

Summary
Cellular senescence is a stress or damage response that causes a permanent proliferative arrest and secretion of numerous factors with potent biological activities. This senescence-associated secretory phenotype (SASP) has been characterized largely for secreted proteins that participate in embryogenesis, wound healing, inflammation and many age-related pathologies. By contrast, lipid components of the SASP are understudied. We show that senescent cells activate the biosynthesis of several oxylipins that promote segments of the SASP and reinforce the proliferative arrest. Notably, senescent cells synthesize and accumulate an unstudied intracellular prostaglandin, 1a,1b-dihomo-15-deoxy-delta-12,14-prostaglandin J2. Released 15-deoxy-delta-12,14-prostaglandin J2 is a biomarker of senolysis in culture and in vivo. This and other prostaglandin D2-related lipids promote the senescence arrest and SASP by activating RAS signaling. These data identify an important aspect of cellular senescence and a method to detect senolysis.
eTOC
Senolytics, transgenic and pharmacological interventions that selectively kill senescent cells, are currently in clinical trials aiming to treat age-related degenerative pathologies. Here, Wiley et al. discover that senescent cells produce multiple signaling lipids known as oxylipins. One oxylipin, dihomo-15d-PGJ2, promotes features of senescence by activating RAS and is released from cells during senolysis, serving as the first biomarker of the process in culture and in vivo.
Graphical Abstract
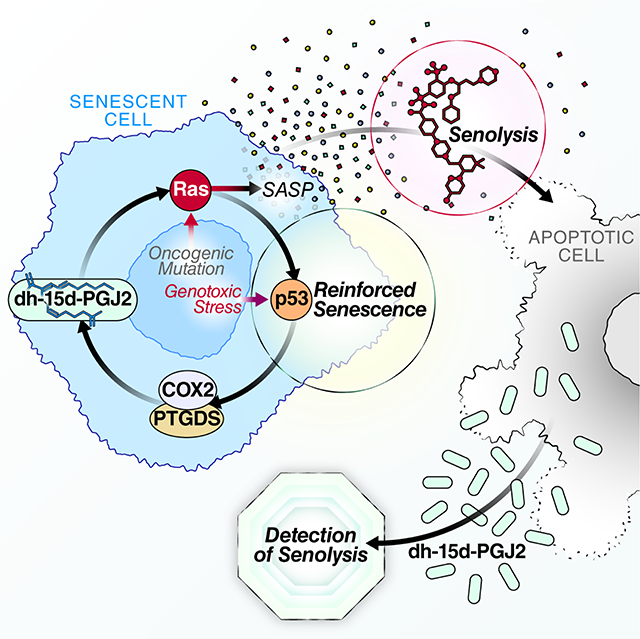
Introduction
Cellular senescence is a multifaceted response to physiological and stress-generated signals, resulting in an essentially permanent arrest of cell proliferation and several phenotypic changes, including cellular hypertrophy, nuclear and epigenetic rearrangements, and metabolic alterations (Kuilman et al., 2010; Wiley and Campisi, 2016). Cell culture and transgenic mouse studies show that senescent cells can drive several age-associated pathologies in part by secreting a myriad of biologically active molecules. These molecules, which include inflammatory cytokines and chemokines, proteases and growth factors, can have potent local – and potentially systemic – effects on tissues (Acosta et al., 2008; Coppe et al., 2008). Senescent cells increase with age in human and mouse tissues (Campisi, 2013; Wiley and Campisi, 2016), which, at least in mice, limits both median life span and health span (Baker et al., 2016).
Thus far, the senescence-associated secretory phenotype (SASP) has been studied primarily with regard to secreted proteins (Coppe et al., 2008; Wiley et al., 2019b). Here we show that senescent cells also synthesize a number of oxylipins – a class of biologically active lipids that arise from oxygenation of polyunsaturated fatty acids (PUFAs). Oxylipins have diverse physiological effects, including inflammation, fever, vasoconstriction and vasodilation, pain, hair loss, asthma and fibrosis (Funk, 2001; Soberman and Christmas, 2003). Senescent cells are known to produce prostaglandin E2 (PGE2) (Cormenier et al., 2018; Dagouassat et al., 2013; Kabir et al., 2016; Martien et al., 2013; Yang et al., 2011; Zdanov et al., 2007), and the leukotriene-producing 5-lipoxygenase activity was shown to increase in replicative and oncogene-induced senescence, where it reinforced senescence-associated phenotypes by increasing reactive oxygen species (Catalano et al., 2005). We show that senescent human cells synthesize a much larger set of oxylipins, including PGD2 and its derivatives, 22-carbon PUFAs and monounsaturated fatty acids (MUFAs). Further, the dihomo-prostaglandins – less-studied derivatives of adrenic acid – are the primary prostaglandins produced by senescent cells, and one of these derivatives, dihomo-15d-PGJ2, is an intracellular biomarker of senescence and its release from cells is a hallmark of senolysis in culture and in vivo.
Results
Cellular senescence activates oxylipin biosynthesis
The senescence arrest and SASP entail significant changes in metabolism (Wiley and Campisi, 2016), including lipid metabolism (Buratta et al., 2017; Cadenas et al., 2012; Catalano et al., 2005; Flor et al., 2017; Quijano et al., 2012). To better understand these changes, we extracted intracellular lipids from cultured human fibroblasts (IMR-90) that were either proliferating (PRO, 10% serum), quiescent (QUI, 0.2% serum) or induced to senesce by ionizing radiation (IR), then cultured in either 10% or 0.2% serum. We also extracted lipids from mitochondrial dysfunction-associated senescent (MiDAS) cells (0.2% serum) (Wiley et al., 2016). Thus, we controlled for differences in growth state (QUI vs PRO vs SEN(IR) and MiDAS) and culture media (0.2% vs 10% serum). We measured the relative abundance of the extracted lipids by mass spectrometry.
The lipid profiles (Table S1) showed that certain subsets of lipids significantly increased or decreased upon senescence. Most notable were striking elevations in the relative abundance of oxylipins: a class of potent signaling lipids derived from 20- and 22-carbon fatty acids such as arachidonic (AA) and adrenic acid (AdA) (Figure 1A). We also observed changes in intracellular ceramides and other lipids (Table S1).
Figure 1. Senescent cells synthesize oxylipins.

A-B. IMR-90 cells were proliferative in 10% serum, made quiescent by incubation for 3 days in 0.2% serum, or made senescent by treatment with 10 Gy X-rays (IR) or ethidium bromide to induce mitochondrial-dysfunction induced senescence (MiDAS). Lipids were extracted from proliferating (PRO – 10%), quiescent (QUI – 0.2%), IR-induced senescent (SEN(IR) – 10% or 0.2% serum), or mitochondrial dysfunction-associated senescent (MiDAS – 0.2%) cells and analyzed by liquid chromatography combined with mass spectrometry (LC-MS). Putative oxylipins (A) and lipid precursors (B) were detected in control and senescent cells. Heat maps indicate the averages of at least 3 experiments (* = p<0.05, 1-way ANOVA). C. RNA was isolated from QUI, SEN(IR), MiDAS and vector or RAS-V12 expressing [SEN(RAS)] cells, reverse transcribed, and mRNAs encoding oxylipin synthesis genes were measured by quantitative (q) PCR. Heat maps indicate the averages of 3 experiments (* = p<0.05, 1-way ANOVA). D. p16–3MR mice were given a single dose (10 mg/kg) of doxorubicin (DOXO) or phosphate-buffered saline (PBS) by intraperitoneal (i.p.) injection. After 5 days, mice were given GCV (25 mg/kg) or vehicle by i.p. injection for 5 consecutive days. Livers were harvested on day 10 and analyzed for oxylipin synthase mRNAs by qPCR. Heat maps indicate averages of at least 5 mice (* = p<0.05, 1-way ANOVA). E. IMR-90 fibroblasts were induced to senesce by MiDAS, IR, or RAS-V12 overexpression. 10 days after induction of senescence, conditioned media were collected and analyzed for PGD2 by ELISA. F-G. HEPG2 (F) and HUVEC (G) cells were induced to senesce by IR. Conditioned media were collected 10 days after IR and analyzed for PGD2 by ELISA. Data show means of at least 3 experiments (* = p<0.05, ** = p<0.01, *** = p<0.001, unpaired, 2-tailed t test). H-I. RNA from (C) was analyzed for expression of SLCO2A1 or PGDH by qPCR. J. HUVECs were induced to senesce by IR, and analyzed at Day 10 for SLCO2A1 and PGDH as in H and I. See also Figure S1 and Table S1.
The most highly elevated senescence-associated prostaglandin was 1a,1b-dihomo-15-deoxy-Δ12,14-prostaglandin J2 (dihomo-15d-PGJ2). However, 1a,1b-dihomo-prostaglandin D2 (PGD2), 1a,1b-dihomo-prostaglandin E2 (PGE2), and other variants of prostaglandins PGD2, PGE2 and PGF2α also increased (Figure 1A). Additionally, senescent cells contained higher levels of specific leukotrienes, notably leukotriene B4 (LTB4) and the related lipoxygenase product 5-hydroxyeicosatetraenoic acid (5-HETE) (Figure 1A). Consistent with these findings, the 20-carbon oxylipin precursors arachidonic acid (AA), eicosapentanoic acid (EPA), and dihomo-gamma-linoleic acid (DGLA) were elevated in senescent cells (Figure 1B).
The 22-carbon lipid AdA, a product of AA elongation (Jump, 2009; Ohno et al., 2010) and precursor of dihomo prostaglandins (Sprecher et al., 1982), also increased in senescent cells (Figure 1B). Further, senescent cells contained higher levels of the 22-carbon PUFAs docosahexanoic acid (DHA) and docosapentanoic acid (DPA) and the 22-carbon MUFA erucic acid, but not the 22-carbon saturated fatty acid behenic acid. Additionally, incubating senescent cells with deuterated AA resulted in an increase in deuterated AdA, confirming elongation of AA to AdA by senescent cells (Figure S1A). Thus, senescent cells show a selective and marked increase in oxylipins and elongated PUFAs and MUFAs.
To understand the mechanism by which senescent cells accumulate these lipids, we used qPCR to measure mRNA levels of several genes that function in oxylipin biosynthesis. We compared cells induced to senesce by ionizing radiation [SEN(IR)], mitochondrial dysfunction (MiDAS) and RAS oncogene activation [SEN(RAS)] (Figure 1C). Among the mRNAs that increased significantly were those encoding prostaglandin synthases PTGS2 (COX-2), PTGDS, PTGES and TBXAS, but not PTGIS, which showed lower expression. mRNAs encoding leukotriene biosynthetic genes, including ALOX5 (5-LO), ALOX15, ALOX5AP, LTC4S and to a lesser extent LTA4H were similarly elevated. We observed similar qPCR results for oxylipin biosynthesis genes using human umbilical cord endothelial cells induced to senesce by IR [SEN(IR) HUVECs] (Figure S1B) and replicatively senescent [SEN(Rep)] IMR-90 fibroblasts (Figure S1C).
To determine whether oxylipin synthesis is elevated in senescent cells in vivo, we used a mouse model in which p16Ink4a-positive senescent cells can be eliminated by administration of ganciclovir (GCV) (p16–3MR mice) (Demaria et al., 2014). We treated p16–3MR mice with doxorubicin (DOXO) - an inducer of widespread senescence in mice (Demaria et al., 2017) - and measured mRNA levels of several oxylipin synthases in livers (Figure 1D). Much like cultured human cells, oxylipin synthase mRNAs increased significantly upon inducing senescence in vivo in mice. Further, eliminating senescent cells with GCV lowered both oxylipin synthase (Figure 1D) and p16Ink4a (Figure S1D) expression, confirming that the elevation was senescence-associated. Kidneys from aged mice also showed elevated levels of oxylipin synthase mRNAs (Figure S1E).
Since many oxylipins are secreted, we collected conditioned media from IMR-90 fibroblasts that were either quiescent or induced to senesce by mitochondrial dysfunction (MiDAS), ionizing radiation [SEN(IR)] or RAS oncogene activation [SEN(Ras)]. Using ELISAs, we measured the levels of PGD2 – a secreted prostaglandin that is also a precursor of 15d-PGJ2 (Figure 1E). All senescent cells showed elevated PGD2 secretion, although the ELISA does not distinguish dihomo-PGD2 from PGD2. The same was true for an epithelial-derived human cancer cell line (HEPG2 – Figure 1F) and primary human umbilical vein endothelial cells (HUVECs – Figure 1G) induced to senesce by IR. Notably, hydrocortisone – a glucocorticoid that prevents AA release from intracellular membranes by activating annexin A1 (Perretti and D’Acquisto, 2009), prevented the senescence-associated release of PGD2 (Figure S1F), suggesting that the primary source of AA for prostaglandin synthesis is likely intracellular membranes. Since glucocorticoids can extend the replicative lifespan of cultured cells (Rosner and Cristofalo, 1979) and attenuate the SASP (Laberge et al., 2012), one or more AA-derived oxylipins might prevent senescence and promote the SASP.
Prostaglandins are inactivated primarily by coordinated import by a prostaglandin transporter (gene name: SLCO2A1) coupled to NAD+-dependent oxidation of the 15(S)-hydroxyl group to an inactive 15-keto-prostaglandin metabolite by 15-hydroxy-prostaglandin dehydrogenase (PGDH) (Nomura et al., 2004). Consistent with increased intracellular levels of prostaglandins (Figure 1A), SLCO2A1 mRNA levels increased >100-fold in human fibroblasts induced to senesce by IR and mitochondrial dysfunction (Figure 1H) and >50-fold in RAS-induced senescent cells (Figure 1I). Smaller increases occurred during replicative senescence (Figure S1) and in IR-induced senescent HUVECs (Figure 1J). However, there was no commensurate increase in PGDH mRNA during any form of senescence (Figures 1H–1J, Figure S1H). Indeed, PDGH mRNA levels decreased in replicatively senescent fibroblasts and IR-induced senescent HUVECs (Figure 1J, Figure S1G). Thus, senescence entails an activation of several oxylipin-synthesis pathways, especially those that generate PGD2-related lipids (Figure S1H). This activation is likely coupled to increased import without inactivation by PGDH, resulting in intracellular accumulation of 15-deoxy-prostaglandins.
Dihomo-15d-PGJ2 is a biomarker of senolysis in culture and in vivo.
Since dihomo-15d-PGJ2 accumulated inside senescent cells (Figure 1A) and was not substantially secreted (Figure 2A–D), we hypothesized that senolysis -- the death of senescent cells – might release dihomo-15d-PGJ2 into the media of cultured cells and plasma of mice. To test this hypothesis, we cultured quiescent (QUI), mitochondrial dysfunction-associated senescent (MiDAS) and IR-induced senescent [SEN(IR)] IMR-90 fibroblasts in the presence of DMSO or the senolytic drug ABT-263 (10 μM) (Chang et al., 2016). After 24 hours, we collected conditioned media. At 10 μM, ABT-263 killed at least 90% of cells, regardless of senescence or quiescence. We then measured 15d-PGJ2 levels in the media by commercial ELISA - which detects both 15d-PGJ2 and dihomo-15d-PGJ2. Since the assay cannot distinguish between these lipids, we refer to the ELISA signal as “(dh)15d-PGJ2” to acknowledge that this signal may come from either lipid. Media from MiDAS cells (which have a low basal level of apoptosis) showed some (dh)15d-PGJ2, but this increased significantly when the cells were treated with ABT-263 (Figure 2A). Similarly, SEN(IR) media contained only small amounts of (dh)15d-PGJ2, but contained much more after ABT-263-mediated senolysis (Figure 2A).
Figure 2. (Dihomo)15d-PGJ2 is a biomarker of senolysis.

A. IMR-90 fibroblasts were induced to senesce by MiDAS or IR. 10 days after induction, cells were treated with DMSO or 10 μM ABT-263, and (dihomo)15d-PGJ2 [(dh)-15d-PGJ2] was measured in conditioned media by ELISA. Data reflect means of at least 3 experiments. B-C. HEPG2 (B) or HUVEC(C) cells were induced to senesce by IR, and conditioned media from DMSO or ABT-263 treated cells were generated as in (A). (dh)15d-PGJ2 was measured by ELISA. D. Representative overlay of dihomo-15d-PGJ2 peaks from extracted ion chromatograms of lipids from media treated as in A. E-I. C57BL/6 mice were injected intraperitoneally with doxorubicin (DOXO; 10 mg/kg) or PBS. 6 weeks later, ABT-263 (50 mg/kg) or vehicle were administered by gavage. Data are presented as box plots for at least 6 mice per condition. E-F. Epididymal fat pads were harvested 3 hours after ABT-263 administration and analyzed for p16INK4a (E) or p21WAF1 (F) mRNA levels by qPCR. G. 3 hours after gavage, blood was collected by cardiac puncture, and (dh)15d-PGJ2 was measured by ELISA. H. 12 hours after gavage, urine was collected, and (dh)15d-PGJ2 was measured by ELISA. I. Representative overlay of dihomo-15d-PGJ2 peaks from extracted ion chromatograms of lipids extracted from plasma of the animals from (G). * = p<0.05, ** = p<0.01, *** = p<0.001, 1-way ANOVA. See also Figure S2.
To determine whether (dh)15d-PGJ2 predicted senolysis in other cell lineages, we measured its release upon ABT-263-induced senolysis in senescent HEPG2 and HUVEC cells. In each case, senolysis increased (dh)15d-PGJ2 (Figures 2B and 2C). Senescent HUVECs showed some (dh)15d-PGJ2 when vehicle-treated (Figure 2C), but, since some HUVECs detach from the plate and undergo anoikis during routine culture, this elevation was expected. Since the ELISA antibody was raised against 15d-PGJ2, not specifically dihomo-15d-PGJ2, we used mass spectrometry to determine whether dihomo-15d-PGJ2 was released by senescent cells. We assayed conditioned media from QUI+DMSO, SEN(IR)+DMSO, QUI+ABT-263, and SEN(IR)+ABT-263 cells. In agreement with Figure 1A and the ELISA data, we detected substantially elevated dihomo-15d-PGJ2 only in SEN(IR)+ABT-263 cells (Figure 2D). By comparison, non-senescent IMR-90s did not release dihomo-15d-PGJ2 when treated with non-selective apoptosis inducers (staurosporine or TRAIL) (Figure S2A), indicating that dihomo-15d-PGJ2 is a distinct feature of senolysis in culture.
To determine whether dihomo-15d-PGJ2 could be used to detect senolysis in vivo, we treated mice with DOXO and allowed them to accumulate senescent cells over 6 weeks, as described (Demaria et al., 2017). We then administered ABT-263 (50 mg/kg) by gavage (Chang et al., 2016). After 3 hours, mRNA levels of p16Ink4a and p21Waf1 declined significantly in the epididymal fat of ABT-263-treated mice (Figures 2E and 2F). Further, ELISAs showed elevated (dh)15d-PGJ2 in the plasma of DOXO + ABT-263 treated mice, but not control mice (Figure 2G). Similarly, 12 hours after ABT-263-induced senolysis, urine (dh)15d-PGJ2 was elevated relative to controls (Figure 2H). Ion chromatograms from mouse plasma from DOXO + ABT-263 mice revealed the 343m/z peak indicative of dihomo-15d-PGJ2 (Figure 2I; Figure S2B), which was absent from plasma from PBS + Vehicle, PBS + ABT-263 and DOXO + Vehicle mice (Figure 2I, Figure S2C). We did not detect a (non-dihomo)15d-PGJ2 peak in plasma from any treatment group examined (Figure S2D). We conclude that senolysis is accompanied by the release of dihomo-15d-PGJ2 from senescent cells in vivo.
Dihomo-15d-PGJ2 was highly elevated and abundant (~1–5 μM) in senescent cells - but is considerably unstudied (detected in one published list of analytes) (Lee et al., 2016). To confirm its identity, we obtained a synthetic dihomo-15d-PGJ2 standard (Jubilant Chemsys; 94% purity) (Figure S3A, Figure S3B), and used it to confirm the identity of the senolysis biomarker. Dihomo-15d-PGJ2 differs from 15-PGJ2 by an additional C2H4 group, resulting in a mass shift of 28 Da (from 315.2 to 343.2 m/z) (Figure S2B). In addition, the MS/MS fragmentation patterns of the standard (Figure 3A) revealed peaks at 299.2 m/z (loss of CO2), 283.3 m/z (loss of CO2 + H2O), 255.2 (loss of CO2 + H2O + C2H4), and 233.2 (loss of H2O + C7H8). Retention times between the dihomo-15d-PGJ2 standard and samples from senolytic-treated culture or plasma samples closely matched (Figure 3B, filled peaks). Samples also showed a second peak at a later time that was not specific for senolysis (Figure 3B, unfilled peaks). Analysis of the senolysis-specific peaks by MS/MS fragmentation revealed characteristic peaks at 283.3 mz, 255.2 m/z and 233.2 m/z (Figure 3C), as well as the 299.2 peak (presented separately in Figure 3D). Together, these peaks confirmed that dihomo-15d-PGJ2 was the senolysis-released lipid detected in our analyses.
Figure 3. Validation of dihomo-15d-PGJ2 as a biomarker of senolysis.

A. A synthesized dihomo-15dPGJ2 standard was analyzed at 3 different voltages for fragmentation patterns. Key identification peaks included 299.2 m/z (C21H31O−: green box), 283.2 m/z (C21H31−: blue box), 255.2 (C19:H27−: red box), 233.2 (C15H21O2−: purple box). B. Extracted ion chromatograms from a dihomo-15d-PGJ2 standard (top), plasma from a DOXO+ABT-263 mouse (middle), and senescent cell extracts (bottom). Filled peaks appeared only in senolysis (middle) or senescence (bottom). C. Fragmentation patterns of filled peaks from B indicating a dihomo-15d-PGJ2 standard (top), plasma from a DOXO+ABT-263 mouse (middle), or senescent cell extracts (bottom). D. Zoomed chromatograms for the 299.2 m/z peak (zoomed area indicated by green boxes on Figure 3C) in DOXO + ABT-263 mouse plasma (upper) or senescent cell extracts (lower). Note that relative abundance of a non-specific peak (unfilled in Figure 3B) required earlier fragmentation in SEN(IR) extracts. See also Figure S3.
Prostaglandins promote senescent phenotypes
To understand the consequence of senescence-associated oxylipin biosynthesis, we considered that cyclooxygenase 2 (Ptgs2) overexpression causes premature aging phenotypes in mice (Kim et al., 2016), and oxylipin synthesis inhibitors extend male mouse life span (Strong et al., 2008). To determine whether prostaglandin synthesis promotes senescent phenotypes, we treated irradiated cells [SEN(IR)] with either of two prostaglandin synthase 2 (PTGS2/COX-2) inhibitors: CAY-10404 (CAY) or NS-398 (NS), which delays replicative senescence (Han et al., 2004). Both inhibitors suppressed secreted PGD2 levels, as determined by ELISA (Figure 4A). Notably, when counting cells for normalization (Figure 4A), SEN(IR) cells exposed to either inhibitor showed increased cell number relative to control cells (Figure S4A). This finding suggests that prostaglandin biosynthesis might reinforce the senescence proliferative arrest.
Figure 4. Prostaglandins promote cellular senescence phenotypes.

A-F. IMR-90 fibroblasts were induced to senesce by IR and treated with DMSO or COX-2 inhibitors CAY-10404 (CAY) or NS-398 (NS) for 10 days. A. Conditioned media were harvested and secreted PGD2 was measured by ELISA. B. 24 h EdU incorporation for irradiated cells in each treatment group. C. SA-Bgal positivity for irradiated cells in each treatment group. D. p21 mRNA levels in each treatment group. E. p16 mRNA levels from each treatment group. F. Conditioned media from (A) were analyzed for IL-6 secretion by ELISA. G. mRNA levels of genes encoding SASP factors from all treatment groups were measured by qPCR. H-N. Proliferating cells were cultured for 10 days in the presence of DMSO or 5 μM 15d-PGJ2 or dihomo-15d-PGJ2. H. 24 h EdU labeling indices for each treatment. I. Example of SA-Bgal positive cells after each treatment. Panels are cropped zooms to larger images, and red saturation has been lowered for all images in order to allow visualization of the blue x-gal staining. J. SA-Bgal positivity for each treatment. K. p21 mRNA levels in extracts from prostaglandin- and DMSO-treated cells. L. p16INK4a mRNA levels in extracts from prostaglandin-and DMSO-treated cells. M. LMNB1 mRNA levels in extracts from prostaglandin- and DMSO-treated cells. N. SASP factor PTGS2, PTGDS and PTGES mRNA levels were measured by qPCR. Normalized to actin. O. Cells were transduced with lentiviruses expressing shRNA targeting either GFP (shGFP; control) or PTGDS (shPTGDS), irradiated as in (A), and mRNA extracted 10 d after irradiation. SASP gene expression was measured by qPCR. Bar graphs indicate averages of at least 3 experiments (* = p<0.05, ** = p<0.01, *** = p<0.001, unpaired, 2-tailed t test). Heat maps indicate averages of 3 experiments (* = p<0.05, 2-way ANOVA). See also Figure S4.
To address this possibility, we irradiated IMR-90 fibroblasts with a lower IR dose (5 Gy vs 10 Gy), which causes a senescence growth arrest in 90% vs >99% of cells, and treated them with CAY or NS. CAY and, to a greater degree NS, significantly increased EdU incorporation (Figure 4B), decreased senescence-associated beta-galactosidase (SA-Bgal) positivity (Figure 4C) and decreased both CDKN1A (p21) and CDKN2A (p16INK4a) mRNA levels (Figures 4D and 4E).
Most prostaglandins and leukotrienes are secreted and many can promote cell proliferation and inflammation (Castellone et al., 2005; Dennis and Norris, 2015; Ricciotti and FitzGerald, 2011). Thus, the senescence-associated oxylipins we detected might drive the inflammatory arm of the SASP. To test this idea, we inhibited prostaglandin synthesis with CAY or NS in SEN(IR) cells and measured secreted levels of the proinflammatory SASP factor interleukin 6 (IL-6) (Figure 4F). Both compounds reduced secreted IL-6 levels (Figure 4F).
To assess effects on other SASP factors, we extracted mRNA from each treatment group and measured gene expression by qPCR. COX-2 inhibition by either compound decreased mRNA levels of all the inflammatory cytokines assayed, as well as MMP3, but had no effect on VEGF – part of a pro-angiogenic subset of SASP factors (Figure 4G). COX-2 inhibition similarly affected cell number, IL-6 secretion, EdU incorporation and colony formation in oncogenic RAS-induced senescent cells (Catalano et al., 2005; Serrano et al., 1997) (Figures S4B–S4F). Thus, prostaglandin synthesis promotes phenotypes associated with senescence, including cell cycle arrest and the expression and secretion of certain SASP factors.
Dihomo-15d-PGJ2 markedly increased in senescent cells, suggesting this lipid and/or similar cyclopentenone prostaglandins might drive senescence phenotypes. To test this idea, we treated non-senescent cells with 15d-PGJ2 or dihomo-15d-PGJ2 and measured markers of senescence. Both lipids reduced EdU incorporation (Figure 4H) and LMNB1 mRNA (Figure 4M), increased SA-Bgal activity (Figure 4I–4J) and CDKN1A/p21 mRNA levels (Figure 4K). Only dihomo-15d-PGJ2 increased CDKN2A/p16 mRNA (Figure 4L). Further, although both lipids induced several SASP factor mRNAs, dihomo-15d-PGJ2 induced higher levels of these mRNAs than 15d-PGJ2 (Figure 4N). Both lipids also induced PTGS2/COX-2, PTGES, and PTGDS mRNAs, suggesting a positive feedback loop maintains prostaglandin synthesis and senescence phenotypes. PGD2 and PGJ2 had similar effects on senescence phenotypes (Figures S4G–S4L), as did PGE2 to a lesser degree, as reported (Martien et al., 2013). In agreement with positive feedback by prostaglandins, PTGS2/COX-2 expression declined upon inhibition of COX-2 with NS or CAY (Figure S4M). Phenotypes induced by PGE2 were unlikely due to senescence, as cells treated with PGE2 - but not PGD2 or PGJ2 - became smaller and resumed proliferation once PGE2 was removed (Figures S4N–S4O). Thus, PGD2 and related metabolites can promote senescence.
To further test this idea, we depleted prostaglandin D synthase (PTGDS) by lentiviral-delivered shRNA (shPTGDS), and measured mRNA levels of SASP factors following induction of senescence by IR. One shRNA efficiently reduced PTGDS expression (Figure S4P), and also reduced several SASP factor mRNAs compared to senescent cells transduced with a control shRNA (shGFP) (Figure 4O). Together with Figures 4H–4N, we conclude that PGD2 and related lipids promote cellular senescence phenotypes.
Extracellular PGD2 signals through two receptors, DP1 (PTGDR) and DP2 (PTGDR2) (Hata and Breyer, 2004; Ricciotti and FitzGerald, 2011); 15d-PGJ2 preferentially binds DP2 (Monneret et al., 2002; Ricciotti and FitzGerald, 2011). To determine if either receptor drives senescence, we treated non-senescent cells with PGD2, a DP1-specific agonist (BW245C) (Town et al., 1983), a DP2-specific agonist (13,14-dihydro-15-keto-PGD2) (Hirai et al., 2001), both agonists or vehicle (DMSO). The DP1 agonist increased cAMP levels, while the DP2 agonist lowered cAMP levels (Figure S5A), confirming receptor activation (Hata and Breyer, 2004; Hirai et al., 2001; Hirata et al., 1994; Ricciotti and FitzGerald, 2011). PGD2 had no effect on cAMP levels (Figure S5A). Both the DP1 agonist and PGD2 reduced EdU incorporation, but only PGD2 elevated SA-Bgal activity (Figure S5B). PGD2 also increased mRNAs encoding p21 and p16INK4a (Figure S5C and D), whereas the DP1 agonist lowered these mRNA levels. The DP2 agonist had no effect. Further, although PTGDR mRNA levels were unchanged between senescent and non-senescent cells, PTGDR2 mRNA was ~50-fold lower in senescent cells, and thus unlikely to affect senescence (Figure S5D). PGD2 induced many mRNAs encoding SASP factors, but the DP1 agonist did not (Figure S5F). Finally, we treated cells with either a DP1 (BW-A868C) (Giles et al., 1989) or DP2 (OC000459) (Pettipher et al., 2012) antagonist for 10 days after inducing senescence by IR. Neither antagonist altered SASP gene expression (Figure S5G). These data indicate that PGD2 and its derivatives do not drive senescence through classic prostaglandin D2 receptor signaling, consistent with requirements for μM concentrations (these receptors typically require nM concentrations). We therefore sought to identify alternative pathways.
15d-PGJ2 is the dehydration product of PGD2 (Forman et al., 1995; Kliewer et al., 1995), and dihomo-15d-PGJ2 is the most highly elevated prostaglandin inside senescent cells (Figure 1A), we therefore tested the possibility that intracellular dihomo-15d-PGJ2 promotes senescence, independent of cell surface receptor signaling. 15d-PGJ2 is a ligand for PPAR-γ (Forman et al., 1995; Kliewer et al., 1995), which is linked to senescence (Gan et al., 2008). We transduced IMR-90 fibroblasts with a baculovirus containing a luciferase PPAR-γ reporter and measured its activity in quiescent and SEN(IR) cells cultured in DMSO or the prostaglandin synthase inhibitor NS-398. PPAR-γ reporter activity increased ~18 fold upon senescence, and NS-398 lowered this activity by ~50% (Figure S5H), indicating that PPAR-γ activation is partially dependent on prostaglandin synthesis in senescent cells. We then cultured cells with two PPAR-γ agonists -- ciglitazone (20 μM) or troglitazone (20 mM) -- for 10 days. Unlike 15d-PGJ2, neither agonist induced senescence, as determine by EdU incorporation and SA-Bgal activity (Figure S5I). Therefore, while prostaglandin synthesis indeed activates PPAR-γ in senescent cells, PPAR-γ does not drive senescence phenotypes.
Prostaglandins reinforce senescence via positive RAS/p53 feedback
H-RAS is activated by exogenous 15d-PGJ2 at 3–5 μM (Oliva et al., 2003) – roughly the dihomo-15d-PGJ2 level in senescent cells, and other cyclopentenone prostaglandins activate other RAS isoforms or similar GTPases (Anta et al., 2016; Renedo et al., 2007; Wall et al., 2015). We hypothesized that constitutive RAS activation might drive senescence in response to the elevated dihomo-15d-PGJ2. Indeed, incubation of quiescent cells with 15d-PGJ2 resulted in RAS activation, as well as phosphorylation of the downstream effector kinases ERK1/2 (Figure 5A; left lanes) (Oliva et al., 2003). Consistent with elevated 15d-PGJ2 levels, SEN(IR) cells showed increased RAS activation (RAS:GTP) and phospho-ERK1/2, which was attenuated by CAY or NS (Figure 5A). Similarly, shRNA-mediated knockdown of PTGDS (shPTGDS) selectively prevented RAS activation (Figure 5A). RAS activation is also associated with loss of intracellular dNTPs due to suppression of RRM2 expression (Aird et al., 2013). A similar loss of RRM2 occurred in IR-induced senescence, which was prevented by CAY or NS (Figure 5B). Importantly, administration of deuterated 15d-PGJ2 resulted in roughly equimolar accumulation of deuterated 15d-PGJ2 inside senescent cells (Figure S5J), demonstrating that exogenously-administered prostaglandins can accumulate intracellularly at levels that approximate those found in senescent cells.
Figure 5. 15d-PGJ2 promotes senescent phenotypes by activating RAS.

A. Extracts from IMR-90 fibroblasts were analyzed for activated RAS (Ras:GTP), total Ras, phospho-ERK1/2, total ERK1/2, and actin by immunoblot. (Left) Quiescent cells were treated with DMSO or 5 μM 15d-PGJ2 for 15 minutes before harvest. (Right) Cells were induced to senesce by 10 Gy IR, and treated with DMSO, CAY-10404, NS-398, or lentiviral shRNAs to GFP (shGFP) or PTGDS (shPTGDS). Cells were cultured for 10 days prior to harvest, and in 0.2% FBS for the last 48 hours. B. Cells were induced to senesce by 5 Gy IR, and cultured in the presence of vehicle (DMSO), CAY or NS for 10 d. RNA was extracted, reverse-transcribed, and analyzed for RRM2 mRNA relative to actin mRNA by qPCR. C. Immunoblot of cells treated with DMSO or 10 μM 15d-PGJ2 for 10 days. Blots were probed for phospho-PRAK-T182, total PRAK, phospho-p53-S37, and total p53, p21 and actin. D. Cells were treated with vehicle or 15d-PGJ2, and DMSO or AZ 628 for 10 d. Protein was extracted and analyzed by immunoblot for phospho-ERK1/2, total ERK1/2, phospho-p53-S37, and total p53, p21 and actin. E. Cells treated as in D were analyzed for SA-Bgal or 24 h EdU incorporation. F. Cells were induced to senesce by 10 Gy IR or continuous culture with 10 μM 15d-PGJ2 for 10 days. DMSO or AZ 628 were concurrently added from Day 0 and present until day 10. Quiescent cells cultured in DMSO served as a control. RNA was then extracted and analyzed by qPCR for mRNAs encoding SASP factors, PTGS2 and PTGDS. G-H. Cells were transduced with lentiviral shRNAs to either GFP (shGFP) or p53 (shp53), mock irradiated or given 10 Gy IR and cultured for 10 days (shGFP) or 3 days (shp53) before RNA extraction and analysis for (G) PTGS2 or (H) PTGDS mRNAs. I. Model for a feedback loop involving 15d-PGJ2, RAS and p53 that reinforces senescent phenotypes. Chemiluminescent western images were captured with either film or by Bio-rad Gel-Doc. Image formats may therefore be inconsistent between lanes within panels. Bar graphs indicate averages of at least 3 experiments (*** = p<0.001, unpaired, 2-tailed t test). Heat maps indicate averages of 3 experiments (* = p<0.05, 2-way ANOVA). See Also Figure S5.
RAS-induced senescence is driven by a mitogen activated kinase (MAPK) cascade, resulting in p53 phosphorylation on serine 37 (p53-S37) by p38-regulated activated kinase (PRAK/MAPKAPK5) (Sun et al., 2007). Indeed, PRAK was phosphorylated on threonine 182 (PRAK-R182), p53 was phosphorylated on serine 37 and p21 protein levels increased in cells induced to senesce by 15d-PGJ2 (Figures 5C–5D). These data suggest that 15d-PGJ2 reinforces senescence though a RAS-driven pathway.
H-RAS knockdown was toxic, so we used instead a RAF inhibitor (AZ 628) (Montagut et al., 2008) to block downstream RAS signaling. AZ 628 (20, 5 or 1 μM) strongly inhibited ERK phosphorylation in response to serum stimulation without affecting basal levels of p53 or p21 (Figure S5K). Further, 1 μM AZ 628 did not alter SA-b-gal activity (Figure S5L–S5M) or EdU labeling (Figure S5N). We therefore used 1 μM for further studies. In response to 15d-PGJ2, AZ 628 blocked ERK1/2 and p53 phosphorylation, thus preventing the rise in p53 and p21 proteins (Figure 5D). AZ 628 also prevented the rise in SA-Bgal and decline in EdU labeling in response to 15d-PGJ2 (Figure 5E). Finally, continuous culture of either SEN(IR) or 15d-PGJ2-treated cells with AZ 628 prevented much of the rise in proinflammatory SASP factor mRNAs, lowered levels of both PTGS2 and PTGDS mRNAs, and thus antagonized the positive feedback initiated by 15d-PGJ2 (Figure 5F). AZ 628 also prevented the rise in mRNAs encoding p21 and p16INK4a and reduction in LMNB1 mRNA in response to 10 Gy IR (Figures S5O–S5Q). Thus, activation of the RAS pathway is a key step in the development of senescent phenotypes, even in the absence of activating mutations.
Since 15d-PGJ2 activated p53 (S37 phosphorylation), and COX-2/PTGS2 inhibitors lowered PTGS2 mRNA levels in senescent cells (Figure S4M), we tested the possibility that p53 is required for the increased expression of prostaglandin synthase enzymes. Indeed, p53 depletion by a lentiviral shRNA (shp53) completely abolished PTGS2 and PTGDS expression in response to IR (Figures 5G–5H). CDKN1A/p21 expression was similarly attenuated, as expected (Figure S5R). By comparison, depletion of the NF-κB subunit RELA using a lentiviral shRNA (shRELA) had no effect on prostaglandin synthase expression (Figures S5S–S5T), despite lowering IL6 expression (Figure S5U), as described (Freund et al., 2011). Thus, activation of p53 or RAS results in a positive feedback loop that promotes senescence and segments of the SASP by activating prostaglandin synthesis (Figure 5I). Given that prostaglandin synthesis is required for senescence, the data indicate that prostaglandin-mediated RAS activation is a common and necessary feature of at least some forms of cellular senescence.
Discussion
Our results indicate that oxylipins are a lipid-based component of the SASP. Secretion of these lipids likely has significant physiological effects, apart from the cell-autonomous role of dihomo-15d-PGJ2. For example, chorioamnion-derived PGE2 and PGF2α are key signals for parturition (Gibb, 1998) and senescent cells appear in the chorioamniotic membranes of women in labor (Behnia et al., 2015; Menon et al., 2016), suggesting that senescent cells may be a source of parturition-inducing prostaglandins.
Importantly, we observe intracellular accumulation of cyclopentenone prostaglandins, such as dihomo-15d-PGJ2. This accumulation was linked to increased expression of prostaglandin transporter, but not prostaglandin dehydrogenase, which might explain the accumulation of 15-deoxy prostaglandins. Increased import without a commensurate change in PGDH might promote cytoplasmic accumulation of dehydrated 15-deoxy prostaglandins, rather than 15-keto prostaglandins.
Given that elevated intracellular dihomo-15d-PGJ2 and its biosynthetic enzymes occurs in several cell types and tissues in response to multiple senescence inducers, we propose that the synthesis of dihomo-15d-PGJ2 and RAS activation are common features of senescent cells. While oncogenic RAS mutations are known to drive cells into senescence (Serrano et al., 1997), our studies identify a role for wild type RAS in senescence. We show that the RAS-RAF signaling pathway is important for many aspects of senescence, including components of the SASP, increased levels of p21 and p16INK4a, and decline in lamin B1 levels.
We previously showed that p53 restrains segments of the SASP by antagonizing NF-kB signaling (Freund et al., 2011; Wiley et al., 2018). Here, we show that p53 is required for prostaglandin biosynthesis, and inhibition of prostaglandin synthesis antagonizes the SASP. These findings explain why p53 does not completely prevent the SASP. Thus, inhibition of prostaglandin synthesis did not return inflammatory cytokine levels to those of non-senescent cells, unlike RELA depletion, suggesting that prostaglandins contribute to, but are not exclusively necessary for, the secretion of proinflammatory factors. The role of p53-dependent prostaglandin synthesis in development of the SASP is likely overshadowed by the loss of NF-kB antagonism when p53 is depleted, resulting in increased inflammatory cytokine secretion in the absence of p53, even when prostaglandin synthesis is lost.
Senescence and the SASP are dynamic phenotypes that change over time (De Cecco et al., 2019; Hernandez-Segura et al., 2017). The lipids described here likely undergo dynamic regulation as well. Indeed, we recently showed that expression of specific lipid synthases is temporally regulated in the context of pulmonary fibrosis, suggesting a transition from acute to more chronic stress phenotypes (Wiley et al., 2019a).
Finally, our finding that released dihomo-15d-PGJ2 can be used as a biomarker for senolysis has several potential applications. Senolytic drugs are increasingly being used in aging and related research and – importantly – have entered early clinical trials (Hickson et al., 2019; Justice et al., 2019). Determining that senolysis is taking place is essential for evaluating these compounds as therapeutic agents. Detection of dihomo-15d-PGJ2 in biological fluids may allow rapid evaluation of the efficacy of these compounds.
Limitations of the study.
Outside of dihomo-15d-PGJ2, most of the factors in Figure 1A and 1B are assigned by m/z, and therefore are only likely assignees. We list these factors in the figure legend as “putative” as a result. This study only features analyses from mice and human cells, and for 3 cell types as representatives of endothelial, epithelial, and mesenchymal lineages. Results may not reflect all lineages or inducers of senescence.
STAR Methods
RESOURCE AVAILABILITY.
Lead contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Judith Campisi (jcampisi@buckinstitute.org).
Material Availability.
This study did not generate unique reagents. The schematic for synthesis of dihomo-15d-PGJ2 will be provided upon request.
Data and Code Availability.
Lipid profiling data is available at the NIH Common Fund’s National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench (https://www.metabolomicsworkbench.org), Project ID #: PR001093. See also the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-PRAK-T182 | Peiqing Sun - Wake Forest Univeristy | |
| Phospho-p53 (Ser37) Antibody | Cell Signaling Technology | #9289 |
| p21 Waf1/Cip1 (12D1) Rabbit mAb | Cell Signaling Technology | #2947 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Antibody | Cell Signaling Technology | #9101 |
| p44/42 MAPK (Erk1/2) Antibody | Cell Signaling Technology | #9102 |
| p38 MAPK Antibody | Cell Signaling Technology | #9212 |
| Anti-Lamin B1 antibody | Abcam | ab16048 |
| Anti-HMGB1 antibody | Abcam | ab18256 |
| Monoclonal Anti-β-Actin antibody | Sigma | A2228 |
| p53 Antibody (DO-1) | Santa Cruz Biotechnology | sc-126 |
| Bacterial and Virus Strains | ||
| shGFP lentivirus | Campisi Lab | |
| shP53 lentivirus | Campisi Lab | |
| shPTGDS lentivirus | Sigma-Aldrich | SHCLNV-NM_000954 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NS-398 | Cayman Chemical Company | 70590 |
| CAY-10404 | Cayman Chemical Company | 70210 |
| Ciglitazone | Cayman Chemical Company | 71730 |
| Troglitazone | Cayman Chemical Company | 71750 |
| Staurosporine | Cayman Chemical Company | 81590 |
| Recombinant Human TRAIL protein | Abcam | ab78818 |
| Arachidonic Acid-d11 | Cayman Chemical Company | 10006758 |
| Prostaglandin A2 | Cayman Chemical Company | 10210 |
| Prostaglandin D2 | Cayman Chemical Company | 12010 |
| Prostaglandin E2 | Cayman Chemical Company | 14010 |
| Prostaglandin F2α | Cayman Chemical Company | 16010 |
| Prostaglandin J2 | Cayman Chemical Company | 18500 |
| 15-deoxy-Δ12,14-Prostaglandin J2 | Cayman Chemical Company | 18570 |
| 1a,1b-dihomo-15-deoxy-Δ12,14-Prostaglandin J2 | Jubilant Chemsys | P1171 |
| 13,14-dihydro-15-keto Prostaglandin D2 | Cayman Chemical Company | 12610 |
| BW A868C | Cayman Chemical Company | 12060 |
| OC000459 | Cayman Chemical Company | 12027 |
| BW 245C | Cayman Chemical Company | 12050 |
| Hydrocortisone | Cayman Chemical Company | 20739 |
| Acetic Acid, glacial | VWR | VW0125–3 |
| Acetonitrile | VWR/BDH | BDH83639.400 |
| Ammonium acetate | Sigma Aldrich | 73594–25G-F |
| LCMS Water | Honeywell Burdick & Jackson | LC365–4 |
| Methanol | Honeywell Burdick & Jackson | LC230–4 |
| Chloroform | JT Baker | 9175–2 |
| Critical Commercial Assays | ||
| Baculoviral PPAR-gamma reporter assay | Signosis | BT-0014 |
| Senescence Detection Kit | BioVision | K320 |
| 15-deoxy-Δ12,14-PGJ2 ELISA Kit | ENZO | ADI-900–023 |
| Prostaglandin D2-MOX Express ELISA Kit | Cayman Chemical Company | 500151 |
| IL-6 AlphaLISA | PerkinElmer | AL223F |
| Ras Pull-down Activation Assay Biochem Kit | Cytoskeleton, Inc | BK008-S |
| Experimental Models: Cell Lines | ||
| HEPG2 | ATCC | HB-8065 |
| IMR-90 | ATCC | CCL-186 |
| HUVEC-C | ATCC | CRL-1730 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory | 000664 |
| p16–3MR | Campisi Laboratory | |
| Teklad global 18% protein | Envigo Teklad | 2918 |
| Oligonucleotides | ||
| See Table S2 | ||
| Software and Algorithms | ||
| MassHunter | Agilent | |
| Other | ||
| Kinetex 5μm EVO C18 100Å 150x2.1mm | Phenomenex | 00F-4633-AN |
| Data accessibility | ||
| Lipid Profiling Dataset | National Metabolomics Data Repository (NMDR) | PR001093 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS.
Cell lines and strains.
All cells were purchased from the American Type Culture Collection (ATCC), see Key Resource Table. IMR-90 human female fetal lung fibroblasts were the primary cell type used in this study, and were used prior to 40 population doublings - other than replicative senescent cells, which were cultured until replicative exhaustion. HEPG2 human adolescent male hepatocellular carcinoma are immortalized, but p53-positive, and therefore no population doublings were recorded. IMR-90 and HEPG2 cells were cultured in Dulbecco’s modified eagle medium (DMEM 4.5 g/L glucose, without sodium pyruvate - Gibco) supplemented with 10% FBS and penicillin/streptomycin (Gibco). HUVEC human female umbilical vein endothelial cells were cultured using the ATCC protocol and culture media and assayed within 40 population doublings. Quiescence was induced by replacing culture media with media containing 0.2% FBS for 72 h before analysis. All cells were cultured at 37°C and 3% O2. All cells were mycoplasma free.
Animals.
Animal experiments were conducted using a protocol approved by the Institutional Animal Care and Use Committee of the Buck Institute. Mice were housed in alternating 12:12 hour light/dark cycles at 68–72°F. General animal health rounds were performed daily, 7 days a week by trained Vivarium staff members. Any noted illness or deaths were reported via the Health Surveillance system to the Vivarium Director and/or the Attending Veterinarian (AV). The Buck Vivarium is an SPF barrier facility and routine health screening is done facility-wide to ensure the health of all colonies. Sentinel mice that were exposed to soiled bedding and byproducts from other mice on the same rack were screened quarterly for common murine viruses as directed by the AV. Internal and external parasitology screening was also done on all sentinel mice. For DOXO treatments, 10–16 wk old male p16–3MR transgenic mice on a C57BL/6 background received one intraperitoneal (i.p.) injection of 10 mg/kg of doxorubicin hydrochloride in PBS, and treated 5 d later with GCV or vehicle. GCV was administered via daily i.p. injections for 5 consecutive days at 25 mg/kg in PBS. Control mice were injected with an equal volume of PBS. Mice were euthanized and tissues collected 10 d after DOXO challenge. For aging studies, 8 male and 8 female C57BL/6 mice were aged for 6 or 24 mo, at which point mice were euthanized and tissues collected for analysis. For the biomarker studies, 10 male C57BL/6 mice per cohort with challenged with DOXO as above, and given either vehicle or ABT-263 6 weeks after DOXO challenge. Whole blood was collected by cardiac puncture into EDTA-tubes (lavender caps – BD Biosciences) and spun at 2000g for 5 minutes. For urine collection, mice were placed in plastic containers prior to euthanasia.
METHOD DETAILS.
Gene expression.
RNA was extracted from cells or tissues using commercially available kits (Isolate II - Bioline for cells; Direct-zol - Zymo for tissues) according to the manufacturer’s instructions. cDNA synthesis was performed using a High Capacity cDNA Reverse Transcription Kit (Thermo Fisher) according to the manufacturer’s instructions. Quantitative PCR was performed on a LightCycler 480 II (Roche) using primers and probes designed for the Universal Probe Library. Primers and probes used for human transcripts are listed in Table S2.
Induction of senescence.
Senescence SEN(IR) was induced by either 5 or 10 Gy ionizing-irradiation (IR). Non-senescent controls (proliferating or quiescent) were placed in the irradiator for an identical period of time but without irradiation. Oncogene-induced senescence was induced using lentiviral-mediated overexpression of HRASV12, as described (Wiley et al., 2016), and the lentiviral backbone (Vector) served as a control. MiDAS was induced by depleting mitochondrial DNA in the presence of 100 ng/mL ethidium bromide, 100 ug/mL sodium pyruvate, and 50 ug/mL uridine for 2 months, followed by removal of pyruvate, as described (Wiley et al., 2016).
Generation of conditioned media.
Conditioned media (CM) were generated by culturing cells in appropriate serum-free media supplemented with penicillin/streptomycin for 24 h before harvest, followed by clarification by centrifugation. Cells were then trypsinized and counted. All quantitative assays (e.g. ELISAs) from CM were normalized to cell number.
Induction of apoptosis or senolysis.
Non-senescent or SEN(IR) cells were cultured in the presence of 10 μM ABT-263 for 24 h. Non-senescent IMR-90 fibroblasts were treated with either 1 μM staurosporine (Cayman) or 100 ng/mL recombinant TRAIL (Abcam) for 24 h before collection. Media were centrifuged twice at 5000xg for 15 minus at 4°C for clearing of apoptotic bodies and stored at −80°C until analysis.
Senescence-associated beta-galactosidase.
SA-Bgal activity was detected as described (Dimri et al., 1995) using a commercial kit (Biovision).
Colony forming assay.
Ras-transduced senescent cells were plated at a density of 1000/well and cultured in the presence of growth media containing either DMSO, CAY-10404, or NS-398 for 7 days. Cells were washed in PBS, fixed in 95% methanol, and incubated for 30 min in 0.2% crystal violet in 2% methanol. Crystal violet was removed and wells were washed in tap water until the solution ran clear, and allowed to dry.
RAS activation assay.
Active RAS (RAS:GTP) was immunoprecipitated from culture lysates using a commercial kit (Cytoskeleton, Inc) following the manufacturer’s instructions. Precipitates were analyzed by immunoblot.
Immunoblots.
Cells were lysed in 5% SDS in 10 mM Tris, pH 7.4, and protein content determined by BCA assay. 20 μg protein was separated by electrophoresis and transferred to PVDF membranes. Membranes were blocked in TBST + 5% BSA, incubated overnight with primary antibody, washed in TBST, incubated with HRP-conjugated secondary antibody for 30 min, and visualized by chemiluminescence – combined with either film or on a BioRAD Hood II Gel Doc universal imager. Antibodies to phosphorylated p53-S37, p21, phospho-ERK1/2, total ERK1/2 and total p38MAPK were from Cell Signaling. Antibodies against LMNB1, HMGB1, and actin were from Abcam, anti-phospho-p38 was from PhosphoSolutions, and anti-PRAK was from Novus. Anti-phospho-PRAK-T182 was a kind gift from Peiqing Sun, and was described in (Yoshizuka et al., 2012). Anti-RAS was provided in the kit provided by Cytoskeleton, Inc (above).
IL-6 ELISA.
3×104 cells in 12-well plates were treated as indicated, and cultured in 0.5–1 ml serum-free DMEM for 24 hr. CM were collected and clarified at 2,000x g for 10 min. Supernatants were transferred to a tube; cells were trypsinized and counted. CM (2.5 μl) were analyzed by bead-based ELISAs (AlphaLISA, Perkin-Elmer) as instructed by the manufacturer and normalized to cell number.
PGD2-MOX ELISA.
CM were isolated as for IL-6 ELISA (above), and analyzed according the manufacturer’s instructions (Cayman Chemical), and quantified as pg/cell.
15d-PGJ2 ELISA.
1×105 cells in 6-well plates were treated as indicated for 10 days, and then cultured in 1 ml of serum-free medium plus DMEM or ABT-263 for 24 h. CM were then clarified by centrifugation at 2,500x g for 10 min. 15d-PGJ2 ELISA was from Enzo Life Sciences, and was performed according to the manufacturer’s instructions. Cell culture supernatant was analyzed directly, and all numbers were normalized to cell number. For in vivo fluids, 250 μL of blood plasma or 50 μL of urine were extracted as in Liquid-liquid extraction (LLE - below) with the following modification – dehydrated lipid samples were resuspended in 250 μL of assay buffer before analysis.
Reporter assays.
Baculoviral PPARγ reporter constructs (Signosis) were transduced into proliferating cells. Following treatment, cells were lysed using Passive Lysis Buffer (Promega) and analyzed for luciferase activity using a commercial kit (Promega) and Perkin-Elmer Victor™X3 luminometer.
Chemicals and standards.
Ammonium acetate was from Sigma Aldrich (St. Louis, MO). HPLC-grade solvents acetonitrile and methanol were from Fisher Scientific (Pittsburgh, PA, USA) and VWR (Radnor, PA, USA). Deionized water was generated in-house. PGA2, PGD2, PGE2, PGF2α, PGJ2, 15d-PGJ2, 15d-PGJ2-d4, NS-398, CAY-10404, ciglitazone, troglitazone and staurosporine were from Cayman Chemical. ABT-263 was from Apex Biotechnology. The dihomo-15d-PGJ2 standard was synthesized by Jubilant Chemsys (Bangalore, India). TRAIL was from Abcam.
Liquid-liquid extraction (LLE) of lipids.
Lipid extraction was performed as reported (Bligh and Dyer, 1959; Folch et al., 1951) with some modifications. Cells were rinsed with PBS and quenched using 1mL 50% methanol; 2 μg/mL 13C1-leucine and 5 ng/mL hexanesulfonic acid were added as internal standards. 2 mL of chloroform with 1 μg/mL heptadecanoic acid was added to each sample and mixed for 10 min at 4° C. Samples were centrifuged at 4,000g for 15 min at 4° C to separate aqueous and organic layers. After centrifugation, 700 μL of the aqueous layer and 1.5 mL of the organic layer were recovered and concentrated by speedvac and N2, respectively. Both aqueous and organic fractions were reconstituted in 100μL 50% methanol and chloroform prior to liquid-chromatography mass spectrometry (LC-MS) analysis.
Solid phase extraction (SPE) of lipids.
Lipid extraction was performed based as reported (Harkewicz et al., 2007) with some modifications. Cells were rinsed with PBS and quenched in 1 mL methanol with 2 μg/mL 13C1-leucine, 1μg/mL heptadecanoic acid, and 5 ng/mL hexanesulfonic acid added as internal standards. 1 mL PBS was added to each sample and mixed for 10 min. Samples were centrifuged at 4,000g for 15 min at 4° C. Following centrifugation, oxylipins were separated on Phenomenex Strata-X polymeric sorbent columns connected to a vacuum manifold. Columns were pre-washed with 2 mL methanol followed by 2 mL water. Samples were loaded onto columns, washed with 2 mL 90:10 water:methanol, then eluted with 1mL 100% methanol. Extracts were concentrated by speedvac and reconstituted in 100μL 100% methanol prior to LC-MS analysis.
High-pressure liquid chromatography quadrupole-time-of-flight mass spectrometry (HPLC-QTOF-MS).
LC-MS analyses were performed on a Agilent 6520 QTOF mass spectrometer coupled to a Agilent 1260 Infinity liquid chromatography system consisting of the following modules: u-degasser (G1322A), binary pump (G1312B), thermostated column compartment (G1330B), and HiPALS auto sampler (G1367E). Chromatographic separation of cellular extracts was performed on a Phenomenex Luna NH2 (2.0mm × 150mm, 3.0μM) column. The mobile phase included A:20mM ammonium acetate and 5% acetonitrile, pH9.5 and B:acetonitrile. The gradient is as follows: 0 to 20min, 95–10%B, 25–30min 10%B, and 30.1–35min 95%B. LC conditions included an auto sampler temperature of 4° C, injection volume of 10μL and solvent flow-rate of 0.3mL/min. Analyses were performed using the following ionization parameters: gas temperature (TEM) 350°C; drying gas, 9L/min; Vcap, 2500V; nebulizer, 35psig; fragmentor, 125V; and skimmer, 65V. MS1 acquisition was operated in the negative ion scanning mode for a mass range of 50–1000 m/z.
LC-MS data was acquired and analyzed using Agilent MassHunter Workstation (B.05.00), Agilent MassHunter Qualitative Analysis Software (B.08.00), Mass Profiler Professional (B.12.0), and Microsoft Excel 2007 (Redmond, WA, USA). Peak areas were assigned using Agilent MassHunter Qualitative Analysis Software in combination with the Find by Formula (FBF) algorithm. Peak areas were normalized to total protein or cell number.
High-pressure liquid chromatography quadropole ion trap mass spectrometry (HPLC-QTRAP-MS).
HPLC was performed using a Shimadzu UFLC prominence system fitted with following modules: CBM-20A (Communication bus module), DGU-A3(degasser), two LC-20AD (liquid chromatography, binary pump), SIL-20AC HT (auto sampler) and connected to a Phenomenex Luna NH2 (2.0mm × 150mm, 3.0μM) column. The solvent system was A=20mM ammonium acetate pH 9.5 with 5% acetonitrile and B=acetonitrile. The starting gradient conditions were 95% B at a flow rate of 0.3mL/min. The following gradient program was used: 0 to 20min, 95–10%B, 25–30min 10%B, and 30.1–35min 95%B. Samples were kept at +4°C, and the injection volume was 10μL. Mass spectrometric analysis was conducted using negative ion electrospray ionization in the multiple reaction monitoring mode (MRM) on an AbSciex 4000 QTRAP (Foster City, CA, USA) mass spectrometer fitted with a TurboV™ ion source. The ionization parameters were set as follows: curtain gas (CAD); 20psi; collision gas: medium; ion spray voltage (IS): −4500V; Temperature (TEM): 550°C; Ion source Gas 1 (GS1); 60psi; and Ion source Gas 2 (GS2): 50psi. The compound parameters were established using appropriate standards. The compound parameters were set as follows: entrance potential (EP): −10.0V; and collision cell exit potential (CXP): −5V. ABSciex Analyst®v1.6.1 was used for all data acquisition and an in-depth analysis of the HPLC-MS data, specifically for calculating the peak areas for the identified features from cellular extracts. Peak areas were normalized to total protein.
Reverse-phase LC-MS method for detection of 15d-PGJ2.
The LC-MS analysis was performed using Agilent 6520 accurate mass quadruple time of flight (Q-ToF) mass spectrometer coupled with Agilent 1260 Infinity Pump (Agilent Technologies, Santa Clara, CA). Ionization was performed in negative mode. Nitrogen was used as a desolvation and collision gas. The separation was carried out using Kinetex EVO C18 100Å 150×2.1, 5 micron (serial #H15173269) from Phenomenex. The autosampler was set at 5°C. The solvent system was composed of Solvent A- 0.1% acetic acid in 95:5 (water: ACN) containing 10 mM ammonium acetate and Solvent B- 0.1% acetic acid in 95:5 (ACN: water) containing 10 mM ammonium acetate. The flow rate was maintained at 0.3 mL/min, and the initial solvent conditions started with 5% solvent B. At 2 min, the percentage of B was increased to 70% B over 14 min, followed by further increase of % B to 95% for 10 mins. At 24 mins % B was maintained at 95% B for 5 mins and then reduced to 5%B and maintained at 5% B for 5 mins. The run time was 35 mins. Drying gas flow and temperature was set at 9.0 L/min and 350 °C, respectively and nebulizer gas pressure was set at 35 psig. The applied capillary voltage was 3000 V. Full scan spectra was acquired from 100–1000 m/z. The instrument was operated with Agilent MassHunter Work station LC/MS Data Acquisition version 05.01, and chromatograms were processed with MassHunter Workstation qualitative software version B.08.00.
Quantitation of intracellular dihomo-15d-PGJ2 concentration.
A standard curve was generated using 100, 50, 25, and 10 nM dihomo-15d-PGJ2, and peak areas were quantified by mass spectrometry. Based upon an average measured concentration of 69 nM in 1 mL of cell lysate representing 5 × 106 senescent cells, and assuming an average volume of 7.5 pL per senescent cell, we estimated a mean intracellular concentration of 1.8 μM by mass spectrometry, or 5.8 μM by ELISA.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Representation and Statistical Analysis.
All data are presented as means +/− SEM, with the exception of box plots, which follow the standard median/quartile structure. All culture datasets represent means of at least 3 experiments, and mouse datasets reflect a minimum of 6 mice. Comparisons between groups were performed using a 2-tailed student t-test, 1- or 2-way ANOVA, as appropriate. No methods were used to determine whether the data met the assumptions of the statistical approaches. Heat maps use p<0.05 for all entities. Significance is indicated by * = p<0.05, ** = p<0.01, and *** = p<0.001. Statistical parameters can be found in Figure legends.
Supplementary Material
Table S1. Abundances of oxylipins and free fatty acids, Related to Figure 1. Lipid profiles were generated as in Figures 1A and 1B. This table reflects raw peak area data for all putative lipids analyzed in this manuscript.
Table S2. Primer and probe sequences used in this manuscript, Related to STAR Methods.
Highlights.
Senescent cells make several oxylipins, dihomo-prostaglandins, and leukotrienes
Dihomo-15d-PGJ2 is intracellular during senescence and is released during senolysis
Dihomo-15d-PGJ2 activates RAS, promoting senescence and the SASP
Positive feedback between prostaglandins, RAS, and p53 maintains senescence
Acknowledgements.
The authors thank Peiqing Sun for the gift of the phospho-PRAK antibody, and Pierre-Yves Desprez for critical reading of this manuscript. This work was supported by grants from the National Institutes of Health (AG051729, AG057353, AG0172442, U2C-DK119886, and T32-AG000266) and the Science and Engineering Research Board of India (DST- SERB SPR/2019/001265).
Footnotes
Declaration of Interests. JC is a founder of Unity Biotechnology, which develops senolytic therapies. CW, AR, and JC are inventors on a patent for detection of oxylipins as biomarkers of senolysis.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Acosta JC, O’Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. (2008). Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018. [DOI] [PubMed] [Google Scholar]
- Aird KM, Zhang G, Li H, Tu Z, Bitler BG, Garipov A, Wu H, Wei Z, Wagner SN, Herlyn M, et al. (2013). Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep 3, 1252–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anta B, Perez-Rodriguez A, Castro J, Garcia-Dominguez CA, Ibiza S, Martinez N, Dura LM, Hernandez S, Gragera T, Pena-Jimenez D, et al. (2016). PGA1-induced apoptosis involves specific activation of H-Ras and N-Ras in cellular endomembranes. Cell Death Dis 7, e2311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnia F, Taylor BD, Woodson M, Kacerovsky M, Hawkins H, Fortunato SJ, Saade GR, and Menon R (2015). Chorioamniotic membrane senescence: a signal for parturition? Am J Obstet Gynecol 213, 359 e351–316. [DOI] [PubMed] [Google Scholar]
- Bligh EG, and Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Buratta S, Urbanelli L, Sagini K, Giovagnoli S, Caponi S, Fioretto D, Mitro N, Caruso D, and Emiliani C (2017). Extracellular vesicles released by fibroblasts undergoing H-Ras induced senescence show changes in lipid profile. PLoS One 12, e0188840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadenas C, Vosbeck S, Hein EM, Hellwig B, Langer A, Hayen H, Franckenstein D, Buttner B, Hammad S, Marchan R, et al. (2012). Glycerophospholipid profile in oncogene-induced senescence. Biochim Biophys Acta 1821, 1256–1268. [DOI] [PubMed] [Google Scholar]
- Campisi J (2013). Aging, cellular senescence, and cancer. Annu Rev Physiol 75, 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellone MD, Teramoto H, Williams BO, Druey KM, and Gutkind JS (2005). Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science 310, 1504–1510. [DOI] [PubMed] [Google Scholar]
- Catalano A, Rodilossi S, Caprari P, Coppola V, and Procopio A (2005). 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. EMBO J 24, 170–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, and Campisi J (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormenier J, Martin N, Desle J, Salazar-Cardozo C, Pourtier A, Abbadie C, and Pluquet O (2018). The ATF6alpha arm of the Unfolded Protein Response mediates replicative senescence in human fibroblasts through a COX2/prostaglandin E2 intracrine pathway. Mech Ageing Dev 170, 82–91. [DOI] [PubMed] [Google Scholar]
- Dagouassat M, Gagliolo JM, Chrusciel S, Bourin MC, Duprez C, Caramelle P, Boyer L, Hue S, Stern JB, Validire P, et al. (2013). The cyclooxygenase-2-prostaglandin E2 pathway maintains senescence of chronic obstructive pulmonary disease fibroblasts. Am J Respir Crit Care Med 187, 703–714. [DOI] [PubMed] [Google Scholar]
- De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. (2019). L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F, Koenig K, Le C, Mitin N, Deal AM, et al. (2017). Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov 7, 165–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, Van Steeg H, Dolle ME, et al. (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31, 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis EA, and Norris PC (2015). Eicosanoid storm in infection and inflammation. Nat Rev Immunol 15, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. (1995). A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 92, 9363–9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor AC, Wolfgeher D, Wu D, and Kron SJ (2017). A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov 3, 17075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Ascoli I, Lees M, Meath JA, and Le BN (1951). Preparation of lipide extracts from brain tissue. J Biol Chem 191, 833–841. [PubMed] [Google Scholar]
- Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, and Evans RM (1995). 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 83, 803–812. [DOI] [PubMed] [Google Scholar]
- Freund A, Patil CK, and Campisi J (2011). p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 30, 1536–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk CD (2001). Prostaglandins and leukotrienes: advances in eicosanoid biology. Science 294, 1871–1875. [DOI] [PubMed] [Google Scholar]
- Gan Q, Huang J, Zhou R, Niu J, Zhu X, Wang J, Zhang Z, and Tong T (2008). PPAR{gamma} accelerates cellular senescence by inducing p16INK4{alpha} expression in human diploid fibroblasts. J Cell Sci 121, 2235–2245. [DOI] [PubMed] [Google Scholar]
- Gibb W (1998). The role of prostaglandins in human parturition. Ann Med 30, 235–241. [DOI] [PubMed] [Google Scholar]
- Giles H, Leff P, Bolofo ML, Kelly MG, and Robertson AD (1989). The classification of prostaglandin DP-receptors in platelets and vasculature using BW A868C, a novel, selective and potent competitive antagonist. Br J Pharmacol 96, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JH, Roh MS, Park CH, Park KC, Cho KH, Kim KH, Eun HC, and Chung JH (2004). Selective COX-2 inhibitor, NS-398, inhibits the replicative senescence of cultured dermal fibroblasts. Mech Ageing Dev 125, 359–366. [DOI] [PubMed] [Google Scholar]
- Harkewicz R, Fahy E, Andreyev A, and Dennis EA (2007). Arachidonate-derived dihomoprostaglandin production observed in endotoxin-stimulated macrophage-like cells. J Biol Chem 282, 2899–2910. [DOI] [PubMed] [Google Scholar]
- Hata AN, and Breyer RM (2004). Pharmacology and signaling of prostaglandin receptors: multiple roles in inflammation and immune modulation. Pharmacol Ther 103, 147–166. [DOI] [PubMed] [Google Scholar]
- Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, and Demaria M (2017). Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr Biol 27, 2652–2660 e2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, Herrmann SM, Jensen MD, Jia Q, Jordan KL, et al. (2019). Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 47, 446–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirai H, Tanaka K, Yoshie O, Ogawa K, Kenmotsu K, Takamori Y, Ichimasa M, Sugamura K, Nakamura M, Takano S, et al. (2001). Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med 193, 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata M, Kakizuka A, Aizawa M, Ushikubi F, and Narumiya S (1994). Molecular characterization of a mouse prostaglandin D receptor and functional expression of the cloned gene. Proc Natl Acad Sci U S A 91, 11192–11196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jump DB (2009). Mammalian fatty acid elongases. Methods Mol Biol 579, 375–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, Prata L, Masternak MM, Kritchevsky SB, Musi N, et al. (2019). Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir TD, Leigh RJ, Tasena H, Mellone M, Coletta RD, Parkinson EK, Prime SS, Thomas GJ, Paterson IC, Zhou D, et al. (2016). A miR-335/COX-2/PTEN axis regulates the secretory phenotype of senescent cancer-associated fibroblasts. Aging (Albany NY) 8, 1608–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Vaish V, Feng M, Field K, Chatzistamou I, and Shim M (2016). Transgenic expression of cyclooxygenase-2 (COX2) causes premature aging phenotypes in mice. Aging (Albany NY) 8, 2392–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, and Lehmann JM (1995). A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 83, 813–819. [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, and Peeper DS (2010). The essence of senescence. Genes Dev 24, 2463–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laberge RM, Zhou L, Sarantos MR, Rodier F, Freund A, de Keizer PL, Liu S, Demaria M, Cong YS, Kapahi P, et al. (2012). Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 11, 569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Mok HJ, Lee DY, Park SC, Ban MS, Choi J, Park CG, Ahn YS, Kim KP, and Kim HD (2016). UPLC-MS/MS-Based Profiling of Eicosanoids in RAW264.7 Cells Treated with Lipopolysaccharide. Int J Mol Sci 17, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martien S, Pluquet O, Vercamer C, Malaquin N, Martin N, Gosselin K, Pourtier A, and Abbadie C (2013). Cellular senescence involves an intracrine prostaglandin E2 pathway in human fibroblasts. Biochim Biophys Acta 1831, 1217–1227. [DOI] [PubMed] [Google Scholar]
- Menon R, Behnia F, Polettini J, Saade GR, Campisi J, and Velarde M (2016). Placental membrane aging and HMGB1 signaling associated with human parturition. Aging (Albany NY) 8, 216–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monneret G, Li H, Vasilescu J, Rokach J, and Powell WS (2002). 15-Deoxy-delta 12,14-prostaglandins D2 and J2 are potent activators of human eosinophils. J Immunol 168, 3563–3569. [DOI] [PubMed] [Google Scholar]
- Montagut C, Sharma SV, Shioda T, McDermott U, Ulman M, Ulkus LE, Dias-Santagata D, Stubbs H, Lee DY, Singh A, et al. (2008). Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res 68, 4853–4861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomura T, Lu R, Pucci ML, and Schuster VL (2004). The two-step model of prostaglandin signal termination: in vitro reconstitution with the prostaglandin transporter and prostaglandin 15 dehydrogenase. Mol Pharmacol 65, 973–978. [DOI] [PubMed] [Google Scholar]
- Ohno Y, Suto S, Yamanaka M, Mizutani Y, Mitsutake S, Igarashi Y, Sassa T, and Kihara A (2010). ELOVL1 production of C24 acyl-CoAs is linked to C24 sphingolipid synthesis. Proc Natl Acad Sci U S A 107, 18439–18444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva JL, Perez-Sala D, Castrillo A, Martinez N, Canada FJ, Bosca L, and Rojas JM (2003). The cyclopentenone 15-deoxy-delta 12,14-prostaglandin J2 binds to and activates H-Ras. Proc Natl Acad Sci U S A 100, 4772–4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perretti M, and D’Acquisto F (2009). Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat Rev Immunol 9, 62–70. [DOI] [PubMed] [Google Scholar]
- Pettipher R, Vinall SL, Xue L, Speight G, Townsend ER, Gazi L, Whelan CJ, Armer RE, Payton MA, and Hunter MG (2012). Pharmacologic profile of OC000459, a potent, selective, and orally active D prostanoid receptor 2 antagonist that inhibits mast cell-dependent activation of T helper 2 lymphocytes and eosinophils. J Pharmacol Exp Ther 340, 473–482. [DOI] [PubMed] [Google Scholar]
- Quijano C, Cao L, Fergusson MM, Romero H, Liu J, Gutkind S, Rovira II, Mohney RP, Karoly ED, and Finkel T (2012). Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 11, 1383–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renedo M, Gayarre J, Garcia-Dominguez CA, Perez-Rodriguez A, Prieto A, Canada FJ, Rojas JM, and Perez-Sala D (2007). Modification and activation of Ras proteins by electrophilic prostanoids with different structure are site-selective. Biochemistry 46, 6607–6616. [DOI] [PubMed] [Google Scholar]
- Ricciotti E, and FitzGerald GA (2011). Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol 31, 986–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner BA, and Cristofalo VJ (1979). Hydrocortisone: a specific modulator of in vitro cell proliferation and aging. Mech Ageing Dev 9, 485–496. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, and Lowe SW (1997). Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88, 593–602. [DOI] [PubMed] [Google Scholar]
- Soberman RJ, and Christmas P (2003). The organization and consequences of eicosanoid signaling. J Clin Invest 111, 1107–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher H, VanRollins M, Sun F, Wyche A, and Needleman P (1982). Dihomo-prostaglandins and -thromboxane. A prostaglandin family from adrenic acid that may be preferentially synthesized in the kidney. J Biol Chem 257, 3912–3918. [PubMed] [Google Scholar]
- Strong R, Miller RA, Astle CM, Floyd RA, Flurkey K, Hensley KL, Javors MA, Leeuwenburgh C, Nelson JF, Ongini E, et al. (2008). Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell 7, 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, Xie C, Chen J, Deng Q, Yamout M, et al. (2007). PRAK is essential for ras-induced senescence and tumor suppression. Cell 128, 295–308. [DOI] [PubMed] [Google Scholar]
- Town MH, Casals-Stenzel J, and Schillinger E (1983). Pharmacological and cardiovascular properties of a hydantoin derivative, BW 245 C, with high affinity and selectivity for PGD2 receptors. Prostaglandins 25, 13–28. [DOI] [PubMed] [Google Scholar]
- Wall SB, Oh JY, Mitchell L, Laube AH, Campbell SL, Renfrow MB, and Landar A (2015). Rac1 modification by an electrophilic 15-deoxy Delta(12,14)-prostaglandin J2 analog. Redox Biol 4, 346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Brumwell AN, Davis SS, Jackson JR, Valdovinos A, Calhoun C, Alimirah F, Castellanos CA, Ruan R, Wei Y, et al. (2019a). Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, and Campisi J (2016). From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab 23, 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Liu S, Limbad C, Zawadzka AM, Beck J, Demaria M, Artwood R, Alimirah F, Lopez-Dominguez JA, Kuehnemann C, et al. (2019b). SILAC Analysis Reveals Increased Secretion of Hemostasis-Related Factors by Senescent Cells. Cell Rep 28, 3329–3337 e3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Schaum N, Alimirah F, Lopez-Dominguez JA, Orjalo AV, Scott G, Desprez PY, Benz C, Davalos AR, and Campisi J (2018). Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep 8, 2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, et al. (2016). Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 23, 303–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HH, Kim C, Jung B, Kim KS, and Kim JR (2011). Involvement of IGF binding protein 5 in prostaglandin E(2)-induced cellular senescence in human fibroblasts. Biogerontology 12, 239–252. [DOI] [PubMed] [Google Scholar]
- Yoshizuka N, Chen RM, Xu Z, Liao R, Hong L, Hu WY, Yu G, Han J, Chen L, and Sun P (2012). A novel function of p38-regulated/activated kinase in endothelial cell migration and tumor angiogenesis. Mol Cell Biol 32, 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zdanov S, Bernard D, Debacq-Chainiaux F, Martien S, Gosselin K, Vercamer C, Chelli F, Toussaint O, and Abbadie C (2007). Normal or stress-induced fibroblast senescence involves COX-2 activity. Exp Cell Res 313, 3046–3056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Abundances of oxylipins and free fatty acids, Related to Figure 1. Lipid profiles were generated as in Figures 1A and 1B. This table reflects raw peak area data for all putative lipids analyzed in this manuscript.
Table S2. Primer and probe sequences used in this manuscript, Related to STAR Methods.
Data Availability Statement
Lipid profiling data is available at the NIH Common Fund’s National Metabolomics Data Repository (NMDR) website, the Metabolomics Workbench (https://www.metabolomicsworkbench.org), Project ID #: PR001093. See also the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-PRAK-T182 | Peiqing Sun - Wake Forest Univeristy | |
| Phospho-p53 (Ser37) Antibody | Cell Signaling Technology | #9289 |
| p21 Waf1/Cip1 (12D1) Rabbit mAb | Cell Signaling Technology | #2947 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) Antibody | Cell Signaling Technology | #9101 |
| p44/42 MAPK (Erk1/2) Antibody | Cell Signaling Technology | #9102 |
| p38 MAPK Antibody | Cell Signaling Technology | #9212 |
| Anti-Lamin B1 antibody | Abcam | ab16048 |
| Anti-HMGB1 antibody | Abcam | ab18256 |
| Monoclonal Anti-β-Actin antibody | Sigma | A2228 |
| p53 Antibody (DO-1) | Santa Cruz Biotechnology | sc-126 |
| Bacterial and Virus Strains | ||
| shGFP lentivirus | Campisi Lab | |
| shP53 lentivirus | Campisi Lab | |
| shPTGDS lentivirus | Sigma-Aldrich | SHCLNV-NM_000954 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NS-398 | Cayman Chemical Company | 70590 |
| CAY-10404 | Cayman Chemical Company | 70210 |
| Ciglitazone | Cayman Chemical Company | 71730 |
| Troglitazone | Cayman Chemical Company | 71750 |
| Staurosporine | Cayman Chemical Company | 81590 |
| Recombinant Human TRAIL protein | Abcam | ab78818 |
| Arachidonic Acid-d11 | Cayman Chemical Company | 10006758 |
| Prostaglandin A2 | Cayman Chemical Company | 10210 |
| Prostaglandin D2 | Cayman Chemical Company | 12010 |
| Prostaglandin E2 | Cayman Chemical Company | 14010 |
| Prostaglandin F2α | Cayman Chemical Company | 16010 |
| Prostaglandin J2 | Cayman Chemical Company | 18500 |
| 15-deoxy-Δ12,14-Prostaglandin J2 | Cayman Chemical Company | 18570 |
| 1a,1b-dihomo-15-deoxy-Δ12,14-Prostaglandin J2 | Jubilant Chemsys | P1171 |
| 13,14-dihydro-15-keto Prostaglandin D2 | Cayman Chemical Company | 12610 |
| BW A868C | Cayman Chemical Company | 12060 |
| OC000459 | Cayman Chemical Company | 12027 |
| BW 245C | Cayman Chemical Company | 12050 |
| Hydrocortisone | Cayman Chemical Company | 20739 |
| Acetic Acid, glacial | VWR | VW0125–3 |
| Acetonitrile | VWR/BDH | BDH83639.400 |
| Ammonium acetate | Sigma Aldrich | 73594–25G-F |
| LCMS Water | Honeywell Burdick & Jackson | LC365–4 |
| Methanol | Honeywell Burdick & Jackson | LC230–4 |
| Chloroform | JT Baker | 9175–2 |
| Critical Commercial Assays | ||
| Baculoviral PPAR-gamma reporter assay | Signosis | BT-0014 |
| Senescence Detection Kit | BioVision | K320 |
| 15-deoxy-Δ12,14-PGJ2 ELISA Kit | ENZO | ADI-900–023 |
| Prostaglandin D2-MOX Express ELISA Kit | Cayman Chemical Company | 500151 |
| IL-6 AlphaLISA | PerkinElmer | AL223F |
| Ras Pull-down Activation Assay Biochem Kit | Cytoskeleton, Inc | BK008-S |
| Experimental Models: Cell Lines | ||
| HEPG2 | ATCC | HB-8065 |
| IMR-90 | ATCC | CCL-186 |
| HUVEC-C | ATCC | CRL-1730 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | The Jackson Laboratory | 000664 |
| p16–3MR | Campisi Laboratory | |
| Teklad global 18% protein | Envigo Teklad | 2918 |
| Oligonucleotides | ||
| See Table S2 | ||
| Software and Algorithms | ||
| MassHunter | Agilent | |
| Other | ||
| Kinetex 5μm EVO C18 100Å 150x2.1mm | Phenomenex | 00F-4633-AN |
| Data accessibility | ||
| Lipid Profiling Dataset | National Metabolomics Data Repository (NMDR) | PR001093 |