

Abstract
Generating combinatorial libraries of specific sets of mutations are essential for addressing protein engineering questions involving contingency in molecular evolution, epistatic relationships between mutations, as well as functional antibody and enzyme engineering. Here we present optimization of a combinatorial mutagenesis method involving template-based nicking mutagenesis, which allows for the generation of libraries with >99% coverage for tens of thousands of user-defined variants. The non-optimized method resulted in low library coverage, which could be rationalized by a model of oligonucleotide annealing bias resulting from the nucleotide mismatch free-energy difference between mutagenic oligo and template. The optimized method mitigated this thermodynamic bias using longer primer sets and faster annealing conditions. Our updated method, applied to two antibody fragments, delivered between 99.0% (32451/32768 library members) to >99.9% coverage (32757/32768) for our desired libraries in 2 days and at an approximate 140-fold sequencing depth of coverage.
Keywords: antibody engineering, combinatorial mutagenesis, enzyme engineering, molecular evolution, nicking mutagenesis
Introduction
Combinatorial mutagenesis is a technique for generating a library of sequences containing defined mutations at specific positions in a DNA sequence. Combinatorial mutagenic libraries can be used for a variety of objectives. For example, directed evolution workflows are enhanced by the ability to screen the combinatorial set of individual beneficial mutations. Combinatorial libraries can also help answer some of our basic scientific questions regarding molecular evolution and mutational epistasis (Starr and Thornton, 2016; Poelwijk et al., 2019). These libraries in conjunction with directed evolution and deep mutational scanning allows for efficient understanding of both single point mutations and permutations of those mutations for enzyme engineering applications (Choi et al., 2019). Our group is specifically interested in combinatorial mutagenesis in order to map the set of possible evolutionary trajectories of antibodies during affinity maturation.
There have been many published techniques for the generation of combinatorial libraries, of which DNA shuffling is perhaps the most ubiquitous (Stemmer, 1994). Researchers have improved upon the inherent randomness of the DNA shuffling protocol by introducing a low rate of associated point mutations (Zhao et al., 1998), although shuffling still requires the user to optimize the DNA polymerization in the PCR reaction and it also fails when mutations are clustered near others. Combinatorial codon mutagenesis is another method that generates targeted, user-defined libraries with little parental DNA background in which the mutation frequency can be tuned by PCR cycles and fragmentation parameters (Belsare et al., 2017). While exciting, this method has successfully been applied to only a handful of positions for a library and the exact composition of the libraries has not been evaluated by deep sequencing. The classical technique of cassette mutagenesis can also produce combinatorial libraries but is limited to spatially proximate regions, like complementarity-determining regions in antibodies, and has a lower probability for adjacent nucleotide substitutions or mutations spread across the length of the gene (Hidalgo et al., 2008). More recently, techniques from Poelwijk et al. (2019) and Choi et al. (2019) have been described in which many gene fragments containing mutations of interest are recombined in controlled reactions. Importantly, a unique barcode is haplotyped to a specific variant, which greatly simplifies deep sequencing workflows. Disadvantages of the above methods include the requirement for many gene fragments (e.g. 34 distinct gene fragments in Poelwijk et al.), being time consuming due to several rounds of bacterial transformation and selection, and the relatively modest size libraries demonstrated to date (between 1000 and 10 000 library members).
An appraisal of the existing techniques led us to seek out a new fast method that (i) could be used to generate large libraries of tens of thousands of user-defined variants, that (ii) includes mutations that are distributed throughout the length of a given gene sequence and that (iii) is easily adapted to new templates. Recently, our group has developed a technique called nicking mutagenesis (NM) (Wrenbeck et al., 2016; Steiner et al., 2020). NM can be used to generate comprehensive single site mutational libraries or to generate defined mutations at multiple sites (‘multi-site NM’). In multi-site NM, mutations are incorporated at defined positions by annealing oligonucleotides containing nucleotide (nt) mismatch(es) to a closed single stranded DNA template. These oligonucleotides are present in the reaction in molar excess to the template and typically contain one or more nucleotide mismatches flanked by ~18 or more complementary nts on both the 5′ and 3′. We reasoned comprehensive combinatorial libraries could be realized by modifying the multi-site NM protocol by using a set of oligos perfectly complementary to the original sequence or encoding a user-defined mismatch(es).
Here we present an optimization of the multi-site NM protocol to generate near-comprehensive combinatorial mutagenesis libraries containing tens of thousands of library variants. We show that, under the original protocol, incorporation of mutations by annealing oligonucleotides is inefficient because of large free energy differences between oligos containing nt mismatches and ones perfectly complementary to the target sequence. We identify several strategies to overcome this thermodynamic constraint and show an updated protocol to successfully produce libraries with >99% coverage of tens of thousands of user-defined variants in 2020 days.
Materials and Methods
Strains
Escherichia coli strain XL1-Blue high-efficiency electrocompetent cells (Agilent Cat # 200228) was used for all results presented here.
Plasmid constructs
Gene fragments were ordered for CR6261, UCA_CR6261, CR9114 and UCA_CR9114 sequences (full sequences in Supplementary Note S1) from Integrated DNA Technologies (IDT) and were cloned into an empty yeast surface display vector pETcon (Addgene plasmid #41522) via standard restriction enzyme cloning. A BbvCI restriction enzyme site, which is necessary for multi-site NM, exists once in each of the gene fragments. The working vectors for all scFv constructs are 6.8 kb in length. Plasmid constructs were sequence verified (Genewiz). pEDA5_GFPmut3_Y66H (Addgene plasmid #80085) was used for all the GFP experiments and is 4.3 kb in length.
Degenerate oligonucleotides
Sequences for degenerate oligonucleotide oligos are shown in Supplementary Table S1 and were purchased from Integrated DNA Technologies. For the libraries, the unmutated common ancestor gene sequence and mature antibody gene sequence were aligned to identify (i) the set of mutations (CR6261:14 positions; CR9114: 15 positions) and (ii) the nt distance between each adjacent mutation. Mutated codons separated by distances of 30 nts or further apart were incorporated into different oligonucleotides, whereas spatially proximal mutations were encoded as degenerate bases in a single oligonucleotide. The oligos used for the construction of the initial CR6261 library had 18 nts homology arms on 5′ and 3′ end. Homology arms used for all subsequent libraries were 30 nts or longer. The specific degenerate nucleotides used to create the libraries correspond to nts encoding for the mature or germline amino acid. Position 77 for CR6261 and 74 for CR9114 had mutations that could not be encoded by a single nt change, which necessitated incorporation of undesired mutations.
The Boltzmann model for mutational incorporation
The free energy for each mutation, ΔGi, in the first round of NM was determined from tabulated data (Allawi and SantaLucia, 1997; Allawi and SantaLucia, 1998a, 1998b) and adjusted to the annealing temperature of 55°C using the following equation where ΔHi is the enthalpy of the mismatch, T is the temperature and ΔSi is the entropy of the mismatch.
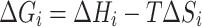 |
The change in free-energy values for a given variant, or ΔΔGi, were calculated from the difference in ΔGi and ΔGwildtype.
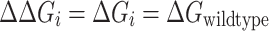 |
ΔΔG values for oligonucleotides with more than one mismatch (ΔΔGi,multiple) were approximated as the sum of the ΔΔGi values for n individual mismatches ΔΔGi:
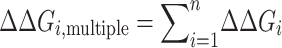 |
These individual free energy values for each mutant i (including WT) were incorporated into a Boltzmann model to determine the predicted frequency of mutant i (freqi):
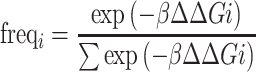 |
The temperature factor β was fit to the experimental data. For the data presented in Figure 1E a β = 0.33 mol/kcal was used.
Fig. 1.
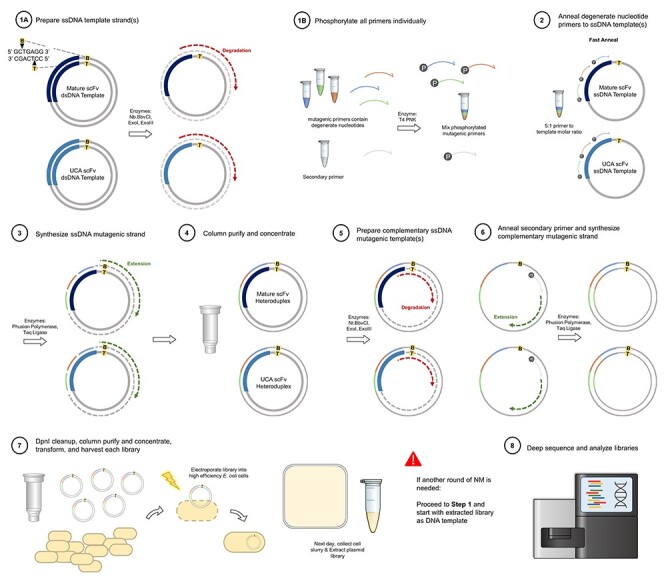
Combinatorial NM Flowchart. Steps 1A & 1B can be performed simultaneously and involve selectively nicking and degrading one strand of dsDNA parental template(s) (1A) and phosphorylating the degenerate nucleotide mutagenic primers and secondary primer (1B). Step 2: the phosphorylated mutagenic primers are annealed to the ssDNA parental template(s) using a fast annealing temperature program. Step 3: the remainder of the ssDNA mutagenic strand is synthesized and ligated. Step 4: the heteroduplex plasmid library is column purified and concentrated. Step 5: the complementary mutagenic strand is generated by selectively nicking and degrading the complementary strand of DNA. Step 6: the phosphorylated secondary primer is annealed to an unmutagenized region of the ssDNA template(s) and the remainder of the strand is synthesized and ligated. Step 7: an enzymatic DpnI cleanup step is followed by another column cleanup and concentration where the plasmid library is then ready for transformation and next-day harvest. At this point if another round of combinatorial NM is required, proceed to step 1 with the extracted library as the dsDNA template. Step 8: The library is deep sequenced and analyzed.
For an example of the calculation from the model, consider the primer MBK-105 which contains the degenerate nucleotides for residues 28 and 30. There are four possible sequences this primer can encode: (1) ‘template-matched’ which encodes no mutations, (2) P28T mutation, (3) R30S mutation, (4) both the P28T and R30S mutation. Thus, the predicted frequency for the P28T mutation is calculated as follows:
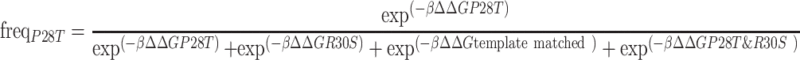 |
The predicted frequencies using this model were calculated for each of the potential mutations (single, double and triple) encoded on a single primer in the first round of multi-site NM (MBK-105, MBK-107, MBK-109) for two replicates. All ΔΔG values and predicted frequencies are given in Supplementary Table S2.
Combinatorial library construction
The final step-by-step protocol generating combinatorial libraries using NM has been published (Kirby and Whitehead, 2021) and a flowchart detailing combinatorial NM is shown in Figure 1. Our initial CR6261 combinatorial libraries were prepared following multi-site NM as follows. All enzymes and reagents were purchased from New England Biolabs (NEB) unless otherwise stated. The mutagenic primers MBK-105, MBK-107, MBK-109 and secondary primer MBK-142 were individually phosphorylated by adding 18 μL nuclease free water (IDT DNA), 3 μL of 10X T4 Polynucleotide Kinase buffer, 1 μL of 10 mM ATP and 1 μL of T4 Polynucleotide Kinase to 7 μL of the 100 μM oligo and incubating at 37°C for 1 h. Mutagenic primers were then diluted 1:20 by adding 2 μL of each phosphorylated primer (MBK-105, MBK-107, MBK-109) to 34 μL nuclease free water (40 μL total). The ssDNA CR6261 parental DNA template is made from freshly prepped (Monarch® Plasmid Miniprep Kit, NEB) plasmid by taking 0.76 picomoles of dsDNA plasmid and adding 2 μL 10X CutSmart Buffer, 1 μL of 1:10 diluted exonuclease III (diluted into 1X CutSmart Buffer, final concentration of 10 U/μL), 1 μL of Nb.BbvCI, 1 μL of exonuclease I and nuclease free water up to 20 μL in a single PCR tube. The PCR tube is placed on a preheated thermocycler (Eppendorf™ Mastercycler™ Pro) at 37°C and the following program is run: 60 min at 37°C, 20 min at 80°C, hold at 4°C. The diluted phosphorylated mutagenic oligos are annealed to the ssDNA template by adding 16.7 μL nuclease free water, 10 μL 5X Phusion HF Buffer and 3.3 μL of the 1:20 diluted oligo mixture to the ssDNA template tube for a total reaction volume of 50 μL. The thermal cycler program for annealing the oligos in our initial library used a slow anneal step where the reaction tube is placed on the thermocycler preheated to 98°C and then held at 98°C for 2 min, then steps down from 98 to 55°C over 15 min before holding at 55°C for a minimum of 5 min followed by an indefinite hold at 55°C. While keeping the reaction tube on the thermal cycler block, the following components were added to the reaction tube: 11 μL nuclease free water, 10 μL 5X Phusion HF Buffer, 20 μL 50 mM DTT, 1 μL 50 mM NAD+, 2 μL 10 mM dNTPs, 5 μL Taq DNA Ligase and 1 μL Phusion HF Polymerase. The following 100 μL reaction stays on the thermal cycler as the temperature programs runs 72°C for 10 min, followed by 45°C for 20 min and holds at 4°C. The reaction is then purified using a Monarch® PCR & DNA Cleanup Kit according to the manufacturer’s instructions (NEB) but with eluting in 15 μL of nuclease free water and waiting 5 min after applying eluant to the column before centrifuging. Next, we degrade the complementary template strand by transferring 14 μL of the purified DNA product to a new PCR tube and adding 2 μL 10X CutSmart Buffer, 2 μL 1:50 diluted exonuclease III (diluted into 1X CutSmart Buffer, final concentration of 2 U/μL), 1 μL 1:10 diluted Nt.BbvCI (diluted into 1X CutSmart Buffer, final concentration of 1 U/μL), and 1 μL exonuclease I for a total reaction volume of 20 μL. The reaction tube is then placed on the thermocycler preheated to 37°C and is kept at 37°C for 1 h, then 20 min at 80°C and held at 4°C. The phosphorylated secondary primer MBK-142 is diluted 1:20 by adding 2 μL of the phosphorylated oligo to 38 μL of nuclease free water. The complementary mutagenic strand is then synthesized by taking the reaction tube off the thermocycler, placing it on ice and adding the following: 27.7 μL nuclease free water, 20 μL Phusion HF Buffer, 3.3 μL 1:20 diluted secondary primer, 20 μL of 50 mM DTT, 1 μL of 50 mM NAD+, 2 μL 10 mM dNTPs, 5 μL Taq DNA Ligase and 1 μL Phusion HF Polymerase for a total reaction volume of 100 μL. The thermal cycler should again be preheated to 98°C, then the reaction tube can be placed on the cycler block and run at 98°C for 30 s, 45 s at 55°C, 10 min at 72°C, 20 min at 45°C and hold at 4°C. The reaction tube can be removed from the thermal cycler and 2 μL of DpnI is added and then the tube is incubated at 37°C for 1 h to degrade methylated and hemimethylated wild-type DNA. The reaction is then purified using a Monarch® PCR & DNA Cleanup Kit according to the manufacturer’s instructions but with eluting in 6 μL of nuclease free water and waiting 5 min after applying eluant to the column before centrifuging. The entire 6 μL purified product is then transformed into E. coli strain XL1 Blue high efficiency cells following standard electrocompetent transformation protocols. After a 1-h recovery of the transformed cells in SOC media, prepare six 10-fold serial dilutions (starting with 10 μL, or a ‘100x’ dilution) on a carbenicillin supplemented agar plate and then add 1 mL SOC to the recovered cells and plate on a large bioassay dish (Corning) also supplemented with carbenicillin. After the plates are dried, they are placed in an incubator at 37°C overnight until the cells are grown. After confirming there were sufficient transformants on the dilution plate (100x the library size, >256 000) the large bioassay dish was scraped, and colonies were resuspended in sterile SOB media. The mutagenic library was then extracted from an aliquot of the cells using the Monarch® Plasmid Miniprep Kit according to the manufacturer’s instructions. The library was ready for the second round of multi-site NM which was performed exactly as the first round but with MBK-106 and MBK-108 as the mutagenic oligos. The large bioassay dish was scraped after confirming >1638 400 colonies (100-fold coverage of the expected number of library variants) on the dilution plate and the library was extracted using the Monarch® Plasmid Miniprep Kit. Plasmid prepped from both the first and second round of multi-site NM were prepared and submitted for deep sequencing.
The updated CR6261 libraries and CR9114 libraries were made similarly but with a few key differences. The first round of multi-site NM for CR6261_pETcon used MBK-137, MBK-138 and MBK-139 as the mutagenic oligos with MBK-142 as the secondary primer. For the parental DNA template UCA_CR6261_pETcon, the first round mutagenic primers were MBK-137, MBK-139 and MBK-153, with MBK-142 as the secondary primer. The first round was performed as previously described except that when annealing the mutagenic primers to the ssDNA parental template, a quick anneal was implemented where the thermal cycler was still preheated to 98°C and the reaction tube was held at 98°C for 2 min but the temperature dropped from 98 to 55°C as quickly as possible and then was held at 55°C for 5 min. For the Eppendorf™ Mastercycler™ Pro thermal cycler used here the reported maximum cooling rate for the silver block is 6C/s, although we did not validate this rate. We used thin polypropylene tubes (VWR, Cat # 53509-304) with a moderate thermal conductivity, so the actual cooling rate is expected to be slightly lower than 6°C/s. For the second round of mutagenesis both the CR6261 and UCA_CR6261 round 1 libraries were prepped and used the mutagenic primers MBK-145 and MBK-146 with MBK-142 again as the secondary primer. Biological replicate libraries were prepared on separate days.
The CR9114_pETcon round 1 and UCA_CR9114_pETcon round 1 of multi-site NM followed the protocol as described with the quick anneal. For CR9114_pETcon, the first round mutagenic primers were MBK-140 and MBK-141, and for UCA_CR9114_pETcon the first round mutagenic primers were MBK-140 and MBK-165. Both used MBK-142 as the secondary primer. After it was confirmed that >6400 colonies were observed on the dilution plates (100-fold coverage of the expected number of library variants), the bioassay dishes were scraped and the plasmids were extracted for multi-site NM round 2. In round 2, the mutagenic primers for both sub-libraries were MBK-149 and MBK-150 with MBK-142 as the secondary oligo. Round 2 implemented the quick anneal and plates were scraped after confirming >3 276 800 transformation efficiency (100-fold coverage of the expected number of library variants). Biological replicate libraries were prepared on separate days.
Multi-site NM optimization using GFP
GFP multi-site optimization experiments all used the multi-site NM protocol as described above either with the slow anneal step or with the quick anneal step where noted. All primers used, listed in Supplementary Table S1, were individually designed as noted in the main text. Template matched and missense oligos were mixed equimolarly before each experiment. All GFP reactions used MBK-130 as the secondary primer. The parental DNA template was first selectively nicked with Nt.BbvCI and nicked with Nb.BbvCI when synthesizing the complementary strand. The experiments to mimic our initial experiment used MBK-125 and MBK-126 as the mutagenic oligos. The experiments with extended primers used MBK-128 and MBK-129 as the mutagenic oligos. Lastly, the experiments with a silent mutation added to match free energies used primers MBK-125 and MBK-127. After mutagenesis, the reaction was transformed and recovered for 1 h before making 10-fold serial dilutions and plating each dilution onto 1 agar plate supplemented with antibiotic. The next day, the number of GFP fluorescent colonies and total colonies were counted. GFP replicate experiments were performed on separate days.
Deep sequencing preparation and data analysis
The combinatorial libraries were prepared for deep sequencing exactly as described in Kowalsky et al., 2015, following ‘method B’ (Kowalsky et al., 2015). Primers used for deep sequencing preparation are included in Supplementary Table S1. MBK-110 and MBK-111 were used for all CR6261 and UCA_CR6261 libraries. MBK-143 and MBK-144 were used for all CR9114 and UCA_CR9114 libraries. The libraries were sequenced on an Illumina MiSeq using 2 × 250 paired end reads by the BioFrontiers Sequencing Core at the University of Colorado, Boulder. The software package PACT (Klesmith and Hackel, 2018), freely available at GitHub (https://github.com/JKlesmith/PACT/), was used to merge the paired end reads and calculate the sequencing counts obtained from raw FASTQ files.
Results
We desired defined combinatorial mutagenesis libraries for synthetic genes encoding single chain variable fragments (scFvs) for the anti-Influenza Hemagglutinin antibodies CR6261 (Ekiert et al., 2009) and CR9114 (Dreyfus et al., 2012). CR6261 and CR9114 are unusual antibodies in which the entire paratope is contained in the variable heavy chain (VH). For CR6261, we identified 14 positions on VH positions 28–104 with different amino acid identities between the mature CR6261 and the CR6261 unmutated common ancestor (UCA_CR6261). Generating a combinatorial library encoding either the UCA or mature CR6261 amino acid at these 14 positions results in a theoretical library size of 16 384 variants (214). Similarly, we selected the 15 amino acid differences between CR9114 and its UCA_CR9114 on VH positions 24–97, resulting in a theoretical library size of 32 768 variants (215). We propose three key metrics that determine the combinatorial library quality. The most important metric is the completeness of the library, while secondary metrics include both the percentage of the library containing desired mutants and the distribution of variants within the library.
Initial library generated with multi-site NM
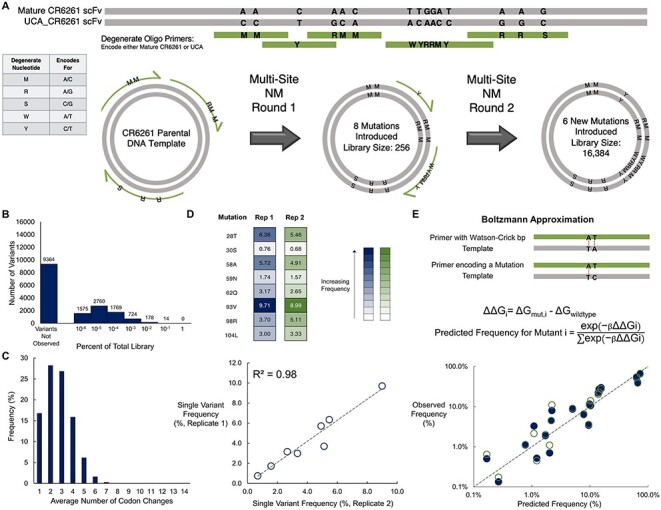
We first attempted to construct these combinatorial libraries using the template-based method multi-site NM (Fig. 2A). For CR6261, the 14 desired mutable positions spanned positions 28–104 on the VH with multiple mutations clustered near another including four codons in a row on positions 74–77. Therefore, we performed two sequential NM reactions. For the first NM round, three non-overlapping oligonucleotides were annealed to the CR6261 template. These oligos contained two or three degenerate nucleotides encoding for a total library diversity of 256 (28) members. The NM reaction product was transformed into E. coli, and the harvested library plasmids were used as a template for the second round of NM using two additional non-overlapping oligonucleotides containing the remaining desired mutations. The libraries generated were sequenced on an Illumina MiSeq and analyzed using PACT (Klesmith and Hackel, 2018). The library from this initial construction contained only 42.8% (7020/16 384) of the desired variants (Fig. 2B). The constructed variants were not uniformly distributed in sequence space—over 87% of the sequences contained four or fewer codon substitutions from the parental CR6261 template, with a mode of two, and essentially no coverage (<0.1%) for eight or more codon substitutions (Fig. 2C).
Fig. 2.
Optimized multi-site NM protocol yields near-comprehensive coverage of combinatorial libraries. Results from the optimized ‘dual directions’ strategy for generating comprehensive combinatorial libraries. (left) Histograms for frequency of the total number of variants and (right) violin plots of the frequency of each variant relative to the number of mutations from CR6261 (replicate 1 A; replicate 2 B) or CR9114 (replicate 1 C; replicate 2 D). For all panels, the violin plots were normalized where a log10 frequency of zero corresponds to a mutation not being observed by deep sequencing. The dashed gray lines indicate the relative frequency for a uniformly distributed library.
We questioned why this library had such low coverage of the desired mutations given each degenerate position on a given oligo had a 50% probability of containing a mismatch or match to the template. If all oligos were annealed equally the resulting uniform distribution for CR6261 would have a mode of seven codon substitutions. Extrinsic factors may be important here, including low numbers of transformants leading to low library complexity. We first speculated that insufficient annealing of primers caused by mismatches with the template could be responsible for the low library complexity. However, enumeration of the melting temperatures for all possible primer sequences revealed melting temperatures with template of at least 24°C above the annealing temperature of 55°C (Supplementary Table S3).
We reasoned that if the frequencies of specific mutations were reproducible between replicates, then an intrinsic factor(s) may be governing these distributions. To further investigate these possibilities, we repeated the NM experiment for round 1 covering 8 positions (256 total variants). The two NM reactions had nearly the same low frequency of incorporation for the individual single mutations (R2 = 0.98), hinting at the low completeness of the library arising from intrinsic factors (Fig. 2D).
Boltzmann thermodynamic model
We noted that some of the individual point mutations were incorporated in much larger frequency in the 256-member library compared to others. Specifically, the library frequency of M93V, encoded by an adenine to guanine transition, is ~9% while R30S, encoded by a more energetically unfavorable adenine to cytosine transversion, is ~0.7% (Fig. 2D). This finding suggests some relationship between the thermodynamic penalty of a given mismatch and the library frequency of the resultant mutation.
Based on this initial supposition, we constructed a simple model to predict the mutational incorporation observed in these combinatorial libraries. First, we used the nearest neighbor model (Allawi and SantaLucia, 1997; Allawi and SantaLucia, 1998b, 1998a, 1998c) to estimate the change in free energy (ΔΔGi) for each annealed mutagenic oligo i relative to the change in free energy of an annealed oligo perfectly complementary to template (Supplementary Table S2). Next, we incorporated these ΔΔGi values into a Boltzmann distribution to predict the frequency of each single, double, and triple mutant encoded by each single degenerate oligo used in the NM reaction (Fig. 2E). The temperature factor β is the only adjustable parameter in the model. Adjusting the temperature factor resulted in a reasonable agreement between the model and experimental results performed in replicate, with at least 88% of the variance in frequencies for specific mutations explained by the model (Fig. 2E).
GFP mutational incorporation experiments
We set out to improve the NM protocol guided by this simple thermodynamic model. To accomplish this optimization, we used a plasmid constitutively expressing a GFP variant with a single-nucleotide polymorphism encoding Y66H, which renders the GFP non-fluorescent. Fluorescence can be recovered using NM with an appropriate H66Y mutagenic oligo (Fig. 3A). In contrast, successful annealing with an H66 oligo recovers the non-fluorescent product (Fig. 3A). The frequency of incorporation of the desired mutation can be determined by counting the percentage of fluorescent colonies after transformation.
Fig. 3.
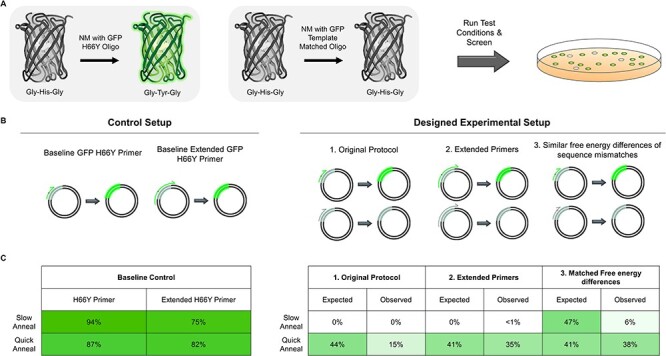
Improving multi-site NM mutational incorporation using a GFP-based screen. (A) Performing NM on pEDA5_GFPmut3_Y66H using an H66Y mutagenic oligo reverts non fluorescent GFP to fluoresce, whereas the oligo encoding the same sequence as the template (‘template matched’) results in non-fluorescent colonies. The mutational efficiency for different experiments is determined by counting the percentage of fluorescent green colonies. (B) Design of experiments for diagnosing and improvingmulti-site NM. A positive control was performed which included only the H66Y oligo that reverts the GFP gene back to fluoresce. A fast anneal and a slow anneal step were performed for all controls and experimental conditions. Experiments were designed to (1.) mimic the initial method, (2.) use oligos with longer regions of homology, and (3.) introduce silent mutations to match the free energy differences of the desired mutation. (C) Results of the GFP experiments. Controls with H66Y oligos show 75–94% incorporation in the absence of a competing ‘template matched’ oligo. Quick annealing resulting in a much higher level of incorporation across all experiments. Both extending oligo 5′ and 3′ homology arms and matching the free energy differences increased mutational incorporation relative to the original protocol. The values reported are averages from two distinct experiments performed on separate days.
We identified three factors which could improve the mutagenic incorporation. Because our original multi-site NM protocol uses a relatively slow annealing step, we rationalized that a much faster annealing step may introduce some kinetic control over the oligonucleotide annealing, thus improving mutational incorporation. We also tested NM using longer oligos with 30 rather than 18 nucleotides on each end, under the assumption that longer oligos may dissociate from template slower than shorter sequences. Finally, we tested whether we could minimize the free-energy difference in annealing between two oligo sequences. To do so, we used an H66 oligo where a silent mutation (G67G; GGT → GGG 7.1 kcal mol−1) is included to match the mismatch free energy of the Y66 oligo (CAT→ TAT 7.0 kcal mol−1) (Fig. 3B). All experiments were performed in replicate on separate days. Individual replicate results are provided in Supplementary Figure S1.
We first determined the mutational incorporation for the H66Y primers alone in NM reactions using (i) slow anneal and the quick anneal steps, and (ii) with original or longer (‘extended’) mutagenic oligo lengths. These experiments resulted in, depending on the experimental condition, an average of 75–95% GFP positive colonies (Fig. 3C). Next, we mimicked the original protocol under the original (‘slow anneal’) and quick anneal conditions using an equimolar mixture of the H66Y and H66 oligos. Based on the thermodynamic model we predicted 0% incorporation under original conditions. Consistent with this model we observed 0% fluorescent colonies. In contrast, addition of the quick annealing step alone led to an average of 15% observed fluorescent colonies. However, equal incorporation of either oligo would lead to 44% incorporation (50% times the 88% incorporation of the positive control oligo), suggesting considerable room for further improvement.
Extending oligo homology arms did not appreciably improve mutagenic incorporation under the slow anneal case. However, we observed 35% fluorescent colonies with the quick anneal protocol. Finally, matching of the free energy differences led to a 6% mutagenic incorporation under the slow anneal and 38% for the quick anneal (Fig. 3C). We conclude from these experiments that modifying the multi-site NM protocol by using a quick annealing step combined with either increased oligonucleotide length or using oligos with minimized differences in free energy would be sufficient to generate relatively unbiased combinatorial libraries. For ease of use, designing primers with silent mutations to match the desired free-energy differences of introducing a mutation can be cumbersome, especially when each designed oligo contains several sets of mutations. Thus, our optimized method uses quick annealing with oligos modified to have increased regions of homology from approximately 18 base pairs (bps) to 33–40 bps.
Improved libraries with updated multi-site NM
We chose to test the updated combinatorial NM protocol using longer primers and on two separate starting points: the CR6261 template and the UCA_CR6261 template. Biological replicates were performed, and the two resulting libraries were pooled to produce the complete combinatorial library. After creating the libraries with UCA_CR6261 as the parental DNA template and pooling with our existing CR6261 libraries, we achieved >99.9% (16 373/16 384; 16 382/16 384) coverage with an average 300-fold depth of coverage for both replicates (Fig. 4A and B and Table I). Library replicates contained on average 74% of specifically encoded variants with fewer than 5% wildtype parental DNA sequences (Table I). Notably, poorly encoded mutations prior to optimization were now present at a much higher abundance and the observed frequencies could be fit to a Boltzmann model with a lower temperature factor (β) than what was required for our unoptimized model (Supplementary Fig. S2). In an ideal uniform distribution of variants, every mutant including the parental DNA templates would be made in the same frequency. In practice, variants with 1 or 2 changes from CR6261 or UCA_CR6261 are oversampled at more than 10-fold higher frequencies than the other library members (Fig. 4A and B). However, these variants represent just a tiny minority of total variants (210/16 384), and this level of oversampling is tolerable for most practical deep mutational scanning workflows. For each mutated position, we ideally want to have 50% of the pooled library sequences contain the mature antibody codon and the other 50% contain the UCA codon. In our libraries we had slightly higher representation of sequences containing the mature CR6261 codons: on average the CR6261 codon was incorporated in 55.9% of sequences for replicate 1 and 53.4% of sequences for replicate 2 (Supplementary Fig. S3).
Table I.
Summary of library statistics
| CR6261 Full Library | CR6261 Full Library | CR9114 Full Library | CR9114 Full Library | |||||
|---|---|---|---|---|---|---|---|---|
| Replicate 1 | Replicate 2 | Replicate 1 | Replicate 2 | |||||
| CR6261 | UCA_CR6261 | CR6261 | UCA_CR6261 | CR9114 | UCA_CR9114 | CR9114 | UCA_CR9114 | |
| Sequencing reads after quality filtering (Fold coverage) | 4 639 427 (283) | 1 133 491 (69) | 2 750 816 (168) | 1 332 944 (81) | 1 373 400 (42) | 3 504 280 (107) | 2 044 245 (62) | 2 247 779 (69) |
| Percentage of reads: | ||||||||
| Wildtype parental DNA | 4.74% | 0.42% | 2.28% | 0.47% | 0.04% | 0.06% | 0.03% | 0.49% |
| Encoded mutations | 74.24% | 74.28% | 73.85% | 75.58% | 76.06% | 65.13% | 78.33% | 77.22% |
| Desired mutations | 55.18% | 52.28% | 53.96% | 54.86% | 40.47% | 32.88% | 39.11% | 40.50% |
| Library coverage (Number of variants made) | 95.89% (15711/16384) | 97.52% (15978/16384) | 96.72% (15846/16384) | 98.81% (16189/16384) | 97.56% (31967/32768) | 98.85% (32390/32768) | 95.20% (31194/32768) | 86.24% (28260/32768) |
| Overall library coverage | 99.93% (16 373/16 384) | 99.99% (16 382/16 384) | 99.97% (32 757/32 768) | 99.03% (32 451/32 768) | ||||
Fig. 4.
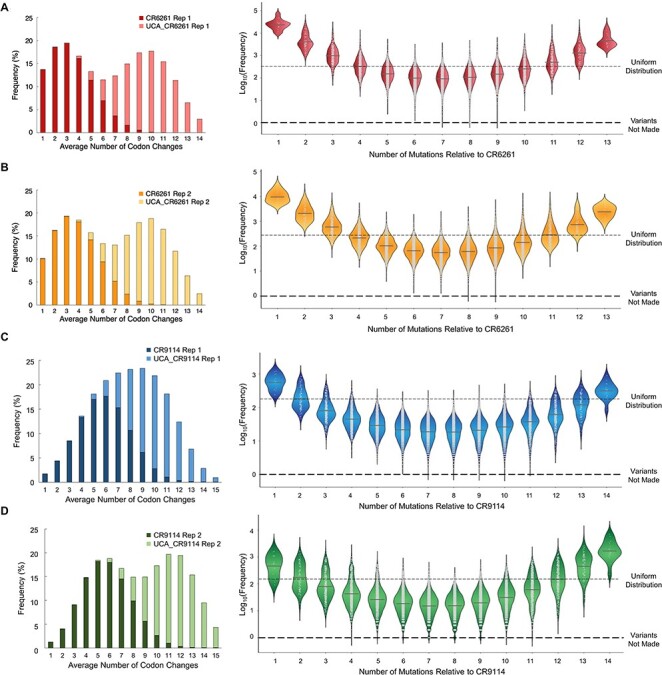
Optimized multi-site NM protocol yields near-comprehensive coverage of combinatorial libraries. Results from the optimized ‘dual directions’ strategy for generating comprehensive combinatorial libraries. (left) Histograms for frequency of the total number of variants and (right) violin plots of the frequency of each variant relative to the number of mutations from CR6261 (replicate 1 A; replicate 2 B) or CR9114 (replicate 1 C; replicate 2 D). For all panels, the violin plots were normalized where a log10 frequency of zero corresponds to a mutation not being observed by deep sequencing. The dashed gray lines indicate the relative frequency for a uniformly distributed library.
To confirm the robustness of the method, we chose to construct the complete combinatorial library of 32 768 variants (215) connecting CR9114 and UCA_CR9114 at VH positions 24–95. We used the optimized combinatorial NM protocol with both CR9114 and UCA_CR9114 as templates. In all, four degenerate oligos were used in two sequential NM rounds (Supplementary Fig. S5). 99.9% (32 753/32 768) and 99.0% (32 446/32 768) library coverage was achieved for these larger replicate libraries (Table I, Fig. 4C and D, Supplementary Fig. S4) at an average 140-fold depth of coverage for the library. The frequency distribution of library members was remarkably similar to those from the CR6261 combinatorial library (Fig. 4C and D). In these updated libraries, fewer than 1% of the library contained parental DNA sequences and on average 74% of the library members were specifically encoded by the mutagenic primer(s) (Table I). The pooled CR9114 libraries had an average of 48.9% sequences containing the CR9114 codon in replicate 1 and 45% for replicate 2 (Supplementary Fig. S3).
Under thermodynamic control, encoding contiguous mutations would be uncommon relative to non-contiguous mutations. Our complete combinatorial libraries contain both contiguous and non-contiguous mutations. To evaluate any bias in our method between contiguous and non-contiguous mutations, we compared the relative frequencies of the two groups using Mann–Whitney U tests (Supplementary Fig. S6). Overall, the libraries contained contiguous mutations and multiple non-contiguous mutations at similar frequencies. However, in some cases, there was a higher incorporation of contiguous mutations than multiple non-contiguous ones at a statistically significant threshold. Still, this effect is modest with at most an average 2-fold change in frequency between groups. We conclude that combinatorial NM can produce libraries containing both contiguous and non-contiguous mutations at similar frequencies.
Discussion
We have demonstrated near-complete coverage of the set of possible mutational combinations between mature and unmutated common ancestor antibody sequences for both the CR6261 and the CR9114 VH antibody fragments. These libraries encode a specific mutation at 14 or 15 sites, resulting in a respective library size of 16 384 or 32 768. For the interested reader, we have published a detailed step-by-step protocol (Kirby and Whitehead, 2021). There are several advantages, opportunities and limitations of this multi-site NM protocol compared with other published techniques.
The advantage of the protocol presented here, compared with competing approaches, is the fast (Supplemental Table S4) generation of relatively unbiased, near-comprehensive combinatorial libraries containing tens of thousands of variants with mutations spread across approximately 250 nucleotides of a gene. The reagent cost for creating the full CR6261 and CR9114 libraries is just over $1000 each for tens of thousands of variants, with the majority of the expense incurred for custom gene and oligonucleotide primer synthesis (Supplemental Table S5). As such, we anticipate a niche for this technique for end-uses requiring the generation of libraries containing two to four residues at ten or more non-contiguous positions. For sets of mutations encoded in near-contiguous regions cassette mutagenesis would be preferable, while combinatorial codon mutagenesis by Belsare et al. may be desirable for encoding saturation mutagenesis libraries at 4–5 distinct positions (Belsare et al., 2017).
There are also several opportunities for methodological improvement. First, we did not establish an upper limit for library size that could be encoded by this technique. We observed >99% coverage of our 32 768 member CR9114 library sequenced at approximately a 140-fold depth of coverage. Multi-site NM has a bottleneck at the level of transformation of the reaction product. In our hands, we typically get ten million transformants per reaction. Together with the depth of coverage determined by deep sequencing, this suggests a practical upper limit of 200 000 library members. While in the present work we only demonstrated at most three or four mutations per codon (CR6261: position 77; CR9114: position 74), we could potentially encode more mutations per codon. Relatedly, we did not fully take advantage of our finding that mutational incorporation could be improved by encoding silent mutations complementary to the wild-type sequence. Both of these opportunities could be addressed using clever oligonucleotide design strategies. We also encoded mutations within an approximate 250 nt window for compatibility with short-read Illumina sequencing. For larger libraries or those spanning longer genes, DNA barcoding (Blundell and Levy, 2014; Davidsson et al., 2016) with a set of unique molecular identifiers could be implemented.
There are also a couple limitations with multi-site NM. First, NM is best performed on a plasmid that is small, without significant secondary structure, and with at least one BbvCI site, or multiple sites present in the same orientation. Thus, for end-uses requiring larger plasmids an intermediate cloning step may be necessary. However, both CR6261 and CR9114 libraries were produced in the larger 6.8 kb working yeast surface display vectors. Additionally, the protocol optimized on a smaller 4.3 kb GFP plasmid showed qualitative agreement with results obtained using the larger 6.8 kb vector. Second, even with the optimization steps performed here we see library overrepresentation of WT and variants with 1–3 codon changes. Still, the distribution of variants shown here is tolerable for most deep sequencing workflows, and the library sizes prepared here are several-fold higher than demonstrated from recent comprehensive combinatorial methods (Poelwijk et al., 2019; Choi et al., 2019).
Conclusion
Here we present an updated combinatorial NM protocol that allows for the successful generation of two unique large combinatorial libraries with greater than 99% completeness for tens of thousands of programmed mutations. This facile method can be adapted by the user in order to produce full combinatorial libraries with relatively even distribution of variants. We anticipate this updated method may prove useful for several applications, including for directed evolution experiments, in mapping the complete set of evolutionary pathways in molecular evolution, and in sequence- and mutational-based prediction of protein structural assembly and function (Bolognesi et al., 2019).
Funding
This work was supported by the National Science Foundation (CBET Award number 2030221 to T.A.W.) and the National Institute of Allergy and Infectious Diseases of the National Institutes of Health (Award number R01AI141452 to T.A.W.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Data Availability
Raw sequencing reads for this work have been deposited in the SRA under accession numbers SAMN20086200 - SAMN20086210.
Author Contributions
Designed research: MBK AVM-C TAW.
Performed research: MBK AVM-C ZTB.
Wrote manuscript: MBK TAW.
Supplementary Material
Acknowledgements
We thank A. Scott at the BioFrontiers sequencing core for technical guidance, and members of the Whitehead lab for intellectual and material support, including P.J. Steiner for helpful discussions on free energy calculations.
Contributor Information
Monica B Kirby, Department of Chemical and Biological Engineering, University of Colorado, Boulder, CO 80305, USA.
Angélica V Medina-Cucurella, Department of Chemical Engineering and Materials Science, Michigan State University, East Lansing, MI 48824, USA; GigaGen Inc., South San Francisco, CA 94080, USA.
Zachary T Baumer, Department of Chemical and Biological Engineering, University of Colorado, Boulder, CO 80305, USA.
Timothy A Whitehead, Department of Chemical and Biological Engineering, University of Colorado, Boulder, CO 80305, USA.
References
- Allawi, H.T. and SantaLucia, J. (1997) Biochemistry, 36, 10581–10594. [DOI] [PubMed] [Google Scholar]
- Allawi, H.T. and SantaLucia, J. (1998a) Biochemistry, 37, 9435–9444. [DOI] [PubMed] [Google Scholar]
- Allawi, H.T. and SantaLucia, J. (1998b) Nucleic Acids Res., 26, 2694–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsare, K.D., Andorfer, M.C., Cardenas, F.S., Chael, J.R., Park, H.J. and Lewis, J.C. (2017) ACS Synth. Biol., 6, 416–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blundell, J.R. and Levy, S.F. (2014) Genomics, 104, 417–430. [DOI] [PubMed] [Google Scholar]
- Bolognesi, B., Faure, A.J., Seuma, M., Schmiedel, J.M., Tartaglia, G.G. and Lehner, B. (2019) Nat. Commun., 10, 4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, G.C.G., Zhou, P., Yuen, C.T.L., Chan, B.K.C., Feng, X., Bao, S., Chu, H.Y. et al. (2019) Nat. Methods, 16, 722–730. [DOI] [PubMed] [Google Scholar]
- Davidsson, M., Diaz-Fernandez, P., Schwich, O.D., Torroba, M., Wang, G. and Björklund, T. (2016) Sci. Rep., 6, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreyfus, C., Laursen, N.S., Kwaks, T., Zuijdgeest, D., Khayat, R., Ekiert, D.C., Lee, J.H. et al. (2012) Sci., 337, 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert, D.C., Bhabha, G., Elsliger, M.-A., Friesen, R.H.E., Jongeneelen, M., Throsby, M., Goudsmit, J. and Wilson, I.A. (2009) Science, 324, 246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo, A., Schließmann, A., Molina, R., Hermoso, J. and Bornscheuer, U.T. (2008) Protein Eng. Des. Sel., 21, 567–576. [DOI] [PubMed] [Google Scholar]
- Kirby, M.B. and Whitehead, T.A. (2021) Methods Mol. Biol. Andrew Currin, PhD and Neil Swainston, PhD (Eds.) in Directed Evolution: Methods and Protocols in press [Google Scholar]
- Klesmith, J.R. and Hackel, B.J. (2018) Bioinformatics, 35, 2707–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalsky, C.A., Klesmith, J.R., Stapleton, J.A., Kelly, V., Reichkitzer, N. and Whitehead, T.A. (2015) PLoS One, 10, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poelwijk, F.J., Socolich, M. and Ranganathan, R. (2019) Nat. Commun., 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, T.N. and Thornton, J.W. (2016) Protein Sci., 25, 1204–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner, P.J., Baumer, Z.T. and Whitehead, T.A. (2020) Bio-Protocol. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer, W.P.C. (1994) Genetics, 91, 10747–10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrenbeck, E.E., Klesmith, J.R., Stapleton, J.A., Adeniran, A., Tyo, K.E.J. and Whitehead, T.A. (2016) Nat. Methods, 13, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H., Giver, L., Shao, Z., Affholter, J.A. and Arnold, F.H. (1998) Nat. Biotechnol., 16, 258–261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing reads for this work have been deposited in the SRA under accession numbers SAMN20086200 - SAMN20086210.