyuridine (CldU) for 48 hr and then immunostained for CldU under non-denaturing conditions. As shown in Figure 2E–F, combined loss of BRCA1 and RAD18 led to a >2-fold increase in CldU foci-positive cells relative to BRCA1- or RAD18-deficient cells, indicating that RAD18 loss induces the accumulation of ssDNA in BRCA1-deficient cells. To monitor the presence of ssDNA gaps on newly synthesized DNA, actively replicating forks in the above cells were labeled with sequential pulses of iodo-2′-deoxyuridine (IdU) and CldU, and then incubated in the presence or absence of the S1 nuclease, which shortens the length of ssDNA-containing fibers by introducing nicks on ssDNA gaps (Quinet et al., 2017; Quinet et al., 2016). As shown in Figures 2G and S2E, siRNA-mediated BRCA1 depletion induced S1 nuclease-dependent shortening of DNA fiber lengths in RAD18 KO cells, revealing that simultaneous loss of BRCA1 and RAD18 results in increased accumulation of ssDNA gaps during DNA replication.
REV1 loss is synthetic lethal with BRCA1/2 deficiency
To determine whether the synthetic lethality induced by RAD18 loss in BRCA1/2-deficient cells is due to TLS deficiency, we investigated the function of REV1, a master regulator of TLS (Vaisman and Woodgate, 2017), by generating REV1 knockout (REV1 KO) and REV1/RAD18 double-knockout U2OS cells (REV1/RAD18 KO) (Figure S2F). Notably, REV1 KO cells, similar to RAD18 KO and REV1/RAD18 KO cells, displayed a >2-fold increase in sensitivity to BRCA1/2 depletion relative to WT cells (Figure 3A), indicating that REV1 and RAD18 act epistatically in maintaining the viability of BRCA1/2-deficient cancer cells.
FIGURE 3. Cellular viability and ssDNA gap formation in BRCA1/2-deficient cells following loss of RAD18, REV1 and/or PRIMPOL.

A) Viability of WT, REV1 KO, RAD18 KO and REV1/RAD18 KO U2OS cells after treatment with the indicated siRNAs. Cell viability is represented as a percentage of viable cells relative to control siRNA-treated cells. Values of individual experiments are indicated as dots. Columns represent the mean ± SEM of independent biological replicates (n = 3). Statistical analysis was conducted by unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
B) Viability of UWB1.289 and MDA-MB-436 cells with and without BRCA1 re-expression after transfection of Cas9 protein in complex with the indicated sgRNAs. Cell viability is represented as a percentage of viable cells relative to control sgRNA-treated cells (n = 4). Graphical representation and statistical analysis were conducted as in (A).
C-D) Schematic of the IdU/CldU pulse-labeling protocol with S1 nuclease treatment (top). Dot plot and median of CldU tract lengths (μm) in PRIMPOL KO cells complemented or not with PRIMPOL WT or mutant cDNAs and subjected to the indicated experimental conditions (bottom). Doxycycline (DOX) induces the expression of Cas9 in cells carrying Cas9 and RAD18 sgRNA constructs. P-values were calculated by Mann-Whitney test (****p < 0.0001).
E) Viability of WT and PRIMPOL KO U2OS cells transfected with the indicated sgRNAs in complex with Cas9 after treatment with the indicated siRNAs. Cell viability is represented as a percentage of viable cells relative to control siRNA-treated cells (n = 4). Graphical representation and statistical analysis were conducted as in (A).
F) Viability of PRIMPOL KO U2OS cells with and without complementation with PRIMPOL WT or mutant cDNAs after treatments with the indicated siRNAs and DOX, as in (C) and (D). Graphical representation and statistical analysis were conducted as in (A).
To determine the relevance of our findings for BRCA1-mutant cancers, we deleted RAD18 and/or REV1 in isogenic pairs of BRCA1-deficient and BRCA1-reconstituted ovarian and breast cancer cell lines. In line with our results in U2OS cells, CRISPR-mediated disruption of RAD18 or REV1 caused a 5-fold reduction in the viability of BRCA1-mutant UWB1.289 ovarian cancer cells (Figures 3B and S2G), which was not further enhanced by combined disruption of RAD18 and REV1 (Figures 3B and S2G). Notably, we observed only a moderate reduction (1.2–1.6 fold) in the viability of BRCA1-reconstituted UWB1.289 cells following RAD18 and/or REV1 loss, confirming the selectivity of the observed phenotype for HR-deficient cells (Figures 3B and S2G). Accordingly, propidium iodide and Annexin V staining revealed a greater increase in apoptotic cells induced by RAD18 and/or REV1 loss in UWB1.289 cells relative to their BRCA1-reconstituted counterparts (Figure S2I–J). Similar results were obtained upon RAD18 and/or REV1 loss in BRCA1-deficient and BRCA1-reconstituted MDA-MB-436 breast carcinoma cells (Figures 3B and S2H). Thus, BRCA1 deficiency generates a dependency on RAD18 and REV1 in breast and ovarian cancer cells.
The synthetic lethal phenotype induced by simultaneous loss of BRCA1/2 and RAD18 is caused by PRIMPOL-dependent ssDNA gap formation
The above observations indicate that ssDNA gaps formed during unperturbed DNA replication in BRCA1/2-deficient cells require RAD18 and REV1 for efficient repair. ssDNA gap formation can result from PRIMPOL-dependent repriming of DNA synthesis downstream of replication fork-blocking lesions (Bai et al., 2020; Bianchi et al., 2013; Garcia-Gomez et al., 2013; Mouron et al., 2013; Quinet et al., 2020; Wan et al., 2013). To determine whether PRIMPOL generates the ssDNA gaps that require TLS-mediated repair in HR-deficient cells, we used the S1 nuclease to measure gap formation in PRIMPOL KO U2OS cells. Remarkably, PRIMPOL deficiency prevented the accumulation of ssDNA gaps induced by combined loss of BRCA1/2 and RAD18 (Figures 3C and S3A–B), and gap formation was restored by re-expression of WT PRIMPOL, but not mutants deficient in its DNA primase (CH) or DNA primase/polymerase (AxA) activities (Mouron et al., 2013) (Figure 3C–D and S3A). Moreover, the number of RAD18 foci induced by BRCA1/2 depletion was significantly reduced in PRIMPOL KO cells (Figure S3C). PRIMPOL deficiency also suppressed the G2/M arrest and loss of viability induced by the combination of RAD18/REV1 disruption and BRCA1/2 depletion (Figures 3E and S3D–F), and the observed synthetic lethal phenotype was re-established upon re-expression of WT PRIMPOL, but not the CH or AxA PRIMPOL mutants (Figure 3F). Together, these findings indicate that ssDNA gaps generated by PRIMPOL-mediated repriming are repaired by RAD18 and REV1 to maintain the viability of BRCA1/2-deficient cells.
The REV1-Polζ inhibitor JH-RE-06 shows preferential cytotoxicity towards BRCA1-deficient cancer cells
Based on the data described above, small molecules that target RAD18, REV1 or the REV1-associated TLS polymerases may be preferentially toxic to BRCA1/2-deficient cells. To test this hypothesis, we measured the effects of the REV1-Polζ inhibitor JH-RE-06 (Wojtaszek et al., 2019) on a panel of breast and ovarian tumor lines. Remarkably, the BRCA1-deficient cancer cell lines (i.e., MDA-MB-436, UWB1.289, HCC1937) displayed an increased dose-dependent sensitivity to JH-RE-06 (Figure 4A) and a >4-fold reduction of its EC50 relative to BRCA1-proficient cells (Figure 4B). Moreover, BRCA1-reconstituted MDA-MB-436 and UWB1.289 cells exhibited resistance to JH-RE-06 (Figure 4C–D), confirming that JH-RE-06 sensitivity is dependent on BRCA1 loss. Of particular importance, JH-RE-06 showed additive toxicity with either the PARP inhibitor (PARPi) olaparib or the DNA-crosslinking agent cisplatin in BRCA1-mutant cells (i.e., MDA-MB-436 and UWB1.289 cells) (Figures 4F–G and S4A–D). Interestingly, HCC1937 cells, which have acquired PARPi resistance despite remaining HR deficient (Du et al., 2016; Zhang et al., 2004) (Figure S4E), as confirmed by their inability to efficiently form RAD51 foci after DNA damage (Figure S4F), displayed marked sensitivity to JH-RE-06 (Figure 4E). In line with our findings in U2OS cells (Figure 1H), 53BP1 depletion promoted JH-RE-06 resistance in HCC1937 cells (Figure S4G–I), confirming that restoration of HR in BRCA1-mutant cells suppresses the toxicity of JH-RE-06. These findings indicate that JH-RE-06 is a potent cytotoxic agent for BRCA1-deficient cancer cells, which can be employed to target HR-deficient but PARPi-resistant BRCA1-mutant cancer cells or combined with cisplatin and PARPi to further improve their therapeutic efficacy.
FIGURE 4. Survival analysis in BRCA1-proficient and BRCA1-deficient cancer cells upon treatment with the REV1-Polζ inhibitor JH-RE-06.
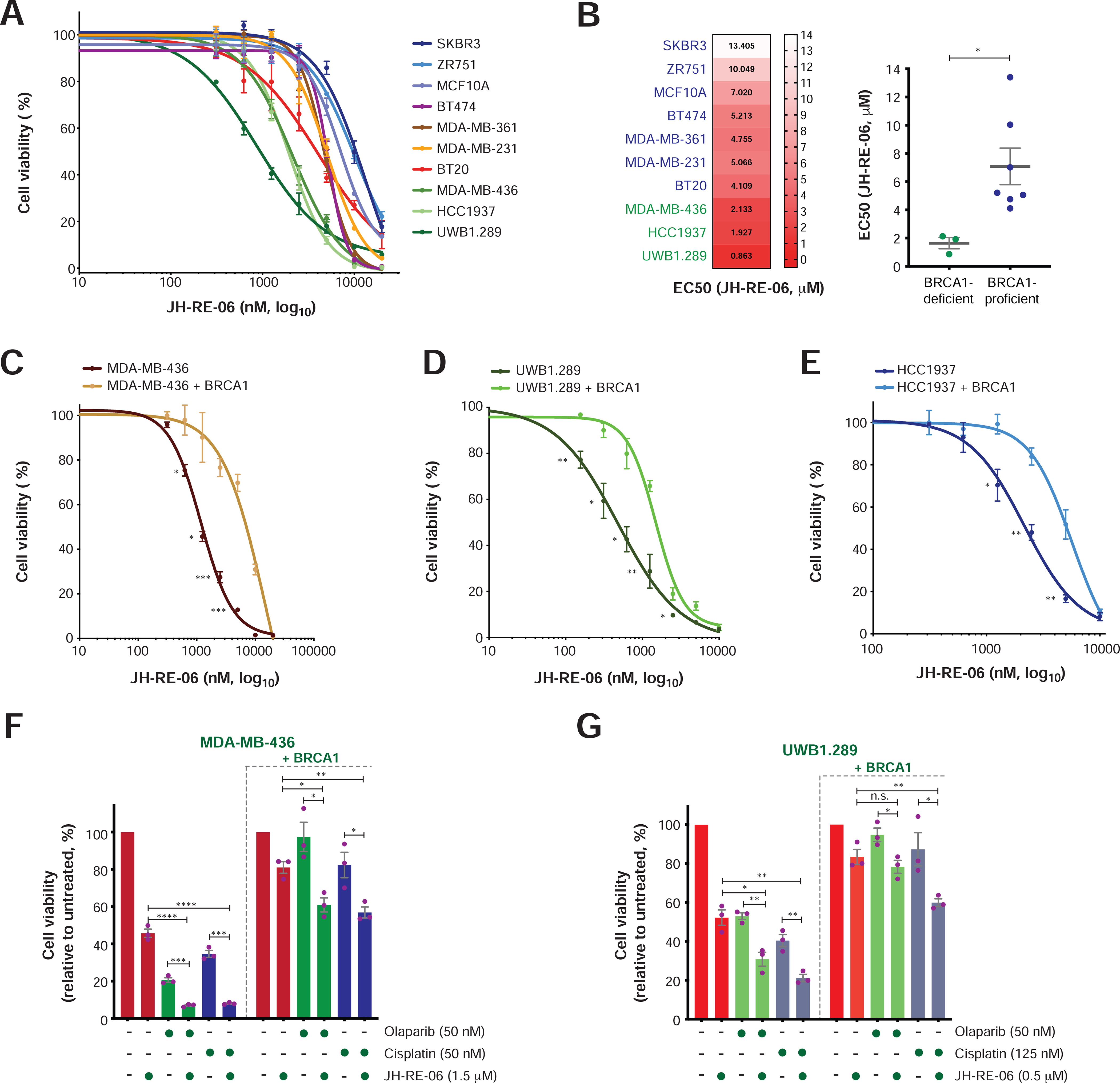
A) Survival analysis in the indicated breast and ovarian cell lines following treatment with JH-RE-06. Cell survival is expressed as percentage relative to the untreated control, and data represent the mean ± SEM of three replicates per condition.
B) EC50 of JH-RE-06 per cell line obtained from the survival curves shown in (A) (left panel). Grouped analysis of JH-RE-06 EC50 values in BRCA1-deficient and BRCA1-proficient cells (right panel). Statistical analysis was conducted by unpaired t-test (*p < 0.05).
C-E) Survival analyses upon JH-RE-06 treatment of the indicated BRCA1-deficient cell lines with or without BRCA1 re-expression. Data analysis and graphical representation were conducted as in (A). Statistical analysis was performed by unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.001).
F-G) Viability of the indicated BRCA1-deficient and BRCA1-reconstituted cell lines upon treatment with olaparib, cisplatin, JH-RE-06 and combinations of olaparib or cisplatin with JH-RE-06 at the indicated doses. Cell survival is expressed as percentage relative to the untreated control, and data represent the mean ± SEM of three replicates per condition. Statistical analysis was conducted by unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
The cytotoxicity of JH-RE-06 in BRCA1-deficient cancer cells is dependent on PRIMPOL and ssDNA accumulation
To investigate the mechanism of JH-RE-06 toxicity, we first monitored the formation of ssDNA gaps using the S1 nuclease assay on DNA fibers isolated from HCC1937 cells. Consistent with our EM observations (Figure 1A–B), the S1 nuclease induced a modest but significant shortening of CldU tract lengths in BRCA1-deficient, but not BRCA1-reconstituted, HCC1937 cells – an effect that was further enhanced by JH-RE-06 treatment (Figures 5A and S5C). Importantly, PRIMPOL depletion restored CldU tract lengths in both untreated and JH-RE-06-treated BRCA1-deficient cells, indicating that the ssDNA gaps formed by PRIMPOL in BRCA1-deficient cells are repaired by the REV1-Polζ complex (Figures 5A and S5A, C). PRIMPOL loss also led to restoration of cellular resistance to JH-RE-06 in both HCC1937 and MDA-MB-436 BRCA1-deficient cells (Figure S5A, D–E, G), and also suppressed JH-RE-06-induced loss of viability and ssDNA gap accumulation in BRCA1-depleted U2OS cells (Figure S5H–I). JH-RE-06 did not further sensitize REV1 KO cells to BRCA1 depletion, indicating that it acts through REV1 inhibition (Figure S5I). In agreement with the above findings, JH-RE-06 treatment induced PRIMPOL-dependent accumulation of replication fork intermediates with >1,000-nt-long ssDNA regions in BRCA1-deficient, but not BRCA1-reconstituted, HCC1937 cells, as determined by EM (Figures 5B–C and S5J). Interestingly, HCC1937 and MDA-MB-436 cells exhibited a marked increase in PRIMPOL mRNA and protein levels relative to their BRCA1-reconstituted counterparts (Figure S5A–B, E–F), suggesting that PRIMPOL-dependent activity might be elevated in BRCA1-mutant tumor cells. Together, these results indicate that the enhanced toxicity of JH-RE-06 towards BRCA1-deficient cells is due to defective repair, and consequent accumulation, of ssDNA gaps generated by PRIMPOL during DNA replication.
FIGURE 5. Characterization of PRIMPOL’s role in the response to JH-RE-06 treatment in BRCA1-mutant cancer cells.

A) Schematic of the IdU/CldU pulse-labeling protocol with or without JH-RE-06 treatment followed by S1 nuclease digestion (top). Dot plot and median of CldU tract lengths (μm) in HCC1937 cells subjected to the indicated experimental conditions (bottom). P-values were calculated by Mann-Whitney test (*p < 0.05, **p < 0.01, ****p < 0.0001).
B) Representative image of a replication fork detected by EM in HCC1937 cells treated with JH-RE-06 (2.5 μM) for 3 hr. dsDNA and ssDNA tracts are indicated.
C) Dot plot and mean of ssDNA tract lengths (nt) detected by EM at replication fork intermediates from HCC1937 cells treated with the indicated siRNAs, with and without JH-RE-06 (2.5 μM), as in (B). P-values were calculated by Mann-Whitney test (*p < 0.05, ****p < 0.0001).
D) Schematic of the CldU/IdU pulse-labeling protocol upon treatment of HCC1937 cells with JH-RE-06 with or without DMSO or the CDC7 inhibitor PHA-767491 (top). Representative image of a DNA replication origin activated during the 1st pulse (CldU; middle) and percentage of origins (1st pulse) relative to the total number of replication intermediates (bottom). Values of individual experiments are indicated as dots. Columns represent the mean ± SEM of independent biological replicates (n = 3). Statistical analysis was conducted by unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.001).
E) Viability of HCC1937 cells treated with the indicated sgRNAs and DMSO or CDC7 inhibitors in the presence of the indicated doses of JH-RE-06. Cell viability under the indicated conditions is shown as percentage relative to the untreated control. Graphical representation and statistical analysis was conducted as in (D).
F) Schematic of the HPRT gene mutation assay (top). Representative images of 6-thioguanine (6-TG)-resistant colonies of HCT116 cells subjected to the indicated treatments (bottom).
G) Fold change in the number of 6-TG-resistant colonies in HCT116 cells treated as in (F) relative to cells treated with control siRNA and DMSO. Graphical representation and statistical analysis were conducted as in (D). The number of 6-TG resistant colonies per 105 cells after background subtraction and normalization to plating efficiency is reported in Table S1.
The restoration of viability induced by PRIMPOL loss in JH-RE-06-treated BRCA1-deficient cells is mediated by increased CDC7-dependent origin firing
PRIMPOL re-initiates DNA synthesis at stalled forks, thus allowing the completion of DNA replication (Joseph et al., 2020). We therefore reasoned that the resistance to JH-RE-06 induced by PRIMPOL loss in HR-deficient cells may depend on the activation of compensatory pathways that ensure the completion of DNA replication. Previous studies have shown that loss of PRIMPOL leads to increased fork reversal events promoted by the SMARCAL1 DNA translocase (Quinet et al., 2020). SMARCAL1 depletion, however, did not resensitize BRCA1-deficient PRIMPOL KO U2OS cells and PRIMPOL-deficient HCC1937 cells to JH-RE-06 treatment (Figure S6A–D), excluding a role for SMARCAL1-mediated fork reversal in promoting resistance to JH-RE-06 in PRIMPOL- and BRCA1-deficient cells.
Fork stalling caused by PRIMPOL deficiency has also been reported to induce compensatory activation of dormant origins (Gonzalez-Acosta et al., 2021; Mouron et al., 2013; Rodriguez-Acebes et al., 2018). In line with these findings, we observed an increase in the percentage of origins and termination events, and a concomitant reduction in ongoing forks, following PRIMPOL depletion in JH-RE-06-treated HCC1937 cells (Figures 5D and S6E). Origin firing is promoted by the CDC7 kinase, which phosphorylates MCM2 and other MCM subunits to initiate DNA replication (Fei and Xu, 2018). Notably, serine 40 (S40) of MCM2, a known CDC7 substrate (Montagnoli et al., 2006), is hyperphosphorylated in PRIMPOL-deficient HCC1937 cells treated with JH-RE-06, indicating hyperactivation of CDC7 (Figure S6F). To determine the consequences of CDC7 hyperactivation, we selected the minimal dose of the CDC7 inhibitor PHA-767491 (Montagnoli et al., 2008) that re-established normal levels of MCM2 S40 phosphorylation and origin firing in PRIMPOL-deficient HCC1937 cells treated with JH-RE-06 (Figures 5D and S6E, G–H). Remarkably, the selected dose of CDC7 inhibitor restored the sensitivity of PRIMPOL-deficient HCC1937 cells to JH-RE-06, while having no effect on HCC1937 control cells (Figure 5E). Similar results were also obtained using the CDC7 inhibitor TAK-931 (Iwai et al., 2019) (Figure 5E and S6G). These findings suggest that CDC7-mediated origin firing protects cells that are deficient in BRCA1-mediated HR and REV1-Polζ-dependent TLS from loss of PRIMPOL-dependent fork repriming by ensuring the completion of DNA replication.
PRIMPOL promotes REV1-Polζ-mediated mutagenesis in BRCA1/2-deficient cells
The above observations indicate that PRIMPOL renders HR-deficient cells dependent on REV1-Polζ for the repair of ssDNA gaps formed during DNA replication. Considering that REV1 and Polζ are error-prone DNA polymerases (Vaisman and Woodgate, 2017; Waters et al., 2009), we hypothesized that REV1-Polζ-mediated repair of ssDNA gaps might lead to elevated rates of spontaneous mutagenesis in BRCA1/2-deficient cells. To test this hypothesis, we depleted BRCA1/2 in HCT116 cells with or without PRIMPOL and/or JH-RE-06 treatment (Figure S6I). BRCA1/2 depletion enhanced the sensitivity of HCT116 cells to JH-RE-06, and this effect was suppressed by PRIMPOL loss, confirming our results in other cell lines (Figure S6J). The rate of DNA mutagenesis in these cells was then assessed using the HPRT gene mutation assay, which measures the number of 6-thioguanine (6-TG)-resistant colonies formed as a result of inactivating mutations in the HPRT gene (Johnson, 2012) (Figure 5F). These studies revealed that BRCA1/2 depletion increased the rate of spontaneous mutations in HCT116 cells by 2–3-fold (Figure 5F–G). Notably, this effect was suppressed by either PRIMPOL depletion or treatment with JH-RE-06 (Figure 5F–G), suggesting that PRIMPOL-dependent ssDNA gaps are repaired by error-prone DNA synthesis mediated by REV1-Polζ, leading to elevated spontaneous mutagenesis in BRCA1/2-deficient cells.
SMUG1 promotes ssDNA gap accumulation and sensitivity to JH-RE-06 in BRCA1-mutant cells
Having established that PRIMPOL-mediated ssDNA gaps undergo REV1-Polζ-dependent mutagenic repair in HR-deficient cells, we next examined the nature of the endogenous DNA lesions that lead to PRIMPOL-mediated repriming in BRCA1-mutant cancer cells. Interestingly, previous studies had implicated PRIMPOL in the repriming of DNA synthesis upon fork stalling induced by abasic sites (Garcia-Gomez et al., 2013; Kobayashi et al., 2016) and showed that REV1 and Polζ promote their bypass (Choi et al., 2010; Shachar et al., 2009; Weerasooriya et al., 2014). Abasic sites can originate upon spontaneous base loss or following base excision by DNA glycosylases (Thompson and Cortez, 2020). To examine whether abasic sites generated by DNA glycosylases render BRCA1-mutant cancer cells dependent on REV1-Polζ, we monitored the survival of JH-RE-06-treated UWB1.289 and HCC1937 cells upon depletion of the uracil glycosylases UNG and SMUG1, the 8-oxoguanine glycosylase OGG1, the adenine glycosylase MUTYH or the N-methylpurine glycosylase MPG (Figure S7A–B). Remarkably, the sensitivity of UWB1.289 cells to JH-RE-06 was significantly reduced by knockdown of the uracil glycosylases UNG and SMUG1, while only SMUG1 depletion suppressed the JH-RE-06 sensitivity of HCC1937 cells (Figure 6A–B). SMUG1 depletion also induced JH-RE-06 resistance in MDA-MB-436 cells, and SMUG1 disruption by CRISPR-Cas9 rendered HCC1937 cells resistant to JH-RE-06, similarly to PRIMPOL loss (Figures 6C–D and S7C–D). Co-depletion of SMUG1 and PRIMPOL did not result in additive effects, suggesting that SMUG1 and PRIMPOL operate in the same pathway that sensitizes BRCA1-mutant cells to JH-RE-06 treatment (Figure 6E–F). Accordingly, ssDNA gap accumulation in JH-RE-06-treated HCC1937 cells was suppressed in a comparable manner by either SMUG1 or PRIMPOL depletion (Figure 6G). These results suggest that SMUG1-mediated DNA lesions induce replication fork stalling and PRIMPOL-dependent repriming, thus leading to the formation of ssDNA gaps that cause lethality in cells defective for both REV1-Polζ-mediated TLS and BRCA1-dependent HR.
FIGURE 6. Cellular viability and ssDNA gap formation upon loss of SMUG1 and/or PRIMPOL in BRCA1-mutant cancer cells.

A-B) Viability of HCC1937 (A) and UWB1.289 (B) cells treated with the indicated siRNAs and JH-RE-06 (2.5 μM and 1.5 μM, respectively). Cell viability is expressed as percentage relative to the untreated control. Values of individual experiments are indicated as dots. Columns represent the mean ± SEM of independent biological replicates (n = 3). Statistical analysis was conducted by unpaired t-test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
C) Viability of MDA-MB-436 cells treated with control and SMUG1 siRNAs at the indicated JH-RE-06 doses. Graphical representation and statistical analyses were conducted as in (A) and (B).
D) Viability of HCC1937 cells treated with control, SMUG1 and PRIMPOL sgRNAs at the indicated JH-RE-06 doses. Graphical representation and statistical analyses were conducted as in (A) and (B).
E) PRIMPOL and SMUG1 levels in HCC1937 following transfection of the indicated siRNAs, as detected by western blot. Tubulin is shown as control.
F) Viability of HCC1937 cells following treatment with the indicated siRNAs and doses of JH-RE-06. Graphical representation and statistical analysis were conducted as in (A) and (B).
G) Schematic of the IdU/CldU pulse-labeling protocol with JH-RE-06 treatment followed by S1 nuclease digestion (top). Dot plot and median of CldU tract lengths (μm) in HCC1937 cells subjected to the indicated experimental conditions (bottom). P-values were calculated by Mann-Whitney test (****p < 0.0001).
REV1-Polζ inhibition impairs BRCA1-deficient tumor growth
To determine whether inactivation of REV1-Polζ could be employed as a targeted therapy for BRCA1-mutant tumors, we first examined the cytotoxic effects induced by JH-RE-06 in BRCA1-deficient and BRCA1-reconstituted cancer cells cultured in three-dimensional conditions (3D-Matrigel) (Weigelt et al., 2014). Strikingly, JH-RE-06 severely diminished the tumor spheroid-forming ability of the BRCA1-deficient cells, while showing no significant deleterious effects on the growth of BRCA1-proficient tumor spheroids (Figure 7A–B).
FIGURE 7. Growth of BRCA1-deficient and BRCA1-reconstituted tumors following JH-RE-06 treatment.

A) Representative images of organoids of BRCA1-deficient and BRCA1-reconstituted UWB1.289 cells treated with DMSO or JH-RE-06 (1 μM) for 20 days. Scale bar = 400 μm.
B) Number of UWB1.289 organoids per well treated as in (A). Values of individual experiments are indicated as dots. Columns represent the mean ± SEM of independent biological replicates (n = 3). Statistical analysis was conducted by unpaired t-test (**p < 0.01).
C and E) Growth curves of xenograft tumors induced by MDA-MB-436 cells with or without BRCA1 re-expression upon treatment with vehicle control or JH-RE-06. Error bars represent SEM of 11 and 5 xenograft tumors per treatment group in (C) and (E), respectively. P-values of tumor volume differences between groups were calculated by unpaired t-test (*p < 0.05).
D and F) Representative images (top panel) and dot plot with mean (bottom panel) of xenograft tumor masses collected at day 46 of the experiments shown in (C) and (E). The reference ruler is in cm. Statistical analysis between treatment groups was conducted using unpaired t-test (*p < 0.05).
Having established the cytotoxicity of JH-RE-06 for BRCA1-mutant cancer cells in 3D-cultures, we then examined whether JH-RE-06 would also inhibit BRCA1-mutant tumor growth in vivo. To this end, BRCA1-deficient and BRCA1-reconstituted MDA-MB-436 breast cancer cells were injected subcutaneously into nude mice treated with vehicle control or JH-RE-06. Remarkably, JH-RE-06 treatment profoundly impaired the growth of BRCA1-deficient (Figure 7C–D), but not BRCA1-reconstituted (Figure 7E–F), tumors. In agreement with these observations, low expression of the TLS genes RAD18 and REV3L significantly correlated with better prognosis in ovarian cancer patients carrying mutations in HR genes (Figure S7E–G). These studies establish REV1-Polζ inhibitors as potential therapeutic agents for the treatment of BRCA1-mutant cancer patients.
DISCUSSION
In this study, we report that TLS suppression by loss of RAD18 or REV1 or by treatment with a REV1-Polζ inhibitor (JH-RE-06) causes accumulation of ssDNA gaps, reduction of DNA mutagenesis and loss of viability in BRCA1/2-deficient cancer cells. Importantly, the observed accumulation of ssDNA gaps and loss of viability depend on the DNA glycosylase SMUG1 and the DNA repriming activity of PRIMPOL. In addition, we show that JH-RE-06 is preferentially toxic towards HR-deficient cancer cells in vitro and in vivo and displays additive cytotoxicity in combination with DNA crosslinking agents and PARPi. Altogether, our studies identify an intrinsic addiction to error-prone REV1-Polζ-dependent TLS in BRCA1/2-deficient cancer cells that could be exploited for cancer therapy.
RAD18 promotes REV1-Polζ-mediated mutagenic repair of ssDNA gaps to maintain the viability of BRCA1/2-deficient cells
Through EM analysis of DNA replication intermediates, we show that BRCA1 deficiency causes the accumulation of spontaneous replication-associated ssDNA gaps (Figure 1A–B), in agreement with our previous observations in Xenopus egg extracts depleted of BRCA2 or RAD51 (Hashimoto et al., 2010; Kolinjivadi et al., 2017). In line with the need for ssDNA gap repair mechanisms to support the viability of BRCA1/2-deficient cancer cells, loss of RAD18, an E3 ligase that promotes the post-replicative repair of ssDNA gaps by both the TLS and template-switching pathways (Branzei and Szakal, 2017; Garcia-Rodriguez et al., 2016), causes synthetic lethality in combination with BRCA1/2 deficiency (Figures 1C–G and S1B–H). The observation that loss of the TLS polymerase REV1 or inhibition of REV1-Polζ by JH-RE-06 phenocopies RAD18 loss in BRCA1/2-deficient cells (Figures 3 and 4) implicates the error-prone REV1-Polζ branch of TLS as the post-replicative repair pathway required for the viability of BRCA1/2-deficient cells. Mechanistically, we find that RAD18 loss or JH-RE-06 treatment results in an increased accumulation of ssDNA intermediates in BRCA1/2-deficient cells, as detected by CldU foci formation, S1 nuclease assay, and EM studies (Figures 2E–G, 5A–C, and S5J). Interestingly, long stretches of ssDNA (>1,000 nt) were detected by EM in BRCA1-mutant cells treated with JH-RE-06 (Figures 5B–C and S5J), possibly due to aberrant gap extension by MRE11 or other nucleases, as observed in RAD51- and BRCA2-depleted Xenopus egg extracts and BRCA2-deficient mammalian cells under hypoxia (Hashimoto et al., 2010; Kolinjivadi et al., 2017; Somyajit et al., 2021). These studies highlight the cytotoxic effects induced by unrepaired ssDNA gaps in JH-RE-06-treated BRCA1/2-deficient cells, in accord with recent findings showing that accumulation of ssDNA gaps following REV1 inhibition causes cancer cell death and that the hypersensitivity of BRCA1/2-deficient cells to genotoxic agents depends on the induction of ssDNA gaps (Cong et al., 2021; Nayak et al., 2020; Panzarino et al., 2021).
Our results also show that REV1-Polζ maintains the viability of BRCA1/2-deficient cells at the expense of DNA mutagenesis. In particular, we observed that BRCA1/2-deficiency causes an increased rate of spontaneous mutations in human cancer cells, as previously observed in chicken cells (Zamborszky et al., 2017), and that this increase is reversed by JH-RE-06 treatment (Figures 5F–G). Consistent with our work, rad51 mutant yeast strains accumulate base substitutions in a manner dependent on Rev1, Rev7 and Rev3 (Loeillet et al., 2020), suggesting a conserved mutagenic role for REV1-Polζ in HR-deficient cells from yeast to humans. Importantly, BRCA1/2-mutant tumors are characterized by elevated SNVs associated with base substitution signature 3 (Alexandrov et al., 2013; Lal et al., 2019; Nik-Zainal et al., 2012; Nik-Zainal et al., 2016), a mutational signature of unknown etiology. Future studies should address whether REV1-Polζ contributes to substitution signature 3 or other mutational signatures of BRCA1/2-mutant tumors.
PRIMPOL-mediated repriming is required for ssDNA gap formation in HR-deficient cells
Our work indicates that the accumulation of ssDNA gaps in BRCA1/2-deficient cells depends on the DNA primase/polymerase PRIMPOL (Figures 3C–D, 5A and S5H). In HR-proficient cells, PRIMPOL-dependent ssDNA gaps are preferentially repaired by HR-mediated mechanisms to prevent TLS-dependent mutagenic processes (Piberger et al., 2020; Pilzecker et al., 2016). However, our results suggest that in HR-deficient cells PRIMPOL-dependent ssDNA gaps are instead repaired by error-prone TLS, as indicated by the observation that PRIMPOL deficiency suppresses the REV1-Polζ-dependent increase in spontaneous mutagenesis displayed by BRCA1/2-deficient cells (Figure 5F–G). Remarkably, the accumulation of ssDNA gaps and the loss of viability induced by disrupting TLS in BRCA1/2-deficient cells is rescued by PRIMPOL loss (Figures 3C, E, 5A and S5D, G–I) and can be re-established upon re-expression of WT PRIMPOL, but not its catalytic mutants (Figure 3C–F), indicating that the repriming activity of PRIMPOL is responsible for the formation of ssDNA gaps that are repaired by REV1-Polζ in HR-deficient cells.
BRCA1-deficient cancer cells have been shown to induce PRIMPOL expression to adapt to recurrent cisplatin treatments (Quinet et al., 2020). Interestingly, our studies indicate that BRCA1 also suppresses the expression of PRIMPOL in cancer cells in unperturbed conditions (Figure S5A–B, E–F). Induction of PRIMPOL in BRCA1-mutant cells was reported to enhance the restart of stalled forks by repriming, thus preventing fork reversal and limiting the degradation of nascent DNA in BRCA1/2-deficient cells (Kolinjivadi et al., 2017; Lemacon et al., 2017; Mijic et al., 2017; Quinet et al., 2020; Taglialatela et al., 2017). Nonetheless, our studies indicate that repriming would be detrimental in BRCA1/2-deficient cells under conditions of RAD18 or REV1-Polζ deficiency due to the accumulation of toxic unrepaired ssDNA gaps (Figures 3C–D and 5A). Although loss of PRIMPOL leads to increased fork reversal (Quinet et al., 2020), impairing fork reversal by SMARCAL1 depletion (Kolinjivadi et al., 2017; Taglialatela et al., 2017) does not restore sensitivity to JH-RE-06 in HR- and PRIMPOL-deficient cells (Figure S6B, D). Instead, these cells display a CDC7-dependent increase in origin firing that circumvents JH-RE-06-induced loss of cell viability (Figures 5D–E and S6E–H). These findings indicate that increased origin firing can compensate for the loss of fork repriming in cells that are deficient for both BRCA1/2-dependent HR and REV1-Polζ-mediated TLS by allowing completion of DNA synthesis and maintenance of cell viability. These studies highlight the plasticity of the replication fork-associated mechanisms by which cancer cells cope with endogenous replication stress.
ssDNA gap formation in HR-deficient cells requires the SMUG1 glycosylase, implicating abasic sites as endogenous DNA lesions that initiate PRIMPOL-mediated repriming
Our observation that ssDNA gaps form at replication fork intermediates in BRCA1-deficient cells (Figure 1A–B) suggests that roadblocks impairing fork progression are the primary cause of ssDNA gap formation in HR-deficient cells. Removal of modified or misincorporated bases by DNA glycosylases generates abasic sites, one of the most frequent endogenous DNA lesions that stall fork progression (Thompson and Cortez, 2020). Our finding that the DNA glycosylase SMUG1 confers sensitivity to JH-RE-06 in multiple BRCA1-mutant cancer cell lines (Figure 6A–D) implies that abasic site formation by SMUG1 is detrimental in cells that are defective for both HR and REV1-Polζ-dependent TLS. SMUG1 is known to preferentially excise 5-hydroxymethyl-uracil (5-hmU) originating from either oxidative damage to thymine or deamination of 5-hydroxymethyl-cytosine (5-hmC) (Bordin et al., 2021; Fugger et al., 2021; Raja and Van Houten, 2021). SMUG1’s activity on 5-hmU was recently reported to enhance PARPi sensitivity in HR-deficient cells (Fugger et al., 2021), suggesting that 5-hmU processing by SMUG1 might play an important role in the generation of DNA lesions in HR-deficient cells. In addition to 5-hmU, SMUG1 can also excise 5-formyluracil, a product of thymine oxidation (Masaoka et al., 2003), and this activity might likewise contribute to the generation of toxic DNA lesions, as recently shown in the context of ALC1 deficiency (Hewitt et al., 2021). Besides excising uracil derivatives, SMUG1 can also act as a backup to UNG for the removal of uracil itself (Alsoe et al., 2017; Kavli et al., 2002; Nilsen et al., 2001). Interestingly, we observed that UNG loss causes JH-RE-06 resistance in UWB1.289, but not HCC1937, cells (Figure 6A–B), suggesting that uracil removal might contribute to loss of viability upon JH-RE-06 treatment in certain HR-deficient cell lines. Identifying the specific SMUG1 substrates that determine the sensitivity of HR-deficient cells to REV1-Polζ inhibition will be critical for defining the endogenous source(s) of DNA damage that cause genomic instability and possibly malignant transformation/progression in BRCA1/2-deficient cells.
Our observations that SMUG1 causes ssDNA gap accumulation and PRIMPOL-dependent loss of viability in JH-RE-06-treated BRCA1-deficient cells (Figure 6F–G) suggest that abasic sites generated by SMUG1 could initiate PRIMPOL-mediated repriming in HR-deficient cells. Consistent with this notion, PRIMPOL can reprime DNA synthesis downstream of abasic sites (Garcia-Gomez et al., 2013; Kobayashi et al., 2016). Recent studies have shown that abasic sites on ssDNA are covalently bound by HMCES (Mehta et al., 2020; Mohni et al., 2019; Thompson et al., 2019), however the role of HMCES-abasic site complexes in PRIMPOL-dependent repriming remains to be defined. Once ssDNA gaps are formed by PRIMPOL, REV1-Polζ could complete DNA synthesis by bypassing the abasic sites, as previously shown (Choi et al., 2010; Haracska et al., 2001; Kim et al., 2011; Nelson et al., 1996; Shachar et al., 2009; Weerasooriya et al., 2014). Under this scenario, REV1-Polζ would act as an alternative to HR for the repair of ssDNA gaps formed at abasic sites. Interestingly, in S. cerevisiae, the Shu complex of Rad51 paralogs was shown to bind abasic sites and initiate HR-mediated gap repair (Rosenbaum et al., 2019). It remains, however, to be established whether a similar function for the Shu complex is conserved in humans. Together with the above studies, our work highlights the complex interplay between base excision repair, repriming of DNA synthesis, HR and TLS during DNA replication.
REV1-Polζ is a promising therapeutic target for the treatment of HR-mutant tumors
Our studies show that inhibition of REV1-Polζ by JH-RE-06 results in enhanced toxicity towards BRCA1/2-deficient cancer cells both in vitro and in vivo (Figures 4A–D and 7), and that this effect is enhanced by either DNA crosslinking agents or PARPi (Figure 4F–G). In addition, we demonstrate that JH-RE-06 can also be employed to treat PARPi-resistant cancer cells, such as BRCA1-mutant HCC1937 breast cancer cells (Figures 4E and S4E–I), which have acquired PARPi resistance despite remaining HR deficient (Du et al., 2016; Zhang et al., 2004). Based on our work (Figures 4A–E and S4E–I), we expect that JH-RE-06 would also sensitize other BRCA1/2-mutant cancer cell lines that have acquired PARPi resistance through HR-independent mechanisms (e.g., restoration of fork protection, downregulation of PARP1 or PARG), while not affecting BRCA1/2-mutant tumors that have acquired resistance by HR restoration (e.g., reactivation of BRCA1/2, loss of 53BP1). These studies suggest that REV1-Polζ inhibitors might represent a therapeutic opportunity for HR-deficient tumors displaying PARPi resistance.
In recent years, different classes of TLS inhibitors have been developed as potential adjuvant anti-cancer agents (Patel et al., 2021; Yamanaka et al., 2017). However, cancer patient subpopulations that might specifically benefit from TLS inhibitors, as well as biomarkers that would predict effective response to TLS inhibition, have not as yet been identified. By demonstrating that BRCA1/2-deficient cancer cells are addicted to REV1-Polζ-mediated TLS, our studies suggest that HR deficiency should serve as a predictive biomarker of response to REV1-Polζ inhibitors. An important implication of this work is that HR-deficient tumors are constantly evolving even in the absence of therapeutic selective pressure, given their reliance on error-prone REV1-Polζ-mediated TLS for ssDNA gap repair and cellular viability. Based on these considerations, therapeutic strategies for BRCA1/2-deficient tumors that include REV1-Polζ inhibitors could act on multiple levels, by inducing cytotoxicity through the accumulation of unrepaired ssDNA gaps, while at the same time reducing mutagenesis-driven cancer heterogeneity and therefore limiting the appearance of therapy-resistant cancer clones. Defining the precise contribution of the REV1-Polζ pathway in promoting resistance to targeted therapies in BRCA1/2-deficient tumors will be of paramount importance for the development of treatments that limit cancer recurrence.
Limitations of study
Our work has been performed using immortalized cancer cell lines as an experimental model. Additional studies using patient-derived primary cancer cells and xenografts will be required to determine the clinical relevance and translational potential of our findings. To more accurately assess the in vivo safety and efficacy of JH-RE-06 treatment, derivatives of JH-RE-06 with enhanced pharmacological properties will be needed, given that the current JH-RE-06 formulation requires intra-tumor injection. Our studies show that the toxicity induced by JH-RE-06 in BRCA1/2-deficient cells is caused by SMUG1, suggesting a possible involvement of SMUG1-induced abasic sites in the response to REV1-Polζ inhibition. It remains, however, to be experimentally determined whether DNA base excision by SMUG1 is required for the toxic effects induced by JH-RE-06 in BRCA1/2-deficient cells and whether additional SMUG1 functions (Pettersen et al., 2007; Raja and Van Houten, 2021) might also regulate the response to REV1-Polζ inhibition.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Alberto Ciccia (ac3685@cumc.columbia.edu).
Materials availability
The plasmids and cell lines generated in this study are available from the Lead Contact upon request.
Data and code availability
Unprocessed western blot and microscopy images have been deposited at Mendeley Data (DOI: 10.17632/rmyvkwbmyn.1) and are publicly available as of the date of publication. Source data for Figure S7E–G are available at https://www.cbioportal.org/. Microscopy data reported in this paper will be shared by the lead contact upon request.
No original code has been generated for this publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T, MDA-MB-231, ZR75–1, MDA-MB-361, BT474, BT20, SKBR3, HCC1937, MDA-MB-436, HCT116 and U2OS cells were grown in DMEM supplemented with 10% Fetalgro, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. UWB1.289 were grown in a 1:1 mixture of RPMI and MEGM BulletKit (Lonza CC-3150) supplemented with 3% Fetalgro, 100 U/ml of penicillin, and 100 μg/ml of streptomycin. MCF10A cells were grown in a 1:1 mixture of DMEM and Ham’s F12 medium (Thermo Fisher Scientific), supplemented with 5% horse serum (Thermo Fisher Scientific), 20 ng/ml of human epidermal growth factor (Peprotech), 100 ng/ml of cholera toxin, 10 μg/ml of insulin and 0.5 μg/ml of hydrocortisone (Sigma-Aldrich), 100 U/ml of penicillin and 100 μg/ml of streptomycin. All cell lines were cultured at 37°C, 5% CO2. The cell lines used were either obtained from ATCC or kindly provided by other laboratories. PRIMPOL KO U2OS cells, UWB1.289 and UWB1.289 reconstituted with BRCA1 were kindly provided by Alessandro Vindigni (Quinet et al., 2020). HCC1937 and HCC1937 reconstituted with BRCA1 were kindly provided by Simon Powell (Fridlich et al., 2015). MDA-MB-436 and MDA-MB-436 reconstituted with BRCA1 were kindly provided by Neil Johnson (Johnson et al., 2013).
Mice
6–8 week-old NU/J mice (nude mice) for xenograft studies were purchased from Jackson laboratories. All mouse experiments were conducted under a protocol approved by the Columbia University Institutional Animal Care and Use Committee (IACUC) (Protocol #AC-AABH6553). All mice were housed in a pathogen-free environment at the Institute of Comparative Medicine animal facility and were handled in strict accordance with the “Guide for the Care and Use of Laboratory Animals.”
METHOD DETAILS
Plasmids
To generate the doxycycline-inducible lentiviral vector for the expression of SpCas9, the pCW-Cas9 (plasmid #50661, Addgene) was modified as follows: SpCas9 was removed using the NheI and BamHI restriction sites and replaced by a multiple cloning site (MluI, HpaI, EcoRI and SalI). The puromycin resistance was removed by digestion with BamHI and XbaI and the blasticidin resistance was synthesized as a gBlock (IDT) and inserted by Gibson assembly. SpCas9 was then reintroduced into the BamHI and NheI restriction sites. sgRNAs targeting RAD18 (TCAGTGTCCAACTTGCTGTG) and REV1 (GTGGCTGTTACAAGTAACAG) were cloned into pLenti-SpBsmBI (plasmid #62205, Addgene). The pMSCV-N-HA-FLAG-RAD18 retroviral plasmid for RAD18 expression was previously described (Nambiar et al., 2019). Plasmids expressing V5-tagged WT PRIMPOL, and the AxA and CH mutants were generously provided by Juan Mendez (Mouron et al., 2013).
Recombinant viral production and infection
Recombinant retroviruses and lentiviruses were generated by co-transfecting helper packaging vectors together with retroviral or lentiviral vectors into HEK293T cells using the TransIT-293 transfection reagent (Mirus). Virus-containing supernatants were collected 48 hr after transfection and utilized to infect target cells in the presence of 8 μg/ml of polybrene. Forty-eight hours after viral addition, successfully infected cells were selected using 1 μg/ml of puromycin, 100 μg/ml of hygromycin or 10 μg/ml of blasticidin for 3–5 days.
siRNA and Cas9-sgRNA transfection
RNAi experiments were carried out by transfection of the indicated siRNAs using Lipofectamine RNAiMAX (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cas9-sgRNA transfections in pooled cell populations were conducted using synthetic sgRNAs and recombinant Cas9 protein (Synthego), which were combined to obtain Cas9-sgRNA RNP complexes according to the manufacturer’s instructions. RNP complexes were transfected using Lipofectamine CRISPRMAX (Thermo Fisher Scientific).
Generation of KO and reconstituted cell lines
RAD18 KO and REV1 KO U2OS cells were obtained by CRISPR-Cas9 technology. In particular, U2OS cells expressing a doxycycline-inducible SpCas9 were obtained by lentiviral infection and blasticidin selection. Doxycycline-inducible SpCas9-expressing cells were further infected with lentiviral sgRNA constructs targeting the gene of interest and selected with hygromycin. Five days after doxycycline-induced SpCas9 expression, cells were plated at single cell density for clonal selection. KO clones for the genes of interest were selected by western blot analysis. RAD18/REV1 KO U2OS cells were obtained by transient transfection of RAD18 KO cells with synthetic sgRNAs targeting REV1 (Synthego) followed by clonal selection and western blot analysis. RAD18 KO cells reconstituted with RAD18 were obtained by pMSCV-N-HA-FLAG-RAD18 lentiviral infection followed by puromycin selection. PRIMPOL KO U2OS cells (obtained as described above) were transfected in 6-well plates with plasmids for transient expression of V5-tagged PRIMPOL WT, CH and AxA mutants using Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Electron microscopy
DNA analysis by EM was performed as previously described (Neelsen et al., 2014) with some modifications. Briefly, for each sample 10–15 × 106 cells were collected. After standard trypsinization, the cells were transferred to 15 ml Falcon tubes and spun down at 600 × g for 5 min. The cell pellet was washed once with 5 ml of ice-cold 1X PBS, resuspended in 10 ml of ice-cold 1X PBS and transferred to a Petri dish, to which 10 μg/ml of TMP (Trimethylpsoralen, Sigma-Aldrich) were added. The Petri dish was incubated in the dark for 5 min on a pre-cooled metal surface and then UV-irradiated for 5 min in a Stratalinker equipped with monochromatic 365 nm lamps. The cycle of TMP addition, dark incubation and irradiation was repeated four times. Cells were then transferred to a 15 ml tube, washed twice with 1 ml of PBS and DNA was purified using chloroform:isoamyl alcohol extraction. The DNA pellet was resuspended in TE buffer. Thirty micrograms of genomic DNA were digested with 150 U of PvuII HF restriction enzyme (New England Biolabs) for 3.5 hr at 37°C. DNA replication intermediates were further purified by Qiagen Genomic-tips 20/G columns according to manufacturer instructions and the final DNA sample was eluted by means of a buffer containing 10 mM Tris/HCl pH 8, 1 M NaCl, 1.8% (W/V) caffeine. DNA was then cleaned using Amicon Ultra-0.5 Centrifugal Filter Unit with Ultracel-100 (Millipore). DNA samples were loaded on a 0.8% agarose gel to determine DNA quality and concentration. The DNA was then subjected to uranyl acetate-positive staining and platinum coating by rotary shadowing as previously described (Neelsen et al., 2014) using a Med20 evaporator (Leica). Images were acquired at the IFOM Electron Microscopy DNA/Single Molecules unit using TECNAI12 electron microscope equipped with a GATAN camera run by Digital Micrograph software. Approximately one hundred replication forks were analyzed per sample/condition per experiment. Each experiment was repeated at least 2 times.
Cell viability assay
Cells were seeded into 6-well plates at 20–30% density 24 hr after siRNA, sgRNA or RNP transfection. After incubation for 5 days, cells were either fixed or passaged (1:6) and incubated for additional 5 days. After incubation for 5 and 10 days, cell viability was assessed by crystal violet staining. Adherent cells were fixed and stained with a solution containing 1% formaldehyde, 1% crystal violet and 10% methanol. The absorbed dye was resolubilized with methanol containing 0.1% SDS, which was then transferred into 96-well plates and measured photometrically (595 nm) in a microplate reader. Following background subtraction, cell viability was calculated by normalizing the absorbance of each sample to the control transfected sample. Cell viability data are shown at day 10 after transfection, unless otherwise indicated, and are representative of three or more independent experiments.
For viability assays upon drug treatment, cells were seeded into 12-well plates at 10–20% density. Twenty-four hours after seeding, the cells were treated or not with the chemical agents described in the text at the indicated concentrations. After incubation for 7–10 days, cell viability was assessed by crystal violet staining. Adherent cells were fixed and stained with a solution containing 1% formaldehyde, 1% crystal violet and 10% methanol. The absorbed dye was resolubilized with methanol containing 0.1% SDS, which was then transferred into 96-well plates and measured photometrically (595 nm) in a microplate reader. Following background subtraction, cell survival was calculated by normalizing the absorbance of the treated samples to the absorbance of untreated controls. Survival curves were generated using the nonlinear regression algorithm of GraphPad Prism software. Data are representative of three or more independent experiments.
HPRT gene mutation assay
HCT116 cells were cultured for 7 days with HAT (hypoxanthine-aminopterin-thymidine) medium for mutant cleansing and released in HT (hypoxanthine-thymidine) medium for 24 hr. Cells (2 × 105) were then seeded in 6-well plates and treated with 6-thioguanine (6-TG; 5 μg/ml) to establish the frequency of background mutants. At the same time, HCT116 cells were transfected with the indicated siRNAs followed by treatment with DMSO or JH-RE-06. After 5 days, HCT116 cells were collected and 2 × 105 of them were seeded for a second round of siRNA transfection followed by DMSO or JH-RE-06 treatment. After 10 days of treatment, HCT116 cells were left to recover in fresh medium for 3 additional days, and 2 × 105 cells were seeded in 6-well plates in duplicate. Two days after seeding, cells were counted to establish the plating efficiency and selected with 6-TG for 12–15 days. 6-TG resistant colonies were fixed and stained by crystal violet. De novo mutation rates were defined after subtraction of background mutants and normalization to plating efficiency. Each experiment was repeated 3 times independently.
S1 nuclease and origin firing assays
Exponentially growing cells were pulse-labeled with 30 μM IdU (10 min), washed with PBS twice and exposed to 150 μM CldU (50 min). When indicated, the cells were treated before and during pulses with JH-RE-06 at 2.5 μM. After exposure to the second nucleotide analog, the cells were collected, washed in 1X PBS and permeabilized with CSK buffer (100 mM NaCl, 10 mM MOPS pH 7, 3 mM MgCl2, 300 mM sucrose and 0.05% Triton X-100 in water) for 10 min on ice and centrifuged at 4,600 × g for 5 min at 4°C. Permeabilized cells were treated with 100 μl of S1 buffer (30 mM sodium acetate pH 4.6, 10 mM zinc acetate, 5% glycerol, 50 mM NaCl in water) with or without the S1 nuclease (18001–016, Thermo Fisher Scientific) at 10 U/ml for 15 min at 37°C. Cells were pelleted at 4,600 × g for 5 min at 4°C, then resuspended in PBS. To evaluate origin firing, exponentially growing cells were treated as indicated and pulse-labeled with 30 μM CldU for 20 min, washed with PBS twice and exposed to 150 μM IdU for 20 min. Labeled cells were harvested and resuspended in PBS at a concentration of 2 × 105 cells/ml. Two microliters of cell suspension were spotted onto a pre-cleaned glass slide and lysed with 10 μl of spreading buffer (0.5% SDS in 200 mM Tris-HCl, pH 7.4 and 50 mM EDTA). After 6 min, the slides were tilted at 15° relative to horizontal, allowing the DNA to spread. Slides were air-dried, fixed in methanol and acetic acid (3:1) for 2 min, rehydrated in PBS for 10 min and denatured with 2.5 M HCl for 50 min at room temperature. Slides were then rinsed in PBS and blocked in PBS + 0.1% Triton X-100 (PBS-T) + 5% BSA for 1 hr at room temperature. Rat anti-BrdU (1:100, Abcam) and mouse anti-BrdU (1:100, Becton Dickinson) were then applied to detect CldU and IdU, respectively. After a 1 hr incubation, slides were washed in PBS and stained with Alexa Fluor 488-labeled goat anti-mouse IgG1 antibody and Alexa Fluor 594-labeled goat anti-rat antibody (1:300 each, Thermo Fisher Scientific). Slides were mounted in Prolong Gold Antifade (Thermo Fisher Scientific) and held at −20°C. Replication tracks were imaged on a Nikon Eclipse 90i microscope fitted with a PL Apo 40X/0.95 numerical aperture (NA) objective and measured using ImageJ software. The length of each tract was measured manually using the segmented line tool on ImageJ software (NIH). The pixel values were converted into μm using the scale bar generated by the microscope software. Size distribution of tract lengths from individual DNA fibers were plotted as scatter dot plot with the line representing the median. In each experiment, 100 or more tracts were measured. Statistical differences in DNA fiber tract length distributions were determined by Mann-Whitney test. Each experiment was repeated at least 2 times independently.
Western blotting
Cells were collected by trypsinization and lysed in SB lysis buffer (37.5 mM Tris-HCl pH 6.8, 1.25% SDS, 10% glycerol, 3% β-mercaptoethanol, 0.002% bromophenol blue). Whole-cell extracts were sonicated and boiled for 5 min at 95°C. Protein lysates were run on precast Mini-PROTEAN TGX 4–15% gels (Bio-Rad). Specifically for BRCA2 detection, protein lysates were run on NuPAGE 3–8% Tris-Acetate Protein Gels (Thermo Fisher Scientific). Following gel electrophoresis and transfer, nitrocellulose membranes were incubated for 1 hr or overnight in blocking buffer (5% milk in TBS + 0.1% tween). Membranes were subsequently incubated with primary antibodies diluted in antibody blocking buffer for 2 hr at room temperature or overnight at 4°C. Anti-BRCA1 (Santa Cruz sc-6954 1:100), anti-BRCA2 (EMD Millipore OP95 1:50), anti-RAD18 (Cell Signaling 9040S 1:5,000–1:20,000), anti-REV1 (Santa Cruz sc-393022 1:100), anti-PCNA (Thermo Fisher MA5–11358 1:5,000), anti-PCNA-Ub (Cell Signaling 13439S 1:2,000), anti-PRIMPOL ((Mouron et al., 2013), 1:500), anti-V5 (Thermo Fisher R960–25 1:5,000), anti-tubulin (Novus Biologicals NB600–506 1:50,000), and anti-vinculin (Sigma V9131 1:100,000) primary antibodies were used. Detection was achieved using appropriate horseradish peroxidase-conjugated secondary antibodies.
Quantitative real-time PCR
Total RNA was isolated using RNeasy kit (Qiagen). Reverse transcription was carried out with 1 μg of total RNA using random hexamer primers and the SuperScript III reverse transcriptase kit (Thermo Fisher Scientific). Equal amounts of cDNA were mixed with the Power SYBR green PCR master mix (Thermo Fisher Scientific) and run on a Stratagene MX3005 Real-Time PCR System. PRIMPOL mRNA levels were determined by comparing threshold cycle values for each experimental condition relative to GAPDH mRNA levels. Each experiment was repeated 3 times independently.
Immunofluorescence
Twenty four hours after siRNA or Cas9/sgRNA RNP transfection, U2OS cells were seeded on black 96-well bottom-glass plates. After 3 additional days of incubation, cells were simultaneously fixed and permeabilized (4% paraformaldehyde, 0.5% Triton X-100) for 10 min at room temperature. Cells were incubated in blocking solution (3% BSA in TBS-Tween 0.1%) for 1 hr and then with primary antibody diluted in blocking solution for 1.5 hr at room temperature or overnight at 4°C. Cells were washed 3 times with TBS-T and then incubated for 1 hr at room temperature with the appropriate secondary antibody, Alexa Fluor 488-labeled anti-rabbit and Alexa Fluor 594-labeled goat anti-mouse at 1:1,000 dilution (A-11008 and A-11005, Thermo Fisher Scientific). After three washes in TBS-T, cells were incubated with DAPI for 5 min at room temperature to counterstain nuclei. Multi-color acquisitions were made using the ImageXpress Nano Automated Imaging System microscope (Molecular Devices) equipped with a 40× Plan Apo objective (0.95 numerical aperture). An integrated imaging software (MetaXpress 6) was used for image analysis. At least 300 cells per experimental point were analyzed, and each experiment was repeated at least 2 times independently.
Cell cycle and apoptosis
After transfection of the indicated siRNAs or Cas9/sgRNA RNP complexes, cells were pulse-labeled with 30 μM BrdU for 30 min, collected and fixed overnight in ice-cold 70% ethanol. DNA denaturation was performed using a solution of 0.2 mg/ml of pepsin (Sigma-Aldrich) in 2 M HCl. BrdU was detected using an anti-BrdU-FITC conjugated antibody (AbD Serotec) and a solution of propidium iodide/RNaseA (10 μg/ml and 0.1 mg/ml, respectively) was used for total DNA staining. Apoptosis was assayed using the Annexin V-FITC Apoptosis Staining kit (Abcam). Cell cycle distribution and apoptosis was determined using the BD LSR Fortessa machine and data processed using FlowJo v10 software. Each experiment was repeated 3 or more times independently.
3D-Matrigel organoid assay
One hundred microliters of Matrigel were added to each well of an 8-well chamber slide (BD Falcon CultureSlide) and spread evenly in the well. The slides were then placed in a cell culture incubator to allow the Matrigel to gel completely (15 min). UWB1.289 cells were collected, resuspended in regular media and seeded on Matrigel at a density of 1,000 cells per well. Cells were grown in the incubator for 20 days and supplemented every 5 days with fresh media plus DMSO or JH-RE-06 (1 μM). UWB1.289 organoids were imaged using the Evos Cell Imaging System and quantified by counting the number of organoids in each well. Each experiment was repeated 3 times independently.
Xenograft studies
BRCA1-deficient and BRCA1-reconstituted MDA-MB-436 cells (2 × 106) were injected subcutaneously into flanks of female, 6–8 week-old NU/J (nude mice from Jackson laboratories) mice. After the tumors grew to a volume of at least 50 mm3, control (vehicle) or JH-RE-06 treatment (2 mg/kg) was administered, with a total volume of 100 μl injected directly into the tumor. Caliper measurements and treatments were carried out every three days starting from day 7 after the first caliper measurement for a period of 46 days. The mice were sedated with isoflurane prior to measurements and treatments. JH-RE-06 was formulated in 10% EtOH, 40% PEG400, and 50% saline. Tumor volumes were calculated by the formula V= (L × W2)/2. Each experiment was repeated two times independently.
Kaplan-Meier analysis
Patients survival analysis was performed on the ovarian serous cystadenocarcinoma TCGA dataset. All 585 cases and subgroups of cases carrying non-silent mutations or homozygous deletions in a core set of HR genes (BARD1, BRCA1, BRCA2, PALB2, RAD51) or an extended set of HR genes (BARD1, BRCA1, BRCA2, CDK12, PALB2, RAD50, RAD51, RAD51B, RAD51D, RAD52, RAD54L, XRCC2) were selected and stratified by normalized REV3L and RAD18 expression. Cases were classified into altered groups with z-scores of REV3L and/or RAD18 expression <−1, while unaltered group includes all the other cases and defines z-scores of REV3L and/or RAD18 expression ≥−1. Kaplan-Meier curves for overall survival were generated using cBioPortal (https://www.cbioportal.org/). P-value analysis by log-rank test of altered vs unaltered groups is shown.
QUANTIFICATION AND STATISTICAL ANALYSIS
All quantitative data are presented as mean ± SEM, as indicated, of at least three independent experiments or biological replicates. Statistical analyses were performed using GraphPad Prism 7 and Microsoft Excel. P-values were calculated as described in the figure legend for each experiment. P < 0.05 was considered to be statistically significant. Data shown from DNA fiber and HPRT gene mutation assays, 3D-organoid and xenograft tumor studies, and immunofluorescence and western blot assays are representative of two or more independent experiments with similar results.
Supplementary Material
TABLE S1, Related to Figure 5. Rate of de novo mutations at the HPRT locus
Average number ± SD of 6-TG resistant colonies per 105 cells. After the indicated treatments (Figure 5F–G), HCT116 cells were seeded in equal numbers and HPRT mutants were selected with 6-TG (5 μg/ml). The number of de novo mutants was calculated by subtraction of the number of background mutants and normalization to plating efficiency for each condition.
TABLE S2, Related to STAR Methods. List of oligonucleotides used in this study
List of oligonucleotides utilized in the study, along with their source and sequence.
Highlights.
ssDNA gaps arise in BRCA1-mutant cancer cells due to PRIMPOL-mediated repriming
BRCA1/2 deficiency leads to mutagenic ssDNA gap repair by REV1-Polζ-dependent TLS
Targeted REV1-Polζ inhibition shows enhanced toxicity in HR-deficient cancer cells
ssDNA gaps formed by SMUG1 and PRIMPOL mediate the toxicity of REV1-Polζ inhibition
ACKNOWLEDGMENTS
The authors would like to thank Shao Anqi, Lillian Lawrence, Lorraine Symington, Chao Lu, Xiao Chen and all members of Alberto Ciccia’s laboratory for helpful suggestions and critical discussions, and assistance in the generation of preliminary data. PRIMPOL KO U2OS cells and UWB1.289 cells, HCC1937 cells, and MDA-MB-436 cells were kindly provided by Alessandro Vindigni, Simon Powell and Neil Johnson, respectively. The PRIMPOL antibody and WT and mutant PRIMPOL constructs were generously provided by Juan Mendez. The doxycycline-inducible SpCas9 construct was generated by Pierre Billion. This work was supported by NIH grants R01CA197774, R01CA227450 and P01CA174653 to A.C., and the Italian Association for Cancer Research (AIRC) AIRC-IG Ref: 21824 and Fondazione Regionale per la Ricerca Biomedica (FIRBB) grants to V.C. G.L. was supported by an Italian Association for Cancer Research (AIRC) post-doctoral fellowship and R.C.-M. by an EMBO Long-Term Fellowship (ALTF 366–2019). These studies used the HICCC Flow Cytometry Shared Resource supported in part by the NIH award S10RR027050 and through Center Grant P30CA013696.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. (2013). Signatures of mutational processes in human cancer. Nature 500, 415–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsoe L, Sarno A, Carracedo S, Domanska D, Dingler F, Lirussi L, SenGupta T, Tekin NB, Jobert L, Alexandrov LB, et al. (2017). Uracil Accumulation and Mutagenesis Dominated by Cytosine Deamination in CpG Dinucleotides in Mice Lacking UNG and SMUG1. Scientific reports 7, 7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou P, and Fostira F (2013). Hereditary breast cancer: the era of new susceptibility genes. Biomed Res Int 2013, 747318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai G, Kermi C, Stoy H, Schiltz CJ, Bacal J, Zaino AM, Hadden MK, Eichman BF, Lopes M, and Cimprich KA (2020). HLTF Promotes Fork Reversal, Limiting Replication Stress Resistance and Preventing Multiple Mechanisms of Unrestrained DNA Synthesis. Mol Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi J, Rudd SG, Jozwiakowski SK, Bailey LJ, Soura V, Taylor E, Stevanovic I, Green AJ, Stracker TH, Lindsay HD, et al. (2013). PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol Cell 52, 566–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordin DL, Lirussi L, and Nilsen H (2021). Cellular response to endogenous DNA damage: DNA base modifications in gene expression regulation. DNA Repair (Amst) 99, 103051. [DOI] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. (2010). 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, and Szakal B (2017). Building up and breaking down: mechanisms controlling recombination during replication. Crit Rev Biochem Mol Biol 52, 381–394. [DOI] [PubMed] [Google Scholar]
- Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, Kyle S, Meuth M, Curtin NJ, and Helleday T (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JY, Lim S, Kim EJ, Jo A, and Guengerich FP (2010). Translesion synthesis across abasic lesions by human B-family and Y-family DNA polymerases alpha, delta, eta, iota, kappa, and REV1. J Mol Biol 404, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong K, Peng M, Kousholt AN, Lee WTC, Lee S, Nayak S, Krais J, VanderVere-Carozza PS, Pawelczak KS, Calvo J, et al. (2021). Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Molecular Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groote FH, Jansen JG, Masuda Y, Shah DM, Kamiya K, de Wind N, and Siegal G (2011). The Rev1 translesion synthesis polymerase has multiple distinct DNA binding modes. DNA Repair (Amst) 10, 915–925. [DOI] [PubMed] [Google Scholar]
- Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, Lin WC, Yu WH, Leonard PG, Lee G.R.t., et al. (2016). Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med 22, 194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Fei L, and Xu H (2018). Role of MCM2–7 protein phosphorylation in human cancer cells. Cell Biosci 8, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng W, and Jasin M (2017). BRCA2 suppresses replication stress-induced mitotic and G1 abnormalities through homologous recombination. Nature communications 8, 525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridlich R, Annamalai D, Roy R, Bernheim G, and Powell SN (2015). BRCA1 and BRCA2 protect against oxidative DNA damage converted into double-strand breaks during DNA replication. DNA Repair (Amst) 30, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugger K, Bajrami I, Silva Dos Santos M, Young SJ, Kunzelmann S, Kelly G, Hewitt G, Patel H, Goldstone R, Carell T, et al. (2021). Targeting the nucleotide salvage factor DNPH1 sensitizes BRCA-deficient cells to PARP inhibitors. Science 372, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii S, Isogawa A, and Fuchs RP (2018). Chronological Switch from Translesion Synthesis to Homology-Dependent Gap Repair In Vivo. Toxicol Res 34, 297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo D, Kim T, Szakal B, Saayman X, Narula A, Park Y, Branzei D, Zhang Z, and Brown GW (2019). Rad5 Recruits Error-Prone DNA Polymerases for Mutagenic Repair of ssDNA Gaps on Undamaged Templates. Mol Cell 73, 900–914 e909. [DOI] [PubMed] [Google Scholar]
- Garcia-Gomez S, Reyes A, Martinez-Jimenez MI, Chocron ES, Mouron S, Terrados G, Powell C, Salido E, Mendez J, Holt IJ, et al. (2013). PrimPol, an archaic primase/polymerase operating in human cells. Mol Cell 52, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Rodriguez N, Wong RP, and Ulrich HD (2016). Functions of Ubiquitin and SUMO in DNA Replication and Replication Stress. Front Genet 7, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogola E, Rottenberg S, and Jonkers J (2019). Resistance to PARP Inhibitors: Lessons from Preclinical Models of BRCA-Associated Cancer. Annual Review of Cancer Biology 3, 235–254. [Google Scholar]
- Gonzalez-Acosta D, Blanco-Romero E, Ubieto-Capella P, Mutreja K, Miguez S, Llanos S, Garcia F, Munoz J, Blanco L, Lopes M, et al. (2021). PrimPol-mediated repriming facilitates replication traverse of DNA interstrand crosslinks. EMBO J 40, e106355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Sonoda E, Tang TS, Parker JL, Bielen AB, Takeda S, Ulrich HD, and Friedberg EC (2006). REV1 protein interacts with PCNA: significance of the REV1 BRCT domain in vitro and in vivo. Mol Cell 23, 265–271. [DOI] [PubMed] [Google Scholar]
- Haracska L, Unk I, Johnson RE, Johansson E, Burgers PM, Prakash S, and Prakash L (2001). Roles of yeast DNA polymerases delta and zeta and of Rev1 in the bypass of abasic sites. Genes Dev 15, 945–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto Y, Ray Chaudhuri A, Lopes M, and Costanzo V (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17, 1305–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G, Borel V, Segura-Bayona S, Takaki T, Ruis P, Bellelli R, Lehmann LC, Sommerova L, Vancevska A, Tomas-Loba A, et al. (2021). Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD. Mol Cell 81, 767–783 e711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, and Jentsch S (2002). RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 419, 135–141. [DOI] [PubMed] [Google Scholar]
- Iwai K, Nambu T, Dairiki R, Ohori M, Yu J, Burke K, Gotou M, Yamamoto Y, Ebara S, Shibata S, et al. (2019). Molecular mechanism and potential target indication of TAK-931, a novel CDC7-selective inhibitor. Sci Adv 5, eaav3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GE (2012). Mammalian cell HPRT gene mutation assay: test methods. Methods Mol Biol 817, 55–67. [DOI] [PubMed] [Google Scholar]
- Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, Wang Y, Capelletti M, Sarosiek KA, Moreau LA, et al. (2013). Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci U S A 110, 17041–17046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph SA, Taglialatela A, Leuzzi G, Huang JW, Cuella-Martin R, and Ciccia A (2020). Time for remodeling: SNF2-family DNA translocases in replication fork metabolism and human disease. DNA Repair (Amst). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavli B, Sundheim O, Akbari M, Otterlei M, Nilsen H, Skorpen F, Aas PA, Hagen L, Krokan HE, and Slupphaug G (2002). hUNG2 is the major repair enzyme for removal of uracil from U:A matches, U:G mismatches, and U in single-stranded DNA, with hSMUG1 as a broad specificity backup. J Biol Chem 277, 39926–39936. [DOI] [PubMed] [Google Scholar]
- Kim N, Mudrak SV, and Jinks-Robertson S (2011). The dCMP transferase activity of yeast Rev1 is biologically relevant during the bypass of endogenously generated AP sites. DNA Repair (Amst) 10, 1262–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Guilliam TA, Tsuda M, Yamamoto J, Bailey LJ, Iwai S, Takeda S, Doherty AJ, and Hirota K (2016). Repriming by PrimPol is critical for DNA replication restart downstream of lesions and chain-terminating nucleosides. Cell Cycle 15, 1997–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenova OV, Daee DL, Mertz TM, and Shcherbakova PV (2015). DNA polymerase zeta-dependent lesion bypass in Saccharomyces cerevisiae is accompanied by error-prone copying of long stretches of adjacent DNA. PLoS Genet 11, e1005110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Techer H, Baldi G, Shen R, Ciccia A, Pellegrini L, et al. (2017). Smarcal1-Mediated Fork Reversal Triggers Mre11-Dependent Degradation of Nascent DNA in the Absence of Brca2 and Stable Rad51 Nucleofilaments. Mol Cell 67, 867–881 e867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Ramazzotti D, Weng Z, Liu K, Ford JM, and Sidow A (2019). Comprehensive genomic characterization of breast tumors with BRCA1 and BRCA2 mutations. BMC Med Genomics 12, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemacon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N, et al. (2017). MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81-dependent fork rescue in BRCA2-deficient cells. Nature communications 8, 860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Ryu KS, Ko J, Sun D, Lim K, Lee JO, Hwang J, Lee ZW, and Choi BS (2013). Insights into the regulation of human Rev1 for translesion synthesis polymerases revealed by the structural studies on its polymerase-interacting domain. J Mol Cell Biol 5, 204–206. [DOI] [PubMed] [Google Scholar]
- Loeillet S, Herzog M, Puddu F, Legoix P, Baulande S, Jackson SP, and Nicolas AG (2020). Trajectory and uniqueness of mutational signatures in yeast mutators. Proc Natl Acad Sci U S A 117, 24947–24956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes M, Foiani M, and Sogo JM (2006). Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol Cell 21, 15–27. [DOI] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2016). BRCAness revisited. Nat Rev Cancer 16, 110–120. [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. (2011). 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol 13, 243–253. [DOI] [PubMed] [Google Scholar]
- Ma W, Westmoreland JW, and Resnick MA (2013). Homologous recombination rescues ssDNA gaps generated by nucleotide excision repair and reduced translesion DNA synthesis in yeast G2 cells. Proc Natl Acad Sci U S A 110, E2895–2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaoka A, Matsubara M, Hasegawa R, Tanaka T, Kurisu S, Terato H, Ohyama Y, Karino N, Matsuda A, and Ide H (2003). Mammalian 5-formyluracil-DNA glycosylase. 2. Role of SMUG1 uracil-DNA glycosylase in repair of 5-formyluracil and other oxidized and deaminated base lesions. Biochemistry 42, 5003–5012. [DOI] [PubMed] [Google Scholar]
- Mehta KPM, Lovejoy CA, Zhao R, Heintzman DR, and Cortez D (2020). HMCES Maintains Replication Fork Progression and Prevents Double-Strand Breaks in Response to APOBEC Deamination and Abasic Site Formation. Cell reports 31, 107705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, et al. (2017). Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nature communications 8, 859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohni KN, Wessel SR, Zhao R, Wojciechowski AC, Luzwick JW, Layden H, Eichman BF, Thompson PS, Mehta KPM, and Cortez D (2019). HMCES Maintains Genome Integrity by Shielding Abasic Sites in Single-Strand DNA. Cell 176, 144–153 e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Brotherton D, Troiani S, Rainoldi S, Tenca P, Molinari A, and Santocanale C (2006). Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J Biol Chem 281, 10281–10290. [DOI] [PubMed] [Google Scholar]
- Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C, et al. (2008). A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat Chem Biol 4, 357–365. [DOI] [PubMed] [Google Scholar]
- Mouron S, Rodriguez-Acebes S, Martinez-Jimenez MI, Garcia-Gomez S, Chocron S, Blanco L, and Mendez J (2013). Repriming of DNA synthesis at stalled replication forks by human PrimPol. Nat Struct Mol Biol 20, 1383–1389. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Chiu JW, Koller BH, and Jasin M (1999). BRCA1 controls homology-directed DNA repair. Mol Cell 4, 511–518. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Pierce AJ, and Jasin M (2001). BRCA2 is required for homology-directed repair of chromosomal breaks. Molec. Cell. 7, 263–272. [DOI] [PubMed] [Google Scholar]
- Naiman K, Pages V, and Fuchs RP (2016). A defect in homologous recombination leads to increased translesion synthesis in E. coli. Nucleic Acids Res 44, 7691–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambiar TS, Billon P, Diedenhofen G, Hayward SB, Taglialatela A, Cai K, Huang JW, Leuzzi G, Cuella-Martin R, Palacios A, et al. (2019). Stimulation of CRISPR-mediated homology-directed repair by an engineered RAD18 variant. Nat Commun 10, 3395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak S, Calvo JA, Cong K, Peng M, Berthiaume E, Jackson J, Zaino AM, Vindigni A, Hadden MK, and Cantor SB (2020). Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci Adv 6, eaaz7808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neelsen KJ, Chaudhuri AR, Follonier C, Herrador R, and Lopes M (2014). Visualization and interpretation of eukaryotic DNA replication intermediates in vivo by electron microscopy. Methods in molecular biology 1094, 177–208. [DOI] [PubMed] [Google Scholar]
- Nelson JR, Lawrence CW, and Hinkle DC (1996). Deoxycytidyl transferase activity of yeast REV1 protein. Nature 382, 729–731. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, et al. (2012). Mutational processes molding the genomes of 21 breast cancers. Cell 149, 979–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. (2016). Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen H, Haushalter KA, Robins P, Barnes DE, Verdine GL, and Lindahl T (2001). Excision of deaminated cytosine from the vertebrate genome: role of the SMUG1 uracil-DNA glycosylase. EMBO J 20, 4278–4286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzarino NJ, Krais JJ, Cong K, Peng M, Mosqueda M, Nayak SU, Bond SM, Calvo JA, Doshi MB, Bere M, et al. (2021). Replication Gaps Underlie BRCA Deficiency and Therapy Response. Cancer Res 81, 1388–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SM, Dash RC, and Hadden MK (2021). Translesion synthesis inhibitors as a new class of cancer chemotherapeutics. Expert Opin Investig Drugs 30, 13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Bade S, Le Guillou M, Burke K, Reed R, Bowman-Colin C, Su Y, Ting DT, Polyak K, Richardson AL, et al. (2014). BRCA1 haploinsufficiency for replication stress suppression in primary cells. Nature communications 5, 5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania S, Nguyen J, Hill SJ, Scully R, Adelmant GO, Marto JA, Feunteun J, and Livingston DM (2011). BRCA1 is required for postreplication repair after UV-induced DNA damage. Mol Cell 44, 235–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen HS, Sundheim O, Gilljam KM, Slupphaug G, Krokan HE, and Kavli B (2007). Uracil-DNA glycosylases SMUG1 and UNG2 coordinate the initial steps of base excision repair by distinct mechanisms. Nucleic Acids Res 35, 3879–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piberger AL, Bowry A, Kelly RDW, Walker AK, Gonzalez-Acosta D, Bailey LJ, Doherty AJ, Mendez J, Morris JR, Bryant HE, et al. (2020). PrimPol-dependent single-stranded gap formation mediates homologous recombination at bulky DNA adducts. Nature communications 11, 5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilzecker B, Buoninfante OA, Pritchard C, Blomberg OS, Huijbers IJ, van den Berk PC, and Jacobs H (2016). PrimPol prevents APOBEC/AID family mediated DNA mutagenesis. Nucleic Acids Res 44, 4734–4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozhidaeva A, Pustovalova Y, D’Souza S, Bezsonova I, Walker GC, and Korzhnev DM (2012). NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase eta. Biochemistry 51, 5506–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pustovalova Y, Magalhaes MT, D’Souza S, Rizzo AA, Korza G, Walker GC, and Korzhnev DM (2016). Interaction between the Rev1 C-Terminal Domain and the PolD3 Subunit of Polzeta Suggests a Mechanism of Polymerase Exchange upon Rev1/Polzeta-Dependent Translesion Synthesis. Biochemistry 55, 2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Carvajal-Maldonado D, Lemacon D, and Vindigni A (2017). DNA Fiber Analysis: Mind the Gap! Methods Enzymol 591, 55–82. [DOI] [PubMed] [Google Scholar]
- Quinet A, Martins DJ, Vessoni AT, Biard D, Sarasin A, Stary A, and Menck CF (2016). Translesion synthesis mechanisms depend on the nature of DNA damage in UV-irradiated human cells. Nucleic Acids Res 44, 5717–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinet A, Tirman S, Jackson J, Svikovic S, Lemacon D, Carvajal-Maldonado D, Gonzalez-Acosta D, Vessoni AT, Cybulla E, Wood M, et al. (2020). PRIMPOL-Mediated Adaptive Response Suppresses Replication Fork Reversal in BRCA-Deficient Cells. Mol Cell 77, 461–474 e469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja S, and Van Houten B (2021). The Multiple Cellular Roles of SMUG1 in Genome Maintenance and Cancer. Int J Mol Sci 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Acebes S, Mouron S, and Mendez J (2018). Uncoupling fork speed and origin activity to identify the primary cause of replicative stress phenotypes. J Biol Chem 293, 12855–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JC, Bonilla B, Hengel SR, Mertz TM, Herken BW, Kazemier HG, Pressimone CA, Ratterman TC, MacNary E, De Magis A, et al. (2019). The Rad51 paralogs facilitate a novel DNA strand specific damage tolerance pathway. Nature communications 10, 3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupp WD, Wilde CE, Reno DL, and Howard-Flanders P (1971). Exchanges between DNA strands in ultraviolet-irradiated Escherichia coli. J. Mol. Biol. 61, 25–44. [DOI] [PubMed] [Google Scholar]
- Schlacher K, Christ N, Siaud N, Egashira A, Wu H, and Jasin M (2011). Double-strand break repair-independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145, 529–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlacher K, Wu H, and Jasin M (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachar S, Ziv O, Avkin S, Adar S, Wittschieben J, Reissner T, Chaney S, Friedberg EC, Wang Z, Carell T, et al. (2009). Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J 28, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Spies J, Coscia F, Kirik U, Rask MB, Lee JH, Neelsen KJ, Mund A, Jensen LJ, Paull TT, et al. (2021). Homology-directed repair protects the replicating genome from metabolic assaults. Developmental cell 56, 461–477 e467. [DOI] [PubMed] [Google Scholar]
- Spies J, Lukas C, Somyajit K, Rask MB, Lukas J, and Neelsen KJ (2019). 53BP1 nuclear bodies enforce replication timing at under-replicated DNA to limit heritable DNA damage. Nat Cell Biol 21, 487–497. [DOI] [PubMed] [Google Scholar]
- Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, Madubata C, Anand R, Levy B, Rabadan R, et al. (2017). Restoration of Replication Fork Stability in BRCA1- and BRCA2-Deficient Cells by Inactivation of SNF2-Family Fork Remodelers. Mol Cell 68, 414–430 e418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakar T, Leung W, Nicolae CM, Clements KE, Shen B, Bielinsky AK, and Moldovan GL (2020). Ubiquitinated-PCNA protects replication forks from DNA2-mediated degradation by regulating Okazaki fragment maturation and chromatin assembly. Nature communications 11, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PS, Amidon KM, Mohni KN, Cortez D, and Eichman BF (2019). Protection of abasic sites during DNA replication by a stable thiazolidine protein-DNA cross-link. Nat Struct Mol Biol 26, 613–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PS, and Cortez D (2020). New insights into abasic site repair and tolerance. DNA Repair (Amst) 90, 102866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich HD, and Walden H (2010). Ubiquitin signalling in DNA replication and repair. Nat Rev Mol Cell Biol 11, 479–489. [DOI] [PubMed] [Google Scholar]
- Vaisman A, and Woodgate R (2017). Translesion DNA polymerases in eukaryotes: what makes them tick? Crit Rev Biochem Mol Biol 52, 274–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villafanez F, Garcia IA, Carbajosa S, Pansa MF, Mansilla S, Llorens MC, Angiolini V, Guantay L, Jacobs H, Madauss KP, et al. (2019). AKT inhibition impairs PCNA ubiquitylation and triggers synthetic lethality in homologous recombination-deficient cells submitted to replication stress. Oncogene 38, 4310–4324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L, Lou J, Xia Y, Su B, Liu T, Cui J, Sun Y, Lou H, and Huang J (2013). hPrimpol1/CCDC111 is a human DNA primase-polymerase required for the maintenance of genome integrity. EMBO Rep 14, 1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters LS, Minesinger BK, Wiltrout ME, D’Souza S, Woodruff RV, and Walker GC (2009). Eukaryotic translesion polymerases and their roles and regulation in DNA damage tolerance. Microbiol Mol Biol Rev 73, 134–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weerasooriya S, Jasti VP, and Basu AK (2014). Replicative bypass of abasic site in Escherichia coli and human cells: similarities and differences. PLoS One 9, e107915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigelt B, Ghajar CM, and Bissell MJ (2014). The need for complex 3D culture models to unravel novel pathways and identify accurate biomarkers in breast cancer. Adv Drug Deliv Rev 69–70, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West SC, Cassuto E, and Howard-Flanders P (1982). Postreplication repair in E. coli: Strand exchange reactions of gapped DNA by RecA protein. Mol. Gen. Genet. 187, 209–217. [DOI] [PubMed] [Google Scholar]
- Wojtaszek JL, Chatterjee N, Najeeb J, Ramos A, Lee M, Bian K, Xue JY, Fenton BA, Park H, Li D, et al. (2019). A Small Molecule Targeting Mutagenic Translesion Synthesis Improves Chemotherapy. Cell 178, 152–159 e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chatterjee N, Hemann MT, and Walker GC (2017). Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet 13, e1006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita YM, Okada T, Matsusaka T, Sonoda E, Zhao GY, Araki K, Tateishi S, Yamaizumi M, and Takeda S (2002). RAD18 and RAD54 cooperatively contribute to maintenance of genomic stability in vertebrate cells. EMBO J 21, 5558–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, and Gao Y (2018). Translesion and Repair DNA Polymerases: Diverse Structure and Mechanism. Annu Rev Biochem 87, 239–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gao Y, Mutter-Rottmayer L, Zlatanou A, Durando M, Ding W, Wyatt D, Ramsden D, Tanoue Y, Tateishi S, et al. (2017). DNA repair factor RAD18 and DNA polymerase Polkappa confer tolerance of oncogenic DNA replication stress. J Cell Biol 216, 3097–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamborszky J, Szikriszt B, Gervai JZ, Pipek O, Poti A, Krzystanek M, Ribli D, Szalai-Gindl JM, Csabai I, Szallasi Z, et al. (2017). Loss of BRCA1 or BRCA2 markedly increases the rate of base substitution mutagenesis and has distinct effects on genomic deletions. Oncogene 36, 746–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, and Xia F (2004). Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol 24, 708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Garg P, Stith CM, Nick McElhinny SA, Kissling GE, Burgers PM, and Kunkel TA (2006). The fidelity of DNA synthesis by yeast DNA polymerase zeta alone and with accessory proteins. Nucleic Acids Res 34, 4731–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1, Related to Figure 5. Rate of de novo mutations at the HPRT locus
Average number ± SD of 6-TG resistant colonies per 105 cells. After the indicated treatments (Figure 5F–G), HCT116 cells were seeded in equal numbers and HPRT mutants were selected with 6-TG (5 μg/ml). The number of de novo mutants was calculated by subtraction of the number of background mutants and normalization to plating efficiency for each condition.
TABLE S2, Related to STAR Methods. List of oligonucleotides used in this study
List of oligonucleotides utilized in the study, along with their source and sequence.
Data Availability Statement
Unprocessed western blot and microscopy images have been deposited at Mendeley Data (DOI: 10.17632/rmyvkwbmyn.1) and are publicly available as of the date of publication. Source data for Figure S7E–G are available at https://www.cbioportal.org/. Microscopy data reported in this paper will be shared by the lead contact upon request.
No original code has been generated for this publication.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.