

Abstract
The main viral protease (3CLpro) is indispensable for SARS-CoV-2 replication. We delineate the human protein substrate landscape of 3CLpro by TAILS substrate-targeted N-terminomics. We identify more than 100 substrates in human lung and kidney cells supported by analyses of SARS-CoV-2-infected cells. Enzyme kinetics and molecular docking simulations of 3CLpro engaging substrates reveal how noncanonical cleavage sites, which diverge from SARS-CoV, guide substrate specificity. Cleaving the interactors of essential effector proteins, effectively stranding them from their binding partners, amplifies the consequences of proteolysis. We show that 3CLpro targets the Hippo pathway, including inactivation of MAP4K5, and key effectors of transcription, mRNA processing, and translation. We demonstrate that Spike glycoprotein directly binds galectin-8, with galectin-8 cleavage disengaging CALCOCO2/NDP52 to decouple antiviral-autophagy. Indeed, in post-mortem COVID-19 lung samples, NDP52 rarely colocalizes with galectin-8, unlike in healthy lungs. The 3CLpro substrate degradome establishes a foundational substrate atlas to accelerate exploration of SARS-CoV-2 pathology and drug design.
Keywords: COVID-19, SARS-CoV-2 3CLpro, SARS-CoV-2 main protease, proteases, subsite specificity, active site structure, substrates, degradomics, proteomics, interactome
Graphical abstract
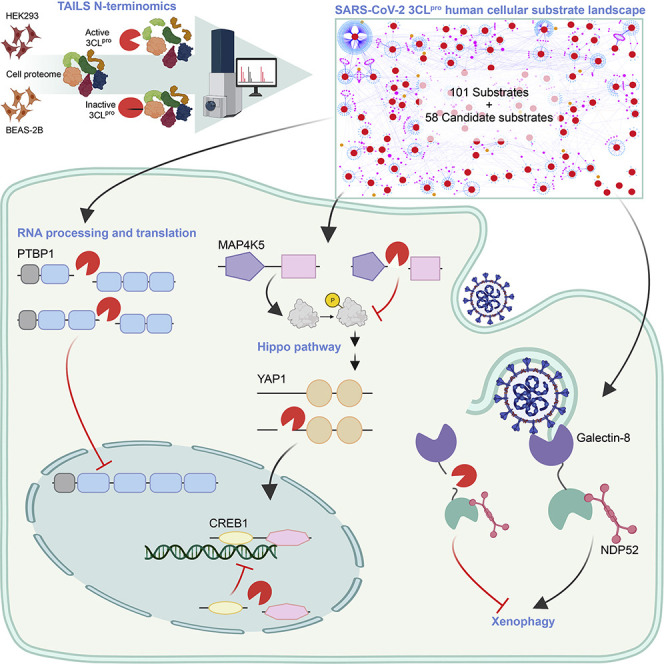
Pablos et al. report diverse SARS-CoV-2 3CLpro host substrates and interactors, providing insights into pathological mechanisms. In addition to blocking viral polyprotein processing, 3CLpro inhibitor-drugs should restore multiple antiviral defenses and intracellular sensing of CoV-2 Spike protein by galectin-8, which triggers protective xenophagy in infection.
Introduction
The current understanding of how SARS-CoV-2 overwhelms the host cell machinery and escapes antiviral defenses is far from complete. Viruses have evolved an ability to maximize a small genome; thus, their proteins are pleiotropic and multifunctional. As multitasking proteins present challenges for drug development (Butler and Overall, 2009), deciphering the pleiotropic roles of viral proteins in host cells will inform the identification of novel drug targets for SARS-CoV-2 and other beta-coronaviruses. Within the two polyproteins encoded by SARS-CoV-2 reside two essential proteases for replication (Kim et al., 2020). Nonstructural protein-5 (NSP5) encodes the main protease, 3-chymotrypsin-like protease (3CLpro) (Dai et al., 2020), and NSP3 encodes papain-like protease (Shin et al., 2020). 3CLpro is a validated drug target that releases 16 NSPs by cleaving at eleven L/FQ↓(S/A/G/N) sites for viral replication complex assembly. In addition, host cell protein cleavage by viral proteases is a critical component of viral pathogenicity (Jagdeo et al., 2018), including diverting cellular processes to viral replication, defeating antiviral responses and immune response modulation. However, determining the repertoire and diversity of proteolytic cell targets is a long-standing challenge, and the pathobiological mechanisms driven by 3CLpro in COVID-19 remain elusive. Substrate cleavage requires that the amino acids flanking the scissile bond on the proximal nonprime (P) side and the distal prime (P’) side fit the protease S and S’ subsites, respectively (Klein et al., 2018). Medicinal chemistry classically focuses on the P-side interface to increase drug potency. However, knowledge of human cellular target proteins would improve the characterization of P’-recognition subsites to guide drug development and decipher infection pathways to understand and predict outcomes of 3CLpro-inhibitor drug therapy of COVID-19.
Many large-scale analyses of the SARS-CoV-2 infected-cell transcriptome (Stukalov et al., 2021), proteome (Stukalov et al., 2021), phosphoproteome (Bouhaddou et al., 2020) and interactomes (Gordon et al., 2020; Stukalov et al., 2021) are described. With only 14 substrates reported in SARS-CoV-2 infection (Meyer et al., 2021; Moustaqil et al., 2021), the 3CLpro human substrate repertoire, also known as the degradome (López-Otín and Overall, 2002), is not well understood. Thus, the opaque contribution of 3CLpro to overwhelming the host cell machinery remains understudied. We addressed this challenge by employing state-of-the-art substrate-targeted proteomics and substrate winnowing analyses to comprehensively profile the human host cell substrates of 3CLpro. Here, we expanded the 3CLpro substrate landscape to over 100 substrates and 58 additional high confidence candidate substrates. In exploring the consequences of 3CLpro cleavage events, we demonstrate the direct binding of galectin-8 to Spike S1 glycoprotein and found this complex is disrupted upon galectin-8 cleavage to impact antiviral-autophagy, also known as xenophagy. Cleavage of four Hippo signaling proteins, including Yes-associated protein-1 (YAP1), cyclic AMP responsive element-binding protein 1 (CREB1) and cyclic AMP-dependent transcription factor 1 (ATF1), as well as cleavage-inactivation of a Hippo pathway regulator—mitogen-activated kinase-kinase-kinase-kinase 5 (MAP4K5), suggests a route to combat antiviral defenses. Our protein-protein interaction analyses of 101 3CLpro substrates reveal extensive disruption of cellular protein interaction networks resulting from viral proteolysis leading to the isolation, or “stranding,” of crucial cellular proteins. Thus, our substrate degradome atlas provides a powerful resource to inspire mechanistic studies of COVID-19 pathobiology.
Results
Deciphering the SARS-CoV-2 3CLpro human substrate landscape
We profiled the substrate repertoire of 3CLpro in human cell proteomes by Terminal Amine Isotopic Labeling of Substrates (TAILS) (Kleifeld et al., 2010), a targeted method to selectively purify neo-N-terminal peptides corresponding to substrate P’-cleavage products (Figure 1 A; Tables 1 and S1–S6). We analyzed 3CLpro cleavages in native proteome extracts from human embryonic kidney (HEK293) cells (N = 3) (Figures 1A and S1A–S1D; Tables S2 and S3A–S3C). Antiviral type I interferons (IFN) induce host cell protection by interferon-stimulated gene (ISG) responses. To seek respiratory cell substrates connected with COVID-19 lung pathobiology and to investigate whether 3CLpro dampens antiviral responses by cleavage of ISG proteins, we treated human lung epithelial (BEAS-2B) cells with IFN-α (N = 3), IFN-β (N = 3), or vehicle (N = 3) (Figures S1E–S1H; Tables S4, S5A–S5C and S7). Following incubation with 3CLpro, whole protein isotopic labeling by heavy [+34 Da]-dimethylation of neo-N-termini exposed by 3CLpro cleavage enabled identification of the P’-sequence of cut-sites by liquid chromatography-tandem mass spectrometry (LC-MS/MS). By quantitative comparison with light [+28 Da]-labeled inactive 3CLpro-C145A-treated control samples, the increased abundance of [+34 Da]-dimethylated neo-N-terminal peptides after cleavage identified candidate 3CLpro substrates.
Figure 1.

3CLpro cleavage sites in human proteins identified by TAILS
(A) Experimental design. Neo-N-termini of 3CLpro-cleaved substrates in HEK293 and BEAS-2B human lung epithelial cell lysates were isolated and identified by TAILS LC-MS/MS. Only those neo-N-termini in ≥ 2/3 HEK293 or ≥ 7/9 BEAS-2B independent cell experiments were considered for further substrate winnowing.
(B) Classification of 1,649 quantified N-termini in N = 12 independent experiments.
(C) 3CLpro candidate substrate cleavage site specificities (n = 292) versus other quantified neo-N-termini (n = 663).
(D) Cellular distribution of high confidence cleavage sites (n = 102) in 101 human substrates after substrate winnowing (Table 1).
(E) Substrate Reactome gene set enrichment by hypergeometric distribution followed by FDR correction. Node radius designates gene enrichment; line widths are proportional to the overlap of shared substrates between connected nodes sharing ≥ 20% genes.
See also Figure S1 and Document S1. Figures S1–S6 and Table S1, Table S2. 3CLpro cleavage sites and substrate proteins identified in human embryonic kidney (HEK293) cells, related to Figures 1B–1D, Table S4. 3CLpro cleavage sites and substrate proteins identified in human bronchial epithelial (BEAS-2B) cells, related to Figures 1B–1D, Table S7. Comparison of interferon-treated BEAS-2B versus control. MSstats multiple sample t test results, related to Figure 1, Table S8. Reactome gene sets enrichment in SARS-CoV-2 3CLpro substrates, related to Figure 1E.
Table 1.
3CLpro cleavage sites and substrate proteins stringently identified in human embryonic kidney (HEK293) and human lung epithelial (BEAS-2B) cells, related to Figure 1
 |
 |
After bioinformatics analysis and substrate winnowing, n = 102 cut sites in n = 101 human protein substrates of 3CLpro were confidently identified. Fields marked as “ ” or “
” or “ ” indicate in which of the N = 12 independent cell experiments that the cleaved neo-N-terminal P′ peptide was found by TAILS LC-MS/MS with an FDR ≤ 0.01 at the peptide level. For protein identification, the TAILS and preTAILS shotgun proteomic analyses were combined in each experiment, with an FDR ≤ 0.01 at the protein level. Cleaved neo-N-terminal peptides of substrates that were reproducibly identified in ≥ 2/3 HEK293 or ≥ 7/9 BEAS-2B experiments were further substrate winnowed by sequence distance score calculation and manual inspection of all MS/MS spectra in order to be considered bona fide substrates.
” indicate in which of the N = 12 independent cell experiments that the cleaved neo-N-terminal P′ peptide was found by TAILS LC-MS/MS with an FDR ≤ 0.01 at the peptide level. For protein identification, the TAILS and preTAILS shotgun proteomic analyses were combined in each experiment, with an FDR ≤ 0.01 at the protein level. Cleaved neo-N-terminal peptides of substrates that were reproducibly identified in ≥ 2/3 HEK293 or ≥ 7/9 BEAS-2B experiments were further substrate winnowed by sequence distance score calculation and manual inspection of all MS/MS spectra in order to be considered bona fide substrates.
∗, Amino acid sequence of the cleavage site and P1′ amino acid position identified from the neo-N-terminal peptide.  , scissile bond. †, Cleavage site confirmed by MALDI-TOF MS analysis of 3CLpro enzyme kinetics of P4 – P4′ spanning peptide cleavage (+). ‡, SRRM2 has two cleavage sites identified in the same protein, one in 12/12 experiments, the other in 9/9 BEAS-2B experiments. §, MCM4 was identified with a sequence distance score below the 10th percentile, but the P4 – P4′ synthetic peptide was cleaved in MALDI-TOF MS analysis. ¶, Substrate found in 2/3 HEK293 cell experiments only (n = 4) or ≤ 6/9 BEAS-2B cell experiments only (n = 3), but with other compelling evidence or biology, including peptide cleavage in MALDI-TOF MS analysis, to be designated as a substrate.
, scissile bond. †, Cleavage site confirmed by MALDI-TOF MS analysis of 3CLpro enzyme kinetics of P4 – P4′ spanning peptide cleavage (+). ‡, SRRM2 has two cleavage sites identified in the same protein, one in 12/12 experiments, the other in 9/9 BEAS-2B experiments. §, MCM4 was identified with a sequence distance score below the 10th percentile, but the P4 – P4′ synthetic peptide was cleaved in MALDI-TOF MS analysis. ¶, Substrate found in 2/3 HEK293 cell experiments only (n = 4) or ≤ 6/9 BEAS-2B cell experiments only (n = 3), but with other compelling evidence or biology, including peptide cleavage in MALDI-TOF MS analysis, to be designated as a substrate.
For definitive identification as a 3CLpro substrate, we required further high stringency conditions to be met. Heavy-labeled neo-N-termini had to be present solely as a “heavy singleton” without the corresponding isotopic light-counterpart from control samples. For confident identification as a biologically relevant cleavage site, these neo-N-termini had to be identified in ≥ 2/3 independent HEK293 or ≥ 7/9 independent BEAS-2B cell experiments. Combining the HEK293 and BEAS-2B datasets, we quantified 1,649 labeled N-termini, including 955 neo-N-termini (Figure 1B; Tables S6A and S6B). Thereby, we identified 292 3CLpro-cleaved neo-N-termini in 229 proteins (Figures 1C, S1D and S1H; Table S6A). The sequence logo of the 292 cleavage sites in native cellular proteins is consistent with the 3CLpro cleavage specificities in the viral polyprotein (Scott et al., 2021), and natural and non-natural amino acid peptide substrates (Rut et al., 2021) (vide infra). Notably, the ‘other’ 663 neo-N-termini winnowed out were found not to start after the SARS-CoV-3CLpro consensus P1-Gln (Figure 1C; Table S6B).
Finally, to select only bona fide substrates, we generated a position-specific scoring matrix (PSSM) using the normalized relative frequency of amino acids in positions P4–P4’ of the 292 deemed as 3CLpro cut-sites (Figure 1C). We then calculated a score for the P4–P4’ sequence of all 955 neo-N-termini to measure similarity relative to the PSSM and selected the 3CLpro sites scoring higher than the 90th percentile of the non-3CLpro cleavage sites (n = 171). All MS/MS spectra of these neo-N-terminal peptides were then manually inspected. Spectra from ragged-protein ends, showing poor fragmentation or noise, and four other sites not validated by synthetic peptide cleavage (STAR Methods) were excluded (n = 69, Table S1).
We conclude that 3CLpro targets at least 101 human substrates at 102 sites (Table 1) that could not be disproven by our substrate winnowing strategy, including 34 proteins identified in both cell lines (Figure 1D), 28 of which were found in all twelve or 11/12 independent experiments. Adding further weight to our analyses, 38 of the 167 cut sites we found in Table 1 and Table S1 were independently reported in a proteomics dataset brief (Koudelka et al., 2021), using in vitro N-terminomics in lung epithelial carcinoma cells (H441) and human pulmonary microvascular endothelial cells. However, no further biochemical or physiological validation was performed. In addition, Meyer et al. (2021) very recently reported cleavage of NUP107 (Table 1) at position Gln35 in SARS-CoV-2–infected A549-ACE2 cells and GOLGA3 at Gln365 (Table S1), and ATAD2 at Gln949 (Table 1), which we also found. In their study, GOLGA3 cleavage was elegantly validated in 3CLpro transfected cells, whereas NUP107 and ATAD2 cleavages were attributed to 3CLpro based on the cleavage logo but without direct evidence. Likewise, our data validate the cut site at position Gln444 of TAB1 (Table 1) that (Moustaqil et al., 2021) inferred from the electrophoretic migration of TAB1 proteolytic fragments and 3CLpro cleavage specificity.
We quantified the relative protein abundance of 45 substrates identified from a total of 2,767 quantified proteins in interferon-treated BEAS-2B cells (STAR Methods). Only galectin-8 increased protein expression in response to type I interferons, whereas YAP1 and VAT1 decreased (Figure S1I and S1J; Table S7). Hence, ISGs are not a significant substrate class of 3CLpro. Overall, 3CLpro cleaves cellular substrates involved in three main processes: (1) RNA splicing, processing, activation, and metabolism; (2) translation; (3) and cell cycle control (Figure 1E; Table S8), affording insight into the processes of cellular subjugation utilized by SARS-CoV-2.
Structure-activity relationships of canonical versus noncanonical 3CLpro cut-sites
Using MALDI-TOF-MS, we calculated the apparent (app) specificity constant, app(k cat/K M), of 3CLpro for synthetic peptides spanning P4–P4’ of all cleavage sites in the 34 common substrates identified in HEK293 and BEAS-2B cells (Figure 1D). In addition, we assayed cleavage-site peptides from 12 candidate substrates with compelling biology. 3CLpro cleaved all peptides from the 34 common substrates and 9/12 peptides from the candidate substrates (Figures 2 A, 2B, and S2A). The app(k cat/K M) of 3CLpro cleaved peptides was consistent with the 3CLpro preferences for small amino acids in P1’, glutamine in P1, and leucine in P2 (Figure 1C), but with surprising yet unequivocal exceptions. The presence in P1 of methionine (T22D2, MAP4K5) or histidine (RBM15, MCM4) did not block cleavage (Figure 2B). Although no previous reports identify the noncanonical Met at P1 in substrates, we also found the same neo-N-terminal peptide for MAP4K5 by data-mining the proteomic dataset report of Koudelka et al. (2021), which had not been designated a candidate substrate as it lacked the P1-Gln.
Figure 2.

Characterization of 3CLpro cleavage specificity
(A and B) MALDI-TOF-MS spectra of synthetic peptides spanning P4–P4’ of protein cleavage sites after incubation with 3CLpro (1:20 molar ratio, E:S). Product generation (red) and substrate consumption (black) were calculated as the peak area normalized to the total peak area in the spectrum. Apparent (app) kcat/KM values for 1 μM 3CLpro to convert 50% of substrate in 5, 15, 30, 60, 120 or 240 min are listed alongside bins of 4 peptides that share similar kinetic values arranged on a row-by-row basis. P4–P4’ sequence alignment using the Shapley color scale. Green protein names had cut sites identified by Edman sequencing of recombinant substrate digests. Boxed peptides, no cleavage.
(C–I) Structures of the highest-ranked of 50,000 models of the active site of 3CLpro protomer 1 (PDB: 6XHM) docked with P4–P4’ peptides from six 3CLpro substrates exhibiting a range of appkcat/KM values (circled in B). (C) 3CLpro dimer. Protomer 1, gray surface or green ribbons with catalytic Cys145 shown. Protomer 2, orange surface with Ser1 shown. Docking models with P4–P4’ peptide of: (C and D) RPAP1 (I_sc = −31.7), (E) IMA4 (I_sc = −30.6), (F) PTBP1 (I_sc = −31.7), (G) RBM15 (I_sc = −39.65), (H) MAP4K5 (I_sc = −28.1), and (I) CREB1 (I_sc = −30.3). Blue and red sticks, P and P’ amino acid residues, respectively. Yellow dashed sticks, hydrogen-bonds.
See also Figure S2.
We also demonstrate similarities and divergence at P2 from the dominant leucine specificity (Figure 1C) previously reported in the SARS-CoV-2 polyprotein (Scott et al., 2021), peptides (Rut et al., 2021), and monkey and human proteins (Koudelka et al., 2021; Meyer et al., 2021). In the polyprotein, P2-Val and P2-Phe each occur once. We too found valine (CREB1, site 2) and phenylalanine (SRRM2), as well as methionine (MCM4) and alanine (CLCB) at P2, which we validated (Figure 2B). Additionally, we established the occurrence of isoleucine (RS21), glutamine (SF3B2, NACAM), and proline (IF4G1, PTBP1-2nd site) in P2, which were previously unreported. The noncanonical P2 residues impaired catalytic efficiency but did not block cleavage. We frequently found glutamine and valine at P3 (e.g., GOLGA2 and CREB1, respectively), and at P4, valine and eight instances of proline (NUP107 and FYCO1, respectively). However, the most significant difference between the specificity logos is the prime-side specificity profile C-terminal to P1’, which has been largely overlooked in the other studies of SARS-CoV-2 3CLpro. Thus, the kinetics analyses confirm the cleavage specificity divergence we found by sequence analysis of cleaved native human proteins identified by TAILS (Figure 1C). These unexpected findings are fundamental to inform drug development and derive from an approach that does not require manual searches based on assumed cleavage site preferences that miss such deviations.
Several structural analyses reported the P-side interactions of peptides or inhibitors with 3CLpro (Vuong et al., 2020; Zhang et al., 2020). However, to our knowledge, only one paper described a P’-side sequence engaged in the 3CLpro-S’ interface, but the autocatalytic NSP5 P1’–P3’ sequence (Ser-Ala-Val) was reported to fit poorly (Lee et al., 2020). Indeed, none of the 101 human substrates display this sequence. Reasoning that human substrate complexes with 3CLpro would reveal biologically relevant structure-activity relationships, we modeled the binding complex of the 3CLpro dimer/cleavage-site peptide of seven human substrates by high-resolution peptide-protein docking. All models displayed highly negative I_sc (Rosetta interface score) values, indicating a favorable 3CLpro and peptide interaction (Figures 2C–2I), and for the P-side interactions, our models resembled published structures. Hydrogen-bond lengths were within 3.5 Å (Kajander et al., 2000), and best-fit models varied due to molecular dynamics.
Even when the P-sequence is optimal, cleavage was affected by the fit of residues in subsites on the P’-side. The most prominent position is P1’ since the S1’ subsite cannot typically accommodate bulky residues due to steric hindrance imposed by Thr25, Leu27, and His41 side-chains. The consensus P1’-Ala/Ser/Gly each fit optimally in S1’. Nevertheless, some substrates are efficiently cleaved despite relatively bulky side-chains at P1’, e.g., LQ78↓N in IMA4 and LQ133↓L in YAP1 (Figure 2B). In IMA4, the P1’-Asn points toward S3’, where the side-chain amide group is within hydrogen-bonding distance of Thr25 (3.2 Å), His41 (2.9 Å), Cys44 (1.6 Å), and Ser46 (3.3 Å) (Figure 2E). Thus, S3’ is dynamic, accommodating residues from other P’-side positions. In RBM15, the P1’–P4’ residues form β-sheet-like hydrogen bonds with Thr24–26 (Figure 2G), contributing significantly to the best P’-side fit ( i.e., lowest I_sc = −39.65) of the modeled substrates.
Our docking simulations provide structural insights into noncanonical P1 substitutions. The side-chain of a P1-His behaves like the amide group of the canonical P1-Gln side-chain where its imidazolyl nitrogen atoms act as both hydrogen bond donor and acceptor according to their protonation state. The protonated Nε2 atom of P1-His donates a 2.0-Å hydrogen bond to the Glu166 Oε1 (Figure 2G), whereas the deprotonated Nε1 acts as a hydrogen-bond acceptor through the interaction with the Ser144 Oγ (3.0 Å) and His163 Nε2 (2.3 Å). Both the noncanonical P1-residue interactions involving the main chain are conserved. We discovered cleavage after a P1-Met in two substrates and P1-His in seven substrates (Table 1; Figure 2B), plus two candidate substrates each (Table S1) ( i.e., ∼10% of substrates). In this case, the main-chain oxygen atom of the P1-His or Met accepts hydrogen bonds from the main-chain nitrogen of Gly143, Ser144, and Cys145 to promote cleavage at the Leu-Met↓Ser and Leu-His↓Ser sites. These noncanonical P1 residues and the dynamic occupancy of S3' were unexpected and can be leveraged for 3CLpro/inhibitor drug development and predictions of off-targets in treatment.
3CLpro cleaves RPAP1 and PTBP1, altering PTBP1 subcellular localization
The subversion of transcription and translation machinery is a recognized strategy to co-opt host cells for optimal viral replication (Walsh and Mohr, 2011). Indeed, the three major gene sets enriched with 3CLpro substrates are proteins involved in these processes (Figure 1E; Table S3). We further characterized two substrates. RNA polymerase II-associated protein 1 (RPAP1) is crucial for optimal RNA polymerase II activity—by binding a protein known as Mediator, RPAP1 couples RNA polymerase II to enhancer elements to elevate transcription (Lynch et al., 2018). Polypyrimidine tract binding protein (PTBP1) binds mRNA and is essential for the sequential phases of viral translation and replication (Florez et al., 2005). RPAP1, cleaved in N = 12/12 experiments, is one of the best substrates for 3CLpro with an app k cat/K M > 1.5 x103 M-1sec-1 and PTBP1 was identified in N = 3/3 HEK293 cell experiments (Figure 3 A). In time-course 3CLpro in vitro cleavage assays, we observed loss of both substrates coincident with sequential cleavage-product generation at molecular weights predicted from the cut-site locations (Figures 3B and S3). Catalytically inactive mutant 3CLpro-C145A or incorporation of a 3CLpro inhibitor, GC376 (Vuong et al., 2020), confirmed 3CLpro cleavage of the substrates. Edman sequencing validated the RPAP1 and PTBP1 neo-N-termini identified by TAILS and identified other cleavage sites, which we supported by peptide cleavage kinetics assays (Figure 3C). For technical reasons, these cleavage products would not have been observable by MS/MS (Figures 3B and S3). In addition, endogenous RPAP1 and PTBP1 were cleaved by 3CLpro in lysates of primary human airway epithelial cells (HAECs) from five donors (Figures 3D and S4B).
Figure 3.

3CLpro cleaves RPAP1 and PTBP1, altering PTBP1 localization
(A) Locations of 3CLpro cleavage sites in RPAP1 and PTBP1 identified by TAILS neo-N-terminal peptides (red) and Edman sequencing (green). Representative MS/MS spectra of cleaved neo-N-terminal peptides.
(B) SDS-PAGE and Edman sequencing of human recombinant RPAP1 and PTBP1 incubated with 3CLpro+/− inhibitor GC376, or 3CLpro-C145A (1:5 mol/mol, E:S). Δ-substrate, no sequence obtained.
(C) MALDI-TOF-MS kinetic analyses of 3CLpro cleavage of synthetic P4–P4’ peptides.
(D) RPAP1 and PTBP1 immunoblots of primary HAECs lysates from 5 donors incubated with 3CLpro or 3CLpro-C145A (1:200 w/w, E:S) for 18 h, 37°C.
(E and F) (E) Immunoblots of 3CLpro, N-protein and (F) PTBP1 of Vero E6 cells infected with SARS-CoV-2 at a MOI 0.1 for 24 (n = 4) and 48 (n = 4) hpi, or mock (n = 3) ∗, unspecific bands.
(G) Subcellular localization of PTBP1 by confocal imaging. SARS-CoV-2-infected Vero E6 cells (N = 5, scale bar 50 μm). Cyan boxes, Spike-negative (S–) uninfected cells. Green boxes, Spike-positive (S+) infected cells. Enlarged detail of mock-infected, S+ and S– cells in the same field, scale bar 10 μm. (H) Nuclear to cytoplasmic ratio of PTBP1 was quantified for mock and Spike-positive SARS-CoV-2 infected cells. Statistical significance was assessed by Student’s t test (mean ± SD, N = 5, n = 51 cells, ∗p ≤ 0.05). β-actin and β-tubulin loading controls.
See also Figures S2–S5.
To confirm cleavage of PTBP1 during infection, we infected Vero E6 cells with SARS-CoV-2 at a multiplicity of infection (MOI) of 0.1 and collected cell lysates at 24 and 48-h post-infection (hpi) (n = 4, each time point). Immunoblots showed the expression of nucleocapsid protein and 3CLpro (Figure 3E). Compared with mock-infected cells, a decrease in intact PTBP1 at 48 hpi coincident with the appearance of cleavage fragments confirmed PTBP1 cleavage in SARS-CoV-2 infection (Figure 3F). Similar results were obtained for PTBP1 in infected Calu-3 human lung epithelial cells (Figure S4C). However, high background in Calu-3 cells made specific band identification challenging.
PTBP1 isoforms 1, 2 and 3 have a 3CLpro cleavage site, AALQ↓AVNS, in the linker between RNA recognition motif (RRM)1 and RRM2 (Figure 3A). In addition, PTBP1 isoforms 2 and 3 have a validated cleavage site, AIPQ↓AAGL, in the linker between RRM2 and RRM3 (Figure S2B). This unusual cleavage sequence, i.e., P2-Pro followed by P1-Gln, is spliced out from isoform-1. Since cleavage at the shared site will remove the nuclear localization sequence from the N terminus of all PTBP1 isoforms (Figure 3A), we examined whether SARS-CoV-2 infection altered the nuclear localization of PTBP1, as previously reported for other coronaviruses (Sola et al., 2011). In uninfected Vero E6 cells, PTBP1 was exclusively located in the nucleus (Figure 3G) with a nuclear to cytosolic ratio of 1.9 (Figure 3H). However, upon SARS-CoV-2 infection, PTBP1 translocated to the cytoplasm with a nuclear/cytosol ratio of 0.3 at 48 hpi (N = 5, n > 50 cells, Figures 3G and 3H and S5). Frequently, the same microscopy fields evidenced nuclear-to-cytosol transit of PTBP1 in infected cells but not in nearby uninfected cells, which is more evident at high magnification (Figures 3G and S5D). Thus, proteolytic removal of the NLS could explain the loss of nuclear localization of PTBP1 in coronavirus infection. Moreover, we showed that IMA4, which is involved in cargo recognition, and TPR and NUP107, which are integral parts of the nuclear pore ring, are all substrates of 3CLpro (Figures S2C–S2E and S4B). These substrates provide evidence for potential mechanisms in the targeted shutdown of nucleocytoplasmic transport by SARS-CoV-2, a viral strategy to repress host cell translation (Caly et al., 2015).
In picornavirus, RNAi-silencing reveals that full-length PTBP1 negatively regulates viral RNA transcription (Florez et al., 2005). Hence, PTBP1 cleavages may relieve an inhibitory effect on SARS-CoV-2 replication. Alternately, poliovirus 3CDpro reportedly cuts PTBP1 and blocks IRES-dependent protein synthesis, switching from viral translation to replication (Back et al., 2002). Notably, knockdown of RPAP1 results in broad reductions in transcription and leads to cell dedifferentiation (Lynch et al., 2018), which is often a feature of viral infection but is poorly understood. Thus, the fragmentation of RPAP1 by 3CLpro, which we hypothesize phenocopies RPAP1 silencing, together with direct cleavage of RNA polymerase I (Table 1), negatively impacts host transcription and translation to reinforce the switch from host to viral transcription and translation, warranting mechanistic investigation.
3CLpro targets the Hippo pathway
The Hippo signaling pathway, which regulates cell morphology, mechanotransduction, tissue growth and regeneration, is not a generally recognized target of viral proteolytic attack (Yalamanchili et al., 1997). Nevertheless, TAILS identified three substrates integral to Hippo signaling: YAP1, CREB1, and ATF1, with a fourth, MAP4K5, involved in the regulation of Hippo/EGFR crosstalk. The phosphorylation of YAP1 by LATS1/2, a downstream phosphorylation target of the MAP4K family, prevents nuclear translocation and transcriptional activity of YAP1 (Rausch and Hansen, 2020). MAP4K5 contains ten Leu-Gln instances with at least three optimal sequences for 3CLpro cleavage, yet none were cut in 9/9 independent BEAS-2B analyses. Instead, TAILS identified a noncanonical SKLM456↓SENT cleavage site between the kinase and the regulatory citron homology domains in all experiments (Figure 4 A). P1-Met was previously unknown to be susceptible to 3CLpro. Therefore, we verified the TAILS cut-site by cleaving the corresponding P4–P4’ synthetic peptide (Figure 4B). Edman sequencing confirmed that product-2 of cleaved recombinant MAP4K5 protein was from scission at Met456↓Ser, with immunoblotting showing the N-terminal origin of product-1 (Figure 4C). Hence, in addition to glutamine and histidine, 3CLpro accommodates methionine in P1 (Figure 2H), which must now be considered integral to its specificity profile.
Figure 4.

Hippo pathway substrate validation
(A) 3CLpro cleavage sites in MAP4K5 and CREB1 identified by TAILS neo-N-terminal peptides (red) and Edman sequencing (green). Representative MS/MS spectra of cleaved neo-N-terminal peptides.
(B) MALDI-TOF-MS kinetic analyses of 3CLpro cleavage of P4–P4’ peptides of MAP4K5 and CREB1.
(C) SDS-PAGE, Edman sequencing (green) and immunoblot validation of human MAP4K5 and CREB1 substrates incubated with 3CLpro+/− inhibitor GC376, or 3CLpro-C145A (1:5 mol/mol, E:S). ΔMAP4K5 or ΔCREB1, no sequence obtained.
(D and H) (D) YAP1, MAP4K5, CREB1 and (H) FYCO1 and FAF1 immunoblots of lysates from primary HAECs incubated with 3CLpro or 3CLpro-C145A (1:200 w/w, E:S) for 18 h, 37°C.
(E and F) (E) YAP1 and (F) MAP4K5 immunoblots of Vero E6 cells at 24 (n = 4) and 48 (n = 4) hpi (MOI 0.1) or mock (n = 3). MAP4K5 activity assay measured as ATP consumption using myelin basic protein as substrate. The area under the curve was calculated and compared by Student’s t test, (mean ± SD, n = 2, ∗∗p ≤ 0.01).
(G, I, and J) (G) Lysates of human Calu-3 lung cells infected with SARS-CoV-2 (MOI 0.1 and 1.0, n = 4, mock n = 2) were immunoblotted for (G) CREB1 48 hpi, (I) FYCO1 24 hpi and (J) FAF1 48 hpi. Statistical analysis of the relative amount of full-length protein (E and F) or proteolytic bands (G, I, and J) identified by molecular weights relative to β-actin was assessed by one-way ANOVA and Dunnett’s multiple comparisons test. Box and whiskers (min to max) plots, ∗∗∗p ≤ 0.001, ∗∗p ≤ 0.01, ∗p ≤ 0.05, ns p > 0.05. β-actin and β-tubulin loading controls.
See also Figures S3 and S4.
After activation by upstream signals, including the Hippo pathway, CREB1 dimerizes with ATF1 to form a competent transcription factor that binds the cAMP-responsive element to promote expression of anti-apoptotic and cell proliferation genes (Persengiev and Green, 2003). Moreover, the Hippo signaling pathway cross-talks with Wnt, Notch, the EGF receptor ERBB4, and the TGFß pathway through SMAD1 and SMAD7 (Dupont et al., 2011). Cleavage at VVVQ243↓AASG in CREB1, identified in 12/12 TAILS experiments (Figure 4A), detaches the N-terminal kinase-inducible domain from the C-terminal basic leucine zipper region. Cleavage of a synthetic P4–P4’ peptide (Figure 4B) and recombinant CREB1 (Figure 4C) occurred at moderate rates, consistent with P2-Val being accommodated but as a nonpreferred amino acid residue (Figure 2I) (Rut et al., 2021). Edman sequencing confirmed cleavage at VVVQ243↓AASG and revealed a 2nd site at TILQ223↓YAQT (product-2, Figure 4C). We mined the TAILS data and found proteomic evidence for this site (n = 2/12, HEK-TAILS2_ACN, MS/MS #46,629). Hence, our stringent substrate winnowing criteria identified substrates with great confidence but at the expense of underestimating substrate numbers. TAILS also identified the identical site, TILQ151↓YAQT, in ATF1 (Table 1), which we confirmed by peptide cleavage (Figure S2A).
YAP1, MAP4K5 and CREB1 in primary HAECs (N = 5) were cleaved by 3CLpro, but not inactive 3CLpro-C145A (Figure 4D), with cleavage of CREB1 and YAP1 dimers also evident (Figure S4A). In Vero E6 cells infected with SARS-CoV-2, we identified reductions in endogenous YAP1 (Figure 4E) and MAP4K5 (Figure 4F). MAP4K5 cleavage products were at the expected apparent molecular weights (Figure 4F) and consistent with the MAP4K5 cleavage products shown in primary HAECs (Figure 4D). In SARS-CoV-2 infection of a second cell type, human Calu-3 cells, antibodies to CREB1 did not show a decrease of the full-length band consistent with the low cleavage rate of the synthetic peptide and the recombinant protein shown by in vitro cleavage assays (Figures 4B and 4C). Higher molecular weight bands (Figure 4G) with similar-size products were observed in bronchial epithelium after cleavage by 3CLpro (Figures S4B and S4C). However, the antibody specificities were not optimal for more definitive conclusions in these cells. We measured kinase activity of MAP4K5 and found that cleavage separation of the Ser/Thr-kinase domain from the citron homology domain by 3CLpro halted kinase activity (Figure 4F). Thus, 3CLpro redundantly targets the transcription arm of the Hippo pathway.
Phosphorylation of Ser381 targets YAP1 for proteasomal degradation, whereas phospho-Ser127 triggers YAP1 binding to 14-3-3 ε, which sequesters YAP1 in the cytosol, preventing transit to the nucleus as a transcriptional coactivator (Rausch and Hansen, 2020). YAP1 cleavage at ASLQ133↓LGAV was observed in 9/9 independent BEAS-2B TAILS experiments, which we confirmed by peptide cleavage kinetic analyses (Figure 2B). Scission at Gln133 could prevent Ser127 phosphorylation, 14-3-3 ε binding and hence nuclear translocation. Truncation of YAP1 at Gln133 generates a C-terminal fragment homologous to the transcriptionally inactive isoform-4 of YAP1, which efficiently inhibits IRF3 translocation and innate antiviral responses (Wang et al., 2017). Thus, the redundant inactivation of YAP1 by removal of the YAP1 Ser127 kinase-activation sequence/14-3-3 ε binding site, the inactivation of an upstream regulator kinase, MAP4K5, together with two downstream transcription factor targets, CREB1 and ATF1, strongly implicate the importance of repressing Hippo-regulated gene transcription and TBK1 activity for optimal SARS-CoV-2 infection.
Diverse 3CLpro targets in viral subjugation of the cell in COVID-19
We validated substrates from other pathways relevant to the viral hijacking of the cell. These include EIF3 (Figure 2A), which blocks binding of SARS-CoV-2 NSP1 to the 40S ribosomal subunit (Lapointe et al., 2021); and FAS-associated factor 1 (FAF1) (Figures 4H and 4J), a positive regulator of type I interferon signaling (Kim et al., 2017). Insulin receptor substrate 2 (IRS2) (Figure S4A), a key phosphorylation target of the insulin receptor (Guo et al., 2006), was also cleaved, as were two integral components of nuclear pore transport—nuclear pore complex protein (NUP107) and importin subunit alpha-4 (IMA4) (Figures S2C–S2E and S4C). Finally, we validated the cleavage of two autophagy adaptors FYVE and coiled-coil domain-containing protein 1 (FYCO1) (Figures 4H and 4I), which is critical for translocation of autophagic vesicles (Cheng et al., 2016), and galectin-8 (Figure 5 ) (Wang et al., 2020).
Figure 5.

3CLpro disrupts galectin-8 binding to Spike in antiviral-autophagy
(A) Immunoblot of human galectin-8 (Gal8) in BEAS-2B cells in response to IFN-α, IFN-β, or vehicle. One way ANOVA and Dunnett's posthoc test (mean ± SD, n = 3 each, *** p ≤ 0.001, * p ≤ 0.05).
(B) MALDI-TOF-MS of intact versus 3CLpro-cleaved synthetic Gal8 P4–P4’ peptide.
(C) SDS-PAGE and Edman sequencing of Gal8 incubated with 3CLpro+/− inhibitor GC376, or 3CLpro C145A (1:5 mol/mol, E:S).
(D) Structural model of Gal8 docked onto 3CLpro. 3CLpro cleavage site identified by the neo-N-terminal peptide (red) in 9/9 independent TAILS analyses.
(E) Gal8 immunoblots of lysates from primary HAECs incubated with 3CLpro or 3CLpro-C145A (1:200 w/w, E:S) for 18 h, 37°C (N = 5).
(F) Gal8 immunoblot of infected Calu-3 cells at 24 (n = 4) and 48 (n = 4) hpi (MOI 1.0, mock n = 3). β-actin and β-tubulin loading controls.
(G) ELISA of SARS-CoV-2 Spike S1 protein binding intact Gal8 or 3CLpro-cleaved (ΔGal8) (mean ± SD, n = 2, N = 2, ∗∗∗∗p ≤ 0.0001, ∗∗p ≤ 0.01, ns p > 0.05, two-way ANOVA with Šídák’s multiple comparison test).
(H) Immunoprecipitation (IP) by α-FLAG agarose-beads of HeLa cell lysates co-transfected with GFP-NDP52, WT-Gal8-FLAG or C-Gal8 (159-317)-FLAG.
(I) Intact or cleaved Gal8 binding immobilized NDP52 detected with anti-C-Gal8 antibody. Student's t test, (mean ± SD, n = 3, N = 2, ns p > 0.05).
(J) NDP52 binding Spike S1-associated intact or cleaved ΔGal8 (mean ± SD, n = 2, N = 2, ∗∗∗∗p ≤ 0.0001, ∗∗∗p ≤ 0.001, ∗p ≤ 0.05, ns p > 0.05, two-way ANOVA with Šídák’s multiple comparison test).
(K) Confocal microscopy of NDP52 and FLAG immunofluorescence after osmotic shock of HEK293 cells transfected with FLAG-tagged (WT) Gal8, N-Gal8 (1-158), or C-Gal8 (159-317). Scale bar, 20 μm. Quantification of NDP52 and Gal8-positive puncta, mean ± SD, n = 30, N = 5, ∗∗∗∗p ≤ 0.0001, ns p > 0.05, one-way ANOVA with Tukey’s multiple comparison test.
(L) Human lung tissue sections from normal subjects (N = 3) and post-mortem COVID-19 patients (N = 4) stained with hematoxylin and eosin (H&E), DAPI (blue) and immunofluorescence on the same sections for Gal8 (green), NDP52 (red), Spike (magenta) and merged image (orange). Scale bar, 50 μm.
(M) Numbers of immunopositive cells for Gal8, NDP52 and Gal8 colocalized with NDP52 in healthy versus COVID-19 lung tissue. Total cell count distribution shown as a scatter dot plot, bar = median.
See also Figure S6.
Galectins are essential in host defense by directly interacting with pathogens and regulating the immune response (Wang et al., 2020). Galectin-8 was the only 3CLpro substrate elevated by type I interferons, consistent with an antiviral role (Figures 5A and S1I; Table S6). Proteolysis of galectin-8 at SDLQ158↓STQA occurred in all nine BEAS-2B cell analyses (Figure S6A), suggesting an alternative viral evasion mechanism to overcome cell resistance to SARS-CoV-2 infection. Cleavage was validated at the peptide (Figure 5B) and protein levels (Figures 5C and S6B) by MALDI-TOF-MS kinetic analyses and Edman sequencing, respectively. The site of 3CLpro scission is in the short linker (Phe153-Pro186) of galectin-8, which dislocates the amino carbohydrate recognition domain (CRD)-1 from the carboxyl CRD2 (Figure 5D). The cleavage site is also in the linker (Phe153-Pro229) of the long galectin-8 isoform, which should also be susceptible to cleavage. Molecular docking simulations revealed unimpeded access of the linker to the 3CLpro active site (I_sc = −19.6), where hydrogen bonding by Gly143 (1.5 Å) and His163 (3.3 Å) stabilize the galectin-8 P1-Gln158 (Figures 5D and S6C). Significant interactions also occur on the P’-side, mainly by Thr21, Thr24 and Thr26. The 3CLpro protomer-2 further stabilizes the 3CLpro/galectin-8 complex by hydrogen bonds between Cys300 (2.7 Å) and Ser301 (3.5 Å) of 3CLpro to Thr168 and Glu169, respectively, of galectin-8.
Galectin-8 binds glycans on the cell surface (Carlsson et al., 2007) and has hemagglutination activity due to its bivalent carbohydrate-binding capacity. We found that 3CLpro cleavage disrupts glycan-binding—separation of CRD1 from CRD2 by 3CLpro prevented hemagglutination of human erythrocytes (Figures S6D and S6E) and surface adhesion of Jurkat-T cells (Figure S6F). In addition, proteolysis of endogenous galectin-8 by 3CLpro, but not inactive 3CLpro-C145A, was observed in primary HAECs (Figures 5E and S6G) and Calu-3 cells infected with SARS-CoV-2 (Figure 5F).
On permeabilized endosomes, intracellular galectin-8 detects exposed glycans normally on the cell exterior, leading to cell resistance to infection [e.g., by S. Typhimurium (Thurston et al., 2012) and picornavirus (Staring et al., 2017)]. Upon exposure of alpha-2, 3-sialylated- and 3’-sulfated glycans to the cytosol (e.g., on membrane damage), galectin-8 recruits an autophagy adaptor, CALCOCO2/nuclear dot protein-52-kDa (NDP52), which binds microtubule-associated protein-1 light chain-3 (MAP1LC3). MAP1LC3-coated autophagosomes are then targeted for lysosomal degradation (Mohamud and Luo, 2019). We hypothesized that in SARS-CoV-2 infection, galectin-8 senses the highly glycosylated Spike S1 protein and activates antiviral-xenophagy, reducing SARS-CoV-2 infection. Of significance for viral entry and potential escape from xenophagy, we demonstrated direct binding of galectin-8 to immobilized Spike S1 protein and Spike S1 to immobilized galectin-8 (Figure S6H). This protein complex was dismantled following 3CLpro cleavage of galectin-8 (Figures 5G and S6J). Decisively, a competitive inhibitor of galectin glycan-binding sites, thiodigalactoside, blocked binding (Figures S6I and S6J), confirming this previously unknown direct interaction between galectin-8 and Spike S1 glycans, which 3CLpro disrupts.
To determine the potential for galectin-8 acting as a cell sensor for SARS-CoV-2, we confirmed glycan independent NDP52 binding to galectin-8 (Kim et al., 2013) (Figure S6K). By immunoprecipitation with α-FLAG antibody, we confirmed NDP52 binds the C domain of galectin-8 generated after 3CLpro cleavage (Figures 5H and S6L), which we also showed by ELISA (Figure 5I) as previously reported by (Li et al., 2013). NDP52 and Spike S1 were not susceptible to 3CLpro cleavage (Figure S6M). We assembled the trimeric complex comprised of NDP52 bound to galectin-8 bound to immobilized Spike S1 protein. Using this complex, we showed that upon galectin-8 cleavage, the indirect tethering of NDP52 to Spike S1 was lost (Figure 5J).
To model the effect of 3CLpro cleavage of galectin-8 on autophagy, we transfected HEK293 cells with galectin-8 or the 3CLpro-cleavage analogs FLAG-N-Gal8 (1–158) and FLAG-C-Gal8 (159–317). Upon disruption of endosomal/lysosomal integrity by osmotic shock, we observed that transfected FLAG-tagged galectin-8 was recruited to damaged vesicles and formed puncta that colocalized with NDP52 (Figure 5K). In contrast, transfected FLAG-tagged cleavage-fragment analogs failed to form puncta or colocalize with NDP52 in HEK293 cells (Figure 5K).
Analysis of human lung autopsy samples from post-mortem COVID-19 patients (N = 4) was insightful. The overall immunofluorescence signal intensities of galectin-8, NDP52 and DAPI were slightly weaker in COVID-19 tissues samples than the healthy lung samples from noninfected subjects (N = 3). This was likely from cytopathic effects on the cells caused by the disease, including massive fluid infusion into the lungs. Lung weights were on average 2.8 times heavier than normal lungs, with significant signs of damaged lung parenchyma with immense cytopathic effects and swollen cells as described in the autopsy collection (Szekely et al., 2021). Despite the slightly weaker staining, this did not affect the colocalization analysis as only cells showing intact nuclei with DAPI staining present were counted. The difference in the expression pattern of NDP52 and galectin-8 was both substantial and consistent for each patient and field of view (n = 30). That is, there was virtually a complete overlap between the two proteins in normal lung (> 95% colocalization) versus in the patient samples, where only 5% of galectin-8 colocalized with NDP52 (Figures 5L and 5M). Hence, our results showing direct binding of galectin-8 to Spike S1 protein and the C-domain of galectin-8 to NDP52 suggests an antiviral autophagy mechanism that SARS-CoV-2 3CLpro counteracts by cleavage of galectin-8 and FYCO1.
Protein-protein interaction landscape of 3CLpro host cell substrates
We reasoned that in addition to direct cleavage of essential host proteins, 3CLpro proteolytic activity could hijack the cellular machinery by indirectly modifying the function of substrate-interacting proteins. To explore this, we constructed a protein-protein interaction network using the 101 substrates of the 3CLpro degradome as seeds (Figure 6 A, red circles). We retrieved 2,202 human proteins from the Imex/Intact database having rigorous experimental evidence for direct interactions or physical associations (Figure 6A). Among the interactors are 16 proteins from Table S6 classified as “candidate” substrates (Figure 6A, orange circles; Table S2), increasing confidence for their future promotion to substrates. The interactome of 3CLpro human substrates is a highly interconnected network where 94 substrates interact directly or via third-party interactors. This connectivity suggests that proteolytic processing of the cellular proteome by 3CLpro sculpts SARS-CoV-2/host interactions by disrupting cellular processes in a concerted and redundant manner, as seen for the Hippo and xenophagy pathways. Notably, the interactome reveals several hub proteins (Figure 6A, magenta circles) left “stranded” by 3CLpro cleavage after losing two or more interactors. Without a scissile bond, we hypothesize that these numerous stranded proteins are opportunistically targeted by cleavage of essential interactors, thereby directly impacting their function to favor viral replication. Three of the processes left isolated by 3CLpro with pertinence to the clinical features of COVID-19 are: (1) the NF-κB signaling pathway, the central regulator of innate and adaptive immunity, including the NEMO subunit NF-κB2 and the negative regulators, RelA and RelB; (2) the proto-oncogene products Myc, Jun, and Fos—all involved in immune cell activation, cytokine expression, and interferon signaling (Casey et al., 2018; Chanda et al., 2003); and (3) NDP52 and PICK1, required for antiviral-autophagy and endosome maturation.
Figure 6.

Protein-protein interaction landscape of 3CLpro cell substrates
(A) Network of direct (solid line) and physically associated (dashed line) interactors (blue dots) of the 101 3CLpro substrates (red circles) generated by the Intact-database (accessed May 2021). Interactors of ≥ 2 substrates are in magenta, only interactors of ≥ 3 substrates are labeled, the number of substrate interactions is indicated by circle size. Orange circles, candidate substrates manually annotated.
(B) Top 5 CORUM protein complexes statistically enriched in the 3CLpro substrate-human interactome.
(C) One-step direct neighborhood protein-protein interaction network of 3CLpro human substrates (n = 74, red circles) that are directly connected to SARS-CoV-2 proteins (n = 26, yellow diamonds) or connected via direct neighbors (n = 197). Assembled from IMex/Intact Coronavirus dataset accessed June 2, 2021. Yellow diamonds, viral proteins. Blue circles, direct interactors connecting viral proteins to substrates. Magenta circles, direct interactors connecting viral proteins to ≥ 2 substrates. Black edge, SARS-CoV-2/human protein interactions.
See also Data S1.
The most significantly enriched protein complexes in the 3CLpro substrate interactome are the spliceosome, the PA700-20S-PA28 proteasome, the EIF3 complex, the anti-HDAC2 complex, and the TNF-α/NF-kB signaling complex (Figure 6B). These complexes are consistent with the functional categories of 3CLpro substrates (Figure 1E) and the cellular processes impacted by stranded proteins. Finally, we show that 26 viral proteins connect to 74 substrates, either by direct interactions (n = 16) in the virus/human-substrate interactome or via a shared interacting partner (Figure 6C). Notably, the substrate PTBP1 is the most connected with seven viral protein interactors, followed by ARPC4 and SMC4, with three each, and PUF60 with two. The high connectivity of substrates with SARS-CoV-2 proteins implicates host protective and viral promoting roles of 3CLpro substrates in the CoV-2 life cycle.
Discussion
Understanding the role of each viral protein in infection is immensely important for the development of antiviral therapies. We have delineated the substrate landscape and cleavage site flexibility of SARS-CoV-2 3CLpro in depth. We show that 3CLpro is a pleiotropic viral factor that proteolytically processes over one hundred host cell proteins involved in essential cellular processes. Proteolytic processing is fundamentally different from degradation to completion via lysosomes and the ubiquitin-proteasome system (Klein et al., 2018). We demonstrated pertinent biological effects of processing with examples of altered protein function and subcellular localization after 3CLpro cleavage. Unlike viral competition for cellular resources, which are reversible, 3CLpro proteolytic processing of host cell substrates is irreversible. Thus, the targeted sculpting of the host cell proteome by viral proteases is one of the few direct ways that a virus, with a limited genome, can subvert the cell at multiple points to enhance replication and infection while rapidly defeating antiviral defenses. Moreover, the effects of 3CLpro proteolysis reverberate through the cell by cleaving interactors of what we term “stranded” proteins that are not cut, effectively isolating essential cofactors and impairing their function or disassembling protein complexes.
We demonstrate that galectin-8, the only ISG we found targeted by 3CLpro, loses the ability to recruit the autophagy adaptor NDP52 to damaged endosomes upon cleavage by 3CLpro. We further showed that galectin-8 functions as an intracellular sensor for SARS-CoV-2-loaded endosomes by recognizing the glycans decorating Spike S1. We suggest that proteolytic processing of galectin-8 and FYCO1 defeats an antiviral mechanism allowing SARS-CoV-2 to escape antiviral xenophagy. Our demonstration of galectin-8 being a direct viral protease target adds to the other substrates also reported in the galectin-8/NDP52/LC3 axis (Herhaus et al., 2020). Histological in situ analysis of post-mortem COVID-19 lung samples showed a strong phenotype where antibody-based imaging revealed virtually no colocalization of galectin-8 with NDP52, dramatically different from the healthy lung samples. This warrants further investigation as a desired antiviral action of 3CLpro inhibitor drugs in development. The transcription of ribosomal pre-rRNA by RNA polymerase I and mRNA by RNA polymerase II are impacted by 3CLpro—RNA polymerase I is a substrate, and cleavage of RPAP1 should disrupt the RPAP1 bridging of RNA polymerase II to gene enhancers. The Hippo-YAP pathway is emerging as a regulator of innate antiviral immunity. YAP/TAZ dimers dampen autophosphorylation of the antiviral mediator TANK binding kinase 1 (TBK1), the activator and trigger for translocation of IRF3 to the nucleus where it induces type I interferon transcription (Zhang et al., 2017). Notably, we also found that the TBK1 activators, TAB1 and TTC4, are 3CLpro substrates, as is FAF1, which also upregulates type I interferon signaling. Thus, the inactivation of anti-apoptotic and cell proliferation proteins by cleavage-deregulation of the Hippo signaling pathway deserves further study in SARS-CoV-2 infection. The functional YAP/TAZ dimer interacts with, regulates, and is regulated by plasma membrane structures. Therefore, deregulation of the Hippo pathway that relays cell shape and plasma membrane status should contribute to the dramatically altered cell morphology in SARS-CoV-2–infected cells and in the lungs of COVID-19 patients.
Due to the structural similarity between SARS-CoV and SARS-CoV-2 3CLpro, it is generally assumed that both enzymes behave with similar substrate preferences and kinetics. Most attention has been devoted to studying nonprime side interactions for drug development. In contrast, our study highlights the role of the substrate prime side and shows that SARS-CoV-2 3CLpro can cleave noncanonical sequences after methionine and histidine. We empirically showed cleavage occurs even with a bulky aliphatic residue in P1’. This can only occur after a significant conformational rearrangement of the substrate cleft, which has implications for the rational design of inhibitor drugs. The mechanistic insight gained from the over 100 substrates we discovered—with the promise of more by mining our data resource—and further exploration of the entire substrate degradome provides a foundational resource for the scientific community.
With many opposing cell mechanisms at play to favor viral translation and viral replication, targeting of essential host proteins by 3CLpro with precise temporal-spatial localization over a range of cleavage rates may synchronize the wave of events in the COVID-19 cellular coup d’état. Thus, our study strengthens the case for 3CLpro inhibition as an attractive therapeutic option to not only block viral polyprotein processing and assembly of the replication complex but also synergistically restore protective antiviral intracellular defense pathways. Our atlas of 101 substrates and the additional 58 candidate substrates provides rational start points for further investigations of the pathobiology of SARS-CoV-2 infection leading to COVID-19, triggered by 3CLpro cleavage of these host proteins. The cleaved substrate neo-termini in our atlas will help assess on-target drug efficacy in vivo. Moreover, clinical translation to detect cleaved substrate neo-N-termini, which more precisely reflect disease stage than the levels of the protein or transcript alone, is a precise diagnostic strategy for infection surveillance of SARS-CoV-2 and future coronavirus outbreaks that infect humans—which is just a matter of time.
Limitations of the study
Like all proteomic analyses, TAILS relies on mass spectrometry with inherent limitations in LC-MS/MS peptide identification and mass spectrometer sensitivity. These contribute to missing low abundance peptides, short or very long peptides, rare peptides from low abundance proteins, and some membrane proteins. However, in TAILS, short semi-tryptic neo-N-terminal peptides resulting from proteolytic cleavage are often lengthened somewhat as in our workflow, lysine amino acid residues are blocked by dimethylation, which trypsin cannot cut. In addition, the polymer enrichment of neo-N-terminal peptides can amplify the detection of low abundance peptides, and we also accurately identified peptides > 30 amino acids in length (e.g., Figure 3A). To generate the most accurate atlas of substrates possible, we employed rigorous substrate winnowing criteria. This means that, although we identified many substrates with high confidence, we likely did not include some bone fide cleavage events. These can be data-mined and followed up in subsequent studies, especially when data from emerging studies can be cross-referenced. Nonetheless, identifying the same cleavage products in many independent experiments heightens the biological relevance of these cleavage events. The use of recombinant 3CLpro cleavage of prepared proteomes to determine the human substrate landscape might be seen as a limitation since it risks identifying cleavage events in proteins that are typically spatially or temporally separated by compartmentalization and altered protease/substrate stoichiometry. Nonetheless, our histological analyses revealed extensive disruption of cell compartments, and high viral mRNA and protein loads in infected cells soothe this potential limitation. Indeed, prioritizing confident substrate identification enables us to attribute cleavages directly to 3CLpro without complications from cleavages by coexpression of SARS-CoV-2 PLpro and host cell proteases, especially those released in viral infection (e.g., damage of cell membranes releases destructive lysosomal cathepsins). Cleavage events in infected cell populations may be masked by cells at different stages of infection and uninfected bystander cells. Compensation for loss of substrates by increased protein expression may also obscure substrate identification. Substrate cleavage events identified using our method can be targeted later in protease-transfected or SARS-CoV-2 infected cells. Thus, despite the above limitations, we consider that our approach was the most appropriate to achieve a high coverage atlas of confident 3CLpro substrates.
STAR★Methods
Key resources table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Alexa Fluor 488 goat anti-rabbit (1:500) | Invitrogen | Cat# A11034; RRID AB_2576217 |
| Alexa Fluor 546 goat anti-mouse (1:500) | Invitrogen | Cat# A11030; RRID: AB_2534089 |
| Alexa Fluor 594 phalloidin (5 μl) | Invitrogen | Cat# A12381; RRID: AB_2315633 |
| Alexa Fluor 680 donkey anti-goat (1:10,000) | Invitrogen | Cat# A21084; RRID: AB_2535741 |
| Alexa Fluor 680 goat anti-mouse (1:10,000) | Invitrogen | Cat# A21057; RRID: AB_2535723 |
| Alexa Fluor 680 goat anti-rabbit (1:10,000) | Invitrogen | Cat# A21109; RRID: AB_2535758 |
| Alexa Fluor Plus 488 goat anti-rabbit (1:1,000) | Invitrogen | Cat# A32731; RRID: AB_2633280 |
| Alexa Fluor Plus 647 goat anti-mouse (1:1,000) | Invitrogen | Cat# A32728; RRID: AB_2633277 |
| Alexa Fluor Plus 800 goat anti-mouse (1:20,000) | Invitrogen | Cat# A32730; RRID: AB_2633279 |
| goat anti-mouse IgG (H+L)-HRP conjugated (1:1,000) | Bio-Rad | Cat# 170-6516; RRID: AB_11125547 |
| goat anti-rabbit IgG (H+L)-HRP conjugated (1:1,000) | Bio-Rad | Cat# 172-1019; RRID: AB_11125143 |
| IRDye 800CW goat anti-rabbit (1:10,000) | Li-COR | Cat# 926-32211; RRID: AB_621843 |
| goat anti-mouse IgG (H+L)-HRP conjugated (1:5,000) | Thermo Fischer Scientific | Cat# 31430; RRID: AB_228307 |
| goat anti-rabbit IgG (H+L)-HRP conjugated (1:3,000) | Cell Signaling Technology | Cat# 7074; RRID: AB_2099233 |
| goat polyclonal anti-Gal8 (1:400) | R&D Systems | Cat# AF1305; RRID: AB_2137229 |
| rabbit polyclonal anti-C-Gal8 antibody (1:500) | Thermo Fisher Scientific | Cat# PA5-19729; RRID: AB_10984508 |
| rabbit monoclonal anti-GFP (D5.1) (1:1,000) | Cell Signaling Technology | Cat# 2956; RRID: AB_1196615 |
| rabbit monoclonal anti-Gal8 (EPR4857) (1:1,000), | Abcam | Cat# ab109519; RRID: AB_10861755 |
| mouse monoclonal anti-CALCOCO2/NDP52 (1:1,000) | Santa Cruz Biotechnology | Cat# sc-376540, F-6; RRID: AB_11150487 |
| mouse monoclonal anti-FLAG M2 (1:10,000) | Sigma | Cat# F3165; RRID: AB_259529 |
| mouse monoclonal anti-His-tag (1:1,000) | Cedarlanelabs | Cat# CLH101AP |
| mouse monoclonal anti-IRS2 (1:300) | R&D Systems | Cat# MAB6347, 676415; RRID: AB_10992928 |
| mouse monoclonal anti-PTBP1 (1:500) | Biolegend | Cat# 630101, 3H7; RRID: AB_2171285 |
| mouse monoclonal anti-PTBP1 (1:66) | Biolegend | Cat# 630101, 3H7; RRID: AB_2171285 |
| mouse monoclonal anti-SARS-CoV-2 nucleocapsid (1:1,000) | Invitrogen | Cat# MA5-29981; RRID: AB_2785780 |
| mouse monoclonal anti-β-actin (1:1,000) | Abcam | Cat# ab8226; RRID: AB_306371 |
| mouse monoclonal anti-β-tubulin (1:2,000) | AbLabs | Cat# 21-0018-00, clone BT7R |
| rabbit anti-CALCOCO2/NDP52 (1:400) | Atlas Antibodies AB | Cat# HPA022989; RRID: AB_1845914 |
| rabbit anti-galectin-8 (1:15) | Atlas Antibodies AB | Cat# HPA030491; RRID: AB_10602345 |
| rabbit anti-goat IgG (H+L)-HRP conjugated (1:1,000) | Bio-Rad | Cat# 172-1034; RRID: AB_11125144 |
| rabbit anti-SARS-CoV-1 3CLpro (1:2000) | Rockland | Cat# 200-401-A51; RRID: AB_828457 |
| rabbit monoclonal anti-FLAG (1:1,000) | Cell Signaling Technology | Cat# 14793S; RRID: AB_2572291 |
| rabbit monoclonal anti-SARS-CoV-2 Spike S1 (1:500) | Sino Biological | Cat# 40150-R007; RRID: AB_2827979 |
| rabbit monoclonal anti-β-actin (1:200) | Abcam | Cat# ab115777; RRID: AB_10899528 |
| rabbit polyclonal anti-CALCOCO2/NDP52 (1:1,000) | Abclonal | Cat# A7358; RRID: AB_2767894 |
| rabbit polyclonal anti-CREB1 (1:1,000) | Abclonal | Cat# A11989; RRID: AB_2758916 |
| rabbit polyclonal anti-FAF1 (1:1,000) | Abclonal | Cat# A2921; RRID: AB_2764739 |
| rabbit polyclonal anti-FYCO1 (1:1,000) | Cusabio | Cat# CSB-PA866262LA01HU; RRID: AB_2892085 |
| rabbit polyclonal anti-KPNA3 (IMA4) (1:1,000) | Abclonal | Cat# A8347; RRID: AB_2770124 |
| rabbit polyclonal anti-MAP4K5 (1:1,000) | Cusabio | Cat# CSB-PA013440DSR2HU; RRID: AB_2892084 |
| rabbit polyclonal anti-NUP107 (1:1,000) | Abclonal | Cat# A13110; RRID: AB_2759959 |
| rabbit polyclonal anti-RPAP1 (1:1,000) | Proteintech | Cat# 15138-1-AP; RRID: AB_2301137 |
| rabbit polyclonal anti-YAP1 (1:1,000) | Abclonal | Cat# A11430; RRID: AB_2758556 |
| Bacterial and virus strains | ||
| SARS COV-2/Canada/VIDO-01/2020 | Sunnybrook Research Institute, Toronto, ON, Canada | Kindly provided by Dr. S. Mubareka |
| SARS-CoV-2 (SARS-CoV-2/SB3) clinical isolate | MacMaster University, Hamilton, ON, Canada | (Banerjee et al., 2020) |
| E. coli BL21(DE3)pLysS | Thermo Fisher Scientific | Cat# C606010 |
| Biological samples | ||
| Primary human airway epithelial cells (HAECs) from five donors (1 female, 57; 4 males, 37, 47, 61, 71 years old) | McMaster University, Hamilton, ON, Canada | protocol #HiREB-5099-T) |
| Normal human lung samples (1 female, 54; 2 males, 15, 45 years old) | Uppsala Biobank | Uppsala Ethical Review Board (Ref # 2002-577, 2005-388). |
| Human blood from a healthy volunteer | University of British Columbia, BC, Canada | University of British Columbia Human Ethics number: H06-00047 |
| COVID-19 human lung tissue samples (64, 97, 60 and 31 years old) | University Hospital, Huddinge, Stockholm | The Swedish Ethical Review Authority DNR 2020-02446 and 2020-04339. |
| Chemicals, peptides, and recombinant proteins | ||
| 14-mer peptides (49) with the sequence AA(X1–X8)YAYR, with X1–X8 being the P4 – P4’ sequence of 46 3CLpro cleavage sites | Genscript | In this paper, Figure 2, Table 1, Figure S2 |
| 0.5-ml Amicon Ultra with a 3-kDa cutoff | Millipore-Sigma | Cat#UFC500396 |
| 15-ml Amicon Ultra with a 10-kDa cutoff | Millipore-Sigma | Cat#UFC901008 |
| 75 μm × 300 mm analytical column filled with ReproSil-Pur C18 (1.8 μm stationary phase) | Dr. Maisch GmbH | https://dr-maisch.com |
| 8-well chamber slide (Lab-Tek II | NalgeNunc | Cat#154534 |
| 8-well chambered cover glass | Thermo Fisher Scientific | Cat#155411 |
| Ac-Abu-Tle-Leu-Gln-ACC (quenched fluorescence specific peptide) | N/A | Kindly provided by Dr. Marcin Drag (Rut et al., 2021) |
| Antibiotic-Antimycotic | GIBCO | Cat#15240062 |
| Benzonase | Sigma | Cat#E1014 |
| bovine serum albumin (BSA) | Sigma | Cat#A7030 |
| calcium-binding and coiled-coil domain-containing protein 2 (CALCOCO2/NDP52), GST-tagged recombinant protein (1 – 446 aa, NP_005822.1) | Abnova | Cat#H00010241-P01 |
| Cover Glass Circle #1 12mm | Fisher Scientific | Cat#12-545-80 |
| cyclic AMP-responsive element-binding protein 1 (CREB1), 6x His-tagged recombinant protein (1-327 aa, NM_004379) | Origene | Cat#TP760318 |
| DAPI | Invitrogen | Cat#3571 |
| DMEM (Dulbecco’s Modified Eagle Medium) | Sigma | Cat#D6429 |
| DMEM/F12 | GIBCO | Cat#11330057 |
| (MEM) Eagle’s Minimum Essential Medium | Sigma | Cat# M4655-500ML |
| Empore SPE C18 disc (for StageTips) | VWR | Cat#76333-132 |
| Eosin | Bio-Optica | Cat#05-10003/L |
| Eppendorf LowBind Protein tubes | Eppendorf | Cat#13-698-794 |
| FBS (Fetal Bovine Serum) | Sigma | Cat#F1051 |
| Fluoroshield with DAPI | Sigma-Aldrich | Cat#F6057 |
| Formaldehyde - “light” | Sigma | Cat#252549 |
| Formaldehyde (20% W/W IN D2O; 13C,99%; D2,98%) - “heavy” | ACP Chemicals | Cat#CDLM4599 |
| Formic acid (MS grade) | Fisher Scientific | Cat#A117-50 |
| galectin-8 (LGALS8) recombinant protein, (1-317 aa, AAF19370.1) | Sino Biological | Cat#10301-HNAE-50 |
| GC376, 3CLpro specific inhibitor | N/A | Kindly provided by Dr. John Vederas (Vuong et al., 2020) |
| HALT Protease Inhibitor Cocktail | Thermo Fisher Scientific | Cat# PI-78442 |
| HisTrap HP column (Cytiva) | Sigma | Cat#GE17-5247-01 |
| HPG-ALD 100K polymer | UBC Flintbox | www.bit.ly/3iHPs8P |
| IFN-α2a (carrier free) | PBL Assay Science | Cat#11101-2 |
| IFN-β1a (carrier free) | PBL Assay Science | Cat#11410-2 |
| Imperial protein stain | Thermo Fisher Scientific | Cat#24617 |
| importin subunit alpha-4 (IMA4), partial 6x His-tagged recombinant protein (3-220 aa, NP_002258.2 | Aviva System Biology | Cat#OPCD04723 |
| Intercept (PBS) Protein-free Blocking Buffer | Li-COR | Cat#92790001 |
| Lipofectamine 2000 | Invitrogen | Cat#11668019 |
| Mayers Htx Plus (Hematoxylin) | Histolab | Cat#01825 |
| MEM-α (Minimum Essential Medium-α) | Thermo Fisher Scientific | Cat#12571063 |
| mitogen-activated protein kinase kinase kinase kinase 5 (MAP4K5), GST/6x His-tagged recombinant protein (1- 846 aa, NP_006566.2) | Sino Biological | Cat#11773-H20B-50 |
| MS grade trypsin protease | Thermo Fisher Scientific | Cat#PI90058 |
| N-ethylmaleimide (NEM) | Sigma | Cat#E3876 |
| Native pig myelin basic protein | Abcam | Cat#ab64311 |
| Opti-MEM | Thermo Fisher Scientific | Cat#31985070 |
| Paraformaldehyde (4%) | Sigma | Cat#158127 |
| paraformaldehyde (methanol free) | Thermo Fischer Scientific | Cat#28909 |
| PERTEX mounting medium | Histolab | Cat#00801-Ex |
| PneumaCult-Ex Plus Media | StemCell Technologies, Inc. | Cat# 05040 |
| polyethylene glycol | Sigma | Cat#P-3265 |
| polypyrimidine-tract binding protein 1 (PTBP1), 6x His-tagged recombinant protein (1-557 aa, NP_002810.1) | Aviva System Biology | Cat#OPCD00681 |
| ProLong Gold antifade mounting media | Invitrogen | Cat#P36930 |
| ProLong™ Glass Antifade Mounting Media | Thermo Fisher Scientific | Cat# P36984 |
| Protease inhibitor cocktail | Bimake.com | Cat#B14002 |
| RIPA buffer | Abcam | Cat#ab156034 |
| RNA polymerase II-associated protein 1 (RPAP1), partial 6x His-tagged recombinant protein (1-351 aa, BC000246) | Proteintech | Cat#AG7856 |
| RPMI-1640 | Sigma | Cat#R8758 |
| SARS-CoV-2 Spike S1, 6x His-tagged recombinant protein (16-685 aa, YP_009724390.1) | Sino Biological | Cat#40591-V08H-100 |
| Scepter 60-μm sensor | Millipore-Sigma | Cat# PHCC60500 |
| Sigmafast OPD tablets (peroxidase substrate o-phenylenediamine dihydrochloride) | Sigma | Cat#P9187 |
| Sodium Cyonoborohydrate (NaCNBH3) | Sigma | Cat#156159 |
| thiodigalactoside (TDG) (inhibitor) | Sigma | Cat#SML2310 |
| UltraAb Diluent | Thermo Fisher Scientific | Cat#TA-125-UD |
| Vacutainer containing sodium citrate | Fisher Scientific | Cat#BD363083 |
| Versene buffer | Thermo Fisher Scientific | Cat#15040066 |
| α-Cyano-4-hydroxycinnamic acid (CHCA MALDI matrix) | Sigma | Cat#2020 |
| β-casein | Sigma | Cat#C6905 |
| Critical commercial assays | ||
| Akoya Biosciences Opal 480 reagent pack | Thermo Fisher Scientific | Cat#FP1500001KT |
| Akoya Biosciences Opal 520 reagent pack | Thermo Fisher Scientific | Cat# FP1487001KT |
| Akoya Biosciences Opal 570 reagent pack | Thermo Fisher Scientific | Cat# FP1488001KT |
| Akoya Biosciences Opal 650 reagent pack | Thermo Fisher Scientific | Cat# FP1496001KT |
| Epridia Lab Vision PT Module Deparaffinization and heat-induced and epitope retrieval Solutions | Thermo Fisher Scientific | Cat#TA-250-PM1X |
| LookOut Mycoplasma PCR Detection Kit | Sigma | Cat#MP0035 |
| Universal Kinase Activity kit | R&D Systems | Cat#EA004 |
| Deposited data | ||
| ProteomeXchange Consortium via the PRIDE partner repository (PXD026797 for HEK293) | ProteomeXchange | http://www.proteomexchange.org/ |
| ProteomeXchange Consortium via the PRIDE partner repository (PXD026815 for BEAS-2B) | ProteomeXchange | https://www.ebi.ac.uk/pride/archive/ |
| Uniport human database (UP000005640_9606)/Byonic | Uniprot | https://www.uniprot.org/ |
| IMEx/IntAct Coronavirus Dataset: SARS-COV-2+human context downloaded from NDEx | NDEx (Perfetto et al., 2020) | http://www.ndexbio.org/#/networkset/4c2268a1-a0f0-11ea-aaef-0ac135e8bacf |
| (pET21b(+)_SARS-CoV-2_3CLpro-Q306A | Addgene | ID: 177334 |
| pET21b(+)_SARS-CoV-2_3CLpro-C145A-Q306A | Addgene | ID: 177335 |
| Proteomics raw data | Mendeley | https://data.mendeley.com//datasets/b97d5nrb72/1 |
| Protein-protein interaction raw data | Mendeley | https://data.mendeley.com//datasets/b97d5nrb72/1 |
| Uncropped SDS-PAGE gels and western blot images | Mendeley | https://data.mendeley.com/datasets/b97d5nrb72/2 |
| Experimental models: Cell lines | ||
| BEAS-2B, human lung bronchus epithelial cells | ATCC | Cat#CRL-9609; RRID: CVCL_0168 |
| Calu-3, human lung adenocarcinoma epithelial cells | ATCC | Cat#HTB-55; RRID: CVCL_0609 |
| HEK293, human embryonic kidney epithelial cells | ATCC | Cat#CRL-1573; RRID: CVCL_0045 |
| Jurkat cells, human immortalized T lymphocytes | ATCC | Cat#TIB-152; RRID: CVCL_0367 |
| HeLa, human cervix adenocarcinoma epithelial cells | ATCC | Cat# CCL-2 RRID: CVCL_0030 |
| Vero E6, monkey kidney epithelial | ATCC | Cat#CRL-1586; RRID: CVCL_0574 |
| Recombinant DNA | ||
| pET21b(+)-SARS-CoV-2 3CLpro-Q306A (NC_45512.2) | Genscript | this paper |
| pET21b(+)-SARS-CoV-2 3CLpro-C145A-Q306A (NC_45512.2) | Genscript | this paper |
| pcDNA3.1-LGALS8-flag tag (human galectin-8, NM_006499.4) | Genscript | Cat#OHu23472 (ORF clone) |
| pcDNA3.1-FLAG-tagged-N-LGALS8 (1-158), custom synthesized based on original ORF clone | Genscript | Cat#OHu23472 (ORF clone) |
| pcDNA3.1-LGALS8 (159-317)-flag tag, custom synthesized based on original ORF clone | Genscript | Cat#OHu23472 (ORF clone) |
| Software and algorithms | ||
| ImageJ 1.53c | (Schneider et al., 2012) | https://imagej.nih.gov/ij/ |
| Prism version 9.0.0. 121 | Graphpad | https://www.graphpad.com/ |
| Compass oTOF control 1.9 | Bruker | https://www.bruker.com |
| Byonic PMI-Byonic-Com:v3.8.13 | Protein Metrics, San Carlos, CA USA | https://proteinmetrics.com/ |
| Rosetta FlexPepDock (3D molecular docking simulations of peptide-protein and protein-protein docking) | Rosetta | www.rosettacommons.org/software |
| ProtParam Tool | ExPASy | https://web.expasy.org/protparam/ |
| IntAct app (v 0.9.7) in Cytoscape (v 3.8.2) | Cytoscape | https://cytoscape.org |
| GLay community clustering algorithm plugin clusterMaker2 (v 1.3.1) in Cytoscape (v 3.8.2) | Cytoscape | https://cytoscape.org |
| Scaffold (v4.11.0) | Proteome Software Inc., Portland, OR, USA | http://www.proteomesoftware.com |
| Skyline (v 20.1.0.155) | MacCoss Lab, UW, Seattle, WA, USA | https://skyline.ms |
| TopFinder (part of TopFind 4.1) | Overall Lab, UBC, Vancouver, BC, Canada | https://topfind.clip.msl.ubc.ca |
| Data Explorer (v 4.5) for 4700 spectra analysis | Applied Biosystem | |
| GelAnalyzer version 19.1. | Istvan Lazar Jr., Ph.D. and Istvan Lazar Sr., Ph.D., C.Sc | www.gelanalyzer.com |
| Image Studio Software version 5.2.5. | Li-Cor | www.licor.com |
| iceLogos | Colaert, N. et al. Nature Methods 6, 786-787 (2009) | https://iomics.ugent.be/icelogoserver/ |
| Other | ||
| Zeiss LSM 880 Inverted Confocal Microscope | Zeiss | www.zeiss.com |
| Leica TCS SP5 II Confocal Microscope | Leica | www.leica-microsystems.com |
| Zeiss Axio Scan.Z1 Slide Scanner equipped with a Zeiss Colibri 7, Type RGB-UV fluorescence light source | Zeiss | www.zeiss.com |
| POLARstar optima microplate reader | BMG LABTECH | discontinued |
| Aperio AT2 slide scanner | Leica | www.leica-microsystems.com |
| TMArrayer | Pathology Devices | https://pathologydevices.com |
| Beecher Instruments Manual Tissue Arrayer MTA-1 | Estigen OÜ | https://estigen.com/ |
| Waterfall Microm HM 355S | Thermo Fisher Scientific | www.thermofisher.com |
| Decloaking chamber | Biocare Medical | https://biocare.net |
| Odyssey-Classic infrared imager (application software 3.0.30) | Li-COR | https://www.licor.com |
| SpectraMax 384 Plus spectrophotometer plate reader | Molecular Devices | www.moleculardevices.com |
| Sonic Dismembrator Model 100 | Fisher Scientific | www.fishersci.ca |
| Scepter handheld automated cell counter | Millipore-Sigma | discontinued |
| Leica DMRA2 microscope | Leica | www.leica-microsystems.com |
| ӒKTAexplorer | Amersham Pharmacia Biotech, now Cytiva | discontinued |
| Easy nLC-1000 (UHPLC) coupled to Impact II | Thermo-Fisher Scientific | www.thermofisher.com |
| Impact II Q-TOF mass spectrometer with a CaptiveSpray ionization interface equipped with a NanoBooster | Bruker-Daltonics | www.bruker.com |
| MALDI-TOF/TOF 4700 Proteomics Analyzer | Applied Biosystems | discontinued |
| ABI 494 Protein Sequencer | Applied Biosystems | discontinued |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Christopher Overall (chris.overall@ubc.ca).
Materials availability
This study generated 49 new synthetic 14-mer peptides spanning substrate P4–P4’ cleavage sites suitable for MALDI-TOF-MS analysis and are available from the lead contact.
This study generated eukaryotic cell expression DNA constructs in plasmids for FLAG-tagged full-length human galectin-8 and FLAG-tagged 3CLpro cleavage-fragment analogs of human galectin-8 designated N-galectin-8 (1-158) and C-galectin-8 (159-317) and are available from the lead contact.
This study generated C-terminal-tagged recombinant wild-type (active) and inactive mutant 3CLpro-C145A plasmids, which have been deposited to Addgene, (pET21b(+)_SARS-CoV-2_3CLpro-Q306A (Addgene, ID 177334) and pET21b(+)_SARS-CoV-2_3CLpro-C145A-Q306A (Addgene, ID 177335).
Experimental model and subject details
Cells lines
HEK293, human embryonic kidney epithelial cells (ATCC, CRL-1573, RRID: CVCL_0045), were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (Sigma) supplemented with 10% (v/v) fetal bovine serum (FBS) (Sigma) and 1x penicillin-streptomycin (Pen-Strep) (GIBCO). BEAS-2B, human lung bronchus epithelial cells (ATCC, CRL-9609, RRID: CVCL_0168), were maintained in DMEM/F12 (GIBCO) supplemented with 10% (v/v) FBS, 1x Pen-Strep, L-glutamine (GIBCO), and 0.1% sodium bicarbonate (Sigma). Calu-3, human lung adenocarcinoma epithelial cells (ATCC, HTB-55, RRID: CVCL_0609) were maintained in Minimum Essential Medium-α (MEM-α) (GIBCO) supplemented with 20% (v/v) FBS, 1x Pen-Strep, 1x Antibiotic-Antimycotic (GIBCO) and 1x GlutaMAX (GIBCO). HeLa, human cervix adenocarcinoma epithelial cells (ATCC, CCL-2, RRID: CVCL_0030), were maintained in Eagle’s Minimum Essential Medium (MEM) (Sigma) supplemented with 10% (v/v) FBS and 1x Pen-Strep. Jurkat cells, human immortalized T lymphocytes (ATCC, TIB-152, RRID: CVCL_0367) were maintained in RPMI 1640 medium (Sigma) supplemented with 10% (v/v) FBS and 1x Pen-Strep. Vero E6, monkey kidney epithelial cells (ATCC, CRL-1586, RRID: CVCL_0574) were maintained in DMEM supplemented with 10% (v/v) FBS and 1x Pen-Strep. All cell lines used in this study were cultured at 37°C and 5% CO2 and tested negative for mycoplasma (LookOut Mycoplasma PCR Detection Kit, Sigma).
Primary cells
Primary human airway epithelial cells (HAECs) collection from five donors (one female, 57 years old and four males, 37, 47, 61, 71 years old) was approved by the University of Hamilton Integrated Research Ethics Board (HiREB) under protocol HiREB-5099-T. HAECs were cultured using PneumaCult-Ex Plus Media (STEMCELL Technologies). HAECs were cultured at 37°C and 5% CO2.
Viruses (McMaster University, ON, Canada)
SARS-CoV-2 experiments were performed at McMaster University’s (Hamilton ON, Canada) Biosafety Level 3 laboratory (BSL3) following regulations from the Public Health Agency of Canada and guidelines from McMaster University. For infection of Calu-3 cells, a clinical isolate of SARS-CoV-2 (SARS-CoV-2/SB3) was propagated on Vero E6 cells and validated by next-generation sequencing (Banerjee et al., 2020). A fresh vial of virus stocks was used for each experiment to avoid repeated freeze-thawing.
Viruses (University of British Columbia, BC, Canada)
SARS-CoV-2 infections of monkey Vero E6 (RRID: CVCL_0574) cells were performed in the University of British Columbia (UBC) BSL3 facility (FINDER) following the Public Health Agency of Canada and UBC FINDER regulations (UBC BSL3 Permit # B20-0099 to EJ). SARS COV-2/Canada/VIDO-01/2020 was kindly provided by Dr. S. Mubareka (Sunnybrook Research Institute, Toronto, ON, Canada).
Human blood collection
Human blood (∼10 ml) was collected from a healthy volunteer donor (male, 24 years old) at the UBC Centre for Blood Research (UBC Human Ethics number: H06-00047) in a Vacutainer (BD) containing sodium citrate.
Human lung tissue collection
Human tissue samples were collected and handled following Swedish laws and regulations. Normal lung samples (N = 3) were obtained from the Clinical Pathology Department, Uppsala University Hospital, Sweden and collected within the Uppsala Biobank organization. The samples were anonymized for personal identity by following the approval and advisory report from the Uppsala Ethical Review Board (Ref # 2002-577, 2005-388). The tissue samples representing one female 54 years old (F54) and two males 15 and 45 years old (M15 and M45) were collected based on hematoxylin-eosin (H&E) stained tissue sections showing representative normal lung histology and quality-controlled by a certified pathologist.
COVID-19 lung tissue samples (N = 4) were collected during clinical autopsies to establish the precise cause of death at the Department of Clinical Pathology/Cytology, Karolinska University Hospital, Huddinge, Stockholm, Sweden, described previously (Szekely et al., 2021). The Swedish Ethical Review Authority approved the study under the registration number DNR 2020-02446 and 2020-04339. Samples from four individuals were used (age 64, 97, 60 and 31), corresponding to cases 1, 8, 9 and 11 with the patient characteristics and clinical parameters described in detail (Szekely et al., 2021).
Method details
SARS-CoV-2 3CLpro expression and purification
The DNA sequence of SARS-CoV-2 main protease (3CLpro, NSP5; NCBI: YP_009725301.1 (protein ID), GenBank: NC_45512.2 (whole SARS-CoV-2 genome) was synthesized and cloned into the expression vector pET-21b (+) using NdeI and BamHI restriction sites. During synthesis, a second NdeI cleavage site in the original sequence was deleted by silent mutation (GenScript). For efficient expression and purification, a Gln306Ala mutation was introduced, eliminating the C-terminal 3CLpro autoproteolytic cleavage site (FQ306↓G). This site was followed by a 3x Gly flexible linker, the Factor Xa cleavage site, 2x Gly linker, 3x FLAG-tag, 2x Gly linker, Myc-tag, 2x Gly linker, and 6x His-tag. The catalytic inactive 3CLpro expression plasmid was constructed by introducing a mutation into the codon for the catalytic cysteine 145 to alanine (3CLpro-C145A).
Active and inactive proteases were expressed in E. coli BL21(DE3)pLysS (Thermo Fisher Scientific). Bacteria were grown at 37°C until expression was induced with 0.4 mM IPTG, after which the cultures were grown at room temperature (RT) for ∼20 h. Bacterial pellets were collected by centrifugation at 5,000g for 20 min and lysed with lysis buffer (300 mM NaCl, 10 mM imidazole, 1 mM DTT, 50 mM Tris-HCl, pH 7.4).
Purification of 3CLpro and 3CLpro-C145A was performed by immobilized metal affinity chromatography using a 1 mL HisTap HP column (Cytiva). A continuous gradient up to 250 mM imidazole eluted the recombinant proteins on an ӒKTAexplorer (Amersham Pharmacia Biotech, now Cytiva). Protein fractions were pooled and dialyzed against assay buffer (150 mM NaCl, 2 mM DTT, 1 mM EDTA, 0.05% Brij 35, 50 mM Tris-HCl, pH 7.2), snap-frozen in liquid N2 and stored at –80°C until use. The activity of the purified protease was confirmed using the quenched fluorescence specific peptide (Ac-Abu-Tle-Leu-Gln-ACC) at 20 μM as described (Rut et al., 2021) and measured with a λex 320 nm and λem 460 nm using a POLARstar optima (BMG LABTECH) microplate reader.
Cell proteome extraction in native condition
HEK293, BEAS-2B and HAECs were maintained as described. To induce interferon-stimulated gene proteins that may be 3CLpro substrates, the BEAS-2B cells were cultured in DMEM/F12 with 10% (v/v) FBS and treated with 104 U/ml carrier-free IFN-α2a or IFN-β1a (PBL Assay Science), or medium (control) for 18 h. Cells were harvested, and lysates were prepared under native conditions taking the necessary steps to reduce any cellular proteolytic activity. All steps were performed on ice. First, cells were washed twice with phosphate-buffered saline (PBS) and lifted with Versene buffer (0.5 mM EDTA, PBS). The cell pellet was washed twice with 150 mM NaCl, 50 mM Tris-HCl, pH 7.2 by centrifugation at 300g for 10 min. Protease inhibitor cocktail (bimake.com) and 5 mM N-ethylmaleimide (NEM) (Sigma) were added to the cell pellet in hypotonic lysis buffer (2 mM MgCl2, 1 μl/ml Benzonase (Sigma), 50 mM Tris-HCl, pH 7.2). For lysis, cells were pushed through a 27-gauge needle for 10 cycles and rested on ice for 1 h with agitations every 10 min. Cell lysates were flash-frozen in liquid N2 and stored at –80°C.
Terminal Amine Isotopic Labeling of Substrates
For 3CLpro substrate profiling, the cell lysates were thawed on ice, ultrasonicated (3 cycles, 20 s, power 3) (Sonic Dismembrator Model 100, Fisher Scientific) and clarified by centrifugation at 400g for 10 min. Buffer exchange to Brij-free 3CLpro assay buffer was performed 3x in a 0.5 mL Amicon Ultra with a 3-kDa cutoff (Millipore). 3CLpro or inactive 3CLpro-C145A (control) at 2.5 μM were incubated with 500 μg native cell protein in their respective 0.5 mL Amicon Ultra filter cartridges, 37°C for 18 h (Figure 1A, panel a). Quenched fluorescent peptide assays (Rut et al., 2021) of the samples before and after incubation confirmed 3CLpro activity. The incubated samples were then analyzed by Terminal Amine Isotopic Labeling of Substrates (TAILS) and preTAILS shotgun proteomics (Figure 1) using a modified protocol from that described before (Kleifeld et al., 2011). One volume (∼140 μl) of 8% (w/v) SDS, 20 mM DTT, 100 mM HEPES, pH 8.0 was added to the samples in the Amicon filters used for digestion and incubated for 1 h at 37°C. Samples were centrifuged (12,000g, 10 min), washed twice with wash buffer (20 mM HEPES, pH 7.0), and cysteines were alkylated by adding one volume of 40 mM NEM in wash buffer followed by a 30-min incubation at RT. After adding additional 10 mM DTT for 10 min at RT, the samples were concentrated by ultrafiltration to ∼100 μl and transferred to Lo-bind Eppendorf tubes. Amicon filters were rinsed 2x with 50 μl of wash buffer and added to the samples (150 μl) for precipitation with methanol/chloroform/H2O (4:1:3) (Wessel and Flügge, 1984).
Protein precipitates were collected and resuspended in 50 μl of 4% (w/v) SDS, 50 mM HEPES, pH 6.8. All protein N-termini, i.e., neo-N-termini generated by 3CLpro cleavage and natural protein starts, were isotopically labeled at the protein level using 2.5 μl of 1 M heavy [+34 Da] (for 3CLpro) or light [+28 Da] (for 3CLpro-C145A) formaldehyde and 2.5 μl of 500 mM NaCNBH3, for 4 h, 42°C (Figure 1A, panel b). Excess formaldehyde was quenched with 5 μl 1 M Tris, pH 6.8 for 1 h. Then, samples were pooled and cleaned by methanol/chloroform/H2O (4:1:3) precipitation, resuspended in 200 μl of 20 mM HEPES, pH 8.0. The labeled protein was then digested with MS grade trypsin protease (Thermo Fisher Scientific), 1:50 enzyme:protein (w/w) overnight, 37°C (Figure 1A, panel c).
For preTAILS, 20 μl of the peptide digest was desalted using C18 StageTips, lyophilized, and stored at –20°C until liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis. The remaining sample was pH-adjusted to 6.5 with HCl. N-terminal peptides were enriched by depleting the tryptic peptides via covalent coupling to our in-house synthesized HPG-ALD 100K polymer (available via UBC Flintbox, bit.ly/3iHPs8P) (Figure 1A, panel c), 5:1 (w:w; polymer:peptide) in the presence of 30 mM NaCNBH3 for 4 h, 42°C. N-terminal blocked peptides were retrieved by ultrafiltration in 10-kDa filters by centrifugation, desalted using C18 StageTips, lyophilized, and stored at –20°C until LC-MS/MS (Figure 1A, panel d).
Liquid chromatography-tandem mass spectrometry
Data-dependent acquisition was performed using UHPLC (Easy nLC-1000, Thermo-Fisher Scientific) coupled to an Impact II Q-TOF mass spectrometer (Bruker-Daltonics) with a CaptiveSpray ionization interface equipped with a NanoBooster. Peptide samples (1 μg) were injected onto a 75 μm × 300 mm analytical column (packed in house) with ReproSil-Pur C18 1.8 μm stationary phase (Dr. Maisch GmbH). Peptides were eluted using a 120-min curved gradient at 250 nl/min from 5% to 24% buffer B (99.9% acetonitrile, 0.1% formic acid), then increased to 34% over 10 min, further increased to 95% buffer B over 5 min and finally held at 95% for 10 min. CaptiveSpray source voltage was set to 1,250 V, the mass spectrometer was operated in positive ion polarity mode, and precursor ions were detected from 150 to 2,250 m/z. MS/MS spectra were acquired using a Top12 selection method with an intensity-adjusted MS/MS summation time (duty cycle 1.3–1.8 s). Acquired precursors were excluded for 14 s before a new acquisition (Compass oTOF control 1.9, Bruker). Samples were measured twice, once using acetonitrile, with a second using methanol as the dopant in the NanoBooster.
Validation of 3CLpro cut-sites by MALDI-TOF-MS
A total of 49 14-mer peptides with the sequence AA(X1–X8)YAYR, with X1–X8 being the P4–P4’ sequence of 46 3CLpro cleavage sites identified by TAILS plus 3 cut sites identified by Edman sequencing, were synthesized (GenScript). Peptides (25 μM) were diluted in 3CLpro assay buffer and incubated with 3CLpro (1:20 molar ratio, E:S) in a 25-μl final volume at 37°C in a humidified chamber for 5, 15, 30, 60, 120, 240 min. At the indicated time points, 0.5 μl of the enzymatic reaction was deposited on a MALDI plate pre-spotted with CHCA matrix. After which, 0.5 μl CHCA matrix was immediately added, and the plate was air-dried. The samples were desalted by immersing the whole plate in ice-cold 0.1% formic acid bath. After air-drying, samples were measured in positive ion mode in a MALDI-TOF/TOF 4700 Proteomics Analyzer (Applied Biosystems). MALDI spectra were analyzed using Applied Biosystem Data Explorer, version 4.5. Estimations of apparent (app)k cat/K M (Starr and Overall, 2009) were done under the assumption of a first-order reaction where:
The other important assumption is that the peak areas of the substrate and product fragments in MALDI-TOF MS spectra are directly proportional to their relative abundance. As this is not always necessarily true, we limited the scope of the (app) k cat/K M calculations to rank the substrates in bins of four according to degradation rate.
3D modeling of 3CLpro docking to substrates
Peptide-protein docking
Peptide-3CLpro molecular docking simulations were performed using the Rosetta FlexPepDock ab-initio protocol (Raveh et al., 2011) implemented within the Rosetta software suite (Leaver-Fay et al., 2011). First, the crystal structure of 3CLpro (PDB: 6XHU) was prepared for docking calculations by running the Rosetta relax application using the flags listed in the Supplementary text. The starting backbone conformation of the peptides (RPAP1: ARLQAMAP; IMA4: AILQNATS; PTBP1: AALQAVNS; RBM15: SRLHSYSS; MAP4K5: SKLMSENT; CREB1: VVVQAASG) were created as a preliminary extended structure, truncated at both N- and C-termini, using the BuildPeptide Rosetta application. For each peptide, a fragment library of trimer and pentamer backbone was generated from known structures available in the PDB, based on the target sequence similarity and its predicted secondary structure. Briefly, FlexPepDock ab-initio simulations started from the extended peptide structure placed at 15 Å apart from the 3CLpro active site. A total of 50,000 models were then generated through a fast low-resolution step. The side-chains are represented as a single centroid sphere, followed by a high-resolution step that uses a full-atom energy function that enables complete flexibility for all peptide and receptor side-chains (Alford et al., 2017). A flat harmonic function was used to penalize models when the Euclidean distance between Cys145 Sγ and Cα of P1 exceeds 4 Å. The 500 lowest-scoring models, based on Rosetta total energy, were selected. The model with the most significant structural similarity within this subset, given by the root-mean-square deviation (RMSD), was chosen as the representative model.
Protein–protein docking
An initial full-length 3D model of galectin-8 was built by comparative modeling using the RosettaCM protocol (Song et al., 2013) and PDB: 4FQZA as the template structure. Before docking simulations, we generated structural ensembles with backbone conformational variations for both the 3CLpro dimer (PDB: 6XHU) and galectin-8 top-ranked full-length models using Normal Mode Analysis, with perturbation steps of 1 Å, through RosettaScripts (Fleishman et al., 2011). The ensembles were used as input structures for the docking simulation between 3CLpro and galectin-8 using the RosettaDock algorithm implemented in the Rosetta macromolecular modeling suite. Constraint was applied to penalize models having Cys145 Sγ atom of 3CLpro and Gln158 Cα of galectin-8 spaced by more than 4 Å. A total of 33,500 models were generated, and the decoy with the greatest structural similarity within the 500 lowest-scoring models was selected as a representative model.
Substrate cleavage assay and Edman sequencing
The recombinant proteins assayed were: RNA polymerase II-associated protein 1 (RPAP1), partial 6x His-tagged (1 – 351 aa, GenBank: BC000246, Proteintech); polypyrimidine-tract binding protein 1 (PTBP1), 6x His-tagged (1 – 557 aa, NCBI: NP_002810.1, Aviva System Biology); mitogen-activated protein kinase kinase kinase kinase 5 (MAP4K5), GST/6x His-tagged (1 – 846 aa, NCBI: NP_006566.2, Sino Biological); cyclic AMP-responsive element-binding protein 1 (CREB1), 6x His-tagged (1 – 327 aa, NCBI: NM_004379, Origene); galectin-8 (LGALS8) (1 – 317 aa, GenBank: AAF19370.1, Sino Biological); SARS-CoV-2 Spike S1, 6x His-tagged recombinant protein, (16 – 685 aa, NCBI: YP_009724390.1, Sino Biological); calcium-binding and coiled-coil domain-containing protein 2 (CALCOCO2/NDP52), GST-tagged recombinant protein (1 – 446 aa, NCBI: NP_005822.1, Abnova); importin subunit alpha-4 (IMA4), partial 6x His-tagged (3 – 220 aa, NCBI: NP_002258.2, Aviva System Biology).
Recombinant proteins were incubated with 3CLpro, 3CLpro-C145A, and 3CLpro inhibited with the specific 3CLpro specific inhibitor, 1 μM GC376 (Vuong et al., 2020), at a molar ratio of 1:5 mol/mol, E:S in time course assays (0.25, 0.5, 1, 2, 4 and 16 h) at 37°C. Protein cleavage was confirmed by SDS-PAGE followed by Imperial protein staining (Thermo Fisher Scientific). Edman sequencing was used to identify the N-terminal sequence of cleaved proteins using an ABI 494 Protein Sequencer (Tufts University Core Facility, Boston, MA, USA) as previously described (Doucet and Overall, 2011). The apparent molecular weights of cleaved protein fragments on SDS gels were calculated using GelAnalyzer version 19.1. (www.gelanalyzer.com by Istvan Lazar Jr., Ph.D. and Istvan Lazar Sr., Ph.D., C.Sc.).
MAP4K5 activity assay
The kinase activity of intact or cleaved (Δ) recombinant MAP4K5 (500 ng) was measured at 1:2 serial dilutions in duplicate using the Universal Kinase Activity kit (R&D systems, EA004) as per manufacturer instructions. Native pig myelin basic protein (Abcam, ab64311) at 5 mM was the acceptor substrate. The ATP consumption (nmol of phosphate) was measured on a POLARstar optima (BMG LABTECH) at 620 nm. Statistical significance was calculated by comparing the area under the curve with Prism version 9.0.0 (121) and a Student’s t test (GraphPad).
Substrate cleavage validation by immunoblots
HAEC lysates were incubated with 3CLpro or 3CLpro-C145A as described above. Vero E6 cells were seeded at ∼0.1 × 106 in 12-well and cultured as described above. SARS-CoV-2 was absorbed to the cells at an MOI 0.1 for 60 min in Opti-MEM, 37°C, washed with PBS, pH 7.4 and then incubated with complete DMEM, 24 and 48 h (N = 4 for each time point). Cells treated with DMEM alone were considered as controls (N = 3, mock). For immunoblot characterization of PTBP1 cleavage at 24- and 48-hpi with SARS-CoV-2, the cells were washed 3x PBS before lysis in 1x Halt Protease Inhibitor Cocktail (Thermo Fisher Scientific) and RIPA buffer (150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 25 mM Tris-HCl pH 7.4). Calu-3 cells were seeded at ∼0.7 × 106 cells/T-25cm2 flask and infected with SARS-CoV-2/SB3 at different multiplicities of infection (MOI) of 0.1 or 1.0, or mock-infected as controls. Cell lysates were collected at 24- and 48-hpi in the presence of Halt Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific) using 1x lysis buffer (2% SDS (w/v), 10% glycerol (v/v), and 1% β-mercaptoethanol (v/v), 160 mM Tris-HCl, pH 6.8) and boiled for 10 min.
Samples of HAEC, Vero E6 and Calu-3 cell lysates (20 μg) were electrophoresed on 12% or 4%–12% gradient NuPAGE Bis-Tris 1.0 mm Mini protein gels at constant 200 V, or 3%–8% gradient NuPAGE Tris-Acetate 1.0 mm gels at 150 V (Invitrogen). Proteins were transferred to PVDF membranes (Immobilon-FL, Millipore-Sigma). After blocking with Intercept (PBS) Protein-free Blocking Buffer (Li-COR) for 1 h membranes were incubated with the primary antibodies listed below in blocking buffer, 0.2% Tween 20 overnight at 4°C. Membranes were then washed 3x with PBST buffer (1x phosphate-buffered saline, 0.1% Tween 20) and incubated with secondary antibodies (listed below) in blocking buffer, 0.2% Tween and 0.01%SDS for 1 h at RT. Membranes were washed 3x with PBST buffer, rinsed with water and imaged on an Odyssey-Classic infrared imager (application software 3.0.30, Li-COR). For densitometric analyses of the immunoblots, we used the Image Studio Software version 5.2.5. Fold change was calculated relative to the corresponding loading control bands, and statistical analyses were performed with Prism version 9.0.0 (121) and one-way ANOVA followed by Dunnett’s multiple comparison test (GraphPad). The predicted molecular weight of protein bands was calculated using ProtParam, ExPASy.
The primary antibodies and dilutions used were: mouse monoclonal anti-SARS-CoV-2 nucleocapsid antibody (1:1,000, Invitrogen, MA5-29981, RRID: AB_2785780); rabbit anti-SARS-CoV-1 3CLpro antibody (1:2000, Rockland, 200-401-A51, RRID: AB_828457); rabbit polyclonal anti-RPAP1 antibody (1:1,000, Proteintech, 15138-1-AP, RRID: AB_2301137); mouse monoclonal anti-PTBP1 antibody (1:500, Biolegend, 630101, 3H7, RRID: AB_2171285); rabbit polyclonal anti-MAP4K5 antibody (1:1,000, Cusabio, CSB-PA013440DSR2HU, RRID: AB_2892084); rabbit polyclonal anti-CREB1 antibody (1:1,000, Abclonal, A11989, RRID: AB_2758916); rabbit polyclonal anti-YAP1 antibody (1:1,000, Abclonal, A11430, RRID: AB_2758556); rabbit polyclonal anti-FYCO1 antibody (1:1,000, Cusabio, CSB-PA866262LA01HU, RRID: AB_2892085); rabbit polyclonal anti-FAF1 antibody (1:1,000, Abclonal, A2921, RRID: AB_2764739); goat polyclonal anti-Gal8 antibody (1:400, R&D Systems, AF1305, RRID: AB_2137229); rabbit polyclonal anti-KPNA3 (IMA4) antibody (1:1,000, Abclonal, A8347, RRID: AB_2770124); rabbit polyclonal anti-NUP107 antibody (1:1,000, Abclonal, A13110, RRID: AB_2759959); mouse monoclonal anti-IRS2 antibody (1:300, R&D Systems, MAB6347, 676415, RRID: AB_10992928); mouse monoclonal anti-FLAG M2 antibody (1:10,000, Sigma, F3165, RRID: AB_259529); mouse monoclonal anti-β-tubulin antibody (1:2000, AbLab, 21-0018-00, clone BT7R); mouse monoclonal anti-β-actin antibody (1:1,000, Abcam, ab8226, RRID: AB_306371); rabbit monoclonal anti-β-actin antibody (1:200, Abcam, ab115777, RRID: AB_10899528).
The secondary antibodies and dilutions used were: IRDye 800CW goat anti-rabbit (1:10,000, Li-COR, 926-32211, RRID: AB_621843); Alexa Fluor Plus 800 goat anti-mouse (1:20,000, Invitrogen, A32730, RRID: AB_2633279); Alexa Fluor 680 goat anti-rabbit (1:10,000, Invitrogen, A21109, RRID: AB_2535758); Alexa Fluor 680 goat anti-mouse (1:10,000, Invitrogen, A21057, RRID: AB_2535723); and Alexa Fluor 680 donkey anti-goat (1:10,000, Invitrogen, A21084, RRID: AB_2535741).
Immunostaining of PTBP1 in virus infected cells
Vero E6 cells (0.1 × 106) were seeded in 12-well plates containing coverslips and cultured as described above. Vero E6 cells were infected with SARS-CoV-2 as described above (MOI 0.1, N = 5) for 48 h. Cells treated with DMEM alone were considered as controls (N = 5). Immunostaining and confocal image acquisition were performed at Wax-it Histological Services, Vancouver, BC, Canada. Coverslips with the attached cells were washed once with PBS followed by fixation and virus inactivation with 4% paraformaldehyde, 30 min with three subsequent PBS washes. Briefly, coverslips were blocked with Wax-it blocking solution for 1 h. Primary antibodies, mouse monoclonal anti-PTBP1 antibody (1:66, Biolegend, 630101, 3H7, RRID: AB_2171285) and rabbit monoclonal anti-SARS-CoV-2 Spike S1 antibody (1:500, Sino Biological, 40150-R007, RRID: AB_2827979) were incubated overnight at 4°C. After 3 washes with Wax-it washing solution, the coverslips were incubated with secondary antibodies, goat anti-rabbit Alexa 488 (1:500, Invitrogen, A11034, RRID AB_2576217) and goat anti-mouse Alexa 546 (1:500, Invitrogen, A11030, RRID: AB_2534089), for 1 h at RT. Coverslips were mounted with ProLong Gold mounting media with DAPI (provided by Wax-it) and imaged by confocal microscopy. One image (63x magnification) per coverslip (mock, N = 5 and infected N = 5) were acquired.
Galectin-8-induced hemagglutination assay
Whole blood was centrifuged at 1,000 g, 5 min to separate red blood cells (RBC) from plasma. After washing with PBS 4x, 50 μl of 4% (v/v) RBCs were mixed in a 96-U-shaped well plate with 50 μl of serial-diluted intact or cleaved (Δ) galectin-8 (50 to 3.1 μg/ml) at RT. 3CLpro at the same concentration used to cleave galectin-8 (0.26 μM) was used as a control. The plate was incubated for 1 h at RT and photographed.
Galectin-8-induced cell adhesion assay
Jurkat T cells were seeded in an 8-well chamber slide (Lab-Tek II, 154534, NalgeNunc) at 2 × 105 cells/well in 150 μl RPMI serum-free medium, 2 h at 37°C in 5% CO2. Intact or cleaved (Δ) galectin-8 at 0.4 μM (n = 2) was added in RPMI serum-free medium for 1 h at 37°C. 3CLpro at the same concentration used to generate ΔGal8 (0.08 μM), and RPMI serum-free medium (n = 2) were used as controls. Non-adherent cells were collected, washed twice with PBS and counted using a Scepter handheld automated cell counter (Millipore-Sigma) with a 60-μm sensor. Adherent cells were washed with PBS and fixed with 4% formaldehyde in PBS at RT for 10 min. Then, the cells were washed with PBS and permeabilized with 0.1% Triton X-100 for 5 min at RT. Staining of F-actin and nuclei was performed in 200 μl PBS with 5 μl Alexa Fluor 594 phalloidin (Invitrogen, A12381, RRID: AB_2315633) and DAPI (1:1,000, Invitrogen, D3571) for 30 min at RT in the dark. The slide was washed twice with PBS and mounted with ProLong Gold antifade mounting media (Invitrogen, P36930). Images were acquired with a 20x and 100x objective lens in a Leica DMRA2 microscope (Leica) from 2 randomly selected fields of view. Adherent cells were counted using 20x images via ImageJ 1.53c. Statistical analyses were performed with Prism version 9.0.0.121 using one-way ANOVA followed by Tukey’s multiple comparisons test (GraphPad).
Galectin-8 binding to Spike protein and NDP52
Galectin-8 binding to SARS-CoV-2 Spike S1 glycoprotein was assessed by a Sandwich ELISA (n = 2). Recombinant galectin-8 (1 – 317 aa, AAF19370.1, Sino Biological) or recombinant SARS-CoV-2 Spike S1-6x His-tagged protein (16 – 685 aa, YP_009724390.1, Sino Biological) at 5 μg/ml were coated on ELISA plate wells at 4°C, overnight. Blocking was with 1% β-casein (Sigma) for 1 h at RT. Next, 0.2 μM SARS-CoV-2 Spike S1 glycoprotein or 0.2 μM galectin-8 were added to the galectin-8 or SARS-CoV-2 Spike S1 glycoprotein-coated wells, respectively, for 2 h at RT. To detect the bound protein, mouse monoclonal anti-His-tag antibody (1:1,000, Cedarlane Labs, CLH101AP) (for Spike S1 glycoprotein) or goat polyclonal anti-Gal8 antibody (1:200, R&D Systems, AF1305, RRID: AB_2137229) and rabbit polyclonal anti-C-Gal8 antibody (1:500, Thermo Fisher Scientific, PA5-19729, RRID: AB_10984508) (for galectin-8) were added as appropriate and incubated for 1 h at RT. Detection was either by goat anti-mouse IgG (H+L)-HRP conjugated (1:1,000, Bio-Rad, 170-6516, RRID: AB_11125547), rabbit anti-goat IgG (H+L)-HRP conjugated (1:1,000, Bio-Rad, 172-1034, RRID: AB_11125144) or goat anti-rabbit IgG (H+L)-HRP conjugated (1:1,000, Bio-Rad, 172-1019, RRID: AB_11125143) for 1 h at RT. The colorimetric assay was developed using the peroxidase substrate o-phenylenediamine dihydrochloride tablets for 20 min at RT. Four washes with PBST buffer followed every step of the binding assay. The colorimetric signal at OD 450 nm was measured on a SpectraMax 384 Plus (Molecular Devices) spectrophotometer plate reader.
Immobilized SARS-CoV-2 Spike S1 glycoprotein binding to 1 μM of galectin-8 was repeated in the presence of 20 mM of the competitive inhibitor thiodigalactoside (TDG, Sigma, SML2310) or ΔGal8 at 0.5 μM (1:2 serial dilutions) (n = 2). For galectin 8 detection, two different antibodies were used (n = 2 with anti-Gal8 antibody and n = 3 with anti-C-Gal8 antibody).
Immobilized CALCOCO2/NDP52 (5 μg/ml) binding to 0.3 μM of intact Gal8, pre-incubated with TDG intact Gal8 or cleaved ΔGal8 (n = 3) was study by ELISA as described above. Rabbit polyclonal anti-C-Gal8 antibody (1:500, Thermo Fisher Scientific, PA5-19729, RRID: AB_10984508) followed by goat anti-rabbit IgG (H+L)-HRP conjugated (1:1,000, Bio-Rad, 172-1019, RRID: AB_11125143) detected the bound galectin-8.
Finally, the trimeric interaction between galectin-8, SARS-CoV-2 Spike S1 glycoprotein and CALCOCO2/NDP52 was studied (n = 2). Recombinant SARS-CoV-2 Spike S1 glycoprotein was coated at 5 μg/ml followed by incubation with recombinant intact or ΔGal8 at 1 μM (1:2 serial dilutions) for 2 h at RT. After 3 PBS washes, 0.2 μM of recombinant CALCOCO2/NDP52 was added and incubated for 2 h at RT. Bound CALCOCO2/NDP52, as the third member of the complex, was detected with anti-CALCOCO2/NDP52 antibody. Statistical analyses were performed with Prism version 9.0.0 (121) (GraphPad). Student’s t test was used to assess statistical significance between two groups, and two-way ANOVA followed by Šídák’s multiple comparisons test was used when the effect of two variables across different groups were analyzed.
Immunoprecipitation
HeLa cells were co-transfected with 1 μg of GPF-NDP52 (Dr. Richard Youle at the National Institute of Neurological Disorders and Stroke, USA) and WT-Gal8-FLAG or C-Gal8 (159-317)-FLAG plasmid constructs engineered and synthesized from the galectin-8 ORF clone OHu23472 (GenScript) were cultured as described above. HeLa cells were harvested in FLAG lysis buffer (150 mM NaCl, 1.0% Triton X-100, 50 mM Tris-HCl pH 7.4, 1 mM EDTA and proteinase inhibitor cocktail). Protein complexes were immunoprecipitated for 16 h with Anti-FLAG M2 Affinity Gel (Sigma, A2220) before washing 3 times. Samples were eluted with 2x SDS sample buffer and separated on 12% polyacrylamide gel and transferred to nitrocellulose membrane for western blotting as described here. The following primary antibodies were used: mouse monoclonal anti-FLAG M2 antibody (1:1,000, Sigma, F3165, RRID: AB_259529), mouse monoclonal anti-CALCOCO2/NDP52 (1:1,000, Santa Cruz Biotechnology, sc-376540, F-6, RRID: AB_11150487), rabbit monoclonal anti-GFP (D5.1) (1:1,000, Cell Signaling Technology, 2956, RRID: AB_1196615) and rabbit monoclonal anti-Gal8 (EPR4857) (1:1,000, Abcam, ab109519, RRID:AB_10861755). Secondary antibodies used: goat anti-mouse IgG (H+L)-HRP conjugated (1:5,000, Thermo Fischer Scientific, 31430, RRID: AB_228307) and goat anti-rabbit IgG (H+L)-HRP conjugated (1:3,000, Cell Signaling Technology, 7074, RRID: AB_2099233).
Galectin-8 puncta assay
Sterile damage to cell vesicles in the puncta assay was performed as previously described (Thurston et al., 2012). Briefly, HEK293 cells were seeded in an 8-well chambered coverglass (Thermo Fisher Scientific, 155411), incubated in DMEM 10% FBS, 16 h and transfected with either FLAG-tagged galectin-8 or 3CLpro-cleavage analogs of galectin-8 using Lipofectamine 2000 (Invitrogen, 11668019). The FLAG-tagged-N-Gal8 (1-158), or C-Gal8 (159-317)-FLAG-tag plasmid constructs engineered and synthesized from the galectin-8 ORF clone OHu23472 (GenScript). After recovery for 24-h culture in medium, the cells were exposed for 10 min to hypertonic medium (0.5 M sucrose (Calbiochem, 8510) and 15% polyethylene glycol (Sigma, P-3265) in PBS (Sigma, D8537). Cells were rinsed twice with PBS and incubated in 60% PBS for 3 min followed by a 20-min recovery period in complete DMEM +10% FBS. The assay was terminated by fixing the osmotically-shocked cells for 15 min in 4% methanol-free paraformaldehyde (Thermo Fischer Scientific, 28909). Cells were rinsed with 100 mM glycine/PBS solution for 15 min and subsequently permeabilized with a 3-min incubation in 0.1% Triton X-100. The fixed and permeabilized cells were blocked for 1 h with 3% bovine serum albumin (Sigma, A7030), followed by an overnight 4°C incubation with primary rabbit monoclonal anti-FLAG (1:1,000, Cell Signaling Technology, 14793S, RRID: AB_2572291) and mouse monoclonal anti-CALCOCO2/NDP52 (1:1,000, Santa Cruz Biotechnology, sc-376540, F-6, RRID: AB_11150487). After 15-min washes with PBS, cells were incubated with fluorescent secondary antibodies, Alexa Fluor Plus 488 goat anti-rabbit (1:1,000, Invitrogen, A32731, RRID: AB_2633280) and Alexa Fluor Plus 647 goat anti-mouse (1:1,000, Invitrogen, A32728, RRID: AB_2633277) for 1 h. After 15-min final washes, the coverslips were mounted using Fluoroshield with DAPI (Sigma-Aldrich, F6057). Confocal images were captured with 63x objective lens (Zeiss LSM 880 Inverted Confocal Microscope) from 5 randomly selected fields (n > 30 cells), and the percentage of cells positive for CALCOCO2/NDP52/Gal8 puncta was manually quantified. Statistical analyses were performed with Prism version 9.0.0.121 and one-way ANOVA followed by Tukey’s multiple comparison test (GraphPad).
Immunofluorescence of human lung tissue
Normal and COVID-19 lung tissue samples were formalin-fixed and paraffin-embedded, followed by generation of tissue microarrays (TMAs). TMAs containing 1-mm cores were generated essentially as previously described (Kampf et al., 2012; Uhlén et al., 2015), using a TMArrayer (Pathology Devices) and the Beecher Instruments Manual Tissue Arrayer MTA-1 (Estigen OÜ). One core each from two of the normal lung samples (F54 and M45) was included in the TMA with the COVID-19 lung samples, thus serving as controls that there were no staining reproducibility issues between sections. The M15 sample was kept as a full block for staining as a large section, ensuring no regional difference in the staining pattern. From each of the COVID-19 lung samples, two representative cores of ten different lung areas were sampled, i.e., in total, n = 20 lung TMA cores for each COVID-19 patient. The two cores from each area represented different regions of the corresponding tissue blocks. If available, regions with different tissue morphology were selected, e.g., areas heavily affected by the disease versus areas with more normal histology.
The TMA block and the M15 lung-tissue block were cut in 4-μm thick sections using waterfall microtomes (Thermo Fisher Scientific, Microm HM 355S), dried at RT overnight and baked at 50°C for 12 – 24 h before multiplex fluorescence immunohistochemistry. The sections were deparaffinized in xylene, hydrated in graded alcohols and blocked for endogenous peroxidase in 30% hydrogen peroxide diluted 1:100 in 95% ethanol, final concentration 0.3%. For antigen retrieval, a Decloaking chamber (Biocare Medical) was used. Slides were immersed and boiled in Antigen Retrieval Buffer (PT Module Buffer 1, 100x Citrate Buffer, pH 6, TA-250-PM1X, Thermo Fischer Scientific) for 4 min at 125°C and then allowed to cool to 90°C. For antibody validation, all antibodies were first tested with brightfield immunohistochemistry (IHC) on a test TMA containing 20 different normal tissue types. Staining intensity across the tested tissues was compared with mRNA expression levels, in line with the orthogonal approach following guidelines of the International Working Group of Antibody Validation (IWGAV) (Sivertsson et al., 2020; Uhlen et al., 2016). IHC was performed essentially as previously described in detail (Kampf et al., 2012).
After evaluating the IHC results and determining the optimal antibody dilution, multiplex fluorescence IHC was performed based on a 3-plex Opal strategy. Thus, antibodies were added one at a time at RT, and insoluble Opal reagents connected to different fluorophores were used for visualization, followed by heating and inactivation of the previous antibody for each staining cycle. Primary antibodies specific for galectin-8 (1:15, Atlas Antibodies AB, HPA030491, RRID: AB_10602345), CALCOCO2/NDP52 (1:400, Atlas Antibodies AB, HPA022989, RRID: AB_1845914), and SARS-CoV-2 Spike S1 glycoprotein (1:500, Sino Biological, 40150-R007, RRID: AB_2827979) were diluted in UltraAb Diluent (Thermo Fisher Scientific). The primary antibodies and secondary HRP polymer were incubated for 30 min each, followed by 10-min development with the Opal FP1500001KT reagents Opal 650 (Cy5/magenta, Spike S1 glycoprotein, first staining cycle), Opal 520 (FITC/green, galectin-8, second staining cycle) and Opal 570 (Cy3/red, NDP52, third staining cycle, red). All incubations were followed by rinsing in wash buffer (Thermo Fisher Scientific). Inactivation between each cycle was performed in Antigen Retrieval Buffer (Thermo Fischer Scientific) by heating the slides to 90°C for 20 min using a Decloaking chamber and then allowed to cool slowly to 80°C. Slides were incubated with DAPI (1:1,000, Invitrogen) for 5 min and mounted using ProLong™ Glass Antifade Mounting Media (Life Technologies, 2157948). Slides from both normal and COVID-19 lung tissue samples were stained simultaneously to avoid bias between runs. Digital fluorescent images were obtained using a Zeiss Axio Scan.Z1 System equipped with a Zeiss Colibri 7, Type RGB-UV fluorescence light source. Exposure times and visualization parameters were set for normal lung using cell types with known positivity based on the initial IHC results and adjusted for each channel to obtain distinct signals with minimal autofluorescence. All parameters were kept consistent between the M15 normal lung large section and the TMA. H&E images of the same sections were obtained after the fluorescence image acquisition by removal of the coverslips, immersing the slides in Antigen Retrieval Buffer (Thermo Fischer Scientific) at 40°C overnight, followed by staining with hematoxylin (Mayers Htx Plus, Histolab 01825) and eosin (Bio-Optica, 05-10003/L). The H&E slides were coverslipped using PERTEX (Histolab) as mounting medium and scanned with Aperio AT2 slide scanner (Aperio) using a 40x objective.
Quantification and statistical analysis
Data were analyzed within Prism version 9.0.0.121 (GraphPad Software Inc., San Diego, CA). The description of specific statistical tests used for each experiment are detailed in the figure legends and the method details section above. All N (independent biological experiments) and n (intra-experimental independent replicates) values are reported in the results for the data presented.
Mass spectrometry data analysis
All MS/MS data were analyzed using Byonic (Protein Metrics, San Carlos, CA USA; version PMI-Byonic-Com:v3.8.13). Byonic was set to search the uniprot_human database (UP000005640_9606) that included the 3CLpro constructs we expressed and common contaminants. An initial limited search was performed using Preview (v3.8.13) to determine m/z errors and derive recalibration parameters for precursor and fragment ions. The main search parameters were: semi-specific N-ragged ArgC, maximum of 2 missed cleavages; mass tolerance was set to 20 ppm for precursor and fragment ions; fragmentation type, QTOF/HCD; and precursors and fragments were recalibrated from Preview. NEM (+125.0477 Da) at Cys and dimethyl light (+ 28.0313 Da) at Lys were set as fixed modifications, heavy-labeled lysine was set as a +6.0318 Da variable modification over the dimethyl-light. Peptide N-terminal dimethyl light (+ 28.0313 Da), dimethyl heavy (34.0631 Da), pyroglutamic acid (–17.0265 Da), Met oxidation (+ 19.9949 Da), and acetylation (+42.0106 Da) of protein N terminus were set as variable modifications. The peptide score cut off was set to automatic, and the protein score cut off was set to a 1% False Discovery Rate (FDR) or 20 hits in the reverse database, whichever was reached last.
Scaffold (version Scaffold_4.11.0, Proteome Software Inc., Portland, OR, USA) validated the MS/MS-based peptide and protein identifications. Peptide identifications were accepted if established at greater than 99.0% probability to achieve an FDR < 1.0% by the Scaffold Local FDR algorithm. Protein identifications were accepted if they could be established at greater than 99.0% probability to achieve an FDR < 1.0%. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. N-termini were annotated using our in-house program TopFinder 4.1 (https://topfind.clip.msl.ubc.ca).
Retention time alignment and MS1-level quantification of all identified peptides were performed via Skyline (v 20.1.0.155). Only quantitative values with an idotp ≥ 0.85 were considered. Fold-changes between heavy and light forms of the peptide were obtained by dividing their respective MS1 peak areas. For singleton heavy peptides, the peak area was considered as fold-change. The inverse of the MS1 area was taken as the fold-change for a singleton light.
To interrogate whether 3CLpro cleaves substrates regulated by type I interferons (IFN-α and IFN-β), we compared the relative protein abundance in type I interferon-stimulated BEAS-2B cells (N = 6) versus unstimulated control cells (N = 3). To do so, we used the MS1 intensity of the respective preTAILS runs acquired with methanol in the nanoBooster. MS1 quantification and statistical analysis were performed using the default settings in the MSstats tool integrated into Skyline. Statistical significance was determined by multiple sample t test, and adjusted p-values were obtained using Benjamini-Hochberg correction.
To validate the treatment strategy, we compared the 49 proteins upregulated by IFN-α and IFN-β with known ISGs and found all were previously reported, including STAT-1, IFIT2, IFIT3, OAS, MX1, among others.
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifiers PXD026797 for HEK293, and PXD026815 for BEAS-2B.
3CLpro substrate winnowing
In our experimental design, the neo-N-termini generated from 3CLpro activity must necessarily and exclusively be labeled with dimethyl heavy (+ 34 kDa). Therefore, we considered as possible 3CLpro substrates only singleton heavy peptides that were identified in ≥ 2/3 HEK293 or ≥ 7/9 BEAS-2B independent biological experiments. A poor-quality or false identification of a light form of the peptide across the whole experiment was sufficient to disqualify the protein as a substrate. In addition, we excluded all peptides that could be explained from residual labeling of tryptic peptides, Met1 removal, or protein N-terminal ragging.
To further increase the confidence of the 3CLpro substrates, a score was derived (using a custom script) that compared the sequence of each identified cleavage site to the normalized relative frequency of amino acids in positions P4–P4’ of all cleavage sites meeting all the criteria described above. To define the confidently identified cleavage sites generated by 3CLpro, the score of the 90th percentile of non-confidently identified cleavage sites was used as the minimum cutoff. Finally, all MS/MS spectra of the winnowed neo-N-terminal peptides were manually inspected, discarding any displaying poor fragmentation, noise, or ragged termini and were not further considered. The iceLogos presented were generated using iceLogo (https://iomics.ugent.be/icelogoserver/) (Colaert et al., 2009).
3CLpro and substrate modeling
Peptide-protein docking
The following flags and command lines were used in the peptide-protein docking simulations:
Prepacking the complex to remove potential internal clashes and guarantee a uniform conformational background in non-interface regions.
$ROSETTA_BIN/FlexPepDocking.mpi.linuxgccrelease @prepack_flags
prepack_flags:
s 3CL_YAP1_a.pdb
ex1
ex2aro
database $ROSETTA_DB_Path
scorefile prepack.score.sc
flexpep_score_only
flexpep_prepack
nstruct 1
out:path:pdb output
out:path:score output
use_truncated_termini
flexPepDocking:receptor_chain A
flexPepDocking:peptide_chain D
Model generation beginning from the prepacked structure
$ROSETTA_BIN/FlexPepDocking.mpi.linuxgccrelease @abinitio_flags
abinitio_flags:
s input/3CL_YAP1_ppk.pdb
lowres_abinitio
pep_refine
flexpep_score_only
ex1
ex2aro
use_truncated_termini
frag3 input/frags/frags.3mers.offset
flexPepDocking:frag5 input/frags/frags.5mers.offset
flexPepDocking:frag5_weight 0.25
constraints:cst_weight 2
constraints:cst_fa_file input/constraint_file
constraints:cst_file input/constraints_file
constraints:cst_fa_weight 2
score:weights ref2015_cst
out:path:pdb output
out:file:silent output/SARS_mono_Isa_peptide1_silent.out
out:file:scorefile output/score_mono_Isa_peptide1.sc
nstruct 50000
flexPepDocking:receptor_chain A
flexPepDocking:peptide_chain D
Given below is the constraint file content and the flat harmonic function used to favor models where the Euclidean distance between Ser145 Sγ and Cα of P1 are less than 4 Å.
Constraint file
AtomPair SG 145A CA 310D FLAT_HARMONIC 2.0 0.25 2.0
Flat Harmonic function
Protein-protein docking
The following command lines were used in the 3CLpro/Gal8 docking simulations:
RosettaCM Hybridize
$ROSETTA_BIN/rosetta_scripts.mpi.linuxgccrelease @rosetta_cm.options
rosetta_cm.options:
in:file:fasta galac.fasta
parser:protocol rosetta_cm.xml
nstruct 15000
out:file:silent output/Isa_galectin8_silent.out
out:file:scorefile output/score_ galectin8_CompCan.sc
relax:minimize_bond_angles
relax:minimize_bond_lengths
relax:jump_move true
default_max_cycles 200
relax:min_type lbfgs_armijo_nonmonotone
relax:jump_move true
score:weights stage3.wts
use_bicubic_interpolation
hybridize:stage1_probability 1.0
where rosetta.xml is:
< ROSETTASCRIPTS >
< TASKOPERATIONS >
< /TASKOPERATIONS >
< SCOREFXNS >
< ScoreFunction name = ”stage1” weights = ”stage1.wts” symmetric = ”0” >
< Reweight scoretype = ”atom_pair_constraint” weight = ”1”/ >
< /ScoreFunction >
< ScoreFunction name = ”stage2” weights = ”stage2.wts” symmetric = ”0” >
< Reweight scoretype = ”atom_pair_constraint” weight = ”0.5”/ >
< /ScoreFunction >
< ScoreFunction name = ”fullatom” weights = ”stage3.wts” symmetric = ”0” >
< Reweight scoretype = ”atom_pair_constraint” weight = ”0.5”/ >
< /ScoreFunction >
< /SCOREFXNS >
< FILTERS >
< /FILTERS >
< MOVERS >
< Hybridize name = ”hybridize” stage1_scorefxn = ”stage1” stage2_scorefxn = ”stage2” fa_scorefxn = ”fullatom” batch = ”1” stage1_increase_cycles = ”1.0” stage2_increase_cycles = ”1.0” linmin_only = ”1” >
< Fragments three_mers = ”4FQZA_3.frags” nine_mers = ”4FQZA_9.frags”/ >
< Template pdb = ”4FQZA.pdb” cst_file = ”AUTO” weight = ”1.000” / >
< /Hybridize >
< /MOVERS >
< APPLY_TO_POSE >
< /APPLY_TO_POSE >
< PROTOCOLS >
< Add mover = ”hybridize”/ >
< /PROTOCOLS >
< /ROSETTASCRIPTS >
Backbone ensembles generation
$ROSETTA_BIN/rosetta_scripts.mpi.linuxgccrelease @ensembles_nma_flag
ensembles_nma_flag:
in:file:s 3CL_6xhm_ppk.pdb
nstruct 150
suffix _nma
parser:protocol nma.xml
where nma.xml is:
< ROSETTASCRIPTS >
< SCOREFXNS >
< ScoreFunction name = ”ref_cart” weights = ”ref2015_cart” / >
< /SCOREFXNS >
< RESIDUE_SELECTORS >
< /RESIDUE_SELECTORS >
< TASKOPERATIONS >
< /TASKOPERATIONS >
< FILTERS >
< /FILTERS >
< MOVERS >
< NormalModeRelax name = ”nma” cartesian = ”true” centroid = ”false” scorefxn = ”ref_cart” nmodes = ”5” mix_modes = ”true” pertscale = ”1.0” randomselect = ”false” relaxmode = ”relax” nsample = ”20” cartesian_minimize = ”false” / >
< /MOVERS >
< APPLY_TO_POSE >
< /APPLY_TO_POSE >
< PROTOCOLS >
< Add mover = ”nma” / >
< /PROTOCOLS >
< OUTPUT scorefxn = ”ref_cart” / >
< /ROSETTASCRIPTS >
Docking Simulation
$ROSETTA_BIN/docking_protocol.mpi.linuxgccrelease @protein_protein_docking_flag
protein_protein_docking_flag:
in:file:s complex_start_3CL_6xhm_galactin8_ppk1.pdb
ensemble1 3CL_ensembles_pdblist
ensemble2 galactin8_ensembles_pdblist
partners BC_A
detect_disulf true
rebuild_disulf true
ex1
ex2aro
overwrite
nstruct 50000
constraints:cst_weight 2
constraints:cst_fa_file constraints.txt
constraints:cst_file constraints.txt
constraints:cst_fa_weight 2
score:weights ref2015_cst
out:file:silent output/silent_3CL_dimer_Isa_galectin8_1.out
out:file:scorefile output/score_3CL_dimer_Isa_gelactin8_1.sc
where constraints.txt is:
AtomPair SG 145B CB 158A FLAT_HARMONIC 2.0
Immunofluorescence analysis nuclear/cytoplasmatic PTBP1 ratio
Nuclear to cytoplasmic (N/C) ratio was calculated using the ImageJ (version 1.46r) intensity ratio nuclei cytoplasm tool (Intensity Ratio Nuclei Cytoplasm Tool, RRID: SCR_018573) on five fields of views for mock and SARS-COV-2 infected cells. Spike-positive cells where manually segmented using anti-Spike and DAPI for total cell and nuclear area, respectively. The intensity of PTBP1 was acquired through ImageJ, and a ratio of nuclear to cytoplasmic intensity was calculated (n = 51 cells across 5 experiments). Statistical analyses were performed with Prism version 9.0.0.121 using Student’s t test (GraphPad).
Immunofluorescence analysis of lung samples
All TMA cores and the whole M15 large section were evaluated by manual inspection to assure that the staining patterns observed were consistent across all samples, TMA cores and fields of view. For quantification and verification of the results observed by manual inspection, the number of macrophages positive for galectin-8, NDP52, or both were counted in 30 COVID-19 TMA cores corresponding to a total area of 23.6 mm2, as well as six fields of view of the M15 section corresponding to a total area of 32.55 mm2. Identification of macrophages was based on histological expertise by comparing the same fields of view with the H&E sections. Only intact macrophages with visible nuclei in the DAPI channel were counted. Each cell was evaluated separately, toggling between the channels to determine if a cell showed positivity of only NDP52, only Gal8, or both. The total number of macrophages counted was 1,924 in COVID-19 lung samples and 1,472 in normal lung samples.
Protein-protein interaction network construction
The human protein-protein interaction network of the human 3CLpro substrates was constructed by using the 101 high-confidence substrates listed in Table 1 as seeds and retrieving all known interactors using the IntAct app (v 0.9.7) (Ragueneau et al., 2021) in Cytoscape v 3.8.2 (Shannon et al., 2003). Then, a filter was applied to retain only human proteins with curated direct interactions or physical associations to the 101 substrates resulting in a network of 2,301 nodes and 2,931 edges. To simplify and better visualize the network, we used the GLay community clustering algorithm in the Cytoscape plugin clusterMaker2 (v 1.3.1) (Morris et al., 2011). The 101 substrates, their direct interactors, and physically associated proteins were then mapped onto the IMEx/IntAct Coronavirus Dataset: SARS-COV-2+human context (Perfetto et al., 2020) downloaded from NDEx (http://www.ndexbio.org/#/networkset/4c2268a1-a0f0-11ea-aaef-0ac135e8bacf ), retrieving a network consisting of SARS-COV-2 proteins that interact with 3CLpro human substrates either directly or via a 3rd common interactor. For visualization, a circular layout was applied to the network using the yFiles Layout plugin. To determine the protein complexes enriched in the network, we performed a functional enrichment analysis with CORUM database complexes using the ShinyGO (Ge et al., 2020) online tool. Visualization was generated using Cytoscape.
Acknowledgments
Edman degradation was performed at the Tufts University Core Facility. This work was supported by the Canada Research Chairs program (950-01-126 to C.M.O. and 950-231595 to J.A.H.), the Canadian Institutes of Health Research (CIHR) Foundation grant program (FDN-148408 to C.M.O.), the CIHR 2019 Novel Coronavirus (COVID-19) Rapid Research grant program (OV3-170636 to E.J. and C.M.O., FRN-170645 to A.B.), the CIHR (OV4-170645 to K.M., PJT-173318 and MM1-174898 to H.L., MOP-130408 and PJT-153270 to J.N.K., 388726 to J.A.H., and a Banting and Best Canada Graduate Doctoral Award 201911 FBD-434976-268115 to P. M.G.), the Canada Foundation for Innovation (31059 to C.M.O.), the Michael Smith Foundation for Health Research (MSRF) (IN-NPG-00105 to C.M.O., and a postdoctoral fellowship 17905 to Y.M.), the Knut and Alice Wallenberg Foundation (C.L.), the Stockholms Läns Landsting (929362 to L.S.), the Natural Sciences and Engineering Research Council (NSERC) (RGPIN-2016-03811 to H.L., RGPIN-2018-03828 to J.N.K., a postdoctoral fellowship 532117-2019 to A.B.), the Polish Medical Research Agency (2020/ABM/SARS/1 to M.D.), the National Science Centre in Poland (2020/01/0/NZ1/00063 to M.D.), and a ALS-Canada/Brain-Canada Doctoral Fellowship (Y. Mohamud).
Author contributions
I.P. performed recombinant 3CLpro expression, designed and performed all experiments, except as listed, analyzed all data, prepared all figures, wrote and edited the paper. Y. Machado designed and performed all TAILS and preTAILS LC-MS/MS, interactome, and MALDI-TOF-MS enzyme kinetics experiments, analyzed all data, prepared figures and tables, wrote and edited the paper. H.C.R. performed molecular simulations, wrote results and prepared figure panels. A.B. performed all SARS-CoV-2 infections of Calu-3 cells, K.M. supervised. Y. Mohamud performed cell transfections, autophagy imaging, data analyses, and quantification of Vero E6 infected cell images, wrote results, H.L. supervised. R.K. designed plasmids, prepared recombinant inactive 3CLpro-C145A, sourced and ordered all chemicals and reagents, and managed the laboratory and COVID-19 safety protocols. C.L. prepared, imaged and analyzed COVID-19 lung samples, wrote results, L.S., A.S. performed autopsies, collected lung samples. P.A.B. performed mutagenesis and cut-site bioinformatics. G.S.B. assisted with substrate cleavage assays and edited the paper. P.M.G. performed immunoblots of pSTAT1 and MAP4K5, assisted in 3CLpro purification. N.S. assisted in M.S. maintenance. Q.T.C., J.P.N. prepared HAEC cell lysates, J.H. supervised. M.V. performed Vero E6 SARS-CoV-2 infections, E.J. supervised. S.A. synthesized the HPG-Polymer, J.N.K. supervised. J.C.V. synthesized and supplied GC376 inhibitor. W.R. synthesized quenched fluorescent peptide for activity assays, M.D. supervised. H.C.R., R.K., P.A.B., G.S.B., N.S., and P.M.G. provided input into experimental design and edited the manuscript. C.M.O. designed experiments and plasmids, analyzed the data, wrote and edited the paper, conceived and supervised the research.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
Published: October 9, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109892.
Supplemental information
(A) HEK293 quantified peptides in TAILS + PreTAILS. (B) Scaffold spectrum report for HEK293 TAILS. (C) Scaffold spectrum report for HEK293 preTAILS.
(A) BEAS-2B quantified peptides in TAILS + PreTAILS. (B) Scaffold spectrum report for BEAS-2B TAILS. (C) Scaffold spectrum report for BEAS-2B preTAILS.
(A) Quantified neo-N-termini in human embryonic kidney (HEK293) cells and human lung epithelial (BEAS-2B) cells. (B) Quantified “other” neo-N-termini in human embryonic kidney (HEK293) cells and human lung epithelial (BEAS-2B) cells.
Data and code availability
The mass spectrometry proteomics data are available via ProteomeXchange with identifiers PXD026797 (HEK293) and PXD026815 (BEAS-2B). The interactive version of PPI networks presented in Figures 6A and 6C are available online in the NDEx repository (https://public.ndexbio.org/#/network/1b9f868d-d391-11eb-b666-0ac135e8bacf?accesskey=f186d2601af5583f94dbd3e32898225e761d0bd5aa7bb885dbe5a8927a6a3962) and (https://public.ndexbio.org/#/network/195436fa-d391-11eb-b666-0ac135e8bacf?accesskey=8d7377ab2f26a9ca4b13276a71e55864cead1543752cd4737d9c803cb4fd6540). All flags and commands lines used to generate the structural models reported in this study are available in this paper’s supplemental information. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Uncropped full-length SDS-PAGE gels and western blots associated with this paper are available from Mendeley Data at (https://data.mendeley.com/datasets/b97d5nrb72/2).
References
- Alford R.F., Leaver-Fay A., Jeliazkov J.R., O’Meara M.J., DiMaio F.P., Park H., Shapovalov M.V., Renfrew P.D., Mulligan V.K., Kappel K. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J. Chem. Theory Comput. 2017;13:3031–3048. doi: 10.1021/acs.jctc.7b00125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back S.H., Kim Y.K., Kim W.J., Cho S., Oh H.R., Kim J.E., Jang S.K. Translation of polioviral mRNA is inhibited by cleavage of polypyrimidine tract-binding proteins executed by polioviral 3C(pro) J. Virol. 2002;76:2529–2542. doi: 10.1128/jvi.76.5.2529-2542.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A., Nasir J.A., Budylowski P., Yip L., Aftanas P., Christie N., Ghalami A., Baid K., Raphenya A.R., Hirota J.A. Isolation, Sequence, Infectivity, and Replication Kinetics of Severe Acute Respiratory Syndrome Coronavirus 2. Emerg. Infect. Dis. 2020;26:2054–2063. doi: 10.3201/eid2609.201495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M., Memon D., Meyer B., White K.M., Rezelj V.V., Correa Marrero M., Polacco B.J., Melnyk J.E., Ulferts S., Kaake R.M. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell. 2020;182:685–712.e19. doi: 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler G.S., Overall C.M. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 2009;8:935–948. doi: 10.1038/nrd2945. [DOI] [PubMed] [Google Scholar]
- Caly L., Ghildyal R., Jans D.A. Respiratory virus modulation of host nucleocytoplasmic transport; target for therapeutic intervention? Front. Microbiol. 2015;6:848. doi: 10.3389/fmicb.2015.00848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson S., Öberg C.T., Carlsson M.C., Sundin A., Nilsson U.J., Smith D., Cummings R.D., Almkvist J., Karlsson A., Leffler H. Affinity of galectin-8 and its carbohydrate recognition domains for ligands in solution and at the cell surface. Glycobiology. 2007;17:663–676. doi: 10.1093/glycob/cwm026. [DOI] [PubMed] [Google Scholar]
- Casey S.C., Baylot V., Felsher D.W. The MYC oncogene is a global regulator of the immune response. Blood. 2018;131:2007–2015. doi: 10.1182/blood-2017-11-742577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S.K., White S., Orth A.P., Reisdorph R., Miraglia L., Thomas R.S., DeJesus P., Mason D.E., Huang Q., Vega R. Genome-scale functional profiling of the mammalian AP-1 signaling pathway. Proc. Natl. Acad. Sci. USA. 2003;100:12153–12158. doi: 10.1073/pnas.1934839100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Wang Y., Gong Y., Li F., Guo Y., Hu S., Liu J., Pan L. Structural basis of FYCO1 and MAP1LC3A interaction reveals a novel binding mode for Atg8-family proteins. Autophagy. 2016;12:1330–1339. doi: 10.1080/15548627.2016.1185590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaert N., Helsens K., Martens L., Vandekerckhove J., Gevaert K. Improved visualization of protein consensus sequences by iceLogo. Nat. Methods. 2009;6:786–787. doi: 10.1038/nmeth1109-786. [DOI] [PubMed] [Google Scholar]
- Dai W., Zhang B., Jiang X.M., Su H., Li J., Zhao Y., Xie X., Jin Z., Peng J., Liu F. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368:1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucet A., Overall C.M. Broad coverage identification of multiple proteolytic cleavage site sequences in complex high molecular weight proteins using quantitative proteomics as a complement to edman sequencing. Mol. Cell. Proteomics. 2011;10 doi: 10.1074/mcp.M110.003533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., Zanconato F., Le Digabel J., Forcato M., Bicciato S. Role of YAP/TAZ in mechanotransduction. Nature. 2011;474:179–183. doi: 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Fleishman S.J., Leaver-Fay A., Corn J.E., Strauch E.M., Khare S.D., Koga N., Ashworth J., Murphy P., Richter F., Lemmon G. RosettaScripts: a scripting language interface to the Rosetta macromolecular modeling suite. PLoS ONE. 2011;6:e20161. doi: 10.1371/journal.pone.0020161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florez P.M., Sessions O.M., Wagner E.J., Gromeier M., Garcia-Blanco M.A. The polypyrimidine tract binding protein is required for efficient picornavirus gene expression and propagation. J. Virol. 2005;79:6172–6179. doi: 10.1128/JVI.79.10.6172-6179.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge S.X., Jung D., Yao R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics. 2020;36:2628–2629. doi: 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Dunn S.L., White M.F. The reciprocal stability of FOXO1 and IRS2 creates a regulatory circuit that controls insulin signaling. Mol. Endocrinol. 2006;20:3389–3399. doi: 10.1210/me.2006-0092. [DOI] [PubMed] [Google Scholar]
- Herhaus L., Bhaskara R.M., Lystad A.H., Gestal-Mato U., Covarrubias-Pinto A., Bonn F., Simonsen A., Hummer G., Dikic I. TBK1-mediated phosphorylation of LC3C and GABARAP-L2 controls autophagosome shedding by ATG4 protease. EMBO reports. 2020;21:e48317. doi: 10.15252/embr.201948317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagdeo J.M., Dufour A., Klein T., Solis N., Kleifeld O., Kizhakkedathu J., Luo H., Overall C.M., Jan E. N-Terminomics TAILS Identifies Host Cell Substrates of Poliovirus and Coxsackievirus B3 3C Proteinases That Modulate Virus Infection. J. Virol. 2018;92:e02211–e02217. doi: 10.1128/JVI.02211-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajander T., Kahn P.C., Passila S.H., Cohen D.C., Lehtiö L., Adolfsen W., Warwicker J., Schell U., Goldman A. Buried charged surface in proteins. Structure. 2000;8:1203–1214. doi: 10.1016/s0969-2126(00)00520-7. [DOI] [PubMed] [Google Scholar]
- Kampf C., Olsson I., Ryberg U., Sjöstedt E., Pontén F. Production of tissue microarrays, immunohistochemistry staining and digitalization within the human protein atlas. J. Vis. Exp. 2012;(63):3620. doi: 10.3791/3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B.-W., Hong S.B., Kim J.H., Kwon D.H., Song H.K. Structural basis for recognition of autophagic receptor NDP52 by the sugar receptor galectin-8. Nat. Commun. 2013;4:1613. doi: 10.1038/ncomms2606. [DOI] [PubMed] [Google Scholar]
- Kim J.H., Park M.E., Nikapitiya C., Kim T.H., Uddin M.B., Lee H.C., Kim E., Ma J.Y., Jung J.U., Kim C.J., Lee J.S. FAS-associated factor-1 positively regulates type I interferon response to RNA virus infection by targeting NLRX1. PLoS Pathog. 2017;13:e1006398. doi: 10.1371/journal.ppat.1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Lee J.Y., Yang J.S., Kim J.W., Kim V.N., Chang H. The Architecture of SARS-CoV-2 Transcriptome. Cell. 2020;181:914–921.e10. doi: 10.1016/j.cell.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleifeld O., Doucet A., auf dem Keller U., Prudova A., Schilling O., Kainthan R.K., Starr A.E., Foster L.J., Kizhakkedathu J.N., Overall C.M. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 2010;28:281–288. doi: 10.1038/nbt.1611. [DOI] [PubMed] [Google Scholar]
- Kleifeld O., Doucet A., Prudova A., auf dem Keller U., Gioia M., Kizhakkedathu J.N., Overall C.M. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 2011;6:1578–1611. doi: 10.1038/nprot.2011.382. [DOI] [PubMed] [Google Scholar]
- Klein T., Eckhard U., Dufour A., Solis N., Overall C.M. Proteolytic Cleavage-Mechanisms, Function, and “Omic” Approaches for a Near-Ubiquitous Posttranslational Modification. Chem. Rev. 2018;118:1137–1168. doi: 10.1021/acs.chemrev.7b00120. [DOI] [PubMed] [Google Scholar]
- Koudelka T., Boger J., Henkel A., Schönherr R., Krantz S., Fuchs S., Rodríguez E., Redecke L., Tholey A. N-Terminomics for the Identification of In Vitro Substrates and Cleavage Site Specificity of the SARS-CoV-2 Main Protease. Proteomics. 2021;21:e2000246. doi: 10.1002/pmic.202000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapointe C.P., Grosely R., Johnson A.G., Wang J., Fernández I.S., Puglisi J.D. Dynamic competition between SARS-CoV-2 NSP1 and mRNA on the human ribosome inhibits translation initiation. Proc. Natl. Acad. Sci. USA. 2021;118 doi: 10.1073/pnas.2017715118. e2017715118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver-Fay A., Tyka M., Lewis S.M., Lange O.F., Thompson J., Jacak R., Kaufman K.W., Renfrew P.D., Smith C.A., Sheffler W. In: Methods in Enzymology. Johnson M.L., Brand L., editors. Academic Press; 2011. Rosetta3: An object-oriented software suite for the simulation and design of macromolecules; pp. 545–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Worrall L.J., Vuckovic M., Rosell F.I., Gentile F., Ton A.-T., Caveney N.A., Ban F., Cherkasov A., Paetzel M., Strynadka N.C.J. Crystallographic structure of wild-type SARS-CoV-2 main protease acyl-enzyme intermediate with physiological C-terminal autoprocessing site. Nat. Commun. 2020;11:5877. doi: 10.1038/s41467-020-19662-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Wandel M.P., Li F., Liu Z., He C., Wu J., Shi Y., Randow F. Sterical hindrance promotes selectivity of the autophagy cargo receptor NDP52 for the danger receptor galectin-8 in antibacterial autophagy. Sci. Signal. 2013;6:ra9. doi: 10.1126/scisignal.2003730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Otín C., Overall C.M. Protease degradomics: a new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 2002;3:509–519. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- Lynch C.J., Bernad R., Calvo I., Nóbrega-Pereira S., Ruiz S., Ibarz N., Martinez-Val A., Graña-Castro O., Gómez-López G., Andrés-León E. The RNA Polymerase II Factor RPAP1 Is Critical for Mediator-Driven Transcription and Cell Identity. Cell Rep. 2018;22:396–410. doi: 10.1016/j.celrep.2017.12.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B., Chiaravalli J., Gellenoncourt S., Brownridge P., Bryne D.P., Daly L.A., Grauslys A., Walter M., Agou F., Chakrabarti L.A. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat. Commun. 2021;12:5553. doi: 10.1038/s41467-021-25796-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamud Y., Luo H. The Intertwined Life Cycles of Enterovirus and Autophagy. Virulence. 2019;10:470–480. doi: 10.1080/21505594.2018.1551010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J.H., Apeltsin L., Newman A.M., Baumbach J., Wittkop T., Su G., Bader G.D., Ferrin T.E. clusterMaker: a multi-algorithm clustering plugin for Cytoscape. BMC Bioinformatics. 2011;12:436. doi: 10.1186/1471-2105-12-436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustaqil M., Ollivier E., Chiu H.P., Van Tol S., Rudolffi-Soto P., Stevens C., Bhumkar A., Hunter D.J.B., Freiberg A.N., Jacques D. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): Implications for disease presentation across species. Emerg. Microbes Infect. 2021;10:178–195. doi: 10.1080/22221751.2020.1870414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- Perez-Riverol Y., Csordas A., Bai J., Bernal-Llinares M., Hewapathirana S., Kundu D.J., Inuganti A., Griss J., Mayer G., Eisenacher M. The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 2019;47(D1):D442–D450. doi: 10.1093/nar/gky1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfetto L., Pastrello C., Del-Toro N., Duesbury M., Iannuccelli M., Kotlyar M., Licata L., Meldal B., Panneerselvam K., Panni S. The IMEx coronavirus interactome: an evolving map of Coronaviridae-host molecular interactions. Database (Oxford) 2020;2020:baaa096. doi: 10.1093/database/baaa096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persengiev S.P., Green M.R. The role of ATF/CREB family members in cell growth, survival and apoptosis. Apoptosis. 2003;8:225–228. doi: 10.1023/A:1023633704132. [DOI] [PubMed] [Google Scholar]
- Ragueneau E., Shrivastava A., Morris J.H., Del-Toro N., Hermjakob H., Porras P. IntAct App: a Cytoscape application for molecular interaction network visualisation and analysis. Bioinformatics. 2021:btab319. doi: 10.1093/bioinformatics/btab319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch V., Hansen C.G. The hippo pathway, YAP/TAZ, and the plasma membrane. Trends Cell Biol. 2020;30:32–48. doi: 10.1016/j.tcb.2019.10.005. [DOI] [PubMed] [Google Scholar]
- Raveh B., London N., Zimmerman L., Schueler-Furman O. Rosetta FlexPepDock ab-initio: simultaneous folding, docking and refinement of peptides onto their receptors. PLoS ONE. 2011;6:e18934. doi: 10.1371/journal.pone.0018934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W., Groborz K., Zhang L., Sun X., Zmudzinski M., Pawlik B., Wang X., Jochmans D., Neyts J., Młynarski W. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 2021;17:222–228. doi: 10.1038/s41589-020-00689-z. [DOI] [PubMed] [Google Scholar]
- Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott B., Lacasse V., Blom D., Tonner P., Blom N. Predicted Coronavirus Nsp5 Protease Cleavage Sites in the Human Proteome: A Resource for SARS-CoV-2 Research. bioRxiv. 2021 doi: 10.1186/s12863-022-01044-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P., Markiel A., Ozier O., Baliga N.S., Wang J.T., Ramage D., Amin N., Schwikowski B., Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020;587:657–662. doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivertsson Å., Lindström E., Oksvold P., Katona B., Hikmet F., Vuu J., Gustavsson J., Sjöstedt E., von Feilitzen K., Kampf C. Enhanced Validation of Antibodies Enables the Discovery of Missing Proteins. J. Proteome Res. 2020;19:4766–4781. doi: 10.1021/acs.jproteome.0c00486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sola I., Galán C., Mateos-Gómez P.A., Palacio L., Zúñiga S., Cruz J.L., Almazán F., Enjuanes L. The polypyrimidine tract-binding protein affects coronavirus RNA accumulation levels and relocalizes viral RNAs to novel cytoplasmic domains different from replication-transcription sites. J. Virol. 2011;85:5136–5149. doi: 10.1128/JVI.00195-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y., DiMaio F., Wang R.Y., Kim D., Miles C., Brunette T., Thompson J., Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staring J., von Castelmur E., Blomen V.A., van den Hengel L.G., Brockmann M., Baggen J., Thibaut H.J., Nieuwenhuis J., Janssen H., van Kuppeveld F.J. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature. 2017;541:412–416. doi: 10.1038/nature21032. [DOI] [PubMed] [Google Scholar]
- Starr A.E., Overall C.M. In: Methods in Enzymology. Abelson J.N., Simon M.I., Colowick S.P., Kaplan N.O., editors. Academic Press; 2009. Characterizing proteolytic processing of chemokines by mass spectrometry, biochemistry, neo-epitope antibodies and functional assays; pp. 281–307. [DOI] [PubMed] [Google Scholar]
- Stukalov A., Girault V., Grass V., Karayel O., Bergant V., Urban C., Haas D.A., Huang Y., Oubraham L., Wang A. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature. 2021;594:246–252. doi: 10.1038/s41586-021-03493-4. [DOI] [PubMed] [Google Scholar]
- Szekely L., Bozoky B., Bendek M., Ostad M., Lavignasse P., Haag L., Wu J., Jing X., Gupta S., Saccon E. Pulmonary stromal expansion and intra-alveolar coagulation are primary causes of COVID-19 death. Heliyon. 2021;7:e07134. doi: 10.1016/j.heliyon.2021.e07134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurston T.L., Wandel M.P., von Muhlinen N., Foeglein A., Randow F. Galectin 8 targets damaged vesicles for autophagy to defend cells against bacterial invasion. Nature. 2012;482:414–418. doi: 10.1038/nature10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlén M., Fagerberg L., Hallström B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- Uhlen M., Bandrowski A., Carr S., Edwards A., Ellenberg J., Lundberg E., Rimm D.L., Rodriguez H., Hiltke T., Snyder M., Yamamoto T. A proposal for validation of antibodies. Nat. Methods. 2016;13:823–827. doi: 10.1038/nmeth.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W., Khan M.B., Fischer C., Arutyunova E., Lamer T., Shields J., Saffran H.A., McKay R.T., van Belkum M.J., Joyce M.A. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020;11:4282. doi: 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D., Mohr I. Viral subversion of the host protein synthesis machinery. Nat. Rev. Microbiol. 2011;9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Xie F., Chu F., Zhang Z., Yang B., Dai T., Gao L., Wang L., Ling L., Jia J. YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKε-mediated phosphorylation. Nat. Immunol. 2017;18:733–743. doi: 10.1038/ni.3744. [DOI] [PubMed] [Google Scholar]
- Wang W.-H., Lin C.-Y., Chang M.R., Urbina A.N., Assavalapsakul W., Thitithanyanont A., Chen Y.-H., Liu F.-T., Wang S.-F. The role of galectins in virus infection—A systemic literature review. J. Microbiol. Immunol. Infect. 2020;53:925–935. doi: 10.1016/j.jmii.2019.09.005. [DOI] [PubMed] [Google Scholar]
- Wessel D., Flügge U.I. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal. Biochem. 1984;138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- Yalamanchili P., Datta U., Dasgupta A. Inhibition of host cell transcription by poliovirus: cleavage of transcription factor CREB by poliovirus-encoded protease 3Cpro. J. Virol. 1997;71:1220–1226. doi: 10.1128/jvi.71.2.1220-1226.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Meng F., Chen S., Plouffe S.W., Wu S., Liu S., Li X., Zhou R., Wang J., Zhao B. Hippo signalling governs cytosolic nucleic acid sensing through YAP/TAZ-mediated TBK1 blockade. Nat. Cell Biol. 2017;19:362–374. doi: 10.1038/ncb3496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) HEK293 quantified peptides in TAILS + PreTAILS. (B) Scaffold spectrum report for HEK293 TAILS. (C) Scaffold spectrum report for HEK293 preTAILS.
(A) BEAS-2B quantified peptides in TAILS + PreTAILS. (B) Scaffold spectrum report for BEAS-2B TAILS. (C) Scaffold spectrum report for BEAS-2B preTAILS.
(A) Quantified neo-N-termini in human embryonic kidney (HEK293) cells and human lung epithelial (BEAS-2B) cells. (B) Quantified “other” neo-N-termini in human embryonic kidney (HEK293) cells and human lung epithelial (BEAS-2B) cells.
Data Availability Statement
The mass spectrometry proteomics data are available via ProteomeXchange with identifiers PXD026797 (HEK293) and PXD026815 (BEAS-2B). The interactive version of PPI networks presented in Figures 6A and 6C are available online in the NDEx repository (https://public.ndexbio.org/#/network/1b9f868d-d391-11eb-b666-0ac135e8bacf?accesskey=f186d2601af5583f94dbd3e32898225e761d0bd5aa7bb885dbe5a8927a6a3962) and (https://public.ndexbio.org/#/network/195436fa-d391-11eb-b666-0ac135e8bacf?accesskey=8d7377ab2f26a9ca4b13276a71e55864cead1543752cd4737d9c803cb4fd6540). All flags and commands lines used to generate the structural models reported in this study are available in this paper’s supplemental information. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. Uncropped full-length SDS-PAGE gels and western blots associated with this paper are available from Mendeley Data at (https://data.mendeley.com/datasets/b97d5nrb72/2).