

Abstract
Lactiplantibacillus plantarum, previously named “Lactobacillus plantarum,” is found in a wide variety of environments exhibiting a high level of intraspecies genetic diversity. To investigate the strain diversity, we performed comparative genomic analyses of the 54 complete genome sequences. The results revealed that L. plantarum subsp. plantarum was split into three lineages, A, B and C. Of the genes beneficial for probiotic activity, only those associated with the biosynthesis of plantaricin (Pln), an L. plantarum-specific bacteriocin, were found to be significantly different among the lineages. The genes related to the biosynthesis of plnE/F were conserved throughout the three lineages, whereas the outgroups did not possess any Pln-producing genes. In lineage C, the deepest and ancestral type branch, plnE/F genes, were well conserved. In lineage B, loss of gene function was observed due to mobile elements in the pln loci. In lineage A, most strains were predicted to produce more than one type of Pln by possessing diverse Pln-encoding genes. These results showed the presence of functional diversity arising from the trifurcating evolution in L. plantarum subsp. plantarum and demonstrated that Pln is an indicator for differentiating the three lineages.
Subject terms: Bacterial evolution, Bacterial genomics
Introduction
“Lactobacillus plantarum” was reclassified as Lactiplantibacillus plantarum1 and currently encompasses two subspecies, namely, L. plantarum subsp. plantarum and L. plantarum subsp. argentoratensis. L. plantarum is widely used as a probiotic because of its health benefits and the history of safe use. Because Lactiplantibacillus is present in the normal human flora and aids in the control of pathogens, its application in the development of microbiome therapy products has increased2. L. plantarum is used in the fermentation of vegetables and dairy products and is found in a variety of fermented products worldwide. As L. plantarum is found in diverse habitats, it has evolved in diverse ways, and as a result, a high level of intraspecies genetic diversity is found in this species3. Diversity in strains may be advantageous for industrial applications but is disadvantageous with regard to the safety of food products. Comparative gene analysis is currently being used to further investigate this thoroughly.
According to previous studies, the L. plantarum genome (3.3 Mb) is larger than the typical genomes of other lactic acid bacteria (LAB; 2–2.7 Mb). The large genome size of L. plantarum suggests a very high level of intraspecies genetic diversity, and this characteristic is thought to be because of the nomadic lifestyle of this species, which lives in a variety of habitats and has a large metabolic diversity4,5. Owing to the high level of intraspecies diversity, it is not easy to categorize L. plantarum strains based on simple traits. Previous comparative genomic analysis studies have consistently demonstrated that the evolution of L. plantarum is not related to the isolation source or the geographical location of the strains belonging to this species5,6. However, differences in few gene clusters have been found among L. plantarum strains. Siezen et al. analyzed six strains in 2011 and reported large differences in prophages, IS elements, transposases, and plantaricin (Pln) biosynthesis genes among the strains and found high variability in capsular polysaccharide and extracellular polysaccharide biosynthesis genes7. In 2016, Martino et al. analyzed the presence/absence of orthologous genes using 54 strains and demonstrated that L. plantarum strains separated into two phenotypic clusters based on their extracellular polysaccharides, secretome, and sugar metabolism5. In 2018, Choi et al. showed that 108 strains could be classified into five phenotypic clusters based on their carbohydrate utilization, virulence, and metabolism6. A comparison of 23 strains by Yu et al. in 2017 showed that few strains have evolved to contain a clustered regularly interspaced short palindromic repeat region, antimicrobial activity, and detoxification activity8. However, most of these studies classified the strains into phenotypic clusters based on certain functional genes; therefore, they did not examine the gene clusters based on phylogenetic relationships.
Bacteriocins are narrow- or broad-spectrum antimicrobial peptides produced by wide range of prokaryotes and are classified into several categories. Many LAB produce bacteriocins which can be applied to food preservation or food safety applications9. Recently, bacteriocins are predicted to have a positive effect on the human intestine by maintaining the balance of bacteria in the gastro-intestinal tract microbiome and affecting the host immune system, which may be of value in disease treatment and health improvement10. Lactiplantibacillus plantarum is known to produce class I and II bacteriocins, including Plns A-Y, NC81F, NC8HK, and NC8βα11. A new L. plantarum strain harboring genes coding for different bacteriocins rather than conventional plantaricins and showing broad inhibitory spectrum is discovered recently12. The bacteriocins produced by L. plantarum are known to reduce the gastrointestinal diseases by inhibiting the growth of pathogens like as Staphylococcus aureus and Listeria13 and also known to possess antagonist effects against food spoilage14. Thus, the production of bacteriocin by L. plantarum is one of the important criteria for probiotic strain selection.
The present study aimed to investigate the evolutionary trend in L. plantarum from a phylogenetic perspective. We first performed phylogenomics analysis to subdivide the species L. plantarum subsp. plantarum solely on the basis of phylogenetic relationships and aimed to determine whether certain functional genes can be used to differentiate among the intraspecies gene clusters. Using only complete genome sequences, we attempted to minimize the analytic errors that occur from the use of low-quality draft genomes. We focused on a variety of functional genes, including those associated with carbohydrate utilization and substance biosynthesis, to determine whether there is a significant difference between the groups and to predict whether this difference can affect the role of each subgroup as a probiotic.
Results
General characteristics of the L. plantarum subsp. plantarum genomes
Examination of the complete genomes of 54 L. plantarum subsp. plantarum strains revealed that the genome size ranged from 2.95 to 3.70 Mb, G + C content ranged from 44.08 to 44.93%, and the number of coding sequences (CDSs) ranged from 2729 to 3478 (Table 1). Of the 54 strains studied, 41 contained plasmids in addition to the chromosomes. The core genome consisted of 2207 orthologous gene families, and the pan-genome consisted of 7323 orthologous gene families (Supplementary Fig. S1A). Estimation of openness based on the Heaps’ Law model showed that the L. plantarum subsp. plantarum pan-genome was open with a parameter (γ) of 0.66 (Supplementary Fig. S1B). It was estimated that approximately 40 new gene families will be added to the pan-genome each time new genomic information on this species is added.
Table 1.
List of the complete genome sequences analyzed in this study.
| Strain | Assembly | Country | Isolation source | Predicted CDSs | Genome size (bp) | GC ratio |
|---|---|---|---|---|---|---|
| LP2 | GCA_002109425.1 | China | Fermented vegetable | 3015 | 3,284,622 | 44.55 |
| ST-III | GCA_000148815.2 | Korea | Fermented kimchi | 3019 | 3,307,936 | 44.48 |
| PC520 | GCA_002576835.1 | China | Fermented vegetable | 3209 | 3,452,904 | 44.33 |
| CAUH2 | GCA_001617525.1 | China | Fermented vegetable | 2989 | 3,274,625 | 44.52 |
| LZ95 | GCA_001484005.1 | China | Infant feces | 3053 | 3,322,458 | 44.47 |
| dm | GCA_002220175.1 | USA | Drosophila melanogaster | 3081 | 3,325,676 | 44.52 |
| BDGP2 | GCA_002290185.1 | USA | Drosophila melanogaster | 3362 | 3,581,586 | 44.2 |
| KP | GCA_001704315.1 | Canada | Drosophila melanogaster | 3466 | 3,692,742 | 44.11 |
| DF | GCA_001704315.1 | Canada | Drosophila melanogaster | 3478 | 3,697,306 | 44.08 |
| KC28 | GCA_002948215.1 | Korea | Fermented vegetable | 3042 | 3,291,849 | 44.5 |
| RI-113 | GCA_001990145.1 | Switzerland | Fermented salami | 3226 | 3,462,990 | 44.32 |
| DSM 20174T | GCA_014131735.1 | Unknown | Fermented cabbage | 2968 | 3,250,154 | 44.50 |
| TMW 1.1623 | GCA_002117245.1 | Germany | Fermented food | 3061 | 3,332,882 | 44.35 |
| JBE245 | GCA_001596095.1 | Korea | Fermented soybean | 2987 | 3,262,611 | 44.48 |
| TMW 1.25 | GCA_002117245.1 | Germany | Fermented sausage | 3112 | 3,351,899 | 44.3 |
| TMW 1.277 | GCA_002117245.1 | Germany | Fermented wine | 3164 | 3,400,131 | 44.22 |
| LM1004 | GCA_002895245.1 | Korea | Fermented vegetable | 2937 | 3,198,690 | 44.59 |
| 5–2 | GCA_001278015.1 | China | Fermented soybean | 2963 | 3,237,652 | 44.66 |
| Zhang-LL | GCA_001581895.1 | China | Fermented rice | 2729 | 2,952,218 | 44.93 |
| LQ80 | GCA_003097595.1 | Japan | Pig feed plant | 3170 | 3,447,624 | 44.34 |
| SRCM100434 | GCA_002174195.1 | Korea | Fermented food | 2907 | 3,223,596 | 44.6 |
| TMW 1.708 | GCA_002117245.1 | Germany | Fermented sausage | 3001 | 3,246,485 | 44.56 |
| 10CH | GCA_002005385.2 | UK | Fermented cheese | 3012 | 3,311,056 | 44.51 |
| TS12 | GCA_001908455.1 | Malaysia | Fermented tofu | 3283 | 3,433,628 | 44.3 |
| WCFS1 | GCA_000203855.3 | England | Human saliva | 3053 | 3,348,624 | 44.42 |
| ZS2058 | GCA_001296095.1 | China | Fermented vegetable | 2900 | 3,198,337 | 44.66 |
| B21 | GCA_000931425.2 | Vietnam | Fermented sausage | 3030 | 3,310,674 | 44.42 |
| BLS41 | GCA_002116955.1 | Korea | Fermented vegetable | 3214 | 3,476,011 | 44.19 |
| CBT LP3 | GCA_002286275.1 | Korea | Fermented kimchi | 3035 | 3,329,954 | 44.44 |
| MF1298 | GCA_001880185.1 | Norway | Fermented sausage | 3272 | 3,564,579 | 44.22 |
| K259 | GCA_002868775.1 | Korea | Fermented vegetable | 3098 | 3,373,076 | 44.49 |
| LB1-2 | GCA_002906875.1 | Philippines | Apis mellifera | 3297 | 3,541,869 | 44.14 |
| DOMLa | GCA_000604105.1 | China | CDC | 2934 | 3,210,111 | 44.64 |
| JDM1 | GCA_000023085.1 | China | Unknown | 2923 | 3,197,759 | 44.66 |
| ATCC 8014 | GCA_002749655.1 | Unknown | Unknown | 3052 | 3,309,473 | 44.47 |
| KC3 | GCA_002868755.1 | Korea | Fermented vegetable | 3123 | 3,330,006 | 44.55 |
| GB-LP1 | GCA_002220815.1 | Korea | Fermented food | 2794 | 3,040,388 | 44.87 |
| X7021 | GCA_002943545.1 | China | Fermented tofu | 3201 | 3,407,054 | 44.38 |
| LPL-1 | GCA_002205775.2 | China | Fermented fish | 2942 | 3,200,572 | 44.64 |
| JBE490 | GCA_002109405.1 | Korea | Fermented rice | 2994 | 3,196,967 | 44.6 |
| ZJ316 | GCA_000338115.2 | China | Infant feces | 3068 | 3,299,755 | 44.49 |
| LZ206 | GCA_001659745.1 | China | Cow milk | 3103 | 3,263,715 | 44.55 |
| LZ227 | GCA_001660025.1 | China | Cow milk | 3230 | 3,425,292 | 44.35 |
| C410L1 | GCA_001874125.1 | China | Environment (mud) | 3182 | 3,392,777 | 44.43 |
| 16 | GCA_000412205.1 | England | Malt production steep water | 3076 | 3,361,015 | 44.34 |
| P-8 | GCA_000392485.2 | China | Fermented vegetable | 3027 | 3,246,630 | 44.55 |
| HFC8 | GCA_001302645.1 | India | Human feces | 3282 | 3,405,709 | 44.31 |
| LY-78 | GCA_001715615.1 | China | Fermented vegetable | 2866 | 3,128,783 | 44.74 |
| K25 | GCA_003020005.1 | China | Fermented milk | 3176 | 3,412,154 | 44.38 |
| CGMCC 1.557 | GCA_001272315.2 | China | Fermented vegetable | 3037 | 3,273,239 | 44.44 |
| HAC01 | GCA_003143915.1 | Korea | Fermented vegetable | 2953 | 3,230,597 | 44.54 |
| CLP0611 | GCA_002024845.1 | Korea | Environment | 2954 | 3,230,754 | 44.54 |
| SRCM102022 | GCA_002173655.1 | Korea | Fermented food | 3084 | 3,331,364 | 44.43 |
| NCU116 | GCA_001672035.1 | China | Fermented vegetable | 3090 | 3,354,689 | 44.36 |
The 54 genome sequences of Lactiplantibacillus plantarum subsp. plantarum available in the public database were included in this study. The type strain genomes of L. plantarum subsp. argentoratensis DSM 16365T (GCA_003641165.1) and L. paraplantarum L-ZS9T (GCA_001443645.1) were used as outgroups but are not presented in this table. The strain information was obtained from the biological descriptions provided in the NCBI database.
Trifurcating evolution of L. plantarum subsp. plantarum
From the 56 genomes studied, including the outgroup, 1884 orthologous single-copy core gene sets were extracted. These were concatenated into a sequence comprising 553,499 amino acids, and the maximum-likelihood tree for the sequence was constructed using the Jones–Taylor–Thornton model. Phylogenetic analysis showed a trifurcation-pattern evolution of L. plantarum subsp. plantarum into lineages A, B, and C (Supplementary Fig. S2). Lineage A consisted of 32 genomes, lineage B consisted of 15 genomes, and lineage C consisted of 7 genomes.
Independent horizontal gene transfer of the three lineages
To evaluate the occurrence of nonvertical evolution in this species, a network tree was generated using a binary matrix in the presence or absence of orthologous genes. The analysis showed that the three lineages were also differentiated in the network tree (Fig. 1). Except for few strains, most strains in each lineage showed intra-lineage levels of horizontal gene transfer, supporting the theory of trifurcating evolution in L. plantarum subsp. plantarum.
Figure 1.

Network tree of Lactiplantibacillus plantarum subsp. plantarum showing the possibility of nonvertical evolution among strains. The network tree was generated using the SplitTree 4 program based on a binary matrix of the presence and absence of gene families generated using the Ortho-MCL program. Reticulations in the tree indicate the possibility of horizontal gene transfer between branches. The red, blue, and green letters indicate the strains belonging to lineages A, B, and C, respectively.
Differences in bacteriocin biosynthesis among the three lineages
The genes encoding Pln were functional indicators for the trifurcating evolution of L. plantarum subsp. plantarum. Pln-related genes were only observed in L. plantarum subsp. plantarum. The sister subspecies, L. plantarum subsp. argentoratensis, or the outgroup, L. paraplantarum, did not possess any Pln-producing genes. Among the diverse pln genes identified in L. plantarum subsp. plantarum, only plnE/F genes were observed throughout the three lineages, suggesting that the common ancestor of L. plantarum subsp. plantarum possessed these genes (Fig. 2).
Figure 2.
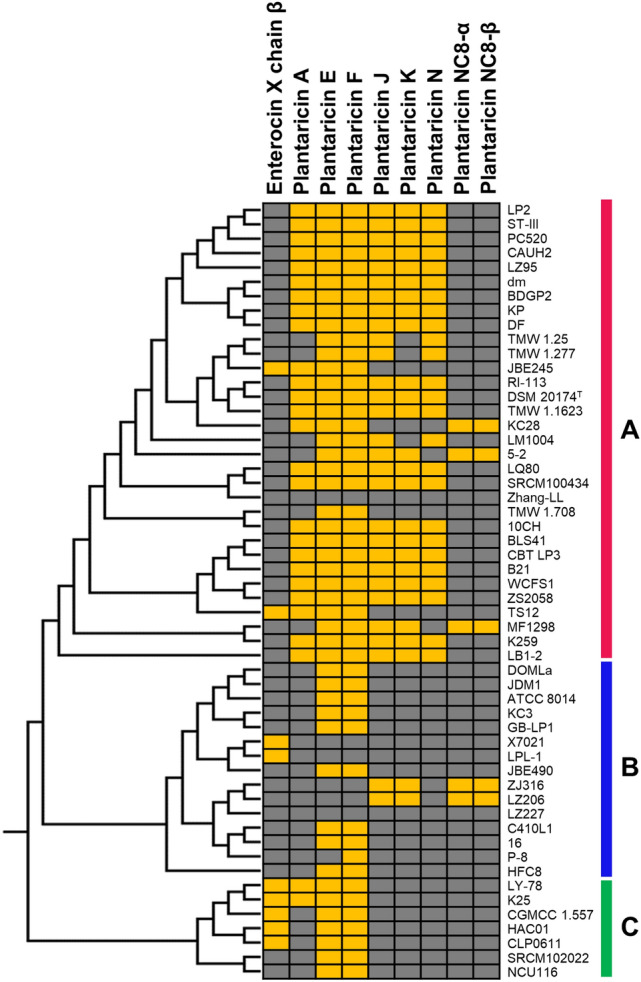
Bacteriocin profiling on the chromosomes of Lactiplantibacillus plantarum subsp. plantarum. The presence (yellow) and absence (gray) of bacteriocin genes are represented in the matrix. The dendrogram was obtained from the phylogenomic tree constructed using RAxML v8.2.4.
In lineage C, the deepest branch, plnE/F genes were well conserved in the plnEFI operon, and no mobile elements were observed in the pln loci of this lineage (Fig. 3). Enterocin X chain β encoding gene was frequently observed in lineage C, but it seemed nonfunctional owing to the lack of chain α-encoding gene. In lineage B, loss of gene function was observed because many pln genes were frameshifted, truncated, or disrupted by mobile elements (transposase and integrase). In lineage A, diverse Pln-encoding genes, including plnA, plnQ, plnE/F, pln J/K, and plnN, were observed. Comparison of the locations of the Pln operon loci in the genomes showed that the Pln biosynthesis genes were organized into operons and well conserved in lineage A. Except for two strains (Zhang-LL and TMW 1.708), all strains belonging to lineage A were predicted to produce more than one type of Pln.
Figure 3.
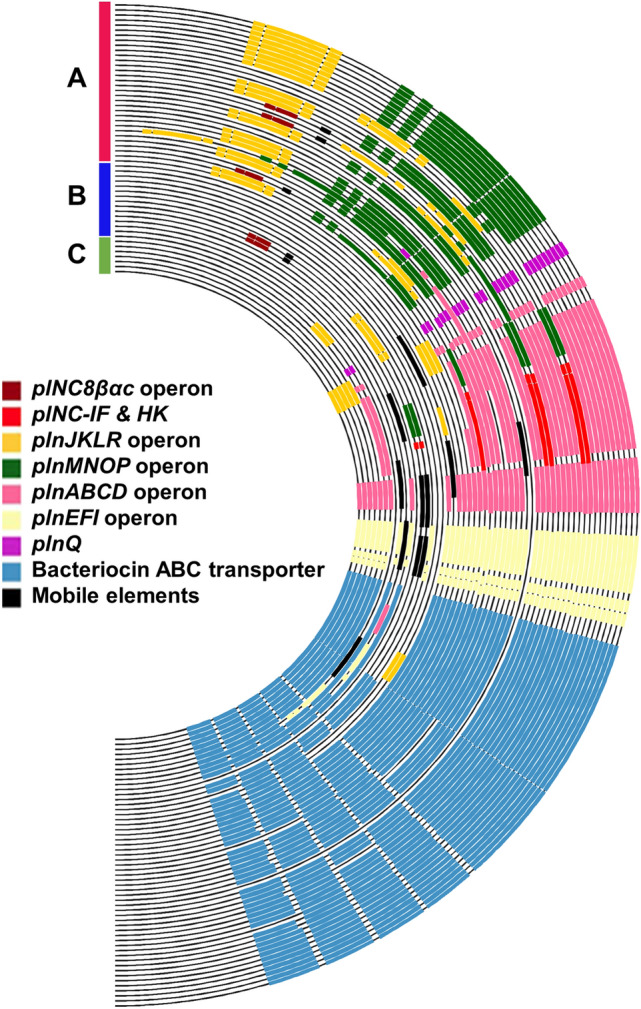
Plantaricin gene (pln) loci observed in the 54 complete genomes of Lactiplantibacillus plantarum subsp. plantarum. The genes related to the biosynthesis and transport systems of plantaricin were analyzed using BLAST and visualized using Circos v0.67. The order of the strains was determined from the results of the phylogenomic tree shown in Supplementary Fig. S1.
The plNC8βαc operon15, which was found in L. plantarum subsp. plantarum NC8, was observed in five strains in which the plnMNOP and plnJKLR operons were deleted, and certain mobile element sequences were found. plNC8-IF and histidine kinase (HK) do not encode Pln but control bacteriocin production by encoding induction factor (IF) and HK16. The aforementioned five strains, but not the other strains, possess the plNC8βαc operon as well as both IF and HK genes. Unlike the pln genes, the plnNC-8 α/β operon and plNC8-IF and HK seemed not originate from a common ancestor but were likely introduced in the genome by mobile elements during the evolutionary process.
Characteristics of other bacteriocins
In addition to the abovementioned Plns, a small number of bacteriocin genes were sporadically observed, independent of lineage. These included genes related to plnNC-8 α/β found in L. plantarum subsp. plantarum NC8 and genes related to enterocin. The plnNC-8 α/β operon was found in five strains, and no difference was found in the frequency among the lineages. In all strains containing the plnNC-8 α/β operon, the plnMNOP and plnJKLR operons were deleted, and few sequences of mobile elements were found downstream of the plnNC-8 α/β operon. This suggests that unlike the pln genes, the plnNC-8 α/β operon did not originate from a common ancestor but from a different species and was recently acquired by this species via lateral gene transfer. Moreover, certain strains contained genes related to the biosynthesis of bacteriocins other than Pln, which were present on plasmids instead of their chromosomes (Supplementary Table S1).
Characteristics of probiotic-associated functional genes
Differences in functional genes of interest associated with the following roles were analyzed among the three lineages: (1) utilization of carbohydrates, (2) utilization of human milk oligosaccharides (HMOs), (3) SCFA biosynthesis, (4) acetoin biosynthesis, (5) vitamin biosynthesis, (6) gamma-aminobutyric acid (GABA) biosynthesis, (7) alcohol degradation activity, (8) antioxidant biosynthesis, (9) non-ribosomal peptide synthetases (NRPS) and polyketide synthases (PKS) biosynthesis, and (10) antibiotic resistance. The results of the analyses showed no differences in these functional genes among the strains of the three lineages.
Utilization of carbohydrates.
The utilization of carbohydrates was found to be unrelated to the source of strain isolation. It was also found that there was no difference in carbohydrate utilization among the strains of the three lineages. All strains included in the analysis possessed the complete genes required for glycolysis and the pentose phosphate pathway. The strains did not carry the genes required for performing the tricarboxylic acid cycle, which is in agreement with the general characteristics of the genus Lactobacillus. In addition, the strains synthesized d/l-lactate from pyruvate, one of the end products of glycolysis, via the fermentation process and also produced acetate or ethanol. Monosaccharides that were used as a carbon source by all strains included glucose, fructose, mannose, and galactose. Disaccharides utilized by all strains included sucrose, maltose, isomaltose, cellobiose, and lactose, and none of the strains could use trehalose. Beta-galactosidase (EC 3.2.1.23), which hydrolyzes lactose, was produced by all strains.
The utilization of rhamnose/rhamnulose varied depending on the strain (Supplementary Table S2). The utilization of sugar alcohol also varied depending on the strain, but this difference was unrelated to the lineage or source of isolation. L. plantarum subsp. plantarum carries genes encoding two types of phosphotransferase systems (PTS) for the utilization of sorbitol. PTS type 1 was found in most of the strains, whereas PTS type 2 was only present in 10 strains. l-iditol 2-dehydrogenase (EC 1.1.1.14), which converts sorbitol into fructose, was produced by all strains, except for three strains (ATCC 8014, KC3, and P-8) in lineage B, indicating that most strains can metabolize sorbitol. Additionally, the sugar alcohol mannitol was utilized by all strains.
Three types of orthologous genes of alpha-galactosidase (α-Gal, EC 3.2.1.22), which are required for the conversion of galactinol and melibitol to galactose, were found in the strains studied. Genes encoding α-Gal type 1 were carried by most strains included in the analyses, except for JBE49. Genes encoding type 2 were present in all strains in lineages B and C, but the percentage was less than half in lineage A, whereas the genes encoding type 3 were present in only 33% of strains in lineage B, but not in other lineages. Taken together, all strains carried one or more α-Gal-encoding genes.
-
2.
Utilization of HMOs.
To break down HMOs, glycosyl hydrolases, such as GH2 (α-galactosidase), GH16 (endo-β-1,4-galactosidase), GH18 (endo-β-N-acetylglycos aminidase), GH20 (β-hexosaminidase/lacto-N-biosidase), GH29 (α-1,3/4-fucosidase), GH33 (sialidase), and GH95 (α-1,2-fucosidase), are required. Our analyses showed that all strains in the present study carried genes encoding the GH2 family. However, genes coding for GH16, GH18, GH29, GH33, and GH95 were absent in all strains. Genes belonging to the GH20 family were carried by some strains of lineages A (81%), B (20%), and C (29%).
-
3.
SCFA biosynthesis.
SCFAs are volatile fatty acids that consist of 2–6 carbon atoms and have been recently reported to be closely related to immune function. All strains analyzed contained the formate C-acetyltransferase (EC 2.3.1.54) gene, which synthesizes formate from pyruvate, and the 2-acetate kinase (EC 2.7.2.1) gene, which synthesizes propanoate from oxobutanoate. However, genes related to the biosynthesis of butyrate, isobutyrate, valerate, and isovalerate were not found in any strain.
-
4.
(R)-2-acetoin biosynthesis.
Certain bacteria produce acetoin in the form of an energy storage molecule, which is an aroma compound with flavor-enhancing effects. Pyruvate, the end product of glycolysis, is converted to acetoin via (S)-2-acetolactate, an intermediate metabolite, by the enzymes acetolactate synthase I/II/III large subunit (EC 2.2.1.6) and acetolactate decarboxylase (EC 4.1.1.5). All strains contained these two genes, suggesting that they used (R)-2-acetoin as an energy storage molecule. Additionally, strains BDGP2 and HFC8 synthesized not only (R)-2-acetoin but also (S)-2-acetoin and contained genes related to meso-butanediol dehydrogenase/(S,S)-butanediol dehydrogenase/diacetyl reductase (EC 1.1.1.-, 1.1.1.76, 1.1.1.304), which converts (S)-2-acetoin into (S,S)-butane-2,3,-diol or meso-butane-2,3,-diol. Strain LZ227 alone utilized an alternative pathway that used the synthesized (R)-2-acetoin to produce meso-butane-2,3,-diol and (R,R)-butane-2,3-diol.
-
5.
Vitamin biosynthesis.
Vitamin biosynthesis in LAB increases their utility as probiotics. The results showed that except for strain WCFS1 in lineage A and strain 16 in lineage B, 51 strains produced thiamin and riboflavin. However, only strain RI-113 in lineage A and strain HFC8 in lineage B synthesized nicotinate. None of the strains synthesized pantothenate, pyridoxal, or biotin.
-
6.
GABA biosynthesis.
GABA is an inhibitory neurotransmitter, and GABAs produced by LAB are used in functional food products. All strains used in this study carried glutamate decarboxylase (EC 4.1.115) genes, which indicates that the strains could produce GABA from glutamate.
-
7.
Alcohol dehydrogenase biosynthesis.
LAB showing alcohol degradation activity are beneficial for alcohol hangover relief and liver health. All strains used in this study carried alcohol dehydrogenase (EC 1.1.1.1) and bifunctional aldehyde-alcohol dehydrogenase (EC 1.2.1.10) genes and therefore had alcohol degradation activity. However, 29–188 amino acids were deleted in the N-terminal region of the protein produced by strain B21 in lineage A and strain LZ206 in lineage B, indicating that these strains could not synthesize the enzyme or that the synthesized enzyme would be inactive.
-
8.
Antioxidant biosynthesis.
LAB strains that remove reactive oxygen species are of interest for industrial applications. All strains used in this study carried genes encoding known antioxidant enzymes, including glutathione peroxidase, glutathione reductase, catalase, and peroxidase (POD). All strains carried only one copy of glutathione peroxidase. Four types of POD (POD types 1–4) genes were present in all strains, whereas type 5 was only found in four strains (HFC8, HAC01, K25, and MF1298).
Glutathione reductase type 1–4-encoding genes were present in all strains. However, strain CBT LP3 in lineage A carried type 5 genes instead of type 4 genes. This was because of a frameshift in the type 4 gene of CBT LP3, which led to the prediction of two CDSs. Glutathione reductase type 6 was found in the plasmids of seven strains (CBT LP3, CAUH2, CGMCC 1.557, LZ206, LZ227, NCU116, and SRCM102022), and no difference was found among the lineages.
-
9.
NRPS-PKS biosynthesis.
NRPS-PKS is a large multienzymatic megasynthase that aids in the defense against pathogenic bacteria by producing a number of antimicrobial substances. Orthologs of the genes related to NRPS synthesis were found in the genomes of strains TMW 1.708, B21, and WCFS1 in lineage A. As gene clusters involved in fatty acid synthesis are present near the genes related to NRPS synthesis, these strains were predicted to be able to synthesize NRPS, and the monomer that can be synthesized was predicted to be (Ala-Ala-Ala-Ser-Gly) + (Ala). The Norine database was used to search for this monomer structure, which was identified as pyoverdine GM, a type of siderophore. Strain LQ80 alone carried two genes (LpLQ80_16120 and LpLQ80_16125) containing a fatty acid-NRPS module in a plasmid.
-
10.
Antibiotic resistance.
CARD was used for the analysis, and the results showed that none of the strains used in this study carried genes related to antibiotic resistance.
Discussion
The present study aimed to investigate the intraspecies functional diversity of L. plantarum subsp. plantarum by performing comparative genomic analysis of strains isolated from a variety of sources. The results showed that the sizes of the core- and pan-genomes showed a similar pattern to that in previous studies that used draft genomes for the analysis5,6. Genomic and phylogenetic analyses supported the trifurcation of L. plantarum subsp. plantarum strains into lineages A, B, and C. The three lineages could not be clearly differentiated by the geographical location of the strains (Supplementary Fig. S3). However, most strains isolated from Western countries belong to lineage A. For the rest, the geographic localization of the strains was observed in several sub-clades with solid clustering. In certain LAB17, the source of isolation correlates with sugar metabolism traits; however, it has no correlation with the species L. plantarum5,6, and the present study also found similar results.
The only difference among the three lineages was the presence of bacteriocin-encoding genes. The two main plantaricins observed in this study are Pln EF (in lineages A and C) and JK (in lineage A). Both plantaricins form pores in the plasma membrane of target bacteria, but show complementary ion selectivity18. Pln EF pores show high conductivity for cations but not for anions, while Pln JK pores conduct anions well but not cations18. Thus, the killing efficiency is enhanced in the strains which possess both complementary bacteriocins. This implies the superior adaptation of lineage A to lineage B or C in the competitive microbial community. Loss of gene function due to mobile genetic elements was observed in lineage B, suggesting the gradual loss of genes related to the biosynthesis of bacteriocins. It is hypothesized that the common ancestor of L. plantarum subsp. plantarum possesses Pln E/F, and this feature is preserved in the deep branch (lineage C). The descendant types further split into two lineages (A and B), and the evolution of these two lineages was clearly differentiated. Lineage A acquired diverse pln genes and GH20 genes for adaptive evolution in competitive environments with HMOs, whereas lineage B was adapted to environments where Pln was not required.
Interestingly, the majority (75%) of lineage A was fermented food origin while the majority (60%) of lineage B was environment or intestinal origin. In addition, strains of lineage C were all originated from fermented food sources. It is known that bacteriocin’s critical role in mediating population- and community-level interactions is ensured when the cell density is high in the environment19. Bacteria use bacteriocin to compete with close cells in dense population, but they do not need to produce bacteriocin in low cell density environments. Thus, it is presumed that lineages A and C strains have evolved and adapted to high cell density of fermenting foods by possessing bacteriocin genes, while lineage B strains which have adapt to natural environments are losing the unnecessary genes.
This study demonstrated that there was no difference in sugar utilization among the three lineages. A previous study found differences in fructooligosaccharide and raffinose utilization between the two groups of L. plantarum that were classified based on a single nucleotide polymorphism phylogenetic tree6. Similarly, a study that performed hierarchical clustering based on sugar gene loci reported a difference in the genes related to sugar metabolism among the clusters5. However, these studies divided the clusters based on the presence or absence of orthologous genes or sugar utilization genes, whereas the phylogenetic tree in the present study was drawn based on whole genomes. Therefore, it was not possible to directly compare our results with those of these studies.
Minor differences in the utilization of HMOs were found among the lineages. HMOs contain a common lactose at their reducing end, which is elongated with various combinations of N-acetyllactosamine (Galβ1-4GlcNAc) or lacto-N-biose I units (Galβ1-3GlcNAc) and then fucosylated and/or sialylated to produce approximately 200 types of oligosaccharides. Therefore, a variety of glycoside hydrolase (GHs) is necessary for the utilization of HMOs. More specifically, GH2 (α-galactosidase), GH16 (endo-β-1,4-galactosidase), GH18 (endo-β-N-acetylglycosaminidase), GH20 (β-hexosaminidase/lacto-N-biosidase), GH29 (α-1,3/4-fucosidase), GH33 (sialidase), and GH95 (α-1,2-fucosidase) are known to be involved in HMO utilization20. The results of this study revealed that almost all L. plantarum subsp. plantarum strains, regardless of their lineage, carried GH2, suggesting their ability to use lactose. In contrast, the GH20 genes were found more frequently in lineage A than in lineage B or C, implying the advanced adaptation of lineage A to HMOs than that of other lineages.
The bacteriocin distribution within a phylogenetic clade has been investigated in several fermenting environments. Gontijo et al. found that the distribution of bacteriocin structural genes is related to phylogenetic clades of LAB species of artisanal cheese, with a higher frequency in some specific clades21. Azevedo et al. reported the occurrence of different classes of bacteriocin among phylogenetic clades of ruminal bacteria and archaea22. Collins et al. reported the bacteriocin gene diversity and complexity across the Lactobacillus genus complex23. Those previous studies accounted the interspecific genetic diversity of bacteriocin production genes, but our study focused on the intraspecific distribution within the commercially important species.
The production of bacteriocins by L. plantarum is an indicator of their potential use as natural food preservatives and as dietary supplements. Therefore, in terms of Pln production, the strains in lineage A seemed to have better bacterial community control power than that of the strains in lineage B and C. Previous comparative genomic studies on L. plantarum have not featured the association between the evolutionary lineages and the source of isolation. The differential distribution of bacteriocin genes and biased ecological origin inherent to the trifurcating lineages imply the habitat adapted evolution occurring in this species.
Methods
Genome sequences
Among the L. plantarum subsp. plantarum genomes available in the GenBank database of the National Centre for Biotechnology Information (Bethesda, MD, USA) as of May 2018, the complete genomes of 54 different strains were selected for comparative genomic analysis (Table 1). Lactobacillus plantarum subsp. argentoratensis strain DKO 22T was obtained from the German Collection of Microorganisms and Cell Cultures GmbH (DSM 16365T), and the complete genome sequence was determined in this study (GCA_003641165.1). The genome of L. paraplantarum L-ZS9 (GCA_001443645.1) was obtained from GenBank and used as an outgroup in the phylogenomic analyses.
Gene prediction and pan- and core-genome analysis
Protein coding sequences (CDSs) were predicted using Prodigal v.2.6.324, and the Rfam database25 was used to search for noncoding RNA genes. Orthologous gene families were analyzed using OrthoMCL26, which utilizes an all-against-all protein–protein basic local alignment search tool (BLAST) and a Markov cluster algorithm, with an inflation value of 2.0. Pan/core-genome curves were drawn using PanGP v.1.0.1.27. The openness of the pan-genome was estimated based on the Heaps’ Law model using the R package micropan28.
Genome-based construction of the phylogenetic tree
A phylogenetic tree was constructed by selecting only single-copy orthologous genes that were included in the core genome. The amino acid sequences of these genes were aligned using MUSCLE v3.8.3129. The aligned positions showing a gap in more than 50% of the 54 genomes were removed using Gblocks v0.9130. The final sequence alignments were concatenated using FASconCAT31. A model test was performed using ProtTest v3.232 to select a suitable evolution model, and a maximum-likelihood tree was constructed using RAxML v8.2.433. A binary matrix of the presence or absence of orthologs was calculated to analyze the degree of non-vertical gene transfer within the groups. SplitTree v4.14.534 was used to construct a network tree. All phylogenetic trees were drawn using Dendroscope v3.2.235.
Functional annotation of gene families
Functional classifications of the gene families including orthologs and singleton sequence orphan open reading frames were performed through BLAST searches using UniProt36, Clusters of Orthologous Groups of proteins37, and Pfam38 databases. Glycoside hydrolase (GH) family annotation was performed using the Carbohydrate-Active enZYmes database39. Metabolic pathways for short-chain fatty acid (SCFA) and vitamin biosynthesis were analyzed using the Kyoto Encyclopedia of Genes and Genomes automatic annotation server40. Gene clusters involved in the biosynthesis of non-ribosomal peptides and polyketides (NRPS/PKS) were predicted using antiSMASH 3.041. Nonribosomal peptides were identified using the Norine database42. The resistance gene identifier of the Comprehensive Antibiotic Resistance Database (CARD)43 was used to identify antibiotic resistance genes.
Bacteriocin analyses
Bacteriocin-encoding genes were identified using Bagel344. The presence or absence and the location of previously identified bacteriocin genes, including the L. plantarum-specific bacteriocin (pln) and Enterococcus-specific bacteriocin (enterocin), were analyzed. The graphical pln loci map of the complete genome was constructed and visualized using Circos v0.6745.
Supplementary Information
Acknowledgements
This study was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2019R1F1A1059925) and by the research grant from the Strategic Initiative for Microbiomes in Agriculture and Food funded by the Ministry of Agriculture, Food and Rural Affairs (918010-4).
Author contributions
H.Y.: Study concept and design; S.C., M.B.: Data analysis and Manuscript preparations, M.C., S.L.: Data acquisition; H.Y.: Manuscript editing; all authors reviewed the manuscript and approved the final version of it. Statement: all methods were carried out in accordance with relevant guidelines and regulations.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
The online version contains supplementary material available at 10.1038/s41598-021-99683-1.
References
- 1.Zheng J, et al. A taxonomic note on the genus Lactobacillus: Description of 23 novel genera, emended description of the genus Lactobacillus Beijerinck 1901, and union of Lactobacillaceae and Leuconostocaceae. Int. J. Syst. Evol. Microbiol. 2020;70:2782–2858. doi: 10.1099/ijsem.0.004107. [DOI] [PubMed] [Google Scholar]
- 2.Hemarajata P, Versalovic J. Effects of probiotics on gut microbiota: Mechanisms of intestinal immunomodulation and neuromodulation. Therap. Adv. Gastroenterol. 2013;6:39–51. doi: 10.1177/1756283X12459294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hammes WP, Hertel C, et al. Lactobacillus. In: Whitman WB, et al., editors. Bergey's Manual of Systematics of Archaea and Bacteria. Wiley, in association with Bergey's Manual Trust; 2015. pp. 1–76. [Google Scholar]
- 4.Siezen RJ, et al. Phenotypic and genomic diversity of Lactobacillus plantarum strains isolated from various environmental niches. Environ. Microbiol. 2010;12:758–773. doi: 10.1111/j.1462-2920.2009.02119.x. [DOI] [PubMed] [Google Scholar]
- 5.Martino ME, et al. Nomadic lifestyle of Lactobacillus plantarum revealed by comparative genomics of 54 strains isolated from different habitats. Environ. Microbiol. 2016;18:4974–4989. doi: 10.1111/1462-2920.13455. [DOI] [PubMed] [Google Scholar]
- 6.Choi S, Jin GD, Park J, You I, Kim EB. Pan-genomics of Lactobacillus plantarum revealed group-specific genomic profiles without habitat association. J. Microbiol. Biotechnol. 2018;28:1352–1359. doi: 10.4014/jmb.1803.03029. [DOI] [PubMed] [Google Scholar]
- 7.Siezen RJ, van Hylckama Vlieg JE. Genomic diversity and versatility of Lactobacillus plantarum, a natural metabolic engineer. Microb. Cell Fact. 2011;10:S3. doi: 10.1186/1475-2859-10-S1-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu J, et al. Comparative genomic analysis of Lactobacillus plantarum GB-LP1 isolated from traditional Korean fermented food. J. Microbiol. Biotechnol. 2017;27:1419–1427. doi: 10.4014/jmb.1704.04005. [DOI] [PubMed] [Google Scholar]
- 9.Cotter PD, Hill C, Ross RP. Bacteriocins: Developing innate immunity for food. Nat. Rev. Microbiol. 2005;3:777–788. doi: 10.1038/nrmicro1273. [DOI] [PubMed] [Google Scholar]
- 10.Cotter PD, Ross RP, Hill C. Bacteriocins—A viable alternative to antibiotics? Nat. Rev. Microbiol. 2013;11:95–105. doi: 10.1038/nrmicro2937. [DOI] [PubMed] [Google Scholar]
- 11.Wang G, Li X, Wang Z. APD3: The antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 2016;44:D1087–D1093. doi: 10.1093/nar/gkv1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tenea GN, Ortega C. Genome characterization of Lactiplantibacillus plantarum strain UTNGt2 originated from Theobroma grandiflorum (white cacao) of ecuadorian amazon: Antimicrobial peptides from safety to potential applications. Antibiotics. 2021;10:383. doi: 10.3390/antibiotics10040383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu HJ, et al. Screening for Lactobacillus plantarum with potential inhibitory activity against enteric pathogens. Ann. Microbiol. 2015;65:1257–1265. doi: 10.1007/s13213-014-0963-3. [DOI] [Google Scholar]
- 14.Arena MP, et al. Use of Lactobacillus plantarum strains as a bio-control strategy against food-borne pathogenic microorganisms. Front. Microbiol. 2016;7:464. doi: 10.3389/fmicb.2016.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nissen-Meyer J, Larsen AG, Sletten K, Daeschel M, Nes IF. Purification and characterization of plantaricin A, a Lactobacillus plantarum bacteriocin whose activity depends on the action of two peptides. J. Gen. Microbiol. 1993;139:1973–1978. doi: 10.1099/00221287-139-9-1973. [DOI] [PubMed] [Google Scholar]
- 16.Maldonado A, Jimenez-Diaz R, Ruiz-Barba JL. Induction of plantaricin production in Lactobacillus plantarum NC8 after coculture with specific gram-positive bacteria is mediated by an autoinduction mechanism. J. Bacteriol. 2004;186:1556–1564. doi: 10.1128/JB.186.5.1556-1564.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Douillard FP, et al. Comparative genomic and functional analysis of 100 Lactobacillus rhamnosus strains and their comparison with strain GG. PLoS Genet. 2013 doi: 10.1371/journal.pgen.1003683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moll GN, et al. Complementary and overlapping selectivity of the two-peptide bacteriocins plantaricin EF and JK. J. Bacteriol. 1999;181:4848–4852. doi: 10.1128/JB.181.16.4848-4852.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riley MA, Wertz JE. Bacteriocin diversity: Ecological and evolutionary perspectives. Biochimie. 2002;84:357–364. doi: 10.1016/S0300-9084(02)01421-9. [DOI] [PubMed] [Google Scholar]
- 20.Marcobal A, Sonnenburg JL. Human milk oligosaccharide consumption by intestinal microbiota. Clin. Microbiol. Infect. 2012;18:12–15. doi: 10.1111/j.1469-0691.2012.03863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Azevedo AC, Bento CB, Ruiz JC, Queiroz MV, Mantovani HC. Distribution and genetic diversity of bacteriocin gene clusters in rumen microbial genomes. Appl. Environ. Microbiol. 2015;81:7290–7304. doi: 10.1128/AEM.01223-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gontijo MTP, Silva JDS, Vidigal PMP, Martin JGP. Phylogenetic distribution of the bacteriocin repertoire of lactic acid bacteria species associated with artisanal cheese. Food Res. Int. 2020;128:108783. doi: 10.1016/j.foodres.2019.108783. [DOI] [PubMed] [Google Scholar]
- 23.Collins FWJ, et al. Bacteriocin gene-trait matching across the complete Lactobacillus pan-genome. Sci. Rep. 2017;7:3481. doi: 10.1038/s41598-017-03339-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hyatt D, et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010;11:119. doi: 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalvari I, et al. Non-coding RNA analysis using the Rfam database. Curr. Protoc. Bioinfom. 2018;62:e51. doi: 10.1002/cpbi.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao YB, et al. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics. 2014;30:1297–1299. doi: 10.1093/bioinformatics/btu017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snipen L, Liland KH. micropan: An R-package for microbial pan-genomics. BMC Bioinform. 2015;16:79. doi: 10.1186/s12859-015-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edgar RC. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 31.Kuck P, Meusemann K. FASconCAT: Convenient handling of data matrices. Mol. Phylogenet. Evol. 2010;56:1115–1118. doi: 10.1016/j.ympev.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 32.Abascal F, Zardoya R, Posada D. ProtTest: Selection of best-fit models of protein evolution. Bioinformatics. 2005;21:2104–2105. doi: 10.1093/bioinformatics/bti263. [DOI] [PubMed] [Google Scholar]
- 33.Stamatakis A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 35.Huson DH, et al. Dendroscope: An interactive viewer for large phylogenetic trees. BMC Bioinform. 2007;8:460. doi: 10.1186/1471-2105-8-460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.The UniProt Consortium UniProt: The universal protein knowledgebase. Nucleic Acids Res. 2017;45:D158–D169. doi: 10.1093/nar/gkw1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000;28:33–36. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Finn RD, et al. Pfam: The protein families database. Nucleic Acids Res. 2014;42:D222–D230. doi: 10.1093/nar/gkt1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cantarel BL, et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37:D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–W185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber T, et al. antiSMASH 3.0—A comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015;43:W237–W243. doi: 10.1093/nar/gkv437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Caboche S, et al. NORINE: A database of nonribosomal peptides. Nucleic Acids Res. 2008;36:D326–D331. doi: 10.1093/nar/gkm792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McArthur AG, et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 2013;57:3348–3357. doi: 10.1128/AAC.00419-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Heel AJ, de Jong A, Montalban-Lopez M, Kok J, Kuipers OP. BAGEL3: Automated identification of genes encoding bacteriocins and (non-)bactericidal posttranslationally modified peptides. Nucleic Acids Res. 2013;41:W448–W453. doi: 10.1093/nar/gkt391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Krzywinski M, et al. Circos: An information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.