

Abstract
Stunning advances have been achieved in addressing the protein folding problem, providing deeper understanding of the mechanisms by which proteins navigate energy landscapes to reach their native states and enabling powerful algorithms to connect sequence to structure. However, the realities of the in vivo protein folding problem remain a challenge to reckon with. Here, we discuss the concept of the “proteome folding problem”—the problem of how organisms build and maintain a functional proteome—by admitting that folding energy landscapes are characterized by many misfolded states and that cells must deploy a network of chaperones and degradation enzymes to minimize deleterious impacts of these off-pathway species. The resulting proteostasis network is an inextricable part of in vivo protein folding and must be understood in detail if we are to solve the proteome folding problem. We discuss how the development of computational models for the proteostasis network’s actions and the relationship to the biophysical properties of the proteome has begun to offer new insights and capabilities.
Keywords: Protein folding, proteome folding, proteostasis, chaperone, energy landscape, computational modeling
The Protein Folding Problem
In 1958, Kendrew et al. published the first three-dimensional model of the structure of a globular protein, sperm whale myoglobin. Although the resolution was only 6 Å, the complexity of the structure was abundantly clear and led the authors to remark: “The arrangement seems to be almost totally lacking in the kind of regularities which one instinctively anticipates, and it is more complicated than has been predicted by any theory of protein structure” [1]. This complexity became even more apparent in later, higher resolution structures of myoglobin [2]. In parallel with these studies, Anfinsen and others were studying the process of protein folding using bovine pancreatic ribonuclease as a model protein. They discovered that the complicated structures reported by Kendrew and others seemed to be the thermodynamically most stable conformations that could be adopted by polypeptide chains. Moreover, they showed that these structures could be attained without assistance from any external biological machinery, despite their complexity [3–6]. These two lines of work immediately suggested that protein primary structures contain all the information needed to produce the rich complexity evident in protein tertiary structures. And so, the protein folding problem was born.
The protein folding problem has dominated protein science for the past 60 years. There are many definitions of it, but we prefer Dill’s three-part formulation [7, 8]: (1) how do we predict a protein’s three-dimensional tertiary structure from its one-dimensional primary structure? (2) how do the various intra- and intermolecular interactions in the system (protein-protein, protein-solvent, and solvent-solvent) determine this three-dimensional structure? and (3) what is (are) the pathway(s) by which it is formed? Tremendous progress has been made on all three aspects of the protein folding problem, and this progress has been accelerating as more powerful computational and experimental methods have been brought to bear [9]. This past year, however, we witnessed an extraordinary advance toward a solution to the protein folding problem in the performance of AlphaFold, an artificial intelligence program developed by DeepMind to predict protein structures. In CASP14 (the 14th running of the Critical Assessment of Structure Prediction competition), AlphaFold was able to predict the three-dimensional structures of an array of proteins with a median accuracy that was as high as that of models built from experimental data [10, 11]. The potential utility of this algorithm was recently powerfully demonstrated by predicting the structures of almost every protein in the human proteome [12]. While AlphaFold cannot be considered a comprehensive solution to the protein folding problem—it addresses only the first of the three questions that make up the protein folding problem as defined above—we believe that it probably will mark a new era in protein science. As such this seems like a suitable moment to pause and consider what challenges might define the next 60 years of protein science the way that the protein folding problem has defined the last 60.
The Proteome Folding Problem
The protein folding problem focuses on one outcome that can occur when an unstructured protein is placed under native conditions: folding to the functional native structure. However, it has long been known that folding is not so straightforward for many (if not most) proteins, and that other, more untoward outcomes are possible [13]. Proteins can become trapped in misfolded conformations that represent local minima on their folding energy landscapes [14, 15]. This is especially true for longer proteins with more complicated topologies, which are underrepresented among proteins for which detailed in vitro folding studies have been done [16]. This situation can be visualized with a folding energy landscape that contains more than one deep minimum, as suggested by Clark in 2004 [15]. Such a folding energy landscape is depicted in Figure 1. If these misfolded conformers are abundant enough and persist for long enough, they can self-associate to form aggregates that are virtually never salutary to a cell and are usually toxic, especially in their early stages of formation [17–20]. Even if misfolded proteins did not aggregate, it is very unlikely that they would be functional, and such a loss-of-function could be devastating [17, 19, 20]. Moreover, many different proteins are present simultaneously in vivo, a feature of the proteome folding problem that introduces major complexity. To mitigate the effects of protein misfolding, all organisms from the time of the last universal common ancestor have had a network of cellular components that is tasked with maintaining protein homeostasis, or “proteostasis”, which is the condition of an organism having enough properly folded proteins to carry out the functions essential to life but not so much misfolded/aggregated protein as to interfere with these functions [21, 22].
Figure 1.
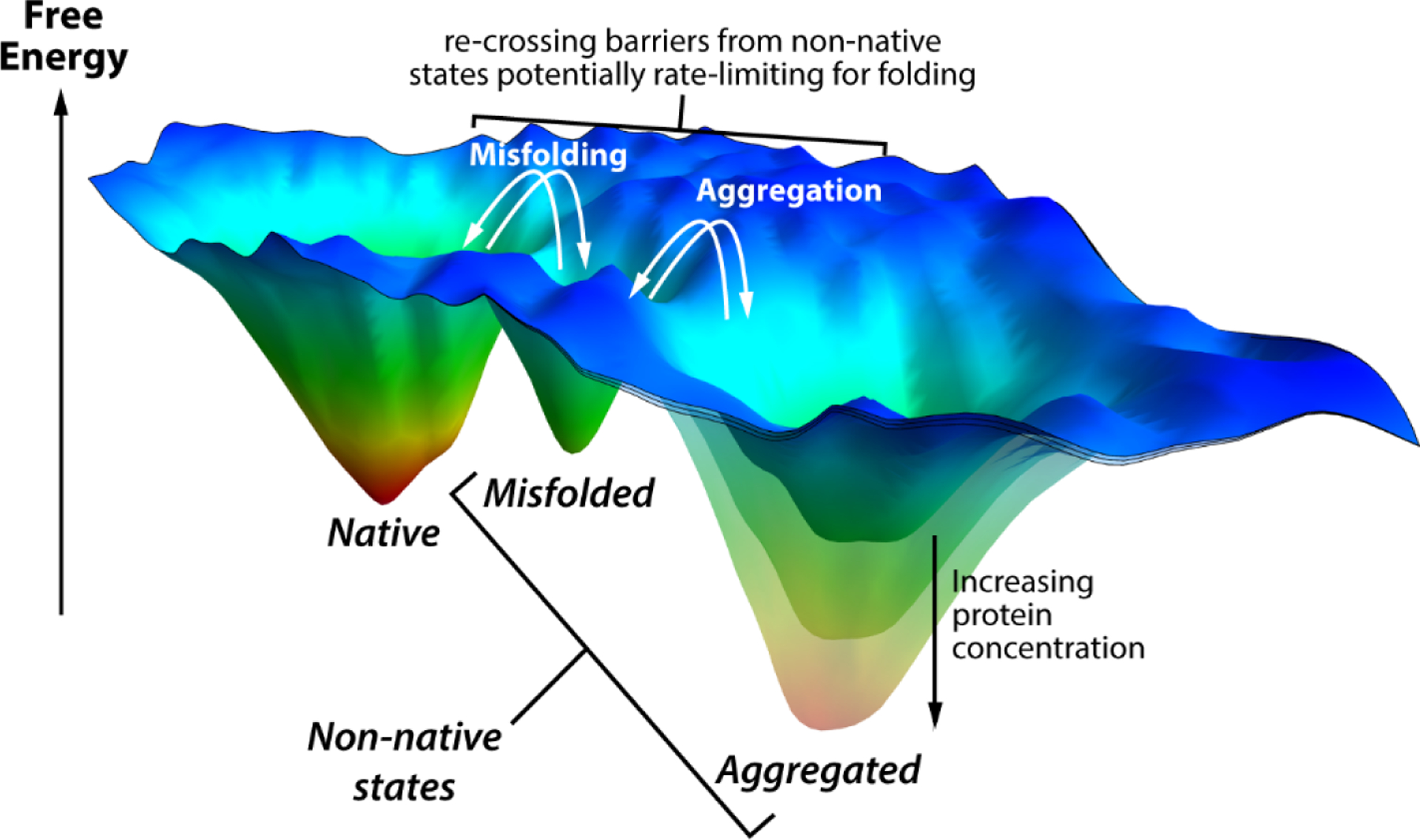
Protein folding energy landscape with multiple minima, following the depiction by Clark [15]. The blue areas represent the unfolded state, while the three energy wells shown represent the native state and two non-native states (a misfolded monomer and an aggregated state, as labeled). The aggregated state becomes more stable as the protein concentration increases. Protein that enters the non-native states may have to cross significant energy barriers to reach the native state, making these steps potentially rate-limiting for folding.
Recognizing that protein folding is not an ideal process and that there is biological machinery that exists to help it along raises a different problem that has been gaining recognition over the past two decades [14–19, 23, 24] and that we believe will become one of the dominant questions in protein science going forward: the proteome folding problem. In our view, the proteome folding problem can be broken into two questions. The first is still intrinsic to protein sequences under a given set of conditions: what alternative states are competitive with the native state, either thermodynamically or kinetically or both, on the protein folding energy landscape as it exists in living cells? This question subsumes the original protein folding problem, but it also requires an understanding of the energetics of non-native states, which would have been considered decoys or incorrect solutions in the context of the original protein folding problem. In addition, it includes issues relating to the environment within cells. Many environmental variables that can profoundly influence protein folding vary not only between cells but also within cells—from organelle to organelle and potentially even between liquid-liquid phase separated domains in a single organelle [25–29]. These variables include but are not limited to: the pH; the concentrations of ions and osmolytes [30–32]; and macromolecular crowding [33–35].
The second question in the proteome folding problem is largely extrinsic to protein sequences: how do components of the biological folding machinery, the “proteostasis network”, interact with proteins to optimize protein folding outcomes, or when folding fails, how do they mitigate the impact of protein folding failures? We examine these questions in the following sections.
Non-native States of Proteins and the Cellular Folding Environment
A useful example of the non-native states of proteins comes from work by the Anfinsen lab on ribonuclease folding [4, 15]. Ribonuclease has eight cysteine residues that can form four disulfide bonds. There are 105 possible pairings of these cysteines as disulfides, only one of which allows native folding and function. Re-folding reduced and denatured ribonuclease under conditions that permit cysteine oxidation to disulfides, but not subsequent disulfide shuffling, produces a mixture in which many, or perhaps even most, of the 104 mispaired combinations of disulfides can be found; these all represent non-native states.
Non-native states would not be a problem for biological protein folding if they were transient. They become a problem only when substantial amounts of protein become trapped in non-native states on time scales comparable to other biological processes like protein synthesis, protein trafficking, cell division, etc. In the case of ribonuclease, even when refolding is performed in a redox buffer so that disulfide interchange is possible, non-native states with mispaired disulfides form quickly but resolve relatively slowly to the native state.[3, 4, 36] As a result, a substantial fraction of ribonuclease is non-native as it reacquires the native state over the course of more than an hour. As Anfinsen observed at the time [37], this situation seems biologically untenable.
Ribonuclease becomes trapped in non-native states because these states are kinetically stabilized by the disulfide bonds between mispaired cysteine residues. Such covalent linkages, however, are not necessary to create long-lived non-native states. The same forces that stabilize protein native states can stabilize misfolded states, perhaps not as efficiently since misfolded states should not be as well-packed as native states, but the energy barrier to “un-misfold” a non-native state could cause a substantial slowing of the overall rate of native folding and lead to a persistent population of non-native protein [15].
Non-native states can also be stabilized by aggregation. In this case, parts of a protein that normally interact intramolecularly in the native state end up on the surface of a non-native state where they can drive self-association via intermolecular interactions [15, 18, 38, 39]. The stability of aggregates formed in this way is concentration dependent. If the aggregation-prone non-native state is below a “critical concentration”, then aggregation will not occur. Above that concentration, aggregation is possible [18, 39, 40]. In addition, aggregation will be faster as the concentration increases further [39, 41]. Native folding is likely to be rate limited by the off-rate for protein molecules dissociating from aggregates, which could be very slow indeed (Figure 1). Even worse, the aggregated state can overtake the native state to become the thermodynamically most stable state, especially if the native state stability is compromised by an environmental factor (temperature, pH, etc.) or mutation, or if the protein is highly concentrated as could occur if it was, for example, preferentially partitioned into a phase separated domain [25].
Answering the first question of the proteome folding problem—what alternative states compete with the native state on the protein folding energy landscape?—requires the ability to predict the properties of non-native states in the cellular environment (i.e., accounting for the pH, solute composition, crowding, etc. inside the cell) from sequence information. This is like the protein folding problem, but non-native states of proteins are much more difficult to study than native states. Non-native states are heterogeneous, and/or amorphous, and/or insoluble; all these qualities interfere with the usual techniques that are used to study proteins. Nevertheless, substantial inroads have been made in the experimental characterization of non-native states, especially the class of fibrillar, and often disease-associated, protein aggregates known as amyloid [42]. Detailed structures of amyloid fibrils have now been obtained by solid-state NMR [43, 44], X-ray and electron diffraction of microcrystals [45, 46], and cryo-electron microscopy [47, 48]. Less structured non-native states have been characterized as well (albeit at much lower resolution) using, for example, single-molecule methods [49–52], small-angle X-ray scattering [53, 54], NMR [55, 56], and other methods [57].
Real progress in determining the sequence-structure-energy relationships in the cellular milieu will of course require much more experimental data of the sort described in the preceding paragraph, but in addition this data will have to be wedded to computational methods so that the underlying physics can be understood. Unfortunately, it seems unlikely that algorithms like AlphaFold, which rely primarily on structural bioinformatics-based methods for native structure prediction, will be useful for this endeavor. The massive databases of non-native protein structures that would be needed do not exist, and probably cannot exist because so many non-native states are heterogeneous and dynamic. Computationally exploring non-native regions of the protein folding energy landscape is likely to be the province of computational molecular physics methods [9]. Such methods operate from first principles and therefore do not need auxiliary databases. As these methods become faster and more reliable, they will become better able to predict protein misfolding as well as protein folding and fill a central role in solving the proteome folding problem.
The Proteostasis Network
If the proteome folding problem were just about characterizing non-native states, it would simply be an extension of the protein folding problem. There would be nothing “proteomic” about such a problem because the structures and energetics of non-native states are still inherent in the primary sequence and the cellular environment, complex as it is. However, the non-native states of cellular proteins must compete for attention from the proteostasis network, and through this competition their folding energy landscapes communicate with each other. Misfolding by one protein means that the proteostasis network has less bandwidth to handle other proteins, which leads to the other proteins misfolding as well; this effect has been compellingly demonstrated in multiple systems [58–61]. Thus, protein misfolding to non-native states and/or aggregates is truly a proteomic problem in a way that native folding is not.
As we mentioned in the previous section, Anfinsen recognized at the time of his original work on ribonuclease folding that ribonuclease’s unassisted folding rate was too slow to be biologically reasonable. To address this, Anfinsen and others searched for, and found, a catalyst for ribonuclease folding in cell extracts: protein disulfide isomerase (PDI) [15, 37, 62–65]. Although this result did not get as much attention as the finding that ribonuclease’s native state was its thermodynamically most stable conformation, we believe it was equally profound. It showed that, although protein folding can spontaneously yield the native state, it nevertheless requires biological intervention to operate reliably enough in living, evolving organisms with the inherent challenges of the in vivo environment and the multitude of simultaneous folding/unfolding/misfolding events.
After the discovery of PDI it still took decades before there was a more-or-less complete accounting of the types of components that make up the proteostasis network. Now we recognize that perhaps the most prominent components of the proteostasis network are the chaperones, including the Hsp70 [66], Hsp60 [67], and Hsp90 systems [68], the small Hsps [69], and the Hsp100s [70] (Hsp = heat shock protein). Also prominent are the ATP-driven proteases that degrade non-native proteins, including AAA+ ATPases like Lon in prokaryotes [71] and larger systems like the ubiquitin-proteasome system [72] and the autophagy-lysosome pathway [73] in eukaryotes. Together, the chaperones and the degradation machinery, along with proto-chaperones like polyphosphate [74]; post-translational modifications like glycosylation and their associated machinery [75]; the effect of co-translational folding [76–79]; folding enzymes like the PDIs [80] and the cis-trans peptidyl-prolyl isomerases [81]; and the signaling pathways that regulate the concentrations of all these components [82–84] act to promote folding, discourage misfolding, disperse aggregates, or degrade non-native proteins to recycle their constituent amino acids.
Answering the second question of the proteome folding problem—how do proteostasis network components optimize protein folding and mitigate the consequences of folding failures?—requires a comprehensive understanding of how proteostasis network components act on their substrates. This includes the kinetics, thermodynamics, and sequence selectivity of substrate binding by proteostasis network components. Some sequence-based predictive algorithms for the energetics of such interactions have already been proposed for some of the more prominent chaperones, like Hsp70 [85–87]. Fundamental understanding of chaperone action also requires an understanding of how substrate conformations are affected by being shuttled through the (frequently ATP-driven) conformational cycles. These cycles have been intensively studied for all the major ATP-driven chaperones and much is known about them; nevertheless, some issues remain unresolved. For example, for Hsp70 chaperones the structural basis for their selective promiscuity of substrate binding is a puzzle: they discriminate incompletely folded proteins from folded but still their clients represent a large fraction of the proteome [88]. How do Hsp70s alter the folding landscape of their substrates? The Hsp70 system includes the Hsp70 itself plus two co-chaperones, a nucleotide exchange factor and a so-called J-domain protein that activates the Hsp70 ATPase and often helps deliver clients [66]. In addition, they frequently hand off their substrates to downstream chaperones such as Hsp90s [89, 90]. Details of these processes are lacking. Lastly, while many properties and functions of Hsp70s are conserved across cellular compartments, species, and kingdoms, the generality of their allosteric properties and functions remains unclear. Efforts to address these questions are being accelerated by the availability of increasingly powerful techniques based on, for example, cryo-electron microscopy and NMR.
The Proteome Folding Problem Can Only Be Solved with Systems Models for In Vivo Protein Folding
The proteome folding problem is not simply the next frontier in protein science. Failures of proteostasis have been implicated in many human diseases, including gain-of-toxicity (Alzheimer’s disease, Parkinson’s disease, the transthyretin amyloidoses, etc.) and loss-of-function diseases (cystic fibrosis, Gaucher’s disease, Fabry’s disease, etc.) [17, 19, 20, 91]. Thus, understanding proteostasis is important at both a fundamental and a practical level. At a fundamental level, understanding proteostasis informs us about the challenges involved in maintaining the thousands to tens of thousands of proteins in typical organismal proteomes in a functional state, which is a clear pre-requisite to understanding how organisms survive and evolve in the face of environmental challenges. At a practical level, many proposed therapies for protein misfolding diseases involve promoting folding or inhibiting aggregation by improving protein folding energetics (through kinetic stabilization or pharmacologic chaperoning of proteins with destabilizing mutations) [92–95] or by manipulating the proteostasis network (through modulating the concentrations or activities of proteostasis network components) [17, 19, 91, 96–101]. Both endeavors require a systems-level appreciation of in vivo protein folding, but how can such an appreciation be achieved?
None of the proteostasis network components operates in isolation. Many of them bind to similar features on unfolded and misfolded proteins (for example, exposed hydrophobic residues) so they likely compete for substrates [85–87, 102–106]. This competition can be especially important when components that favor folding (like chaperones) compete with the degradation machinery. Different chaperones can also collaborate with each other by binding to substrates and acting on them sequentially [89, 90, 107, 108]. We believe that the simultaneous presence of the full array of proteostasis network components and any emergent properties that arise from their joint activities are too complex to be understood by intuition alone. They can only be understood by developing models that account for this complexity [109–121], an example of which is FoldEco [110], a model for proteostasis in E. coli (Figure 2).
Figure 2.
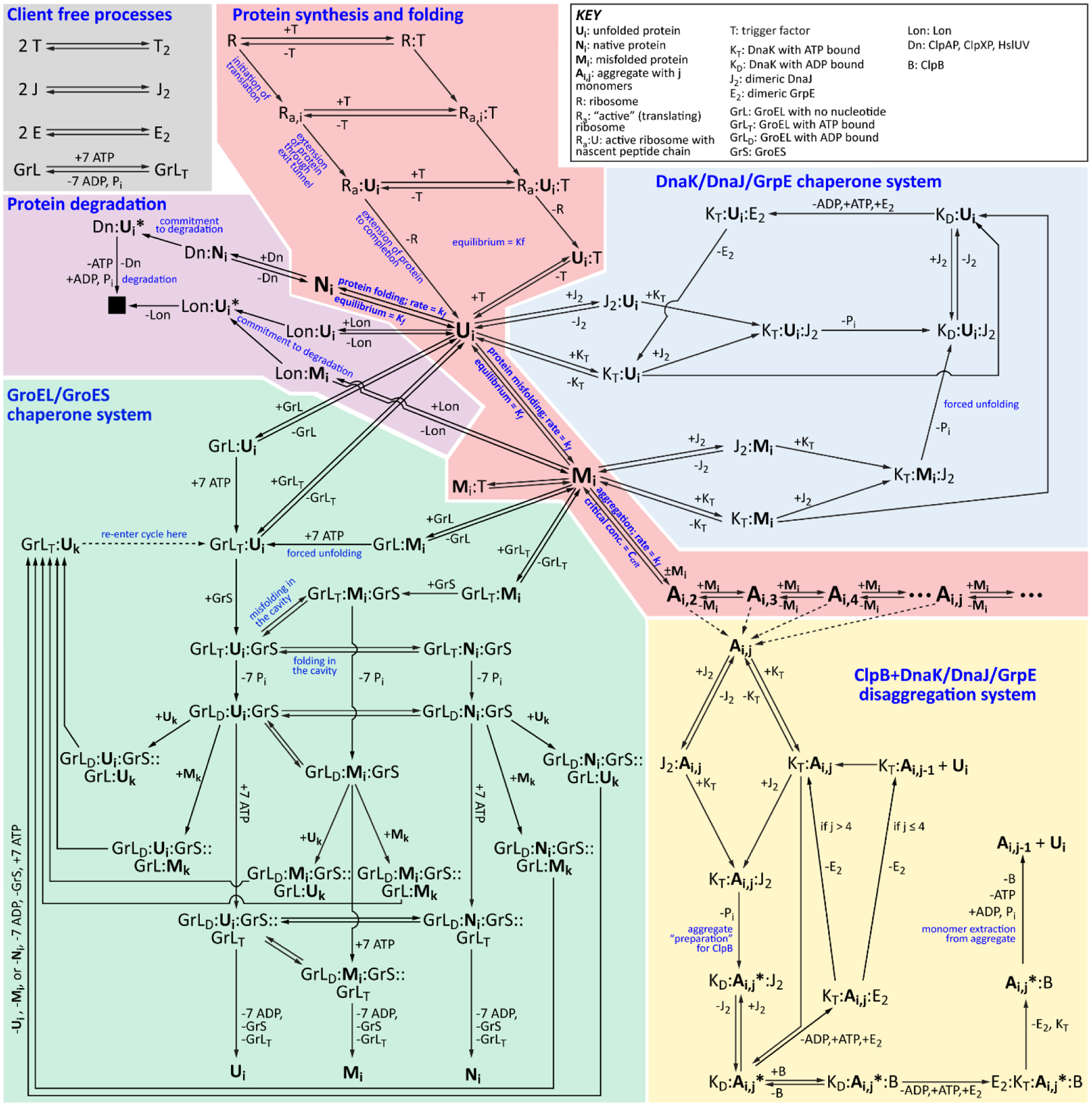
The FoldEco Model for Protein Folding in E. coli. The five subsystems in FoldEco are as follows: protein synthesis and folding (light red), the DnaK/DnaJ/GrpE chaperone system (light blue), the GroEL/GroES chaperone system (light green), the ClpB+DnaK/DnaJ/GrpE disaggregation system (light yellow), and protein degradation (light purple). Also included are several client-free processes, such as DnaJ dimerization (gray). The free unfolded (Ui), misfolded (Mi), native (Ni), and aggregated (Ai,j) states of the client protein are shown in large bold font. Complexes between proteostasis network components and the client protein are denoted by abbreviations for the species in the complex (see key in upper right) separated by colons. When the states of the cis and trans cavities of GroEL are both defined, they are separated by two colons. The subscripts “T” and “D” refer to the ATP- and ADP-bound states of chaperones. Figure and legend reproduced from Powers et al. [110].
These models consist of systems of coupled differential rate equations, one for each species in the system, in which the variables are species concentrations, and each term represents a process in model. The coefficients of the terms in the rate equations are the rate constants for the corresponding process. Although analytical solutions of simplified systems and steady states are often illuminating, these sets of rate equations are generally solved numerically [110, 111]. This process requires that every parameter in the system be given a numerical value, including the initial concentrations of each species and the rate constants for every process, from the unimolecular conformational transitions involved in protein folding/misfolding, to the bimolecular chaperone-substrate recognition events, among many other processes (Figure 2). Thus, the primary challenge with developing a model for in vivo protein folding is parameterizing it. We have argued that literature estimates of these parameters based on in vitro experiments are suitable for processes like associations between chaperones and co-chaperones that do not directly involve the substrate [110]. The parameters for processes that involve individual members of the proteomes, however, should differ from protein to protein, in principle requiring thousands or tens of thousands of parameters for the model.
A computational model of the sort described in the preceding section that could accurately predict these parameters may one day solve this parameterization problem. In the meantime, we are fortunate that near-proteome-wide estimates of many biophysical properties have been made for E. coli proteins using either experimental or computational methods, including native state stability [122, 123], folding kinetics [124], and solubility/aggregation [125]—which together define much of a protein’s folding energy landscape—as well as chaperone efficacy for the Hsp70 and Hsp60 systems [126]. We recently used these estimates to create a coarse-grained representation of the entire proteome for use with a new version of FoldEco called FoldEcoSlim [111]. FoldEcoSlim is simpler than FoldEco because it does not represent the internal steps of chaperone cycles in as much detail as the original, but more complicated in that it accounts for the background proteome, the dynamic response of chaperone concentrations to protein folding stress, and cell growth [111].
These models of proteome folding have already proven valuable for understanding, rationalizing, and/or predicting experimental results about proteostasis. For example, they have been used to analyze how organisms maintain (or fail to maintain) their proteomes under heat [119] or oxidative stress [116], or as they age [117], or as their proteomes evolve [111, 112]. In addition, we have been able to use proteome folding models to understand the behaviors of a variety of specific proteins. For example, we used a very simple model to understand the effects of ligand binding on dihydrofolate reductase expression in E. coli and α-galactosidase A trafficking in human cells [121]. We used FoldEco to understand the effect of the synthesis rate and co-translational folding on luciferase in E. coli [110]. We used FoldEcoSlim to relate the expression efficiencies of cellular retinoic acid binding protein 1 variants to their folding kinetics and thermodynamics. Finally, we used FoldEco in a reverse sense to understand protein folding biophysics based on in vivo protein folding outcomes, rather than understanding in vivo protein folding outcomes based on folding biophysics. In this case, we examined how the extent of aggregation of several proteins changed as the concentrations of proteostasis network components were varied and used this information to infer the proteins’ folding and misfolding energetics [127].
Outlook
Our efforts to solve the proteome folding problem are in their infancy. The models that have been developed for this purpose are still crude with respect to their representation of the processes involved in proteostasis and underdetermined with respect to their parameterizations. These problems will only become more acute as organisms more complicated than single-celled bacteria are addressed. Much more complicated representations of chaperone systems will be needed for proteome folding models of higher organisms since the numbers of chaperone genes scale linearly with genome size [21], to say nothing of the sprawling ubiquitin-proteasome system [72] and the autophagy-lysosome pathway [73]. It is useful to remember, however, that our efforts to solve the protein folding problem were similarly overmatched in its early days. Perhaps the most important contributions that can be made at this point to solving the proteome folding problem are (1) continuing to elucidate chaperone mechanisms; (2) assessing protein folding biophysics on proteomic scales [122, 123, 125], which will help refine computational molecular physics models for sequence-based estimates of protein folding, misfolding, and aggregation energetics; and (3) assessing the effects of chaperones and co-translational folding on folding outcomes on proteomic scales, which will help to define the interactions of proteostasis network components with their substrates. Once these pieces of the puzzle begin to fall into place, we expect rapid progress to be made on revealing the secrets of the proteome folding problem.
Highlights.
The concept of a “proteome folding problem” is discussed–with the complexities of misfolded states that exist on a folding energy landscape and the cellular machinery that contends with them.
A brief discussion is offered of the physiological pressures for folding to occur such that functional, folded proteins are maintained in adequate amounts for life.
The importance of proteostasis networks for folding to occur in vivo is described.
Examples are given of computational modeling of folding in the presence of proteostasis components.
Acknowledgements:
This work was supported in part by grants R35 GM118161 (LMG) and P01 AG054407 (ETP) from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- [1].Kendrew JC, Bodo G, Dintzis HM, Parrish RG, Wyckoff H, Phillips DC. A three-dimensional model of the myoglobin molecule obtained by x-ray analysis. Nature. 1958;181:662–6. [DOI] [PubMed] [Google Scholar]
- [2].Kendrew JC, Dickerson RE, Strandberg BE, Hart RG, Davies DR, Phillips DC, et al. Structure of myoglobin: A three-dimensional Fourier synthesis at 2 A. resolution. Nature. 1960;185:422–7. [DOI] [PubMed] [Google Scholar]
- [3].Anfinsen CB, Haber E, Sela M, White FH Jr. The kinetics of formation of native ribonuclease during oxidation of the reduced polypeptide chain. Proc Natl Acad Sci U S A. 1961;47:1309–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Haber E, Anfinsen CB. Side-chain interactions governing the pairing of half-cystine residues in ribonuclease. J Biol Chem. 1962;237:1839–44. [PubMed] [Google Scholar]
- [5].Sela M, White FH Jr., Anfinsen CB. Reductive cleavage of disulfide bridges in ribonuclease. Science. 1957;125:691–2. [DOI] [PubMed] [Google Scholar]
- [6].White FH Jr. Regeneration of native secondary and tertiary structures by air oxidation of reduced ribonuclease. J Biol Chem. 1961;236:1353–60. [PubMed] [Google Scholar]
- [7].Dill KA, MacCallum JL. The protein-folding problem, 50 years on. Science. 2012;338:1042–6. [DOI] [PubMed] [Google Scholar]
- [8].Dill KA, Ozkan SB, Shell MS, Weikl TR. The protein folding problem. Annu Rev Biophys. 2008;37:289–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Brini E, Simmerling C, Dill K. Protein storytelling through physics. Science. 2020;370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lupas AN, Pereira J, Alva V, Merino F, Coles M, Hartmann MD. The breakthrough in protein structure prediction. Biochem J. 2021;478:1885–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tunyasuvunakool K, Adler J, Wu Z, Green T, Zielinski M, Zidek A, et al. Highly accurate protein structure prediction for the human proteome. Nature. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Anson ML. Protein Denaturation and the Properties of Protein Groups. Adv Protein Chem. 1945;2:361–86. [Google Scholar]
- [14].Gershenson A, Gierasch LM, Pastore A, Radford SE. Energy landscapes of functional proteins are inherently risky. Nat Chem Biol. 2014;10:884–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Clark PL. Protein folding in the cell: reshaping the folding funnel. Trends Biochem Sci. 2004;29:527–34. [DOI] [PubMed] [Google Scholar]
- [16].Braselmann E, Chaney JL, Clark PL. Folding the proteome. Trends Biochem Sci. 2013;38:337–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Balch WE, Morimoto RI, Dillin A, Kelly JW. Adapting proteostasis for disease intervention. Science. 2008;319:916–9. [DOI] [PubMed] [Google Scholar]
- [18].Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. 2014;15:384–96. [DOI] [PubMed] [Google Scholar]
- [19].Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–91. [DOI] [PubMed] [Google Scholar]
- [20].Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27:336–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Powers ET, Balch WE. Diversity in the origins of proteostasis networks--a driver for protein function in evolution. Nat Rev Mol Cell Biol. 2013;14:237–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem. 2013;82:323–55. [DOI] [PubMed] [Google Scholar]
- [23].Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–8. [DOI] [PubMed] [Google Scholar]
- [24].Frydman J Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–47. [DOI] [PubMed] [Google Scholar]
- [25].Babinchak WM, Surewicz WK. Liquid-Liquid Phase Separation and Its Mechanistic Role in Pathological Protein Aggregation. J Mol Biol. 2020;432:1910–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Edun DN, Flanagan MR, Serrano AL. Does liquid-liquid phase separation drive peptide folding? Chem Sci. 2021;12:2474–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Choi JM, Holehouse AS, Pappu RV. Physical Principles Underlying the Complex Biology of Intracellular Phase Transitions. Annu Rev Biophys. 2020;49:107–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bentley EP, Frey BB, Deniz AA. Physical Chemistry of Cellular Liquid-Phase Separation. Chemistry. 2019;25:5600–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Auton M, Rosgen J, Sinev M, Holthauzen LM, Bolen DW. Osmolyte effects on protein stability and solubility: a balancing act between backbone and side-chains. Biophys Chem. 2011;159:90–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Davis-Searles PR, Saunders AJ, Erie DA, Winzor DJ, Pielak GJ. Interpreting the effects of small uncharged solutes on protein-folding equilibria. Annu Rev Biophys Biomol Struct. 2001;30:271–306. [DOI] [PubMed] [Google Scholar]
- [32].Ferreon AC, Moosa MM, Gambin Y, Deniz AA. Counteracting chemical chaperone effects on the single-molecule alpha-synuclein structural landscape. Proc Natl Acad Sci U S A. 2012;109:17826–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhou HX, Rivas G, Minton AP. Macromolecular crowding and confinement: biochemical, biophysical, and potential physiological consequences. Annu Rev Biophys. 2008;37:375–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Miklos AC, Sarkar M, Wang Y, Pielak GJ. Protein crowding tunes protein stability. J Am Chem Soc. 2011;133:7116–20. [DOI] [PubMed] [Google Scholar]
- [35].Schlesinger AP, Wang Y, Tadeo X, Millet O, Pielak GJ. Macromolecular crowding fails to fold a globular protein in cells. J Am Chem Soc. 2011;133:8082–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Epstein CJ, Anfinsen CB, Young DM, Goldberger RF. A Study of Factors Influencing Rate Extent of Enzymic Reactivation during Reoxidation of Reduced Ribonuclease. Arch Biochem Biophys. 1962:223–31.14477774 [Google Scholar]
- [37].Goldberger RF, Epstein CJ, Anfinsen CB. Acceleration of reactivation of reduced bovine pancreatic ribonuclease by a microsomal system from rat liver. J Biol Chem. 1963;238:628–35. [PubMed] [Google Scholar]
- [38].Kelly JW. Alternative conformations of amyloidogenic proteins govern their behavior. Curr Opin Struct Biol. 1996;6:11–7. [DOI] [PubMed] [Google Scholar]
- [39].Chi EY, Krishnan S, Randolph TW, Carpenter JF. Physical stability of proteins in aqueous solution: mechanism and driving forces in nonnative protein aggregation. Pharm Res. 2003;20:1325–36. [DOI] [PubMed] [Google Scholar]
- [40].Pappu RV, Wang X, Vitalis A, Crick SL. A polymer physics perspective on driving forces and mechanisms for protein aggregation. Arch Biochem Biophys. 2008;469:132–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Michaels TCT, Saric A, Habchi J, Chia S, Meisl G, Vendruscolo M, et al. Chemical Kinetics for Bridging Molecular Mechanisms and Macroscopic Measurements of Amyloid Fibril Formation. Annu Rev Phys Chem. 2018;69:273–98. [DOI] [PubMed] [Google Scholar]
- [42].Eisenberg DS, Sawaya MR. Structural Studies of Amyloid Proteins at the Molecular Level. Annu Rev Biochem. 2017;86:69–95. [DOI] [PubMed] [Google Scholar]
- [43].Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R. Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell. 2013;154:1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Walti MA, Ravotti F, Arai H, Glabe CG, Wall JS, Bockmann A, et al. Atomic-resolution structure of a disease-relevant Abeta(1–42) amyloid fibril. Proc Natl Acad Sci U S A. 2016;113:E4976–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rodriguez JA, Ivanova MI, Sawaya MR, Cascio D, Reyes FE, Shi D, et al. Structure of the toxic core of alpha-synuclein from invisible crystals. Nature. 2015;525:486–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sawaya MR, Sambashivan S, Nelson R, Ivanova MI, Sievers SA, Apostol MI, et al. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 2007;447:453–7. [DOI] [PubMed] [Google Scholar]
- [47].Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, et al. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature. 2017;547:185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schmidt M, Wiese S, Adak V, Engler J, Agarwal S, Fritz G, et al. Cryo-EM structure of a transthyretin-derived amyloid fibril from a patient with hereditary ATTR amyloidosis. Nat Commun. 2019;10:5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Banerjee PR, Deniz AA. Shedding light on protein folding landscapes by single-molecule fluorescence. Chem Soc Rev. 2014;43:1172–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Borgia MB, Borgia A, Best RB, Steward A, Nettels D, Wunderlich B, et al. Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature. 2011;474:662–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yu H, Dee DR, Liu X, Brigley AM, Sosova I, Woodside MT. Protein misfolding occurs by slow diffusion across multiple barriers in a rough energy landscape. Proc Natl Acad Sci U S A. 2015;112:8308–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rhoades E, Gussakovsky E, Haran G. Watching proteins fold one molecule at a time. Proc Natl Acad Sci U S A. 2003;100:3197–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bowman MA, Riback JA, Rodriguez A, Guo H, Li J, Sosnick TR, et al. Properties of protein unfolded states suggest broad selection for expanded conformational ensembles. Proc Natl Acad Sci U S A. 2020;117:23356–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Riback JA, Bowman MA, Zmyslowski AM, Knoverek CR, Jumper JM, Hinshaw JR, et al. Innovative scattering analysis shows that hydrophobic disordered proteins are expanded in water. Science. 2017;358:238–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Alderson TR, Kay LE. Unveiling invisible protein states with NMR spectroscopy. Curr Opin Struct Biol. 2020;60:39–49. [DOI] [PubMed] [Google Scholar]
- [56].Whitley MJ, Xi Z, Bartko JC, Jensen MR, Blackledge M, Gronenborn AM. A Combined NMR and SAXS Analysis of the Partially Folded Cataract-Associated V75D gammaD-Crystallin. Biophys J. 2017;112:1135–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Srivastava KR, Lapidus LJ. Prion protein dynamics before aggregation. Proc Natl Acad Sci U S A. 2017;114:3572–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–4. [DOI] [PubMed] [Google Scholar]
- [59].Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, et al. Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell. 2011;144:67–78. [DOI] [PubMed] [Google Scholar]
- [60].Park SH, Kukushkin Y, Gupta R, Chen T, Konagai A, Hipp MS, et al. PolyQ proteins interfere with nuclear degradation of cytosolic proteins by sequestering the Sis1p chaperone. Cell. 2013;154:134–45. [DOI] [PubMed] [Google Scholar]
- [61].Yu A, Shibata Y, Shah B, Calamini B, Lo DC, Morimoto RI. Protein aggregation can inhibit clathrin-mediated endocytosis by chaperone competition. Proc Natl Acad Sci U S A. 2014;111:E1481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].De Lorenzo F, Goldberger RF, Steers E Jr., Givol D, Anfinsen B. Purification and properties of an enzyme from beef liver which catalyzes sulfhydryl-disulfide interchange in proteins. J Biol Chem. 1966;241:1562–7. [PubMed] [Google Scholar]
- [63].Fuchs S, De Lorenzo F, Anfinsen CB. Studies on the mechanism of the enzymic catalysis of disulfide interchange in proteins. J Biol Chem. 1967;242:398–402. [PubMed] [Google Scholar]
- [64].Venetianer P, Straub FB. The enzymic reactivation of reduced ribonuclease. Biochim Biophys Acta. 1963;67:166–8. [DOI] [PubMed] [Google Scholar]
- [65].Venetianer P, Straub FB. The Mechanism of Action of the Ribonuclease-Reactivating Enzyme. Biochim Biophys Acta. 1964;89:189–90. [DOI] [PubMed] [Google Scholar]
- [66].Mayer MP, Gierasch LM. Recent advances in the structural and mechanistic aspects of Hsp70 molecular chaperones. J Biol Chem. 2019;294:2085–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Horwich AL, Fenton WA. Chaperonin-mediated protein folding: using a central cavity to kinetically assist polypeptide chain folding. Q Rev Biophys. 2009;42:83–116. [DOI] [PubMed] [Google Scholar]
- [68].Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nat Rev Mol Cell Biol. 2017;18:345–60. [DOI] [PubMed] [Google Scholar]
- [69].Carra S, Alberti S, Arrigo PA, Benesch JL, Benjamin IJ, Boelens W, et al. The growing world of small heat shock proteins: from structure to functions. Cell Stress Chaperones. 2017;22:601–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Doyle SM, Genest O, Wickner S. Protein rescue from aggregates by powerful molecular chaperone machines. Nat Rev Mol Cell Bio. 2013;14:617–29. [DOI] [PubMed] [Google Scholar]
- [71].Mahmoud SA, Chien P. Regulated Proteolysis in Bacteria. Annu Rev Biochem. 2018;87:677–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Amm I, Sommer T, Wolf DH. Protein quality control and elimination of protein waste: the role of the ubiquitin-proteasome system. Biochim Biophys Acta. 2014;1843:182–96. [DOI] [PubMed] [Google Scholar]
- [73].Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, et al. Mammalian Autophagy: How Does It Work? Annu Rev Biochem. 2016;85:685–713. [DOI] [PubMed] [Google Scholar]
- [74].Gray MJ, Wholey WY, Wagner NO, Cremers CM, Mueller-Schickert A, Hock NT, et al. Polyphosphate is a primordial chaperone. Mol Cell. 2014;53:689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Hebert DN, Lamriben L, Powers ET, Kelly JW. The intrinsic and extrinsic effects of N-linked glycans on glycoproteostasis. Nat Chem Biol. 2014;10:902–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Evans MS, Sander IM, Clark PL. Cotranslational folding promotes beta-helix formation and avoids aggregation in vivo. J Mol Biol. 2008;383:683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Nilsson OB, Nickson AA, Hollins JJ, Wickles S, Steward A, Beckmann R, et al. Cotranslational folding of spectrin domains via partially structured states. Nat Struct Mol Biol. 2017;24:221–5. [DOI] [PubMed] [Google Scholar]
- [78].Stein KC, Frydman J. The stop-and-go traffic regulating protein biogenesis: How translation kinetics controls proteostasis. J Biol Chem. 2019;294:2076–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Walsh IM, Bowman MA, Soto Santarriaga IF, Rodriguez A, Clark PL. Synonymous codon substitutions perturb cotranslational protein folding in vivo and impair cell fitness. Proc Natl Acad Sci U S A. 2020;117:3528–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Matsusaki M, Kanemura S, Kinoshita M, Lee YH, Inaba K, Okumura M. The Protein Disulfide Isomerase Family: from proteostasis to pathogenesis. Biochim Biophys Acta Gen Subj. 2020;1864:129338. [DOI] [PubMed] [Google Scholar]
- [81].Gothel SF, Marahiel MA. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol Life Sci. 1999;55:423–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Plate L, Wiseman RL. Regulating Secretory Proteostasis through the Unfolded Protein Response: From Function to Therapy. Trends Cell Biol. 2017;27:722–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Akerfelt M, Morimoto RI, Sistonen L. Heat shock factors: integrators of cell stress, development and lifespan. Nat Rev Mol Cell Biol. 2010;11:545–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17:1374–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Rudiger S, Germeroth L, Schneider-Mergener J, Bukau B. Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. EMBO J. 1997;16:1501–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Rudiger S, Schneider-Mergener J, Bukau B. Its substrate specificity characterizes the DnaJ co-chaperone as a scanning factor for the DnaK chaperone. EMBO J. 2001;20:1042–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Van Durme J, Maurer-Stroh S, Gallardo R, Wilkinson H, Rousseau F, Schymkowitz J. Accurate prediction of DnaK-peptide binding via homology modelling and experimental data. PLoS Comput Biol. 2009;5:e1000475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Calloni G, Chen T, Schermann SM, Chang HC, Genevaux P, Agostini F, et al. DnaK functions as a central hub in the E. coli chaperone network. Cell Rep. 2012;1:251–64. [DOI] [PubMed] [Google Scholar]
- [89].Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell. 2014;157:1685–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wegele H, Wandinger SK, Schmid AB, Reinstein J, Buchner J. Substrate transfer from the chaperone Hsp70 to Hsp90. J Mol Biol. 2006;356:802–11. [DOI] [PubMed] [Google Scholar]
- [91].Hipp MS, Park SH, Hartl FU. Proteostasis impairment in protein-misfolding and - aggregation diseases. Trends Cell Biol. 2014;24:506–14. [DOI] [PubMed] [Google Scholar]
- [92].Germain DP, Fan JQ. Pharmacological chaperone therapy by active-site-specific chaperones in Fabry disease: in vitro and preclinical studies. Int J Clin Pharmacol Ther. 2009;47 Suppl 1:S111–7. [PubMed] [Google Scholar]
- [93].Parenti G, Andria G, Valenzano KJ. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol Ther. 2015;23:1138–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher disease. Proc Natl Acad Sci U S A. 2002;99:15428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Powers ET, Kelly JW. From uncovering the mechanism of transthyretin aggregation to the drug tafamidis for ameliorating neurodegeneration and cardiomyopathy. In: Smith RA, Kaspar BK, Svendsen CN, editors. Neurotherapeutics in the Era of Translational Medicine. London, United Kingdom: Academic Press; 2021. p. 65–103. [Google Scholar]
- [96].Eisele YS, Monteiro C, Fearns C, Encalada SE, Wiseman RL, Powers ET, et al. Targeting protein aggregation for the treatment of degenerative diseases. Nat Rev Drug Discov. 2015;14:759–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Calamini B, Morimoto RI. Protein homeostasis as a therapeutic target for diseases of protein conformation. Curr Top Med Chem. 2012;12:2623–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Calamini B, Silva MC, Madoux F, Hutt DM, Khanna S, Chalfant MA, et al. Small-molecule proteostasis regulators for protein conformational diseases. Nature Chemical Biology. 2012;8:185–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Grandjean JMD, Wiseman RL. Small molecule strategies to harness the unfolded protein response: where do we go from here? J Biol Chem. 2020;295:15692–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, Komiyama T, et al. Activation of Hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nat Chem Biol. 2013;9:112–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Aoki K, Motojima F, Taguchi H, Yomo T, Yoshida M. GroEL binds artificial proteins with random sequences. J Biol Chem. 2000;275:13755–8. [DOI] [PubMed] [Google Scholar]
- [103].Clerico EM, Tilitsky JM, Meng W, Gierasch LM. How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol. 2015;427:1575–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Landry SJ, Gierasch LM. The chaperonin GroEL binds a polypeptide in an alpha-helical conformation. Biochemistry. 1991;30:7359–62. [DOI] [PubMed] [Google Scholar]
- [105].Wang Z, Feng H, Landry SJ, Maxwell J, Gierasch LM. Basis of substrate binding by the chaperonin GroEL. Biochemistry. 1999;38:12537–46. [DOI] [PubMed] [Google Scholar]
- [106].Tartaglia GG, Dobson CM, Hartl FU, Vendruscolo M. Physicochemical determinants of chaperone requirements. J Mol Biol. 2010;400:579–88. [DOI] [PubMed] [Google Scholar]
- [107].Langer T, Lu C, Echols H, Flanagan J, Hayer MK, Hartl FU. Successive action of DnaK, DnaJ and GroEL along the pathway of chaperone-mediated protein folding. Nature. 1992;356:683–9. [DOI] [PubMed] [Google Scholar]
- [108].Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med (Maywood). 2003;228:111–33. [DOI] [PubMed] [Google Scholar]
- [109].Wiseman RL, Powers ET, Buxbaum JN, Kelly JW, Balch WE. An adaptable standard for protein export from the endoplasmic reticulum. Cell. 2007;131:809–21. [DOI] [PubMed] [Google Scholar]
- [110].Powers ET, Powers DL, Gierasch LM. FoldEco: a model for proteostasis in E. coli. Cell Rep. 2012;1:265–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Pobre KFR, Powers DL, Ghosh K, Gierasch LM, Powers ET. Kinetic versus thermodynamic control of mutational effects on protein homeostasis: A perspective from computational modeling and experiment. Protein Sci. 2019;28:1324–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Bershtein S, Mu W, Serohijos AW, Zhou J, Shakhnovich EI. Protein quality control acts on folding intermediates to shape the effects of mutations on organismal fitness. Mol Cell. 2013;49:133–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Dickson A, Brooks CL 3rd. Quantifying chaperone-mediated transitions in the proteostasis network of E. coli. PLoS Comput Biol. 2013;9:e1003324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Royle K, Kontoravdi C. A systems biology approach to optimising hosts for industrial protein production. Biotechnol Lett. 2013;35:1961–9. [DOI] [PubMed] [Google Scholar]
- [115].Hall D On the nature of the optimal form of the holdase-type chaperone stress response. FEBS Lett. 2020;594:43–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Santra M, Dill KA, de Graff AMR. How Do Chaperones Protect a Cell’s Proteins from Oxidative Damage? Cell Syst. 2018;6:743–51 e3. [DOI] [PubMed] [Google Scholar]
- [117].Santra M, Dill KA, de Graff AMR. Proteostasis collapse is a driver of cell aging and death. Proc Natl Acad Sci U S A. 2019;116:22173–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].de Graff AM, Mosedale DE, Sharp T, Dill KA, Grainger DJ. Proteostasis is adaptive: Balancing chaperone holdases against foldases. PLoS Comput Biol. 2020;16:e1008460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Chen K, Gao Y, Mih N, O’Brien EJ, Yang L, Palsson BO. Thermosensitivity of growth is determined by chaperone-mediated proteome reallocation. Proc Natl Acad Sci U S A. 2017;114:11548–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Santra M, Farrell DW, Dill KA. Bacterial proteostasis balances energy and chaperone utilization efficiently. Proc Natl Acad Sci U S A. 2017;114:E2654–E61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Hingorani KS, Metcalf MC, Deming DT, Garman SC, Powers ET, Gierasch LM. Ligand-promoted protein folding by biased kinetic partitioning. Nat Chem Biol. 2017;13:369–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Leuenberger P, Ganscha S, Kahraman A, Cappelletti V, Boersema PJ, von Mering C, et al. Cell-wide analysis of protein thermal unfolding reveals determinants of thermostability. Science. 2017;355. [DOI] [PubMed] [Google Scholar]
- [123].Mateus A, Bobonis J, Kurzawa N, Stein F, Helm D, Hevler J, et al. Thermal proteome profiling in bacteria: probing protein state in vivo. Mol Syst Biol. 2018;14:e8242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Zou T, Williams N, Ozkan SB, Ghosh K. Proteome folding kinetics is limited by protein halflife. PLoS One. 2014;9:e112701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Niwa T, Ying BW, Saito K, Jin W, Takada S, Ueda T, et al. Bimodal protein solubility distribution revealed by an aggregation analysis of the entire ensemble of Escherichia coli proteins. Proc Natl Acad Sci U S A. 2009;106:4201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Niwa T, Kanamori T, Ueda T, Taguchi H. Global analysis of chaperone effects using a reconstituted cell-free translation system. Proc Natl Acad Sci U S A. 2012;109:8937–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Cho Y, Zhang X, Pobre KF, Liu Y, Powers DL, Kelly JW, et al. Individual and collective contributions of chaperoning and degradation to protein homeostasis in E. coli. Cell Rep. 2015;11:321–33. [DOI] [PMC free article] [PubMed] [Google Scholar]