

Abstract
The pioneer event in eukaryotic DNA replication is binding of chromosomal DNA by the origin recognitioncomplex (ORC). The ORC-DNA complex directs the formation of origins, the specific chromosomal regions where DNA synthesis initiates. In all eukaryotes, incompletely understood features of chromatin promote ORC-DNA binding. Here, we uncover a role for the Fkh1 (Forkhead homolog) protein and its forkhead associated (FHA) domain in promoting ORC-origin binding and origin activity at a subset of origins in Saccharomyces cerevisiae. Several of the FHA-dependent origins examined required a distinct Fkh1 binding site located 5′ of and proximal to their ORC sites (5′-FKH-T site). Genetic and molecular experiments provided evidence that the Fkh1-FHA domain promoted origin activity directly through Fkh1 binding to this 5′ FKH-T site. Nucleotide substitutions within two relevant origins that enhanced their ORC-DNA affinity bypassed the requirement for their 5′ FKH-T sites and for the Fkh1-FHA domain. Significantly, assessment of ORC-origin binding by ChIPSeq provided evidence that this mechanism was relevant at ∼25% of yeast origins. Thus, the FHA domain of the conserved cell-cycle transcription factor Fkh1 enhanced origin selection in yeast at the level of ORC-origin binding.
INTRODUCTION
Eukaryotic chromosomes rely on multiple spatially and temporally distributed DNA replication origins for their efficient and accurate duplication. Each origin has a distinct probability of activation. Thus, some origins act more efficiently and earlier in S-phase than others. These differences in origin activation probabilities generate a spatiotemporal pattern of genome duplication with relevance to genome stability and cell function (1–5). Because chromatin heterogeneity is essential for the structure and function of chromosomes, each individual origin must act within a distinct local chromatin environment. While it is clear that differences in chromatin structure can affect the probability of origin activation, the specific features of chromatin and the molecular mechanisms by which they control origins are incompletely defined.
Every yeast origin requires the same core proteins and sequential series of protein-DNA mediated reactions for its activation (6). First, the ORC (origin recognition complex) selects origins by binding to the underlying chromosomal DNA (7). In G1-phase, this ORC–DNA complex initiates a a series of molecular interactions culminating in the assembly of an inert form of the DNA replicative helicase, the MCM (mini chromosome maintenance) complex, onto chromosomal DNA (origin licensing) (8). The position of the MCM complex on the chromosomal DNA determines the position of the origin because, in S-phase, multiple proteins convert the loaded MCM complex into two bidirectional replicative helicases that unwind the DNA, allowing for the initiation of DNA synthesis (origin firing or activation). Given the origin-defining role of ORC-DNA interactions in this cycle, it is not surprising that chromatin’s effect on DNA accessibility plays a role in chromosomal origin distribution, with both ORC binding and origin activation being more prevalent within open, transcriptionally active chromatin regions of the genome (2,9). However, direct interactions between ORC and nucleosomes or non-histone chromatin-associated proteins also help recruit ORC to chromosomes, potentially promoting the formation of origin-competent ORC-DNA complexes in less receptive chromatin environments (10–14).
Saccharomyces cerevisiae (yeast) is a powerful model for addressing the role of chromatin in controlling origins because so much is known about this organism’s ∼400 origins through multiple high-resolution genome-scale studies. Data are available about ORC binding, origin efficiency (fraction of cell cycles that an origin fires in a dividing population of yeast cells) and replication time (the minutes after S-phase begins when an origin is replicated) (14–21). High-resolution data describing nucleosome occupancy and modification status as well as binding locations of relevant non-histone chromatin-associated proteins are also available (22,23). Because yeast ORC, unlike ORCs from other model organisms, binds to a defined DNA sequence element that is essential though not sufficient for origin function, the precise position of the origin-determining ORC–DNA interface for most yeast origins is known. Although it may seem paradoxical, the requirement for a specific, discrete ORC-DNA interface has the potential to facilitate defining chromatin-mediated mechanisms relevant to functional ORC-origin binding. In particular, while the ORC site is conserved in yeast origins, its precise sequence and affinity for ORC varies considerably between individual origins (14,17,24). Thus yeast origins can be parsed by the contribution the essential ORC-DNA interface makes to their levels of ORC–origin binding in vivo. Origins with notably weak ORC binding sites can then serve as tools to examine how local chromatin features promote functional ORC–origin interfaces.
Previously, we identified two distinct collections of yeast origins that bind ORC using clearly contrasting mechanisms (24). Specifically, these origins were first selected by their strong affinity for ORC in vivo, as defined by retention of a strong ORC ChIP signal even in orc2-1 cells (17,24). Next, these origins were examined for their ORC-DNA affinity in vitro. Approximately 20 origins were assigned to the positive-DNA cohort (originally called DNA-dependent origins), because they contained high-affinity ORC sites (apparent Kds ranging from ∼4 to 14 nM), whereas another 20 origins were assigned to the positive-chromatin cohort (originally called chromatin-dependent origins), because their ORC sites had a low affinity for ORC in vitro (apparent Kds ranging from ∼30 to > 300 nM) that could not account for their strong association with ORC in vivo. We hypothesized that positive-chromatin origins used a feature(s) of their local chromatin environment to promote ORC–DNA binding (Figure 1).
Figure 1.

Model to explain the ORC-binding differences between two distinct yeast origin cohorts (24). Origins in the positive-DNA cohort rely on direct interactions between ORC (orange crescent) and the the origin’s ORC binding site to achieve normal levels of ORC binding, while origins in the positive-chromatin cohort require additional interactions with an origin-adjacent protein(s) to achieve normal levels of ORC binding to their intrinsically weak ORC sites. The weaker ORC–DNA interactions at positive-chromatin origins are symbolized with open ORC. In this simple model, a sequence-specific DNA binding protein (green cylinder) binds near the origin and promotes ORC binding (arrow). The arrow is not meant to imply a specific mechanism by which the green factor promotes ORC binding.
Here, we applied genetic, molecular and bioinformatic approaches to compare positive-chromatin and positive-DNA origins and uncovered a role for the Fkh1 (forkhead homolog) protein in promoting ORC-origin binding at a subset of yeast origins. The N-terminal FHA (forkhead associated) domain of Fkh1, and more precisely its conserved phosphothreonine-specific protein–protein interaction function, was required by the majority (75%) of positive-chromatin origins for full activity. FHA-dependent origins were twice as likely as the FHA-independent origins to contain a FKH site in a 5′ to 3′ T-rich orientation (5′-FKH-T) positioned within 250 bp and 5′ of their ORC site. Relevant mutations assayed singly and in combination provided evidence that this 5′ FKH-T site contributed to origin activity and accounted for these origins’ FHA-dependence. Converting the low-affinity ORC binding sites within two FHA-dependent positive-chromatin origins to high-affinity ORC sites eliminated their dependence on the Fkh1-FHA domain, supporting the conclusion that the Fkh1-FHA domain promoted ORC binding to these origins. Analyses of ORC-origin binding by ORC ChIPSeq provided evidence that FHA-dependent positive-chromatin origins and many origins outside of this strictly defined cohort required the Fkh1-FHA domain for full ORC binding in vivo.
MATERIALS AND METHODS
Yeast strains and plasmids
Yeast strains were derivatives of W303, and complete genotypes are listed in supplemental Supplementary Table S1. All ARS plasmids were made by cloning yeast origin fragments into the Not1 site of pCF1897, a derivative of the pARS plasmid used in (25). ARS sequences are listed in Supplementary Table S2. The ars1529.5-FKHΔ mutation was engineered into the native S. cerevisiae ARS1529.5 locus by standard one-step integration. First, the Kluyveromyces lactis (Kl) URA3 gene was used to replace the native the S. cerevisiaeARS1529.5 locus from positions -500 to +536, numbering relative to the start ‘0’ nucleotide of the T-rich strand of the ARS1529.5 ORC binding site, to create ars1529.5Δ::URA3kl. This strain was transformed with an ars1529.5-FKHΔ::Sal1 mutant fragment containing nucleotides –550 to +586, and 5-FOA counter-selection was used to select for URA3 replacement events. Successful integration of the ars1529.5-FKHΔ::Sal1 mutant fragment was verified by PCR (oCF4665 and oCF4666) and Sanger sequencing.
Plasmid loss (ARS) assays
Plasmid loss assays were performed as described previously (26–28) with the exception that yeast colonies were grown in 96-well plates as described for the ‘tadpoling’ assay (29). Yeast transformed with an ARS were inoculated into 5 ml of selective media (lacking uracil) and grown at 25°C for ∼20 h, until cultures reached OD600 1.0–1.5. The culture was diluted 1000-fold into 5 ml non-selective media and the diluted culture grown ∼25 h at 25°C. After each round of growth in liquid media, the culture was serially diluted into uracil-lacking or uracil-supplemented media in a 96-well plate (seven 10-fold dilutions). Yeast were grown at 25°C for 36 h, after which colonies were counted. Plasmid loss rates (PLRs) were determined using the formula PLR = 1 – (F/I)n where I (Initial) is the percentage of live cells that contain a plasmid after 20 h of selective growth, F (Final) is the percentage of live cells that contain a plasmid after ∼25 h of non-selective growth and n is the number of cell generations. The values presented are means of all biological replicates for the indicated ARS (≥3 independent yeast transformants) ± standard error.
Basal ARS function
To determine whether ARS1529.5 constructs in Figure 2 D conferred basal ARS activity in wildtype (CFY145) or orc2-1 (CFY266) yeast, four ODs of yeast were transformed with 100 ng of the indicated ARS and cells were grown on solid selective media at 23°C, the permissive growth temperature for orc2-1 cells.
Figure 2.
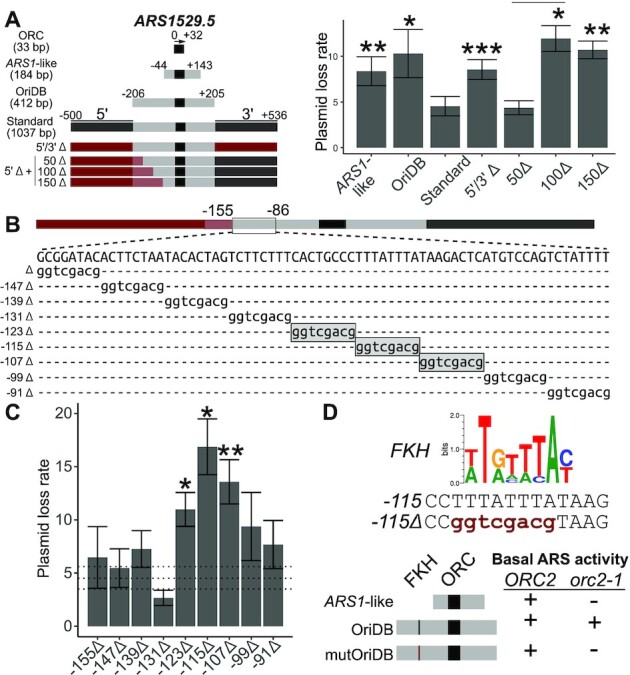
The positive-chromatin origin ARS1529.5 required a 5′ proximal FKH binding site for optimal function. (A) The indicated ARS1529.5 chromosomal fragments (left) were cloned into a plasmid and the corresponding plasmid loss rates (PLRs; mean ± SE) were determined (right). Asterisks here and in all subsequent figures indicate P-values for the relevant comparisons (***P < 0.001, **P < 0.01 and *P < 0.05). P-values are derived from two-tailed Student’s t-tests comparing constructs to ARS1529.5-standard. The sequences that were used to substitute for native ARS1529.5 sequences to create the indicated Δ clones are derived from KANMX and are denoted in red, or red shading when they overlapped OriDB regions (B) DNA sequences between positions –155 and –86 of the ARS1529.5 5′Δ + 50Δ ARS construct were substituted with the 8 bp expanded Sal1 restriction site, gGTCGACg, as indicated. Mutations that significantly raised PLRs (P-values < 0.05) relative to ARS1529.5-standard are boxed in gray shading. (C) PLRs of Sal1 linker-containing ARS1529.5 mutants in (B). Horizontal dashed lines represent the PLR mean ± se of the wild-type ARS1529.5 50Δ fragment in A. (D) (Top) An FKH consensus motif (23). Sequence of the wild-type region of ARS1529.5 containing the FKH site (top), and the SalI linker substitution in ars1529.5-FKHΔ::Sal1. Basal ARS activity is defined as whether colonies were recovered when yeast were transformed with the indicated ARSs. The indicated yeast cells were transformed with the ARS1-like version, OriDB version, or OriDB version containing the FKHΔ::Sal1 substitution (mutOriDB) of ARS1529.5. ‘+’ indicates that colonies were recovered. ‘−’ indicates that no colonies were recovered.
Cell-cycle arrest and flow cytometry
To prepare genomic DNA samples for copy-number analyses, MATa cells were grown in 25 mL YPD at 30°C to an OD600 of ∼0.3 at which time α factor was added to a concentration of 5 μg/ml. Cultures continued to incubate at 30°C for 100 min to allow the majority of cells to reach G1-arrest. Cultures were then collected on a Whatman Nylon membrane (GE Healthcare #7404-004) using a Nalgene filtering apparatus (Thermo Scientific #300-4050) and washed with 100 ml YPD pre-warmed to 30°C. The cells were then immediately transferred to fresh pre-warmed 25 ml YPD supplemented with 200 μg/ml of pronase. 2.5 ml of culture was harvested at time 0 (G1-arrested sample) and at 35 min (mid S-phase) after release into YPD-pronase. Genomic DNA was purified from Zymolase-treated yeast pellets using phenol–chloroform–isoamyl alcohol, followed by ethanol-based extraction. DNA was quantified on a Quantus fluorimeter. To determine cell-cycle distribution of each sample, cells were fixed with ethanol, stained with Sytox Green and analyzed by flow cytometry (BD Accuri C6 Flow Cytometer).
Droplet digital PCR (ddPCR)
ddPCR was performed following a published protocol for examining yeast DNA replication (30) using Evagreen based chemistry (Droplet oil and Supermix; BioRad #1864005 and #1864033, respectively). ddPCR was performed on the indicated origins or non-origin control loci. Primers are listed in Supplementary Table S3.
Apparent Kds for ORC-origin binding
Recombinant yeast ORC purified from Sf9 cells was used in binding reactions with radioactively labeled origin DNA fragments as substrates as described previously (24). The reactions were analyzed using electrophoretic mobility shift (EMSA) assays, and radioactive ORC–DNA and free DNA complexes were imaged using a GE Typhoon SLA9000 phosphoimager. Images were analyzed in ImageJ, and apparent Kd values were derived by fitting data to a one-site binding hyperbola and constraining the Bmax to 1 in Graphpad Prism 4.0. Each reaction was performed in triplicate and the mean apparent Kd was normalized to that measured for the high-affinity internal control ARS317 (HMR-E) probe (appKd 7 nM).
ORC ChIP-qPCR
ORC ChIP-qPCR was performed using the XChIP protocol (31). 50 ml yeast cultures of three biological replicates per strain (wildtype CFY3533 and ars1529.5-FKHΔ::Sal1 CFY4479 ) were grown to mid-log phase (OD600 ∼0.5 OD) and cross-linked with 1% formaldehyde for 15 min. Chromatin was purified and fragmented with MNase (Worthington Biochem #LS004797) at a ratio of 0.5 MNase units/OD yeast until a 1:1 ratio of 150:300 bp fragments was reached (indicating a 1:1 ratio of mono- to dinucleosomes). Digested chromatin was briefly sonicated and then clarified. Ten percent of the sheared chromatin was reserved to represent starting material (Input), while the remainder was incubated with a cocktail of monoclonal antibodies against ORC as described previously (17). Quantitative PCR was performed in a BioRad C1000 Touch thermocycler using GoTaq mix (Promega #A6001) (32). Primer pairs for target loci are listed in Supplementary Table S3. Primers were validated in a standard curve assay, demanding E and R2 values ≥ 0.8 and ≥ 0.95, respectively. IP and Input samples were diluted 1.2- and 2.5-fold, respectively, and technical duplicate reactions were performed. The mean of the resulting Cq values from technical duplicates were calculated, demanding technical errors ≤1.5% (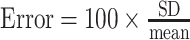 ). Technical means were further normalized by the relative dilution factor using the following equation: normalized means = mean Cq − log 2(DF). Finally, percent input values were calculated for each target measured in each biological replicate for each experiment:
). Technical means were further normalized by the relative dilution factor using the following equation: normalized means = mean Cq − log 2(DF). Finally, percent input values were calculated for each target measured in each biological replicate for each experiment:
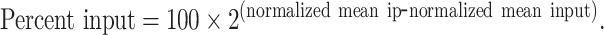 |
ORC ChIPSeq
ORC ChIPSeq was performed using the XChIP protocol (31). Crosslinked chromatin was isolated from 50 ml cultures of FKH1 or fkh1R80A cells at 0.5 OD/ml. Chromatin was fragmented as described and subjected to ORC directed IP as in (14,17). Libraries for next generation sequencing were prepared using the Lucigen NxSeq UltraLow DNA Library Kit v2 (#15012-2). Three independent replicates of each genotype were sequenced. The ORC signal was determined for the IP and starting material and per nucleotide coverages were assessed. Coverages from like replicates were summed and then normalized for sequencing breadth and depth as described (31). Once normalized, per nucleotide IP/input ratios were determined and mapped to 2001 bp origin fragments centered and oriented to the T-rich start of ORC site matches (n = 393). Mapped ratios from either FKH1 or fkh1R80A cells at each locus were internally scaled by taking the median ratio measured within distal, flanking positions within the fragment (–1000 to –700 and +700 to +1000), and dividing each ratio within the fragment as in (31).
Accessing genomic data
The raw data for the ORC ChIPSeq experiment can be accessed at BioProject PRJNA69402 and for the Fkh1/2 ChIPchip at GEO GSE165464.
RESULTS
Nomenclature for origin fragments
The ARS assay (autonomous replicating sequence) assay is useful for defining chromosomal DNA sequences required for yeast origin activity (25,27,33). In an ARS assay, a chromosomal fragment is cloned into a bacterial plasmid backbone and plasmid replication efficiency in yeast is measured as a plasmid loss rate (PLR). An efficient origin generates a low PLR (≤5%/generation) . To compare positive-chromatin and positive-DNA origins in a systematic manner, we measured ARS activity of standard origin fragments from 32 origins - sixteen each from the positive-chromatin and postive-DNA cohorts. The standard fragment was defined as a 1037 bp chromosomal region centered on the origin’s ORC site (Figure 2 A). The start of the ORC site’s T-rich strand was designated position ‘0’, and nucleotides 5′ and 3′ of ‘0’ were assigned negative or positive numbers, respectively. Thus the standard ARS fragment contained 500 bp 5′ and 536 bp 3′ of the ‘0’ position of the T-rich strand of the origin’s ORC site (i.e. the standard origin fragment spanned nucleotides –500 to +536). The yeast DNA replication origin database (www.OriDB.org) systematically summarizes information about each yeast chromosomal origin, including annotating the chromosomal regions that can function as ARSs as reported in the published literature (34). For ARS1529.5, a positive-chromatin origin, the annotated oriDB ARS fragment is 412 bp (–206 to +205) and was used in some experiments. For reference, the paradigmatic yeast origin, ARS1, defined as the –44 to +143 chromosomal region centered on ARS1’s ORC site, is considerably smaller than the standard origin fragments and the ARS1529.5-OriDB origin fragment used here (25).
The positive-chromatin origin ARS1529.5 required a 5′ proximal FKH binding site for optimal activity
ARS1529.5, like most positive-chromatin origins, is an early and efficient origin (24). Thus, the expectation was that the ARS1529.5-oriDB fragment would be an efficient ARS. However, ARS1529.5-OriDB and ARS1529.5-ARS1-like chromosomal regions produced plasmids that generated relatively high PLRs of ∼10% (Figure 2 B). In contrast, ARS1529.5-standard generated a low PLR of 5%. A construct containing a mutation of the ARS1529.5 ORC site abolished function of ARS1529.5-standard, indicating that enhanced activity of this fragment was not caused by a second cryptic ORC site. The enhanced activity of ARS1529.5-standard was not due to insulating this ARS from inhibitory plasmid sequences because replacement of the flanking chromosomal regions with KANMX sequences reduced ARS1529.5 activity back to that of ARS1529.5-OriDB (5′/3′Δ, Figure 2 A).
The paradigm for yeast origin structure places key regulatory DNA elements 3′ of the T-rich strand of the essential ORC site (25). However, a few yeast origins contain important DNA sequences 5′ of the ORC site, termed domain C, though the precise nature of these elements and their modes of action are unclear (35,36). Therefore, we were motivated to explore the requirement of the 5′ region of ARS1529.5 more closely. ARS assays of additional ARS1529.5 constructs lacking 5′ origin-adjacent sequences indicated that chromosomal DNA between nucleotides -151 and -102 contained element(s) that promoted ARS activity (Figure 2A). Sal1 substitutions were introduced throughout the region, and ARS assays were performed on the resulting mutants (Figure 2B). Three adjacent Sal1 substitutions reduced the function of ARS1529.5, with the central of these (–115Δ:Sal1) causing the most severe defect (Figure 2C). The -115Δ:Sal1 substitution mutated a sequence that matched the consensus DNA binding motif of the paralog cell-cycle transcription factors Fkh1 and Fkh2 (henceforth called an FKH motif). Thus, this result raised the possibility that either the Fkh1 or Fkh2 or both proteins bound 5′ of the T-rich strand of this origin’s ORC site to promote ARS1529.5 activity (Figure 2D).
The screen that defined ARS1529.5 as a positive-chromatin origin demanded that ARS1529.5 retain ORC binding in vivo even under conditions where the level of functional ORC was reduced substantially by the orc2-1 mutation (24). If the 5′ FKH site identified above was required to promote ORC binding to ARS1529.5’s weak ORC site in vivo, then it should be critical for ARS1529.5 function in orc2-1 mutant yeast. To test this prediction, we performed a basal ARS activity assay, which simply assesses whether yeast can be transformed with a plasmid (Figure 2D). Wildtype but not orc2-1 yeast could be transformed with the ARS1-like version of ARS1529.5. In contrast, both wildtype and orc2-1 yeast could be transformed with ARS1529.5-OriDB, indicating that the additional chromosomal regions present on this fragment contained a sequence that allowed for ARS activity even when ORC function was reduced by the orc2-1 mutation. While wildtype could be transformed with ARS1529.5-OriDB containing the FKH site mutation (–115Δ::Sal1 or ars1529.5-FKHΔ), orc2-1 mutant yeast could not. Therefore, the 5′ FKH binding site promoted ARS1529.5 function under conditions of limiting ORC, meeting a criterion for a postulated accessory DNA element that promotes ORC binding to positive-chromatin origins (Figure 1).
The Fkh1-FHA domain promoted ARS activity through the FKH site positioned 5′ of the ARS1529.5 ORC site
Fkh1 contains two conserved domains, a DNA binding domain and a forkhead associated (FHA) domain (Figure 3A). FHA domains are conserved protein-binding modules with specificity for phosphorylated threonines (37,38). A conserved arginine (R) residue within FHA domains is essential for phosphothreonine binding. A mutant allele of FKH1, fkh1R80A, produces normal levels of Fkh1 protein but abolishes the phosphothreonine binding function of Fkh1 (39,40).
Figure 3.
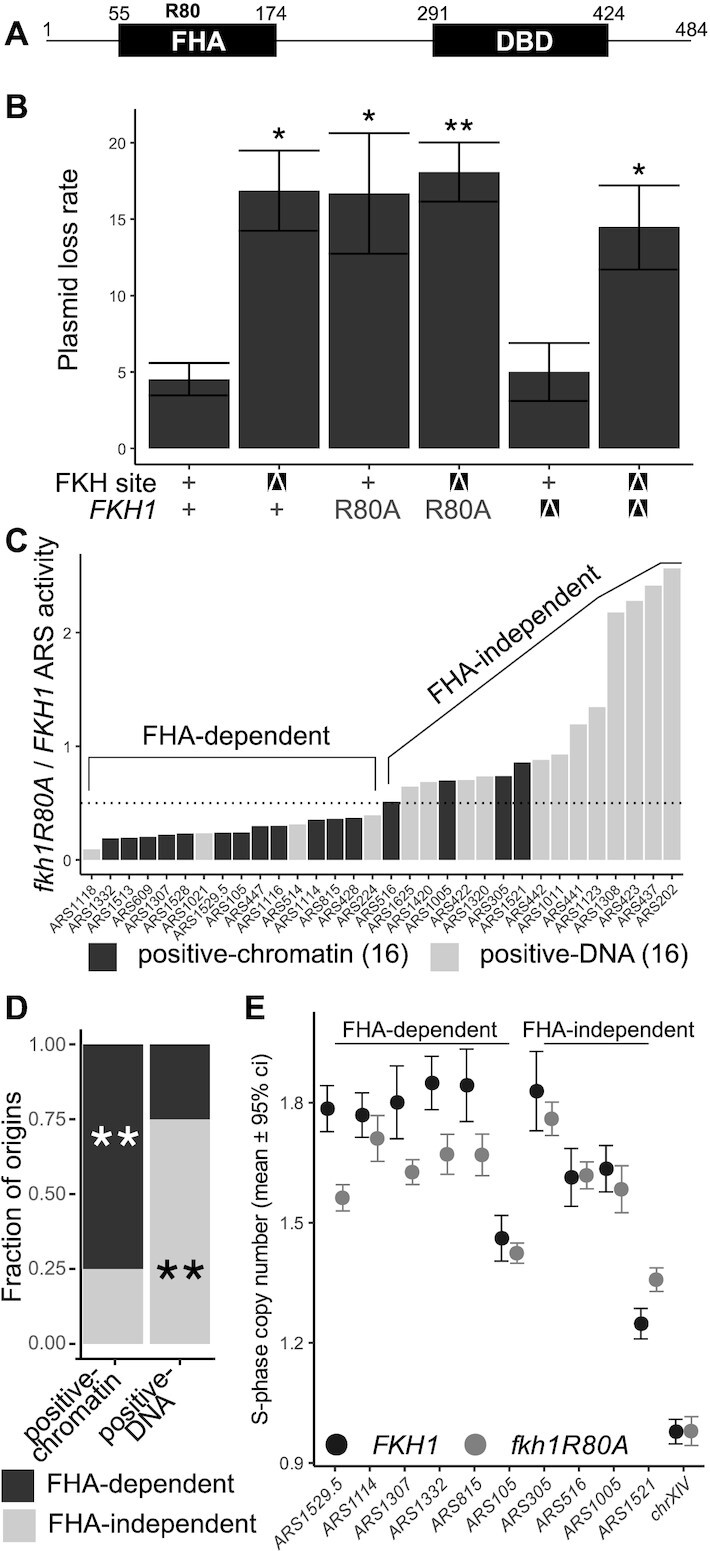
The Fkh1-FHA domain functioned through the FKH site in ARS1529.5 and contributed to the ARS function of the majority of positive-chromatin origins. (A) Diagram of the Fkh1 protein and relevant protein domains. Residue 80 of Fkh1 is a conserved arginine essential for the threonine-phosphopeptide-binding activity of FHA domains (44–46)(38). (B) PLRs measured for the indicated ARS1529.5 plasmids in FKH1, fkh1R80A, or fkh1Δ cells. P-values are derived from two-tailed Student’s t-tests comparing constructs to ARS1529.5-standard. (C) ARS activities (inverse of plasmid loss rates) were determined for the indicated ARSs, cloned as standard fragments (see Figure 2A), in fkh1R80A and FKH1 cells. The data are expressed as ratios of ARS activity measured in fkh1R80A cells to that measured in FKH1 cells. The ARSs are ordered from most sensitive (FHA-dependent) to least affected (FHA-independent). (D) Enrichment of FHA-dependent ARSs (fkh1R80A-sensitive) within the positive-chromatin and positive–DNA origin groups. P-values were derived from a hypogeometric functon comparing the distribution of FHA-dependent or -independent origins in each cohort to their distribution in the entire collection of 32 origins. Significance is denoted as in Figure 2. (E) Chromosomal S-phase copy numbers as measured by ddPCR for six FHA-dependent and four FHA-independent ARSs in C. For each locus, 6 (for wild-type) or 12 (for fkh1R80A mutant strain) independent S-phase cell samples were assessed and the mean ± 95% confidence interval determined (see Supplementary Figure S1). Asterisks indicate P-values from the relevant tests in panels B and D (***P < 0.001, **P < 0.01 and *P < 0.05).
To test whether the Fkh1-FHA domain contributed to ARS1529.5 function, wild-type and fkh1R80A yeast were transformed with plasmids that were replicated by either wild-type ARS1529.5 or ars1529.5-FKHΔ. The ARS function of wild-type ARS1529.5 was reduced in fkh1R80A mutant cells to a level that was indistinguishable from that of ars1529.5-FKHΔ in wild-type cells, revealing that the trans-acting fkh1R80A mutation phenocopied the cis-acting FKH site mutation’s effect on ARS1529.5 activity (Figure 3B). In addition, the activity of ars1529.5-FKHΔ was not further reduced by the fkh1R80A mutation, indicating that the 5′ FKH-T site and the Fkh1-FHA domain did not have independent roles in promoting ARS1529.5 function. These data provided in vivo evidence that the Fkh1-FHA domain promoted ARS1529.5 activity by binding to the ARS1529.5 5′ FKH site.
Due to their substantial sequence similarity, the paralogs Fkh1 and Fkh2 can substitute for each other’s functions in cell-cycle control of transcription (41,42). In addition, the majority of early-acting yeast origins that were previously defined as Fkh1/2-activated origins show altered activity only in the absence of both Fkh1 and Fkh2 (43). However, the ARS data in Figure 3B indicated that the Fkh1-FHA domain function could explain ARS1529.5’s requirement for its 5′ FKH site, indicating that this defective version of Fkh1 could not be rescued by Fkh2. The simplest explanation for this outcome was that mutant fkh1R80A protein remain bound to the ARS1529.5 5′ FKH site. If this explanation were correct, then fkh1Δ FKH2 cells should show no defect in ARS1529.5 activity because, in the complete absence of Fkh1, Fkh2 should now be able to bind the 5′ FKH site. Consistent with this explanation, fkh1Δ cells replicated ARS1529.5 as efficiently as wild-type cells. In addition, this ARS activity required the same 5′ FKH site that Fkh1 required (Figure 3B). Thus, in wild-type cells, the relevant ARS1529.5 FKH site was bound by Fkh1, not Fkh2. Moreover, while the fkh1R80A mutant protein could bind to this site and prevent Fkh2 from binding, it could not perform a post-Fkh-binding function required for full ARS1529.5 activity.
The majority of positive-chromatin origins required the Fkh1-FHA domain for full activity
ARS1529.5 was one of the 20 positive-chromatin origins identified (24). To test whether the Fkh1-FHA domain was required by other positive-chromatin or by any positive-DNA origins, the standard versions of sixteen ARSs from each cohort were assessed for ARS activity in wild-type and fkh1R80A mutant cells. The PLRs for each ARS were converted to ARS activities (ARS activity = 1/PLR) and the ratio of ARS activity for each origin in fkh1R80A to wild-type yeast was plotted in Figure 3 C. The fkh1R80A mutation caused a >2-fold reduction in ARS activity for twelve of the sixteen positive-chromatin origins. In contrast, only four of the sixteen positive-DNA ARSs showed a >2-fold reduction in ARS activity in fkh1R80A mutant yeast. Thus, while the positive-chromatin origin cohort was enriched for Fkh1FHA-dependent ARSs, the positive-DNA cohort was instead enriched for Fkh1FHA-independent ARSs (Figure 3D).
To address whether the Fkh1-FHA domain contributed selectively to the normal replication of FHA-dependent ARSs within their native chromosomal locus, we used droplet digital PCR (ddPCR) as a sensitive measure of DNA copy number as in (30). If the activity of a specific origin is reduced by fkh1R80A, either through altering its efficiency or delaying its activation time, then the duplication of that origin locus should be delayed, resulting in a reduction in its DNA copy number in S-phase relative to control loci. For these experiments, genomic DNA was isolated from S-phase cells after release from a G1 alpha-factor arrest (Supplementary Figure S1). The S-phase copy number of 10 positive-chromatin chromosomal origins was assessed (Figure 3E); six of these showed FHA-dependent ARS activity and four did not (see Figure 3C). Four of the six FHA-dependent positive-chromatin ARSs showed significant reductions in S-phase DNA copy number in fkh1R80A yeast, as expected if their origin activity in their native chromosomal context was reduced (30). In contrast, none of the FHA-independent ARSs showed replication defects. Notably, some of the individual positive-chromatin ARSs assessed here were previously identified as Fkh1/2-activated origins, yet only a subset of these showed significant reductions in S-phase DNA copy number in fkh1R80A yeast (e.g. ARS305 is a Fkh1/2-activated origin. Yet, ARS305 is an FHA-independent ARS (Figure 3 C), and the fkh1R80A mutation did not affect ARS305 chromosomal function (Figure 3 E)). These data provided evidence that the FHA-dependent mechanism was distinct from the previously documented Fkh1/2-activation mechanism (23,47,48). In support of this conclusion, additional bioinformatic analyses of positive-chromatin and positive-DNA origins revealed that the differences between these two origin cohorts were more tightly linked to their proposed differences in ORC-origin binding used to classify them than to either differences in their origin activation time or mode of Fkh1/2-regulation (Supplementary Figure S2).
FHA-dependent origins were enriched for T-rich-oriented FKH motifs positioned 5′ of their ORC binding sites (5′-FKH-T sites)
Previous analyses of Fkh1/2-regulated origins revealed that Fkh1/2-activated and -repressed origin groups show significant quantitative differences in Fkh1/2 binding (23). This result is consistent with the model that Fkh1/2 act as recruitment factors to enhance the local concentration of the S-phase kinase that modifies the MCM complex and triggers origin firing (47). However, while we could recapitulate this observation in a genome-scale Fkh1 binding experiment, the FHA-dependent and -independent origin cohorts defined in this report behaved differently, generating similar levels of Fkh1/2-association (Supplementary Figure S3). Thus, while differential binding of Fkh1/2 could account for the distinct behaviors of Fkh1/2-activated and -repressed origins, this explanation was insufficient to account for the differences between FHA-dependent and FHA-independent cohorts.
To explore other possible explanations, the positions and orientations of the FKH motif matches were mapped across a 600 bp chromosomal region containing the FHA-dependent or -independent origins as defined in Figure 3 C (Figure 4A and B, Supplementary Figure S4). The total number of FKH motifs was similar for the FHA-dependent and -independent origin cohorts (44 motifs within the 16 FHA-dependent origin fragments versus 40 motifs among the 16 FHA-independent origin fragments). However, the two cohorts differed in how their FKH motifs were oriented and distributed relative to their ORC sites (Figure 4B and C). Specifically, origins within the FHA-dependent cohort were more likely to contain an FKH-T motif 5′ of their ORC site (5′ region; 5′-FKH-T), while origins within the FHA-independent cohort were more likely to contain an FKH-T motif 3′ to their ORC site (3′ region; 3′-FKH-T). To examine these FKH motif distributions quantitatively, the fraction of origins within the indicated cohort containing the indicated FKH motif orientation match (FKH-T or FKH-A, as in Figure 4A) was determined for the 5′ and 3′ regions of the indicated origin groups (Figure 4C). These analyses confirmed that the FHA-dependent and -independent cohorts differed from each other most strikingly in terms of the position and orientation of their FKH motifs relative to their ORC sites.
Figure 4.

FHA-dependent ARSs were enriched for T-rich oriented FKH motifs positioned 5′ of their ORC binding sites. (A) FHA-dependent and -independent origin fragments were examined for FKH site matches in either the T-rich (FKH-T) or the A-rich (FKH-A) orientation relative to the T-rich ORC site as depicted. Origin fragments were defined as chromosomal regions with approximately 300 nucleotides 5′ of the ORC site start of ‘0’ (–300 to –1, 5′ region) and 3′ of the end of the ORC site ‘+32’ (+33 to +300, 3′ region). (B) Positions of FKH-T and FKH-A motifs in FHA-dependent (left) and FHA-independent (right) origins. Vertical dotted lines through each graph demarcate the T-rich ORC site boundaries (0 to +32) used to align each origin. (C) Fraction of origins in the indicated cohorts (x-axis) with at least one of the indicated FKH motifs as defined in A (see also Supplementary Figure S3). Asterisks indicate P-values from the Hypergeometric Distribution function comparing a cohort’s fraction with that measured for all origins (dashed line) (***P < 0.001, **P < 0.01 and *P < 0.05). (D) The 5′ FKH-T sites were replaced with an expanded Sal1 restriction site, gGTCGACg, as with ARS1529.5 in Figure 2. The positions of the substituted sites are indicated with a red X. E) Plasmid loss rates for the indicated ARSs were determined in FKH1 or fkh1R80A cells.
Because the FHA-dependent origin cohort was enriched for origins with a 5′-FKH-T motif, we further examined the potential relevance of this motif. First, analyses of Fkh1/2-binding data revealed that the FHA-dependent cohort was enriched for origins that generated Fkh1-ChIP peak apexes 5′ of their ORC sites (Supplementary Figure S3). Second, direct substitution of the 5′-FKH-T motif as for ARS1529.5 in Figure 2 in four additional FHA-dependent ARSs revealed that they also required 5′-FKH-T motifs for Fkh1-FHA dependent ARS function (Figure 4E). Note that the activity of ARS1114 was only mildly reduced by mutation of its 5′-FKH-T motif or by the fkh1R80A mutation, consistent with the chromosomal replication data for ARS1114 documented in Figure 3E. Together, these observations revealed a link between a 5′-FKH-T site and FHA-dependent origin activity within the subset of origins that comprised the positive-chromatin cohort.
A high-affinity ORC site could bypass the requirement of a 5′-FKH-T site for ARS function
The majority of positive-chromatin origins showed Fkh1-FHA-dependent ARS activity (12/16, 75%) (Figure 3C). Positive-chromatin origins were defined by the inability of their low-affinity ORC sites to explain their efficient ORC-origin association in vivo (24). Therefore, we postulated that converting the low-affinity ORC sites within positive-chromatin origins to high-affinity ORC sites might bypass these origins’ requirement for the Fkh1-FHA domain for full activity (Figure 5). To address this possibility, specific nucleotide substitutions were engineered within the ORC sites of two different positive-chromatin FHA-dependent ARSs with the goal of enhancing their intrinsic affinity for ORC (defined as the affinity that ORC has for a purified origin fragment in vitro as measured by in vitro gel shift assays (24) (Figure 5A, Supplementary Figure S5). For ARS1529.5, two different ORC-site-mutations were engineered: ARS1529.5-Δ4, where nucleotide substitutions were used to make the ORC site more similar to the consensus ORC binding motif, and ARS1529.5-Δ514, where the entire ARS1529.5 ORC site (apparent Kd = 192 ± 18 nM) was replaced with the high-affinity ORC site from ARS514 (apparent Kd = 4.1 ± 0.7 nM) (24). For ARS1114 (apparent Kd = 33.3 ± 2 nM), two independent ORC site mutations were generated: ARS1114-C1T, and ARS1114-C30T, with the goal of making the ARS1114 ORC site more similar to the consensus ORC binding motif. In vitro ORC-DNA binding assays were performed as described previously and the binding affinity of these new ORC sites relative to a reference high-affinity ORC site (ARS317 (HMR-E), apparent Kd = 7.2 ± 0.4 nM) was determined (Supplementary Figure S5). Three of the ORC site variants, ARS1529.5-Δ4, ARS1529.5-Δ514 and ARS1114-C30T bound ORC with a high-affinity, while one variant, ARS1114-C1T, failed to enhance the affinity of this ORC site for ORC in vitro (Figure 5 B).
Figure 5.
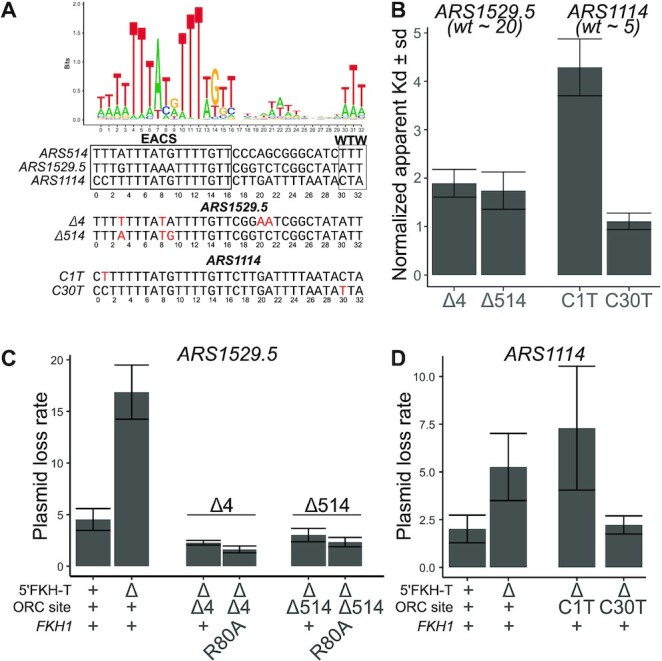
High-affinity ORC sites bypassed requirements for the 5′ FKH-T site and the FHA domain. (A) Top: Web Logo for ORC consensus site derived from 393 chromosomal origins. Bottom: ORC binding sites for three indicated origins, and below that, the nucleotide substitutions, indicated in red, used to generate high-affinity mutant ORC binding sites. (B) The Kds measured by gel shift assays with purified ORC and origin DNA fragments were normalized by dividing by the Kd measured for the tight-binding ORC binding site from ARS317 (see Supplementary Figure S5). The normalized Kds for wildtype ARS1529.5 and ARS1114 are indicated in parentheses below the text of those origins (24). (C) Plasmid loss rates for wildtype and the indicated mutant versions of ARS1529.5 in either FKH1 or fkh1R80A cells. (D) Plasmid loss rates for wild-type and the indicated mutant versions of ARS1114 in FKH1 cells.
The ARS activity of each of these mutant origins was determined (Figure 5 C). Both of the ARS1529.5 mutants containing high-affinity ORC sites functioned efficiently even in the absence of a 5′-FKH-T site or the absence of a functional Fkh1-FHA domain. The ARS1114-C30T mutant that enhanced affinity for ORC also bypassed this ARS’s dependence on its 5′ FKH-T site. However, the ARS1114-C1T mutation that did not enhance ORC-origin binding affinity in vitro still required the 5′-FKH-T site of ARS1114 for wild-type ARS1114 activity. Thus, a high-affinity ORC site could bypass the requirement of the 5′-FKH-T site for the efficient ARS function of these FHA-dependent positive-chromatin origins.
The Fkh1-FHA domain promoted ORC-origin interactions at a substantial fraction of yeast chromosomal origins
The data presented thus far supported the model in Figure 1 for FHA-dependent positive-chromatin origins, wherein Fkh1 binds to a 5′-FKH-motif adjacent to these origins’ ORC sites and uses its FHA domain to promote the essential ORC-DNA interaction. To test whether the 5′-FKH-T motif promoted ORC binding to chromosomal ARS1529.5, the locus was precisely replaced with a ars1529.5-FKHΔ mutant, and ORC association with ARS1529.5 was assessed by ORC-directed ChIP and qPCR (Figure 6A). The ORC ChIP signal generated by the ars1529.5-FKHΔ mutant cells was 2.5-fold lower than the signal generated by the wild-type ARS1529.5 cells, while the control locus was unaffected. These data provided evidence that the 5′-FKH-T motif in ARS1529.5 promoted ORC binding to this origin within its native chromosomal context.
Figure 6.
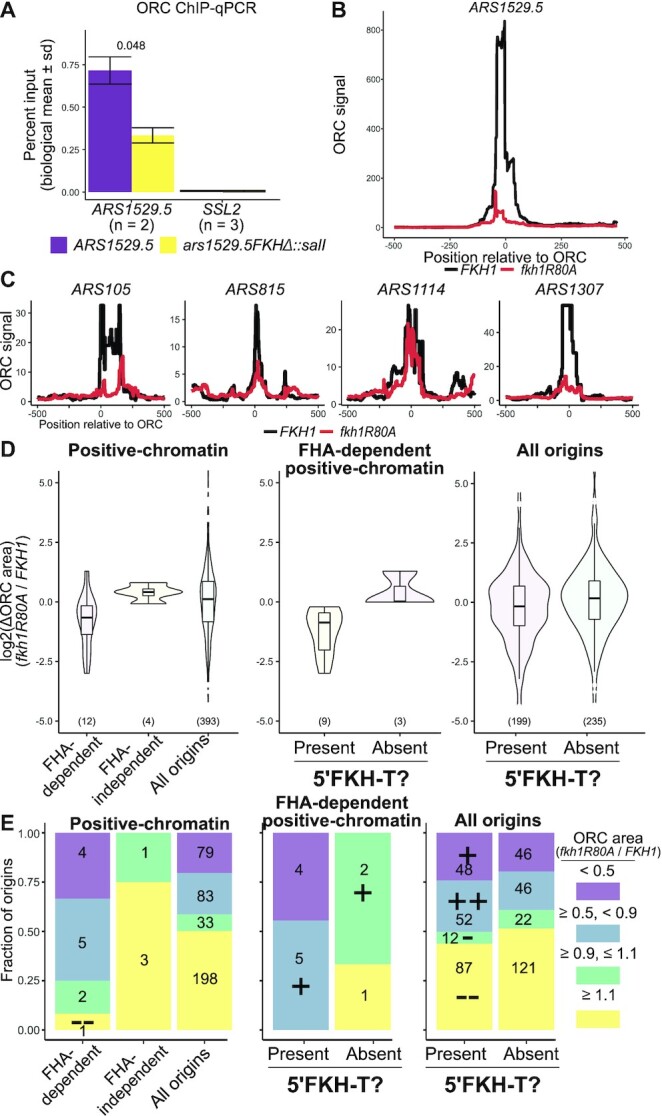
The Fkh1-FHA domain promoted ORC-origin interactions at a substantial fraction of yeast chromosomal origins. (A) ORC signals of ARS1529.5 and ars1529.5-FKHΔ in ORC ChIP-qPCR. (B) ORC signal measured at ARS1529.5 by ORC ChIPSeq in FKH1 and fkh1R80A cells. (C) ORC signals as in B at four additional FHA-dependent positive-chromatin origins identified in Figure 3 C and with 5′ FKH matches validated in ARS assays in Figure 4D and E. (D) Distributions of ratios of fkh1R80A/FKH1 ORC binding areas spanning nucleotides –100 to +100. Distributions of ratios are plotted as box-and-whisker plots overlayed on violin plots. (E) Enrichment analysis of distinct groups of yeast origins defined by their ratios of fkh1R80A/FKH1 ORC binding areas as defined in D. Enrichments (indicated with a ‘+’) or depletion (indicated with a ‘−’) of categories of fkh1R80A/FKH1 scaled ORC ratios were challenged against distributions of those same categories within all confirmed origins with the Hypergeometric Distribution function. ++/−−, P value < 0.01; +/−, P value < 0.05.
To investigate the Fkh1-FHA domain and the 5′-FKH-T site in ORC-origin binding more broadly, ORC ChIPSeq experiments were performed in FKH1 and fkh1R80A cells. A comparison of the internally scaled ORC ChIPSeq signals at the chromosomal ARS1529.5 locus revealed that ARS1529.5’s association with ORC relative to the background signal for this locus was lower in fkh1R80A compared to FKH1 cells (Figure 6B). The same analyses applied to other FHA-dependent positive-chromatin origins produced similar results, though we note that the relative ORC binding signal over ARS1114 was only mildly reduced relative to background in fkh1R80A cells compared to FKH1 cells (Figure 6C). This outcome was consistent with the relatively mild effects of fkh1R80A on ARS1114 origin activity observed in Figures 3E and 4E. To extend the examination of fkh1R80A/FKH1 ORC ChIPSeq signals to all confirmed origins (i.e. all origins), as defined in the OriDB, the fkh1R80A/FKH1 ORC ChIPSeq signal ratio over the chromosomal regions spanning the –100 to +100 nucleotides centered over each origin’s ORC site was determined. Based on these analyses, the fkh1R80A allele led to a relative reduction in ORC association at a fraction of yeast origins. The majority of origins, (59%, 231/393), generated similar or even enhanced fkh1R80A/FKH1 ORC signal ratios (Figure 6D, left panel). Moreover, the fkh1R80A allele did not reduce ORC-chromatin stability as measured by a chromatin fractionation experiment (Supplementary Figure S6). Taken together, these data provided evidence that reduced fkh1R80A/FKH1 ORC signal ratio at FHA-dependent positive-chromatin origins in Figure 6B and C was not solely a consequence of a non-specific genome-wide reduction in ORC-chromosome binding. Instead, the fkh1R80A allele altered the distribution of ORC-origin complexes, with some origins, such as the majority of FHA-dependent positive-chromatin origins, competing less effectively for ORC than they were able to in FKH1 cells.
Next the fkh1R80A/FKH1 ORC signal ratio was used to compare selected groups of origins to each other (Figures 6D and E). The sixteen positive-chromatin origins examined in Figure 3C were parsed by their requirement for the Fkh1-FHA domain for ARS activity, and the log2(fkh1R80A/FKH1) ORC signal for origins within each sub-cohort depicted in violin plots (Figure 6D, left panel). The twelve positive-chromatin origins that showed FHA-dependent ARS activity in Figure 3 behaved differently in terms of relative ORC binding from the remaining four positive-chromatin origins that showed FHA-independent ARS activity (Figure 6D, left panel). Specifically, the majority of the FHA-dependent positive-chromatin origins showed FHA-dependent ORC binding, while none of the FHA-independent positive-chromatin origins did. The 393 confirmed origins generated a large range of fkh1R80A/FKH1 ORC signal ratios, indicating that many origins outside of the experimentally defined positive-chromatin showed some dependence on the Fkh1-FHA domain for their ability to compete effectively for ORC.
To view these data in a different manner, the origins were divided into four distinct categories based on their fkh1R80A/FKH1 ORC signal ratios, with origins generating ratios of ≥0.9 interpreted as binding ORC through a Fkh1-FHA-independent mechanism (green and yellow), and origins generating ratios of <0.9 interpreted as binding ORC through a Fkh1-FHA-dependent mechanism (purple and blue) (Figure 6E). Using these cut-offs, the difference between FHA-dependent and FHA-independent positive-chromatin origins for relative ORC binding was stark, and both of these small groups behaved differently compared to all confirmed origins (Figure 6E, left panel). A key conclusion was that most of the positive-chromatin origins that showed FHA-dependent ARS activity in Figure 3C also showed FHA-dependent ORC association.
The FHA-dependent positive-chromatin origins were further analyzed to probe the relationship between a 5′-FKH-T site and Fkh1-FHA dependent ORC binding. In particular, even though the majority (75%, 9/12) of FHA-dependent positive-chromatin origins showed FHA-dependent ORC association, a substantial minority did not (25%, 3/12). The twelve positive-chromatin origins that generated FHA-dependent ARS activity in Figure 3C were parsed into two sub-cohorts by the presence of a 5′-FKH-T motif within 250 bp 5′ of the ORC site (Figure 6 D and E, middle panels). Strikingly, 100% of FHA-dependent positive-chromatin origins that contained a 5′-FKH-T motif showed FHA-dependent ORC binding, while none of the three remaining FHA-dependent positive-chromatin origins that lacked a 5′-FKH-T motif did. Thus, Fkh1 binding to a 5′-FKH-T site was the dominant feature among positive-chromatin origins that depended on the Fkh1-FHA domain to compete for ORC. Even within the stringently defined origin cohort of positive-chromatin origins, FHA-dependent ARS activity was not obligatorily linked to FHA-dependent alterations in relative ORC binding, suggesting ORC-independent mechanisms by which the Fkh1-FHA domain might affect the activity of some origins. Because the number of positive-chromatin origins that were experimentally investigated, n = 16, was low, the P-value cut-offs for many of the differences noted above relative to the behavior of the 393 confirmed origins did not reach < 0.05. Therefore, as an independent challenge to these outcomes, the analysis scheme was applied to additional origin groups: the positive-DNA cohort, which is comprised of origins where ORC’s affinity for the essential ORC site is sufficient to explain ORC binding in vivo (Figure 5, (24)) and two larger groups of Fkh1/2-regulated origins where Fkh1/2 exerts control at the S-phase MCM complex activation step, i.e. recruitment of an essential origin activation S-phase kinase (43,47) (Supplementary Figure S7). Because the positive-DNA origins, by definition, use a DNA-dependent ORC–origin binding mechanism (Figure 1), the prediction was that this origin cohort would not behave like the positive-chromatin origin cohort, and this outcome was observed (Supplementary Figure S5A). In addition, the Fkh1/2-regulated origins, either Fkh1/2-activated or Fkh1/2-repressed, were no more or less likely than all confirmed origins to contain origins that showed relative FHA-dependent ORC association (Supplementary Figure S7B). Together these analyses supported the conclusion that the FHA-dependent positive-chromatin cohort was distinct in its enrichment for origins that used an Fkh1-FHA-mechanism to promote ORC binding. The data presented above provided evidence that Fkh1-FHA-dependent ORC binding was a predominant mechanism within the positive-chromatin cohort. However, this cohort was intentionally restricted to a small number of yeast origins ( 5%) that met arbitrary stringent cut-offs for ORC binding behavior in vivo and in vitro (24). It was clear that many more confirmed origins showed relative reductions in the fkh1R80A/FKH1 ORC ChIPSeq ratios. Specifically, 42% (162/393) of confirmed origins fell into the FHA-dependent ORC binding category based on the criteria used in these analyses (Figure 6D, left panel). To address whether a link to a 5′-FKH-T site and FHA-dependent ORC binding could be uncovered by examining all confirmed origins, these origins were parsed by the presence of a 5′-FKH-T motif existing within 250 bp of the origin’s ORC site (Figure 6D and E, right panels). Notably, the fraction of confirmed origins that contained at least one 5′-FKH-T motif was significantly enriched for origins that showed relative FHA-dependent ORC binding. These observations suggested that the mechanism identified at the majority of FHA-dependent positive-chromatin origins might operate at other yeast origins.
DISCUSSION
Budding yeast ORC binds to a specific DNA element within yeast origins that is essential for origin activity, a distinct feature of yeast origins that has made this organism so useful for identifying the core origin-binding proteins in eukaryotic cells, including ORC (6). However, while sequence-specific binding by ORC is important for defining yeast origins, several lines of evidence indicate that yeast, like other eukaryotes, also uses incompletely defined features of origin-adjacent chromatin to promote ORC-origin binding (14,16,24,49,50). The experiments in this study revealed that Fkh1 was an origin-adjacent chromatin-associated protein that promotes ORC-origin binding and origin activity at a subset of origins. This Fkh1-dependent mechanism required the conserved Fkh1-FHA domain and a distinct Fkh1 binding site located 5′ of the essential ORC site. Thus, stabilization of the essential ORC-DNA interaction was a mechanism by which Fkh1 promoted the formation of chromosomal origins in budding yeast.
Fkh1 promotes origin activity at multiple steps in the origin cycle
Fkh1 and its paralog Fkh2 positively regulate ∼20% of yeast chromosomal origins, and the majority of these Fkh1/2-activated origins (n = 84) are among the earliest replicating origins in this organism (43,51). Several lines of evidence indicated that the Fkh1-FHA-dependent mechanism described here was distinct from the mechanism operating at Fkh1/2-activated origins. In particular, Fkh1/2-activated origins are regulated primarily at the S-phase activation step by Fkh1/2 proteins bound near origins and recruiting the limiting S-phase kinase, DDK (Dbf4-dependent kinase) (47,48). In contrast, the Fkh1-FHA-dependent mechanism could be explained by enhanced binding of ORC to origin DNA. Evidence supporting this conclusion included direct assessment of ORC-origin binding in vivo and the conversion of FHA-dependent positive-chromatin ARSs to FHA-independent ARSs by engineering mutant ORC sites with enhanced affinities for ORC. In addition, the FHA-dependent mechanism was more tightly linked to ORC-origin binding mechanisms (e.g. positive-chromatin compared to the control/contrast collection of positive-DNA origins) than to either origin activation time or modes of Fkh1/2-regulation. The Fkh1/2-activated mechanism is achieved through a high local concentration of origin-adjacent FKH sites that bind Fkh1/2 that in turn establish a high local concentration of the limiting DDK that triggers origin firing. Consistent with this model, Fkh1/2-activated origins show significantly greater association with Fkh1/2 proteins than Fkh1/2-repressed origins do, accounting for their enhanced competitiveness for the DDK (23). In contrast, FHA-dependent and FHA-independent origins contained a similar number of origin-adjacent FKH motifs and showed similar levels of association with Fkh1, providing evidence that a quantitative difference in Fkh1 binding could not easily account for differences in these origins’ dependencies on the Fkh1-FHA domain. However, while the Fkh1/2-activated and Fkh1-FHA-dependent mechanisms represent distinct mechanisms for how Fkh1 is used to enhance origin activity, these mechanisms are not mutually exclusive. Indeed, while ARS305 is an Fkh1/2-activated but Fkh1-FHA-independent origin, ARS1529.5 is both Fkh1/2-activated and Fkh1-FHA-dependent. Thus, Fkh1 bound to ARS1529.5 may both stabilize ORC and help recruit the DDK, whereas at other origins it may perform only one of these functions.
A distinct structural class of yeast origins: the Fkh1-FHA domain acted through an FKH site positioned 5′ of the essential origin ORC site
An ORC site is an essential but not a sufficient element for yeast origin activity. An influential paradigm for yeast origin structure positions the accessory elements required for normal levels of origin activity 3′ of the T-rich strand of the essential ORC site (25). These 3′ accessory elements are AT-rich and often include near matches to the ORC binding site in the A-rich orientation (i.e. orientation opposite to that of the essential ORC site). Because the Fkh1/2 core binding motif is also AT-rich, these 3′ accessory elements also often contain matches to FKH motifs. The potentially overlapping biochemical functions of these 3′ accessory elements can make assigning their definitive roles at origins challenging. For example, molecular and biochemical studies provide evidence that a match to a reverse ORC site can enhance binding of a second ORC that aids in the loading of the second MCM hexamer during the formation of the double-hexamer MCM complex (52,53). Mutational analyses of a few Fkh1/2-activated origins indicate that FKH motifs positioned 3′ of the essential T-rich ORC site are required for their activation, consistent with these sites acting as Fkh1/2 binding sites in vivo (43,54). However, it is difficult to discern whether mutations of these FKH motifs reduce origin activity because they abolish Fkh1/2 binding or because they reduce the binding of a second ORC or both. Either effect could conceivably alter the sensitivity of the origin to Fkh1/2 activation.
In this report, a definitive role for the 5′-FKH-T motif in several Fkh1-FHA-dependent positive-chromatin origins could be assigned because the fkh1R80A allele precisely abolished the established function of the FHA domain in phosphothreonine peptide binding while leaving Fkh1’s DNA binding domain intact (39,40,46). Therefore, we were able to test whether the Fkh1-FHA domain and the 5′ FKH-T site contributed additively to origin function. Specfically, the activity of a model Fkh1-FHA-dependent origin, ARS1529.5, was reduced substantially in fkh1R80A or in FKH1 cells containing a substitution of the 5′-FKH-T site in ARS1529.5. Moreover, yeast containing both mutations, a fkh1R80A allele and the 5′-FKH-T site substitution, did not reduce the activity of ARS1529.5 further than either mutation alone. Consistent results were observed with other Fkh1-FHA-dependent origins. While studies of a small number of origins indicate that some yeast origins contain element(s) 5′ of their ORC sites that contribute to origin activity, a role for these elements has not been assigned (35,36). The data presented here provide evidence that a FKH site can be an important 5′ origin-accessory element in yeast.
While experimental data provided evidence that the majority (56%, 9/16) of positive-chromatin origins used the 5′-FKH-T, Fkh1-FHA-dependent mechanism reported here, these origins represent only about two percent of confirmed yeast origins. However, the genome-scale analyses of ORC binding provided evidence that the Fkh1-FHA domain was relevant to competitive levels of ORC binding at ∼40% origins, suggesting this domain might be used broadly across the genome to enhance ORC-origin selection. Moreover, a significant link between an origin-adjacent 5′-FKH-T motif and Fkh1-FHA-dependent ORC binding was revealed by analyses of all 393 confirmed origins, suggesting that the less familiar origin structure and mechanism uncovered by a study of positive-chromatin origins could operate more generally, affecting 25% of yeast origins. This value may be an underestimate, as FKH motif searches were confined to 250 bp 5′ of the ORC site. Thus, taken together with what is known about the mechanism that controls Fkh1/2-activated origins, this study reveals that Fkh1, and possibly Fkh2, may contribute directly to origin regulation by influencing several distinct steps of the origin cycle in both G1- and S-phase. A clearer picture of the roles for Fkh proteins in origin control and the relationship of this control to other genomic structures and processes will require higher-resolution molecular information about Fkh1-chromosome and Fkh1-protein interactions.
Abundant evidence indicates that ORC-DNA binding in human cells is particularly sensitive to chromatin (55). While a few non-ORC factors have been implicated in ORC-origin binding in human cells, it is likely that many are yet to be discovered. Human cells encode fifty different FOX proteins, many of which promote cell proliferation (56). One FOX protein, FOXO3, interacts with Cdt1 and promotes the G1/S transition(57). It will be interesting to learn whether additional FOX proteins direct ORC binding or other steps in origin licensing as part of their cell proliferation roles in more complex eukaryotic cells.
DATA AVAILABILITY
The raw data for the ORC ChIPSeq experiment can be accessed at BioProject PRJNA69402 and for the Fkh1/2 ChIPchip at GEO GSE165464
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful for the help from Christen Geyer and the multiple undergraduate researchers who participated in the Team Origin Undergraduate Research Project (Supplementary Figure S8). We thank Laura Vanderploeg (MediaLab, UW-Madison Biochemistry Department) for Figure 1. We also thank Mike Lodes and Rob Brazas (Lucigen Corporation, Madison, WI) for generating and sequencing the ORC ChIP libraries, and Xiaolan Zhao (Memorial Sloan Kettering Cancer Center) and Erika Shor (Center for Discovery and Innovation, Hackensack Meridian Health) for helpful suggestions on the manuscript.
Contributor Information
Timothy Hoggard, Department of Biomolecular Chemistry, School of Medicine and Public Health, University of Wisconsin, Madison, WI 53706, USA.
Allison J Hollatz, Department of Biomolecular Chemistry, School of Medicine and Public Health, University of Wisconsin, Madison, WI 53706, USA; Integrated Program in Biochemistry, University of Wisconsin, Madison, WI 53706, USA.
Rachel E Cherney, Department of Biomolecular Chemistry, School of Medicine and Public Health, University of Wisconsin, Madison, WI 53706, USA.
Melissa R Seman, Department of Biomolecular Chemistry, School of Medicine and Public Health, University of Wisconsin, Madison, WI 53706, USA.
Catherine A Fox, Department of Biomolecular Chemistry, School of Medicine and Public Health, University of Wisconsin, Madison, WI 53706, USA; Integrated Program in Biochemistry, University of Wisconsin, Madison, WI 53706, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH R01 grant (NIGMS 056890 to C.A.F.]; Teaching and Learning Innovation Award (Team Origin Undergraduate Research Project) from UW Madison. Funding for open access charge: NIGMS [056890].
Conflict of interest statement. None declared.
REFERENCES
- 1. Debatisse M., Le T. B., Letessier A., Dutrillaux B., Brison O.. Common fragile sites: mechanisms of instability revisited. Trends Genet. 2012; 28:22–32. [DOI] [PubMed] [Google Scholar]
- 2. Miotto B., Ji Z., Struhl K.. Selectivity of ORC binding sites and the relation to replication timing, fragile sites, and deletions in cancers. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:E4810–E4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van B. A., Buchanan C., Charboneau E., Fangman W., Brewer B.. An origin-deficient yeast artificial chromosome triggers a cell cycle checkpoint. Mol. Cell. 2001; 7:705–713. [DOI] [PubMed] [Google Scholar]
- 4. Müller C., Nieduszynski C.. DNA replication timing influences gene expression level. J. Cell Biol. 2017; 216:1907–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rivera-Mulia J., Kim S., Gabr H., Chakraborty A., Ay F., Kahveci T., Gilbert D.. Replication timing networks reveal a link between transcription regulatory circuits and replication timing control. Genome Res. 2019; 29:1415–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bell S., Labib K.. Chromosome duplication in Saccharomyces cerevisiae. Genetics. 2016; 203:1027–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bell S., Stillman B.. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992; 357:128–134. [DOI] [PubMed] [Google Scholar]
- 8. Remus D., Diffley J.. Eukaryotic DNA replication control: lock and load, then fire. Curr. Opin Cell Biol. 2009; 21:771–777. [DOI] [PubMed] [Google Scholar]
- 9. Lubelsky Y., Prinz J., DeNapoli L., Li Y., Belsky J., MacAlpine D.. DNA replication and transcription programs respond to the same chromatin cues. Genome Res. 2014; 24:1102–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Thomae A., Pich D., Brocher J., Spindler M., Berens C., Hock R., Hammerschmidt W., Schepers A.. Interaction between HMGA1a and the origin recognition complex creates site-specific replication origins. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:1692–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Norseen J., Thomae A., Sridharan V., Aiyar A., Schepers A., Lieberman P.. RNA-dependent recruitment of the origin recognition complex. EMBO J. 2008; 27:3024–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hayashi M., Masukata H.. Regulation of DNA replication by chromatin structures: accessibility and recruitment. Chromosoma. 2011; 120:39–46. [DOI] [PubMed] [Google Scholar]
- 13. Shen Z., Sathyan K., Geng Y., Zheng R., Chakraborty A., Freeman B., Wang F., Prasanth K., Prasanth S.. A WD-repeat protein stabilizes ORC binding to chromatin. Mol. Cell. 2010; 40:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Müller P., Park S., Shor E., Huebert D., Warren C., Ansari A., Weinreich M., Eaton M., MacAlpine D., Fox C.. The conserved bromo-adjacent homology domain of yeast Orc1 functions in the selection of DNA replication origins within chromatin. Genes Dev. 2010; 24:1418–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wyrick J., Aparicio J., Chen T., Barnett J., Jennings E., Young R., Bell S., Aparicio O.. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science. 2001; 294:2357–2360. [DOI] [PubMed] [Google Scholar]
- 16. Eaton M., Galani K., Kang S., Bell S., MacAlpine D.. Conserved nucleosome positioning defines replication origins. Genes. Dev. 2010; 24:748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shor E., Warren C., Tietjen J., Hou Z., Müller U., Alborelli I., Gohard F., Yemm A., Borisov L., Broach J.et al.. The origin recognition complex interacts with a subset of metabolic genes tightly linked to origins of replication. PLoS Genet. 2009; 5:e1000755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McGuffee S., Smith D., Whitehouse I.. Quantitative, genome-wide analysis of eukaryotic replication initiation and termination. Mol. Cell. 2013; 50:123–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yabuki N., Terashima H., Kitada K.. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells. 2002; 7:781–789. [DOI] [PubMed] [Google Scholar]
- 20. Raghuraman M., Winzeler E., Collingwood D., Hunt S., Wodicka L., Conway A., Lockhart D., Davis R., Brewer B., Fangman W.. Replication dynamics of the yeast genome. Science. 2001; 294:115–121. [DOI] [PubMed] [Google Scholar]
- 21. Feng W., Collingwood D., Boeck M., Fox L., Alvino G., Fangman W., Raghuraman M., Brewer B.. Genomic mapping of single-stranded DNA in hydroxyurea-challenged yeasts identifies origins of replication. Nat. Cell Biol. 2006; 8:148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Weiner A., Hsieh T., Appleboim A., Chen H., Rahat A., Amit I., Rando O., Friedman N.. High-resolution chromatin dynamics during a yeast stress response. Mol. Cell. 2015; 58:371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ostrow A., Nellimoottil T., Knott S., Fox C., Tavaré S., Aparicio O.. Fkh1 and Fkh2 bind multiple chromosomal elements in the S. cerevisiae genome with distinct specificities and cell cycle dynamics. PLoS One. 2014; 9:e87647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hoggard T., Shor E., Müller C., Nieduszynski C., Fox C.. A Link between ORC-origin binding mechanisms and origin activation time revealed in budding yeast. PLoS Genet. 2013; 9:e1003798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Marahrens Y., Stillman B.. A yeast chromosomal origin of DNA replication defined by multiple functional elements. Science. 1992; 255:817–823. [DOI] [PubMed] [Google Scholar]
- 26. Palzkill T., Oliver S., Newlon C.. DNA sequence analysis of ARS elements from chromosome III of Saccharomyces cerevisiae: identification of a new conserved sequence. Nucleic Acids Res. 1986; 14:6247–6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang F., May C., Hoggard T., Miller J., Fox C., Weinreich M.. High-resolution analysis of four efficient yeast replication origins reveals new insights into the ORC and putative MCM binding elements. Nucleic Acids Res. 2011; 39:6523–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoggard T., Liachko I., Burt C., Meikle T., Jiang K., Craciun G., Dunham M., Fox C.. High throughput analyses of budding yeast ARSs reveal new DNA elements capable of conferring centromere-independent plasmid propagation. G3 (Bethesda). 2016; 6:993–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Welch A., Koshland D.. A simple colony-formation assay in liquid medium, termed ’tadpoling’, provides a sensitive measure of Saccharomyces cerevisiae culture viability. Yeast. 2013; 30:501–509. [DOI] [PubMed] [Google Scholar]
- 30. Batrakou D., Heron E., Nieduszynski C.. Rapid high-resolution measurement of DNA replication timing by droplet digital PCR. Nucleic Acids Res. 2018; 46:e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Skene P., Henikoff S.. A simple method for generating high-resolution maps of genome-wide protein binding. Elife. 2015; 4:e09225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Radman-Livaja M., Ruben G., Weiner A., Friedman N., Kamakaka R., Rando O.. Dynamics of Sir3 spreading in budding yeast: secondary recruitment sites and euchromatic localization. EMBO J. 2011; 30:1012–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Palzkill T., Newlon C.. A yeast replication origin consists of multiple copies of a small conserved sequence. Cell. 1988; 53:441–450. [DOI] [PubMed] [Google Scholar]
- 34. Siow C., Nieduszynska S., Müller C., Nieduszynski C.. OriDB, the DNA replication origin database updated and extended. Nucleic Acids Res. 2012; 40:D682–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Newlon C., Theis J.. The structure and function of yeast ARS elements. Curr. Opin. Genet. Dev. 1993; 3:752–758. [DOI] [PubMed] [Google Scholar]
- 36. Raychaudhuri S., Byers R., Upton T., Eisenberg S.. Functional analysis of a replication origin from Saccharomyces cerevisiae: identification of a new replication enhancer. Nucleic Acids Res. 1997; 25:5057–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Durocher D., Henckel J., Fersht A., Jackson S.. The FHA domain is a modular phosphopeptide recognition motif. Mol. Cell. 1999; 4:387–394. [DOI] [PubMed] [Google Scholar]
- 38. Reinhardt H., Yaffe M.. Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol. 2013; 14:563–580. [DOI] [PubMed] [Google Scholar]
- 39. Dummer A., Su Z., Cherney R., Choi K., Denu J., Zhao X., Fox C.. Binding of the Fkh1 forkhead associated domain to a phosphopeptide within the Mph1 DNA helicase regulates mating-type switching in budding yeast. PLoS Genet. 2016; 12:e1006094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li J., Coïc E., Lee K., Lee C., Kim J., Wu Q., Haber J.. Regulation of budding yeast mating-type switching donor preference by the FHA domain of Fkh1. PLoS Genet. 2012; 8:e1002630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hollenhorst P., Bose M., Mielke M., Müller U., Fox C.. Forkhead genes in transcriptional silencing, cell morphology and the cell cycle. Overlapping and distinct functions for FKH1 and FKH2 in Saccharomyces cerevisiae. Genetics. 2000; 154:1533–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hollenhorst P., Pietz G., Fox C.. Mechanisms controlling differential promoter-occupancy by the yeast forkhead proteins Fkh1p and Fkh2p: implications for regulating the cell cycle and differentiation. Genes. Dev. 2001; 15:2445–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Knott S., Peace J., Ostrow A., Gan Y., Rex A., Viggiani C., Tavaré S., Aparicio O.. Forkhead transcription factors establish origin timing and long-range clustering in S. cerevisiae. Cell. 2012; 148:99–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Durocher D., Smerdon S., Yaffe M., Jackson S.. The FHA domain in DNA repair and checkpoint signaling. Cold Spring Harb. Symp. Quant. Biol. 2000; 65:423–431. [DOI] [PubMed] [Google Scholar]
- 45. Durocher D., Jackson S.. The FHA domain. FEBS Lett. 2002; 513:58–66. [DOI] [PubMed] [Google Scholar]
- 46. Durocher D., Taylor I., Sarbassova D., Haire L., Westcott S., Jackson S., Smerdon S., Yaffe M.. The molecular basis of FHA domain:phosphopeptide binding specificity and implications for phospho-dependent signaling mechanisms. Mol. Cell. 2000; 6:1169–1182. [DOI] [PubMed] [Google Scholar]
- 47. Fang D., Lengronne A., Shi D., Forey R., Skrzypczak M., Ginalski K., Yan C., Wang X., Cao Q., Pasero P.et al.. Dbf4 recruitment by forkhead transcription factors defines an upstream rate-limiting step in determining origin firing timing. Genes Dev. 2017; 31:2405–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang H., Petrie M., He Y., Peace J., Chiolo I., Aparicio O.. Dynamic relocalization of replication origins by Fkh1 requires execution of DDK function and Cdc45 loading at origins. Elife. 2019; 8:e45512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hu Y., Tareen A., Sheu Y., Ireland W., Speck C., Li H., Joshua-Tor L., Kinney J., Stillman B.. Evolution of DNA replication origin specification and gene silencing mechanisms. Nat. Commun. 2020; 11:5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lee C., Cheung M., Li J., Zhao Y., Lam W., Ho V., Rohs R., Zhai Y., Leung D., Tye B.. Humanizing the yeast origin recognition complex. Nat. Commun. 2021; 12:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Aparicio O. Location, location, location: it’s all in the timing for replication origins. Genes. Dev. 2013; 27:117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Coster G., Diffley J.. Bidirectional eukaryotic DNA replication is established by quasi-symmetrical helicase loading. Science. 2017; 357:314–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miller T., Locke J., Greiwe J., Diffley J., Costa A.. Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature. 2019; 575:704–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reinapae A., Jalakas K., Avvakumov N., Lõoke M., Kristjuhan K., Kristjuhan A.. Recruitment of Fkh1 to replication origins requires precisely positioned Fkh1/2 binding sites and concurrent assembly of the pre-replicative complex. PLoS Genet. 2017; 13:e1006588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Smith O., Aladjem M.. Chromatin structure and replication origins: determinants of chromosome replication and nuclear organization. J. Mol. Biol. 2014; 426:3330–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Jackson B., Carpenter C., Nebert D., Vasiliou V.. Update of human and mouse forkhead box (FOX) gene families. Hum Genomics. 2010; 4:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zhang Y., Xing Y., Zhang L., Mei Y., Yamamoto K., Mak T. W., You H.. Regulation of cell cycle progression by forkhead transcription factor FOXO3 through its binding partner DNA replication factor Cdt1. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:5717–5722. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data for the ORC ChIPSeq experiment can be accessed at BioProject PRJNA69402 and for the Fkh1/2 ChIPchip at GEO GSE165464