

Abstract
Polintons (also known as Mavericks) were initially identified as a widespread class of eukaryotic transposons named for their hallmark type B DNA polymerase and retrovirus-like integrase genes. It has since been recognized that many polintons encode possible capsid proteins and viral genome-packaging ATPases similar to those of a diverse range of double-stranded DNA viruses. This supports the inference that at least some polintons are actually viruses capable of cell-to-cell spread. At present, there are no polinton-associated capsid protein genes annotated in public sequence databases. To rectify this deficiency, we used a data-mining approach to investigate the distribution and gene content of polinton-like elements and related DNA viruses in animal genomic and metagenomic sequence datasets. The results define a discrete family-like clade of viruses with two genus-level divisions. We propose the family name Adintoviridae, connoting similarities to adenovirus virion proteins and the presence of a retrovirus-like integrase gene. Although adintovirus-class PolB sequences were detected in datasets for fungi and various unicellular eukaryotes, sequences resembling adintovirus virion proteins and accessory genes appear to be restricted to animals. Degraded adintovirus sequences are endogenized into the germlines of a wide range of animals, including humans.
Keywords: recombination, polinton, Maverick, transposon, adenain, polB, capsid
1. Introduction
Analyses based on conserved protein structural features have increasingly revealed commonalities between families of eukaryotic viruses with double-stranded DNA (dsDNA) genomes. A current model places a loosely defined group known as polinton-like viruses at the center of a network of evolutionary relationships (Koonin, Dolja, and Krupovic 2015; Koonin, Krupovic, and Yutin 2015). Polintons (also known as Mavericks) are defined by the presence of a type B DNA polymerase (PolB) and a retrovirus-like integrase gene. Although polintons were initially suspected to be transposons, the observation that many of them encode predicted virion proteins supports the later proposal that most elements initially designated as polinton transposons are actually integrated proviruses that may remain capable of infectious cell-to-cell spread (Krupovic, Bamford, and Koonin 2014; Krupovic and Koonin, 2015 ).
Adenoviruses, poxviruses, and baculoviruses are familiar groups of animal-tropic viruses that encode genes distantly similar to polinton PolB and virion proteins (Koonin, Dolja, and Krupovic 2015). An emerging group of viruses known as virophages, which are named for their ability to parasitize megaviruses that infect unicellular eukaryotes, also encode polinton-like PolB and virion protein genes as well as, in some cases, retrovirus-like integrase genes (Duponchel and Fischer 2019).
Although polintons have been widely recognized in animal genomics and transcriptomics datasets (Krupovic, Bamford, and Koonin 2014), the proposed capsid genes of these elements are not currently annotated in public sequence databases. This has led to confusion. For instance, a recent study detected two ‘Maverick transposons’ in insect cell cultures but failed to annotate the capsid genes that identify them as likely viruses (Geisler 2018). In another example, a set of classic polinton PolB gene fragments detected in mouse fecal samples appear in GenBank with annotations incorrectly indicating that they are parvovirus structural proteins (Williams et al. 2018). A primary goal of this study is to develop a coherent classification system for animal-tropic viruses with polinton-like genes and to facilitate further discovery by rendering annotated examples of these viruses searchable in public databases.
2. Materials and methods
2.1 Detection and analysis of viral sequences
Adomavirus LO8 (Adenain) sequences were initially used for TBLASTN searches of the NCBI TSA and WGS databases. The relationship between adomavirus and adintovirus virion proteins is the subject of a separate manuscript (Welch et al. 2020). The Adenain sequences of Nephila orb-weaver spider contig (GFKT014647032) or a Parasteatoda spider contig (AOMJ02256338) were arbitrarily chosen for further TBLASTN searches of eukaryotic datasets in TSA and WGS databases. Adenain-bearing contigs 4–50 kb in length were further searched (using CLC Genomics Workbench) for BLASTP-detectable PolB homologs. Contigs were inspected for the presence of nearly overlapping arrays of large (>100 AA) open reading frames. Contigs with inverted repeats flanking the ORF cluster were favored, but this was not a strict sorting criterion. Selected contigs of interest were initially annotated using DELTA-BLAST searches of GenBank nr or HHpred analyses of single or aligned protein sequences against PDB_mmCIF70, COG_KOG, Pfam-A, and NCBI_CD databases (Altschul et al. 1997, 2005, Soding 2005; Hildebrand et al. 2009; Gerlt et al. 2015; Meier and Soding 2015; Zimmermann et al. 2017). Protein sequences were extracted from the contigs using getORF (http://bioinfo.nhri.org.tw/cgi-bin/emboss/getorf) (Rice, Longden, and Bleasby 2000). Extracted protein sequences were clustered using EFI-EST (https://efi.igb.illinois.edu/efi-est/) (Gerlt et al. 2015; Zallot, Oberg, and Gerlt 2018) and displayed using Cytoscape v3.7.1 (Shannon et al. 2003). Multiple sequence alignments were constructed using MAFFT 7 (https://toolkit.tuebingen.mpg.de/#/tools/mafft). Contigs were annotated using Cenote-Taker (Tisza et al. 2020) with an iteratively refined library of conserved adintovirus protein sequences. Compiled protein sequences are provided as a zipped set of fasta-format text files in Supplementary File S2. Maps were drawn using MacVector 17 software. Phylogenetic analyses were performed using MAFFT 7 (https://mafft.cbrc.jp/alignment/server)/ (Kuraku et al. 2013, Katoh, Rozewicki, and Yamada 2019) and displayed using FigTree 1.4.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Selected contigs for which SRA datasets were available were subjected to reference-guided re-assembly using Megahit 1.2.9 (Li et al. 2015, 2016) and/or the map reads to reference function of CLC Genomics Workbench. Annotated maps were submitted to GenBank as third-party annotation assemblies (TPA_asm). Graphical examples of the annotation process are depicted in Supplementary Fig. S1.
3. Results
3.1 Classification of animal-associated contigs with polinton-like PolB genes
TBLASTN searches using the inferred virion maturational protease (Adenain) of an arbitrarily chosen Parasteatoda spider contig (AOMJ02256338) identified hundreds of >10kb contigs of interest in NCBI’s whole genome shotgun (WGS) and transcriptome shotgun assembly (TSA) databases, as well as in de novo assemblies of various datasets of interest from the Sequence Read Archive (SRA). In animal datasets, a great majority of the larger adenain-bearing elements were found to encode either an archetypal polinton-like PolB (pfam03175) or a divergent PolB <30% identical to the pfam03175 type. Graphical illustrations of the gene detection and annotation process are shown in Supplementary Fig. S1. Both PolB classes were found encode a distinctive N-terminal domain with predicted structural similarity to the ovarian tumor superfamily of ubiquitin-specific proteases (OTU). Adenovirus PolB sequences lack the OTU domain. In this study, we refer to the OTU-pfam03175 PolB class as Alpha and the more divergent OTU-PolB class as Beta. In RepBase (https://www.girinst.org/), polinton groups 1, 2, 3, 4, and 9 each contain both Alpha and Beta PolB genes. Alpha PolB genes have previously been binned with hybrid virophages, ungrouped polinton-like viruses, and Polintons group 2, while Beta PolB genes have been binned with ungrouped polinton-like viruses, plant and fungal mitochondrial plasmids, and Polintons group 1 (Moriyama et al. 2008; Yutin, Raoult, and Koonin 2013; Yutin, Kapitonov, and Koonin 2015; Yutin et al. 2015).
In BLASTP searches, Alpha PolB sequences give strong hits (E-values ∼1e-60) for an emerging family of bipartite parvovirus-like viruses called bidnaviruses (Krupovic and Koonin 2014; Koonin et al. 2020). Use of the DELTA-BLAST algorithm (Boratyn et al. 2012) yields stronger hits (E-values <1e-100) for adenoviruses. Beta PolB sequences typically do not yield bidnavirus hits in BLASTP searches and instead give moderate hits (E-value ∼1e-15) for the PolB proteins of megaviruses (e.g., faustoviruses and klosneuvirus) as well as various bacteriophages (Fig. 1). Neither of the two PolB classes detects known virophage PolB sequences in BLASTP or DELTA-BLAST searches.
Figure 1.
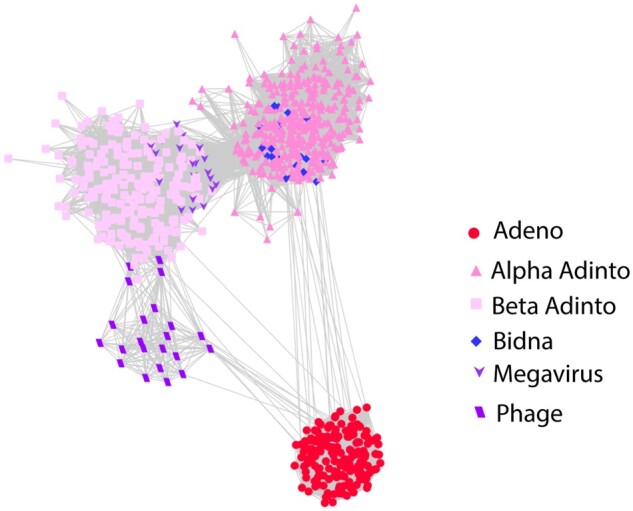
PolB BLASTP relationships. PolB protein sequences were subjected to all-against-all sequence similarity network analysis with a BLASTP E-value cut-off of 1e-06. Supplementary File S1 presents an interactive version of Fig. 1 that can be viewed using Cytoscape software (https://cytoscape.org). Supplementary File S2 contains sequence compilations for PolB and other proteins.
In addition to Adenain and PolB, nearly all >10 kb contigs from the WGS and TSA surveys encode a retrovirus-like integrase (protein family rve) as well as a protein similar to a group of FtsK/HerA-type nucleoside triphosphatases (FtsK) that are thought to mediate the packaging of viral genomes into virions (Iyer et al. 2004).
Alignments of selected contigs back to parent read datasets showed that coverage depth fell to zero near the ends of some contigs. An example is shown graphically on page 6 of Supplementary Fig. S1. In some cases, such as a Mayetiola destructor (barley midge) read dataset, a single predominant apparently free-ended sequence could be assembled but the dataset also contained a range of lower-coverage variant reads near the termini, some of which extended into inverted terminal repeats (ITRs) and host genomic DNA sequences. The observation suggests that the integrase gene is functional and mediates integration events akin to those observed in virophages that encode rve integrases (Fischer and Hackl 2016).
Based on the similarities to adenoviruses and the presence of integrase and virus genome-packaging genes, we suggest that this group of animal-associated elements could be referred to as ‘adintoviruses’. Graphical maps of reference Alpha and Beta adintoviruses are shown in Fig. 2. Maps of additional adintovirus genomes are shown in Supplementary Fig. S2.
Figure 2.

Genome maps of two representative adintoviruses. See main text for descriptions of gene names. Black bars denote inferred termini. Supplementary Table S1 presents accession numbers and full Linnaean designations of animal hosts.
HHpred searches confirmed the presence of ORFs with high-probability predicted structural similarity to the double-jellyroll major capsid proteins (hexons) and single-jellyroll vertex capsomers (pentons, also known as penton bases) of adenoviruses, virophages, megaviruses, or poxviruses (Supplementary Fig. S1). As expected, contigs with Beta PolB genes encode Hexon and Penton proteins that occupy discrete clusters that encompass the Terrapene Beta adintovirus cognates (Supplementary Fig. S3). Although most contigs with Alpha PolB genes encode Hexon and Penton proteins that cluster with the Mayetiola cognates, some Alpha PolB contigs unexpectedly encode virion proteins that are interspersed within the Terrapene cluster. Similar results were observed in analyses using traditional phylogenetic trees. The results suggest the existence of distinct Alpha and Beta adintovirus lineages, but with some examples reflecting horizontal transfer of the virion protein module of the Beta PolB lineage into an Alpha PolB background. We have previously proposed a similar intra-family horizontal gene transfer scenario for some species of polyomaviruses (Buck et al. 2016).
3.2 Other adintovirus genes
Adintoviruses encode three classes of proteins with predicted structures resembling known membrane-active proteins. A previously noted class (Yutin, Raoult, and Koonin 2013) is similar to the phospholipase A2 (PLA2) domain of parvovirus VP1 virion proteins (see page 7 of Supplementary Fig S1). In parvoviruses, the domain is thought to be involved in membrane disruption during the infectious entry process. The PLA2-like genes, which are characteristic of Mayetiola-class (Alpha) virion protein modules, include a C-terminal domain similar to adenovirus virion core precursor protein ten (pX). We suggest the gene name PLA2X.
Beta adintoviruses, as well as Alpha PolB adintoviruses with Terrapene-class (Beta) virion protein modules, encode homologs of the C-terminal regulatory domain of gasdermins, a group of pore-forming proteins that serve as executioners in pyroptosis (a form of inflammatory programmed cell death) (Dubois et al. 2019). Like PLA2X, adintovirus gasdermin homologs typically encode a pX-like domain near the C-terminus. Apparent homologs of a membrane-active spider venom protein known as cupiennin were also observed in Beta-class virion protein modules. The pairing of hallmark Beta-class virion accessory genes (GasderminX, Cupiennin) with a subset of Alpha PolB adintoviruses (Supplementary Fig. S2) supports the hypothesis that some adintovirus species arose through horizontal gene transfer between the Alpha and Beta adintovirus lineages.
Some classes of predicted protein sequences were conserved among adintoviruses but did not show clear hits for known proteins in BLASTP or HHpred searches. We assigned these groups of adintovirus-conserved proteins of unknown function numbered “Adintoc” names.
Small DNA tumor viruses (adenoviruses, polyomaviruses, and papillomaviruses (Pipas 2019)) encode proteins harboring conserved LXCXE motifs that that are known to engage cellular retinoblastoma (Rb) and related tumor suppressor proteins (de Souza, Iyer, and Aravind 2010). Adenovirus E1A, papillomavirus E7, polyomavirus LT, and parvovirus NS3 oncoproteins typically encode the Rb-binding motif just upstream of a consensus casein kinase 2 acceptor motif ((ST)XX(DE)). Some oncogenes, such as E1A, encode an additional conserved region ((DEN)(LIMV)XX(LM)(FY)), referred to as CR1, that binds the groove containing the A and B cyclin folds within the Rb pocket domain (Pipas 1992; Gouw et al. 2018). In general, these predicted Rb-interacting motifs are adjacent to potential zinc- or iron-sulfur-binding motifs (typically, paired CXXC). Open reading frames encoding combinations of these short linear motifs were observed in adintovirus contigs. We refer to these predicted proteins, which typically occupy a region upstream of the PolB gene, as ‘Oncoid’ genes, connoting their similarities to the known oncogenes of small DNA tumor viruses. Adintovirus homologs of anti-apoptotic proteins, such as Bcl2 and IAP, were also observed (Supplementary Figs S1 and S2).
3.3 Distribution of adintovirus-like PolB sequences in eukaryotic WGS datasets
The conserved catalytic core PolB sequences of either the Mayetiola barley midge Alpha adintovirus or Terrapene box turtle Beta adintovirus were used separately as baits in TBLASTN searches of WGS databases for eukaryotes. Retrieved protein sequences were trimmed to 80% similarity and subjected to clustering with an alignment score threshold of 60 (Shannon et al. 2003; Li and Godzik 2006; Huang et al. 2010; Fu et al. 2012; Zallot, Oberg, and Gerlt 2018). The clustering segregated away Beta adintovirus-like PolB sequences encoded by plant and fungal mitochondria (e.g., EU365401, AF061244). The filtered sequences were subjected to phylogenetic analyses (Fig. 3).
Figure 3.

Phylogenetic trees comprised of WGS hits for Alpha or Beta adintovirus PolB sequences (left and right panels, respectively). Hits from insect datasets are colored yellow, hits from tetrapod datasets are red, and fungus-associated hits are blue. All other types of eukaryotes are represented by black lines. Annotated branches show two Alpha adintovirus sequences associated with bovine (Bos) lung samples clustering with adintovirus sequences from insect datasets, suggesting an environmental insect source. In contrast, exemplar Mayetiola and Terrapene adintoviruses cluster with sequences found in other insect or terrestrial vertebrate datasets, respectively. Similarly, adintovirus PolB-like sequences from Powellomyces and Rhizophagus fungi cluster with sequences from other types of fungi. Supplementary Files S3 and S4 are Nexus-format versions of the figure that can be viewed interactively using FigTree software (http://tree.bio.ed.ac.uk/software/figtree/).
Two complete Alpha adintovirus-like contigs (NKLS02000104, NKLS02001728) were observed in assemblies of a PacBio-based WGS survey of bovine lung tissue. Sequences outside the inferred proviral ITRs in the two sequences were highly diverse and mostly unidentifiable, but in a few reads the extra-proviral host sequences showed BLASTN similarity to genomic DNA sequences of various beetles, including Tribolium castaneum (a flour beetle that commonly infests cattle feed). Furthermore, the Bos lung-associated PolB sequences occupy phylogenetic clades comprised of insect-associated PolB sequences (Fig. 3). These observations suggest that the two Alpha adintovirus sequences in the bovine datasets are insect-derived environmental contaminants, rather than mammal-tropic viruses. Similarly, several Beta adintovirus-like contigs (e.g., AANG04004209) found in a housecat oral swab sample show close phylogenetic affinity for adintovirus sequences observed in salmon WGS datasets. In another example, integrated adintoviruses found in a genomic dataset for olive trees (Olea europaea) showed insect-like sequences outside the inferred ITRs and showed phylogenetic affinity with PolB sequences from insect WGS datasets. Other adintovirus-like sequences found in plant datasets resembled adintovirus PolB sequences associated with nematode datasets. It thus appears that adintovirus sequences in some datasets are derived from environmental sources, as opposed to a productive infection of the organism that was the target of the sequencing effort.
Although there are examples of apparent environmental contamination, most adintovirus sequences form discrete clades that recapitulate the phylogeny of the host organisms that were the subjects of the WGS surveys. For example, a distinct clade of Alpha adintovirus PolB sequences was observed in datasets for multiple related species of venomous snakes. Several distinct clades of Beta adintoviruses were observed in datasets for amphibians and reptiles, including the well-populated clade that houses the exemplar Terrapene adintovirus. The exemplar Mayetiola adintovirus likewise occupies a clade exclusively populated by sequences found in insect WGS datasets.
TBLASTN searches against Terrapene box turtle Beta adintovirus PolB and Hexon protein sequences both yielded weak hits (E-value ∼1e-05) for a locus on human chromosome 7. An adintovirus GasderminX sequence was also detected at the locus. Alignments to Terrapene adintovirus protein sequences were used to assign pseudogene annotations (Fig. 4). The detected element is a highly disrupted endogenized Beta adintovirus. Homologous nucleotide sequences were detected in the genomes of primates, rodents, shrews, afrotherians, and xenarthrans but not in datasets for ungulates, carnivores, bats, marsupials, or prototherians. Endogenized adintovirus sequences observed in amphibian and reptile genomes do not share recognizable nucleotide similarity with placental mammal-endogenized adintovirus sequences. It is unclear whether a single adintovirus endogenization event affected an early placental mammal and the endogenized virus was then lost in non-shrew Laurasiatherians or whether multiple distinct endogenization events occurred in separate placental mammal lineages. Identification of extant examples of placental mammal adintoviruses could help resolve this question.
Figure 4.

An endogenized Beta adintovirus relic found on human chromosome 7. Degraded pseudogenes interrupted by nonsense and frameshift mutations were reconstructed based on alignments to the protein sequences of Terrapene box turtle adintovirus. Question marks indicate that the reconstructed gene does not yield hits in BLAST searches of GenBank’s viruses taxon. Tentative gene assignments are based on synteny with the Terrapene adintovirus. The reconstructed Hexon, GasderminX, and PolB protein sequences yield DELTA-BLAST hits with E-values of 1e-21, 4e-05, and 3e-25, respectively. Supplementary File S5 presents an annotated GenBank-format nucleotide map of the human chromosome 7 endogenized adintovirus.
3.4 Viruses with adinto-like genes in non-animal eukaryote datasets
Eukaryotic viruses with midsize (10–50 kb) linear dsDNA genomes show a remarkable degree of genomic modularity (Koonin, Dolja, and Krupovic 2015; Yutin, Kapitonov, and Koonin 2015; Yutin et al. 2015). The apparently promiscuous horizontal gene transfer and lack of any single defining gene for these viruses makes the group taxonomically challenging. We propose the collective acronym MELD (midsize eukaryotic linear dsDNA) virus for the dizzyingly polyphyletic category. The name, which would encompass adenoviruses and adintoviruses, is intended to fill a gap between other operationally defined umbrella groups, such as circular Rep-encoding single-stranded DNA (CRESS) viruses, small DNA tumor viruses, nucleocytoplasmic large DNA viruses, and megaviruses.
Datasets for Blastocystis hominis (a diatom-related unicellular eukaryote that commonly inhabits the human gut) contain MELD virus sequences that unite Alpha adintovirus-like PolB and integrase genes with inferred virion proteins whose primary sequences are not recognizably similar to known virion proteins (Fig. 5). Gene identities for the Blastocystis virus were inferred based on HHpred results. Comparable MELD viruses were confirmed in rumen metagenomic datasets for sheep (Yutin, Kapitonov, and Koonin 2015) and cattle, as well as in WGS datasets for green algae and fungi. In phylogenetic analyses, the PolB sequences of these viruses occupy long branches that are distant from animal-associated PolB clades (Fig. 3).
Figure 5.

MELD viruses (and related elements) with adintovirus-like PolB genes. Greek letters indicate genes similar to Alpha or Beta adintoviruses in BLASTP or DELTA-BLAST searches. “Ad-” indicates similarity to adenovirus sequences. Fiber, predicted structural or primary sequence similarity to bacteriophage tail fibers or coiled-coil proteins; Lectin, predicted structural similarity to galactose-binding domains; SET, sequence similarity to the S-adenosyl methionine-binding pocket of cellular histone-lysine methyltransferases; TRAF, predicted structural similarity to TNF receptor-associated factor 3; UCH, predicted structural similarity to ubiquitin C-terminal hydrolases; IVa2, sequence similarity to adenovirus IVa2 viral genome-packaging ATPases; Dcm, predicted structural similarity to cytosine DNA methyltransferases; TBP, similar to TATA binding proteins; Matrix/Capsid/NC/Pro, similarities to retroviral Gag and retropepsin; S3H, poxvirus D5-like superfamily 3 helicase.
Contigs encoding Alpha adintovirus-like PolB and integrase genes were found in metagenomics datasets for bioreactor-cultured human feces, human urine samples, and human oral swab samples (Santiago-Rodriguez et al. 2015). This group of closely related sequences was only detected in datasets from a single laboratory and not in other human metagenomics surveys. Divergent variants of predicted proteins from the feces-associated virus were found in contigs from datasets for Cyanophora paradoxa, a species of glaucophyte algae (e.g., QPMI01000557), suggesting that the human feces-associated adintovirus-like sequences were derived from an environmental source.
Six MELD virus genomes assembled from a single Powellomyces SRA dataset unite sequences resembling adenovirus vertex fiber proteins with either a Beta adintovirus-like PolB (Fig. 5) or a surprising variety of non-PolB DNA replicases (Fig. 6). MELD virus genomes encoding genes similar to Alpha adintovirus virion proteins (E-values ∼1e-7 to 1e-21) were assembled from datasets for Capsaspora owczarzaki, Monosiga brevicollis, and Trichoplax H2 (unicellular eukaryotes that are thought to be closely related to animals). Aside from the abovementioned insect- and nematode-associated adintovirus sequences found in datasets for plants, adintovirus-like virion protein sequences were not detected in datasets for other non-animal eukaryotes. Capsaspora MELD virus 1 and the Trichoplax MELD virus both encode superfamily 1 helicase (S1H) genes instead of a PolB gene. Various megaviruses and bacteriophages encode similar S1H genes, as does a MELD virus observed in Physarum polycephalum slime mold and Powellomyces MELD virus 4. Full-length S1H replicase genes of this class were not detected in animal WGS datasets, with the exception of seemingly endogenized degraded virus-like contigs in datasets for several coral and jellyfish species and a helitron-like element found in Branchiostoma lancelets (e.g., RDEB01009762, ABEP02037959).
Figure 6.

MELD viruses with other replicases. Greek letters indicate genes with sequences similar to Alpha or Beta adintoviruses in BLASTP searches. Sequences similar to adenoviruses are marked with “Ad-.” Yellow flags represent predicted tRNA genes. dUTPase, similar to poxvirus deoxy-UTP diphosphatases; Fiber, similarity to bacteriophage tail fibers or other coiled-coil proteins; Tapemeasure, similarity to phage tail tape measure proteins; S1H, RecD/Pif1-like superfamily 1 helicase; YRec, homolog of phage tyrosine recombinases; DnaA-like, sequence distantly similar to DnaA and DnaB-like helicases; VI, similar to adenovirus virion core protein six; PolA, DNA polymerase family A (Pfam : 00476); S3H superfamily 3 helicase similar to those observed in virophages and megaviruses; S2H, superfamily 2 helicase similar to DEAD-box helicase of Yellowstone Lake virophage 7 (YP_009177696); SLATT, homolog of host SMODS and SLOG-associating 2TM effector domain proteins; VLTF, homolog of mimivirus VLTF3-like transcription factor. See main text for information about other gene names.
In WGS searches for sequences resembling human adenovirus type 5 PolB, we did not detect any contigs resembling full-length viruses in non-animal datasets. The searches did reveal the complete ITR-bounded genome of a typical mastadenovirus in a dataset for Dipodomys ordii (a type of kangaroo rat) as well as apparently complete MELD viruses in datasets for Hydra oligactis (brown hydra) and Anelosimus studiosus (a type of tangle-web spider). Like known adenoviruses, the Hydra and Anelosimus MELD viruses do not encode integrase genes and their PolB genes do not encode detectable OTU domains.
3.5 Proposal of a new virus group, bidnaparvoviruses
BLASTP searches using Alpha adintovirus PolB sequences return high-likelihood matches (E-values <1e-80) for the PolB genes of viruses in the recently established family Bidnaviridae (Krupovic and Koonin 2014; Koonin et al. 2020) (Fig. 1). Like adintoviruses, bidnavirus PolB genes encode an N-terminal OTU domain. Contigs with bidnavirus-like PolB genes were detected in datasets for the gut contents of African termites (Cubitermes ugandensis), dog (Canis lupus familiaris) feces, the silk glands of a false wolf spider (Tengella perfuga), and Tasmanian devil (Sarcophilus harrisii) feces (Fig. 7). The dog feces PolB sequence is 53% similar to a ‘structural protein’ fragment of Fresh Meadows ‘densovirus’ 3 reported in mouse (Mus musculus) feces (AWB14611) (Williams et al. 2018).
Figure 7.

Genome maps for non-animal parvoviruses, bidnaviruses, and proposed bidnaparvoviruses. GIIM, similarity to group II intron maturases; tailspike, similarity to bacteriophage short tail fibers.
In known bidnaviruses, the termini of each of the two genome segments have matching nucleotide sequences. To search for second segments, we probed assemblies for examples of other contigs with termini similar to the ITRs of the initially observed bidnavirus-like contigs. The datasets were also searched for possible second segments with sequences similar to the proteins of known bidnaviruses. Second segments were not detected, suggesting that the five new bidna-like viruses are monopartite. We propose that the apparently monopartite viruses be referred to as ‘bidnaparvoviruses’, highlighting their shared similarities with the two previously established families.
Searches for examples of parvovirus NS1-like sequences did not reveal clear examples outside of multicellular animal datasets. A marginal exception was a group of sequences found in datasets for Abeoforma whisleri and Ichthyophonus hoferi, two unicellular eukaryotes that are thought to be closely related to multicellular animals. The observations suggest an early-animal origin for parvoviruses that may have involved acquisition of genes from Alpha adintoviruses.
4. Discussion
We have identified a coherent family-like grouping of animal viruses that we call adintoviruses, connoting their hallmark adenovirus-like virion protein genes and retrovirus-like integrase genes. Adintovirus sequences are detectable either as apparently free linear DNA molecules or as endogenized integrants in WGS datasets representing all eumetazoan phyla. Although sequences resembling the PolB proteins of Alpha and Beta adintoviruses can also be found in datasets for non-animal eukaryotes, the sequences of adintovirus virion proteins appear to be restricted to animals.
We imagine that the related Alpha and Beta adintovirus-like lineages might have infected early eukaryotes and the two lineages gradually co-evolved with major divisions of eukaryotes, including multicellular animals. In this model, the sequences of the virion protein genes presumably evolved more rapidly than the conserved catalytic core of PolB, resulting in distinctive mutually unrecognizable virion protein sequences specific to each major division of eukaryotes. The model suggests that adenoviruses could be thought of as a related sister lineage that also arose in or before the first animals. Although adenoviruses are currently only known to infect vertebrates, the idea that the lineage long predates the emergence of vertebrates is consistent with our identification of distantly adenovirus-like sequences in hydra and spider datasets (Figs 5 and 6).
In non-animal eukaryote datasets, adintovirus-like PolB sequences can be found in a wide range of sequence contexts, ranging from elements with no obvious virion proteins to the genomes of megaviruses. Conversely, it appears that adintovirus-like PolB genes can readily be replaced with other types of DNA replicase genes (Fig. 6). This presumably reflects the previously proposed rampant horizontal gene transfer among virus lineages that infect unicellular eukaryotes (Koonin, Dolja, and Krupovic 2015). Although similar horizontal gene transfer events appear to have occurred between various animal-tropic virus families, including adintoviruses and parvoviruses (Fig. 7), adomaviruses and polyomaviruses (Mizutani et al. 2011, Dill et al. 2018, Welch et al. 2020) and papillomaviruses and polyomaviruses (Woolford et al. 2007), each of these cases appears to represent single ancient event. It may be that the evolution of distinct tissues and organs—or the development of cell-mediated immunity in multicellular animals—placed limits on the likelihood that different virus lineages can co-infect a single cell and productively recombine. From this view, the distinctive gene combinations seen in adintoviruses and adenoviruses might simply be bottlenecked examples of the much larger range of gene combinations observed in MELD viruses of unicellular eukaryotes (Yutin, Kapitonov, and Koonin 2015, Yutin et al. 2015).
It has generally been assumed that the functionally similar oncogenes found in adenoviruses, papillomaviruses, parvoviruses, and polyomaviruses arose through convergent evolution or through horizontal gene transfer between virus families (de Souza, Iyer, and Aravind 2010). Although small DNA tumor virus oncogenes show low overall sequence similarity, they can be roughly defined based on the presence of short linear motifs. Many adintoviruses encode candidate ‘Oncoid’ proteins with these motifs. Bombyx silkworm bidnavirus NS3, which we have designated as a candidate Oncoid (Fig. 7), has previously been shown to be similar to a baculovirus protein of unknown function (Krupovic and Koonin 2014). We note that many of the proposed baculovirus homologs (e.g., YP_009506034) also share potential zinc-coordinating cysteine residues as well as a C-terminal LXCXE/CK2 site, qualifying the baculovirus proteins as candidate Oncoids as well. Surprisingly, candidate Oncoids were also observed in MELD viruses of unicellular eukaryotes (Figs 5 and 6). The predicted Oncoid2 gene of Capsaspora protist MELD virus 2 detects polyomavirus Large T oncogenes in DELTA-BLAST searches (E-value 4e-16). It is interesting to imagine that oncogenes in a broad range of animal DNA viruses might share an ancestry that pre-dates the emergence of multicellular animals.
Adintoviruses encode a number of accessory genes that appear to be homologs of membrane-active proteins found in animal venom. These include bee and snake venom PLA2 and melittin, as well as a spider venom protein called cupiennin. Interestingly, venom PLA2 and melittin (which shows similarity to adenovirus protein X in HHpred searches) act in concert (Vogt et al. 1970), suggesting the speculative hypothesis that these venom genes might have arisen from a captured viral PLA2X-like gene.
In unpublished work, our group used a standard baculovirus-based expression system (ThermoFisher) to generate a virus-like particle (VLP) vaccine against BK polyomavirus (BKV) (Peretti et al. 2018). The project provided an inadvertent natural experiment. Recombinant baculoviruses were generated in Sf9 cells and bulk protein expression was performed using the Trichoplusia ni cell line High Five. BKV VLPs were purified according to previously reported methods (Cardone et al. 2014) involving ultracentrifugation through density gradients, nuclease digestion, and size exclusion chromatography. Deep sequencing of DNA extracted from the purified VLP preparation shows high-depth coverage of Spodoptera adintovirus genomes alongside incomplete patchy coverage of endogenized Trichoplusia-specific homologs of the two Spodoptera viruses (Supplementary File S6). It appears that Sf9-derived adintoviruses infected the High Five cells and this led to the production of adintovirus virions that co-purified with the recombinant BKV VLPs. The results suggest that standard insect cell cultures could serve as a laboratory model for productive adintovirus infection.
A Beta adintovirus was detected in transcriptomic and WGS datasets for Mexican blind tetra cavefish (Astyanax mexicanus). Adintovirus transcripts were most abundant in head, kidney, and intestine samples and least abundant in muscle and whole embryo samples (Supplementary Table S2). Analysis of the WGS dataset showed that adintovirus DNA reads outnumbered reads for a single copy host gene (gamma tubulin, NW_019172896) by a factor of 25. At both an RNA and DNA level, the Astyanax sequence showed a high degree of uniformity, suggesting a clonal infection. In contrast, pet store samples of a different species of tetra, Gymnocorymbus ternetzi (SRR2040422), showed such a complex range of adintovirus sequence variants that assembly of contigs representing complete viral genomes was challenging. These observations suggest that tetras might serve as a tractable laboratory model for adintovirus infection.
Adintoviruses have a number of features that could make them useful as recombinant gene transfer vectors. Their genome size is substantially larger than commonly used retroviral and parvoviral vectors. In contrast to adenovirus- and baculovirus-based vector systems, adintovirus genomes are small enough to be manipulated entirely in the setting of standard plasmids. An intriguing feature of the adintovirus integrase gene is the presence of a predicted chromodomain that, in LTR retrotransposons, is believed to influence integration site specificity (Kordis 2005). This could theoretically offer an advantage over retroviral vectors, which show little integration site specificity. Another potential practical use for adintoviruses might be as biocontrol agents for pest organisms, such as Mayetiola destructor barley midges or chytrid fungi that parasitize amphibians.
An important implication of this study is that there may be additional unappreciated families of animal viruses hiding in plain sight in sequence databases. Adintoviruses may have been relatively easy quarry because they are able to integrate into host genomes, such that they are detectable in WGS datasets of randomly sampled animals that did not happen to be suffering from an active infection. In contrast to the hundreds of adintovirus-like contigs detected in our initial WGS survey, focused searches for adenoviruses (which do not encode integrases) detected only a single complete adenovirus genome. For future discovery efforts, it will be important to develop higher throughput methods using sensitive structure-guided searches to identify divergent new examples of viral hallmark genes in sequence datasets representing many individuals, including subjects suffering from disease. A key goal will be to understand which combinations of genes tend to co-occupy single genomes. Recently reported bioinformatics pipelines, such as Cenote-Taker (Tisza et al. 2020) and Mash Screen (Ondov et al. 2019), should be useful for these purposes. Deposition of annotated viral genome sequences into publicly searchable databases will be critical for further expanding our understanding of the eukaryotic virome.
Data availability
GenBank accession numbers for sequences deposited in association with this study are: BK010888BK010889BK010890BK010893BK010894BK010998BK010999BK011000BK011001BK011002BK011003BK011004BK011005BK011006BK011007BK011008BK011009BK011010BK011011BK011022BK011023BK011024BK011025BK011026BK012042BK012043BK012044BK012045BK012046BK012047BK012048BK012049BK012050BK012051BK012052BK012053BK012054BK012055BK012056BK012057BK012058BK012059BK012060BK012061BK012062BK012063BK012064BK012084BK012085BK012086.
Supplementary data
Supplementary data are available at Virus Evolution online.
Supplementary Material
Acknowledgements
The authors are indebted to Eugene Koonin and Natalya Yutin for their generous guidance and for the spirited discussions that inspired us to pursue this study. We thank Karl Münger and Jim Pipas for their extensive advice about oncogene sequence motifs and Chris Bellas for useful discussions and for critical evaluation of the manuscript. We are also grateful to Patrick McTamney for guiding the production of BK polyomavirus VLP stocks. This work utilized the computational resources of the NIH HPC Biowulf cluster. (http://hpc.nih.gov).
Funding
This work was funded by the NIH Intramural Research Program, with support from the Center for Cancer Research, NCI.
Conflict of interest: None declared.
References
- Altschul S. F. et al. (1997) ‘Gapped BLAST and PSI-BLAST: A New Generation of Protein Database Search Programs’, Nucleic Acids Research, 25: 3389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F. et al. (2005) ‘Protein Database Searches Using Compositionally Adjusted Substitution Matrices’, FEBS Journal, 272: 5101–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boratyn G. M. et al. (2012) ‘Domain Enhanced Lookup Time Accelerated BLAST’, Biology Direct, 7: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck C. B. et al. (2016) ‘The Ancient Evolutionary History of Polyomaviruses’, PLoS Pathogens, 12: e1005574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone G. et al. (2014) ‘Maturation of the Human Papillomavirus 16 Capsid’, mBio, 5: e01104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Souza R. F., Iyer L. M., Aravind L. (2010) ‘Diversity and Evolution of Chromatin Proteins Encoded by DNA Viruses’, Biochimica et Biophysica Acta (BBA)—Gene Regulatory Mechanisms, 1799: 302–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dill J. A. et al. (2018) ‘Microscopic and Molecular Evidence of the First Elasmobranch Adomavirus, the Cause of Skin Disease in a Giant Guitarfish, Rhynchobatus djiddensis’, mBio, 9: e00185–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois H. et al. (2019) ‘Nlrp3 Inflammasome Activation and Gasdermin D-Driven Pyroptosis Are Immunopathogenic upon Gastrointestinal Norovirus Infection’, PLoS Pathogens, 15: e1007709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duponchel S., Fischer M. G. (2019) ‘Viva Lavidaviruses! Five Features of Virophages That Parasitize Giant DNA Viruses’, PLoS Pathogens, 15: e1007592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer M. G., Hackl T. (2016) ‘Host Genome Integration and Giant Virus-Induced Reactivation of the Virophage Mavirus’, Nature, 540: 288–91. [DOI] [PubMed] [Google Scholar]
- Fu L. et al. (2012) ‘CD-HIT: Accelerated for Clustering the Next-Generation Sequencing Data’, Bioinformatics, 28: 3150–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler C. (2018) ‘A New Approach for Detecting Adventitious Viruses Shows Sf-Rhabdovirus-Negative Sf-RVN Cells Are Suitable for Safe Biologicals Production’, BMC Biotechnology, 18: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A. et al. (2015) ‘Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST): A Web Tool for Generating Protein Sequence Similarity Networks’, Biochimica et Biophysica Acta (BBA)—Proteins and Proteomics, 1854: 1019–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouw M. et al. (2018) ‘The Eukaryotic Linear Motif Resource—2018 Update’, Nucleic Acids Research, 46: D428–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand A. et al. (2009) ‘Fast and Accurate Automatic Structure Prediction with HHpred’, Proteins: Structure, Function, and Bioinformatics, 77: 128–32. [DOI] [PubMed] [Google Scholar]
- Huang Y. et al. (2010) ‘CD-HIT Suite: A Web Server for Clustering and Comparing Biological Sequences’, Bioinformatics, 26: 680–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer L. M. et al. (2004) ‘Comparative Genomics of the FtsK-HerA Superfamily of Pumping ATPases: Implications for the Origins of Chromosome Segregation, Cell Division and Viral Capsid Packaging’, Nucleic Acids Research, 32: 5260–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Rozewicki J., Yamada K. D. (2019) ‘MAFFT Online Service: Multiple Sequence Alignment, Interactive Sequence Choice and Visualization’, Briefings in Bioinformatics, 20: 1160–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V., Dolja V. V., Krupovic M. (2015) ‘Origins and Evolution of Viruses of Eukaryotes: The Ultimate Modularity’, Virology, 479–480: 2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V. et al. (2020) ‘Global Organization and Proposed Megataxonomy of the Virus World’, Microbiology and Molecular Biology Reviews, 84: e00061–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E. V., Krupovic M., Yutin N. (2015) ‘Evolution of Double-Stranded DNA Viruses of Eukaryotes: From Bacteriophages to Transposons to Giant Viruses’, Annals of the New York Academy of Sciences, 1341: 10–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kordis D. (2005) ‘A Genomic Perspective on the Chromodomain-Containing Retrotransposons: Chromoviruses’, Gene, 347: 161–73. [DOI] [PubMed] [Google Scholar]
- Krupovic M., Bamford D. H., Koonin E. V. (2014) ‘Conservation of Major and Minor Jelly-Roll Capsid Proteins in Polinton (Maverick) Transposons Suggests That They Are Bona Fide Viruses’, Biology Direct, 9: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M., Koonin E. V. (2014) ‘Evolution of Eukaryotic Single-Stranded DNA Viruses of the Bidnaviridae Family from Genes of Four Other Groups of Widely Different Viruses’, Scientific Reports, 4: 5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupovic M., Koonin E. V. (2015) ‘Polintons: A Hotbed of Eukaryotic Virus, Transposon and Plasmid Evolution’, Nature Reviews Microbiology, 13: 105–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuraku S. et al. (2013) ‘ALeaves Facilitates On-Demand Exploration of Metazoan Gene Family Trees on MAFFT Sequence Alignment Server with Enhanced Interactivity’, Nucleic Acids Research, 41: W22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D. et al. (2015) ‘MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph’, Bioinformatics, 31: 1674–6. [DOI] [PubMed] [Google Scholar]
- Li D. et al. (2016) ‘MEGAHIT v1.0: A Fast and Scalable Metagenome Assembler Driven by Advanced Methodologies and Community Practices’, Methods, 102: 3–11. [DOI] [PubMed] [Google Scholar]
- Li W., Godzik A. (2006) ‘Cd-Hit: A Fast Program for Clustering and Comparing Large Sets of Protein or Nucleotide Sequences’, Bioinformatics, 22: 1658–9. [DOI] [PubMed] [Google Scholar]
- Meier A., Soding J. (2015) ‘Automatic Prediction of Protein 3D Structures by Probabilistic Multi-Template Homology Modeling’, PLoS Computational Biology, 11: e1004343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani T. et al. (2011) ‘Novel DNA Virus Isolated from Samples Showing Endothelial Cell Necrosis in the Japanese Eel, Anguilla aponica’, Virology, 412: 179–87. [DOI] [PubMed] [Google Scholar]
- Moriyama T. et al. (2008) ‘Purification and Characterization of Organellar DNA Polymerases in the Red Alga Cyanidioschyzon merolae’, FEBS Journal, 275: 2899–918. [DOI] [PubMed] [Google Scholar]
- Ondov B. D. et al. (2019) ‘Mash Screen: High-Throughput Sequence Containment Estimation for Genome Discovery’, Genome Biology, 20: 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretti A. et al. (2018) ‘Characterization of BK Polyomaviruses from Kidney Transplant Recipients Suggests a Role for APOBEC3 in Driving In-Host Virus Evolution’, Cell Host & Microbe, 23: 628–35 e627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipas J. M. (1992) ‘Common and Unique Features of T Antigens Encoded by the Polyomavirus Group’, Journal of Virology, 66: 3979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipas J. M. (2019) ‘DNA Tumor Viruses and Their Contributions to Molecular Biology’, Journal of Virology, 93: e01524–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice P., Longden I., Bleasby A. (2000) ‘EMBOSS: The European Molecular Biology Open Software Suite’, Trends in Genetics, 16: 276–7. [DOI] [PubMed] [Google Scholar]
- Santiago-Rodriguez T. M. et al. (2015) ‘Chemostat Culture Systems Support Diverse Bacteriophage Communities from Human Feces’, Microbiome, 3: 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P. et al. (2003) ‘Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks’, Genome Research, 13: 2498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soding J. (2005) ‘Protein Homology Detection by HMM-HMM Comparison’, Bioinformatics, 21: 951–60. [DOI] [PubMed] [Google Scholar]
- Tisza M. J. et al. (2020) ‘Discovery of Several Thousand Highly Diverse Circular DNA Viruses’, eLife, 9: e51971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt W. et al. (1970) ‘Synergism between Phospholipase a and Various Peptides and SH-Reagents in Causing Haemolysis’, Naunyn-Schmiedebergs Archiv für Pharmakologie, 265: 442–54. [DOI] [PubMed] [Google Scholar]
- Welch N. L. et al. (2020). “Identification of Adomavirus Virion Proteins,” bioRxiv: 341131.
- Williams S. H. et al. (2018) ‘Viral Diversity of House Mice in New York City’, mBio, 9: e0135417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolford L. et al. (2007) ‘A Novel Virus Detected in Papillomas and Carcinomas of the Endangered Western Barred Bandicoot (Perameles bougainville) Exhibits Genomic Features of Both the Papillomaviridae and Polyomaviridae’, Journal of Virology, 81: 13280–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutin N. et al. (2015) ‘A Novel Group of Diverse Polinton-Like Viruses Discovered by Metagenome Analysis’, BMC Biology, 13: 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutin N., Kapitonov V. V., Koonin E. V. (2015) ‘A New Family of Hybrid Virophages from an Animal Gut Metagenome’, Biology Direct, 10: 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yutin N., Raoult D., Koonin E. V. (2013) ‘Virophages, Polintons, and Transpovirons: A Complex Evolutionary Network of Diverse Selfish Genetic Elements with Different Reproduction Strategies’, Virology Journal, 10: 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R., Oberg N. O., Gerlt J. A. (2018) ‘Democratized' Genomic Enzymology Web Tools for Functional Assignment’, Current Opinion in Chemical Biology, 47: 77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann L. et al. (2017) ‘A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at Its Core’, Journal of Molecular Biology, 430: 2237–43. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GenBank accession numbers for sequences deposited in association with this study are: BK010888BK010889BK010890BK010893BK010894BK010998BK010999BK011000BK011001BK011002BK011003BK011004BK011005BK011006BK011007BK011008BK011009BK011010BK011011BK011022BK011023BK011024BK011025BK011026BK012042BK012043BK012044BK012045BK012046BK012047BK012048BK012049BK012050BK012051BK012052BK012053BK012054BK012055BK012056BK012057BK012058BK012059BK012060BK012061BK012062BK012063BK012064BK012084BK012085BK012086.