

Abstract
IL-33 has emerged as a central mediator of immune, inflammatory, and fibrotic responses. Many studies have focused on mature IL-33, but elevated expression of the precursor, full-length IL-33 (FLIL33), has also been implicated in a spectrum of diseases, including tissue fibrosis. We previously reported and now confirmed that overexpression of FLIL33 induced phosphorylation of the key profibrotic signaling mediator of TGF-β, Smad3, in primary human lung fibroblasts from healthy donors and idiopathic pulmonary fibrosis patients. Presently, we demonstrate that FLIL33-induced Smad3 phosphorylation was not abrogated by anti-TGF-β antibody but was abrogated by ALK5/TGFBR1-specific and Smad3-specific inhibition, indicating that FLIL33 effect was independent of TGF-β but dependent on its receptor, TGFBR. Western blotting analyses revealed that FLIL33 overexpression increased levels, but did not affect subcellular distribution, of the AP2A1 and AP2B1 subunits of the adaptor protein complex 2 (AP2), a known TGFBR binding partner. siRNA-mediated inhibition of these subunits blocked FLIL33-induced Smad3 phosphorylation, whereas AP2 subunit overexpression induced Smad3 phosphorylation even in the absence of FLIL33. RNA-Seq transcriptomic analyses revealed that fibroblast stimulation with TGF-β induced major changes in expression levels of numerous genes, whereas overexpression of FLIL33 induced modest expression changes in a small number of genes. Furthermore, qRT-PCR tests demonstrated that despite inducing Smad3 phosphorylation, FLIL33 did not induce collagen gene transcription and even mildly attenuated TGF-β-induced levels of collagen I and III mRNAs. We conclude that FLIL33 induces Smad3 phosphorylation through a TGF-β-independent but TGF-β receptor- and AP2- dependent mechanism and has limited downstream transcriptomic consequences.
Keywords: IL-33, Smad3, AP2, Fibroblasts, Fibrosis
1. Introduction
Interleukin (IL)-33 has emerged as a central regulator of immune and non-immune, physiological and pathological, processes [1,2]. The majority of its known activities are mediated by the proteolytically matured (MIL33) form acting through the cell-surface receptor termed ST2, also known as T1 or IL-1RL1 [3,4]. Some of the functions are associated with the nuclear localization of the IL-33 precursor, full-length IL-33 (FLIL33), which acts in a cell-surface receptor-independent fashion [5-16]. Tissue and organ fibrosis, including pulmonary fibrosis, is among pathological processes that are regulated by IL-33 [11,12,17-19]. The multifaceted pathophysiology of pulmonary fibrosis—an often debilitating and in some cases deadly malady—remains incompletely understood. Again, the predominant focus of the previous studies has been on ST2-dependent effects of MIL33 [17-19], which is consistent with the known vigorous activation of type 2 immune mechanisms by MIL33 [3,4] combined with the potent profibrotic effects of type 2 immunity [20]. However, in the most severe forms, including connective tissue disease-associated and idiopathic pulmonary fibroses, the manifestations of Th2 activation are not overt; the accumulation of eosinophils, goblet cell hyperplasia with overproduction of mucus, and skewing of the cytokine milieu towards Th2 may be only minimally present, never to the extent seen in a classical type 2 immunity-mediated disease such as atopic asthma.
These considerations suggested that IL-33 may also contribute to pulmonary fibrosis in its precursor form, FLIL33. Consistent with this notion, we observed that FLIL33 is the predominantly expressed form in the lungs of patients with idiopathic pulmonary fibrosis (IPF) and scleroderma [systemic sclerosis (SSc)]-associated pulmonary fibrosis [11,12]. Overexpression of FLIL33 in the lungs in vivo induced a mild non-Th2 lymphocytic infiltration, consistent with that seen in human patients with IPF and SSc [11], and strongly potentiated the profibrotic effect of bleomycin through several mechanisms not involving type 2 cytokines [12]. Our observations suggested that IL-33 expression is elevated in pulmonary fibroblasts of patients with IPF and SSc [11,12], and analysis of publicly available data from transcriptomic studies [21-23] reveal that human lung fibroblasts indeed express IL-33 mRNA, although to a lesser extent than endothelial or epithelial cells. Considering the central role of fibroblasts in tissue fibrosis, we overexpressed FLIL33 in primary cell cultures of normal human lung fibroblasts (NHLFs) from adult donors and observed a strong stimulating effect on phosphorylation of Smad3, which is the central canonical mediator of the profibrotic effect of the most potent inducer of fibrosis, TGF-β [12]. In light of the defining role of Smad3-mediated signaling in the fibrotic process, the aim of the current study was to mechanistically and functionally follow up on the observation of IL-33 gene delivery-induced Smad3 phosphorylation.
2. Materials and methods
2.1. Cell culture
Deidentified NHLFs derived from healthy adult volunteers were purchased from Lonza (Walkersville, MD) or cultured from deidentified normal lung explants initially intended, but ultimately not used, for lung transplantation. Primary lung fibroblasts from patients with IPF were established from lung explants as previously described [11,24]. The majority of the experiments were performed in primary fibroblast cultures from at least two different donors. Overall, NHLFs from eight different donors were used throughout the course of this study. Specific numbers of different NHLF cultures involved in each experiment and the numbers of experimental repeats in each case are indicated in the Results section. The cultures were maintained in T75 culture flasks (NEST Biotechnology, Rahway, NJ) in a humidified atmosphere of 5% CO2 at a temperature of 37° C in DMEM supplemented with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate, 10% calf bovine serum (CBS), minimal essential medium nonessential amino acids, and antibiotic–antimycotic mixture (all from Thermo Fisher Scientific, Waltham, MA) to a final concentration of 100 units/ml of penicillin, 100 μg/ml of streptomycin, and 0.25 μg/ml of amphotericin B. For experimental testing, the cells were harvested by trypsinization at passages 3–6, washed, counted, and seeded on 6-well culture plates (NEST Biotechnology) at a density of 5 × 105 cells/well. Fibroblasts were then pre-cultured overnight in 0.5% dialyzed fetal bovine serum (FBS)-containing medium and then tested at this low serum concentration. NIH3T3, HEK293T, A549, and MRC-5 cell lines were purchased from the American Type Culture Collection (Manassas, VA) and cultured per the supplier’s recommendations, as were primary adult human small airway epithelial cells (SAEC), which were purchased from Lonza. qRT-PCR analyses for collagen chains were performed using pre-validated RT2 primers from SABiosciences Qiagen as previously described [11,12,25,26].
2.2. Gene delivery in cell culture
Gene delivery was achieved using electroporation of cells with recombinant plasmids or siRNA constructs utilizing the Amaxa Nucleofector (Lonza) with Primary Fibroblast kit (catalog no. VPI-1002). All recombinant plasmids on the VQAd5CMVK-NpA backbone (ViraQuest, North Liberty, IA) encoding C-terminally HA-tagged human FLIL33 and other proteins, as well as the non-coding (NULL) plasmid vehicle control, were previously generated, purified, validated, and used for gene delivery in cell culture exactly as described [11,12,25-27]. Additionally, a human FLIL33 construct tagged on the C-terminus with monomeric Neon Green [28] fluorescent protein (FLIL33mNG) was similarly cloned and used. In each reaction, 5 × 105−1 × 106 cells and 0.5–2.0 μg of plasmid were used, based on preliminary experiments, to optimize expression of each delivered recombinant protein. Overexpression of the plasmid-encoded proteins of interest was confirmed by western blotting and/or by ELISA as previously reported [11,12,25-27]. AP2A1 and AP2B1 RNA interference was performed using FlexiTube siRNA (Qiagen, Frederick, MD). The cells were transfected with siRNA or AllStars negative control siRNA (Qiagen) at a concentration of 300 nM using electroporation as described above and incubated for 48 h in a 75-cm2 flask. A second transfection was performed with the recombinant plasmid, and the cells were incubated an additional 48 h in 6-well plates. AP2A1 and AP2B1 knockdown was confirmed by western blotting.
2.3. ELISA
The concentrations of IL-33 protein in cell lysates were tested in ELISAs (R&D Systems, Minneapolis, MN) 48 h post transfection. Subsequent tests were performed, and data reported only if ELISA-measured levels of IL-33 in all experimental cell lysates within a sample set exceeded 200 pg/ml; alternatively, the entire experimental sample set was discarded. In all cases, ELISA tests were also used to confirm that the levels of IL-33 of cell lysates of NULL-transfected cultures did not exceed 20 pg/ml as previously reported [25,26].
2.4. Fluorescent microscopy
For fluorescent microscopy, NHLFs were transfected with the plasmid encoding FLIL33mNG (green fluorescence) and, 48 h after transfection, fixed and stained immunohistochemically for AP2B1 (red fluorescence). Nuclei were stained with DAPI (blue fluorescence). Fluorescent images were accumulated using a Keyence (Itasca, IL) BZ-X700 fluorescent microscope.
2.5. Western blotting
Western blotting assays were performed using the Novex (Thermo Fisher) system with Tris-glycine gels. In some experiments, separation of cytoplasmic and cell membrane fractions was performed using the Mem-PER Plus membrane protein extraction kit (Thermo Fisher, catalog no. 89842) and the fractions were analyzed by western blotting. Wet transfers to PVDF membranes (Bio-Rad laboratories, Hercules, CA) were performed using XCell II (Thermo Fisher). The membranes were blocked and incubated with antibodies from Cell Signaling Technology (Danvers, MA) against GAPDH (catalog no. 5174), phospho-Smad2/3 (catalog no. 8828), phospho-Smad2 (catalog no. 3108), total Smad2/3 (catalog no. 8685), phospho-ERK1/2 (catalog no. 4370), total ERK1/2 (catalog no. 4695), and α-tubulin (catalog no. 2144). Antibodies from Abcam (Cambridge, UK) against HA (catalog no. ab18181), AP2A1 (catalog no. ab170955), AP2B1 (catalog no. ab129169) were also used. Antibodies from Santa Cruz Biotechnology (Dallas, TX) were against Smad4 (catalog no. sc7966) and Smad7 (catalog no. sc365846). The antibody against pan-cadherin was from Thermo Fisher (catalog no. 71-7100). Secondary antibodies used were goat anti-rabbit from EMD Millipore (Billerica, MA) (catalog no. 12–348) and goat anti-mouse from Santa Cruz Biotechnology (catalog no. sc-2005). Membranes were developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) and the Azure c500 imaging system (Azure Biosystems, Dublin, CA 94568). Stripping was performed using ReBlot Plus strong antibody stripping solution (EMD Millipore).
2.6. TGF-β inhibition
Anti-pan-TGF-β (clone 1D11) blocking antibody from R&D Systems was used as previously described [29-32]. To assess the effect of TGF-β blockade on FLIL-33 overexpression-induced changes, NHLFs were transfected with FLIL33-encoding or NULL plasmids as described above. Cells were allowed to recover for 24 h in 10% CBS-containing cell culture medium. The medium was then replaced with fresh 0.5% FBS-containing DMEM described above for 2 h, followed by another replacement of such fresh medium with added 10 μg/ml of anti-TGF-β antibody. After an additional 24 h of culture, cells were analyzed for Smad3 phosphorylation. To confirm the functional activity of this blocking antibody, NHLF were pre-incubated with 10 μg/ml of the antibody for 1 h. Then, cells were stimulated with 1 ng/ml of rhTGF-β for 45 min and analyzed for Smad3 phosphorylation. To attenuate downstream signaling from the TGF-β receptor (TGFBR), pharmacological inhibition was utilized. A small-molecule inhibitor of ALK5, SB431542 (Sigma-Aldrich, St. Louis, MO), was used as previously described [29], at 5 μM concentration, with pre-treatment times similar to those used for anti-TGF-β. A Smad3-specific inhibitor, SIS3, was purchased from Selleck Chemicals (Houston, TX), and used similarly.
2.7. Plasminogen activator inhibitor-1/luciferase assay for active TGF-β
Mink lung epithelial cells stably transfected with an expression construct containing a truncated plasminogen activator inhibitor-1 promoter fused to the firefly luciferase reporter gene (PAIL cells) were a kind gift of Dr. Daniel B. Rifkin, New York University School of Medicine. The cells were maintained and used as previously described [33]. Stimulation with recombinant human (rh) transforming growth factor (TGF)-β (R&D Systems) was used as a positive control, revealing a dose-dependent increase in luminescence in the concentration range of 0.05–1.00 ng/ml. PAIL cells were used to measure active TGF-β in NHLF supernatants by transferring serial dilutions of NHLF conditioned medium. Separately, PAIL cells were co-cultured with NHLF in 1:1 ratio at 80% overall cell confluence to detect NHLF-produced active TGF-β in the immediate cell vicinity. Luminescence was measured using a luciferase assay system (Promega, Madison, WI), according to the manufacturer’s recommendations.
2.8. RNA-Seq analysis
Fibroblasts were transfected by electroporation with the FLIL33-encoding (OriGene) or the matching non-coding control plasmid vehicle (NULL) from the same supplier, and cells were allowed to recover for 24 h. Then, the cultures were or were not stimulated with rhTGF-β (R&D Systems) for an additional 24 h or 48 h. RNA extraction and sequencing, as well as data analyses were performed exactly as previously described [34]. Briefly, total RNA was isolated using TRIzol reagent (Invitrogen Thermo Fisher) and shipped on dry ice to Otogenetics (Atlanta, GA) for the integrity and purity assessment, cDNA library preparation, and short-read sequencing generating 100–125-bp pairedend libraries with an average of 40 million paired reads per sample. The resulting raw RNA-Seq reads (fastq files) were subjected to bioinformatics analyses in-house as described [34]. Briefly, transcript abundance was estimated as counts per million (cpm) values using the htseqcount script of the open source Python package HTSeq 0.6.1p2, and the data deposited in the Gene Expression Omnibus database (accession number GSE130348; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE130348). Pairwise comparisons between the differentially treated fibroblast cultures were performed using the DESeq2 package of Bioconductor.
2.9. Statistical analysis
Experimental data, other than RNA-Seq, were expressed utilizing parametric (mean ± standard deviation) or non-parametric (median, interquartile range) descriptors. Pairwise comparisons of groups were performed either parametrically, utilizing Student’s t-test, or non-parametrically, using Mann-Whitney U test. Multiple groups were analyzed using one-way ANOVA or Kruskal-Wallis test, as indicated for specific results.
3. Results
3.1. Overexpression of FLIL33 in primary fibroblasts induces Smad3 phosphorylation in a TGF-β ligand-independent, TGF-β receptor-dependent fashion
We have previously reported that FLIL33 overexpression in normal human lung fibroblast (NHLF) primary cultures strongly induced phosphorylation of Smad3 [12]. Three cultures from separate healthy adults were initially tested, all showing such response [12]. This FLIL33-induced Smad3 phosphorylation has been additionally observed in five more NHLF cultures, each derived from a separate healthy donor (Suppl. Fig. 1, Fig. 1A). Of note, western blotting for total Smad2/3 indicated the predominant expression of Smad3 (lower band) compared with Smad2 (upper band) in lung fibroblasts, and phosphorylation was observed predominantly for Smad3 and to a substantially lesser degree for Smad2 (Suppl. Fig. 1, Fig. 1A-D). Additional experiments with anti-phospho-Smad2-specific antibody reveal that, indeed, phosphorylation of Smad2 in response to FLIL33 overexpression was minimal and inconsistent (Suppl. Fig. 2). The effect on Smad3 phosphorylation remained consistent at 24, 48, and, to a lesser extent, 72 h after FLIL33 gene delivery (Fig. 1B). Overexpression of the precursor, i.e., FLIL33, induced Smad3 phosphorylation, whereas overexpression of its N-terminal or C-terminal (MIL-33) fragments, or of control proteins, full-length or mature IL-37, did not (Fig 1C). This effect was observed in primary human fibroblasts but not in the immortalized mouse embryonic fibroblast cell line (NIH3T3), the transformed human embryonic kidney cell line (HEK293), the human pulmonary adenocarcinoma epithelial cell line (A549), or primary human small airway epithelial cells (Fig. 1D). Primary pulmonary fibroblasts from patients with IPF demonstrated responses similar to those of NHLF, whereas the responsiveness to FLIL33 overexpression was minimal in an embryonic human lung fibroblast cell line MRC-5 (Fig. 1D). It appears that this phenomenon is restricted to the effect of FLIL33 on Smad3 phosphorylation in primary fibroblasts, whether derived from healthy control individuals or patients with IPF. The non-canonical TGF-β signaling through ERK1/2 was not induced by FLIL33 overexpression in primary fibroblasts (Fig. 1E). Overexpression of FLIL33 had a limited impact on the protein levels of the co-Smad, Smad4, and the inhibitory Smad, Smad7 (Fig. 1F). Considering the central role of Smad3 phosphorylation in TGF-β-induced intracellular signaling, it was somewhat unexpected to find no increase in TGF-β mRNA or protein levels in FLIL33-overexpressing cells in any of the tested NHLF cultures. Furthermore, cocultures of FLIL33-overexpressing NHLFs with PAIL cells [a kind gift from Dr. Daniel B. Rifkin, New York University School of Medicine, [33]], which are highly sensitive to active TGF-β, showed no increase in TGF-β activation. Moreover, isolation of cell-membrane fractions of FLIL33-overexpressing and control NHLFs with subsequent western blotting for TGF-β showed no increase in the membrane-bound form of the cytokine. Consistent with the lack of increase in TGF-β, blocking this cytokine with a specific neutralizing antibody (1D11, R&D Systems, catalog no. MAB1835) did not attenuate Smad3 phosphorylation (Fig. 2A, three separate experiments were performed with similar results). Although the Smad3 phosphorylation-inducing effect of FLIL33 overexpression did not appear to depend on autocrine TGF-β (Fig 2A), pharmacological inhibition of ALK5 (TGF-β receptor kinase) with SB431542 completely blocked this effect of FLIL33 on Smad3 in three independent experiments, one of which is shown in Fig. 2B. Similarly, Smad3-specific inhibition with SIS3 attenuated Smad3 phosphorylation as shown in Fig. 2C. Thus, elevated FLIL33 expression selectively stimulates Smad3 phosphorylation in a TGFBR-dependent yet, somewhat surprisingly, TGF-β-independent fashion. Similar regulation was reported in response to other stimuli, although the mechanisms of intrinsic, cognitive ligand-independent activity of TGFBR need to be better understood [35-38].
Fig. 1.
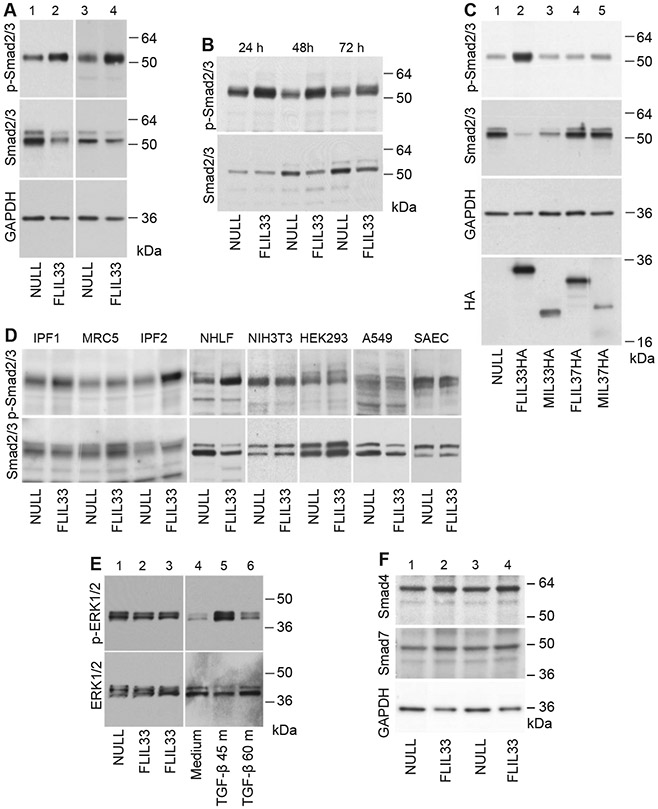
Smad3 phosphorylation is induced by FLIL33 overexpression in primary fibroblasts. A. NHLFs from two different healthy donors (lanes 1, 2 and 3, 4) were electroporated with FLIL33-encoding (lanes 2, 4) or a control NULL (lanes 1, 3) plasmids, as indicated. After 48 h, cells were lysed, and western blotting analyses were performed with anti-phospho-Smad2/3 antibody. The membranes were then stripped and re-developed for total Smad2/3 and GAPDH as indicated. Note the pronounced Smad3 phosphorylation induced by FLIL33 overexpression. The FLIL33 overexpression-induced increases in Smad3 phosphorylation were statistically significant (Suppl. Fig. 1). Such stimulation of Smad3 phosphorylation by FLIL33 overexpression was consistent in all NHLFs tested on at least two, and in some cases more than ten, independent occasions in each of the three previously described [12] and additional five (Suppl. Fig. 1) studied primary cultures. B. FLIL33 overexpression activates Smad3 phosphorylation in a time-dependent fashion. NULL-transfected and FLIL33-overexpressing NHLFs were analyzed at 24 h, 48 h, and 72 h, as indicated. C. Only FLIL33 overexpression but not similar overexpression of MIL33 (aa 112–270), FLIL37, or MIL37 (aa 46–218) induces Smad3 phosphorylation. Western blots were developed for phospho-Smad2/3; the membrane was serially stripped and re-developed with the subsequent antibodies in the order shown for total Smad2/3, GAPDH, and HA (the C-terminal tag in all constructs), as indicated. D. The effect of FLIL33 overexpression on Smad2/3 phosphorylation is pronounced in primary NHLF and fibroblast from patients with IPF, but not observed in NIH3T3, HEK293, A549 cells, or SAEC. E. Phosphorylation of ERK1/2 is not affected by FLIL33 overexpression but is induced by TGF-β. F. FLIL33 overexpression has a limited impact on the levels of Smad4 and Smad7. NHLFs from two different healthy donors (lanes 1, 2 and 3, 4) were electroporated with FLIL33-encoding (lanes 2, 4) or a control NULL (lanes 1, 3) plasmids, as indicated. After 48 h, cells were lysed, and western blotting analyses were performed with anti-Smad4 antibody. The membranes were then stripped and re-developed for total Smad7 and GAPDH as indicated.
Fig. 2.
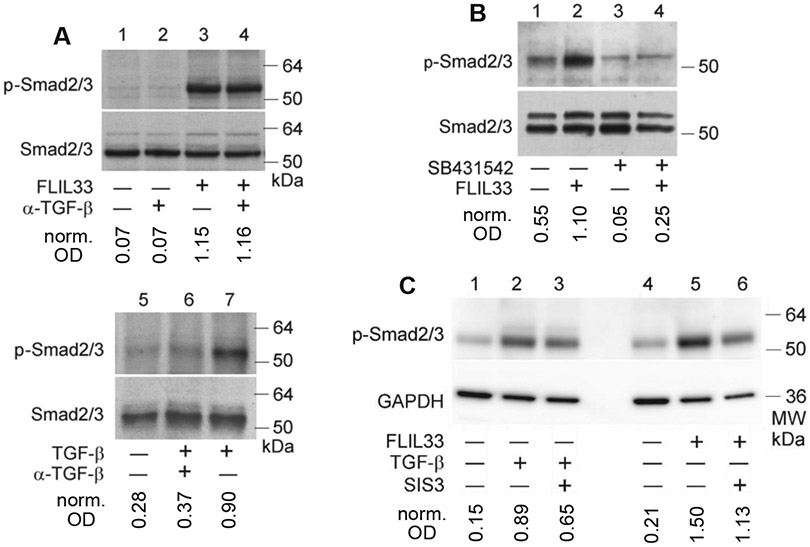
Neutralization of TGF-β with a blocking Ab does not attenuate the FLIL33 effect on Smad3 phosphorylation (A), whereas inhibition of ALK5 with SB431542 (B) or of Smad3 with SIS3 (C) does. rhTGF-β was used at 1 ng/ml, anti-TGF-β Ab at 10 μg/ml, and SB431542 and SIS3 at 5 μM concentration each. Optical densities of the Smad3 bands in each lane were normalized to the corresponding loading controls.
3.2. Subunits of adaptor protein complex 2 (AP2) mediate FLIL33-driven Smad3 phosphorylation
Considering that FLIL33 is nearly, if not exclusively, an intranuclear molecule, its Smad3-activating effect in a cell surface-expressed TGFBR-dependent yet apparently TGF-β-independent fashion needs to be mechanistically explained. Pilot liquid chromatography–mass spectrometry (LC-MS/MS) analyses of FLIL33-overexpressing and NULL-transfected fibroblast lysates were performed as previously described [26] and suggested that the levels of AP2 protein subunits α1 (AP2A1) and β1 (AP2B1) were elevated in FLIL33-overexpressing cells. AP2 is critical for clathrin-mediated endocytosis [39], which is an important aspect of TGFBR biology [40-42]. Further, the AP2B1 subunit can bind directly to TGFBR [43]. Subsequent western blotting analyses confirmed increases in AP2A1 and AP2B1 protein levels in FLIL33-overexpressing compared with control cells; such increases were readily attenuated with specific small interfering (si) RNAs (Suppl. Fig. 3, Fig. 3A). Such inhibition of either AP2A1 or AP2B1 abrogated the stimulating effect of FLIL33 overexpression on Smad3 phosphorylation (Fig. 3B). Furthermore, overexpression of AP2B1 by electroporation with encoding plasmid was sufficient to stimulate Smad3 phosphorylation in the absence of elevated FLIL33 (Fig. 3C). Fluorescence microscopy analyses were performed of NHLF overexpressing FLIL33 that was tagged on the C-terminus with monomeric Neon Green fluorescent protein (FLIL33mNG). AP2B1 was simultaneously immunostained to fluoresce in the red channel. In all cases, FLIL33 was exclusively nuclear, whereas AP2B1 was present in clusters on the cell surface and in the cytoplasm, based on immunocytofluorescence and fluorescent optical sectioning (Fig 3D). Of note, the FLIL33-overexpressing cells appeared to have subcellular AP2 distribution similar to that observed in non-transfected cells. In the bottom row of Fig. 3D, three cells are shown (based on DAPI staining of the nuclei). The cell on the left expresses high levels of FLIL33 (based on the intensity of green florescence of the mNG tag), the upper cell on the right expresses low levels of FLIL33, and the lower cell on the right does not appear to express IL-33 at all. Despite such diverse levels of IL-33 expression, the nature of subcellular distribution of AP2 appears similar across these three cells. To further assess the possibility that FLIL33 overexpression may induce AP2 internalization, cell membrane and cytoplasmic fractions of FLIL33-overexpressing and NULL-transfected NHLFs were separated, the quality of separation confirmed with western blotting for α-tubulin (cytoplasmic marker) and pan-cadherin (cell-surface membrane marker), and the levels of AP2B1 assessed by western blotting. Although the levels of AP2B1 were higher in FLIL33-overexpressing cells, the relative distribution of AP2B1 between the cell membrane and the cytoplasm remained similar to that observed in control cells (Fig. 3E). These findings are consistent with the known involvement of the AP2 complex in potentiating TGFBR signaling [40-42] including through direct binding of AP2B1 to TGFBR [43]. Thus, FLIL33-driven elevation in AP2 subunits is necessary and, in combination with intrinsic activity of ALK5, sufficient to mediate the Smad3 phosphorylation-inducing effect of FLIL33 without inducing AP2 internalization.
Fig. 3.

Subunits of the AP2 adaptor complex mediate the effect of elevated FLIL33 on Smad3 phosphorylation. A. Western blotting of NHLFs reveals that overexpression of FLIL33 elevates the levels of AP2A1 and AP2B1 subunits, and this effect is readily abrogated by specific indicated siRNAs but not scrambled siRNA. Repeated in three additional NHLF cultures from separate donors with similar results. B. AP2A1 or AP2B1 siRNAs, but not scrambled control siRNA, abrogate FLIL33 overexpression-induced Smad3 phosphorylation. Repeated in a separate NHLF culture from a different donor with similar results. C. Plasmid-mediated overexpression of AP2B1 is sufficient to induce Smad3 phosphorylation in the absence of FLIL33 overexpression. D. Fluorescence (top row) and optical sectioning (bottom row) microscopy of NHLFs overexpressing FLIL33mNG (green fluorescence) and immunostained for AP2B1 (red fluorescence); nuclei were stained with DAPI (blue fluorescence). In the bottom panel, the two shown subsections were 2 μm apart, showing that AP2B1 is not localized in the nucleus but is abundant on the cell surface and in the cytoplasm. Repeated in two additional NHLF cultures from separate donors, with similar results. E. Western blotting of cytoplasmic (C) and membrane (M) fractions of NHLFs transfected with the control NULL plasmid or FLIL33-encoding plasmid, as indicated. The indicated levels of IL-33 were measured by ELISA in total cell lysates.
3.3. FLIL33 overexpression and stimulation with TGF-β differentially regulate the fibroblast transcriptome
Considering that ALK5-dependent Smad3 phosphorylation is central to the canonical TGF-β signaling, we assessed the effects of FLIL33 overexpression, stimulation with rhTGF-β, and the combination of this stimuli on the transcriptome of a NHLF culture utilizing the RNA-Seq approach. Cells were electroporated with FLIL33-encoding or non-coding control (NULL) plasmid and, 24 h later, either stimulated or not with 5 ng/ml of TGF-β for an additional 24 or 48 h. Thus, a total of eight cultures were tested, including four cultures at 24 h and four cultures at 48 h of stimulation with TGF-β. At each of these time points, the cultures included: NULL-transfected with no TGF-β stimulation, FLIL33-overexpressing with no TGF-β stimulation, NULL-transfected and stimulated with TGF-β, and FLIL33-overexpressing and stimulated with TGF-β. In Fig. 4, the top row of scatterplots (Fig. 4A-D) shows pairwise comparisons between such samples at 24 h, whereas the bottom row (Fig. 4E-H) shows similar comparisons at 48 h of stimulation with TGF-β. These analyses revealed that the transcriptomic changes were more pronounced at 48 h than at 24 h, both in the number of the affected genes as well as the magnitude of the changes. Importantly, FLIL33-induced changes, either in unstimulated (Fig. 4A, E) or TGF-β-stimulated (Fig. 4D, H) cells were relatively modest, both in the number of the affected mRNAs and in the magnitude of their expression level changes. By contrast, stimulation with TGF-β induced profound transcriptomic changes in both NULL-transfected (Fig. 4B, F) and FLIL33-overexpressing (Fig. 4C, G) cells. These findings indicated that despite their shared capacity for inducing Smad3 phosphorylation, the downstream transcriptional effects of FLIL33 overexpression were different from those of stimulation with TGF-β, possibly because FLIL33 selectively affected phosphorylation of Smad3 but not activation of other, including non-canonical, signaling mediators in the TGF-β pathway. To assess the validity of this notion more specifically and in relation to a pathophysiologically important question of regulation of fibrosis, we assessed the changes in the expression of collagen genes in this dataset (Fig. 4I, J). Stimulation with TGF-β, a potent inducer of Smad3 phosphorylation, led to a significant elevation in the steady-state levels of multiple collagen mRNAs. By contrast, despite the induction of overt Smad3 phosphorylation, FLIL33 overexpression moderately decreased (Fig. 4I), or tended to decrease (Fig. 4J), the levels of collagen mRNAs, either basal or TGF-β-induced, including those for the structural type I and type III collagens.
Fig. 4.

RNA-Seq transcriptomic profiling of NHLFs overexpressing FLIL33 vs transfected with a control non-coding vehicle plasmid (NULL). Additionally, 24 h later these cells were or were not activated with rhTGF-β, as indicated, for an additional 24 h (A–D, I) or 48 h (E–H, J). In panels A–H, log base 2 normalized counts are plotted for the indicated pair-wise comparisons at 24 h (A–D) and 48 h (E–H) of stimulation with rhTGF-β. The colorized dots represent genes with substantially different gene expression levels. The genes with 5-fold higher (red), 2- to 5-fold higher (green), 2- to 5-fold lower (blue), and 5-fold lower (magenta) expression levels are shown, with the similarly colored numbers representing the counts of the corresponding genes. In panels I and J (24 h and 48 h of stimulation with rhTGF-β, respectively), normalized counts for each of the indicated collagen genes are plotted relative to the corresponding count in NULL-transfected cells that were not stimulated with TGF-β. The lines connecting the data points designate the indicated collagen genes. The p-values were calculated using two-tailed paired t-test.
Additional RNA-Seq data analyses focused on the genes expression of which was consistently affected by FLIL33 gene delivery (Suppl. Tables 1, 2). Pairwise comparisons of FLIL33-overexpressing vs NULL-transfected NHLFs were performed at 24 h and 48 h, with and without stimulation with TGF-β at each of these timepoints. A number of genes were identified that similarly changed in response to FLIL33 overexpression in at least two of the four tested conditions (Suppl. Tables 1, 2). Several of such genes, including CXCL10/IP-10, SIRT6, ENG, MMP14/MT1-MMP, PLXNB2, CCN4/WISP1, and other genes shown in Suppl. Fig. 4 are known for their involvement in the regulation of connective tissue homeostasis and fibrosis. However, the overall connective tissue-related regulation by FLIL33 was different from and in some cases opposite to that induced by fibroblast stimulation with TGF-β, including effects on the expression mRNAs for α-smooth muscle actin/ACTA2, CCN1/CYR61, CCN2/CTGF, CCN3/NOV, FN1, ELN1, FBN1, numerous MMPs, and various players in the TGF-β signaling pathway (Suppl. Fig. 4).
3.4. Overexpression of FLIL33 attenuates TGF-β-driven transcriptional upregulation of collagen expression
The data outlined above indicate that activation with TGF-β and overexpression of FLIL33 each induce phosphorylation of a potent profibrotic transcriptional regulator, Smad3. In both cases, such induction is TGFBR-dependent, with FLIL33-driven Smad3-phosphorylation being entirely AP2-dependent. The transcriptomic data suggest that despite similarly inducing Smad3 phosphorylation, the functional differences between stimulation with TGF-β vs increased FLIL33 are notable, especially at the level of collagen mRNA transcripts. To assess this notion more rigorously, primary fibroblasts cultures from multiple donors were either activated with rhTGF-β, electroporated with FLIL33-encoding or control plasmid, or subjected to the combination of these stimuli similar to cell treatments in the transcriptomic tests described above. In all cases, stimulation with TGF-β induced mRNA levels of structural collagen genes whereas overexpression of FLIL33 mildly suppressed or tended to suppress the levels of these mRNAs in both control and TGF-β-stimulated cells (Fig. 5, Suppl. Fig. 5). These observations indicate that the typically profibrotic, TGF-β-induced, Smad3 phosphorylation is not profibrotic, but instead mildly antifibrotic at the transcriptional level when induced by elevated FLIL33. Further supporting the notion of discordant regulation of Smad3 phosphorylation and transcriptional regulation by elevated FLIL33, ALK5 inhibition with SB431542 or siRNA silencing of the AP2 complex notably attenuated Smad3 phosphorylation (Fig. 2B, Fig. 3B) but had limited effect on collagen gene transcription (Suppl. Fig. 6).
Fig. 5.
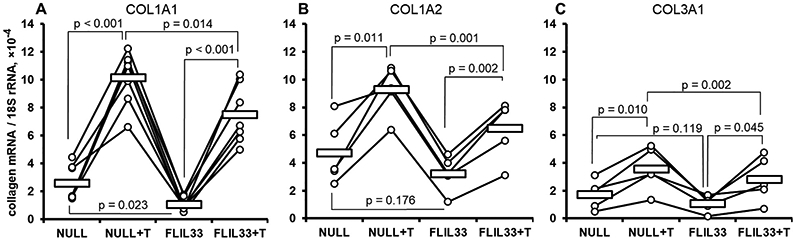
qRT-PCR analyses of NHLFs for steady-state levels of mRNAs for COL1A1 (A, n = 7 separate cultures from different donors), COL1A2 (B, n = 5), and COL3A1 (C, n = 5), all normalized to 18S rRNA that was used as the reference PCR target. Cells were electroporated with the FLIL33-encoding or control non-coding vehicle (NULL) plasmids and, 24 h later, either activated or not with TGF-β (T), as indicated, for an additional 24 h. The mean values in each group are shown with horizontal bars. The lines connecting the data points designate primary cell cultures derived from separate donors. The indicated p-values were calculated using two-tailed paired t-test. One-way ANOVA test revealed p < 0.001 for COL1A1 (A), p < 0.001 for COL1A2 (B), and p = 0.032 for COL3A1 (C). More detailed statistical analyses are presented in Suppl. Fig. 3.
4. Discussion
In this work we followed up, on the cellular level in primary adult lung fibroblasts, on the previous observations that the levels of full-length IL-33 (FLIL33) are elevated in the lungs of patients with pulmonary fibrosis [11,12], that gene delivery of FLIL33 to mouse lungs in vivo elicits mild non-Th2 pulmonary inflammation [11], that FLIL33 activates Smad3 phosphorylation in NHLFs in culture, and that it synergizes with bleomycin injury in inducing overt lung inflammation and, notably, fibrosis [12]. Considering the biomedical importance of pulmonary fibrosis and the central role of TGF-β-driven Smad3 phosphorylation in profibrotic signaling, further experiments were performed in primary fibroblast cultures with the goal of uncovering functional effects of FLIL33-induced Smad3 phosphorylation.
We first confirmed that intracellular FLIL33-induced Smad3 phosphorylation consistently occurs in NHLFs (Suppl. Fig. 1, Fig. 1A) in a time-dependent fashion (Fig. 1B), and that MIL33, FLIL37, or MIL37 do not elicit a similar effect (Fig. 1C). Such regulation was specific to primary lung fibroblasts and was not observed in cell lines or primary airway epithelial cells (Fig. 1D). FLIL33 did not induce non-canonical TGF-β signaling through ERK1/2 (Fig. 1E) and did not have a notable effect on the levels of Smad4 or Smad7 (Fig. 1F). Such FLIL33-induced canonical TGF-β signaling through Smad3 phosphorylation was, somewhat surprisingly, independent of TGF-β itself (Fig. 2A) but dependent on TGFBR (ALK5, Fig. 2B). In primary human lung fibroblasts, Smad2 did not appear to be involved in such regulation (Suppl. Fig. 2, Fig. 2C). Others have also reported TGF-β-independent, TGFBR-dependent Smad3 phosphorylation, that regulates cellular responses to stimuli, although the mechanistic details of such intrinsic signaling by TGFBR that is not driven by the interaction with the cognate ligand remain elusive [35-38]. Such signaling is unlikely to be driven by a direct interaction between TGFBR and FLIL33, because the former is localized on the cell surface whereas the latter is nearly exclusively intranuclear.
The literature suggests two possible groups of mechanisms that might explain the effect of FLIL33 on TGFBR-controlled Smad3 phosphorylation independently of TGF-β. Several reports described ligand-independent TGFBR transactivation, but did so without identifying specific molecular intermediaries that could transactivate the TGFBR serine/threonine kinase domain [36,44-47]. A better supported, potentially relevant mechanism is ligand-independent, clathrin-mediated TGFBR internalization and recycling [40-42]. This process does not by itself induce TGFBR signaling, but may potentiate it (unlike caveola-mediated TGFBR endocytosis, which is inhibitory) [40-42]. Clathrin-mediated endocytosis takes place through the formation of clathrin-coated pits, of which AP2—a heterotetramer of AP2A1 (α adaptin), AP2B1 (β2 adaptin), AP2M1 (μ2 adaptin), and AP2S1 (σ2 adaptin)—is a critical functional component [39]. In addition to centrally contributing to clathrin-mediated endocytosis, AP2B1 can also bind directly to TGFBR [43]. We serendipitously found, and then systematically confirmed, that overexpression of FLIL33, in addition to causing Smad3 phosphorylation, also induced elevations in AP2A1 and AP2B1, an effect that could be abrogated by siRNA-mediated inhibition (Suppl. Fig. 3, Fig. 3A). The previous studies of AP2 involvement in TGF-β signaling were performed in a generalized, disease-unrelated fashion; centered around Smad2 activation with limited attention to Smad3; focused on the fetal mink epithelial cell line Mv1Lu but not on primary human mesenchymal cells; and have not suggested that an unrelated molecule, such as FLIL33, may highjack this process [40-42]. Our work is directly relevant to human health because FLIL33 induces Smad3 phosphorylation only in adult primary fibroblasts (Fig. 1D), in which FLIL33 overexpression led to a pronounced elevation in the levels of AP2A1 and AP2B1, whereas siRNA-mediated inhibition of these AP2 subunits inhibited FLIL33-induced Smad3 phosphorylation (Fig. 3B). Furthermore, overexpression of AP2B1 in NHLFs was by itself sufficient to induce Smad3 phosphorylation even in the absence of elevated FLIL33 (Fig. 3C). The mediation of Smad3 phosphorylation by elevated FLIL33, while dependent on the increase in the levels of AP2A1 and AP2B1, did not involve a change in the subcellular distribution of AP2 between the cell membrane and the cytoplasm (Fig 3D, E).
Thus, Smad3 phosphorylation is induced in the TGFBR-dependent fashion by either activation of cells with TGF-β or by elevation in FLIL33. However, unlike TGF-β that activates TGFBR, FLIL33 relies on intrinsic, cognate ligand-independent, TGFBR activity. Unlike TGF-β, FLIL33 does not activate the non-canonical signaling through ERK1/2. Furthermore, unlike the modifying effect of AP2 on TGF-β-induced Smad-mediated signaling [40-42], the FLIL33-driven Smad3 phosphorylation depends entirely on the elevation in the expression levels of AP2A1 and AP2B1, which are not internalized. How elevated FLIL33 induces the elevation in these AP2 subunits remains unknown, but a contribution from transcriptional regulation is possible (Suppl. Fig. 4). Since phosphorylation of Smad3 is central to the transcriptional regulation control by TGF-β, we tested whether FLIL33 induces transcriptomic changes similar to those induced by TGF-β. Notable differences in the transcriptomic changes induced by these two factors were observed (Fig. 4A-H, Suppl. Tables 1 and 2, Suppl. Fig. 4), and the effects of FLIL33 on the levels of collagen gene transcripts were mild, but opposite to those of TGF-β (Fig. 4I, J).
We therefore observed, for the first time, a functional discordance between, on the one hand, pronounced Smad3 phosphorylation and, on the other hand, elevation in collagen gene mRNA levels. In contrast to the critical role of Smad3 activation in the TGF-β-induced elevation of collagen mRNA, FLIL33 induces Smad3 activation without elevating collagen mRNA levels. Supporting this notion, pharmacological inhibition in FLIL33-overexpression NHLFs notably attenuated Smad3 phosphorylation (Fig. 2B, C), but the suppressive effect of such inhibition on collagen mRNA levels was modest (Suppl. Fig. 6A, B). Furthermore, silencing of the AP2 subunits attenuated FLIL33-induced Smad3 phosphorylation (Fig. 3B), and overexpression of AP2B1 induced it (Fig. 3C). By contrast, silencing of the AP2 subunits only mildly attenuated collagen mRNA levels (Suppl. Fig 6C, D). It can be argued that Smad3 phosphorylation does not by itself define transcriptional regulation. It is likely that differences in the regulation of the non-canonical TGF-β signaling as well as in AP2 involvement contribute to the observed differences in the transcriptional effects of TGF-β and FLIL33. In relevance to pulmonary fibrosis, qRT-PCR experiments in multiple primary NHLF cultures suggested that Smad3 phosphorylation is associated with a mild antifibrotic regulation by FLIL33 at the transcriptional level (Fig. 5, Suppl. Fig. 5, Suppl. Fig. 6).
The observed transcriptomic effects suggest that the profibrotic synergism of FLIL33 and TGF-β on the organismal level in vivo [12] is not mediated at the level of transcriptional regulation. Additional regulatory mechanisms likely contribute at the posttranscriptional level, a possibility that is being pursued in our current work on this report. At the organismal level, contributions to fibrosis from FLIL33-driven inflammatory mechanism and from elevated FLIL33 expression in cells other than fibroblasts, such as epithelial and endothelial cells, cannot be excluded.
Although several relevant questions remain unanswered as outlined above, this report adds important mechanistic information to the existing body of knowledge. Specifically, we demonstrate that intracellular FLIL33 induces Smad3 phosphorylation through a mechanism that requires the intrinsic activity of ALK5/TGFBR1 and is driven by elevated AP2, which is known to potentiate TGFBR signaling. Despite similarly inducing Smad3 phosphorylation, elevated FLIL33 differs from stimulation with TGF-β in the regulation of global gene expression, including notable differences in the transcriptional regulation of collagen genes, with FLIL33 mildly suppressing their mRNA levels independently and against the stimulating effect of TGF-β.
Supplementary Material
Acknowledgements
The authors thank Ms. Mariah V. Salcedo for her support with bioinformatics analyses.
Funding
This study was supported by NIH NHLBI R01HL126897 (to S. P. A.), VA I01CX000101 (to I. G. L.), and VA I01BX002499 (to S. P. A.).
Footnotes
Conflict of interests
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or Department of Veterans Administration.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.cellimm.2020.104203.
References
- [1].Liew FY, Girard JP, Turnquist HR, Interleukin-33 in health and disease, Nat. Rev. Immunol 16 (2016) 676–689. [DOI] [PubMed] [Google Scholar]
- [2].Molofsky AB, Savage AK, Locksley RM, Interleukin-33 in tissue homeostasis, Injury Inflamm. Immunity 42 (2015) 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Cayrol C, Duval A, Schmitt P, Roga S, Camus M, Stella A, Burlet-Schiltz O, Gonzalez-de-Peredo A, Girard JP, Environmental allergens induce allergic inflammation through proteolytic maturation of IL-33, Nat. Immunol 19 (2018) 375–385. [DOI] [PubMed] [Google Scholar]
- [4].Licona-Limon P, Kim LK, Palm NW, Flavell RA, TH2, allergy and group 2 innate lymphoid cells, Nat. Immunol 14 (2013) 536–542. [DOI] [PubMed] [Google Scholar]
- [5].Serrels B, McGivern N, Canel M, Byron A, Johnson SC, McSorley HJ, Quinn N, Taggart D, Von Kreigsheim A, Anderton SM, Serrels A, Frame MC, IL-33 and ST2 mediate FAK-dependent antitumor immune evasion through transcriptional networks, Sci. Signal 10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Oshio T, Komine M, Tsuda H, Tominaga SI, Saito H, Nakae S, Ohtsuki M, Nuclear expression of IL-33 in epidermal keratinocytes promotes wound healing in mice, J. Dermatol. Sci 85 (2017) 106–114. [DOI] [PubMed] [Google Scholar]
- [7].Shan J, Oshima T, Wu L, Fukui H, Watari J, Miwa H, Interferon gamma-induced nuclear interleukin-33 potentiates the release of esophageal epithelial derived cytokines, PLoS ONE 11 (2016) e0151701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Shan J, Oshima T, Muto T, Yasuda K, Fukui H, Watari J, Nakanishi K, Miwa H, Epithelial-derived nuclear IL-33 aggravates inflammation in the pathogenesis of reflux esophagitis, J. Gastroenterol 50 (2015) 414–423. [DOI] [PubMed] [Google Scholar]
- [9].Ni Y, Tao L, Chen C, Song H, Li Z, Gao Y, Nie J, Piccioni M, Shi G, Li B, The deubiquitinase USP17 regulates the stability and nuclear function of IL-33, Int. J. Mol. Sci 16 (2015) 27956–27966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Shao D, Perros F, Caramori G, Meng C, Dormuller P, Chou PC, Church C, Papi A, Casolari P, Welsh D, Peacock A, Humbert M, Adcock IM, Wort SJ, Nuclear IL-33 regulates soluble ST2 receptor and IL-6 expression in primary human arterial endothelial cells and is decreased in idiopathic pulmonary arterial hypertension, Biochem. Biophys. Res. Commun 451 (2014) 8–14. [DOI] [PubMed] [Google Scholar]
- [11].Luzina IG, Pickering EM, Kopach P, Kang PH, Lockatell V, Todd NW, Papadimitriou JC, McKenzie AN, Atamas SP, Full-length IL-33 promotes inflammation but not Th2 response in vivo in an ST2-independent fashion, J. Immunol 189 (2012) 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Luzina IG, Kopach P, Lockatell V, Kang PH, Nagarsekar A, Burke AP, Hasday JD, Todd NW, Atamas SP, Interleukin-33 potentiates bleomycin-induced lung injury, Am. J. Respir. Cell Mol. Biol 49 (2013) 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Choi YS, Park JA, Kim J, Rho SS, Park H, Kim YM, Kwon YG, Nuclear IL-33 is a transcriptional regulator of NF-kappaB p65 and induces endothelial cell activation, Biochem. Biophys. Res. Commun 421 (2012) 305–311. [DOI] [PubMed] [Google Scholar]
- [14].Ali S, Mohs A, Thomas M, Klare J, Ross R, Schmitz ML, Martin MU, The dual function cytokine IL-33 interacts with the transcription factor NF-kappaB to dampen NF-kappaB-stimulated gene transcription, J. Immunol 187 (2011) 1609–1616. [DOI] [PubMed] [Google Scholar]
- [15].Masamune A, Watanabe T, Kikuta K, Satoh K, Kanno A, Shimosegawa T, Nuclear expression of interleukin-33 in pancreatic stellate cells, Am. J. Physiol. Gastrointest. Liver Physiol 299 (2010) G821–832. [DOI] [PubMed] [Google Scholar]
- [16].Carriere V, Roussel L, Ortega N, Lacorre DA, Americh L, Aguilar L, Bouche G, Girard JP, IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo, Proc. Natl. Acad. Sci. U. S. A 104 (2007) 282–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Morimoto Y, Hirahara K, Kiuchi M, Wada T, Ichikawa T, Kanno T, Okano M, Kokubo K, Onodera A, Sakurai D, Okamoto Y, Nakayama T, Amphiregulin-producing pathogenic memory T helper 2 cells instruct eosinophils to secrete osteopontin and facilitate airway fibrosis, Immunity 49 (2018) 134–150 e136. [DOI] [PubMed] [Google Scholar]
- [18].Zhao Y, De Los Santos FG, Wu Z, Liu T, Phan SH, An ST2-dependent role of bone marrow-derived group 2 innate lymphoid cells in pulmonary fibrosis, J. Pathol 245 (2018) 399–409. [DOI] [PubMed] [Google Scholar]
- [19].Gao Q, Li Y, Pan X, Yuan X, Peng X, Li M, Lentivirus expressing soluble ST2 alleviates bleomycin-induced pulmonary fibrosis in mice, Int. Immunopharmacol 30 (2016) 188–193. [DOI] [PubMed] [Google Scholar]
- [20].Gieseck RL 3rd, Wilson MS, Wynn TA, Type 2 immunity in tissue repair and fibrosis, Nat. Rev. Immunol 18 (2018) 62–76. [DOI] [PubMed] [Google Scholar]
- [21].Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen CI, Ren Z, Verma R, Abdala-Valencia H, Nam K, Chi M, Han S, Gonzalez-Gonzalez FJ, Soberanes S, Watanabe S, Williams KJN, Flozak AS, Nicholson TT, Morgan VK, Winter DR, Hinchcliff M, Hrusch CL, Guzy RD, Bonham CA, Sperling AI, Bag R, Hamanaka RB, Mutlu GM, Yeldandi AV, Marshall SA, Shilatifard A, Amaral LAN, Perlman H, Sznajder JI, Argento AC, Gillespie CT, Dematte J, Jain M, Singer BD, Ridge KM, Lam AP, Bharat A, Bhorade SM, Gottardi CJ, Budinger GRS, Misharin AV, Single-cell transcriptomic analysis of human lung provides insights into the pathobiology of pulmonary fibrosis, Am. J. Respir. Crit. Care Med (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lindahl GE, Stock CJ, Shi-Wen X, Leoni P, Sestini P, Howat SL, Bou-Gharios G, Nicholson AG, Denton CP, Grutters JC, Maher TM, Wells AU, Abraham DJ, Renzoni EA, Microarray profiling reveals suppressed interferon stimulated gene program in fibroblasts from scleroderma-associated interstitial lung disease, Respir. Res 14 (2013) 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Peng R, Sridhar S, Tyagi G, Phillips JE, Garrido R, Harris P, Burns L, Renteria L, Woods J, Chen L, Allard J, Ravindran P, Bitter H, Liang Z, Hogaboam CM, Kitson C, Budd DC, Fine JS, Bauer CM, Stevenson CS, Bleomycin induces molecular changes directly relevant to idiopathic pulmonary fibrosis: a model for “active” disease, PLoS ONE 8 (2013) e59348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wyman AE, Noor Z, Fishelevich R, Lockatell V, Shah NG, Todd NW, Atamas SP, Sirtuin 7 is decreased in pulmonary fibrosis and regulates the fibrotic phenotype of lung fibroblasts, Am. J. Physiol. Lung Cell. Mol. Physiol 312 (2017) L945–L958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kopach P, Lockatell V, Pickering EM, Haskell RE, Anderson RD, Hasday JD, Todd NW, Luzina IG, Atamas SP, IFN-gamma directly controls IL-33 protein level through a STAT1-and LMP2-dependent mechanism, J. Biol. Chem 289 (2014) 11829–11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Clerman A, Noor Z, Fishelevich R, Lockatell V, Hampton BS, Shah NG, Salcedo MV, Todd NW, Atamas SP, Luzina IG, The full-length interleukin-33 (FLIL33)-importin-5 interaction does not regulate nuclear localization of FLIL33 but controls its intracellular degradation, J. Biol. Chem 292 (2017) 21653–21661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Luzina IG, Clerman A, Fishelevich R, Todd NW, Lockatell V, Atamas SP, Identification of the IL-33 protein segment that controls subcellular localization, extracellular secretion, and functional maturation, Cytokine 119 (2019) 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J, A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum, Nat. Methods 10 (2013) 407–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Luzina IG, Tsymbalyuk N, Choi J, Hasday JD, Atamas SP, CCL18-stimulated upregulation of collagen production in lung fibroblasts requires Sp1 signaling and basal Smad3 activity, J. Cell. Physiol 206 (2006) 221–228. [DOI] [PubMed] [Google Scholar]
- [30].Luzina IG, Todd NW, Nacu N, Lockatell V, Choi J, Hummers LK, Atamas SP, Regulation of pulmonary inflammation and fibrosis through expression of integrins alphaVbeta3 and alphaVbeta5 on pulmonary T lymphocytes, Arthritis Rheum. 60 (2009) 1530–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mozaffarian A, Brewer AW, Trueblood ES, Luzina IG, Todd NW, Atamas SP, Arnett HA, Mechanisms of oncostatin M-induced pulmonary inflammation and fibrosis, J. Immunol 181 (2008) 7243–7253. [DOI] [PubMed] [Google Scholar]
- [32].Nacu N, Luzina IG, Highsmith K, Lockatell V, Pochetuhen K, Cooper ZA, Gillmeister MP, Todd NW, Atamas SP, Macrophages produce TGF-beta-induced (beta-ig-h3) following ingestion of apoptotic cells and regulate MMP14 levels and collagen turnover in fibroblasts, J. Immunol 180 (2008) 5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB, An assay for transforming growth factor-beta using cells transfected with a plasminogen activator inhibitor-1 promoter-luciferase construct, Anal. Biochem 216 (1994) 276–284. [DOI] [PubMed] [Google Scholar]
- [34].Luzina IG, Salcedo MV, Rojas-Pena ML, Wyman AE, Galvin JR, Sachdeva A, Clerman A, Kim J, Franks TJ, Britt EJ, Hasday JD, Pham SM, Burke AP, Todd NW, Atamas SP, Transcriptomic evidence of immune activation in macroscopically normal-appearing and scarred lung tissues in idiopathic pulmonary fibrosis, Cell. Immunol 325 (2018) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].DeMaio L, Buckley ST, Krishnaveni MS, Flodby P, Dubourd M, Banfalvi A, Xing Y, Ehrhardt C, Minoo P, Zhou B, Crandall ED, Borok Z, Ligand-independent transforming growth factor-beta type I receptor signalling mediates type I collagen-induced epithelial-mesenchymal transition, J. Pathol 226 (2012) 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen G, Chen X, Sukumar A, Gao B, Curley J, Schnaper HW, Ingram AJ, Krepinsky JC, TGFbeta receptor I transactivation mediates stretch-induced Pak1 activation and CTGF upregulation in mesangial cells, J. Cell Sci 126 (2013) 3697–3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Perez-Gomez E, Villa-Morales M, Santos J, Fernandez-Piqueras J, Gamallo C, Dotor J, Bernabeu C, Quintanilla M, A role for endoglin as a suppressor of malignancy during mouse skin carcinogenesis, Cancer Res. 67 (2007) 10268–10277. [DOI] [PubMed] [Google Scholar]
- [38].Feng XH, Derynck R, Ligand-independent activation of transforming growth factor (TGF) beta signaling pathways by heteromeric cytoplasmic domains of TGF-beta receptors, J. Biol. Chem 271 (1996) 13123–13129. [DOI] [PubMed] [Google Scholar]
- [39].Mettlen M, Chen PH, Srinivasan S, Danuser G, Schmid SL, Regulation of Clathrin-Mediated Endocytosis, Annu. Rev. Biochem 87 (2018) 871–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Mitchell H, Choudhury A, Pagano RE, Leof EB, Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11, Mol. Biol. Cell 15 (2004) 4166–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Penheiter SG, Mitchell H, Garamszegi N, Edens M, Dore JJ Jr., Leof EB, Internalization-dependent and -independent requirements for transforming growth factor beta receptor signaling via the Smad pathway, Mol. Cell. Biol 22 (2002) 4750–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL, Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover, Nat. Cell Biol 5 (2003) 410–421. [DOI] [PubMed] [Google Scholar]
- [43].Yao D, Ehrlich M, Henis YI, Leof EB, Transforming growth factor-beta receptors interact with AP2 by direct binding to beta2 subunit, Mol. Biol. Cell 13 (2002) 4001–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chung H, Ramachandran R, Hollenberg MD, Muruve DA, Proteinase-activated receptor-2 transactivation of epidermal growth factor receptor and transforming growth factor-beta receptor signaling pathways contributes to renal fibrosis, J. Biol. Chem 288 (2013) 37319–37331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Little PJ, Burch ML, Getachew R, Al-aryahi S, Osman N, Endothelin-1 stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by endothelin receptor transactivation of the transforming growth factor-[beta] type I receptor, J. Cardiovasc. Pharmacol 56 (2010) 360–368. [DOI] [PubMed] [Google Scholar]
- [46].Burch ML, Ballinger ML, Yang SN, Getachew R, Itman C, Loveland K, Osman N, Little PJ, Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease-activated receptor-1 transactivation of the transforming growth factor beta type I receptor, J. Biol. Chem 285 (2010) 26798–26805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cabello-Verrugio C, Cordova G, Vial C, Zuniga LM, Brandan E, Connective tissue growth factor induction by lysophosphatidic acid requires transactivation of transforming growth factor type beta receptors and the JNK pathway, Cell. Signal 23 (2011) 449–457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.