

Abstract
While small molecule internal strain is crucial to molecular docking, using it in evaluating ligand scores has remained elusive. Here, we investigate a technique that calculates strain using relative torsional populations in the Cambridge Structural Database, enabling fast precalculation of these energies. In retrospective studies of large docking screens of the dopamine D4 receptor and of AmpC β-lactamase, where close to 600 docking hits were tested experimentally, including such strain energies improved hit rates by preferentially reducing the ranks of strained high-scoring decoy molecules. In a 40-target subset of the DUD-E benchmark, we found two thresholds that usefully distinguished between ligands and decoys: one based on the total strain energy of the small molecules and another based on the maximum strain allowed for any given torsion within them. Using these criteria, about 75% of the benchmark targets had improved enrichment after strain filtering. Relying on precalculated population distributions, this approach is rapid, taking less than 0.04 s to evaluate a conformation on a standard core, making it pragmatic for precalculating strain in even ultralarge libraries. Since it is scoring function agnostic, it may be useful to multiple docking approaches; it is openly available at http://tldr.docking.org.
Graphical Abstract
INTRODUCTION
In large library docking screens, hundreds of millions to billions of molecules, each in multiple conformations, are sampled for complementarity to a protein binding site. While low-energy conformations predominate,1,2 inevitably some high-energy conformations are sampled. In a foundational study, Tirado-Rives and Jorgensen showed that it was often possible to find high-energy conformations of small molecules in docking and that the relative energies of these conformations were difficult to rank, even with fairly high-level quantum mechanics (QM).3 The authors concluded that there were inherent errors in docking—here from ligand strain but also from other terms that were outside the limits of binding affinity ranges, docking was likely to sample among its hits (from mM to mid-nM or about 6 kcal/mol). This made accurate rank ordering in docking unlikely, in general, and, in particular, made the improvement of docking scores by the addition of higher-order terms, here ligand strain, problematic.
While docking will remain, for the foreseeable future, a method that cannot reliably rank-order molecules from a large library screen, we wondered if ligand strain could nevertheless improve docking in one of its primary goals: as a categorizing technique that separates a small fraction of plausible new ligands from a much larger library of molecules unlikely to bind.4–7 High-ranking docked molecules can adopt strained conformations, typically owing to high-energy torsion angles. Ideally, such strained conformations should not be sampled, but the need to explore many conformations to achieve favorable fits, combined with necessarily approximate conformational energies and the screening of large, diverse libraries, has often meant that such strained conformations are in fact modeled. Perversely, these strained conformations can often score better in a protein site than a lower energy, unstrained conformation.8 This, in turn, can crowd out unstrained, more favorable molecules, lowering the ability of docking to separate true ligands from false positives. Were such strained conformations removed, it would improve docking hit rates, even if including strain, per se, in the energy score might not measurably improve rank ordering by affinity, as argued by Tirado-Rives and Jorgensen.
Unfortunately, few methods for calculating conformational energies now meet the demands of large library docking, where hundreds of millions to billions of molecules must be assessed,9–11 and several hundred thousands must be evaluated even after the initial docking campaign has completed (postfiltering). The current approaches for strain energy assessment can be organized into three groups: quantum mechanical (QM), molecular mechanics (MM), and database-torsional methods. In ab initio calculations,12–15 torsional energies are calculated using one of several QM basis sets. For example, Rai et al. generated torsion scan profiles of neutral fragments using density functional theory (DFT) (B3LYP).16 Based on the QM calculations at discrete torsion angles, they interpolated the values into a continuous function to estimate the torsion strain energy. This is not yet feasible at scale, and so the method was converted into a semi-knowledge-based approach. Even so, the method still demands ab initio calculation of torsional terms for many new molecules and remains time consuming. In molecular mechanics17 approaches, force fields including OPLS, CHARMM, and AMBER18–22 are used to calculate small molecule strain energies; this can often demand the calculation of new parameters, especially for large and diverse docking libraries. Molecular mechanics methods are less accurate than QM methods since they involve further approximation in the energy equations (for example, in assigning static partial charges to the molecules).15,23 This lack of accuracy is balanced by its much higher speed. Even so, it is unclear that it is suited to the scale of the new ultralarge docking libraries.9–11 A third approach leverages databases of small molecule crystal structures to predict torsion strain using populations of observed angles.24,25 Predecessors to these studies are the MIMUMBA and Mogul systems,26,27 and in a recent study, Groom and colleagues systematically inferred torsion-based ligand strain from the Cambridge Structure Database (CSD)28 and also from the Protein Data Bank (PDB).29 They compiled histograms of observed dihedral angles for each torsion pattern (the sequence of four atoms defining the dihedral angle, encoded in the SMARTS format).30 The torsion patterns were organized hierarchically so that a user can match each torsion pattern in a molecule to the patterns compiled with increasing specificity. They also developed TorsionAnalyzer, which is an interactive graphical tool for strain energy analysis.31
Here, we used this statistical approach to address ligand strain energy in docking, focusing exclusively on terms derived from the CSD, which are more accurate. Based on histograms of each torsion pattern, we calculated the torsion energy using a canonical ensemble approach. We converted counts observed in the data sets for different angle measurements for each torsion pattern into torsion energy units (TEUs). Since we had a filtering strategy in mind, where torsional strain would be either acceptable or unacceptable but would not be added to a docking score per se, it was unimportant how these TEUs related to docking energies; the two would not be merged. With all counts in the same energy scale, we can compare and add conformational strain energy across different torsion patterns for the entire molecule. We then use the total torsional energy of the molecule, and the maximum individual torsional energy within that molecule, to evaluate the strain of a molecule’s conformation. To determine a threshold for applications, we studied two targets with extensive experimental measurements of docking-predicted ligands and also 40 systems from the DUD-E benchmark. We find that at certain thresholds, when used as a filter to remove strained molecules, this method can improve docking hit rates, at least retrospectively. The software is relatively fast, capable of calculating strain for half a million molecules on a small cluster in less than 10 minutes, and is mechanically reliable (few molecules fail to return an energy), both of which make it suitable for large library applications. It is openly available to the community at http://tldr.docking.org.
METHODS
Torsion Library Generation.
We represent every sequence of four atoms defining a dihedral angle by a torsion pattern using the SMARTS line notation.30 We adapted the torsion library of Rarey et al.24,25 to build our own library, which has 514 torsion patterns with the same hierarchical organization as the original. Each torsion pattern has a histogram of the observed counts for each possible dihedral angle measurement in the CSD and PDB. For each torsion pattern’s histogram, if the total count is less than 100, we use an approximate approach. The original torsion library allows “tolerances” about each peak in the histogram, where the observed frequency drops below a certain value. Our approximate approach flags any degree difference between a conformation’s dihedral angle and the histogram’s peak that is larger than the maximum tolerance in the original library. If the total count is larger than 100, we convert the histogram frequencies into torsion energy units (TEUs) by applying the Boltzmann equation (Figure S1), which we term the “exact approach”. To avoid infinite energies from zero counts in the histograms, we add the minimum positive count from each histogram to any zero counts. We assume that the original measurements from the CSD and PDB exist in a canonical ensemble at 298 K, meaning the temperature of the hypothetical ensemble is a tunable parameter in creating the strain energy library. We therefore use TEU instead of kcal/mol to reflect the fact that our energy scale is artificial and based on the databases’ sampling of the hypothetical ensemble. We modeled this unit on Rosetta’s use of Rosetta energy units in its scoring function for protein structure conformational energy.32,33
Workflow of the Software.
The current version can handle two types of input files: mol2 and db2 (a format for DOCK3.7).34 With the energy profile for each torsion pattern precalculated in the library, we only need to look up the energy estimates for the torsion patterns present in the molecule and not calculate them from scratch. Using the Chem submodule in the Python module RDKit,35 we find all of the torsion patterns present in the molecule based on their SMARTS patterns, calculate their dihedral angles, and then extract the relevant energy estimates from the torsion library. We keep the information matching the most specific (least general) torsion pattern in the library’s hierarchy. Then, we can sum the energy estimates for all of the torsion patterns to get the estimate for the molecule’s conformation or simply find the torsion patterns with energy estimates above the desired cutoff (Figure 1).
Figure 1.
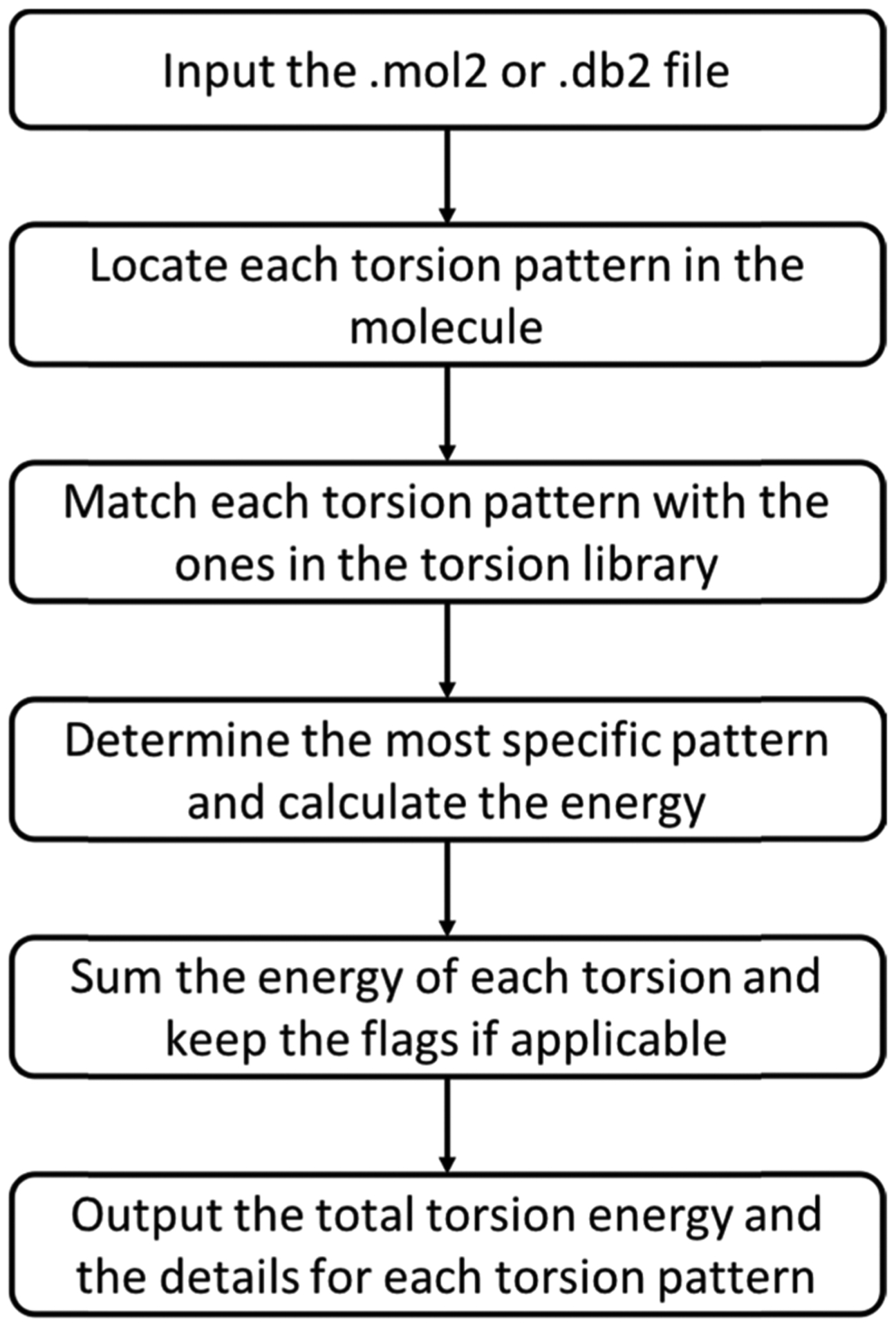
Flowchart for strain energy filtering. The program first locates each torsion pattern in the molecule and calculates its dihedral angle. It then matches each torsion pattern in the molecule with the patterns in the torsion library. There will be multiple such matches since the torsion library contains a hierarchy of patterns. For each match, the program calculates the energy for the observed dihedral angle and determines any flags. For each torsion pattern in the molecule, it keeps only the information from the most specific torsion pattern rule from the library. Ultimately, the program reports the estimated energy for each torsion pattern, the sum for all of the patterns in the molecule, and any flagged patterns.
DUD-E Benchmark Docking.
The full list of DUD-E systems and the corresponding PDB IDs used are listed in Table S1. For docking, the crystallographic ligands were used to generate matching spheres for each target. To prepare the protein in docking, we precalculated energy grids for an AMBER-based van der Waals potential,36 a Poisson–Boltzmann electrostatic potential, using QNIFFT,37,38 and for ligand desolvation using an adapted generalized-Born approach.39 We used DOCK3.734 to dock all of the hits and decoys for each DUD-E target.
Quantum Mechanical Calculation of Strain.
For 20 representative dihedrals, we calculated the strain at the 6–31G** basis set level using the B3LYP-D3 method in Jaguar (Schrodinger, New York). For each torsion, we compared the QM-calculated strain energy to that calculated by the population-based method at angles of both 30 and 60° off the minimum.
RESULTS
D4 Dopamine Receptor Case Study.
We began by investigating the effects of ligand strain on the experimental results of a large library docking campaign against the D4 dopamine receptor.9 Here, 549 docking-ranked compounds, across a wide range of ranks, were tested in the same assay in the same lab, providing an unusually large set of comparable experiments for a diverse set of compounds. We collected 256 of these molecules that had high DOCK3.7 ranks, with scores less (better) than −60 kcal/mol.9 Of these, 62 were found experimentally to bind, with EC50 values ranging from 6 μM to 180 pM, while the other 194 were docking decoys (i.e., high-ranking molecules that did not bind on experimental testing). In the calculation that led to these molecules, no strain energy filter was applied. We can calculate the torsion-based ligand strain for each of these experimental binders and nonbinders, comparing the docked conformation of the molecules to that of their ground state (by torsion strain) (Table S2). If it is true that the decoys tend to be more strained than the binders, then applying a strain filter should remove more of the experimental nonbinders than the true binders and correspondingly, the hit rates, the number-active/number-tested, should increase. These values can be calculated at different strain thresholds (Figure 2). Here, “total” refers to the total strain energy of a compound, adding up all torsions, while “single” means the maximum strain energy of any individual torsion within the molecule. We deem a molecule’s docked pose “strained” if its total or maximum individual torsion strain is greater than the threshold.
Figure 2.
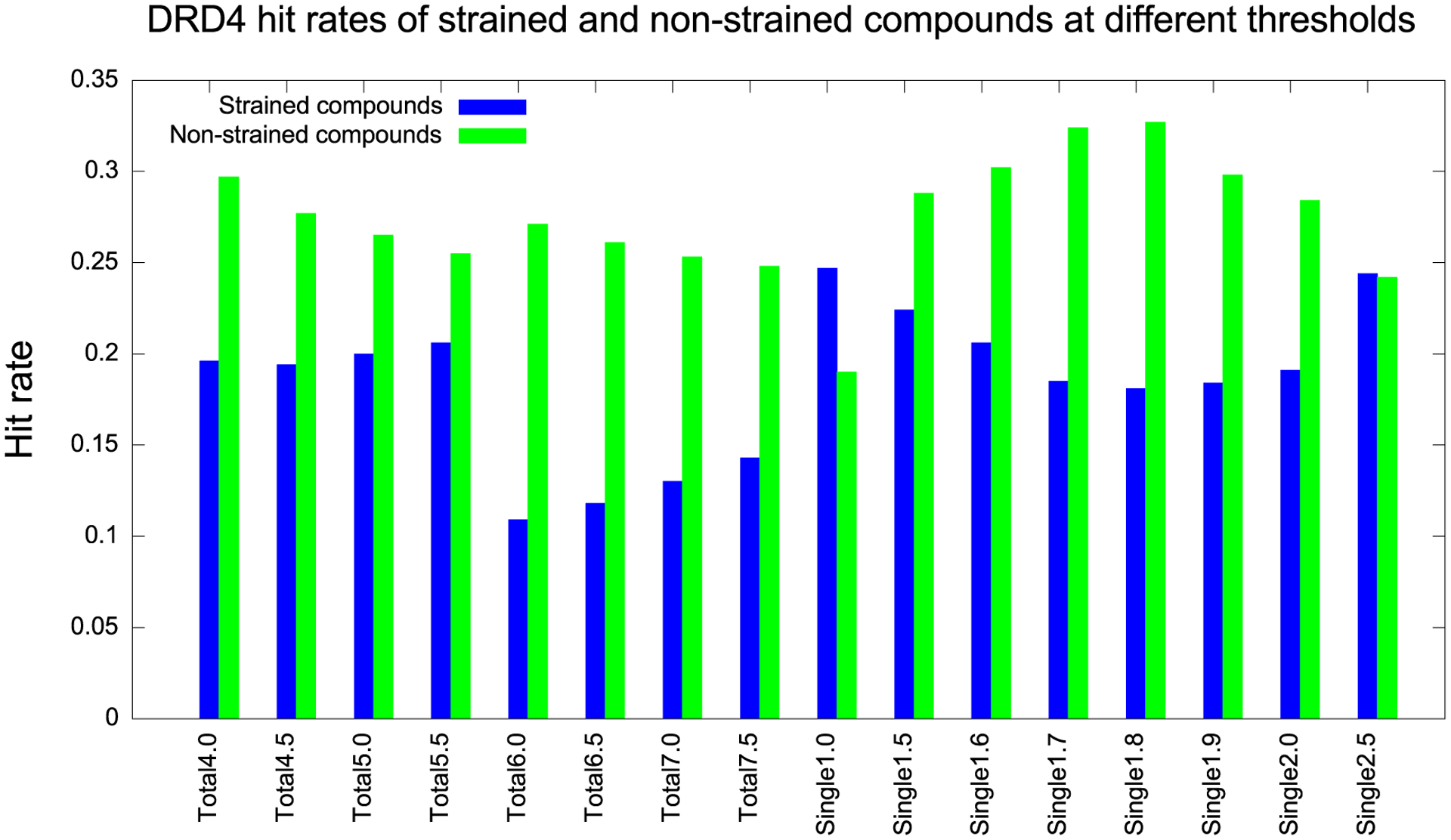
D4 receptor hit rates of strained (blue) and nonstrained (green) compounds at different thresholds, measured in TEU. For the total energy category, 6.0 TEU appears to be a good threshold. For the maximum single energy category, 1.8 TEU appears to be a good choice.
A satisfactory strain energy correction should maximize the hit rate of nonstrained compounds while minimizing that of the strained ones. The hit rate of nonstrained compounds may be calculated as the number of true positives (true ligands correctly labeled as nonstrained) divided by the total number of nonstrained compounds, while the hit rate of strained compounds is the number of false negatives (true ligands wrongly labeled as strained) divided by the total number of strained compounds. In the case study of the D4 receptor, we maximize the difference in hit rates using a total strain energy threshold of 6.0 TEU and a maximum single strain energy threshold of 1.8 TEU.
Inspection of the compounds filtered out or retained affords insight into this process (Figure 3). High-ranking decoys like ZINC000067715288 and ZINC000666614106 break planarity about an amide bond, with an angle of about 30° or more, imparting single energy strains of 4.0 and 5.6 TEUs, respectively, and total strains of 10.4 and 5.8 TEUs, respectively. Molecules like ZINC000377646411 and ZINC000248536951 have strained torsions that break conjugation of an exocyclic group with an aromatic ring. These strained torsions allow these molecules to make favorable interactions with the receptor that they otherwise could not (Figure 3). Conversely, for high-ranking binders like ZINC000480496068 and ZINC000155719879, their torsional violations never exceed 1.2 TEUs for any single angle nor do they add up to more than 4.2 in total.
Figure 3.
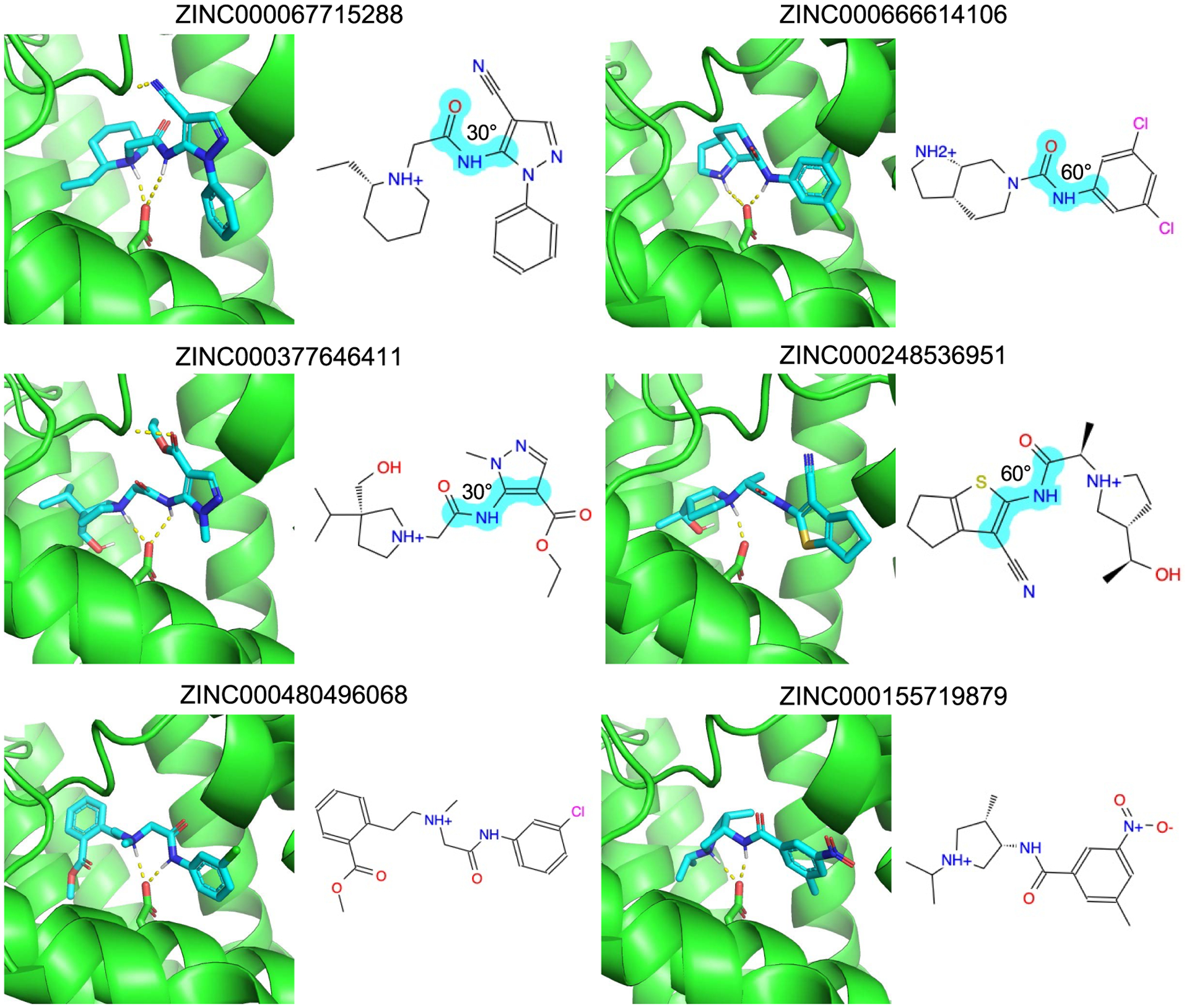
Docking poses of strained decoys (top four) and unstrained binders (bottom two) in DRD4. Two-dimensional (2D) images of the molecules are also shown. The strained torsions are indicated in cyan and labeled with their degrees out of optimum.
We looked at the effects of strain energy filtering on the docking-prioritized D4 receptor compounds, and on the true ligands that emerged from them, using a total strain of 6.0 TEU and a maximum single strain of 1.8 TEU (Table 1). Using the total strain filter alone (columns 3 and 4), the hit rates of strained and nonstrained compounds, i.e., above and below our threshold, are 0.109 and 0.271, respectively. A two-sample proportion Z-test indicates that this difference is significant (p-value 0.010). Using the maximum single strain filter (columns 5 and 6), the hit rates of strained and nonstrained compounds are 0.181 and 0.327, respectively, which is also a significant difference (p-value 0.004). The p-value for the hit rate comparison with and without the maximum single strain filter (columns 6 and 2) is 0.048, which is significant. On the other hand, the p-value for the hit rate comparison with and without the total strain filter (columns 4 and 2) is 0.236. While this p-value is not significant, an even more stringent criterion was explored, which is to classify compounds as strained if they fulfill either of the two conditions (columns 7 and 8). Under these criteria, the hit rate of nonstrained compounds was further improved to 0.340.
Table 1.
DRD4 Hit Rates before and after Strain Filtering at Two Chosen Thresholds
| all (no strain filters) | total strain ≥6 TEU | unstrained (total strain <6 TEU) | max single strain ≥1.8 TEU | unstrained max single strain <1.8 TEU | total strain ≥6 TEU OR max single strain ≥1.8 TEU | unstrained (total strain <6 TEU AND max single strain <1.8 TEU) | |
|---|---|---|---|---|---|---|---|
| number of hits | 62 | 5 | 57 | 27 | 35 | 27 | 35 |
| sample size | 256 | 46 | 210 | 149 | 107 | 153 | 103 |
| hit rate | 0.242 | 0.109 | 0.271 | 0.181 | 0.327 | 0.176 | 0.340 |
Although each threshold is the best choice in its category (with this level of granularity in testing thresholds), the outcomes are different. At a total strain of 6.0 TEU, only 46 compounds are filtered out vs 149 at a maximum single strain of 1.8 TEU; the percentage of remaining compounds is 82 vs 42%. This difference suggests that if the investigator prefers to keep as many hits as possible (conservative in deeming a molecule “strained”), they should use the total strain threshold. To maximize stringency, ensuring that most compounds will be “unstrained”, they should use the maximum single strain threshold.
Case Study of AmpC β-Lactamase.
We applied the same calculations and analyses on the AmpC β-lactamase data set, another target for which we have a substantial number of true actives and experimentally measured decoys from a previous large-scale docking campaign. Of 44 experimentally tested molecules with a DOCK score less than −60,9 5 were true inhibitors (39 were high-ranking decoys) (Table S3 and Figure 4).
Figure 4.
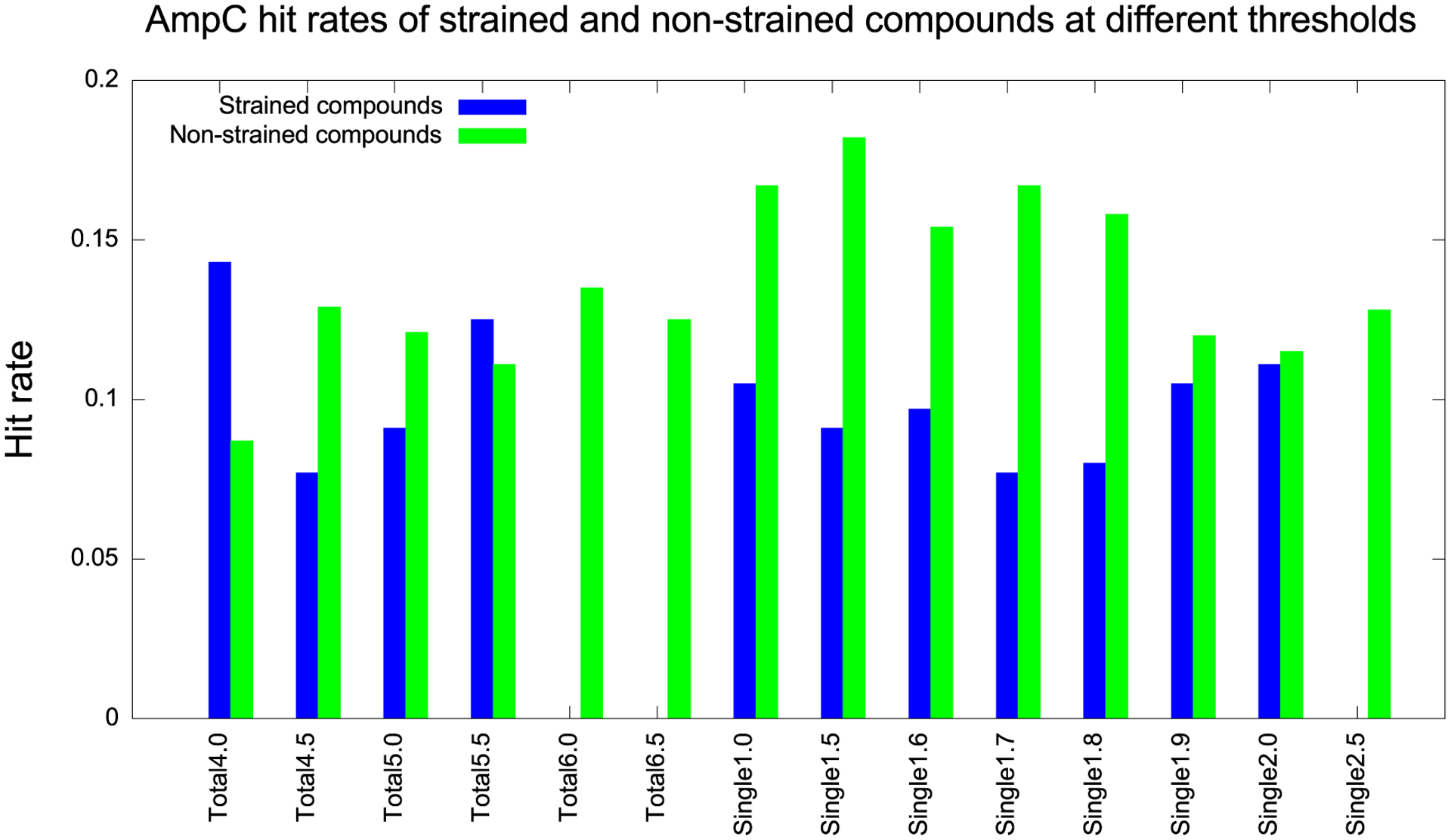
AmpC hit rates of strained (blue) and nonstrained (green) compounds at different thresholds, measured in TEU. For the total energy category, 6.0 TEU is a good choice of threshold. For the maximum single energy category, 1.7 TEU is a good choice.
As with the D4 case, we maximize the difference in hit rates between strained and nonstrained molecules for the AmpC molecules. Here, the maximum difference between the two comes with a total strain energy threshold of 6.0 TEU and a maximum single strain energy threshold of 1.7 TEU (Table 2). Using the total strain energy filter (columns 3 and 4), the hit rates of strained and nonstrained compounds are 0.000 and 0.135, respectively (i.e., none of the true ligands are found to be strained). Using the maximum single strain energy filter (columns 5 and 6), the hit rates of strained and nonstrained compounds are 0.077 and 0.167, respectively. The two-sample proportion Z-test failed to show statistical significance for these differences, as the total sample size of 44 was too small. We also explored an even more stringent criterion, which is to classify compounds as strained if they fulfill either of the two conditions (columns 7 and 8). However, the result did not improve. Still, overall, we observed similar results for AmpC as we did for D4. At a total strain of 6.0 TEU, only 7 compounds are filtered out compared to 26 at a single maximum strain of 1.7 TEU. The percentage of remaining compounds is 84 vs 41%. As we will see, this trend continues when we turn to the DUD-E benchmarks.
Table 2.
AmpC Hit Rates before and after Strain Filtering at Two Chosen Thresholds
| all (no strain filters) | total strain ≥6 TEU | unstrained (total strain <6 TEU) | max single strain ≥1.7 TEU | unstrained max single strain <1.7 TEU | total strain ≥6 TEU OR max single strain ≥1.7 TEU | unstrained (total strain <6 TEU AND max single strain <1.7 TEU) | |
|---|---|---|---|---|---|---|---|
| number of hits | 5 | 0 | 5 | 2 | 3 | 2 | 3 |
| sample size | 44 | 7 | 37 | 26 | 18 | 26 | 18 |
| hit rate | 0.114 | 0.000 | 0.135 | 0.077 | 0.167 | 0.077 | 0.167 |
Here too, we can inspect the conformations adopted by the compounds filtered out by strain (Figure 5). High-ranking decoys like ZINC000188308020 and ZINC000261754704 break planarity about an amide bond, with an angle of about 30°, imparting single energy strains of 2.1 and 2.1 TEUs, respectively, and total strains of 6.8 and 6.6 TEUs, respectively. Molecules like ZINC000317738578 and ZINC000274934818 have strained sulfonamide with nitrogen in a ring. These strained torsions allow these molecules to make favorable interactions with the enzyme that they otherwise could not (Figure 5). Conversely, for high-ranking binders like ZINC000184991516 and ZINC000547933290, their torsional violations never exceed 1.7 TEUs for any single angle nor do they add up to more than 4.5 in total; nevertheless, they make favorable fits with β-lactamase, comfortably placing a phenolate in the oxyanion hole and hydrogen bonding with the key recognition Asn152 of the enzyme.
Figure 5.
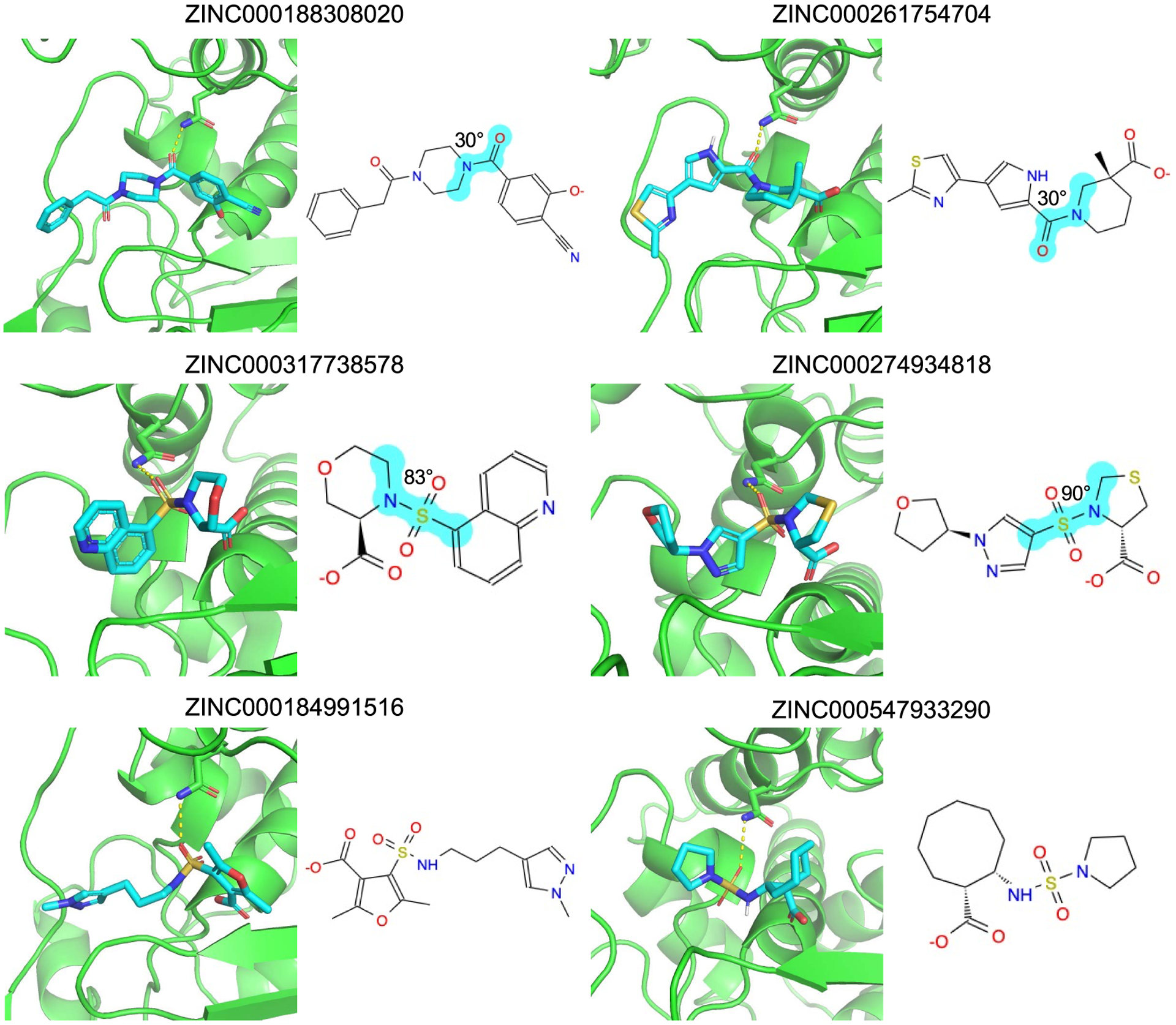
Docking poses of strained decoys (top four) and unstrained binders (bottom two) in AmpC. 2D images of the molecules are also shown. The strained torsions are indicated in cyan and labeled with their degrees out of optimum.
DUD-E Benchmark Tests.
After the case studies of the D4 receptor and AmpC, we investigated the performance against targets from the DUD-E benchmark.40 Here, the decoys are not, as in D4 and AmpC, experimentally measured nonbinders but are based on topological differences from the known ligands. The DUD-E database is a widely used benchmark to test docking.40,41 It includes 102 targets with an average of 224 ligands each and 50 property-matched decoys for each ligand. Compared to AmpC and the D4 receptor, DUD-E targets have the disadvantage of depending on presumed nonbinders vs the experimentally determined nonbinders afforded to us by AmpC and D4. This is balanced by the many DUD-E targets, spanning a wide range of chemotypes, and their wide use in the field. In this DUD-E benchmark test, there were in total 3 127 362 torsions, among which 6493 (~0.2%) were evaluated by the approximate method.
Accordingly, we docked the ligands and decoys for 40 DUD-E systems against their targets and calculated the adjusted Log AUC42 for integrated ligand enrichment before and after strain filtering (Log AUC measures the area under the enrichment curve over the database docked, weighting different order-of-magnitude regions equally, with the range of 0.1–1% of molecules docked weighted the same as 1–10 and 10–100%, thereby upweighting the high-ranking region; the method is adjusted by subtracting the Log AUC expected at random). We used the change in adjusted Log AUC (ΔLog AUC), the Log AUC after strain filtering minus the Log AUC before strain filtering, to measure method performance: a positive ΔLog AUC indicates improvement by filtering for molecular strain.
We measured the effect of different strain energy filter values on integrated enrichment on the DUD-E benchmark (Figure 6), with the left Y-axis the ΔLog AUC at different thresholds (blue bars). As we decrease the value of the strain energy below which molecules are filtered out (stringency increases), ΔLog AUC increases. Said another way, as one filters out compounds with greater and greater stringency (lower and lower strain energy allowed), enrichment improves. For instance, filtering out any compound with a total strain greater than 4 TEU improves ΔLog AUC more than only filtering out compounds with a total strain greater than 8 TEU. When we filter on the single largest torsion energy, the effect is less monotonic, with 1.5 TEU returning the largest ΔLog AUC, followed by 1.0 and 1.8 TEUs. These effects must be balanced against the number of compounds remaining after strain filtering. At thresholds of 1.0 and 1.5 TEUs for the single torsional strain, the percentage of the total remaining compounds fall to only 9.6 and 22.1%, respectively (Figure S2). Accordingly, we preferred a maximum single strain threshold of 1.8 TEU over 1.0 or 1.5 TEU.
Figure 6.
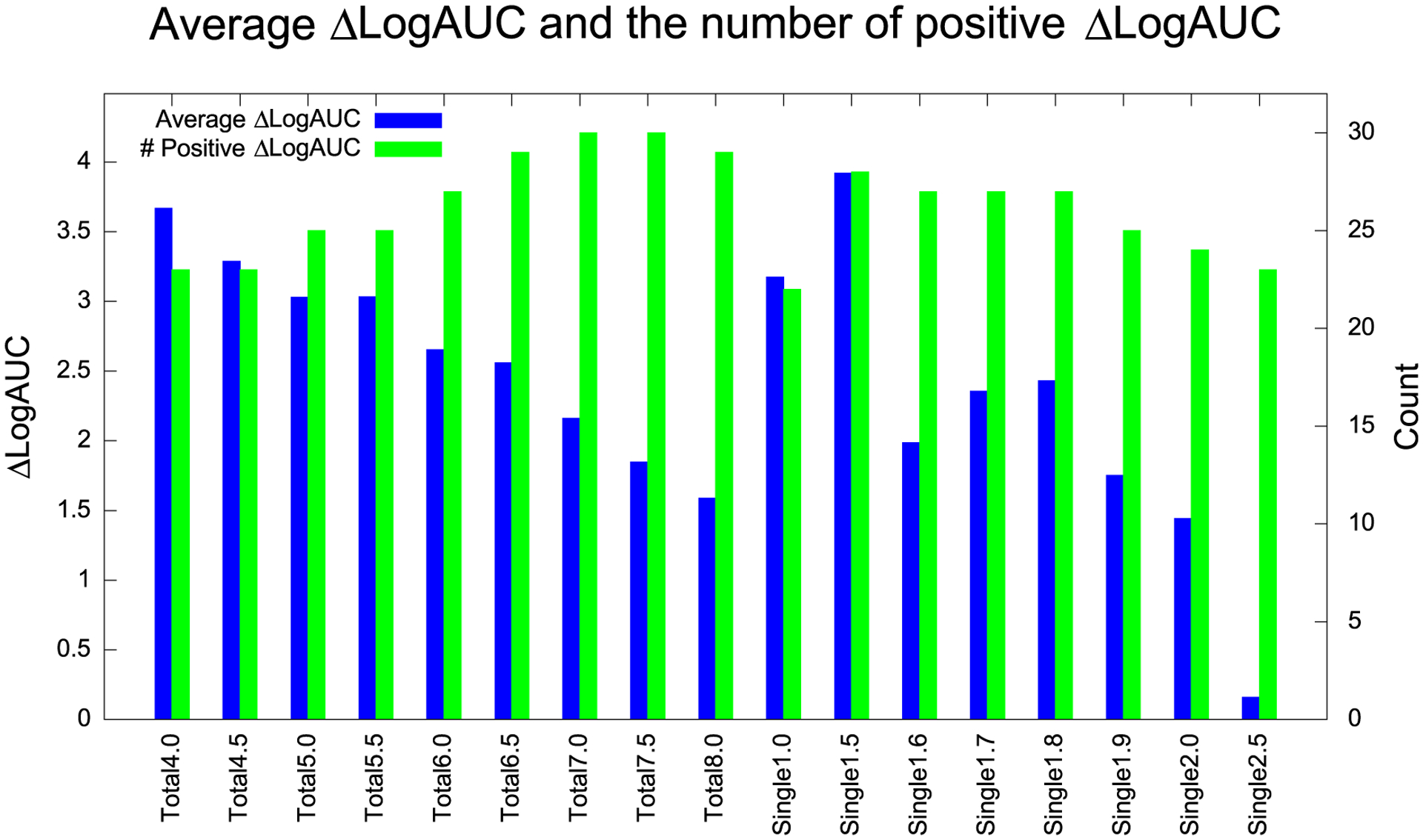
DUD-E benchmark tests at different thresholds (in TEU). Blue is the average ΔLog AUC and green is the number of positive ΔLog AUC among 40 systems.
The effect of the strain energy filters may also be evaluated by the number of systems where integrated enrichment improved (green bars in Figure 6, right Y-axis, over 40 systems). Here, the trend was almost opposite to that for the average ΔLog AUC—as stringency diminished, the total number of systems that improved increased (even though the overall average dropped). Filtering by a total strain energy at 7.0 and 7.5 TEUs saw the highest number of total systems showing improvement, with 30 targets overall, 75% of the systems evaluated, showing improved enrichment. Filtering by the single larger torsional energy in the molecules, 28 systems improved at 1.5 TEU, while filtering at 1.6, 1.7, and 1.8 TEUs all led to improvements in 27 systems. If we take a total of 7.0 TEU and a maximum single strain of 1.8 TEU as filters, an average of 71.4 and 37.1% compounds survives these filters, respectively. This is consistent with what we observed with the D4 receptor and with AmpC β-lactamase: filtering by total strain energy retains more compounds, while filtering by single maximum torsional strain is more stringent. How best to use and combine these filters may depend on context. In large-scale docking, for instance, one is often confronted with an embarrassment of riches, with too many high-scoring molecules to choose; here, the more stringent maximum single torsional filter may be the better choice. Strategies that combine the two criteria may also be imagined.
The results may be broken down by the individual protein in the 40-target benchmark for simplicity, just using a total strain of 7.0 and a maximum single torsional strain of 1.8 TEU (Figure 7). Using the maximum strain filter, 30 targets had improved Log AUC, while at the maximum single torsional strain, 27 targets did. For some targets like renin (RENI) and tryptase β−1 (TRYB1), strain filtering greatly boosted the Log AUC. The ligands of these targets are usually large with multiple rotatable bonds. In Figure S3, we plot ΔLog AUC vs average number of ligand dihedral angles for all of the targets, observing a positive correlation between the ΔLog AUC and the number of ligand- and decoy-rotatable bonds.
Figure 7.
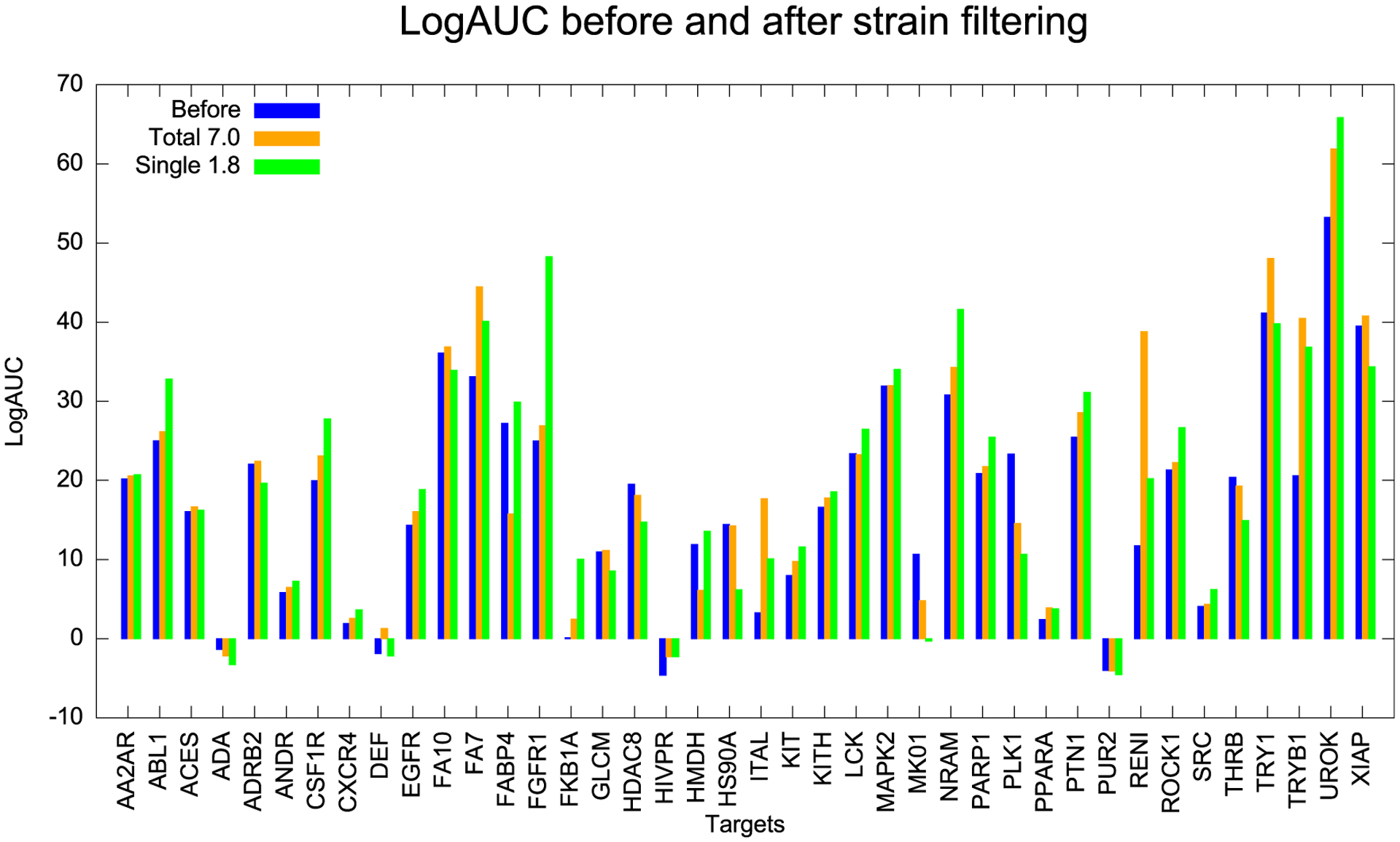
Log AUC before and after strain filtering of 40 systems. Blue is the data before strain filtering. Thirty targets improved at a total strain threshold of 7.0 TEU (orange), while 27 improved at a maximum single strain threshold of 1.8 TEU (green).
As with the D4 and AmpC case studies, we can inspect the conformations adopted by the compounds filtered out by strain for the DUD-E targets (Figure 8). High-ranking decoys like C26299839 and C40966046 break planarity about an amide bond, with an angle of about 30° or more, imparting single energy strains of 2.1 and 3.4 TEUs, respectively, and total strains of 8.4 and 9.5 TEUs, respectively. Molecules like C38700416 and C09344195 have strained torsions that disrupt conjugation of an extra-cyclic group with an aromatic ring. These strained torsions allow these molecules to make favorable interactions with the targets that they otherwise could not (Figure 8). We note that for several systems, using either the total strain energy or a maximum value for a single torsion has different impacts on enrichment, and for several, both led to reduced enrichments. For proteins binding large, flexible ligands, like fatty acid binding protein (FABP4) and HMG-CoA reductase (HMDH), the single torsional filter can lead to better enrichments than does the total strain energy. For docking more flexible molecules, the single torsional filter may be more useful, owing to accumulation of errors as the numbers of torsions increase. For two kinases, both polo-like kinase 1 (PLK1) and mitogen-activated kinase (MK01) strain filters reduced enrichment. For both, a single torsion accounted for most of the calculated strain ([*:1]~[CX3:2]!@[OX2:3]~[*:4] and [*:1]~[CX3:2]!@[CX3:3]~[*:4], respectively). In both cases, the minima populated in the CSD for each torsion were at 0 and 180°, but the preferred docked conformation occurred 60° off these minima, imposing large penalties. The ligands for both targets, however, had large aryl groups at the end of the torsion, something not found in most CSD molecules, and when these torsional energies were calculated quantum-mechanically for the docked molecules, the minima close to the docked conformation or the energies were substantially smaller than implied by the CSD populations. This is a case where the molecules sampled in the CSD are different enough from those docked, around this torsion angle, to make the quantum mechanical approach more accurate when it is applied to the specific molecules in question (something that, given its calculation costs, see below, seems currently impractical for large libraries).
Figure 8.
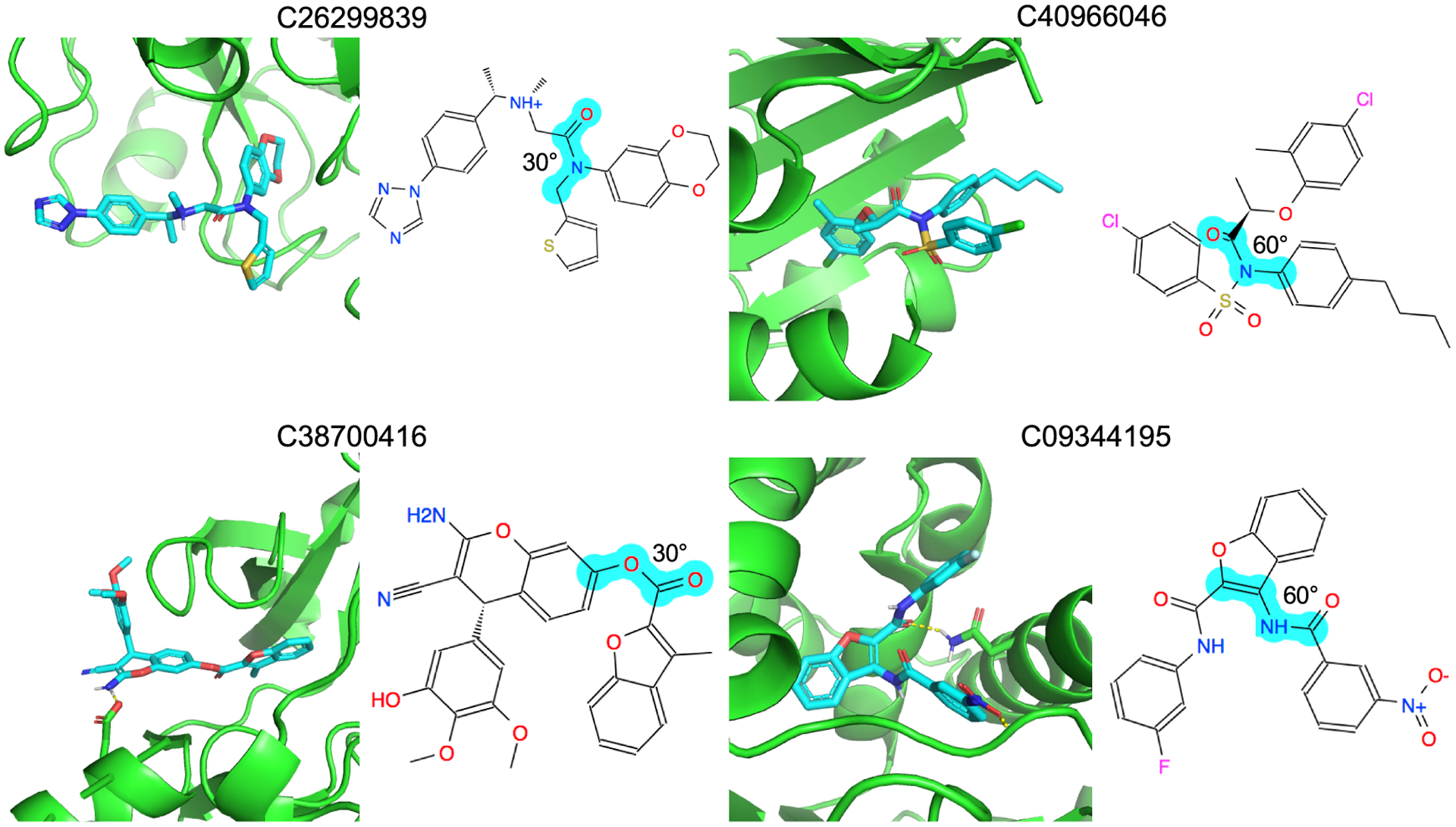
Docking poses of four strained decoys: C26299839 in FA10, C40966046 in ITAL, C38700416 in ABL1, and C09344195 in HS90A. 2D images of the molecules are also shown. The strained torsions are indicated in cyan and labeled with their degrees out of optimum.
Speed and Robustness.
To investigate speed and robustness, we calculated strain energies on three sets of 500 000 compounds each. On a desktop of i5-8400 CPU@2.80 GHz, 16 GB RAM, and CentOS Linux 7.6.1810, it took an average of 319 min to calculate the strain energy for the half-million compounds or less than 0.04 s for each compound. This is fast enough to be used as a postdocking filter. We further performed a parallel test on our cluster, splitting the 500 000 compounds into 100 jobs, each with 5000 compounds. On an Intel Xeon Silver 4210 CPU@2.20 GHz, the running time of each job ranged from 301 to 463 s, with an average of 390. With even a small cluster of 100 cores, this strain energy calculation can be conducted for half-million molecules in 8 min. While this project has focused on using strain as a filter for docking results at the end of a large library campaign, the speed of this calculation suggests that it may be possible to use for library generation, even for the >1 billion molecule ultralarge libraries. Speaking of robustness, in the three tests, the failure rate was <5 per million compounds; almost all of the provided compounds could be properly processed. The ones that failed are usually compounds without any dihedrals, like benzopyrene.
Comparison to Quantum Mechanics Calculated Strain.
To compare the population-based method with a quantum mechanical (QM)-based evaluation of torsional strain, we calculated the strain in 20 representative dihedrals at torsion angles of both 30 and 60° off the minimum (see the Methods section). In the case of certain apparently strained torsions in PLK1 and MK01 inhibitors, the strain was also calculated by QM (see above). Among the 40 energies for the representative dihedrals, the QM strain energy and the population-based one were positively correlated, with an R2 of 0.56 (Figure S4). The slope of 0.22 kcal/mol suggests that the energies in the gas phase in which the QM calculations are undertaken are substantially higher than those observed in the condensed phase of the small molecule crystals in which the populations are observed. We note that it took an average of 33 s to calculate the strain per conformation per core on an Intel i5–1038NG7@2.0 GHz, about 800 times slower than the population-based method, and impractical should we wish to expand it to ultralarge, multiconformer libraries.
DISCUSSION
Applying a strain energy filter to docked compounds improves docking hit rates and integrated enrichment for most targets, at least retrospectively. This is true in cases where both ligands and “decoys” derive from prospective docking screens where the molecules have been shown to bind or not bind by experiment, respectively (as with AmpC and the D4 dopamine receptor), and for cases where the putative nonbinders are property-matched decoys of the sort found in the DUD-E benchmarks. The strain filter improves hit rates and enrichments because the decoy molecules, both experimental and calculated, often find their best-fitting conformations from among those that are relatively strained, compared to the true ligands that can more often complement the receptor in unstrained conformations.
In their seminal paper on conformational strain in docking, Tirado-Rives and Jorgensen argued that the errors in both the strain calculations and overall docking scores were so high, over the 6 kcal/mol or so that defined the range of docking binding energies, as to make even rank ordering a dubious proposition. While this likely remains true, our results may be reconciled with theirs by distinguishing between rank ordering, the focus of their study, and enrichment, the goal of this one. To improve docking enrichment, a strain energy does not need to be even included in the overall docking score; it can be used to filter out molecules that only fit well because they adopt strained conformations. Because these fall more often among decoys than the true ligands, this filter may typically improve docking results.
Several caveats merit airing. Most importantly, this study is retrospective, and the true value of a new docking tool will often only emerge in prospective studies. Mechanically, we calculated Log AUC using the compounds that remained after strain filtering, while those filtered out were not included. Another way of calculating Log AUC would be to include all of the compounds, both ligands and decoys, whether filtered or not. We chose the former to be consistent with the real practice of docking, where one only tests compounds that survive filters and are prioritized by high ranks. Naturally, a strain filter is only needed because library molecules have been calculated in strained conformations.43,44 If these were recognized before docking, the need for this filter would disappear. Indeed, Omega (OpenEye Software, Santa Fe), which we use to precalculate the docking flexibase,45 offers the opportunity to do just that, using a similar statistical potential on which we draw here. In our hands, implementing this at the time of conformation generation undersampled receptor-competent conformations, reducing retrospective hit rates. It may be possible to overcome this in subsequent implementations. We note that rather than using energy strain values as filters, one could simply count violations of allowed ranges. Whereas this would have the advantage of not claiming too much from what are necessarily approximate strain values, it would not easily sum different levels of strain encountered among the different torsions, something to which an energy is well suited. That said, owing to different assumptions and conditions underlying the strain calculation (including no set temperature over the crystals in the CSD, sampling biases in what is crystallized, the effects of crystal packing, the difficulties of modeling rare torsion angles, and not considering differences in local torsional preferences between charged and neutral compounds), we represent strain in “torsion energy units” (TEUs) and do not add them to the overall docking score. Finally, the ligand conformational strain used here could be added directly into the entire docking library, where it could be applied at the time of docking, and not used as a postdocking filter. This would have important advantages and could conceivably be done with methods similar to that used here. Whether this will be pragmatic for ultralarge libraries demands further exploration and is outside of the scope of this study.
These caveats should not obscure the principle observations of this work: despite ongoing difficulties with rank ordering in docking,3 a strain energy filter can improve docking enrichment, the first and principal goal of library screening. Especially in the era of ultralarge libraries, where one suffers from problems of abundance, stringency filters like strain energy can be a great advantage by removing lower likelihood candidates; other such filters can also be imagined. Drawing on a database torsion angle approach,24,25 the filter we describe is fast and mechanically reliable, able to treat millions of molecules in minutes; even from a highly diverse library, only 1 in 200 000 molecules fails to have its torsions matched and an energy calculated. Applying such a filter to docked conformations may increase prospective hit rates by eliminating nonbinders that rank well only by adoption of high-energy conformations. The software is openly available to the community (http://tldr.docking.org).
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by R35GM122481 (to B.K.S.) and GM71896 (to J.J.I.). The authors thank the contributors for RDKit, on which we drew heavily, Schrödinger, Inc. for Jaguar and related tools, and OpenEye scientific for Omega. The authors thank members of the Shoichet Lab for testing the software and for helpful discussion and Stefan Gahbauer and Brian Bender for reading this manuscript.
ABBREVIATIONS
- ABL1
tyrosine-protein kinase ABL
- FA10
coagulation factor X
- FABP4
fatty acid binding protein adipocyte
- FGFR1
fibroblast growth factor receptor 1
- FKB1A
FK506-binding protein 1A
- HMDH
HMG-CoA reductase
- HS90A
heat shock protein HSP 90-α
- ITAL
leukocyte adhesion glycoprotein LFA-1 α
- MK01
mitogen-activated protein kinase
- PLK1
polo-like kinase 1
- RENI
renin
- TRYB1
tryptase β−1
Footnotes
The authors declare no competing financial interest.
The strain software developed here is freely available to all at http://tldr.docking.org. DOCK3.7 is available from the authors and is free for academic research (http://dock.compbio.ucsf.edu/DOCK3.7/). The DUD-E systems used in this study are described in Table S1; the DUD-E database itself is freely available at http://dude.docking.org/. The docking scores and strain values for the ligands from the D4 and AmpC screens, with ligand identities, may be found in Tables S2 and S3, respectively.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jcim.1c00368.
Example of torsion energy profile of a SMARTS pattern (Figure S1); average percent of remaining compounds after strain filtering at different conditions (Figure S2); correlation between ΔLog AUC and the ligands’ average number of dihedrals at a threshold total strain of 7.0 TEU (Figure S3); and correlation between torsional energy calculated by QM and by our population-based method (Figure S4) (PDF)
DUD-E Log AUC results before and after strain filtering at different thresholds (Table S1); ligand docking score and strain energy in D4 receptor (Table S2); ligand docking score and strain energy in AmpC (Table S3) (XLSX)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jcim.1c00368
Contributor Information
Shuo Gu, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94143-2550, United States.
Matthew S. Smith, Department of Pharmaceutical Chemistry, University of California, San Francisco, San Francisco, California 94143-2550, United States; Program of Biophysics, University of California, San Francisco, San Francisco, California 94143-2550, United States.
REFERENCES
- (1).Peach ML; Cachau RE; Nicklaus MC Conformational energy range of ligands in protein crystal structures: the difficult quest for accurate understanding. J. Mol. Recognit 2017, 30, No. e2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Boström J; Norrby P-O; Liljefors T Conformational energy penalties of protein-bound ligands. J. Comput.-Aided Mol. Des 1998, 12, 383. [DOI] [PubMed] [Google Scholar]
- (3).Tirado-Rives J; Jorgensen WL Contribution of conformer focusing to the uncertainty in predicting free energies for protein–ligand binding. J. Med. Chem 2006, 49, 5880–5884. [DOI] [PubMed] [Google Scholar]
- (4).Cecchini M; Kolb P; Majeux N; Caflisch A Automated docking of highly flexible ligands by genetic algorithms: a critical assessment. J. Comput. Chem 2004, 25, 412–422. [DOI] [PubMed] [Google Scholar]
- (5).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JJ; Mainz DT; Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (6).Murphy RB; Repasky MP; Greenwood JR; Tubert-Brohman I; Jerome S; Annabhimoju R; Boyles NA; Schmitz CD; Abel R; Farid R; Friesner RA WScore: A Flexible and Accurate Treatment of Explicit Water Molecules in Ligand-Receptor Docking. J. Med. Chem 2016, 59, 4364–4384. [DOI] [PubMed] [Google Scholar]
- (7).Lam PC; Abagyan R; Totrov M Ligand-biased ensemble receptor docking (LigBEnD): a hybrid ligand/receptor structure-based approach. J. Comput.-Aided Mol. Des 2018, 32, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mobley DL; Dill KA Binding of small-molecule ligands to proteins:“what you see” is not always “what you get”. Structure 2009, 17, 489–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Lyu J; Wang S; Balius TE; Singh I; Levit A; Moroz YS; O’Meara MJ; Che T; Algaa E; Tolmachova K; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Stein RM; Kang HJ; McCorvy JD; Glatfelter GC; Jones AJ; Che T; Slocum S; Huang XP; Savych O; Moroz YS; Stauch B; Johansson LC; Cherezov V; Kenakin T; Irwin JJ; Shoichet BK; Roth BL; Dubocovich ML Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature 2020, 579, 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Gorgulla C; Boeszoermenyi A; Wang ZF; Fischer PD; Coote PW; Padmanabha Das KM; Malets YS; Radchenko DS; Moroz YS; Scott DA; Fackeldey K; Hoffmann M; Iavniuk I; Wagner G; Arthanari H An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Allen FH; Harris SE; Taylor R Comparison of conformer distributions in the crystalline state with conformational energies calculated by ab initio techniques. J. Comput.-Aided Mol. Des 1996, 10, 247–254. [DOI] [PubMed] [Google Scholar]
- (13).Butler KT; Luque FJ; Barril X Toward accurate relative energy predictions of the bioactive conformation of drugs. J. Comput. Chem 2009, 30, 601–610. [DOI] [PubMed] [Google Scholar]
- (14).Wei W; Champion C; Barigye SJ; Liu Z; Labute P; Moitessier N Use of Extended-Hückel Descriptors for Rapid and Accurate Predictions of Conjugated Torsional Energy Barriers. J. Chem. Inf. Model 2020, 60, 3534–3545. [DOI] [PubMed] [Google Scholar]
- (15).Sellers BD; James NC; Gobbi A A Comparison of Quantum and Molecular Mechanical Methods to Estimate Strain Energy in Druglike Fragments. J. Chem. Inf. Model 2017, 57, 1265–1275. [DOI] [PubMed] [Google Scholar]
- (16).Rai BK; Sresht V; Yang Q; Unwalla R; Tu M; Mathiowetz AM; Bakken GA Comprehensive Assessment of Torsional Strain in Crystal Structures of Small Molecules and Protein-Ligand Complexes using ab Initio Calculations. J. Chem. Inf. Model 2019, 59, 4195–4208. [DOI] [PubMed] [Google Scholar]
- (17).Perola E; Charifson PS Conformational analysis of drug-like molecules bound to proteins: an extensive study of ligand reorganization upon binding. J. Med. Chem 2004, 47, 2499–2510. [DOI] [PubMed] [Google Scholar]
- (18).Harder E; Damm W; Maple J; Wu C; Reboul M; Xiang JY; Wang L; Lupyan D; Dahlgren MK; Knight JL; et al. OPLS3: a force field providing broad coverage of drug-like small molecules and proteins. J. Chem. Theory Comput 2016, 12, 281–296. [DOI] [PubMed] [Google Scholar]
- (19).Kim S; Lee J; Jo S; Brooks CL III; Lee HS; Im W CHARMM-GUI ligand reader and modeler for CHARMM force field generation of small molecules. J. Comput. Chem 2017, 38, 1879–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang J; Wolf RM; Caldwell JW; Kollman PA; Case DA Development and testing of a general amber force field. J. Comput. Chem 2004, 25, 1157–1174. [DOI] [PubMed] [Google Scholar]
- (21).Greenidge PA; Kramer C; Mozziconacci JC; Sherman W Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. J. Chem. Inf. Model 2014, 54, 2697–2717. [DOI] [PubMed] [Google Scholar]
- (22).Lee M-L; Aliagas I; Feng JA; Gabriel T; O’donnell T; Sellers BD; Wiswedel B; Gobbi A chemalot and chemalot_knime: Command line programs as workflow tools for drug discovery. J. Cheminf 2017, 9, No. 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Fu Z; Li X; Merz KM Jr. Accurate assessment of the strain energy in a protein-bound drug using QM/MM X-ray refinement and converged quantum chemistry. J. Comput. Chem 2011, 32, 2587–2597. [DOI] [PubMed] [Google Scholar]
- (24).Schärfer C; Schulz-Gasch T; Ehrlich HC; Guba W; Rarey M; Stahl M Torsion Angle Preferences in Druglike Chemical Space: A Comprehensive Guide. J. Med. Chem 2013, 56, 2016–2028. [DOI] [PubMed] [Google Scholar]
- (25).Guba W; Meyder A; Rarey M; Hert J Torsion Library Reloaded: A New Version of Expert-Derived SMARTS Rules for Assessing Conformations of Small Molecules. J. Chem. Inf. Model 2016, 56, 1–5. [DOI] [PubMed] [Google Scholar]
- (26).Sadowski J; Boström J MIMUMBA revisited: Torsion angle rules for conformer generation derived from X-ray structures. J. Chem. Inf. Model 2006, 46, 2305–2309. [DOI] [PubMed] [Google Scholar]
- (27).Bruno IJ; Cole JC; Kessler M; Luo J; Motherwell WS; Purkis LH; Smith BR; Taylor R; Cooper RI; Harris SE; Orpen AG Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci 2004, 44, 2133–2144. [DOI] [PubMed] [Google Scholar]
- (28).Groom CR; Bruno IJ; Lightfoot MP; Ward SC The Cambridge structural database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater 2016, 72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The protein data bank. Nucleic Acids Res 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).James C; Weininger D; Delany J Daylight Theory Manual; Daylight Chemical Information Systems: Laguna Niguel, CA, 2000. [Google Scholar]
- (31).Schärfer C; Schulz-Gasch T; Rarey M TorsionAnalyzer: exploring conformational space. J. Cheminf 2013, 5, No. P3. [Google Scholar]
- (32).Kellogg EH; Leaver-Fay A; Baker D Role of conformational sampling in computing mutation-induced changes in protein structure and stability. Proteins: Struct., Funct., Bioinf 2011, 79, 830–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dunbrack RL Jr.; Cohen FE Bayesian statistical analysis of protein side-chain rotamer preferences. Protein Sci 1997, 6, 1661–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Coleman RG; Carchia M; Sterling T; Irwin JJ; Shoichet BK Ligand pose and orientational sampling in molecular docking. PLoS One 2013, 8, No. e75992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Landrum G Rdkit Documentation, 2013, Vol. 1, pp 1 79. [Google Scholar]
- (36).Meng EC; Shoichet BK; Kuntz ID Automated docking with grid-based energy evaluation. J. Comput. Chem 1992, 13, 505–524. [Google Scholar]
- (37).Sharp KA; Friedman RA; Misra V; Hecht J; Honig B Salt effects on polyelectrolyte–ligand binding: Comparison of Poisson–Boltzmann, and limiting law/counterion binding models. Biopolymers 1995, 36, 245–262. [DOI] [PubMed] [Google Scholar]
- (38).Gallagher K; Sharp K Electrostatic contributions to heat capacity changes of DNA-ligand binding. Biophys. J 1998, 75, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Mysinger MM; Shoichet BK Rapid context-dependent ligand desolvation in molecular docking. J. Chem. Inf. Model 2010, 50, 1561–1573. [DOI] [PubMed] [Google Scholar]
- (40).Mysinger MM; Carchia M; Irwin JJ; Shoichet BK Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Lagarde N; Zagury JF; Montes M Benchmarking Data Sets for the Evaluation of Virtual Ligand Screening Methods: Review and Perspectives. J. Chem. Inf. Model 2015, 55, 1297–1307. [DOI] [PubMed] [Google Scholar]
- (42).Fan H; Schneidman-Duhovny D; Irwin JJ; Dong G; Shoichet BK; Sali A Statistical potential for modeling and ranking of protein–ligand interactions. J. Chem. Inf. Model 2011, 51, 3078–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kothiwale S; Mendenhall JL; Meiler J BCL:: Conf: small molecule conformational sampling using a knowledge based rotamer library. J. Cheminf 2015, 7, No. 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Cole JC; Korb O; McCabe P; Read MG; Taylor R Knowledge-based conformer generation using the Cambridge structural database. J. Chem. Inf. Model 2018, 58, 615–629. [DOI] [PubMed] [Google Scholar]
- (45).Lorber DM; Shoichet BK Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem 2005, 5, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.