

Abstract
Brugada syndrome (BrS) is a complex genetic cardiac ion channel disease that causes a high predisposition to sudden cardiac death. Considering that its heterogeneity in clinical manifestations may result from genetic background, the application of patient-specific induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs) may help to reveal cell phenotype characteristics underlying different genetic variations. Here, to verify and compare the pathogenicity of mutations (SCN5A c.4213G>A andSCN1B c.590C>T) identified from two BrS patients, we generated two novel BrS iPS cell lines that carried missense mutations inSCN5A or SCN1B, compared their structures and electrophysiology, and evaluated the safety of quinidine in patient-specific iPSC-derived CMs. Compared to the control group, BrS-CMs showed a significant reduction in sodium current, prolonged action potential duration, and varying degrees of decreased Vmax, but no structural difference. After applying different concentrations of quinidine, drug-induced cardiotoxicity was not observed within 3-fold unbound effective therapeutic plasma concentration (ETPC). The data presented proved that iPSC-CMs with variants in SCN5A c.4213G>A orSCN1B c.590C>T are able to recapitulate single-cell phenotype features of BrS and respond appropriately to quinidine without increasing incidence of arrhythmic events.
Keywords: human iPSCs, Brugada syndrome, disease modeling, quinidine, SCN5A, SCN1B
Introduction
Brugada syndrome (BrS) is an inheritable cardiac condition characterized by a typical electrocardiogram of right bundle branch block with ST-segment elevation in V1-V3[1]. BrS is associated with a high incidence of ventricular arrhythmia, which is prone to sudden cardiac death, especially in young men[2]. The implantation of a cardiac defibrillator (ICD) is the only established treatment[3]. However, shock from ICD is painful and may cause post-ICD shock stress reactions[4]. Alternatively, drugs that counteract the ionic current imbalance may be efficient. Currently, quinidine is applied in BrS patients to prevent the electrical storm of ICD, but its effectiveness and safety are controversial[5].
Genetic studies have revealed that BrS is associated with variations in multiple genes, including SCN5A, SCN1B, CACNA1C, and KCND3[6]. The most commonly involved gene in BrS is SCN5A, which encodes the α-subunit of the voltage-gated cardiac sodium channel (Nav1.5)[7] and has been reported in more than 25% of BrS patients[8]. Recently, we reported that a single nucleotide variant (A197V) in SCN1B, encoding the β1-subunit of Nav1.5, was responsible for BrS susceptibility[9]. Such loss of function of the sodium channel resulted in impaired depolarization and a prominent transient-outward current (Ito)[10]. However, cardiac phenotype caused by SCN1B variation has been less reported using patient-specific induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs). Also, the characterization of cardiomyocytes with genetic variations in different subunits of Nav1.5 remains to be compared.
Conventional heterologous expression provided insights into single-channel function, but fails to reflect the changes in whole membrane potential and electro-mechanical coupling. Thus, human cardiomyocytes, especially patient-specific cardiomyocytes, are needed to explore the mechanisms underlying the ion channel diseases. Indeed, human iPSCs provide a promising tool for recapitulating the phenotype of cardiac disease and for evaluating the efficiency of drug treatments[11–13]. In recent years, case studies demonstrated that BrS patient-specific iPSC-CMs are accurate disease models in vitro[14–15]. However, their drug responses to quinidine have not been fully elucidated.
In the present study, we recruited two Brugada patients who carried SCN5A c.4213G>A andSCN1B c.590C>T, respectively. We generated patient-specific iPS cell lines and differentiated them into ventricular cardiomyocytes. Through structural and electrophysiological assessments, we investigated the pathological properties of BrS at the cellular level. By evaluating the drug responses to quinidinein vitro, we highlighted the efficacy and safety of quinidine treatment in BrS-CMs.
Materials and methods
Generation of human iPS cell lines
Human iPS cell lines used in this study were derived from peripheral blood mononuclear cells of two Brugada patients and one healthy volunteer. The approval of the study was provided by the Bioethics Committee of the First Affiliated Hospital of Nanjing Medical University (2014-SR-090). The iPSC lines were generated using a CytoTune-iPS 2.0 Sendai reprogramming kit (Thermo Fisher Scientific, USA), which employed four Yamanaka factors (OCT4, KLF4, SOX2, and cMYC), following the manufacturer's instructions with subtle adjustments. Reprogrammed PBMCs were replated onto Matrigel (Corning, USA) coating plates, and the medium was changed to mTeSR1 (STEMCELL Technologies Inc., Canada) after transduction. Individual colonies were identified on day 7, picked 20 days later, and maintained in mTeSR1 medium. Another control iPS cell line was purchased from Wicell (iPS-DF6-9-9T, WiCell Research Institute, USA).
Genome sequencing
To confirm the presence of the variations in the two iPS cell lines, patient-derived iPSCs were examined by Sanger sequencing. Total DNA was isolated using a TIANamp genomic DNA kit (Tiangen Biotech, China), and the coding regions of SCN5A and SCN1B, including each variation site, were amplified by PCR. The PCR products were purified using a DNA purification kit (Tiangen Biotech) and then were sequenced. The results were compared with the SCN5A and SCN1B reference sequences NM_001099404 and NM_001037.
Differentiation of iPSC-CMs
Briefly, iPSCs were allowed to grow to over 90% confluence in mTeSR1 medium and then were switched to RPMI 1640 medium (Thermo Fisher Scientific) containing B-27 supplement without insulin (Thermo Fisher Scientific). On day 0 to 1, additional CHIR-99021 (6 µmol/L, Selleckchem, USA) was added to induce mesodermal differentiation. On day 3 to 4, IWR-1 (5 µmol/L, Sigma-Aldrich, Germany) was added to the medium to inhibit Wnt signaling and induce cardiogenesis. From day 5 to 7, BMS493 (1 µmol/L, Sigma-Aldrich) was added for directly differentiating into ventricular cardiomyocytes. From day 8 onwards, cells were cultured in RPMI 1640 containing B27 complete supplement (Gibco), and the medium was refreshed every other day. Beating cardiomyocytes were observed on day 8 to 10. Besides, to obtain enriched cardiomyocyte populations, glucose-free DMEM supplemented with 4 mmol/L lactate medium (Sigma-Aldrich) and 3.6 mmol/L HEPES (Sigma-Aldrich) were applied for 4 days. Cells for further functional and electrophysiological analyses were cultured for at least 60 days and dissociated using 0.25% trypsin-EDTA.
Flow cytometry analysis
Cells were treated with the appropriate dissociation solution (Accutase for iPS cells, 0.25% trypsin for CMs), fixed with 4% paraformaldehyde, and permeated with 0.1% Triton X-100 at room temperature. Pluripotent stem cells were counted and stained using phycoerythrin conjugated antibodies against SSEA-4 (1:50 dilution; Miltenyi Biotec, Germany) for 30 minutes at 4 °C shielded from light. Cardiomyocytes were co-incubated with anti-cTnT-FITC (1:50 dilution; Miltenyi Biotec) and anti-MLC2v-PE (1:50 dilution; Miltenyi Biotec) in PBS at 4 °C for 30 minutes. REA Control-FITC (1:50 dilution; Miltenyi Biotec) and REA Control-PE (1:50 dilution; Miltenyi Biotec) were used as isotype controls. The cells were measured using FACSCalibur (BD, USA) and subsequent analyses were performed using FlowJo software (Tree Star, version 10.5.3).
Immunofluorescence staining
Cells were seeded on a coverslip and cultured for 3 to 5 days before being fixed in 4% paraformaldehyde for 20 minutes. The cells were permeabilized with 0.1% Triton X-100 for 5 minutes when needed. Cells were then blocked in 5% BSA in PBS for 30 minutes; all these procedures were performed at room temperature. Cells were then incubated overnight at 4 °C with primary antibodies. For pluripotent stem cells, anti-SSEA4 (1:200 dilution; Abcam, UK) and anti-Nanog (1:200 dilution; Abcam) were applied. For cardiomyocytes, anti-α-actinin (1:200 dilution; Sigma-Aldrich), anti-cardiac troponin I (1:200 dilution; Abcam), and anti-Nav1.5 (1:100 dilution; Abcam) were used. The following secondary antibodies included Alexa Fluor 488 goat anti-rabbit IgG (1:800 dilution; Abcam) and Alexa Fluor 555 goat anti-mouse IgG (1:800 dilution; Abcam) were incubated with samples at room temperature for 30 minutes. The nuclei were stained with DAPI (Life Technologies, USA). Then, the cells were rinsed with PBS and observed under a fluorescence microscope (Axio Imager A2, Carl Zeiss, Germany). Images were analyzed and merged with ImageJ (NIH, version 1.8.0_77).
Transmission electron microscopy
CMs were dissociated into single cells with 0.25% trypsin. Samples were then fixed with 2.5% glutaraldehyde (Sigma-Aldrich), postfixed with 1% osmium tetroxide, and rinsed with PBS. After dehydration with a series of ethanol solutions and embedding into resin, ultrathin sections were cut with an ultramicrotome (EM UC7; Leica, Germany) and stained with lead citrate. Visualization was performed under a transmission electron microscope (JEM-1010, Jeol Ltd., Japan) equipped with a CCD camera operated at 75 kV.
Real-time reverse transcriptase PCR
Total RNA was prepared from CMs using TRIzol (Invitrogen) following the manufacturer's protocol. RNA (500 to 1000 ng) was reverse-transcribed into cDNA using iScript cDNA Synthesis Kit (Bio-Rad, USA). cDNA was then determined by the applied biosystems 7900HT fast real-time PCR system (ABI) with Hieff qPCR SYBR green master mix (Yesen-bio, China). The mRNA expression levels were normalized to the housekeeping gene GAPDH and LMNA. Primers used are summarized in Supplementary Table 1 (available online).
Patch-clamp analysis
Cells at day 60 in independent differentiation batches and different culture dishes were used to evaluate electrophysiological properties. Three to 4 days after dissociation, single-cell patch-clamp experiments were performed using an Axopatch 200B amplifier (Axon Instruments, USA) and analyzed with Clampfit 10.3 software (Molecular Devices). Glass pipettes were pulled using borosilicate glass (Sutter Instrument, USA) and a micropipette puller (Model P-100, Sutter Instruments). Pipettes with resistance ranging from 2 to 4 MΩ were perfused with specified intracellular solutions and were used.
Na measurement
The sodium currents (INa) of cardiomyocytes were recorded using a whole-cell patch-clamp technique at room temperature. Data were filtered at 2 kHz and acquired at 20 kHz. INa was examined using the following intracellular solution designed to eliminate the outward potassium current: 10 mmol/L NaCl, 60 mmol/L CsCl, 60 mmol/L CsF, 10 mmol/L TEA-Cl, 1 mmol/L CaCl2, 1 mmol/L MgCl2, 10 mmol/L EGTA, 10 mmol/L glucose, and 10 mmol/L HEPES (pH 7.2 with CsOH). The external bath solution composition was as follows: 130 mmol/L choline chloride, 10 mmol/L NaCl, 1 mmol/L TEA-Cl, 1.8 mmol/L MgCl2, 1.8 mmol/L CaCl2, 0.2 mmol/L NiCl, 10 mmol/L glucose, and 10 mmol/L HEPES (pH 7.2 with NaOH). Nifedipine was added to exclude inward calcium current. The protocol for recording INa ranged from –80 to +40 mV in 10 mV increments that occurred every 50 milliseconds. Cell capacitance was measured to normalize INa by applying a 10 mV voltage step from –100 mV, and current density was calculated as peak current divided by cell capacitance (pA/pF).
Voltage clamp recordings of Ito currents
Ito regulates phase 1 repolarization to modulate the early AP plateau. Here, Ito currents were recorded in extracellular solution that contained the following: 140 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 1 mmol/L MgCl2, 0.33 mmol/L NaH2PO4, 10 mmol/L HEPES, and 10 mmol/L glucose (pH 7.4 with NaOH). The pipettes were filled with an internal solution that contained the following: 90 mmol/L K-aspartate, 45 mmol/L KCl, 5 mmol/L Na-pyruvate, 5 mmol/L MgATP, 5 mmol/L EGTA, 10 mmol/L HEPES, and 10 mmol/L glucose (pH 7.2 with KOH). Ito was determined by increasing the testing potential stepwise from –40 to 40 mV in 10 mV steps from a holding potential of –90 mV with a 20-millisecond pre-pulse to –35 mV to inactivate INa.
Action potential measurement
Action potentials were recorded at 37 °C in Tyrode's solution. Data were filtered at 5 kHz and digitized at 40 kHz. Pipettes were filled with the following internal solution (in mmol/L): 150 KCl, 5 NaCl, 2 CaCl2, 5 Mg ATP, 5 HEPES, 5 EGTA, and 10 glucose (pH 7.2 with KOH). Extracellular solution contained: 135 mmol/L NaCl, 5.4 mmol/L KCl, 1.8 mmol/L CaCl2, 1 mmol/L MgCl2, 0.33 mmol/L NaH2PO4, 10 mmol/L HEPES, and 10 mmol/L glucose, adjusted to pH 7.4 with NaOH. APs were characterized by resting membrane potential (RMP), maximum AP amplitude (APA), maximum upstroke velocity (Vmax), and average action potential duration (APD) at 90%, 50%, and 30% repolarization (APD90, APD50, and APD30, respectively). Each average data was from 10 consecutive recorded APs.
Calcium transient
Cells were loaded with the calcium-sensitive dye fluo-4 AM (Life Technologies) at a working concentration of 5 µmol/L for 60 minutes at 37 °C. Having been rinsed the iPSC-CMs, they were maintained in Tyrode's solution. The spontaneous calcium fluorescence signal was recorded by an inverted fluorescence microscope (Axio Vert A1, Carl Zeiss, Germany) equipped with an sCMOS camera (Tucsen, Dhyana95, China). The excitation and emission wavelengths of Fluo-4 were 488 nm and 505 nm, respectively. Recordings were obtained at the acquisition rate of 25 frames/second for a 15-second duration. To calibrate the cell-to-cell variability and background disturbance, the fluorescence intensity in the region of interest was determined based on the formula: (F-F0)/F0. Herein, F0 represents the baseline intensity, while F represents the intensity at any point in time. The parameters that describe and evaluate the calcium transient include the spontaneous beat rate, amplitude, and arrhythmic events that are EAD-like or fibrillation-like. Data were analyzed using ImageJ.
Drug response
Quinidine (Selleckchem) was dissolved in dimethyl sulfoxide to prepare a 100 mmol/L stock solution. Working solutions were freshly prepared in cell-specific medium. Final concentration was set as 0.1-fold to 10-fold the ETPC (unbound effective therapeutic plasma concentration) of quinidine (0.3, 1, 3, 10, and 30 μmol/L). Dilution ratio up to 1000 had no effect on cell apoptosis or cardiac electrical activity.
Baseline AP was recorded for each cell before the treatment. Then, quinidine was sequentially applied from the lowest to highest concentration at 10-minute intervals for washout to obtain a steady state. For each concentration, 10 recorded APs from each cell were averaged for analysis. The following parameters were quantitated as mentioned above: RMP, APA, Vmax, APD30, APD50, and APD90. Recordings at each concentration were normalized to their baseline.
Statistical analysis
Data with a normal distribution were presented as the mean±SEM. Student's t-tests were then used in comparisons where there were two groups. The Chi-square test was used for categorical variables. For pharmaceutical experiments, the post-drug effects were evaluated by comparing to the baseline using a paired Student's t-test. All statistical analyses were performed with SigmaPlot 12.5 and GraphPad Prism 8 (GraphPad Software, USA) and significance was determined at a P<0.05.
Results
Clinical and genetic profiles
Two BrS patients and one healthy volunteer were recruited. The diagnosis of Brugada syndrome was made based on the 2005 consensus diagnostic criteria[16]. Patient 1 (BrS-SCN5A) was a 42-year-old man presenting with unstable ventricular tachycardia after multiple episodes of recurrent syncope. His resting ECG showed a characteristic type 1 Brugada pattern (Fig. 1A). Moreover, pedigree investigation showed that his brother died from sudden cardiac death. Patient 2 (BrS-SCN1B) was a 46-year-old man with a history of recurrent syncope. The resting ECG exhibited typical type 1 Brugada ECG pattern (Fig. 1B). Both patients received ICD implantation and close follow-up. Genetic testing revealed a SCN5A c.4213G>A (V1405M) variation in BrS-SCN5A and aSCN1B c.590C>T (A197V) variation in BrS-SCN1B. The corresponding positions of each variation on the protein structure of the Nav1.5 sodium channel α subunit or β-1 subunit are illuminated in Fig. 1C.
Figure 1.
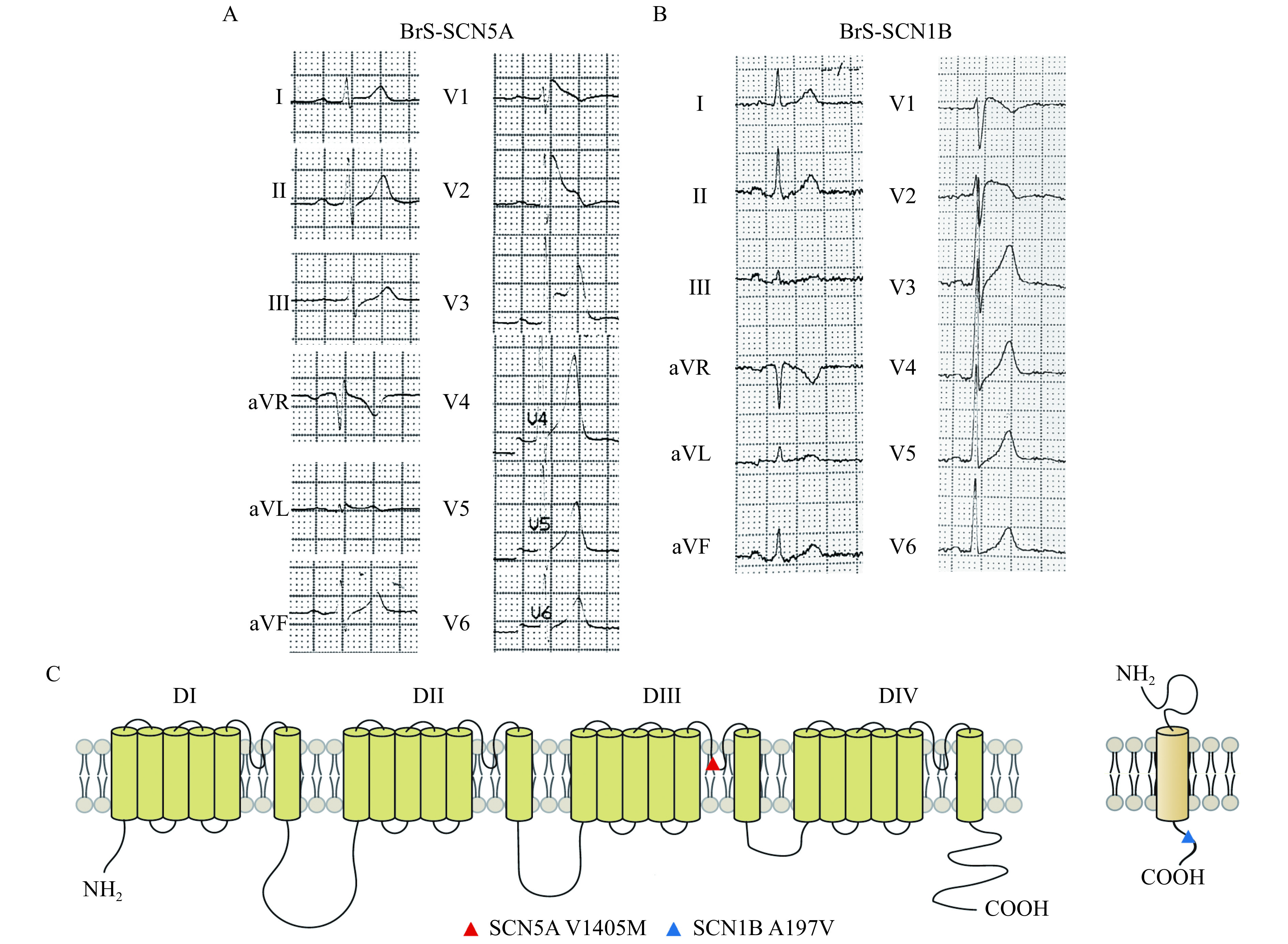
Clinical and genetical profile of the two Brugada syndrome patients.
A and B: Electrocardiograms of both patients show the typical ST segment elevation, a typical pattern of BrS. C: The corresponding positions of V1405M mutation on the protein structure of voltage-gated sodium-channel α subunit gene (left panel), and A197V mutation on the ß1 subunit gene (right panel). BrS: Brugada syndrome.
Our healthy control subject (Control 1) was a 44-year-old young woman with normal ECGs and without personal or family history of sudden death. Whole-genome sequencing indicated no disease-causing variants in any cardiovascular-related genes. Another control cell line (Control 2) was generated from a healthy male.
Characterization of iPS cell lines
Human iPSCs were generated from PBMCs derived from the two patients and the healthy control. To verify the pluripotency of each iPS cell line, undifferentiated stem cells were fixed and stained. As expected, the cells in both the BrS and control groups expressed the robust pluripotency markers NANOG and SSEA4 (Fig. 2A and Supplementary Fig. 1A, available online). Further flow cytometry analysis revealed a high proportion (>95%) of SSEA-4-positive cells in each group (Fig. 2B and (Supplementary Fig. 1B, available online). Genetic testing confirmed the variation sites of SCN5A c.4213G>A andSCN1B c.590C>T in the corresponding iPSC lines, indicating that reprogramming maintained the genetic variations found in the BrS patients (Fig. 2C). Also, relevant variations in control lines were not observed via Sanger sequencing (Supplementary Fig. 1C).
Figure 2.
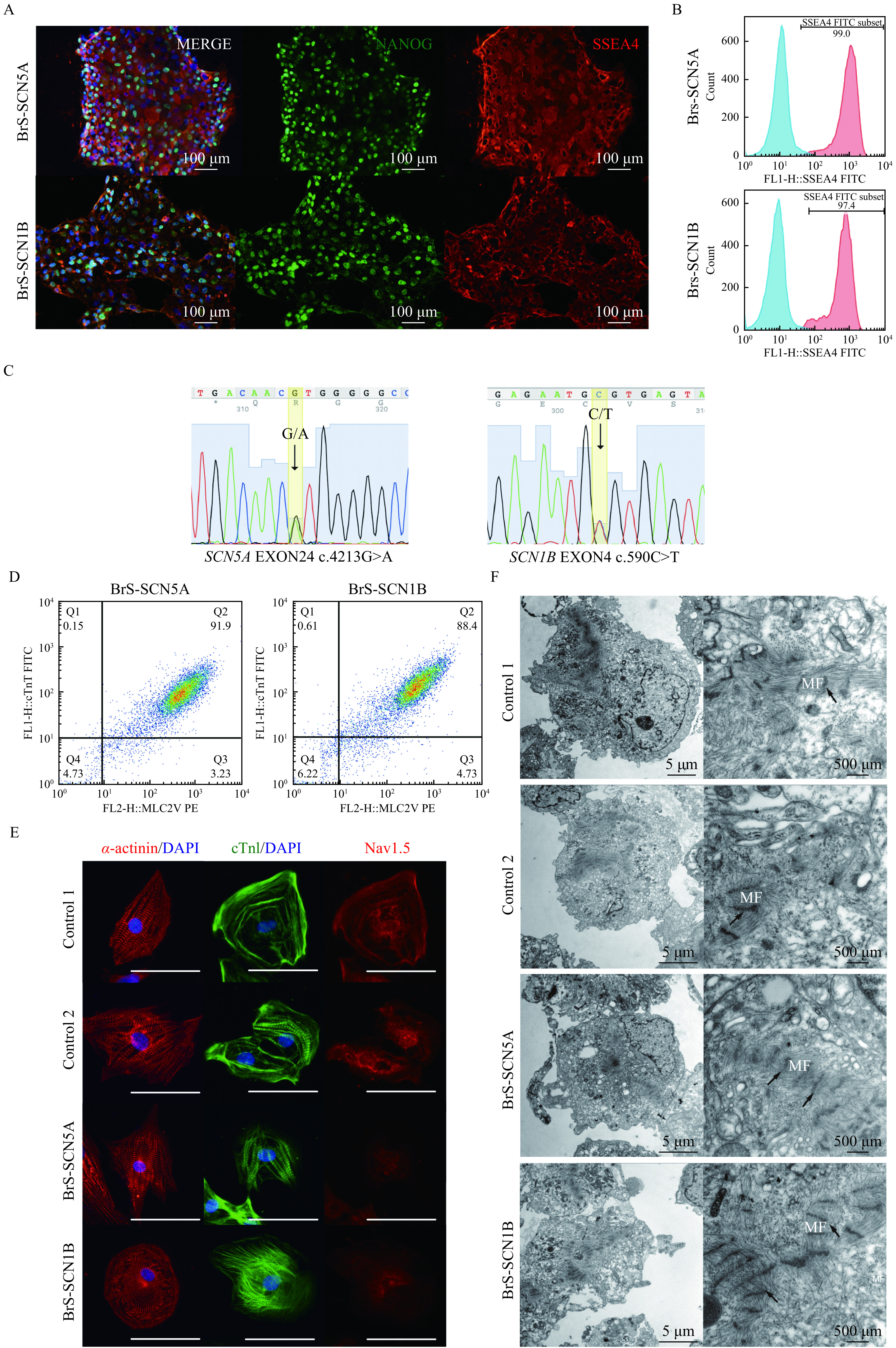
Characterization of iPSC lines and differentiated cardiomyocytes.
A: Images of iPS cells stained with pluripotency markers NANOG (green), SSEA4 (red) for BrS-SCN5A and BrS-SCN1B iPS cell lines. Nuclei were stained with DAPI (blue). Scale bars, 100 μm. B: Flow cytometry analysis of pluripotency markers (SSEA4) in generated BrS iPSC lines. C: Genotyping of BrS-SCN5A and BrS-SCN1B iPSC lines focusing on the loci around each mutation confirmed by DNA sequencing. D: Representative flow cytometry confirming the high percentage of ventricular myocytes in differentiated cells. E: Immunostaining of sarcomeric α-actinin, cTnI, and Nav1.5 at 60 days post differentiation. Scale bars, 50 μm. F: TEM images of myofibrillar organization in control and BrS iPSC-CMs. Black arrow indicates z-lines. iPSC: induced pluripotent stem cell; TEM: transmission electron microscopy.
Structural analysis of BrS-CMs
Highly efficient cardiac differentiation was conducted. FACS analysis of the cardiac-specific marker cTnT and ventricular cardiomyocyte marker MLC2 confirmed the purity of ventricular myocytes to be over 90% (Fig. 2D). Considering the immaturity of newly differentiated cardiomyocytes, prolonged cultivation up to 60 days was applied. To compare the morphological properties of the cells, we stained age-matched cells for cardiac structural proteins α-actinin and cTnI. All cells exhibited robust expression of these markers with organized striation patterns of sarcomeres (Fig. 2E). Besides, the staining of Nav1.5 indicated the expression of Nav1.5 was markedly reduced in BrS cells (Fig. 2E). Furthermore, we examined the cardiac ultrastructure using transmission electron microscopy. In both BrS and control iPSC-CMs, myofibrils were well organized with aligned linear Z-discs (Fig. 2F). Abundant mitochondria and sarcoplasmic reticulum could be seen around the sarcomere. Given the above findings, we reported no significant discrepancy in morphology between control and both BrS cardiomyocytes.
BrS-CMs showed impaired Na+ current
To compare the impact of different variations in the Nav1.5 subunits on sodium current, we measured the sodium current using voltage clamp experiments. Fig. 3A plots representative INa traces for BrS and control cardiomyocytes at varying potentials. It demonstrated a loss of function of the sodium channel current in both the BrS-SCN1B and BrS-SCN5A groups compared to control-derived cardiomyocytes (Fig. 3B). Further analysis demonstrated that peak INa density was reduced by 80% in BrS-SCN5A and 70% in BrS-SCN1B compared to their control counterparts (P<0.001 for both,Fig. 3C), which was in line with the downregulation in relevant gene expression (P<0.001 for both,Fig. 3D and E). Notably, the peak INa density of BrS-SCN5A cardiomyocytes was even lower than that of the BrS-SCN1B group (P=0.02). In addition, we assessed the transient outward current (Ito), which was judged as another potential contributing factor of BrS. The results demonstrated that Ito currents were not significantly changed in either BrS cell line compared with the control (Supplementary Fig. 2
Figure 3.
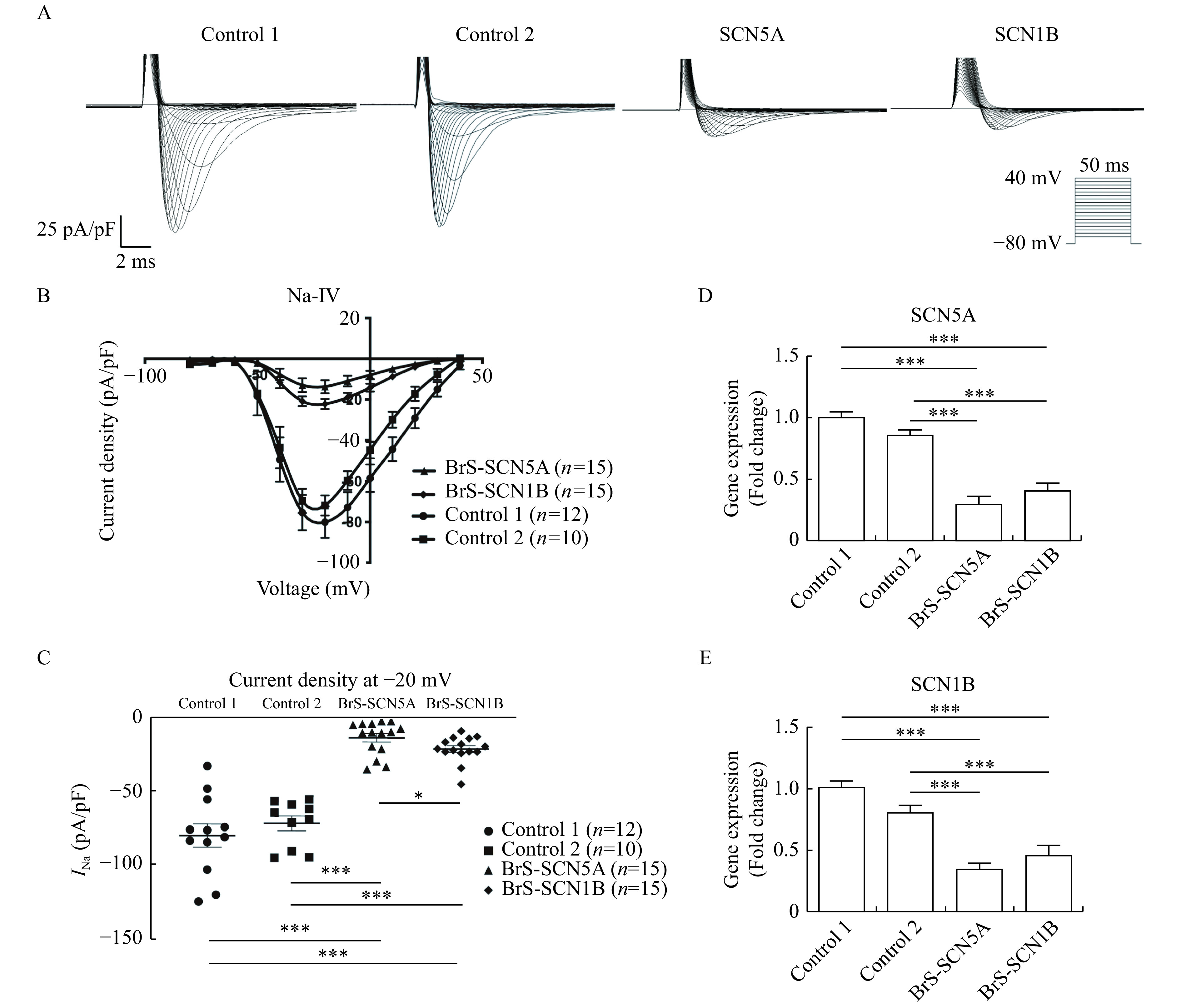
iPSC-CMs carrying SCN5A or SCN1B mutation showed impaired INa properties.
A: Typical INa curve measured by the voltage clamp protocol. Traces for pulses from −80 to −40 mV are shown. B: Current-voltage relationships of INa from Control 1 (n=12), Control 2 (n=10), BrS-SCN5A (n=15), and BrS-SCN1B (n=15) normalized to the cell capacitance. C: Comparison of INa density at −20 mV between control lines and the BrS lines. D and E: SCN5A and SCN1B expression evaluated by real-time RT-PCR (n=6 for each line). Data are presented as mean±SEM. Comparisons between two groups were analyzed using Student's t-test. ***P<0.001,*P<0.05. iPSC-CMs: induced pluripotent stem cell-derived cardiomyocytes;INa: sodium current.
BrS-CMs exhibited abnormal action potential
To investigate whether Nav1.5 variations would affect the whole membrane potential, we next recorded action potentials by patch-clamp. Only ventricular cardiomyocytes were studied in the present study (Fig. 4A)[13]. In line with our sodium current data, APA (Fig. 4B) and Vmax (Fig. 4C) were significantly decreased in the BrS groups compared to the control group (APA: P<0.001 for each comparison,Vmax: P<0.05 for each comparison). Action potential durations (APD 30, 50, and 90) were significantly longer in both BrS cell groups than they were in the healthy control (P<0.001 for all groups,Fig. 4D). To sum up, these results indicated that BrS iPSC-CMs exhibited notably aberrant AP phenotypes. Meanwhile, the action potentials from the BrS-SCN1B and BrS-SCN5A groups presented the same pattern. All the AP characterizations are summarized in Supplementary Table 2 (available online).
Figure 4.
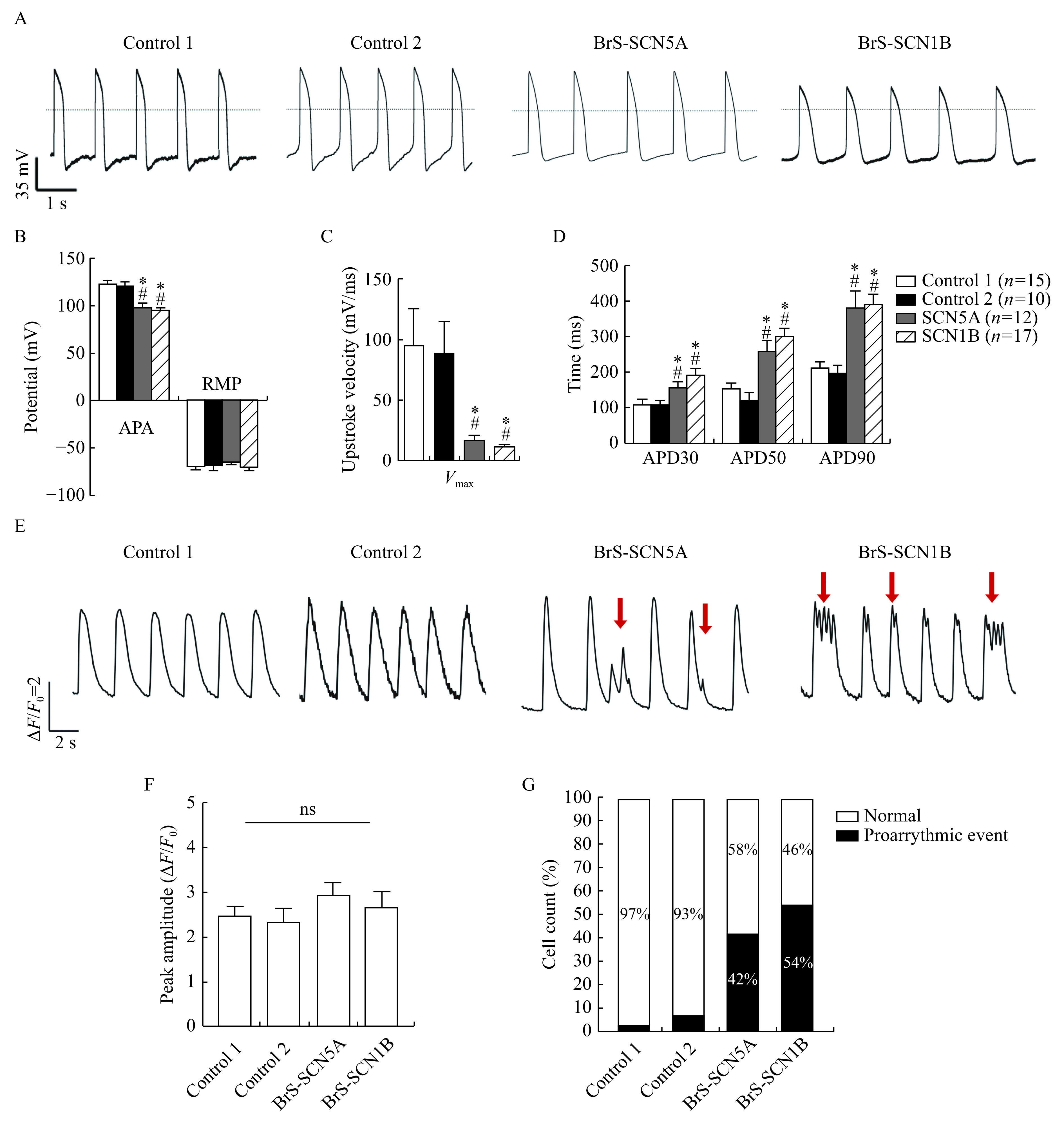
Both BrS iPSC-CMs exhibited prolonged action potential durations, decreased Vmax and abnormal Ca2+ handling.
A: Representative spontaneous AP traces in control and BrS myocytes using current-clamp recording. Dotted line indicates 0 mV. B–D: Mean values for APA, RMP, Vmax, and APD at 30%, 50%, and 90% repolarization (APD30, APD50, and APD90). Control1: n=15; Control2: n=10; BrS-SCN5A: n=12; BrS-SCN1B: n=17. *P<0.01 vs. Control 1; #P<0.01 vs. Control 2. Data are presented as mean±SEM. Comparisons between two groups were analyzed using two-sided Student's t-test. E: Representative spontaneous Ca2+ transients, red arrow indicates irregular Ca2+ waveforms. F: Peak amplitude of spontaneous Ca2+ transients in control and BrS iPSC-CMs (n=30 for each line, Student's t-test). Ns: No significance (P>0.05). G: Proportions of cells showing proarrhythmic events in each line (n=30 for each line, Chi-square test). iPSC-CMs: induced pluripotent stem cell-derived cardiomyocytes; APA: action potential amplitude; RMP: resting membrane potential; Vmax: maximum upstroke velocity; APD: action potential duration.
BrS-CMs exhibited altered Ca2+ handling
Considering that calcium is a critical mediator of excitation-contraction coupling in cardiac cells, we then examined the changes in intracellular calcium concentration using calcium probes. The representative curves of Ca2+ handling from each group are shown in Fig. 4E. Both BrS iPSC-CMs exhibited similar peak amplitudes, compared to control cells (Fig. 4F). Nevertheless, the kinetic stability of calcium handling was disturbed in the BrS groups. As shown in Fig. 4E, BrS cells exhibited more proarrhythmic events, including delayed afterdepolarization-like and early afterdepolarization-like events, than controlled CMs. The percentages of cells showing proarrhythmic events in BrS-SCN5A, BrS-SCN1B, Control 1 and Control 2 lines were 58%, 46%, 3%, and 7%, respectively, P<0.001 for Chi-square test (Fig. 4G).
Cardiac safety evaluation of quinidine using BrS-CMs
Quinidine is a class 1A antiarrhythmic drug with significant Ito blocking properties. It was once expected to be efficient based on positive results in limited series of BrS patients[17] but other studies failed to demonstrate its beneficial effects[18–19]. In our cases, oral quinidine therapy was applied to both BrS-SCN5A and BrS-SCN1B patients after their ICD implantation. During follow-up, there was no report of ICD discharge in these two patients. Meanwhile, quinidine also blocked potassium channels in phase 3. Recent studies reported that quinidine-induced QT prolongation increased the risk of fatal arrhythmia[20]. Hence, further investigation of drug safety is warranted before the prescription of lifetime drug use.
Human iPSC-CMs are considered as precise tools for cardiac safety drug screening in vitro[21]. To further confirm the drug safety of quinidine in cells with these two variations, iPSC cardiomyocytes were subjected to quinidine at concentrations ranging from 0.3 to 30 µmol/L. The results demonstrated that quinidine had no impact on cell viability when examined by CardioExcyte 96 (Nanion Technologies, USA;Supplementary Fig. 3 , available online). Action potential was further analyzed to detect drug effects on cellular electrophysiology. As shown in Fig. 5A–G and Supplementary Table 3 (available online), quinidine decreased the Vmax, APA, and RMP in a concentration-dependent manner among the three groups. Interestingly, APD90 was prolonged in all cell lines as quinidine concentration increased, and then showed a downward trend when the concentration reached 3-fold the ETPC (10 µmol/L) or higher. Besides, the control group had a greater prolongation of APD90 than both BrS groups (P=0.014 for BrS-SCN5A, P<0.001 for BrS-SCN1B). All the groups showed similar mild changes in both APD30 and APD50. In addition, compared to the control group, both BrS groups displayed a higher incidence of arrhythmic events at baseline, including early afterdepolarizations (EADs) or delayed afterdepolarizations (DADs) (0 out of 10 for control, 3 out of 10 for BrS-SCN5A, 2 out of 9 for BrS-SCN1B). After treated with quinidine within 3-fold the ETPC, both BrS-CMs eliminated spontaneously occurring triggered events (Fig. 5H and I). These results highlighted that quinidine had positive effects in BrS-CMs with insurance of safety.
Figure 5.
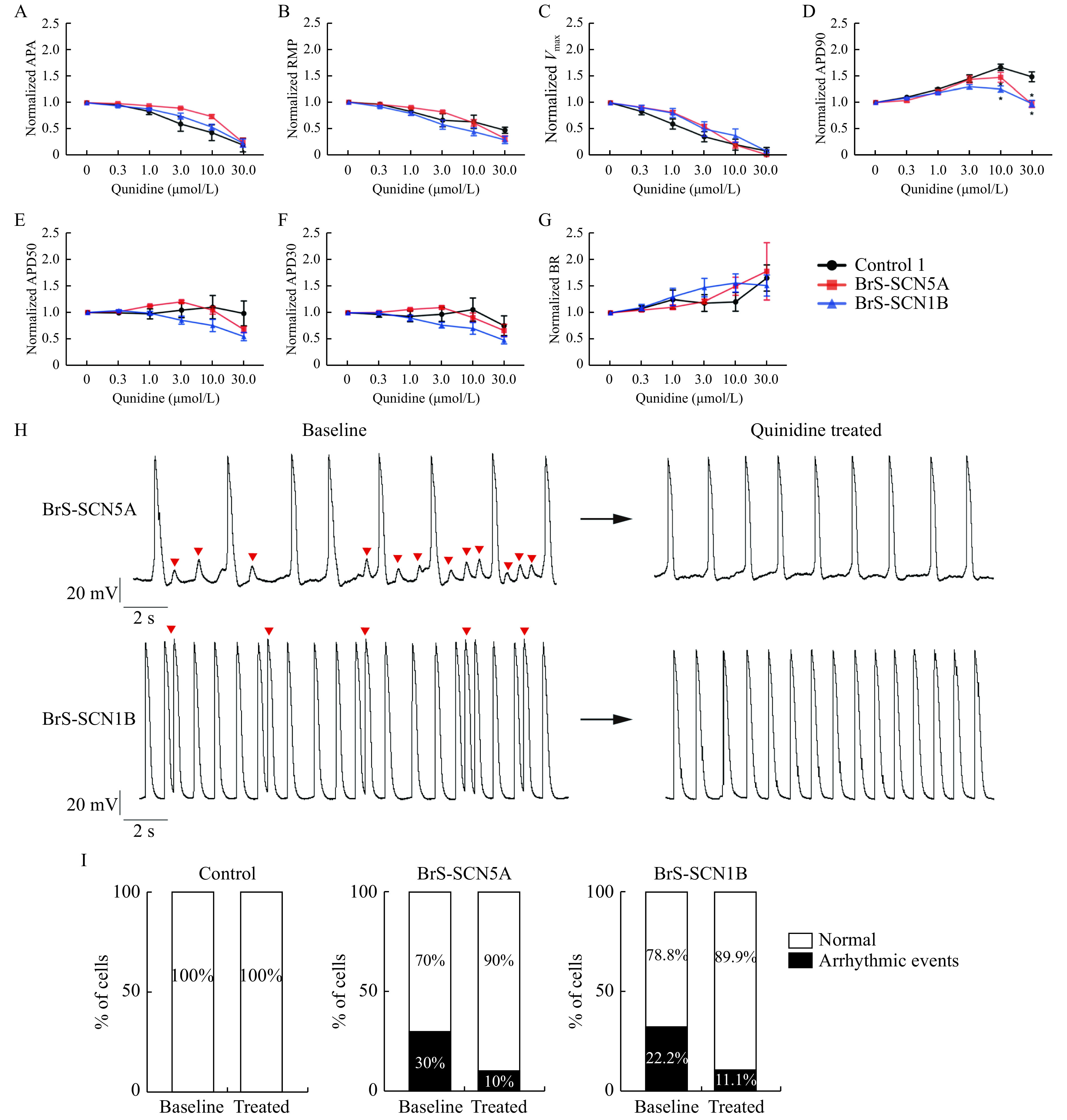
iPSC-CMs in response to escalating concentrations of quinidine.
A–G: Quinidine-treated CMs exhibited the reduction of APA (A), RMP (B), Vmax (C), and the prolongation of APD90 (D) after standardization, while changes in APD50 (E) and APD30 (F) were modest. All the groups had comparable beating rate (BR) (G). Control: n=10; BrS-SCN5A: n=10; BrS-SCN1B: n=9. Values given are mean±SEM. *P<0.05 vs. control (by paired t-test). H: Representative spontaneous AP traces. Left panels: BrS iPSC-CMs AP baseline with EADs and DADs (indicated by red arrows). Right panels: quinidine within 3-fold the ETPC can abolish baseline EADs. I: Percentage of cardiomyocytes displaying arrhythmic events at baseline and after treating with 10 µmol/L quinidine (Control: n=10; BrS-SCN5A: n=10; BrS-SCN1B: n=9, Chi-square test). iPSC-CMs: induced pluripotent stem cell-derived cardiomyocytes; APA: action potential amplitude; RMP: resting membrane potential; Vmax: maximum upstroke velocity; APD: action potential duration; AP: Action potential; EADs: early after depolarizations; DADs: delayed after depolarizations.
Discussion
BrS is an inheritable ion channelopathy associated with a propensity to fetal ventricular arrhythmias. Herein, we identified two novel mutations (SCN5A c.4213G>A andSCN1B c.590C>T) from two BrS patients, verified and compared their pathogenic phenotypes. Our results demonstrated that iPSC-CMs derived from different genotypes of BrS patients showed similar patterns, including the prolongation of APD, impairment ofINa, and high prevalence of proarrhythmic events. Although many patient-specific iPS-CM models have been generated to recapitulate Brugada syndrome at the cellular level (Supplementary Table 4 , available online), none has assessed the cellular response to quinidine, a controversial drug for BrS treatment. Here, our study provided a comprehensive evaluation of quinidine on BrS-CMs, supporting the safety of the drug for lifelong use.
Relatively mature phenotype by prolonged cultivation of iPSC-CMs
It is well known that newly differentiated iPSC-CMs are more fetal-like in phenotype than adult cardiomyocytes, which has impeded the widespread use of iPSC-CMs for scientific studies[22–23]. Currently, prolonged culturing is the most accessible and easy approach for prompting cardiac maturation[24]. In the present study, the cells were cultivated for 60 days in vitro, till differentiation into a mature state. Our morphological analysis indicated that the structures of BrS-CMs were normal. On the other hand, the organized sarcomeres together with ultra-micro aligned Z-disc structures indicated that the D60 cardiomyocytes exhibited a high degree of maturity. We have reported that ion channel function relies on the iPSC-CM maturation status[25]. Thus, applying such relatively mature CMs for disease modeling would be highly reliable.
Electrophysiological abnormalities caused by Nav1.5 gene variations
Among BrS patients, SCN5A mutations are found in a quarter of patients[26]. Meanwhile, other gene mutations including SCN1B have been rarely reported[27–28]. Herein, iPSC-CMs carrying the SCN1B missense mutation (c.590C>T) also had a significant decrease in sodium current, although the decline was not as obvious as that ofSCN5A mutant BrS-CMs. This could be explained by the fact that SCN1B encodes only the accessory subunit of the sodium channel[29].
Compared to INa, action potential can better reflect the impact of variants on the entire cellular electrophysiology, including the compensation of other ion channels to the sodium current impairment. Our study illuminated the abnormality in AP profiles of BrS-CMs, which is in line with previous studies[30]. The BrS-CMs also presented a prolonged AP duration. These characteristics are similar to those of iPS-CMs carrying SCN5A p.1795insD, which causes overlap between the syndromes of BrS and long QT phenotypes[27]. Moreover, BrS-CMs exhibited multiple abnormal calcium-related activities, including EAD-like and DAD-like events, which are thought to be a surrogate for polymorphic VT[31–32]. Collectively, both Nav1.5 gene mutants in our study showed impaired electrophysiological function, triggering adverse events related to BrS clinical manifestations.
Pharmacologic responses to quinidine
The pharmacologic basis of treating BrS is aimed at rebalancing outward and inward currents of the epicardium, either by reducing the outward Ito current or increasing the inward ICa current[33]. Quinidine, a class IA antiarrhythmic agent with significant Ito blocking properties, is reported to reduce electrical heterogeneity, realize synchrony, and terminate phase 2 reentrant activity[34]. However, the efficacy of quinidine is controversial as it may induce prolongation of the QT interval, leading to ventricular arrhythmia[35–36]. Since BrS is a genetic condition, the discrepancy in drug responses may result from the different genetic backgrounds. In our study, we evaluated the effects of quinidine on BrS-CMs with different sodium channel gene variations. We found that all the cell lines showed depressed phase 0 depolarization after quinidine treatment. APD90 presented with a concentration-dependent prolongation caused by quinidine, which is the result of inhibition of sodium channel in phase 0 and potassium channel in phase 1 and 3. It is worth mentioning that the prolongation was more profound in the control cells. The downward trend appeared when the concentration reached 10 µmol/L, suggesting that the L-type calcium channel was blocked by quinidine at such a concentration[37]. There was no prominent effect on APD30 or APD50. These findings are in line with the drug experiment on human ventricular trabeculae[38] or drug tests using optical mapping[39], indicating the pronounced blockade of potassium current especially hERG. Besides, we observed that adverse events were reduced or at least not added in the range of 3-fold the ETPC of quinidine in BrS-CMs. These findings support the idea that quinidine is an effective and safe drug for BrS patients with genetic risk.
Limitation
First, genotype might not equal pathogenesis. A low prevalence of mutation carriers leads to an ambiguous relationship between genotype and phenotype. Patients in the absence of any mutation may have an anatomic structural variant in the right ventricular outflow tract that is sufficient for pathogenesis[40]. However, we believe that the genetic basis is an important contributor to the pathogenesis of BrS. It is reported that the expression heterogeneity in Nav1.5 expression could determine the conduction heterogeneities in different regions[41]. Second, our present model can enable study on the cellular level, but not on the organ or tissue level. Previous studies performed in a canine ventricular wedge model successfully captured the BrS ECG pattern[42]. However, it is unable for iPSC-CMs from monolayer culture to achieve such potential difference in vitro. Hopefully, the pathophysiological changes recapitulated here can be coupled with new techniques for engineering heart tissue.
Conclusion
In summary, our study verified the pathogenicity of two novel mutations in diverse subunits of sodium channel (SCN5A c.4213G>A andSCN1B c.590C>T) using patient-specific iPSC-CMs. Both iPSC-CMs recapitulated similar phenotypes of impaired sodium channel on the cellular level, including deleteriousINa, abnormal action potential parameters, increased arrhythmia events, and cardiotoxicity-free quinidine usage within safe dose. Our findings help to understand the mechanisms of BrS at the cellular level and support patient-specific iPSC-CMs as a promising platform for therapeutic target exploration.
The work was supported by the Natural Science Foundation of Jiangsu Province of China (Grant No. BK20160134 to LW and BK20191071 to CC), National Natural and Science Foundation of China (Grant No. 81900295 to CC), and Special Foundation for Clinical Science and Technology of Jiangsu Province (Grant No. BE2017754 to HWC).
Acknowledgments
The work was supported by the Natural Science Foundation of Jiangsu Province of China (Grant No. BK20160134 to LW and BK20191071 to CC), National Natural and Science Foundation of China (Grant No. 81900295 to CC), and Special Foundation for Clinical Science and Technology of Jiangsu Province (Grant No. BE2017754 to HWC).
Footnotes
CLC number: R541.7, Ducument code: A
The authors reported no conflict of interests.
References
- 1.Brugada P, Brugada J Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20(6):1391–1396. doi: 10.1016/0735-1097(92)90253-J. [DOI] [PubMed] [Google Scholar]
- 2.Antzelevitch C Brugada syndrome. Pacing Clin Electrophysiol. 2006;29(10):1130–1159. doi: 10.1111/j.1540-8159.2006.00507.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brugada J, Brugada R, Brugada P Pharmacological and device approach to therapy of inherited cardiac diseases associated with cardiac arrhythmias and sudden death. J Electrocardiol. 2000;33 Suppl 1:41–47. doi: 10.1054/jelc.2000.20322. [DOI] [PubMed] [Google Scholar]
- 4.Nordkamp LRAO, Postema PG, Knops RE, et al Implantable cardioverter-defibrillator harm in young patients with inherited arrhythmia syndromes: a systematic review and meta-analysis of inappropriate shocks and complications. Heart Rhythm. 2016;13(2):443–454. doi: 10.1016/j.hrthm.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 5.Sieira J, Brugada P Management of Brugada syndrome 2016: should all high risk patients receive an ICD? All high-risk patients should receive an implantable cardiac defibrillator. Circ Arrhythm Electrophysiol. 2016;9(11):e004195. doi: 10.1161/CIRCEP.116.004195. [DOI] [PubMed] [Google Scholar]
- 6.Gourraud JB, Barc J, Thollet A, et al The Brugada syndrome: a rare arrhythmia disorder with complex inheritance. Front Cardiovasc Med. 2016;3:9. doi: 10.3389/fcvm.2016.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schulze-Bahr E, Eckardt L, Breithardt G, et al Sodium channel gene (SCN5A) mutations in 44 index patients with Brugada syndrome: different incidences in familial and sporadic disease. Hum Mutat. 2003;21(6):651–652. doi: 10.1002/humu.9144. [DOI] [PubMed] [Google Scholar]
- 8.Risgaard B, Jabbari R, Refsgaard L, et al High prevalence of genetic variants previously associated with Brugada syndrome in new exome data. Clin Genet. 2013;84(5):489–495. doi: 10.1111/cge.12126. [DOI] [PubMed] [Google Scholar]
- 9.Wang L, Han Z, Dai J, et al Brugada syndrome caused by sodium channel dysfunction associated with a SCN1B variant A197V. Arch Med Res. 2020;51(3):245–253. doi: 10.1016/j.arcmed.2020.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Yan GX, Antzelevitch C Cellular basis for the Brugada syndrome and other mechanisms of arrhythmogenesis associated with ST-segment elevation. Circulation. 1999;100(15):1660–1666. doi: 10.1161/01.CIR.100.15.1660. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, Liang P, Lan F, et al Genome editing of isogenic human induced pluripotent stem cells recapitulates long QT phenotype for drug testing. J Am Coll Cardiol. 2014;64(5):451–459. doi: 10.1016/j.jacc.2014.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jans D, Callewaert G, Krylychkina O, et al Action potential-based MEA platform for in vitro screening of drug-induced cardiotoxicity using human iPSCs and rat neonatal myocytes . J Pharmacol Toxicol Methods. 2017;87:48–52. doi: 10.1016/j.vascn.2017.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Matsa E, Rajamohan D, Dick E, et al Drug evaluation in cardiomyocytes derived from human induced pluripotent stem cells carrying a long QT syndrome type 2 mutation. Eur Heart J. 2011;32(8):952–962. doi: 10.1093/eurheartj/ehr073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Selga E, Sendfeld F, Martinez-Moreno R, et al Sodium channel current loss of function in induced pluripotent stem cell-derived cardiomyocytes from a Brugada syndrome patient. J Mol Cell Cardiol. 2018;114:10–19. doi: 10.1016/j.yjmcc.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang P, Sallam K, Wu HD, et al Patient-specific and genome-edited induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of Brugada syndrome. J Am Coll Cardiol. 2016;68(19):2086–2096. doi: 10.1016/j.jacc.2016.07.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Antzelevitch C, Brugada P, Borggrefe M, et al Brugada syndrome: report of the second consensus conference. Heart Rhythm. 2005;2(4):429–440. doi: 10.1016/j.hrthm.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 17.Priori SG, Wilde AA, Horie M, et al HRS/EHRA/APHRS expert consensus statement on the diagnosis and management of patients with inherited primary arrhythmia syndromes: Document endorsed by HRS, EHRA, and APHRS in May 2013 and by ACCF, AHA, PACES, and AEPC in June 2013. Heart Rhythm. 2013;10(12):1932–1963. doi: 10.1016/j.hrthm.2013.05.014. [DOI] [PubMed] [Google Scholar]
- 18.Belhassen B, Viskin S, Fish R, et al Effects of electrophysiologic-guided therapy with class IA antiarrhythmic drugs on the long-term outcome of patients with idiopathic ventricular fibrillation with or without the Brugada syndrome. J Cardiovasc Electrophysiol. 1999;10(10):1301–1312. doi: 10.1111/j.1540-8167.1999.tb00183.x. [DOI] [PubMed] [Google Scholar]
- 19.Probst V, Gourraud JB Quinidine in Brugada syndrome: still a long way to go…. Circ Arrhythm Electrophysiol. 2015;8(6):1309–1310. doi: 10.1161/CIRCEP.115.003576. [DOI] [PubMed] [Google Scholar]
- 20.Malhi N, Cheung CC, Deif B, et al Challenge and impact of quinidine access in sudden death syndromes: a national experience. JACC: Clin Electrophysiol. 2019;5(3):376–382. doi: 10.1016/j.jacep.2018.10.007. [DOI] [PubMed] [Google Scholar]
- 21.Blinova K, Dang Q, Millard D, et al International multisite study of human-induced pluripotent stem cell-derived cardiomyocytes for drug proarrhythmic potential assessment. Cell Rep. 2018;24(13):3582–3592. doi: 10.1016/j.celrep.2018.08.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lieu DK, Fu JD, Chiamvimonvat N, et al Mechanism-based facilitated maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Arrhythm Electrophysiol. 2013;6(1):191–201. doi: 10.1161/CIRCEP.111.973420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poon E, Yan B, Zhang S, et al Transcriptome-guided functional analyses reveal novel biological properties and regulatory hierarchy of human embryonic stem cell-derived ventricular cardiomyocytes crucial for maturation. PLoS One. 2013;8(10):e77784. doi: 10.1371/journal.pone.0077784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuppusamy KT, Jones DC, Sperber H, et al Let-7 family of microRNA is required for maturation and adult-like metabolism in stem cell-derived cardiomyocytes. Proc Natl Acad Sci U S A. 2015;112(21):E2785–E2794. doi: 10.1073/pnas.1424042112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang J, Cui C, Nan H, et al Graphene sheet-induced global maturation of cardiomyocytes derived from human induced pluripotent stem cells. ACS Appl Mater Interfaces. 2017;9(31):25929–25940. doi: 10.1021/acsami.7b08777. [DOI] [PubMed] [Google Scholar]
- 26.Kapplinger JD, Tester DJ, Alders M, et al An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Portero V, Casini S, Hoekstra M, et al Anti-arrhythmic potential of the late sodium current inhibitor GS-458967 in murine Scn5a-1798insD+/- and human SCN5A-1795insD+/- iPSC-derived cardiomyocytes . Cardiovasc Res. 2017;113(7):829–838. doi: 10.1093/cvr/cvx077. [DOI] [PubMed] [Google Scholar]
- 28.El-Battrawy I, Müller J, Zhao Z, et al Studying Brugada syndrome with an SCN1B variants in human-induced pluripotent stem Cell-derived cardiomyocytes. Front Cell Dev Biol. 2019;7:261. doi: 10.3389/fcell.2019.00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Malhotra JD, Chen C, Rivolta I, et al Characterization of sodium channel α- and β-subunits in rat and mouse cardiac myocytes. Circulation. 2001;103(9):1303–1310. doi: 10.1161/01.CIR.103.9.1303. [DOI] [PubMed] [Google Scholar]
- 30.Kosmidis G, Veerman CC, Casini S, et al Readthrough-promoting drugs gentamicin and PTC124 fail to rescue nav1.5 function of human-induced pluripotent stem cell-derived cardiomyocytes carrying nonsense mutations in the sodium channel gene SCN5A. Circ Arrhythm Electrophysiol. 2016;9(11):e004227. doi: 10.1161/CIRCEP.116.004227. [DOI] [PubMed] [Google Scholar]
- 31.Broyles CN, Robinson P, Daniels MJ Fluorescent, bioluminescent, and optogenetic approaches to study excitable physiology in the single cardiomyocyte. Cells. 2018;7(6):51. doi: 10.3390/cells7060051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kopljar I, Lu HR, Van Ammel K, et al Development of a human iPSC cardiomyocyte-based scoring system for cardiac hazard identification in early drug safety de-risking. Stem Cell Rep. 2018;11(6):1365–1377. doi: 10.1016/j.stemcr.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sieira J, Dendramis G, Brugada P Pathogenesis and management of Brugada syndrome. Nat Rev Cardiol. 2016;13(12):744–756. doi: 10.1038/nrcardio.2016.143. [DOI] [PubMed] [Google Scholar]
- 34.Belhassen B, Glick A, Viskin S Efficacy of quinidine in high-risk patients with Brugada syndrome. Circulation. 2004;110(13):1731–1737. doi: 10.1161/01.CIR.0000143159.30585.90. [DOI] [PubMed] [Google Scholar]
- 35.Vicente J, Johannesen L, Mason JW, et al Comprehensive T wave morphology assessment in a randomized clinical study of dofetilide, quinidine, ranolazine, and verapamil. J Am Heart Assoc. 2015;4(4):e001615. doi: 10.1161/JAHA.114.001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hai JJ, Wong CK, Chan PH, et al Quinidine for Brugada syndrome: panacea or poison? HeartRhythm Case Rep. 2016;2(6):486–490. doi: 10.1016/j.hrcr.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nishida A, Takizawa T, Matsumoto A, et al Inhibition of ATP-sensitive K+ channels and L-Type Ca2+ channels by amiodarone elicits contradictory effect on insulin secretion in MIN6 cells . J Pharmacol Sci. 2011;116(1):73–80. doi: 10.1254/jphs.10294FP. [DOI] [PubMed] [Google Scholar]
- 38.Page G, Ratchada P, Miron Y, et al Human ex-vivo action potential model for pro-arrhythmia risk assessment. J Pharmacol Toxicol Methods. 2016;81:183–195. doi: 10.1016/j.vascn.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pfeiffer-Kaushik ER, Smith GL, Cai B, et al Electrophysiological characterization of drug response in hSC-derived cardiomyocytes using voltage-sensitive optical platforms. J Pharmacol Toxicol Methods. 2019;99:106612. doi: 10.1016/j.vascn.2019.106612. [DOI] [PubMed] [Google Scholar]
- 40.Szél T, Antzelevitch C Abnormal repolarization as the basis for late potentials and fractionated electrograms recorded from epicardium in experimental models of Brugada syndrome. J Am Coll Cardiol. 2014;63(19):2037–2045. doi: 10.1016/j.jacc.2014.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martin CA, Guzadhur L, Grace AA, et al Mapping of reentrant spontaneous polymorphic ventricular tachycardia in a Scn5a+/- mouse model . Am J Physiol Heart Circ Physiol. 2011;300(5):H1853–H1862. doi: 10.1152/ajpheart.00034.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishida K, Fujiki A, Mizumaki K, et al Canine model of Brugada syndrome using regional epicardial cooling of the right ventricular outflow tract. J Cardiovasc Electrophysiol. 2004;15(8):936–941. doi: 10.1046/j.1540-8167.2004.04041.x. [DOI] [PubMed] [Google Scholar]