

Abstract
Brain energy failure is an early pathological event associated with synaptic dysfunction in Alzheimer’s disease (AD). Thus, mitigation or enhancement of brain energy metabolism may offer a therapeutic avenue. However, there is uncertainty as to what metabolic process(es) may be more appropriate to support or augment since metabolism is a multiform process such that each of the various metabolic precursors available is utilized via a specific metabolic pathway. In the brain, these pathways sustain not only a robust rate of energy production but also of carbon replenishment. Triheptanoin, an edible odd-chain fatty acid triglyceride, is uncommon in that it replenishes metabolites in the tricarboxylic acid cycle (TCA) cycle via anaplerosis in addition to fueling the cycle via oxidation, thus potentially leading to both carbon replenishment and enhanced mitochondrial ATP production. To test the hypothesis that triheptanoin is protective in AD, we supplied mice with severe brain amyloidosis (5×FAD mice) with dietary triheptanoin for four and a half months. This had minimal impact on systemic metabolism and brain amyloidosis as well as tauopathy while attenuating brain ATP deficiency and mitochondrial dysfunction including respiration and redox balance. Synaptic density, a disease hallmark, was also preserved in hippocampus and neocortex despite profound amyloid deposition. None of these effects took place in treated control mice. These findings support the energy failure hypothesis of AD and justify investigating the mechanisms in greater depth with ultimate therapeutic intent.
Keywords: Alzheimer’s disease, Triheptanoin, anaplerosis, mitochondrial function, amyloid beta
Introduction:
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by synaptic failure and neuronal loss, culminating in progressive cognitive decline [1]. Amyloid beta (Aβ) deposition and tauopathy are two defining brain pathologies that accompany AD [1, 2]. However, it is increasingly recognized that these may represent intermediate or late phenomena given the limited effectiveness of current Aβ- and tau-targeting therapies, thus justifying the search for safe, alternative treatments capable of meaningfully modifying disease course [3–5]. One such early and potentially central or meaningful context is altered brain metabolism. In the past decades, clinical studies have consistently documented a close relationship between brain energy deficiency and cognitive impairment in patients with mild cognitive impairment (MCI) and AD [6–9]. In view of the importance of a sufficient ATP provision for synaptic activity, brain energy deficiency has been proposed to represent a driving force (rather than an epiphenomenon or a late phenomenon) for the synaptic failure that underlies cognitive deficits in AD [10, 11]. Of note, neurons mostly rely on mitochondrial glucose oxidation as the major source of ATP [12]. In this context, disrupted mitochondrial function and decreased glucose metabolism are features of the brain of patients with AD and animal models [6, 9, 10, 13]. Therefore, avenues to energize mitochondria seem to hold promise for the treatment of AD [14, 15].
There is one additional aspect to metabolism: Anaplerotic molecules capable of supplying net carbon replenish the pool of catalytic intermediates of the tricarboxylic acid (TCA) cycle, enhancing the production of electron transfer cofactors [16]. By virtue of their capacity to promote mitochondrial ATP generation, anaplerotic substrates have been tested in clinical trials for the treatment of neurological disorders and metabolic diseases [16]. Triheptanoin is an odd-chain anaplerotic triglyceride (including three 7-carbon fatty acids C7:0) that can be used as a dietary supplement [17]. The main metabolic product of triheptanoin, heptanoate passes across the blood-brain barrier (BBB) and enters mitochondria to increase succinyl-CoA abundance [18]. In addition, heptanoate can be converted to five-carbon (C5-) ketone bodies or glucose via gluconeogenesis in the liver to replenish the TCA cycle in the brain [19]. We and others have used triheptanoin in clinical trials for the treatment of neurological disorders including glucose transporter type 1 deficiency (G1D) [17], refractory epilepsy [20] and Huntington’s disease (HD) [18, 21] among other diseases. Since triheptanoin is fully and avidly consumed in the course of normal metabolism, in addition to its therapeutic benefit, triheptanoin is devoid of toxicity and exhibits minimal adverse effects at standard treatment doses [18]. Although triheptanoin has not been tested in clinical studies of AD and other dementias, a previous study on a mouse model with AD-like pathology (APP/PS1 mice) reported a preventive effect of triheptanoin in combination with a high-protein ketogenic diet on mouse cognitive deficits and astrogliosis [22]. In view of the potential risks of a long-term high-protein diet including renal and bone integrity [23, 24], we reason that it would be of interest to elucidate whether triheptanoin supplementation alone is protective against amyloid beta (Aβ)-mediated dysfunction.
In this study, we employed a mouse model of AD-like brain amyloidosis (5×FAD mice) at 3.5 months old, when they demonstrate mild brain Aβ deposition mimicking AD at the early stage [25–28]. The mice received dietary supplementation of triheptanoin until 8 months of age. At the end of the treatment period, long-term dietary triheptanoin supplementation showed minimal effect on mouse body weight, food intake, and levels of fasting blood glucose and triglycerides. Moreover, long-term triheptanoin-containing diet did not affect cell density in the liver, suggesting no discernible hepatotoxicity. In contrast to control diet-treated 5×FAD littermates, 5×FAD mice receiving triheptanoin treatment exhibited rescued brain ATP content and elevated brain mitochondrial nicotinamide adenine dinucleotide (NADH) abundance in addition to enhanced brain mitochondrial function including mitochondrial respiration and mitochondrial redox balance. Despite an unchanged brain Aβ load and tau phosphorylation, triheptanoin treatment preserved synaptic density in the hippocampal CA1 region and entorhinal cortex in 5×FAD mice. Our findings suggest a therapeutic benefit of triheptanoin against Aβ-mediated brain pathology in AD mice model following long-term use. This study constitutes the groundwork for further investigation to elucidate the role of brain energy failure in AD pathogenesis while providing preclinical evidence to support the use of triheptanoin supplementation as a disease-modifying therapy for the treatment of AD.
Methods and Materials:
Animals and treatment:
Animal studies were approved by the University of Texas at Dallas Animal Care and Use Committee (IACUC) and were performed in accordance with the National Institutes of Health guidelines for animal care. 5×FAD mice [B6SJL-Tg (APP-SwFlLon, PSEN1*M146L*L286 V) 6799Vas/Mmjax] were obtained from Jackson Laboratory. All mice were using maintenance diet (Lab diet, Formulab Diet 5008) before 3.5 months of age. Both nonTg and 5×FAD mice were randomly assigned to receive either a basal diet (82.9g/100g) (Harlan, TD.130888) supplemented with triheptanoin (17.1g/100g) (SASOL Germany GmbH), or an isocaloric control diet enriched with hydrogenated coconut oil (Sigma-Aldrich). The compositions of the basal diet are described in supplementary table. The mice were fed triheptanoin-based diet or control diet until 8 months of age. The body weight of the mice was measured before euthanasia.
Food intake measurement:
The mice fed with control or triheptanoin diet were single housed in metabolic cages (Tecniplast™) for 24 hours. The diets offered to mice were pre-weighed and food intake was calculated by weighing all remnants of the pellet and subtracting it to the initial weight.
Fasting blood glucose measurement:
Blood glucose in tail-prick samples was measured using the Accu-Chek blood glucose monitoring system (Roche Diagnostics Corp). Blood glucose levels were measured following 8-hour fasting.
Fasting plasma triglycerides measurement
Blood samples were obtained from the orbital sinus after 8 hours of fasting, then collected into pre-cooled heparinized tubes to separate plasma. Plasma triglyceride levels were measured with a Serum Triglyceride Determination kit (Sigma-Aldrich) according to the manufacturer instructions.
Luminescent ATP assay:
ATP synthesis from freshly dissected mice brain was measured using Luminescence ATP detection Assay Kit (Abcam) according to manufacturer’s instructions. Luminescence was detected using a microplate reader (Biotek) controlled by Gen5 software. Standard curve was prepared using ATP as substrate and luminescence readings were expressed in fold change.
NADH measurement:
Brain tissue or isolated mitochondria were homogenized with extraction buffer provided from NAD/NADH Quantification Kit (Sigma -Aldrich), followed by centrifugation at 14,000 × g for 5 minutes to remove insoluble material. The resultant supernatant was heated to 60°C for 30 minutes to decompose NAD. NADH was detected according to manufacturer instructions. The absorbance at 450nm was measured using a microplate reader (Biotek) controlled by Gen5 software.
Brain mitochondrial isolation:
Brain mitochondria were prepared as previously described [27]. Cortices were dissected from mouse brain and homogenized in ice-cold isolation buffer (225 mM mannitol, 75 mM sucrose, 2 mM K2PO4, 0.1% BSA, 5 mM Hepes, 1 mM EGTA (pH 7.2) with a Wheaton dounce homogenizer. After a centrifugation at 1,300 × g for 5 minutes to remove blood and cell debris, the supernatant was layered on 15% Percoll (GE) and centrifuged at 125,000 × g for 10 minutes. The pellets were collected and resuspended in isolation buffer with 0.02% digitonin (Sigma-Aldrich), followed by centrifugation at 8,000 x g for another 10 minutes. The pellets were then washed by additional centrifugation step in ice-cold isolation buffer without EGTA for experiments. Protein concentrations were measured using Bradford assay for protein detection (BioRad).
Mitochondrial respiration assay:
Freshly isolated brain mitochondria were used to perform mitochondrial respiration assays using a temperature temperature-regulated Clark-type oxygen electrode (Oxytherm, Hansatech) as described previously[29]. Isolated mitochondria were added into respiration buffer [225 mM mannitol, 75 mM sucrose, 2 mM K2PO4, 0.1% BSA, 5 mM Hepes, and 1 mM EGTA, (pH 7.2)], followed by 10 passages through a 27 ½ g needle (BD). The homogenate was added to a magnetically stirred chamber and energized with 5 mM glutamate/5 mM malate (Sigma-Aldrich) or succinate (5mM) to allow the mitochondria to respire via complex I or complex II, respectively. 250 µM ADP was added and state III was measured. State IV respiration was measured once the ADP was used up. The ratio of state III respiration over state IV respiration was defined as the Respiration Control Ratio (RCR).
4-hydroxynonenal (4-HNE) measurement:
Isolated brain mitochondria were homogenized with PBS containing 1% triton X-100, followed by centrifugation at 10,000 × g for 10 minutes at 4°C. The resultant supernatants were adsorbed onto a 96-well plate for 2 hours at 37°C. Levels of 4-hydroxynonenal (HNE)-protein adducts content were measured using Oxiselect HNE-His Adduct ELISA Kit (Cell Biolabs) according to the manufacturer instructions [30].
Glutathione measurement:
Reduced glutathione (GSH), oxidized glutathione (GSSG) and total glutathione were measured following the manufacturer instructions (glutathione detection kit, catalog #ADI-900-160, Enzo Life Sciences). In brief, isolated brain mitochondria were homogenized in ice-cold 5% (w/v) meta-phosphoric acid (20 ml/g tissue) (Sigma-Aldrich), followed by centrifugation at 12,000 × g for 10 min at 4°C. The resultant supernatant was collected for glutathione detection. In order to measure GSSG, reduced glutathione was derivatized by adding 2 M 4-vinylpyridine (Sigma-Aldrich) at a dilution of 1:50 (v/v), followed by incubation for 1 hour at room temperature. Total glutathione was measured directly using reaction buffer. A microplate reader (Biotek) was used for the data recording. The amounts of GSH were calculated by subtracting the amounts of GSSG from total glutathione.
Aβ ELISA assay:
Aβ amounts in mouse cortical or mitochondria samples were measured by using human Aβ40 and Aβ42 ELISA kits (Thermo Fisher Scientific, KHB3481 for Aβ40, KHB3441 for Aβ42) following the manufacturer instructions. Tissues were homogenized thoroughly with cold 5 M guanidine HCl/50 mM Tris HCl. The homogenates were incubated at room temperature for 4 hours. The samples were diluted with cold reaction buffer (Dulbecco’s phosphate buffered saline with 5% BSA and 1×protease inhibitor cocktail) and centrifuged at 16,000 × g for 20 minutes at 4°C. The supernatants were diluted with standard diluent buffer provided in the kit and quantified by ELISA kits. Aβ amounts were normalized to total protein content in the samples.
Immunoblotting:
Immunoblotting of mice brain homogenates were performed as previously described [31]. Mouse brains were homogenized in RIPA buffer (150 mM NaCl, 5 mM EDTA pH 8.0, 50 mM Tris-Base pH 8.0, 0.5% sodium deoxycholate, 10% SDS) + 1 × protease inhibitor (Millipore, #20-201). After denaturation at 100°C for 5min, the brain homogenate was separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked with 5% milk for 1 hour at room temperature and probed with the following primary antibodies: rabbit anti-Synaptophysin (Cell Signaling Technology, 5461, 1:2,000), rabbit anti-Aβ (Cell Signaling Technology, 8243, 1: 1,000), mouse anti-β-actin (Sigma-Aldrich, A5441, 1:10,000), mouse monoclonal anti-Phospho-Tau (Ser202, Thr205) (Thermo Fisher, #MN1020, 1:1,000), mouse monoclonal anti-Phospho-Tau (Ser396) (CST, #9632, 1: 5,000), rabbit monoclonal anti-Phospho-Tau (Ser404) (CST, #20194, 1:5,000), mouse monoclonal anti-T-Tau (Tau46) (CST, #4019, 1: 1,000), rabbit anti-PSD95 (CST, #3450, 1:5,000), Total OXPHOS Rodent WB Antibody Cocktail (abcam, #ab110413, 1:2,000), anti-APP C-Terminal Fragment Antibody (BioLegend, # SIG-39150, 1:1,000), mouse anti-amyloid precursor protein (BioLegend, #803002, 1:2000), followed by incubation with the appropriate secondary antibodies, HRP conjugated goat anti-mouse IgG (H + L) (Invitrogen, 626520) or HRP-conjugated goat anti-rabbit IgG (H+L) (Invitrogen, 656120). Membranes were images on Bio-Rad Chemidoc Imaging System. The blots were analyzed using Image J software (NIH).
mouse anti-PSD95 antibody (Cell Signaling Technology, 36233, 1:5,000), Immunofluorescent staining:
Mouse brains were dissected and immediately fixed in 4% paraformaldehyde (PFA) (Sigma-Aldrich) for 24 hours at 4 °C. The frozen tissue sections were prepared as previously described [32]. The slices were blocked with blocking buffer (5% goat or donkey serum, 0.3% Triton X-100 in PBS, pH 7.4) for 1 hour, then incubated with primary antibodies at room temperature overnight. The following primary antibodies were used: mouse anti-PSD95 antibody (Cell Signaling Technology, 36233, 1:400), rabbit anti-Synaptophysin (Cell Signaling Technology, 5461, 1:500), rabbit anti-Aβ (Cell Signaling Technology, 8243, 1: 1,000). After washing with PBS, the slices were then probed with appropriate cross-adsorbed secondary antibodies conjugated to Alexa Fluor 488, Alexa Fluor 594 (Thermo Fisher Scientific, 1:500). Images were collected under a Nikon confocal microscope followed by three-dimensional reconstruction and analysis using Nikon-Elements advanced Research software. Aβ-plaque occupied area were analyzed using “Threshold” operation to choose plaque then divided by the area of the brain. The synapses were defined by colocalization of synaptophysin and PSD95 which was analyzed by using “AND” operation in “binary operation” dialog of NIS element software to overlap two binary layers. The number of synapses was counted and divided by total volume of the image stack.
H&E staining:
Mice liver sections were freshly dissected and fixed in 4% paraformaldehyde (PFA) overnight at 4°C and then frozen sectioned. Hematoxylin-eosin (H&E) staining was performed following standard procedures: Slices were air dried overnight, then rehydrated with 100%, 95%, 80%, 75% and 50% ethanol. The slices were then stained with hematoxylin (Sigma-Aldrich, MHS-16) for 10 minutes followed by 5 minutes water rinsing. Next, slices were immersed in eosin (Sigma-Aldrich, HE110316) for 1 minute and then dipped into double-distilled water. Dehydration with reversed order of ethanol as cited for the rehydration was performed after staining. Images were collected on an Olympus upright microscope. Cell density was counted using Image J software (NIH).
Statistics:
Statistical comparisons were performed using GraphPad Prism 5 software. Two-way ANOVA or unpaired Student’s t-test followed by Numbers of replicates and P values are stated in each figure legend. All data were expressed as the mean ± sem. Significance was concluded when the P value was less than 0.05. Significance was indicated by symbols including * (P < 0.05), ** (P < 0.010), *** (P < 0.001). NS (not significant) denotes P > 0.05.
Results:
Triheptanoin treatment rescues brain ATP content in 5×FAD mice
Previous studies reported that 5×FAD mice display brain ATP deficiency and brain mitochondrial dysfunction accompanying the development of brain amyloidosis [26, 27]. To determine the effect of triheptanoin supplementation on brain energy content, we fed 3.5 month old nontransgenic (nonTg) mice and their 5×FAD littermates with control or triheptanoin-containing diet for a total treatment time of 4.5 months. At the end of the treatment, the resulting four groups of mice exhibited no difference in body weight (Supplementary Fig. 1A) or food intake (Supplementary Fig. 1B). In addition, no difference in the levels of fasting blood glucose (Supplementary Fig. 2A) and fasting plasma triglycerides (Supplementary Fig. 2B) was detected across the four groups. The results indicate negligible or minimal impact of long-term triheptanoin treatment on systemic metabolic profile, in agreement with previous reports [17, 33, 34]. In addition, long-term dietary supplementation of triheptanoin has no effect cell density in the liver (Supplementary Fig. 3A&B), implicating minimal or absent hepatotoxicity after long-term use.
Brain ATP content was measured by luminescent ATP assay [27]. Under the control diet, 5×FAD mice exhibited decreased brain ATP content as compared with their nonTg littermates (Fig. 1A); whereas this 5×FAD phenotypic change was substantially mitigated by triheptanoin supplementation (Fig. 1A). Importantly, further measurement of nicotinamide adenine dinucleotide (NADH) using brain tissues (Fig. 1B) and isolated brain mitochondria (Fig. 1C) from the four groups of mice showed that the reduction of NADH content in 5×FAD mice was significantly mitigated by triheptanoin supplementation. Of note, triheptanoin treatment did not affect baseline levels of brain ATP (Fig. 1A) or NADH (Fig. 1B&C) in nonTg mice. Therefore, the elevated abundance of this critical mitochondrial electron transfer cofactor suggests that rescued brain ATP in triheptanoin-treated 5×FAD mice is, at least, in part a result from enhanced TCA cycle activity.
Fig. 1. Triheptanoin treatment attenuates brain ATP deficiency in 5×FAD mice.
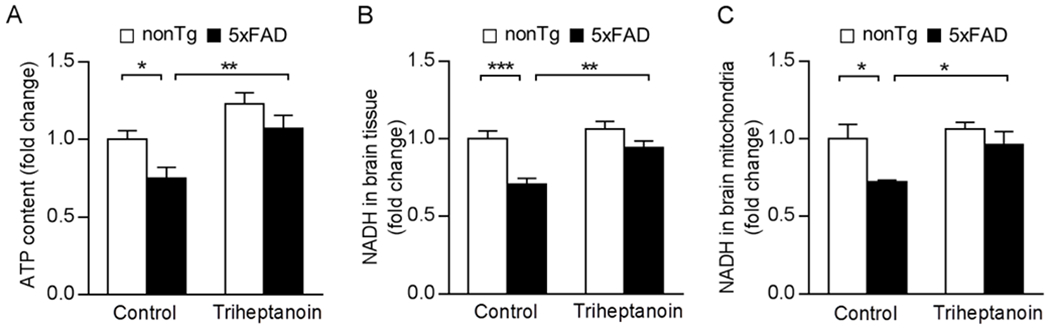
(A) ATP content in brain from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 6-7 mice per group. * P < 0.05, ** P < 0.01. (B&C) NADH levels in Brain (B) and mitochondrial (C) from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 6-7 mice per group. * P < 0.05, ** P < 0.01, *** P < 0.001.
Triheptanoin treatment protects brain mitochondria in 5×FAD mice
Because previous studies have suggested a beneficial effect of triheptanoin on brain mitochondria [35–37], we then examined brain mitochondrial function by measuring respiration using a Clark electrode. Isolated brain mitochondria from all the groups of mice were energized by glutamate/malate or succinate to reflect mitochondrial complex I/III/IV or II/III/IV respiration, respectively. When compared with their control (nonTg) counterparts, 5×FAD mice fed control diet exhibited decreased mitochondrial complex I/III/IV (Fig. 2A1) and II/III/IV (Fig. 2A2) respiration as demonstrated by lowered mitochondrial respiration control ratio (RCR). In contrast, brain mitochondrial complex I/III/IV (Fig. 2A1) and II/III/IV (Fig. 2A2) respiration were rescued in triheptanoin-treated 5×FAD mice, indicating a protective effect of triheptanoin on brain mitochondrial bioenergetics. To determine the potential effect of triheptanoin treatment on mitochondrial oxidative phosphorylation (OXPHOS) complexes, we subjected the isolated brain mitochondria from the four groups of mice to the detection of the relative levels of OXPHOS complexes by immunoblotting for their core subunits including NADH:Ubiquinone oxidoreductase subunit B8 (NDUFB8, complex I), succinate dehydrogenase complex iron sulfur subunit B (SDHB, complex II), ubiquinol-cytochrome C reductase core protein 2 (UQCRC2, complex III), mitochondrially encoded cytochrome C oxidase I (MT-CO1, complex IV) and F1Fo ATP synthase alpha subunit (ATP5A, complex V). Densitometry analysis of the immunoreactive bands showed no influence of triheptanoin on the tested subunits in either nonTg or 5×FAD mice (Supplementary Fig. 4A–F). Moreover, the disrupted mitochondrial redox state of 5×FAD mice was effectively rescued by triheptanoin supplementation as demonstrated by lowered levels of 4-hydroxynonenal (HNE), a sensitive indicator of lipid oxidation [38] (Fig. 2B) and restored levels of reduced glutathione (GSH) (Fig. 2C1) with no change in the levels of total glutathione (Fig. 2C2). The results suggest an antioxidant effect of triheptanoin supplementation, which corroborates previous studies [37]. Triheptanoin treatment had no effect on mitochondrial function in nonTg mice (Fig. 2A–C). Taken together, our findings suggest that dietary triheptanoin supplementation preserves brain mitochondrial function, mitigating brain ATP deficiency in 5×FAD mice.
Fig. 2. Triheptanoin treatment rescues brain mitochondrial function in 5×FAD mice.
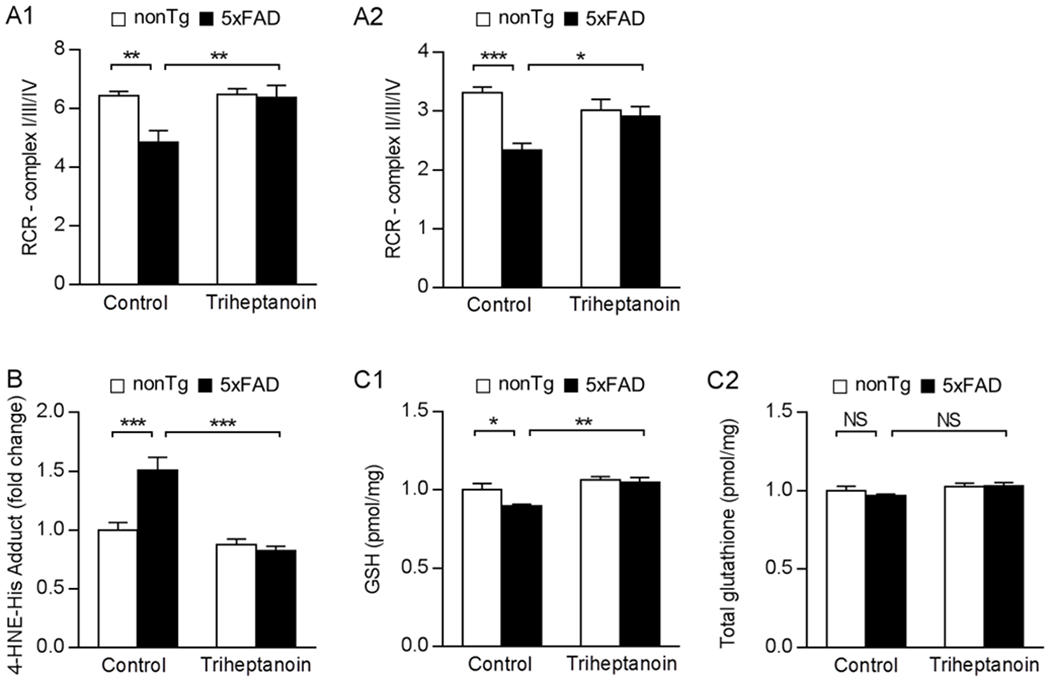
(A) Measurement of glutamate/malate-driven complex I respiration (A1) or succinate-driven complex II respiration (A2) in brain mitochondria isolated from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 5-7 mice per group. * P < 0.05, ** P < 0.01, *** P < 0.001. (B) Measurement of 4-HNE-his adduct in brain mitochondria from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 6-7 mice per group. *** P < 0.001. (C) Reduced glutathione (GSH) (C1) or total glutathione (C2) in brain mitochondria from nonTg and 5×FAD mice control or triheptanoin diet treatment. n = 6-7 mice per group. * P < 0.05, *** P < 0.001; NS, not significant.
Triheptanoin treatment does not affect brain amyloidosis or tau phosphorylation in 5×FAD mice
Aβ toxicity is the driving force of brain ATP deficiency in 5×FAD mice. To determine whether triheptanoin treatment affects brain Aβ production, we first performed ELISA assays for Aβ1-40 and 1-42 levels in cortical homogenates. In comparison with their control-treated counterparts, triheptanoin-treated 5×FAD mice displayed a slight decrease in both Aβ1-40 and 1-42 levels devoid of statistical significance (Fig. 3A). The results were further confirmed by immunoblotting for Aβ in brain homogenates (Fig. 3B1&2). We next sought to examine the impact of triheptanoin on brain Aβ deposition. To this end, we subjected brain slices to immunostaining for Aβ plaque. Regardless of triheptanoin treatment, 5×FAD mice exhibited similar Aβ plaque-occupied area in the cortex and hippocampus (Fig. 3C1&2). In addition, further immunoblotting assays showed no effect of triheptanoin treatment on amyloid precursor protein (APP) processing in 5×FAD mice demonstrated by unchanged expression of total APP (Fig. 3D1&4) or its cleavage productions including C83 (Fig. 3D2&4) and C99 (Fig. 3D3&4). The results suggest little or no impact of triheptanoin on brain Aβ production and deposition in 5×FAD mice, which is in agreement with a previous study using triheptanoin/high-protein diet to treat APP/PS1 mice [22]. Moreover, we extended our study and performed immunoblotting to examine total Tau (T-Tau) and the well-accepted AD-related Tau phosphorylation motifs, including S202/T205/S396/S404 [28, 39]. Analysis of the immunoreactive bands showed no significant difference in the expression levels of total Tau or any of the tested Tau phosphorylation motifs between the drug-treated 5xFAD mice and their vehicle-treated counterparts (Supplementary Fig. 5A–E).
Fig. 3. Triheptanoin treatment has no effect on 5×FAD mice Aβ level.
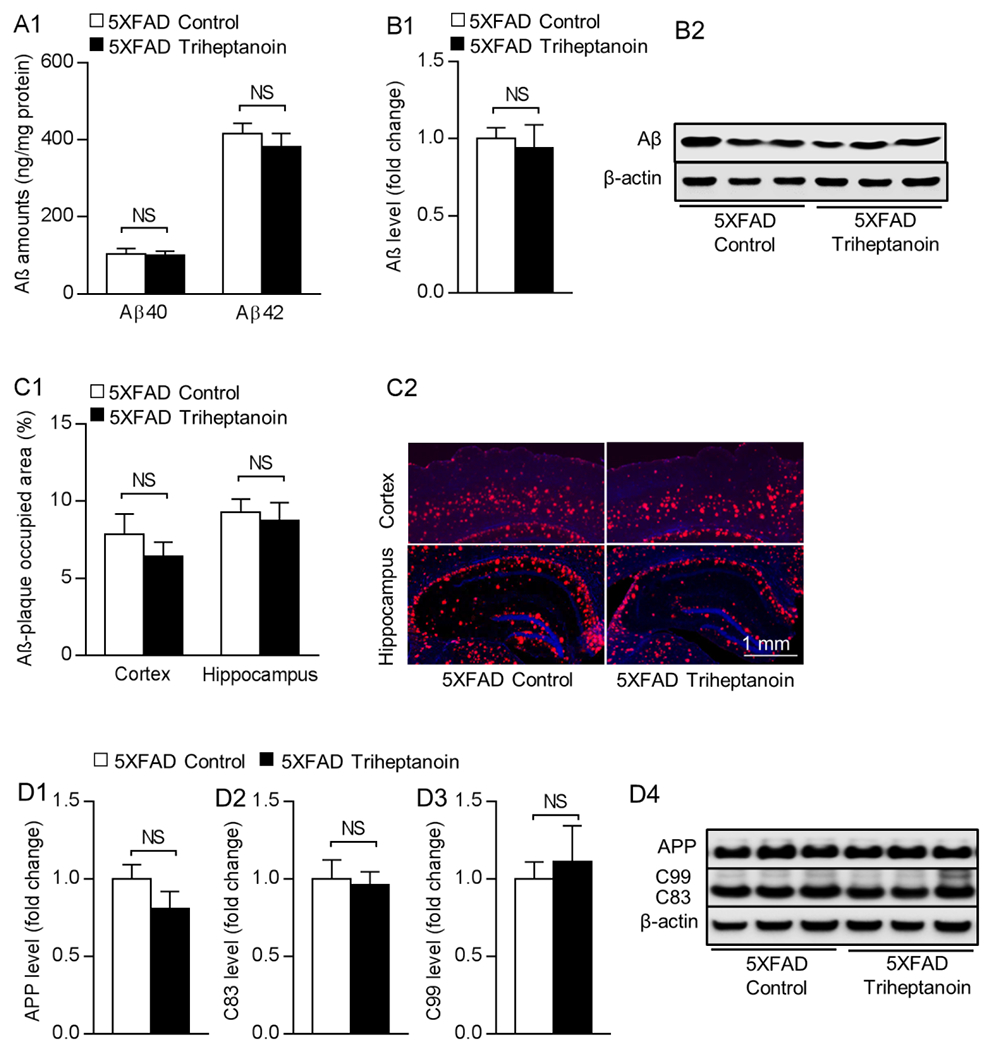
(A) Measurement of cortical Aβ 1-40 and 1-42 levels from nonTg and 5×FAD mice with control or triheptanoin diet treatment using ELISA. Unpaired student’s t-test. n = 5-6 mice per group. NS, not significant. (B1) Immunoblotting of brain Aβ from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Unpaired student’s t-test. n = 6 mice per group. NS, not significant. (B2) are the representative bands of immunoblotting. (C1) Measurement of Aβ deposition using immunostaining in cortex and hippocampus from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Unpaired student’s t-test. n = 6-7 mice per group. NS, not significant. (C2) are the representative images, scale bar = 1 mm. (D1-D4) Immunoblotting of APP (D1) and its cleavage productions includingC83 (D2), C99 (D3) in entorhinal cortex from 5×FAD mice with control or triheptanoin diet treatment. Unpaired student’s t-test. n = 5 mice per group. NS, not significant. (D4) are the representative bands of immunoblotting.
Triheptanoin treatment ameliorates synaptic loss in 5×FAD mice
Sufficient ATP provision is pivotal for synaptic activity. To determine whether triheptanoin diet could attenuate synaptic loss in 5×FAD mice, we subjected brain slices from the four groups of mice to co-staining for synaptophysin, a presynaptic marker, and PSD95, a postsynaptic marker to reflect synaptic density in mouse hippocampal CA1 region and entorhinal cortex, which are AD sensitive brain regions. Under control diet, 5×FAD mice displayed a substantial decrease in their synaptic density in the hippocampal CA1 region (Fig. 4A1&2) and the entorhinal cortex (Fig. 4B1&2) when compared with their nonTg counterparts. In contrast, triheptanoin-treated 5×FAD mice exhibited preserved synaptic density in the tested brain areas (Fig. 4A&B), paralleled with restored expression levels of synaptophysin (Fig. 4C1&2) determined by immunoblotting. Of note, the expression of PSD95 was unchanged across all groups (Fig. 4D1&2). The results indicate the beneficial effect of triheptanoin against synaptic loss in the AD model.
Fig. 4. Triheptanoin treatment preserves synaptic density in 5×FAD mice.
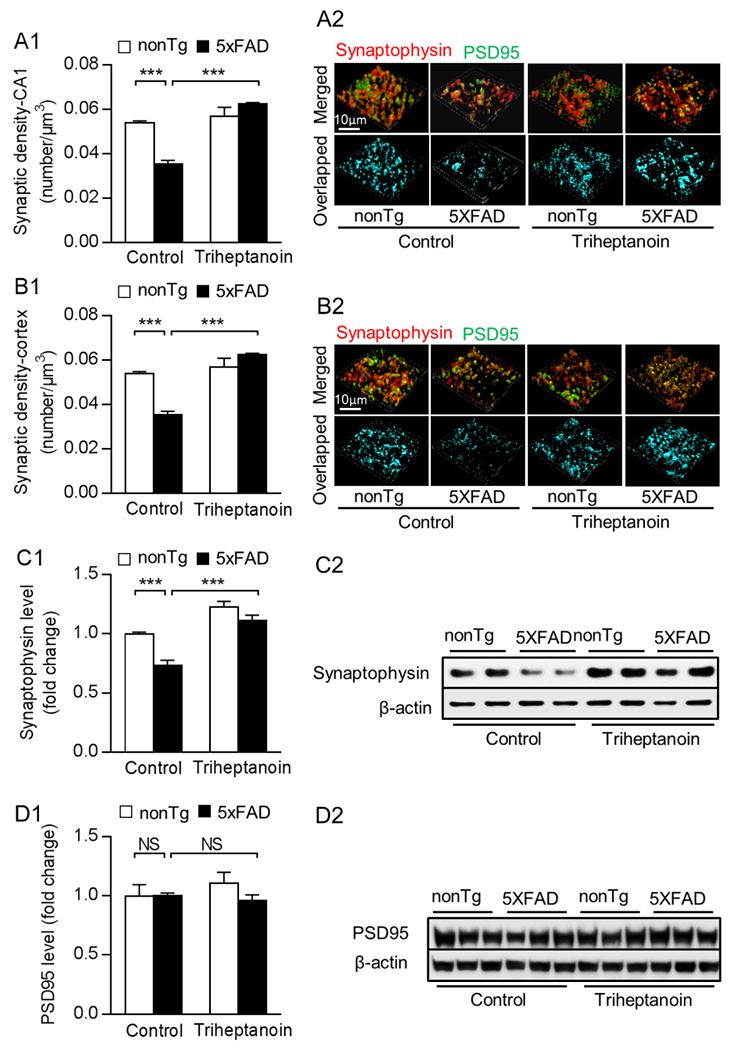
(A1&B1) Analysis of synaptic density in the hippocampal CA1 region (A1) and entorhinal cortex (B1) from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 5 mice per group. *** P < 0.001. (A2&B2) Representative 3D-reconstructed images of synapse staining. Synaptophysin (red) and PSD95 (green) were used to visualize pre- and post-synaptic components, respectively. The overlaid staining (light blue) of synaptophsyin and PSD95 indicates synapses. scale bar =10µm. (C1&C2) Immunoblotting of synaptophysin (C1) in entorhinal cortex from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 5 mice per group. ** P < 0.01, *** P < 0.001; NS, not significant. (C2) are the representative bands of immunoblotting. (D1&D2) Immunoblotting of PSD95 (D1) in entorhinal cortex from nonTg and 5×FAD mice with control or triheptanoin diet treatment. Two-way ANOVA followed by Bonferroni post hoc analysis. n = 3-4 mice per group. NS, not significant. (D2) are the representative bands of immunoblotting.
Discussion:
Brain energy failure is a key pathology underlying synaptic dysfunction in brain aging and Alzheimer’s disease (AD) [40]. In addition to decreased cerebral perfusion [41] and other forms of metabolic dysregulation not as well understood [42], impaired mitochondrial bioenergetics is considered critical in the setting of the reduced brain glucose metabolism that characterizes patients with AD [10]. As a result of compromised aerobic glucose oxidation, a metabolic shift to alternative fuel sources such as fatty acid and glutamine is manifest in AD brains [13, 43, 44]. Despite its benefit to compensate energy deficiency, such a metabolic adaption may exacerbate brain lipid and amino acid dysmetabolism in AD brains, promoting neuroinflammation, oxidative stress and ammonia toxicity, culminating in white matter degeneration and neuronal stress [13, 44]. In this context, anaplerotic therapy may demonstrate a unique advantage over other forms of metabolic therapy. Triheptanoin is a potent anaplerotic substance. It replenishes the loss of metabolites in the TCA cycle with minimal impact on systemic glycemic response [17, 45, 46]. In agreement with previous studies [17, 21, 22, 33], we have found that dietary supplementation of triheptanoin rescued brain ATP deficiency with enhanced production of NADH in brain mitochondria and preserved mitochondrial respiration in 5×FAD mice with severe cerebral amyloidosis and mitochondrial dysfunction. The results suggest a promoting effect of triheptanoin on brain energy supply probably through augmented mitochondrial function. In agreement with our observations of improved mitochondrial respiration through both complex I and II pathways, previous studies reported that triheptanoin benefits mitochondrial complex II and IV activities in stressed mitochondria, resulting in preserved mitochondrial respiration and ATP production [35]. Although the detailed mechanisms underlying triheptanoin-related protection on mitochondrial respiration remain unclear, enhanced NADH production might play a role. First, the restored NADH level reflects that of TCA cycle intermediates, which are required to maintain the rate of the cycle. The principal function of the cycle is the oxidation of acetyl-CoA to CO2, a central process to the generation of NADH and FADH2 for use in the electron transport chain. In principle, the reactions of the TCA cycle could replenish or recycle all of the cycle intermediates. However, most can also be consumed in the course of other metabolic reactions following efflux from mitochondria via specific transporters, leading, if uncompensated, to a net carbon deficit inside mitochondria. This process is balanced via the supply of carbon to the TCA cycle via anaplerotic reactions, which maintain the levels of cycle intermediates required for optimal cycle activity[47]. The anaplerotic, carbon replenishing process is particularly important in tissues with elevated metabolic rate such as the brain, where TCA cycle intermediate abundance is low relative to the robust cycle velocity that the intermediates normally sustain [48]. Moreover, the antioxidant capacity of NADH protects the integrity and activity of mitochondrial respiratory enzymes. This is supported by rescued mitochondrial redox balance in response to triheptanoin treatment in our and others’ studies [37, 49] and the beneficial effects of ROS scavengers on mitochondrial function [50]. Therefore, our findings support the effect of triheptanoin supplementation in improving mitochondrial metabolic function, culminating in enhanced ATP synthesis and reduced oxidative stress. Indeed, we could not rule out the contribution of other triheptanoin-related mechanisms such as regulation of cerebral perfusion and modification of glial metabolic remodeling to rescued brain energy deficits. These questions will be addressed in our further study.
Another benefit of triheptanoin-rich diet is its little or no impact on systemic metabolic profile. Consistent with previous studies [17, 20, 51, 52], long-term treatment of triheptanoin did not affect mouse body weight and food intake. In addition, triheptanoin-treated 5×FAD mice did not show significant change in their circulating glucose and triglycerides. These observations are in agreement with previous clinical studies [17, 20] and animal experiments [34, 52]. In addition, in comparison with common ketogenic diets, triheptanoin treatment necessitates less restriction of the amount and type of carbohydrate consumed [20]. Indeed, and in contrast with a previous study using triheptanoin in combination with a high-protein ketogenic diet on another AD mouse model [22], our findings suggest that triheptanoin alone has a protective effect in correcting brain energy deficiency and preserving synaptic density in mice mimicking AD-like brain pathology. In this context, we postulate that triheptanoin supplementation may be a feasible, effective and well-tolerated therapy for AD patients, especially for those who have poor tolerance to ketogenic diets or suffer from other metabolic disorders such as diabetes or obesity. Moreover, long-term treatment of triheptanoin had no effect on the baseline levels of metabolic profile and mitochondrial function in nontransgenic (nonTg) mice. These results are in agreement with other reports [37], suggesting the safety of long-term triheptanoin treatment. Of note, triheptanoin was ineffective in palliating cardiac injury in a mouse model with very long-chain acyl-CoA dehydrogenase deficiency [52], which may be due to organ type-dependent utilization of triheptanoin. Based on our study, long-term use of triheptanoin alone is effective in protecting brain pathology in 5×FAD mice with little or no hepatotoxicity. However, the potential for organ type-related triheptanoin metabolism should be considered in those clinical studies where this phenomenon may be clinically relevant. Further preclinical studies including any effect on other organs may help refine optimized therapeutic strategies that rely on triheptanoin-based diets.
Lastly, our results showed a marginal effect of long-term triheptanoin treatment on brain amyloidosis and APP processing, which is in agreement with a previous report [22]. Interestingly, previous studies showed that ketogenic diets attenuate brain energy deficiency in association with a significant reduction in brain Aβ aggregation in animals, probably through increased Aβ degradation due to an unknown effect on metabolic state [53, 54]. Such a discrepancy of the effect on brain amyloidosis may arise from the different nature of the treatments. Ketogenic diets have a similar metabolic effect as caloric restriction (CR) regimes, which modify insulin sensitivity and ameliorate insulin resistance, and this may contribute to suppress amyloidogenesis [55]. Another possible reason is the difference in animal models. We administrated triheptanoin to 5×FAD mice in this study while the other study used a mouse model (APP/V717I) with much less aggressive brain Aβ production rate [53]. Of note, previous studies targeting mitochondrial dysfunction, brain energy deficits and the downstream cascades showed protective effects on neuronal stress in different AD mouse models without affecting brain amyloidosis and/or tau phosphorylation [28, 56, 57]. A very recent piece of evidence is that manipulation of AMP-activated protein kinase (AMPK) in Tg19959 mice, which mimic AD like brain amyloidosis, substantially improves synaptic function without affecting brain Aβ load or tau pathology [56]. This supports the complexity of AD pathogenesis and further implicates the contribution of “Aβ-independent” mechanisms of synaptic stress in AD.
In summary, our results indicate a protective effect of triheptanoin supplementation against Aβ-mediated brain energy deficiency and mitochondrial dysfunction, resulting in preserved synaptic density. The protective effect may result from the replenishment of metabolites in the TCA cycle and the rebalancing of mitochondrial redox state. However, several limitations of this study should be noted. For example, the systemic administration of triheptanoin may also impact energy metabolism in glial cells. Therefore, triheptanoin may also be protective against glial cell activation and resultant neuroinflammation in Aβ-rich milieus. Moreover, it remains unclear whether triheptanoin affects cerebral perfusion in 5×FAD mice, thereby improving brain glucose supply to mitigate brain energy failure. Lastly, for future translational study, the effect of long-term use of triheptanoin on cognition in control mice and AD mouse models should be evaluated in a sophisticated manner. These questions constitute the groundwork for future studies. Nevertheless, to the best of our knowledge, this is the first study of the long-term use of triheptanoin for the treatment of AD mouse models with severe brain pathology. Our findings support a key role of brain energy failure in the pathogenesis of AD and suggest the potential of anaplerotic therapy using triheptanoin or other anaplerotic substances for the treatment of this devastating neurological disorder.
Supplementary Material
Acknowledgement:
This work was supported by research funding from Alzheimer’s Association (NIRG-12-242803 to LG, AARG-16-442863 to HD) and National Institutes of Health (NS077015, NS094257 and NS102588 to JMP).
Footnotes
Conflict of interest: The authors have no conflict of interest to claim.
References:
- [1].Querfurth HW, LaFerla FM (2010) Alzheimer’s disease. N Engl J Med 362, 329–344. [DOI] [PubMed] [Google Scholar]
- [2].Cummings JL (2004) Alzheimer’s disease. N Engl J Med 351, 56–67. [DOI] [PubMed] [Google Scholar]
- [3].Honig LS, Vellas B, Woodward M, Boada M, Bullock R, Borrie M, Hager K, Andreasen N, Scarpini E, Liu-Seifert H, Case M, Dean RA, Hake A, Sundell K, Poole Hoffmann V, Carlson C, Khanna R, Mintun M, DeMattos R, Selzler KJ, Siemers E (2018) Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N Engl J Med 378, 321–330. [DOI] [PubMed] [Google Scholar]
- [4].Selkoe DJ (2019) Alzheimer disease and aducanumab: adjusting our approach. Nat Rev Neurol 15, 365–366. [DOI] [PubMed] [Google Scholar]
- [5].Congdon EE, Sigurdsson EM (2018) Tau-targeting therapies for Alzheimer disease. Nat Rev Neurol 14, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].van der Velpen V, Teav T, Gallart-Ayala H, Mehl F, Konz I, Clark C, Oikonomidi A, Peyratout G, Henry H, Delorenzi M, Ivanisevic J, Popp J (2019) Systemic and central nervous system metabolic alterations in Alzheimer’s disease. Alzheimers Res Ther 11, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pagani M, Nobili F, Morbelli S, Arnaldi D, Giuliani A, Oberg J, Girtler N, Brugnolo A, Picco A, Bauckneht M, Piva R, Chincarini A, Sambuceti G, Jonsson C, De Carli F (2017) Early identification of MCI converting to AD: a FDG PET study. Eur J Nucl Med Mol Imaging 44, 2042–2052. [DOI] [PubMed] [Google Scholar]
- [8].Toledo JB, Arnold M, Kastenmuller G, Chang R, Baillie RA, Han X, Thambisetty M, Tenenbaum JD, Suhre K, Thompson JW, John-Williams LS, MahmoudianDehkordi S, Rotroff DM, Jack JR, Motsinger-Reif A, Risacher SL, Blach C, Lucas JE, Massaro T, Louie G, Zhu H, Dallmann G, Klavins K, Koal T, Kim S, Nho K, Shen L, Casanova R, Varma S, Legido-Quigley C, Moseley MA, Zhu K, Henrion MYR, van der Lee SJ, Harms AC, Demirkan A, Hankemeier T, van Duijn CM, Trojanowski JQ, Shaw LM, Saykin AJ, Weiner MW, Doraiswamy PM, Kaddurah-Daouk R, Alzheimer’s Disease Neuroimaging I, the Alzheimer Disease Metabolomics C (2017) Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement 13, 965–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hoyer S (2004) Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. Adv Exp Med Biol 541, 135–152. [DOI] [PubMed] [Google Scholar]
- [10].Perez Ortiz JM, Swerdlow RH (2019) Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br J Pharmacol 176, 3489–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Young-Collier KJ, McArdle M, Bennett JP (2012) The dying of the light: mitochondrial failure in Alzheimer’s disease. J Alzheimers Dis 28, 771–781. [DOI] [PubMed] [Google Scholar]
- [12].Kann O, Kovacs R (2007) Mitochondria and neuronal activity. Am J Physiol Cell Physiol 292, C641–657. [DOI] [PubMed] [Google Scholar]
- [13].Dong Y, Brewer GJ (2019) Global Metabolic Shifts in Age and Alzheimer’s Disease Mouse Brains Pivot at NAD+/NADH Redox Sites. J Alzheimers Dis 71, 119–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wilkins JM, Trushina E (2017) Application of Metabolomics in Alzheimer’s Disease. Front Neurol 8, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Butterfield DA, Halliwell B (2019) Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci 20, 148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Brunengraber H, Roe CR (2006) Anaplerotic molecules: current and future. J Inherit Metab Dis 29, 327–331. [DOI] [PubMed] [Google Scholar]
- [17].Pascual JM, Liu P, Mao D, Kelly DI, Hernandez A, Sheng M, Good LB, Ma Q, Marin-Valencia I, Zhang X, Park JY, Hynan LS, Stavinoha P, Roe CR, Lu H (2014) Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol 71, 1255–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Mochel F, Duteil S, Marelli C, Jauffret C, Barles A, Holm J, Sweetman L, Benoist JF, Rabier D, Carlier PG, Durr A (2010) Dietary anaplerotic therapy improves peripheral tissue energy metabolism in patients with Huntington’s disease. Eur J Hum Genet 18, 1057–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Marin-Valencia I, Good LB, Ma Q, Malloy CR, Pascual JM (2013) Heptanoate as a neural fuel: energetic and neurotransmitter precursors in normal and glucose transporter I-deficient (G1D) brain. J Cereb Blood Flow Metab 33, 175–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Borges K, Kaul N, Germaine J, Kwan P, O’Brien TJ (2019) Randomized trial of add-on triheptanoin vs medium chain triglycerides in adults with refractory epilepsy. Epilepsia Open 4, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Adanyeguh IM, Rinaldi D, Henry PG, Caillet S, Valabregue R, Durr A, Mochel F (2015) Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 84, 490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Aso E, Semakova J, Joda L, Semak V, Halbaut L, Calpena A, Escolano C, Perales JC, Ferrer I (2013) Triheptanoin supplementation to ketogenic diet curbs cognitive impairment in APP/PS1 mice used as a model of familial Alzheimer’s disease. Curr Alzheimer Res 10, 290–297. [DOI] [PubMed] [Google Scholar]
- [23].Shilpa J, Mohan V (2018) Ketogenic diets: Boon or bane? Indian J Med Res 148, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Cuenca-Sanchez M, Navas-Carrillo D, Orenes-Pinero E (2015) Controversies surrounding high-protein diet intake: satiating effect and kidney and bone health. Adv Nutr 6, 260–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Eimer WA, Vassar R (2013) Neuron loss in the 5XFAD mouse model of Alzheimer’s disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang L, Guo L, Lu L, Sun H, Shao M, Beck SJ, Li L, Ramachandran J, Du Y, Du H (2016) Synaptosomal Mitochondrial Dysfunction in 5xFAD Mouse Model of Alzheimer’s Disease. PLoS One 11, e0150441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Beck SJ, Guo L, Phensy A, Tian J, Wang L, Tandon N, Gauba E, Lu L, Pascual JM, Kroener S, Du H (2016) Deregulation of mitochondrial F1FO-ATP synthase via OSCP in Alzheimer’s disease. Nat Commun 7, 11483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tian J, Guo L, Sui S, Driskill C, Phensy A, Wang Q, Gauba E, Zigman JM, Swerdlow RH, Kroener S, Du H (2019) Disrupted hippocampal growth hormone secretagogue receptor 1alpha interaction with dopamine receptor D1 plays a role in Alzheimer’s disease. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Heng Du LG, Fang Fang, Chen Doris, Sosunov Alexander A, McKhann Guy M, Yan Yilin, Wang Chunyu, Zhang Hong, Molkentin Jeffery D, Gunn-Moore Frank J, Vonsattel Jean Paul, Arancio Ottavio, Chen John Xi, Yan Shi Du (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nature Medicine 14, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nyosha Alikhani LG, Yan hiqiang, Du Heng, Pinho Catarina Moreira, Chen John Xi, Glaser Elzbieta, Yan Shirley ShiDu (2012) Decreased Proteolytic Activity of the Mitochondrial Amyloid-β Degrading Enzyme, PreP Peptidasome, in Alzheimer’s Disease Brain Mitochondria. Journal of Alzheimers Disease 27, 75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jing Tian LG, Sui Shaomei, Driskill Christopher, Phensy Aarron, Wang Qi, Gauba Esha, Zigman Jeffrey M., Swerdlow Russell H., Du Sven Kroener Heng (2019) Disrupted hippocampal growth hormone secretagogue receptor 1α interaction with dopamine receptor D1 plays a role in Alzheimer′s disease. Science Translational Medicine 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lin Lu LG, Gauba Esha, Tian Jing, Wang Lu, Tandon Neha, Shankar Malini, Beck Simon J., Du Yifeng, Du Heng (2015) Transient Cerebral Ischemia Promotes Brain Mitochondrial Dysfunction and Exacerbates Cognitive Impairments in Young 5xFAD Mice. PLoS One 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tefera TW, Wong Y, Barkl-Luke ME, Ngo ST, Thomas NK, McDonald TS, Borges K (2016) Triheptanoin Protects Motor Neurons and Delays the Onset of Motor Symptoms in a Mouse Model of Amyotrophic Lateral Sclerosis. PLoS One 11, e0161816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Park MJ, Aja S, Li Q, Degano AL, Penati J, Zhuo J, Roe CR, Ronnett GV (2014) Anaplerotic triheptanoin diet enhances mitochondrial substrate use to remodel the metabolome and improve lifespan, motor function, and sociability in MeCP2-null mice. PLoS One 9, e109527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Schwarzkopf TM, Koch K, Klein J (2015) Reduced severity of ischemic stroke and improvement of mitochondrial function after dietary treatment with the anaplerotic substance triheptanoin. Neuroscience 300, 201–209. [DOI] [PubMed] [Google Scholar]
- [36].Thomas NK, Willis S, Sweetman L, Borges K (2012) Triheptanoin in acute mouse seizure models. Epilepsy Res 99, 312–317. [DOI] [PubMed] [Google Scholar]
- [37].Tan KN, Simmons D, Carrasco-Pozo C, Borges K (2018) Triheptanoin protects against status epilepticus-induced hippocampal mitochondrial dysfunctions, oxidative stress and neuronal degeneration. J Neurochem 144, 431–442. [DOI] [PubMed] [Google Scholar]
- [38].Zhong H, Yin H (2015) Role of lipid peroxidation derived 4-hydroxynonenal (4-HNE) in cancer: focusing on mitochondria. Redox Biol 4, 193–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Mandelkow EM, Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2, a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pathak D, Berthet A, Nakamura K (2013) Energy failure: does it contribute to neurodegeneration? Ann Neurol 74, 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Austin BP, Nair VA, Meier TB, Xu G, Rowley HA, Carlsson CM, Johnson SC, Prabhakaran V (2011) Effects of hypoperfusion in Alzheimer’s disease. J Alzheimers Dis 26 Suppl 3, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Neth BJ, Craft S (2017) Insulin Resistance and Alzheimer’s Disease: Bioenergetic Linkages. Front Aging Neurosci 9, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Craft S (2009) The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol 66, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yao J, Rettberg JR, Klosinski LP, Cadenas E, Brinton RD (2011) Shift in brain metabolism in late onset Alzheimer’s disease: implications for biomarkers and therapeutic interventions. Mol Aspects Med 32, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kovac S, Abramov AY, Walker MC (2013) Energy depletion in seizures: anaplerosis as a strategy for future therapies. Neuropharmacology 69, 96–104. [DOI] [PubMed] [Google Scholar]
- [46].Yudkoff M, Daikhin Y, Melo TM, Nissim I, Sonnewald U, Nissim I (2007) The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu Rev Nutr 27, 415–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Owen OE, Kalhan SC, Hanson RW (2002) The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277, 30409–30412. [DOI] [PubMed] [Google Scholar]
- [48].Hassel B (2000) Carboxylation and anaplerosis in neurons and glia. Mol Neurobiol 22, 21–40. [DOI] [PubMed] [Google Scholar]
- [49].Borges K, Sonnewald U (2012) Triheptanoin--a medium chain triglyceride with odd chain fatty acids: a new anaplerotic anticonvulsant treatment? Epilepsy Res 100, 239–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Atamna H, Nguyen A, Schultz C, Boyle K, Newberry J, Kato H, Ames BN (2008) Methylene blue delays cellular senescence and enhances key mitochondrial biochemical pathways. FASEB J 22, 703–712. [DOI] [PubMed] [Google Scholar]
- [51].Gillingham MB, Heitner SB, Martin J, Rose S, Goldstein A, El-Gharbawy AH, Deward S, Lasarev MR, Pollaro J, DeLany JP, Burchill LJ, Goodpaster B, Shoemaker J, Matern D, Harding CO, Vockley J (2017) Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis 40, 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Tucci S, Floegel U, Beermann F, Behringer S, Spiekerkoetter U (2017) Triheptanoin: long-term effects in the very long-chain acyl-CoA dehydrogenase-deficient mouse. J Lipid Res 58, 196–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Van der Auwera I, Wera S, Van Leuven F, Henderson ST (2005) A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr Metab (Lond) 2, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kashiwaya Y, Takeshima T, Mori N, Nakashima K, Clarke K, Veech RL (2000) D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc Natl Acad Sci U S A 97, 5440–5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhao WQ, Townsend M (2009) Insulin resistance and amyloidogenesis as common molecular foundation for type 2 diabetes and Alzheimer’s disease. Biochim Biophys Acta 1792, 482–496. [DOI] [PubMed] [Google Scholar]
- [56].Zimmermann HR, Yang W, Kasica NP, Zhou X, Wang X, Beckelman BC, Lee J, Furdui CM, Keene CD, Ma T (2020) Brain-specific repression of AMPKalpha1 alleviates pathophysiology in Alzheimer’s model mice. J Clin Invest. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, Yan Y, Wang C, Zhang H, Molkentin JD, Gunn-Moore FJ, Vonsattel JP, Arancio O, Chen JX, Yan SD (2008) Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat Med 14, 1097–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.