

Abstract
The unbound concentrations of 14 commercial drugs, including five non‐efflux/uptake transporter substrates—Class I, five efflux transporter substrates—class II and four influx transporter substrates—Class III, were simultaneously measured in rat liver, muscle, and blood via microanalysis. K puu,liver and K puu,muscle were calculated to evaluate the membrane transport activity and cell metabolism on the unbound drug concentrations in the skeletal muscle and liver. For Class I compounds, represented by antipyrine, unbound concentrations among liver, muscle and blood are symmetrically distributed when compound hepatic clearance is low. And when compound hepatic clearance is high, unbound concentrations among liver, muscle and blood are asymmetrically distributed, such as Propranolol. For Class II and III compounds, overall, the unbound concentrations among liver, muscle, and blood are asymmetrically distributed due to a combination of hepatic metabolism and efflux and/or influx transporter activity.
Keywords: asymmetrically distributed, cell metabolism, membrane transport, microanalysis, unbound drug concentration
Unbound concentrations of of 14 commercial drugs in rat liver, muscle, and blood

Abbreviations
- ACD
acid citrate dextrose
- CL
clearance
- C m , blood
unbound concentration in blood measured by microdialysis
- C m , brain
unbound concentration in brain measured by microdialysis
- C m , liver
unbound concentration in liver measured by microdialysis
- C m, muscle
unbound concentration in muscle measured by microdialysis
- C ss
steady‐state concentration
- C u ,brain
unbound concentration in brain measured by equilibrium dialysis
- CYP
cytochrome
- DMSO
dimethyl sulfoxide
- HPLC/MS/MS
high‐performance liquid chromatography combined with tandem mass spectrometry
- HP‐β‐CD
hydroxypropyl‐β‐cyclodextrin
- h
hour
- IACUC
institutional animal care and use committee
- K puu liver
ratio of unbound liver concentration to unbound blood concentration
- K puu muscle
ratio of unbound muscle concentration to unbound blood concentration
- K puu
ratio of unbound tissue concentration to unbound blood concentration
- OATP
organic anion transporting polypeptides
- P app
apparent permeability
- P‐gp
p‐glycoprotein
- PK
pharmacokinetics
- T 1/2
half‐life
- V ss
volume of distribution at steady state
1. INTRODUCTION
One of the most important concepts in clinical pharmacology is the free drug hypothesis. This concept stipulates that only the unbound (free) drug is able to distribute from the blood circulation across cell membranes to tissues and that only unbound drug can interact with target receptors at the site of action in the target tissue. 1 , 2 , 3 , 4 , 5
As demonstrated in our previous study, the free drug hypothesis is not universally applicable for all drugs, but only for drugs with good permeability that are not substrates of efflux transporters. 6 The unbound concentration of an efflux transporter drug in tissues where efflux transporters are highly expressed is expected to be lower than that in blood. Similarly, the unbound concentration of a drug in an eliminating organ (such as liver) is also expected to be lower than that in blood when the drug is subject to an extensive cell metabolism. In contrast, the unbound concentration of a drug in a given tissue is expected to be higher than blood, if the drug is a substrate of influx transporters that are present in the tissue. 7 , 8
Numerous studies have been conducted for the comparison of unbound drug concentrations in tissues and blood, and most of the studies have focused on the brain. This is because brain is a unique sanctuary organ that is protected by the blood–brain barrier (BBB) where various drug efflux and influx transporters are highly expressed. In addition, brain is not considered a major site of drug metabolism, even though low levels of CYPs enzymes are present.
Kinetic behavior of unbound drug concentrations in other organs and tissues, such as skeletal muscle and liver, has been less explored. Skeletal muscle is the largest tissue of the body, comprising of approximately 40% of total body weight. Like the brain, skeletal muscle is not a major site of drug metabolism, even though low levels of mRNAs of CYPs are detected in skeletal muscle. 9 On the other hand, liver plays a key role in breaking down or modifying toxic substances to make them less harmful to the body. To perform these vital functions, various drug metabolizing enzymes and influx/efflux transporters (P‐gp, BCRP, and MRPs) are expressed in liver. Given the fact that skeletal muscle is the largest tissue of body, it is important to understand the kinetic behavior of unbound drug concentration in the skeletal muscle. Similarly, it is also important to assess the complex interplay effects of drug metabolizing enzymes and drug transporters on the kinetic behavior of unbound drug concentration in liver.
A number of techniques have been developed and used to determine unbound concentrations of drugs in the tissues and blood. Among these, the most common and popular method is to use the plasma and tissue binding unbound fractions for measuring plasma and tissue unbound drug concentrations. 10 , 11 , 12 , 13 However, drug binding in tissue homogenates may not accurately reflect the binding in intact tissue in vivo, because tissue homogenates do not take into account the fact that drug binding may differ between interstitial fluid, cells and subcellular organelles. Therefore, the unbound drug concentrations indirectly measured by using tissue unbound fraction may not accurately reflect the unbound drug concentration in tissues.
Microdialysis is a sampling technique that is used for continuous measurement of unbound drug concentrations in the tissues. 14 , 15 The microdialysis technique requires the insertion of a small microdialysis probe with a semipermeable hollow fiber membrane at its tip into the tissue of interest. Although microdialysis is a minimally‐invasive sampling technique which requires some specialized skills involved time consuming procedures, it is a reliable method to quantify the unbound drug concentrations in target tissue, and has contributed to our current knowledge and understanding of drug distribution in different tissues of the body.
The objective of our study is to assess the effects of transport activity of drug transporters and cell metabolism o on the unbound drug concentrations in the skeletal muscle and liver. Since microdialysis provides a reliable measurement of unbound drug concentrations in tissues and blood, the method of microdialysis is applied in this rat study for direct and simultaneous measurement of unbound drug concentrations in the skeletal muscle, liver, and blood.
2. MATERIALS AND METHODS
2.1. Chemicals and reagents
In this study, 14 compounds with different physiochemical properties were selected. Antipyrine, ofloxacin, lamotrigine, digoxin, quinidine, atenolol, carbamazepine, propranolol, diltiazem, memantine, diphenhydramine, pyrilamine, and gabapentin were purchased from MedChemExpress (Monmouth Junction). Fexofenadine was purchased from Tokyo Chemical Industry. The other chemicals and reagents were of the HPLC grade or better.
2.2. Animals
Healthy male Sprague‐Dawley rats (8–10 weeks, 250–350 g) were purchased from Sibeifu Biotechnology Co., Ltd. The animals were acclimatized to the laboratory environment for at least 1 week before the study and were housed in a 12‐h light/12‐h dark cycle environment with free access to food and water. All studies were approved by Pharmaron's Institutional Animal Care and Use Committee (IACUC).
2.3. Rat liver, muscle, and blood microdialysis studies
Unbound concentrations of the 14 compounds in rats were measured by microdialysis method. Liver, muscle, and blood microdialysis of each compound was simultaneously conducted in male Sprague‐Dawley rats (n = 3). All animals were acclimated to laboratory environment for 3–4 days prior to microdialysis study. At 12 h before dosing, animals were anesthetized and placed into an individual system, and then liver, muscle, and blood microdialysis probes (CMA/20, 10 mm, CMA) were implanted into hepatic lobe, skeletal muscle of the left thigh, and jugular vein respectively. All probes were perfused with blank acid citrate dextrose (ACD; 3.5 mmol/L citric acid, 7.5 mmol/L sodium citrate, 13.6 mmol/L dextrose) solution at a rate of 1 μL/min for 16 h, respectively. On the day of experiment, each animal received an intravenous bolus loading dose followed by an infusion dose via femoral vein. In this study, the loading dose was used to quickly achieve the target steady‐state concentration (Css ), meanwhile following a constant rate of intravenous infusion in rats. The loading dose and infusion rate of each compound can be calculated by the following Equation (1) and (2), respectively, using the kinetic parameters and the pre‐determined desired Css .
| (1) |
| (2) |
The detailed dose level and formulation composition of each compound were listed in Table 1. Liver, muscle, and blood microdialysis dialysates were collected at 0.5 h intervals up to 6 h post‐dose. The animals were subsequently euthanized at 6 h. All samples were stored at −75 ± 15°C before analysis.
TABLE 1.
Loading doses, infusion rates, formulations, and microdialysis probe recoveries used for the 14 compounds in rat microdialysis study (n = 3, mean ± SD)
| Compound | Loading dose (mg/kg) | IV Infusion rate (mg/kg/h) | Formulation a | Probe recovery (liver) (%) | Probe recovery (muscle) (%) | Probe recovery (blood) (%) |
|---|---|---|---|---|---|---|
| Antipyrine | 0.5 | 0.333 | A | 41.2 ± 4.8 | 41.3 ± 4.4 | 19.3 ± 1.7 |
| Lamotrigine | 1 | 0.0833 | B | 53.0 ± 7.8 | 48.5 ± 6.9 | 79.6 ± 6.1 |
| Carbamazepine | 1 | 1.67 | B | 74.7 ± 6.6 | 66.8 ± 1.7 | 75.4 ± 4.8 |
| Propranolol | 5 | 5 | B | 63.3 ± 8.2 | 44.0 ± 7.0 | 59.0 ± 15.0 |
| Diltiazem | 3 | 2 | B | 65.1 ± 3.3 | 42.8 ± 8.8 | 58.4 ± 5.8 |
| Ofloxacin | 3 | 3.33 | B | 27.7 ± 5.3 | 26.7 ± 4.7 | 48.6 ± 9.2 |
| Atenolol | 6 | 4.17 | B | 19.0 ± 10.1 | 8.87 ± 0.89 | 31.9 ± 10.0 |
| Quinidine | 8 | 3.33 | B | 52.6 ± 8.1 | 38.4 ± 1.5 | 45.2 ± 9.0 |
| Fexofenadine | 10 | 16.7 | B | 24.5 ± 4.9 | 18.6 ± 13.6 | 55.5 ± 9.6 |
| Digoxin | 1 | 1 | B | 60.5 ± 6.8 | 54.8 ± 4.9 | 37.7 ± 3.8 |
| Memantine | 8 | 3.33 | A | 45.7 ± 5.4 | 39.6 ± 13 | 50.6 ± 9.3 |
| Diphenhydramine | 5 | 3.33 | A | 64.6 ± 2.6 | 58.3 ± 2.6 | 81.2 ± 19.1 |
| Pyrilamine | 3.75 | 1.88 | A | 52.8 ± 8.6 | 19.6 ± 7.7 | 58.8 ± 22.3 |
| Gabapentin | 5 | 1.67 | A | 21.9 ± 1.4 | 22.2 ± 9.6 | 50.1 ± 12.1 |
Formulation A: Saline. Formulation B: 5% DMSO in ‘10%HP‐β‐CD'.
2.4. In vivo recovery studies
The recovery of compounds in liver, muscle, and blood microdialysis probes was determined by an in vivo retrodialysis method. Liver, muscle, and blood probes were implanted into animals following the procedure in a previous microdialysis study. 300 ng/ml of test compounds in ACD were constantly perfused into blood and brain probes for 5 h, and dialysates were collected at 0.5 h intervals from 2 to 5 h Recovery can be calculated by the following equation:
| (3) |
where C in is the concentration in perfusates, while C out is the average concentration of dialysates collected from 2 to 5 h. All samples were stored at −75 ± 15°C before analysis. The recovery data of the microdialysis probes are listed in Table 1.
2.5. Sample analysis
All samples were analyzed on HPLC/MS/MS systems, which consist of Shimadzu LC‐30AD pumps (Shimadzu), a rack changer II autosampler (Shimadzu), and either a Shimadzu 8060 (Shimadzu) or an AB Sciex API 4000/5500 (AB Sciex) mass spectrometer. Ten μl of dialysates samples were mixed with 100 μl or 200 μl of acetonitrile containing internal standard. The supernatants were diluted with appropriate volumes of water before analysis on HPLC/MS/MS.
2.6. Data analysis
Pharmacokinetic parameters were calculated with WinNolin 8.2 (Pharsight Corporation) by employing a non‐compartmental analysis. The kinetics parameters, the volume of distribution at steady state (V ss), clearance (CL), and terminal half‐life (T½), were determined following intravenous administration. The dose administered was input to the program as mg/kg. Nominal times of blood collection were used for the calculation of parameters.
For the microdialysis study, the steady state unbound drug concentrations in the liver, muscle, and blood were calculated as the average of unbound concentrations from 2 h up to 6 h time points. Graphs and statistical analysis were performed in Graphpad Prism 8.0. T‐test was used to determine the statistical difference of steady‐state unbound concentrations in blood, liver, and muscle. A correlation analysis was performed to evaluate the relationship of the steady‐state unbound concentration among liver, muscle, and blood.
3. RESULTS
For calculation of loading dose and infusion rate for the microdialysis study, the pharmacokinetic parameters, plasma clearance (CL), volume of distribution at steady state (V ss), and plasma half‐life (T 1/2) were determined in rats following a single intravenous administration. The kinetic parameters of antipyrine, ofloxacin, lamotrigine, digoxin, quinidine, atenolol, carbamazepine, propranolol, and diltiazem were obtained from our previous study, 6 while those of memantine, diphenhydramine, pyrilamine, and gabapentin were obtained from the present study (Table 2). In order to quickly achieve desirable steady state unbound drug concentrations, loading doses and infusion rates were calculated for all of the 14 test compounds according to Equations (1) and (2) as described in the methods section.
TABLE 2.
Pharmacokinetic parameters of the 14 compounds following an IV single bolus (n = 3; mean ± SD) (n = 3, mean ± SD)
| Compound | Dose (mg/kg) | CL (ml/min/kg) | V ss (L/kg) | T 1/2 (h) |
|---|---|---|---|---|
| Antipyrine 6 | 5.00 | 4.81 ± 0.77 | 0.724 ± 0.051 | 1,99 ± 0.43 |
| Lamotrigine 6 | 5.00 | 1.48 ± 0.21 | 1.86 ± 0.18 | 15.6 ± 3.0 |
| Carbamazepine 6 | 5.00 | 18.1 ± 2.9 | 0.745 ± 0.081 | 0.993 ± 0.115 |
| Propranolol 6 | 4.38 | 82.8 ± 12.5 | 4.64 ± 0.70 | 0.943 ± 0.062 |
| Diltiazem | 1.00 | 96.4 ± 24.0 | 2.93 ± 0.53 | 0.805 ± 0.201 |
| Ofloxacin 6 | 5.00 | 32.5 ± 4.9 | 5.08 ± 2.9 | 6.32 ± 3.34 |
| Atenolol 6 | 5.00 | 24.5 ± 3.7 | 2.88 ± 0.44 | 2.76 ± 0.77 |
| Quinidine 6 | 5.00 | 76.4 ± 3.5 | 3.80 ± 0.31 | 1.01 ± 0.03 |
| Fexofenadine 6 | 5.00 | 50.9 ± 6.1 | 1.56 ± 0.38 | 1.12 ± 0.09 |
| Digoxin 6 | 5.00 | 9.10 ± 1.30 | 0.376 ± 0.013 | 1.30 ± 0.26 |
| Memantine | 1.00 | 60.5 ± 9.9 | 7.36 ± 1.58 | 1.72 ± 0.03 |
| Diphenhydramine | 1.00 | 152 ± 11 | 5.36 ± 0.48 | 0.664 ± 0.005 |
| Pyrilamine | 1.00 | 167 ± 24 | 12.6 ± 1.6 | 2.16 ± 0.63 |
| Gabapentin | 1.00 | 1.53 ± 0.31 | 0.451 ± 0.001 | 3.58 ± 0.87 |
As shown in Table 2, propranolol, diltiazem, quinidine, memantine, diphenhydramine, and pyrilamine showed high plasma clearance, which approached or exceeded the reported rat hepatic blood flow of 75–90 ml/min/kg. 16 , 17 The mean values of plasma CL were 97.7, 82.8, 76.4, 60.5, 152, and 167 ml/min/kg, respectively, for diltiazem, propranolol, quinidine, memantine, diphenhydramine, and pyrilamine in rats in this study. The observed plasma CL values of these compounds are in good agreement with the reported plasma CL by other investigators. The reported plasma CL of propranolol was in the range of 70–90 ml/min/kg, while the reported plasma CL values of diltiazem, quinidine, memantine, and diphenhydramine were 90, 74, 69, and 115 ml/min/kg. 18 , 19 , 20 , 21 , 22 , 23 In contrast, antipyrine, lamotrigine, digoxin, and carbamezapine showed low plasma CL (<20 ml/min/kg), while atenolol, ofloxacin and fexofenadine showed moderate plasma CL (20–50 ml/min/kg). The plasma Cl values of antipyrine, lamotrigine, digoxin, and carbamezapine (4.81, 1.48, 9.1, and 18.1 ml/min/kg, respectively) were consistent with the reported plasma CL values by other investigators (4.80, 0.830, 17.3, and 15.0 ml/min/kg, respectively). 24 , 25 , 26 , 27
As shown in our previous study, 6 antipyrine, lamotrigine, carbamezapine, propranolol and diltiazem are not substrates of efflux transporters and classified as Class I compounds, while atenolol, ofloxacin, fexofenadine, quinidine, and digoxin are substrates of efflux transporters and classified as Class II compounds. From the literature, memantine, diphenhydramine, pyrilamine and gabapentin are known to be substrates of influx transporters. 21 , 28 , 29 , 30 Thus, these compounds are classified as Class III compounds. The unbound drugs concentrations of Class I, II and III compounds in blood, liver and skeletal muscle were simultaneously measured by microdialysis following loading doses and constant infusion. The unbound drug concentration time profiles of Class I compounds in blood, liver, and muscle are shown in Figure 1, while the unbound drug concentration time profiles of Class II and III compounds are presented in Figures 2 and 3, respectively.
FIGURE 1.
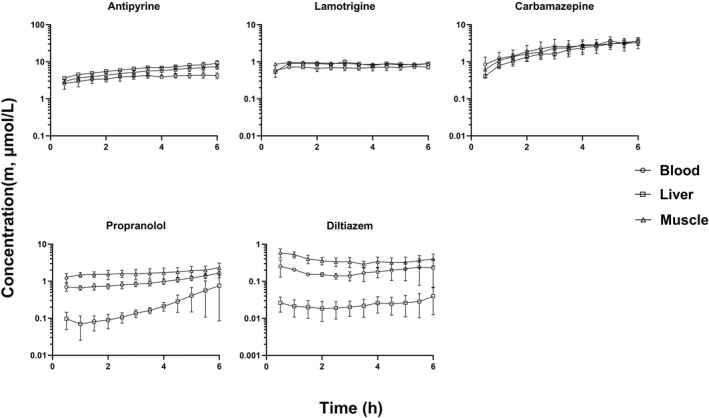
Rat unbound drug concentration time profiles of Class I compounds in liver, muscle, and blood with good membrane permeability that are not substrates of efflux and uptake transporters (mean ± SD, n = 3). The unbound drug concentration was simultaneously measured by microdialysis in liver, muscle, and blood
FIGURE 2.
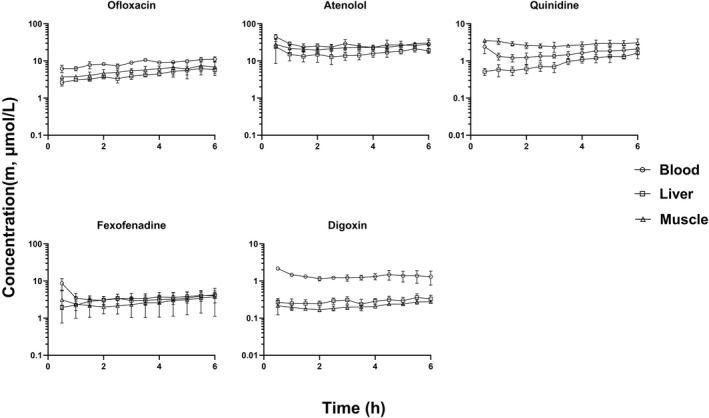
Rat unbound drug concentration time profiles of Class II compounds in liver, muscle, and blood that are substrates of efflux transporters (mean ± SD, n = 3). The unbound drug concentration was simultaneously measured by microdialysis in liver, muscle, and blood
FIGURE 3.
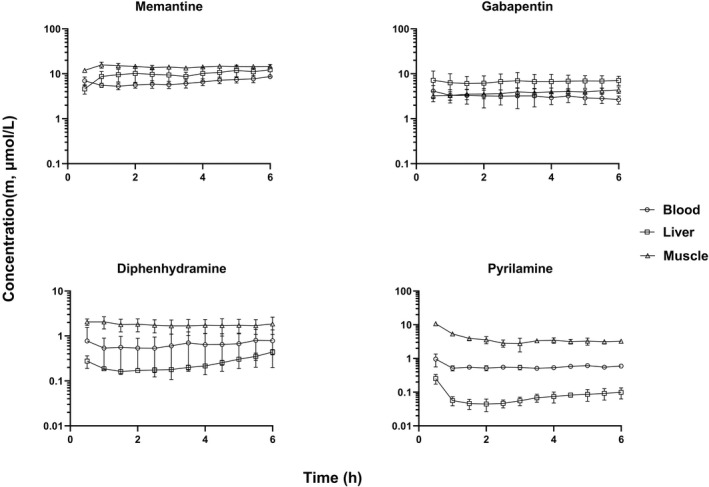
Rat unbound drug concentration time profiles of Class III compounds in liver, muscle, and blood that are substrates of uptake transporters (mean ± SD, n = 3). The unbound drug concentration was simultaneously measured by microdialysis in liver, muscle, and blood
With the exception of propranolol unbound concentrations in liver, the unbound drug concentrations of all compounds reached a steady state in blood, liver and skeletal muscle within 2 h after the start of infusion. Thus, the steady state unbound concentrations were calculated as the average of unbound concentrations from 2 h up to 6 h time points. The steady state unbound concentrations as well as the unbound drug concentration ratios between liver (or skeletal muscle) and blood (K puu) are listed in Tables 3, 4, 5. The steady state K puu ratio gives a direct quantitative description of how the cell membrane handles the drug movement into and out of the cell regarding passive transport and active influx/efflux. When a drug has a K puu close to 1, it is reasonable to conclude that the drug crosses the cell membrane by simple passive diffusion and is not a substrate for efflux or influx transporters. On the other hand, drugs that have a K puu, of <1 are subject to extensive cell metabolism and/or are substrates for efflux transporters, while compounds with a K puu of >1 are likely to be substrates for influx transporters. 5
TABLE 3.
Steady‐state unbound drug concentrations in liver, muscle, and blood of Class I compounds with good membrane permeability that are not efflux and uptake transporter substrates (n = 3, mean ± SD)
| Compound | C m, liver (µmol/L) | C m, muscle (µmol/L) | C m, blood (µmol/L) | K puu liver | K puu muscle |
|---|---|---|---|---|---|
| Antipyrine | 7.16 ± 1.38 | 5.89 ± 1.45 | 7.76 ± 0.68 | 0.932 ± 0.178 | 0.771 ± 0.215 |
| Lamotrigine | 0.879 ± 0.078 | 0.879 ± 0.059 | 0.700 ± 0.092 | 1.28 ± 0.25 | 1.28 ± 0.22 |
| Carbamazepine | 2.41 ± 0.94 | 2.64 ± 0.80 | 2.77 ± 1.08 | 0.913 ± 0.169 | 1.02 ± 0.27 |
| Propranolol | 0.303 ± 0.328* | 1.79 ± 0.51 | 1.07 ± 0.35 | 0.382 ± 0.277 | 1.67 ± 0.36 |
| Diltiazem | 0.0250 ± 0.0139* | 0.342 ± 0.096 | 0.184 ± 0.087 | 0.145 ± 0.085 | 1.96 ± 0.29 |
T‐test was performed for C m, liver versus C m, blood and C m, muscle versus C m, blood.
p < .05.
TABLE 4.
Steady‐state unbound drug concentrations in liver, muscle, and blood of Class II compounds that are efflux transporter substrates (n = 3, mean ± SD)
| Compound | C m, liver (µmol/L) | C m, muscle (µmol/L) | C m, blood (µmol/L) | K puu liver | K puu muscle |
|---|---|---|---|---|---|
| Ofloxacin | 4.72 ± 1.28* | 6.02 ± 1.92 | 9.49 ± 1.52 | 0.501 ± 0.108 | 0.641 ± 0.206 |
| Atenolol | 16.4 ± 4.2 | 24.3 ± 4.7 | 26.1 ± 5.8 | 0.657 ± 0.268 | 0.950 ± 0.173 |
| Quinidine | 1.06 ± 0.41 | 2.76 ± 0.54 | 1.65 ± 0.48 | 0.677 ± 0.27 | 1.75 ± 0.64 |
| Fexofenadine | 3.39 ± 0.79 | 2.78 ± 1.53 | 3.60 ± 1.04 | 1.02 ± 0.44 | 0.764 ± 0.274 |
| Digoxin | 0.296 ± 0.07* | 0.220 ± 0.042* | 1.31 ± 0.31 | 0.232 ± 0.041 | 0.175 ± 0.044 |
T‐test was performed for C m, liver versus C m, blood and C m, muscle versus C m, blood.
p < .05.
TABLE 5.
Steady‐state unbound drug concentrations in liver, muscle, and blood of Class III compounds that are uptake transporter substrates (n = 3, mean ± SD)
| Compound | C m, liver (µmol/L) | C m, muscle (µmol/L) | C m, blood (µmol/L) | K puu liver | K puu muscle |
|---|---|---|---|---|---|
| Memantine | 10.5 ± 3.0 | 14.3 ± 0.9* | 6.78 ± 1.37 | 1.56 ± 0.48 | 2.13 ± 0.25 |
| Diphenhydramine | 0.255 ± 0.094 | 1.74 ± 0.53* | 0.658 ± 0.413 | 0.514 ± 0.266 | 3.08 ± 0.94 |
| Pyrilamine | 0.0713 ± 0.0274* | 3.21 ± 0.64* | 0.556 ± 0.055 | 0.127 ± 0.033 | 5.78 ± 1.09 |
| Gabapentin | 6.80 ± 2.28 | 3.95 ± 0.68 | 3.05 ± 0.94 | 2.23 ± 0.34 | 1.39 ± 0.51 |
T‐test was performed for C m, liver versus C m, blood and C m, muscle versus C m, blood.
p < .05.
As shown in Figure 1, the unbound drug concentrations of antipyrine, lamotrigine and carbamazepine in the liver and skeletal muscle were quantitatively similar to that in blood. There were no statistically significant differences in steady state unbound drug concentrations between blood (C m, blood), liver (C m, liver) and skeletal muscle (C m, muscle) for these three compounds, and their K puu,liver and K puu,muscle values were close to 1 (Table 3). Although the K puu,muscle values of propranolol and diltiazem appeared to be higher than 1, there were no statistically significant differences in the unbound concentrations of these two compounds between blood and skeletal muscle (Table 3). In contrast, the unbound drug concentrations of propranolol and diltiazem were significantly lower in the liver than blood (p < .05). The values of K puu,liver of propranolol and diltiazem were 0.382 and 0.145, respectively, significantly lower than 1 (Table 3).
As aforementioned, all of the 5 Class II compounds are substrates of efflux transporters based on in vitro cell assays. 6 The unbound drug concentrations of ofloxacin, digoxin atenolol and quinidine in liver were lower than that in blood, and the K puu,liver values of these 4 compounds tended to be lower than 1 (Figure 2 and Table 4). However, statistically significant differences between C m, liver and C m, blood were only observed for ofloxacin and digoxin, but not for atenolol and quinidine (Table 4). On the other hand, the unbound concentrations of Class II compounds in skeletal muscle were quantitatively similar to that in blood, with the exception of digoxin (Figure 3). The unbound drug concentrations of digoxin in skeletal muscle were significantly lower than that in blood (p < .05), and the K puu,muscle was 0.175.
All of the 4 Class III compounds, memantine, diphenhydramine, pyrilamine and gabapentin, are known to be substrates of influx transporters. Memantine, diphenhydramine and pyrilamine are substrates of a proton‐coupled organic cation antiporter, while gabapentin is a substrate of large amino acid transporter. 21 , 28 , 29 , 30 The unbound drug concentrations of Class III compounds appeared to be higher in the skeletal muscle than blood except gabapentin (Figure 3), and the K puu,muscle values were significantly higher than 1 (Table 5). Similarly, the unbound concentrations of gabapentin in liver appeared higher than that in blood, even though there was no statistically significant difference between C m, liver and C m, blood (Table 5). In contrast, the unbound concentrations of diphenhydramine and pyrilamine in liver were much lower than that in blood, and the K puu,liver values were less than 1 (Table 5). However, a statistically significant difference was only observed for pyrilamine, but not diphenhydramine.
4. DISCUSSION
All of the five Class I compounds, antipyrine, lamotrigine, carbamazepine, propranolol, and diltiazem are not substrates of efflux transporters based on in vitro cell assays. 6 These 5 compounds have good membrane permeability (P app). The P app values of antipyrine, lamotrigine, carbamazepine, propranolol, and diltiazem were 31.1, 27.2, 27.5, 11.0, and 18.4 × 10−6 cm/s, respectively, based on the parental MDCK cell assay. 6 Given the fact that these five Class I compounds are lipophilic and have good membrane permeability, the unbound drug concentrations of these compounds in the liver and skeletal muscle are expected to be quantitatively similar to that in blood, and the values of K puu,liver and K puu,muscle are expected to be close to 1.
As expected, the unbound drug concentrations of Class I compounds in liver and skeletal muscles were quantitatively similar to that in blood, and values of K puu,liver and K puu,muscle were close to 1, with the exceptions of propranolol and diltiazem (Table 3). Although the unbound concentrations of propranolol and diltiazem were somewhat higher in muscle than that in blood, the differences were not statistically significant. In contrast, the unbound concentrations of propranolol and diltiazem were statistically lower than that in blood (p < .05), and the K puu,liver values of these two compounds were 0.382 and 0.145, respectively, significantly less than 1. The observation that the K puu,liver values were less than 1 for propranolol and diltiazem is most likely due to their extensive metabolism in the liver. As shown in Table 2, the CL of propranolol and diltiazem in rats were about 83 and 92 ml/min/kg, respectively. In rats, propranolol and diltiazem are completely and exclusively metabolized in the liver. Propranolol is mainly oxidized to 4‐hydroxypropranolol and 5‐hydroxypropranolol by rat CYP2D enzyme. 31 A rat study has revealed that diltiazem was subject to an extensive hepatic first‐pass metabolism, and only about 30% of the dose reached the systemic circulation, when it was injected directly into the portal vein in rats. 23 Diltiazem undergoes extensive phase I metabolism including desacetylation, N‐demethylation, and O‐demethylation in rats. 32 Kinetically, the unbound drug concentration of a drug in an eliminating organ (such as liver) is also expected to be lower than that in blood, when the drug is subject to an extensive cell metabolism. 3 , 5
Although antipyrine, lamotrigine and carbamazepine are also largely metabolized in the liver, the K puu,liver values of these compounds are close to 1 because these compounds have low CL (4.81, 1.48 and 18.1 ml/min/kg, respectively) and are not subject to an extensive hepatic metabolism in rats. Antipyrine is almost completely metabolized in liver forming three major metabolites 3‐OH‐, 4‐OH‐ and norantipyrine. 33 Glucuronidation is the major route of metabolism of carbamazepine. In a rat study, the N‐glucuronide of carbamazepine accounted for about 20%–30% of the administered dose. 34 Similarly, lamotrigine is mainly eliminated by UDP‐glucuronosyltransferases leading to the formation of two major metabolites: N‐2 glucuronide and N‐5 glucuronide. 35 No significant differences between the C m, liver and C m, blood for antipyrine, lamotrigine, and carbamazepine are most likely due to their low and slow hepatic metabolism.
As demonstrated in our previous study, 6 all of the Class II compounds, ofloxacin, atenolol, quinidine, fexofenadine and digoxin are substrates of efflux transporters. Ofloxacin, atenolol, quinidine, and fexofenadine showed moderate to high plasma CL, while digoxin had a low CL in rats. The plasma CL values of ofloxacin, atenolol, quinidine, and digoxin were 32.5, 24.5, 76.4 and 9.1 ml/min/kg, respectively, in rats (Table 2). The plasma CL values of these compounds observed in this study are in good agreement with the reported CL values by other investigators. The reported plasma CL values of ofloxacin, atenolol, quinidine and digoxin were 27.5, 26.0, 64.8 and 17.3 ml/min/kg, respectively. 20 , 26 , 36 , 37
Since ofloxacin, atenolol, quinidine, fexofenadine, and digoxin are substrates of efflux transporters, hepatic metabolism, and transporter‐mediated efflux from hepatocyte canalicular membrane into the bile as well as efflux from hepatocytes returning to the blood circulation may play a significant role in the elimination clearance of these compounds. Thus, lower unbound drug concentrations in liver are expected for these Class II compounds. As expected, the unbound concentrations of the Class II compounds in liver were lower than that in blood with the exception of fexofenadine. However, statistically significant differences between C m, liver and C m, blood were only observed for ofloxacin and digoxin, but not for atenolol and quinidine (Table 4). The K puu,liver was about 0.501 and 0.232, respectively, for ofloxacin and digoxin.
The lower unbound concentration of ofloxacin in the liver is likely due to a combination of hepatic metabolism as well as transporter‐mediated efflux from hepatocyte canalicular membrane into the bile. In a rat study, the renal CL of ofloxacin was determined to be about 15 ml/min/kg, accounting 55% of total CL (27.5 ml/min/kg), while the non‐renal CL (biliary excretion + metabolism) accounted for about 45% of total CL. 36 In an in vitro study, ofloxacin was shown to be rapidly metabolized by rat liver microsomes to form three major metabolites, O‐acylglucuronidation, N‐demethylation and N‐oxidation. 38 In another rat study, approximately 30% of radioactivity was recovered from bile following oral administration of S‐14C‐ofloxacin, even though the identity of efflux transporter of ofloxacin biliary excretion remains unknown. 39 These results strongly suggest that hepatic metabolism and biliary excretion play a significant role in the removal of ofloxacin from hepatocytes resulting in lower unbound drug concentrations in the liver.
Like ofloxacin, the lower unbound concentration of digoxin in the liver is also likely due to a combination of hepatic metabolism as well as transporter‐mediated efflux from hepatocytes into the bile and the circulation. In a rat study, renal CL accounted for 20%–30% of total CL of digoxin, while the non‐renal CL (biliary excretion + hepatic metabolism) was about 70%–80%. 26 In rat liver microsomes, digoxin is sequentially metabolized to form digoxigenin bis‐digitoxoside, digoxigenin mono‐digitoxoside and digoxigenin. 40 Given the fact that digoxin is a substrate of P‐gp, the lower unbound concentration of digoxin in the liver may be due to a combination of hepatic metabolism, efflux from hepatocytes into the bile as well as the circulation.
The unbound concentration of atenolol in liver was somewhat lower than that in blood with a K puu,liver of 0.657. This may be due to its moderate rate of hepatic metabolism. Although atenolol is predominantly eliminated via the kidneys in humans (>95%), the compound is evenly eliminated by the liver and the kidneys in rats. 37 , 41 In a rat study, atenolol was shown to be evenly eliminated evenly by hepatic metabolism and renal excretion. The hepatic CL and renal CL of atenolol were determined to be about 14 and 13 ml/min/kg, respectively. 37 In vitro microsomal study has revealed that hydroxylation at the methylene carbon of the carbamoylmethyl group is the major route of metabolism. 42 Atenolol also underwent glucuronidation to form a conjugate metabolite.
The unbound drug concentrations of ofloxacin, atenolol, quinidine and fexofenadine in skeletal muscle were comparable to that in blood and the values were close to 1 (Figure 2 and Table 4). These results suggest that no metabolism or transporter‐mediated efflux occurred in muscle cells for these compounds. However, unexpectedly, the unbound drug concentrations of digoxin in skeletal muscle were much lower than that in blood with a mean K puu,muscle value of 0.175 (Table 4). Since skeletal muscle is not a major site of drug metabolism, the observation that the unbound drug concentrations of digoxin in the muscle were lower than that in blood suggests that digoxin may be subject to a significant efflux transport from muscle cells. It is possible that digoxin may be actively pumped out of muscle cells by other efflux transporters, rather than P‐gp.
With the exception of gabapentin, the unbound drug concentrations of Class III compounds in skeletal muscle were higher than that in blood (Figure 3). The K puu,muscle values of memantine, diphenhydramine and pyrilamine were 2.13, 3.08 and 5.78, respectively (Table 5). The higher unbound drug concentrations of memantine, diphenhydramine, and pyrilamine in skeletal muscle are very likely due to active influx transport. Memantine, diphenhydramine and pyrilamine are known to be a substrate of proton‐coupled organic cation antiporter. 21 , 28 , 29 The notion of active influx transport of these three compounds is supported by other investigators. In a rat microdialysis study, significant higher unbound drug concentrations of these 3 compounds in the brain than blood were observed. 8 The reported K puu,brain values of memantine, diphenhydramine, pyrilamine were 1.80, 3.85 and 2.50, respectively. These strongly results suggest the expression of proton‐coupled organic cation antiporter in muscle cells.
Similarly, the unbound concentrations of gabapentin in the liver were higher than that in blood with a K puu,liver of 2.23, even though the difference between C m, liver and C m, blood was not statistically significant (p = .058) (Figure 3 and Table 4). The higher unbound concentration of gabapentin in the liver is also likely due to active influx. Gabapentin is a substrate of L‐type amino acid transporter (LAT 1), which is responsible for cellular uptake of essential amino acids in various tissues of the body, including the liver. 30 , 43 , 44
The unbound drug concentrations of diphenhydramine and pyrilamine in liver were much lower than that in blood, and the K puu,liver values were 0.514 and 0.127, respectively (Figure 3 and Table 5). Although diphenhydramine and pyrilamine are substrates of a proton‐coupled influx transporter, these two compounds are also subject to extensive hepatic metabolism. Consequently, the transporter‐mediated influx may be offset by an extensive hepatic metabolism, resulting in lower unbound drug concentrations in the liver. As shown in Table 5, a statistically significant difference between C m, liver and C m, blood was observed for pyrilamine, while there was no statistically significant difference for diphenhydramine (Table 5). As shown in Table 1, both diphenhydramine and pyrilamine had high plasma CL (152 and 167 ml/min/kg, respectively). Diphenhydramine undergoes extensive first‐pass metabolism and nearly all the available drug is metabolized by the liver in rats. The major metabolic pathways of diphenhydramine are N‐dealkylation and N‐oxidation forming two major metabolites of N‐demethyl‐diphenhydramine and diphenhydramine N‐oxide. 45 Similarly, pyrilamine is largely metabolized in the rat liver forming two major metabolites of O‐demethyl‐pyrilamine and pyrilamine‐N‐oxide. 46 These results suggest that the hepatic metabolism rate is greater than active influx transport rate for both diphenhydramine and pyrilamine, and the net difference between hepatic metabolism and active influx transport is smaller for diphenhydramine as compared to pyrilamine.
In contrast to diphenhydramine and pyrilamine, the unbound drug concentrations of memantine were somewhat higher in the liver than blood with a K puu,liver of 1.56. Although memantine had a high plasma CL (60.5 ml/min/kg), this compound was not completely metabolized by the liver. In a rat study, the plasma CL of memantine was determined to be about 45 ml/min/kg, while the renal CL was about 13 ml/min/kg. 47 This means that hepatic metabolism accounts for approximately 70% of the memantine CL, unlike diphenhydramine and pyrilamine which are almost completely metabolized by liver. Since memantine is a substrate of proton‐coupled influx transporter, the somewhat higher unbound concentration of memantine in the liver (K puu,liver of 1.56) may reflect that the rate of active influx transport is somewhat greater than the rate of hepatic metabolism.
Although microdialysis is a useful sampling technique used for continuous measurement of unbound drug concentration in tissues, the unbound drug concentration in a given tissue measured by microdialysis represents extracellular (interstitial) unbound concentration, rather than intracellular unbound drug concentration. Due to the lack of proper tools and techniques, direct measurement of intracellular unbound drug concentration is impossible at the current time. Therefore, the intracellular unbound drug concentration is indirectly measured by microdialysis with an assumption that extracellular unbound drug concentration is equal to intracellular unbound concentration. According to the free drug hypothesis, unbound concentrations in all aqueous compartments (blood, extracellular, intracellular) are considered identical at steady state in the absence of active transport and cell metabolism. The assumption is valid for the Class I compounds that are not subject to extensive cell metabolism and are not substrates of drug transporters.
The free drug hypothesis is not universally applicable for all drugs. It is not applicable for drugs that are subject to extensive cell metabolism and are substrates of efflux and influx transporters. The intracellular unbound concentration is expected to be lower than extracellular unbound concentration for the Class II compounds that are substrates of efflux transporters and/or subject to extensive cell metabolism, while the intracellular unbound concentration is expected to be higher than extracellular unbound concentration for the Class III compounds that are substrates of influx transporters. If blood unbound concentration is higher than extracellular unbound concentration of the Class II compounds, it is reasonable to conclude that the blood unbound concentration is greater than tissue unbound concentration. Conversely, if blood unbound concentration is lower than extracellular unbound concentration of the Class III compounds, it is reasonable to conclude that blood unbound concentration is lower than tissue unbound concentration. Therefore, even for the Class II and III compounds, the extracellular unbound drug concentration measured in a given tissue by microdialysis can be used as a reliable surrogate for the unbound drug concentration in the tissue in relation to blood unbound concentration.
The data from this study has not only provided evidence to support the free drug hypothesis, but also contributed to our understanding of the effects of membrane transport activity and cell metabolism on the kinetic behavior of unbound drug concentrations in liver and skeletal muscle. An important lesson learned from this study is that the free drug hypothesis is also not applicable for drugs that are subject to extensive cell metabolism. Propranolol is a good example. Although propranolol is not a substrate of influx and efflux drug transporters, the drug is subject to extensive hepatic metabolism. The unbound concentration of propranolol is much lower in the liver, even though there is no difference in the unbound concentration between the brain (or muscle) and blood. Similar to propranolol, the level of glucose in the brain is much lower than that in blood. Glucose, an important energy source, cannot readily across the BBB due to its polarity. Thus, glucose is delivered to the brain via influx transporters, Na+‐dependent glucose transporters (SGLTs) and glucose transporters (GLUTs). The glucose levels were reported to be about 2.4 and 7.6 mM, respectively, for the brain and plasma in rats, 48 while the corresponding values were 0.82 and 5.5 mM in humans. 49 The much lower glucose level in the brain is due mainly to its extensive metabolism in the brain.
Finally, although it is generally believed that the skeletal muscle is not a major site of drug metabolism, little is known about the expression of drug transporters on the membranes of muscle cells. Recently, it has been speculated that statin‐induced myopathy may be caused by the influx transport activity of OATP2B1 expressed in muscle cells. 50 , 51 Given the clinical importance of statin toxicity, further research of drug transporter‐mediated drug accumulation in skeletal muscle is required.
DISCLOSURE
All authors have no conflict of interest.
AUTHOR CONTRIBUTIONS
Participated in research design: Shuyao Wang. Chun Chen, Chi Guan, Tao Wang. Conducted experiments: Shuyao Wang. Chun Chen, Liping Qiu, Lei Zhang, Shaofeng Zhang, Hongyu Zhou. Performed data analysis: Shuyao Wang. Chun Chen, Chi Guan, Liping Qiu, Lei Zhang, Shaofeng Zhang, Hongyu Zhou, Hongwen Du, Chen Li, Yaqiong Wu, Hang Chang. Wrote or contributed to the writing of the manuscript: Shuyao Wang. Chun Chen, Chi Guan.
ACKNOWLEDGEMENTS
The authors are extremely grateful to Dr Jiunn Lin for his useful and thoughtful scientific discussion. And the authors also thank the Pharmaron in life and bioanalytical groups.
Wang S, Chen C, Guan C, et al. Effects of membrane transport activity and cell metabolism on the unbound drug concentrations in the skeletal muscle and liver of drugs: A microdialysis study in rats. Pharmacol Res Perspect. 2021;9:e00879. doi: 10.1002/prp2.879
DATA AVAILABILITY STATEMENT
All data generated or used during the study appear in the submitted article.
REFERENCES
- 1. Smith DA, Di L, Kerns EH. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat Rev Drug Discov. 2010;9(12):929‐939. doi: 10.1038/nrd3287 [DOI] [PubMed] [Google Scholar]
- 2. Lin JH. Tissue distribution and pharmacodynamics: a complicated relationship. Curr Drug Metab. 2006;7(1):39‐65. doi: 10.2174/138920006774832578 [DOI] [PubMed] [Google Scholar]
- 3. Smith DA, Rowland M. Intracellular and intraorgan concentrations of small molecule drugs: theory, uncertainties in infectious diseases and oncology, and promise. Drug Metab Dispos. 2019;47(6):665‐672. doi: 10.1124/dmd.118.085951 [DOI] [PubMed] [Google Scholar]
- 4. Hammarlund‐Udenaes M, Friden M, Syvanen S, Gupta A. On the rate and extent of drug delivery to the brain. Pharm Res. 2008;25(8):1737‐1750. doi: 10.1007/s11095-007-9502-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang D, Hop CECA, Patilea‐Vrana G, et al. Drug concentration asymmetry in tissues and plasma for small molecule‐related therapeutic modalities. Drug Metab Dispos. 2019;47(10):1122‐1135. doi: 10.1124/dmd.119.086744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen C, Zhou H, Guan C, et al. Applicability of free drug hypothesis to drugs with good membrane permeability that are not efflux transporter substrates: a microdialysis study in rats. Pharmacol Res Perspect. 2020;8(2):e00575. doi: 10.1002/prp2.575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shimomura K, Okura T, Kato S, et al. Functional expression of a proton‐coupled organic cation (H+/OC) antiporter in human brain capillary endothelial cell line hCMEC/D3, a human blood‐brain barrier model. Fluids Barriers CNS. 2013;10(1):8. doi: 10.1186/2045-8118-10-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kitamura A, Okura T, Higuchi K, Deguchi Y. Cocktail‐dosing microdialysis study to simultaneously assess delivery of multiple organic‐cationic drugs to the brain. J Pharm Sci. 2016;105(2):935‐940. doi: 10.1002/jps.24691 [DOI] [PubMed] [Google Scholar]
- 9. Molina‐Ortiz D, González‐Zamora JF, Camacho‐Carranza R, et al. Xenobiotic‐metabolizing enzymes in skeletal muscle of children and adolescents. Pharmacol Pharm. 2013;4:231‐239. [Google Scholar]
- 10. Summerfield SG, Zhang Y, Liu H. Examining the uptake of central nervous system drugs and candidates across the blood‐brain barrier. J Pharmacol Exp Ther. 2016;358(2):294‐305. doi: 10.1124/jpet.116.232447 [DOI] [PubMed] [Google Scholar]
- 11. Kalvass JC, Maurer TS, Pollack GM. Use of plasma and brain unbound fractions to assess the extent of brain distribution of 34 drugs: comparison of unbound concentration ratios to in vivo p‐glycoprotein efflux ratios. Drug Metab Dispos. 2007;35(4):660‐666. doi: 10.1124/dmd.106.012294 [DOI] [PubMed] [Google Scholar]
- 12. Doran AC, Osgood SM, Mancuso JY, Shaffer CL. An evaluation of using rat‐derived single‐dose neuropharmacokinetic parameters to project accurately large animal unbound brain drug concentrations. Drug Metab Dispos. 2012;40(11):2162‐2173. doi: 10.1124/dmd.112.046391 [DOI] [PubMed] [Google Scholar]
- 13. Liu X, Smith BJ, Chen C, et al. Evaluation of cerebrospinal fluid concentration and plasma free concentration as a surrogate measurement for brain free concentration. Drug Metab Dispos. 2006;34(9):1443‐1447. doi: 10.1124/dmd.105.008201 [DOI] [PubMed] [Google Scholar]
- 14. de Lange EC, Danhof M, de Boer AG, Breimer DD. Methodological considerations of intracerebral microdialysis in pharmacokinetic studies on drug transport across the blood‐brain barrier. Brain Res Brain Res Rev. 1997;25(1):27‐49. doi: 10.1016/s0165-0173(97)00014-3 [DOI] [PubMed] [Google Scholar]
- 15. Hammarlund‐Udenaes M. Microdialysis as an important technique in systems pharmacology‐a historical and methodological review. AAPS J. 2017;19(5):1294‐1303. doi: 10.1208/s12248-017-0108-2 [DOI] [PubMed] [Google Scholar]
- 16. Boxenbaum H. Interspecies variation in liver weight, hepatic blood flow, and antipyrine intrinsic clearance: extrapolation of data to benzodiazepines and phenytoin. J Pharmacokinet Biopharm. 1980;8(2):165‐176. doi: 10.1007/BF01065191 [DOI] [PubMed] [Google Scholar]
- 17. Chanteux H, Staelens L, Mancel V, et al. Cross‐species differences in the preclinical pharmacokinetics of CT7758, an alpha4beta1/alpha4beta7 integrin antagonist. Drug Metab Dispos. 2015;43(9):1381‐1391. doi: 10.1124/dmd.115.064436 [DOI] [PubMed] [Google Scholar]
- 18. Terao N, Shen DD. Alterations in serum protein binding and pharmacokinetics of l‐propranolol in the rat elicited by the presence of an indwelling venous catheter. J Pharmacol Exp Ther. 1983;227(2):369‐375. [PubMed] [Google Scholar]
- 19. Iwamoto K, Watanabe J, Araki K, Deguchi N, Sugiyama H. Effect of age on the hepatic clearance of propranolol in rats. J Pharm Pharmacol. 1985;37(7):466‐470. doi: 10.1111/j.2042-7158.1985.tb03041.x [DOI] [PubMed] [Google Scholar]
- 20. Fremstad D, Jacobsen S, Lunde KM. Influence of serum protein binding on the pharmacokinetics of quinidine in normal and anuric rats. Acta Pharmacol Toxicol. 1977;41(2):161‐176. doi: 10.1111/j.1600-0773.1977.tb02136.x [DOI] [PubMed] [Google Scholar]
- 21. Sadiq MW, Borgs A, Okura T, et al. Diphenhydramine active uptake at the blood‐brain barrier and its interaction with oxycodone in vitro and in vivo. J Pharm Sci. 2011;100(9):3912‐3923. doi: 10.1002/jps.22567 [DOI] [PubMed] [Google Scholar]
- 22. Beconi MG, Howland D, Park L, et al. Pharmacokinetics of memantine in rats and mice. PLoS Curr. 2011;3:RRN1291. doi: 10.1371/currents.RRN1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee Y‐H, Lee M‐H, Shim C‐K. Pharmacokinetics of diltiazem and deacetyldiltiazem in rats. Int J Pharm. 1991;76(1‐2):71‐76. doi: 10.1016/0378-5173(91)90345-O [DOI] [Google Scholar]
- 24. Slordal L, Hoyem‐Johansen T, Aarbakke J. Microsomal metabolism of antipyrine in rats treated with antineoplastic drugs. Pharmacology. 1983;26(2):95‐99. doi: 10.1159/000137790 [DOI] [PubMed] [Google Scholar]
- 25. Castel‐Branco MM, Falcao AC, Figueiredo IV, Caramona MM. Lamotrigine pharmacokinetic/pharmacodynamic modelling in rats. Fundam Clin Pharmacol. 2005;19(6):669‐675. doi: 10.1111/j.1472-8206.2005.00380.x [DOI] [PubMed] [Google Scholar]
- 26. Harrison LI, Gibaldi M. Pharmacokinetics of digoxin in the rat. Drug Metab Dispos. 1976;4(1):88‐93. [PubMed] [Google Scholar]
- 27. Remmel RP, Sinz MW, Cloyd JC. Dose‐dependent pharmacokinetics of carbamazepine in rats: determination of the formation clearance of carbamazepine‐10,11‐epoxide. Pharm Res. 1990;7(5):513‐517. doi: 10.1023/a:1015872901523 [DOI] [PubMed] [Google Scholar]
- 28. Okura T, Hattori A, Takano Y, et al. Involvement of the pyrilamine transporter, a putative organic cation transporter, in blood‐brain barrier transport of oxycodone. Drug Metab Dispos. 2008;36(10):2005‐2013. doi: 10.1124/dmd.108.022087 [DOI] [PubMed] [Google Scholar]
- 29. Higuchi K, Kitamura A, Okura T, Deguchi Y. Memantine transport by a proton‐coupled organic cation antiporter in hCMEC/D3 cells, an in vitro human blood‐brain barrier model. Drug Metab Pharmacokinet. 2015;30(2):182‐187. doi: 10.1016/j.dmpk.2014.12.006 [DOI] [PubMed] [Google Scholar]
- 30. Dickens D, Webb SD, Antonyuk S, et al. Transport of gabapentin by LAT1 (SLC7A5). Biochem Pharmacol. 2013;85(11):1672‐1683. doi: 10.1016/j.bcp.2013.03.022 [DOI] [PubMed] [Google Scholar]
- 31. Hung DY, Siebert GA, Chang P, et al. Hepatic pharmacokinetics of propranolol in rats with adjuvant‐induced systemic inflammation. Am J Physiol Gastrointest Liver Physiol. 2006;290(2):G343‐G351. doi: 10.1152/ajpgi.00155.2005 [DOI] [PubMed] [Google Scholar]
- 32. Choi J‐S, Yang J‐S, Choi D‐H. Effects of ticlopidine on the pharmacokinetics of diltiazem and its main metabolite, desacetyldiltiazem. Rats Biomol Ther. 2011;19(2):255‐260. [Google Scholar]
- 33. Buters JT, Zysset T, Reichen J. Metabolism of antipyrine in vivo in two rat models of liver cirrhosis. Its relationship to intrinsic clearance in vitro and microsomal membrane lipid composition. Biochem Pharmacol. 1993;46(6):983‐991. doi: 10.1016/0006-2952(93)90662-g [DOI] [PubMed] [Google Scholar]
- 34. Madden S, Maggs JL, Park BK. Bioactivation of carbamazepine in the rat in vivo. Evidence for the formation of reactive arene oxide(s). Drug Metab Dispos. 1996;24(4):469‐479. [PubMed] [Google Scholar]
- 35. Argikar UA, Senekeo‐Effenberger K, Larson EE, Tukey RH, Remmel RP. Studies on induction of lamotrigine metabolism in transgenic UGT1 mice. Xenobiotica. 2009;39(11):826‐835. doi: 10.3109/00498250903188985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Foote EF, Halstenson CE. Effects of probenecid and cimetidine on renal disposition of ofloxacin in rats. Antimicrob Agents Chemother. 1998;42(2):456‐458. doi: 10.1128/AAC.42.2.456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mehvar R, Gross ME, Kreamer RN. Pharmacokinetics of atenolol enantiomers in humans and rats. J Pharm Sci. 1990;79(10):881‐885. doi: 10.1002/jps.2600791007 [DOI] [PubMed] [Google Scholar]
- 38. Sudo K, Okazaki O, Tsumura M, Tachizawa H. Isolation and identification of metabolites of ofloxacin in rats, dogs and monkeys. Xenobiotica. 1986;16(8):725‐732. doi: 10.3109/00498258609043563 [DOI] [PubMed] [Google Scholar]
- 39. Okazaki O, Kurata T, Tachizawa H. Stereoselective metabolic disposition of enantiomers of ofloxacin in rats. Xenobiotica. 1989;19(4):419‐429. doi: 10.3109/00498258909042283 [DOI] [PubMed] [Google Scholar]
- 40. Salphati L, Benet LZ. Metabolism of digoxin and digoxigenin digitoxosides in rat liver microsomes: involvement of cytochrome P4503A. Xenobiotica. 1999;29(2):171‐185. doi: 10.1080/004982599238722 [DOI] [PubMed] [Google Scholar]
- 41. Kirch W, Gorg KG. Clinical pharmacokinetics of atenolol–a review. Eur J Drug Metab Pharmacokinet. 1982;7(2):81‐91. doi: 10.1007/BF03188723 [DOI] [PubMed] [Google Scholar]
- 42. Reeves PR, Barnfield DJ, Longshaw S, McIntosh DA, Winrow MJ. Disposition and metabolism of atenolol in animals. Xenobiotica. 1978;8(5):305‐311. doi: 10.3109/00498257809060955 [DOI] [PubMed] [Google Scholar]
- 43. Scalise M, Galluccio M, Console L, Pochini L, Indiveri C. The human SLC7A5 (LAT1): the intriguing histidine/large neutral amino acid transporter and its relevance to human health. Front Chem. 2018;6:243. doi: 10.3389/fchem.2018.00243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poncet N, Mitchell FE, Ibrahim AFM, et al. The catalytic subunit of the system L1 amino acid transporter (slc7a5) facilitates nutrient signalling in mouse skeletal muscle. PLoS One. 2014;9(2):e89547. doi: 10.1371/journal.pone.0089547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ali B, Gupta KP, Kumar A, Bhargava KP. Differential stimulation of diphenhydramine, pethidine, morphine and aniline metabolism by chronic methaqualone treatment. Pharmacology. 1980;20(4):181‐187. doi: 10.1159/000137363 [DOI] [PubMed] [Google Scholar]
- 46. Kelly DW, Slikker W Jr. The metabolism and elimination of pyrilamine maleate in the rat. Drug Metab Dispos. 1987;15(4):460‐465. [PubMed] [Google Scholar]
- 47. Choi YA, Song IS, Choi MK. Pharmacokinetic drug‐drug interaction and responsible mechanism between memantine and cimetidine. Pharmaceutics. 2018;10(3):119. doi: 10.3390/pharmaceutics10030119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Silver IA, Erecinska M. Extracellular glucose concentration in mammalian brain: continuous monitoring of changes during increased neuronal activity and upon limitation in oxygen supply in normo‐, hypo‐, and hyperglycemic animals. J Neurosci. 1994;14(8):5068‐5076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Abi‐Saab WM, Maggs DG, Jones T, et al. Striking differences in glucose and lactate levels between brain extracellular fluid and plasma in conscious human subjects: effects of hyperglycemia and hypoglycemia. J Cereb Blood Flow Metab. 2002;22(3):271‐279. doi: 10.1097/00004647-200203000-00004 [DOI] [PubMed] [Google Scholar]
- 50. Knauer MJ, Urquhart BL, Meyer zu Schwabedissen HE, et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ Res. 2010;106(2):297‐306. doi: 10.1161/CIRCRESAHA.109.203596 [DOI] [PubMed] [Google Scholar]
- 51. Turner RM, Statin‐Related PM. Statin‐related myotoxicity: a comprehensive review of pharmacokinetic, pharmacogenomic and muscle components. J Clin Med. 2020;9(1):22. doi: 10.3390/jcm9010022 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or used during the study appear in the submitted article.