

Abstract
Objective
Obesity is associated with low-grade adipose tissue inflammation and locally elevated levels of several glycoprotein 130 (gp130) cytokines. The conversion of white into brown-like adipocytes (browning) may increase energy expenditure and revert the positive energy balance that underlies obesity. Although different gp130 cytokines and their downstream targets were shown to regulate expression of the key browning marker uncoupling protein 1 (Ucp1), it remains largely unknown how this contributes to the development and maintenance of obesity. Herein, we aim to study the role of gp130 cytokine signaling in white adipose tissue (WAT) browning in the obese state.
Methods
Protein and gene expression levels of UCP1 and other thermogenic markers were assessed in a subcutaneous adipocyte cell line, adipose tissue depots from control or adipocyte-specific gp130 knockout (gp130Δadipo) mice fed either chow or a high-fat diet (HFD), or subcutaneous WAT biopsies from a human cohort of lean and obese subjects. WAT browning was modeled in vitro by exposing mature adipocytes to isoproterenol after stimulation with gp130 cytokines. ERK and JAK-STAT signaling were blocked using the inhibitors U0126 and Tofacitinib, respectively.
Results
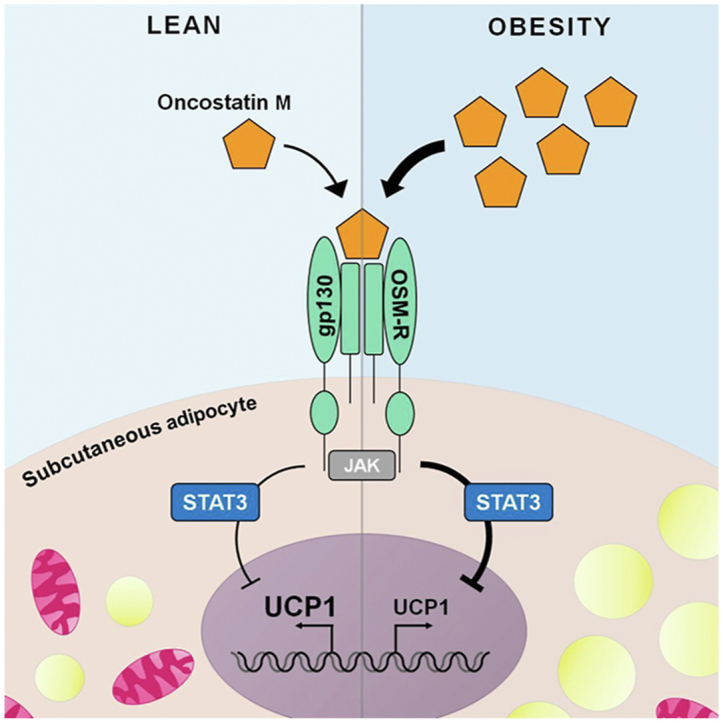
Inguinal WAT of HFD-fed gp130Δadipo mice exhibited significantly elevated levels of UCP1 and other browning markers such as Cidea and Pgc-1α. In vitro, treatment with the gp130 cytokine oncostatin M (OSM) lowered isoproterenol-induced UCP1 protein and gene expression levels in a dose-dependent manner. Mechanistically, OSM mediated the inhibition of Ucp1 via the JAK-STAT but not the ERK pathway. As with mouse data, OSM gene expression in human WAT positively correlated with BMI (r = 0.284, p = 0.021, n = 66) and negatively with UCP1 expression (r = −0.413, p < 0.001, n = 66).
Conclusions
Our data support the notion that OSM negatively regulates thermogenesis in WAT and thus may be an attractive target for treating obesity.
Keywords: Browning, High-fat diet, Obesity, Oncostatin M, UCP1, White adipose tissue
Abbreviations: BAT, brown adipose tissue; CM, complete medium; epi, epididymal; ERK, extracellular signal-regulated kinase; gp130, glycoprotein 130; gp130Δadipo, adipocyte-specific glycoprotein 130 knockout; HFD, high-fat diet; ing, inguinal; JAK, janus kinase; OSM, oncostatin M; OSMRΔadipo, adipocyte-specific oncostatin M receptor knockout; SOCS3, suppressor of cytokine signaling 3; STAT, signal transducer and activator of transcription; SVF, stromal vascular fraction; TBS-T, tris-buffered saline with 0.1% Tween; UCP1, uncoupling protein 1; WAT, white adipose tissue
Graphical abstract
Highlights
-
•
OSM is regulated under obesity and negatively correlates with UCP1 in WAT.
-
•
OSM suppresses isoproterenol-induced UCP1 in subcutaneous adipocytes.
-
•
OSM signals through the gp130-STAT3 pathway to lower UCP1 expression.
-
•
Obese mice lacking gp130 in adipocytes exhibit increased WAT browning.
1. Introduction
Obesity manifests upon a sustained positive energy balance, throughout which excess energy is stored in white adipose tissue (WAT) depots. The key to reversing obesity therefore lies in attaining a negative energy balance. Brown adipocytes can dissipate energy through non-shivering thermogenesis, an essential process for maintaining core temperature that requires uncoupling protein 1 (UCP1). Besides cold exposure–mediated stimulation of β-adrenergic receptors, the main physiological stimulus of brown adipose tissue (BAT) activation, a large number of alternative stimuli, including exercise and specific diets, have been reported [1,2]. However, since obese individuals seem to have rather small amounts of functional BAT [[3], [4], [5]], it may be more promising to target their WAT depots instead. In particular, WAT can burn rather than store energy by converting white into brown-like adipocytes (browning). Consequently, the focus of obesity research on WAT browning has gained substantial momentum over the past decade [6]. Despite huge efforts, only a small number of drugs with proven or presumed effects on WAT browning are presently available in clinic [7], making the search for better alternatives imperative.
The expansion of adipose tissue depots is paralleled by an invasion of immune cells that concomitantly leads to a state of low-grade inflammation [8]. Immune cells communicate with adipocytes and modulate their metabolic functions via the secretion of pro-inflammatory cytokines. The glycoprotein 130 (gp130) cytokine family comprises a main subgroup of such cytokines that consists of interleukin (IL)-6, IL-11, IL-27, IL-35, cardiotrophin-1, cardiotrophin-like cytokine, ciliary neurotrophic factor, leukemia inhibitory factor and oncostatin M (OSM). Although each of these cytokines has a unique receptor on the surface of the adipocyte to mediate intracellular signaling, each receptor consists of at least one gp130 subunit making gp130 the common signal transducer for all family members [9,10]. The two key intracellular signaling cascades activated by all gp130 cytokines are the extracellular signal-regulated kinase (ERK) and Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathways [11]. As such, gp130 cytokines can influence a plethora of metabolic processes. For instance, making use of mice that lacked the gp130 protein in adipocytes, specifically (gp130Δadipo mice), we previously unveiled that gp130 cytokine signaling regulates free fatty acid and leptin release in WAT, thereby indirectly affecting insulin release from pancreatic islets [12,13]. Interestingly, several signaling molecules upstream as well as downstream of the gp130 signaling complex have been demonstrated to be related to WAT browning. For example, activation of both the ERK and the JAK-STAT pathway in adipocytes was associated with either positive or negative changes in WAT browning [[14], [15], [16], [17]]. Furthermore, the gp130 cytokines IL-6 and OSM were linked to more and less UCP1 expression in WAT, respectively [[18], [19], [20]]. Although they are both elevated in WAT in obesity [20,21], their role in WAT browning in the latter condition remains largely unknown. Herein, we explored the role of gp130 signaling in WAT browning in the obese state using previously characterized gp130Δadipo mice. Moreover, by performing experiments in cultured white adipocytes and human WAT, we aimed to shed light on the opposing claims of up- and downstream players of gp130 in this process.
2. Materials and methods
2.1. Human samples
We included 159 individuals with a wide range of BMI (19–73 kg/m2), type 2 diabetes (n = 84), and normal glucose metabolism (n = 75). Paired abdominal subcutaneous adipose tissue biopsies were taken during elective sleeve gastrectomy, Roux-en-Y gastric bypass, and hernia or cholecystectomy surgeries and processed as previously described [22]. The study was approved by the Ethics Committee of the University of Leipzig (approval no: 159-12-21052012) and performed in accordance with the declaration of Helsinki. All subjects gave written informed consent before taking part in this study. RNA from adipose tissue was extracted using RNeasy Lipid tissue Mini Kit (Qiagen, Hilden, Germany). Quantity and integrity of RNA was monitored with NanoVue plus Spectrophotometer (GE Healthcare, Freiburg, Germany). 1 μg total RNA from subcutaneous adipose tissue was reverse transcribed with standard reagents (Life technologies, Darmstadt, Germany). cDNA was then processed for TaqMan probe-based quantitative real-time polymerase chain reaction (qPCR) using the QuantStudio 6 Flex Real-Time PCR System (Life technologies, Darmstadt, Germany). Expression of OSM, UCP1, and IL-6 were calculated by standard curve method and normalized to the expression of hypoxanthine guanine phosphoribosyltransferase 1 (HPRT1) as a housekeeping gene. The probes (Life technologies, Darmstadt, Germany) for OSM (Hs00171165_m1), UCP1 (Hs00222453_m1), IL-6 (Hs00985639_m1), and HPRT1 (Hs01003267_m1) spanned exon–exon boundaries to improve the specificity of the qPCR. For analysis, only subjects in whom all three genes were detected (n = 66) were included.
2.2. Mouse samples
To obtain gp130Δadipo mice, gp130 F/F mice [23] were crossed with Adipoq-Cre mice (The Jackson Laboratory, Bar Harbor, ME, USA) as described [12]. Littermate mice with floxed gp130 but absent Cre-recombinase (Cre) expression were used as controls (gp130 F/F). Every animal was considered an experimental unit. Animals were housed in groups of 2–5 in a specific pathogen-free, temperature-controlled environment (±22 °C) with a 12-h light/12-h dark cycle and crinkles as bedding material. Mice received standard chow diet (ProvimiKliba, Kaiseraugst, Switzerland) or were switched to HFD (D12331; Research Diets, New Brunswick, NJ, USA) at the age of 6 weeks. Animals were allocated to diets based on body weight (similar mean ± SD in starting body weight between the groups). Water was provided ad libitum. Experimenters were not blinded to the identity of a specific mouse. Adipose tissue samples were harvested from 18-week-old male mice after a 5-hour fasting period. Samples were snap-frozen and stored at −80 °C. Experiments were performed according to Swiss animal protection laws and approved by the cantonal veterinary office in Zurich, Switzerland.
2.3. Cell culture and treatments
Previously characterized murine subcutaneous white pre-adipocytes [24] or 3T3-L1 cells were plated in 12- or 24-well plates coated with 0.1% wt/vol. gelatin (Bio-Rad 170-6537). Cells were maintained in complete medium (CM) consisting of DMEM (25 mmol/L glucose) supplemented with 10% vol./vol. fetal bovine serum and 1% vol./vol. penicillin/streptomycin (all from Invitrogen, Basel, Switzerland) that was changed every 2–3 days. Differentiation towards mature adipocytes started two days after reaching confluency. In the first 72 h, CM was supplemented with 3-isobutyl-1-methylxanthine (500 μmol/L), dexamethasone (1 μmol/L), insulin (1.7 μmol/L), and rosiglitazone (1 μmol/L). For the next 2 days, cells were cultured in CM with insulin (0.5 μmol/L). Experiments with subcutaneous adipocytes commenced after another 2 days in CM without further supplements. Differentiation of 3T3-L1 cells, cultivation of RAW 264.7 cells, and isolation of adipocytes and stromal vascular fractions (SVF) was performed as previously described [25,26].
Fully differentiated adipocytes were treated with vehicle or various concentrations of OSM (495-MO, R&D systems, Zug, Switzerland), IL-6 (AG-40B-0108, AdipoGen, Liestal, Switzerland), or leptin (498-OB, R&D systems, Zug, Switzerland) for up to 24 h as indicated in the figure legends. Browning was induced by 3- or 6-hour treatment with 1 or 10 μmol/L isoproterenol as indicated. For inhibitor experiments, cells were pre-treated for 1 h with 50 μmol/L U0126 (ab120241, Abcam, Cambridge, UK), 2 μmol/L Tofacitinib (CP-690550, LuBioScience GmbH, Zurich, Switzerland), or a vehicle. Isolated SVF cells or cultured RAW cells were treated with 10 ng/L lipopolysaccharides (LPS; Sigma–Aldrich, Buchs, Switzerland) for 6 h 3T3-L1 cells were treated with 100 nM human insulin (Actrapid, Novo Nordisk Pharma AG, Zurich, Switzerland) for 10 min. Cells were harvested after a brief wash in ice-cold PBS. Cell culture data represent at least two biological replicates, and each condition was generally performed in duplicates. Cells were not tested for mycoplasma contamination. Experimenters were not blinded to the identity of a specific sample.
2.4. Protein lysis and western blot analysis
Tissues or cells were lysed in a buffer containing 150 mmol/L NaCl, 50 mmol/L Tris–HCl (pH 7.5), 1 mmol/L EGTA, 1% vol./vol. NP-40, 0.25% wt/vol. sodium deoxycholate, 1 mmol/L sodium pyrophosphate, 1 mmol/L sodium vanadate, 1 mmol/L NaF, 10 mmol/L sodium β-glycerolphosphate, 0.2 mmol/L PMSF, and 0.1% vol./vol. protease inhibitor cocktail (Sigma–Aldrich, Buchs, Switzerland). Protein concentrations were determined by BCA assay (Pierce, Rockford, IL, USA). Equal amounts of protein were resolved using SDS-PAGE and electro-transferred onto 0.2-μm nitrocellulose membranes (BioRad, Reinach, Switzerland). Equal loading of proteins was verified by Ponceau S staining. Blots were blocked with 5% wt/vol. dry fat milk dissolved in tris-buffered saline (150 mmol/L NaCl, 50 mmol/L Tris–HCl) containing 0.1% vol./vol. Tween (TBS-T). Membranes were incubated overnight at 4 °C with primary antibody diluted in TBS-T with 5% wt/vol. bovine serum albumin and 0.02% wt/vol. sodium azide, followed by 1 h incubation at room temperature with a secondary antibody diluted in TBS-T with 5% wt/vol. dry fat milk. Membranes were developed using the ECL system and antibody–antigen complexes were detected with a ChemiDoc™ MP Imaging System (Biorad, Reinach, Switzerland). Primary antibodies were used at a dilution of 1:1000 unless specified and include: UCP1, PA1-24894 (Thermofisher Scientific, Waltham, MA, USA); pAkt (Thr308), #13038; Akt, #9272; pERK1/2, #9101; ERK1/2, #9211; pSTAT3, #9145; STAT3, #9132 (Cell Signaling, Danvers, MA, USA); PPARγ, sc-7196 (1:200, Santa Cruz Biotechnology, Santa Cruz, CA, USA); and Actin, MAB1501 (Millipore, Darmstadt, Germany). Molecular weight/pattern of bands were compared to existing literature when possible. Details of antibodies used, such as data sheets, citations, and reviews, can be found via the above-listed catalogue numbers. Secondary antibodies, diluted 1:5000, are goat-anti-rabbit, ab6721 and mouse-anti-rabbit, ab6789 (Abcam, Cambridge, UK).
2.5. RNA extraction and RT-qPCR analysis
Total RNA was extracted from homogenized tissues with the NucleoSpin® RNA Set for NucleoZOL or from cells with NucleoSpin® RNA isolation kit (Macherey–Nagel, Düren, Germany). RNA concentration was determined via NanoDrop® (ThermoFisher Scientific, Waltham, MA, USA). Equal amounts of RNA (50–500 ng) were reverse transcribed with the GoScript™ Reverse Transcription System (Promega, Madison, WI, USA) and cDNA was amplified by real-time PCR using the following probes/primers: Ucp1, Mm0124486_m1; Cidea, Mm00432554_m1; Ppargc1a, Mm01208835_m1; Prdm16, Mm00712556_m1; Dio2, Mm00-515664_m1; Pparg, Mm00440940_m1; Ppara, Mm00627-559_m1; Tbx1, Mm00448949_m1; Tmem26, Mm01173641_m1; Osm, Mm01193966_m1; gp130 (Il6st), Mm00439665_m1; 18s, 4352930 (Applied Biosystems, Rotkreuz, Switzerland). Autotaxin (Enpp2), Fw: GACCCTAAAGCCATTATTGCTAA, Rv: GGGAAGGTGCTGTTTCATGT [27]; Probe: AAACCAGATCAGCACTTTAAGCC (Microsynth, Balgach, Switzerland). Relative gene expression values were obtained after normalization to 18s using the 2−ΔΔCt method. Outliers, defined as values exceeding 2 SD from the mean, were excluded.
2.6. Hematoxylin-eosin and Oil-Red-O staining
Inguinal snap-frozen tissues were fixed overnight in 4% buffered formalin, dehydrated, and embedded in paraffin blocks. 4-μm-thick sections were cut and stained for hematoxylin and eosin. For Oil-Red-O staining, cells were fixed in 4% wt/vol. paraformaldehyde solution (AppliChem, Darmstadt, Germany) for 30 min on ice, rinsed in dH2O, and dehydrated in 60% vol./vol. isoproterenol. Cells were stained with a solution of 0.3% wt/vol. Oil-Red-O (O0625; Sigma–Aldrich, Buchs, Switzerland) dissolved in 100% isoproterenol that was freshly diluted to 60% vol./vol. in dH2O. After 5–10 min, cells were rinsed twice in dH2O. Tissue slices or cells were imaged using an Axio Observer microscope platform (Zeiss, Jena, Germany). Tile scans were analyzed with ImageJ software to calculate the Oil-Red-O positive signal per well.
2.7. Data analysis
Data are presented as mean ± SE. Statistics were calculated using GraphPad Prism 8.00 (GraphPad Software, San Diego, CA, USA) by means of a paired or unpaired two-tailed Student's t-test (for normally distributed data), Mann–Whitney test (for not-normally-distributed data), one-way ANOVA with Tukey multiple comparisons correction, or linear regression analysis. A p-value < 0.05 was considered statistically significant. Power calculation analysis was not performed. Sample size was determined based on previous experiments performed in our laboratory.
2.8. Data and resource availability
The data supporting the findings of this study are available within the article, its Supplementary Information files, or from the corresponding author upon reasonable request. No applicable resources were generated or analyzed during the current study.
3. Results
3.1. Increased browning in ingWAT of HFD-fed gp130Δadipo mice
To test the hypothesis that gp130 plays a role in WAT browning, we examined the levels of UCP1 in white fat depots obtained from our previously characterized chow and high-fat diet (HFD)-fed sedentary and exercised gp130Δadipo mice [12,13,28]. The level of UCP1 was unchanged in inguinal (ing) WAT of chow-fed mice and undetectable in the less browning-prone epididymal (epi) WAT irrespective of diet and genotype (Supplementary Figure 1B–D). However, UCP1 protein levels were significantly elevated in the ingWAT of HFD-fed gp130Δadipo mice compared to littermate controls (Figure 1A–B). Similarly, UCP1 protein levels and the number of multi-locular cells were increased in the ingWAT of exercised HFD-fed gp130Δadipo mice, as illustrated by western blot and histological analysis, respectively (Supplementary Figure 1E–F). As expected, 12 weeks of HFD feeding markedly lowered Ucp1 mRNA and UCP1 protein levels in ingWAT of control mice (Supplementary Figure 1G–H). The above data infer that HFD-fed gp130Δadipo mice are partly protected from such decline in UCP1. To corroborate the latter observation, we performed complementary gene expression analysis of ingWAT depots from HFD-fed mice and found increased Ucp1 mRNA levels in knockout compared to control mice. Moreover, the transcripts of additional browning-related markers, including Cidea, Pgc-1α (Ppargc1a), and Tbx1 were significantly elevated (Figure 1C). Of note, no significant difference in UCP1 protein level was detected when comparing BAT depots of gp130Δadipo and gp130 F/F mice fed either regular chow (Supplementary Figure 1A) or HFD [28]. Together, these results indicate that while gp130 depletion may not affect the thermogenic potential of classical BAT, it protects from the HFD-induced reduction of WAT browning.
Figure 1.

Increased browning in ingWAT of HFD-fed gp130Δadipomice. (A) Representative Western blot of respective targets in ingWAT from HFD-fed gp130 F/F and gp130Δadipo mice. (B) Quantification of UCP1 protein levels in ingWAT of HFD-fed gp130 F/F (n = 11) and gp130Δadipo (n = 13) mice. (C) Gene expression levels of respective targets in ingWAT from HFD-fed gp130 F/F (n = 8–11) and gp130Δadipo mice (n = 9–14). (D) Body weight of HFD-fed gp130 F/F and gp130Δadipo mice at time of sacrifice and corresponding Ucp1 gene expression level in ingWAT (n = 22). (E–F) UCP1 and corresponding p-ERK1/2 or p-STAT3 protein levels in ingWAT from HFD-fed gp130 F/F and gp130Δadipo mice (n = 21). Open circles/squares represent individual gp130 F/F and gp130Δadipo ingWAT samples, respectively. ∗, p < 0.05; ∗∗, p < 0.01. Panel B, Mann–Whitney test; panel C, Student's t-test; panels D, E, and F, Linear Regression analysis. Error bars represent S.E.
Although we previously reported that neither body weight nor energy expenditure was significantly different between HFD-fed gp130Δadipo and gp130 F/F mice [12,28], we hitherto analyzed 17 HFD-fed litters of both genotypes (n = 43 mice for gp130 F/F; n = 35 mice for gp130Δadipo) and observed that the average body weight per litter was significantly reduced in gp130Δadipo mice (−4.3 ± 1.7% body weight; p = 0.02). Importantly, body weight negatively correlated to Ucp1 expression in ingWAT of HFD-fed mice (Figure 1D), suggesting that the observed increase in browning contributed to reduced weight gain.
We next assessed levels of phosphorylated ERK1/2 (p-ERK1/2) and STAT3 (p-STAT3), both representing key proteins in the signaling cascades downstream of the gp130 receptor complex [11]. Levels of p-ERK1/2 and p-STAT3 were distributed relatively heterogeneously in ingWAT of HFD-fed gp130Δadipo and gp130 F/F mice (Figure 1A). Correlative analysis showed that p-STAT3 but not p-ERK1/2 was negatively associated with UCP1 (Figure 1E–F), suggesting a possible involvement of the STAT3 signaling pathway in the regulation of Ucp1 transcription.
3.2. OSM dose-dependently lowers isoproterenol-induced UCP1 levels in cultured adipocytes
As outlined above, the gp130 cytokine family members IL-6 and OSM are elevated in obesity and may impact UCP1 levels [[18], [19], [20],29]. Moreover, the adipokine leptin, which showed reduced secretion and circulating levels in HFD-fed gp130Δadipo mice [12], may modulate WAT browning [[30], [31], [32]]. Expression of the gp130 downstream mediator and potential suppressor of WAT browning autotaxin (gene name Enpp2) [33,34] was not different between genotypes in ingWAT (Figure 1C) and thus unlikely contributes to elevated UCP1 in ingWAT of HFD-fed gp130Δadipo mice. The impacts of the three other candidates on browning were evaluated using a mouse-derived subcutaneous white adipocyte cell line. Basal Ucp1 mRNA and UCP1 protein levels were low and remained unchanged upon stimulation with either OSM or IL-6 (Supplementary Figure 2A and 2B). Subsequent induction of browning by the β3-adrenoreceptor agonist isoproterenol strongly induced UCP1 expression, as expected. Intriguingly, whilst no change in isoproterenol-mediated induction of UCP1 level was noted when adipocytes were stimulated with IL-6 or leptin, a dose-dependent reduction was observed upon incubation with OSM (Figure 2A–B; Supplementary Figure 2C). OSM elicited comparable effects in a second adipocyte cell line (3T3-L1 cells) by dose-dependently lowering Ucp1 gene expression induced by isoproterenol treatment (Figure 2C). These results indicate that OSM suppresses isoproterenol-induced UCP1 levels in cultured white adipocytes, thus suggesting that elevated WAT browning in HFD-fed gp130Δadipo mice can at least in part be attributed to the absence of OSM signaling.
Figure 2.

OSM dose-dependently lowers isoproterenol-induced UCP1 levels in cultured adipocytes. Normalized Ucp1 gene expression and/or UCP1 protein levels of subcutaneous adipocytes (A–B) or 3T3-L1 adipocytes (C) cultured in the presence of a vehicle or different concentrations of OSM or IL-6 for 24 h followed by 6 h of isoproterenol stimulation (n = 4–8). ∗∗, p < 0.01; ∗∗∗, p < 0.001 compared to vehicle-treated control. Panels A and B, One-way ANOVA. Error bars represent S.E.
Within the adipose tissue depot, immune cells are the main source of OSM and may be responsible for its increased levels under obesity [20,35]. In accordance with this, we found higher Osm gene expression in the immune cell with SVF compared to adipocytes isolated from wild-type mice. Moreover, Osm expression tended to be elevated in samples from HFD-fed compared to chow-fed mice (Supplementary Figure 3A). In contrast, no major differences were observed in gp130 mRNA levels between the two cell fractions, irrespective of diet (Supplementary Figure 3B). We hypothesized that an inflammatory milieu, as commonly present in fat depots under obesity, may trigger OSM production in local immune cells. To test our hypothesis, we stimulated SVF cells isolated from WAT of wild-type mice with LPS. Indeed, LPS-treated SVF cells exhibited significantly higher Osm gene expression compared to vehicle-treated cells (Supplementary Figure 3C). An even stronger LPS-induced upregulation of Osm was found in RAW cells, a macrophage cell line (Supplementary Figure 3D), rendering macrophages a plausible source of increased OSM production in WAT under obesity.
3.3. Inhibition of STAT3 activation prevents OSM-mediated reduction in Ucp1 expression
OSM was shown to inhibit pre-adipocyte differentiation [36] and induce dedifferentiation of mature adipocytes [37], possibly providing an alternative explanation for the OSM-mediated reduction in UCP1 expression. Therefore, we assessed the amount of lipid droplets by means of Oil-Red-O staining and determined levels of the adipocyte differentiation marker PPARγ in cultured white adipocytes after OSM treatment. As depicted in Figure 3A and Supplementary Figure 4A–B, no changes in lipid droplet content and PPARγ protein and mRNA levels were observed in OSM-treated subcutaneous adipocytes compared to vehicle-treated control cells. Of note, in 3T3-L1 adipocytes, PPARγ mRNA and protein levels were reduced after incubation with 100 ng/mL, but not with lower doses of OSM (Supplementary Figure 4C–F). Moreover, OSM treatment blunted insulin-induced phosphorylation of Akt in 3T3-L1 cells (Supplementary Figure 4E and G), suggesting that OSM impacts insulin signaling in white adipocytes. Collectively, these data indicate that a high OSM concentration may induce adipocyte dedifferentiation as previously described [37].
Figure 3.

Inhibition of STAT3 activation prevents OSM-mediated reduction in Ucp1 expression. (A–C) Western blot of subcutaneous white adipocytes stimulated with OSM or vehicle for indicated time periods and quantifications of phosphorylated ERK1/2 and STAT3 protein levels (n = 4). (D) Western blot of subcutaneous white adipocytes stimulated with vehicle (control), U0126 (50 μM), or Tofacitinib (2 μM) for 1 h. (E–F) Ucp1 gene expression levels of subcutaneous white adipocytes that were subsequently stimulated with either U0126 or Tofacitinib (1 h), OSM (3 h), and isoproterenol (3 h), displayed relative to a negative control (left panel) or as a percentage of the internal control with and without inhibitor (right panel; n = 6–8). ∗, p < 0.05; ∗∗, p < 0.01; ∗∗∗, p < 0.001. One-way ANOVA (E–F, left panel) or Student's t-test (E–F, right panel). Error bars represent S.E.
To elucidate whether ERK1/2 and/or STAT3 activation may contribute to OSM-mediated reduction in UCP1 expression, p-ERK1/2 and p-STAT3 abundance was assessed in mature subcutaneous adipocytes treated with OSM for different durations of time. As shown in Figure 3A–C, OSM induced ERK1/2 phosphorylation after 15 min but continuously activated STAT3 at all time points over the course of 24 h. To independently study the contributions of the ERK and the STAT pathway to the action of OSM, we used the MEK inhibitor U0126 and the JAK inhibitor Tofacitinib, respectively. As expected, U0126 successfully prevented ERK1/2 phosphorylation, while Tofacitinib completely blocked STAT3 activation (Figure 3D). Pre-incubating subcutaneous adipocytes with U0126 had no effect on the OSM-mediated blunting of Ucp1 mRNA levels (Figure 3F). Conversely, Tofacitinib partially restored Ucp1 mRNA expression (Figure 3F). Thus our data suggests that OSM suppresses browning in subcutaneous adipocytes JAK-STAT3 dependently.
3.4. OSM positively correlates with BMI, but negatively correlates with UCP1 in human subcutaneous WAT
To ascertain the clinical relevance of our mouse data, we measured OSM gene expression in subcutaneous fat biopsies from a cohort of lean and obese human subjects. A moderate but significant positive correlation between OSM mRNA levels and BMI was found (Figure 4A). As expected, IL-6 mRNA level also showed a similar positive correlation, whereas UCP1 gene expression correlated negatively with BMI (Figure 4B, C). Furthermore, while no linear relationship between UCP1 and IL-6 gene expression levels was observed, UCP1 correlated negatively with OSM expression (Figure 4D, E). These results support our mouse data and indicate that elevated levels of OSM associated with obesity may suppress browning of WAT in human.
Figure 4.

OSM positively correlates with BMI but negatively with UCP1 in human subcutaneous WAT. (A–E) Correlations between BMI and OSM, IL-6 or UCP1 mRNA level, and between UCP1 and OSM or IL-6 mRNA levels (n = 66). Gene expression data was obtained from subcutaneous human WAT biopsies. Samples with undetectable gene expression levels were excluded from analysis.
4. Discussion
Impaired WAT thermogenesis (i.e., the capacity of WAT to undergo browning) may contribute to the development and/or maintenance of obesity [38]. Herein, we provide evidence that OSM may contribute to blunted WAT browning. In both mice and humans, increased adiposity is associated with a surge of proinflammatory cytokines, including OSM in fat depots [20,39]. We simulated this in vivo observation by cultivating subcutaneous adipocytes in the presence of different OSM concentrations and observed a dose-dependent blunting of the main thermogenic marker UCP1. Conversely, expression of UCP1 and other browning markers was enhanced in ingWAT depots of HFD-fed gp130Δadipo mice with blocked OSM signaling. Clearly, we cannot exclude the possibility that other metabolic changes and/or gp130 cytokines other than OSM contributed directly or indirectly to the observed changes in ingWAT browning. Our human data confirm a previous small-scale study that indicated that OSM is increased in subcutaneous WAT of people with obesity [20] and suggests that, in conjunction with our mouse data, obesity-induced OSM impairs WAT browning. In support of the physiological significance of such finding, Ucp1 expression in WAT correlated negatively with the body weight and BMI of mice and men, respectively, suggesting that elevated OSM levels contribute to increased body weight in both species. Of note, the gp130Δadipo mouse exhibits a complex phenotype that, in addition to direct effects on adipocytes, includes indirect metabolic changes in the gut, pancreatic islets, liver, and hypothalamus [12,13,28]. In particular, knockout mice revealed reduced circulating levels of insulin and leptin, two substances that are critically involved in the regulation of energy expenditure and/or body weight [40,41]. Accordingly, such changes may have prevented a clearer effect on body weight and/or energy expenditure in these mice [12,28]. Moreover, when feeding mice an HFD, body weight gain is heterogeneous, with some cohorts/litters being more resistant than others. In the current re-analysis, we included about twice as many litters compared to our initial analysis and found that the average body weight per litter was significantly reduced in HFD-fed knockout mice, supporting the notion that increased browning in knockout mice reduces body weight.
A more selective model for the study of OSM signaling in vivo is the adipocyte-specific OSM receptor knockout mouse (OSMRΔadipo) [35]. The OSM receptor dimerizes with a gp130 subunit to form the OSM receptor complex. Of note, the OSMRΔadipo mouse blocks signaling of not only OSM, but also IL-31, as the latter also contains an OSMR subunit in its receptor complex [42,43]. Whether the OSMRΔadipo mouse also exhibits increased browning in ingWAT has not been reported. However, OSMRΔadipo mice are more sensitive to diet-induced obesity and display increased adipose tissue inflammation and insulin resistance [35]. Hence, this phenotype clearly differs from the one of gp130Δadipo mice, which may be expected from the molecular difference between the two models. While OSM action is prevented in either model, it is accompanied by the blocking of IL-31 signaling in the OSMRΔadipo mouse and interference with gp130 signaling in the gp130Δadipo mouse.
Using an in vitro model of WAT browning, we observed no direct effects from IL-6 and leptin on UCP1 expression. Considering that both IL-6 and leptin, like OSM, can signal via the JAK-STAT3 pathway, it remains unclear why OSM but not IL-6 or leptin mediates Ucp1 transcription. One possibility is that their common negative regulator SOCS3 acts differently depending on whether it is activated by OSM or by the other two factors. While SOCS3 was shown to prevent IL-6–mediated STAT3 phosphorylation after two hours [44], we observed herein that OSM promotes sustained activation of STAT3 for up to 24 h, suggesting a reduced inhibitory effect of SOCS3 at this OSM concentration. Other explanations may include differential cross-talk with other cytokines [45] or differences in localization or composition of the receptors. It should be noted that, in contrast to OSM, IL-6 and leptin are generally considered positive regulators of WAT browning. For instance, more WAT browning was found after systemic IL-6 treatment [18] or in mouse models of cancer, where IL-6 was upregulated under conditions of cachexia [19]. Leptin likely promotes WAT browning indirectly by signaling through the hypothalamus [30,31]. Because systemic leptin concentrations were lower in gp130Δadipo mice, we consider it improbable that leptin mediated the observed induction of ingWAT browning. Initially, we also hypothesized that the adipocyte-derived enzyme autotaxin might have contributed to elevated browning in gp130Δadipo mice, especially since an obesity-associated increase in gp130 signaling upregulated autotaxin in adipose tissue [34] and mice that transgenically overexpressed autotaxin displayed reduced browning in WAT [33]. Yet the observation that autotaxin levels in ingWAT were not different between HFD-fed gp130Δadipo and gp130 F/F mice infers an insignificant role of autotaxin on the browning phenotype in our mouse model.
The present study confirms previous work [20,29] showing that Osm levels are elevated in adipose tissue of obese mice, likely due to altered gene expression in the non-adipocyte fraction. We further find that an LPS-mediated inflammatory response induced Osm gene expression in SVF cells as well as cultured macrophages, providing one potential mechanism driving such increase. Despite the putatively higher Osm level in all fat depots, the unchanged UCP1 expression in BAT of HFD-fed gp130Δadipo mice would suggest that the inhibitory role of OSM is limited to ingWAT. This notion is underpinned by a recent study by Sanchez-Infantes et al., who found that cold-induced upregulation of Ucp1 was blunted in ingWAT of mice after local OSM infusion, while no such effect was observed when OSM was infused in supraclavicular BAT [29]. Besides higher OSM levels, increased OSM signaling may be effectuated by elevated OSM sensitivity or augmented expression of gp130 and/or OSM receptor (OSMR). However, while OSM was shown to induce gp130 and Osmr levels in 3T3-L1 cells [35], we did not find increased gp130 mRNA levels in adipocytes of HFD-compared to chow-fed mice. Collectively, these data suggest that lowering of local OSM secretion and/or signaling in WAT could be a potential therapeutic strategy to promote browning and, thus, combat obesity. In contrast, a recent study reported that systemic treatment with OSM improved the metabolic phenotype of obese mice [46]. This underscores the importance of understanding the differential role of OSM in various tissues.
In the current study, we employed Tofacinib to block OSM-mediated STAT3 signaling in cultured adipocytes. This novel JAK2 inhibitor successfully induced UCP1 mRNA in human adipocytes [16] and promoted WAT browning in HFD-fed mice [47]. However, we did not observe induction of Ucp1 by Tofacitinib in cultured adipocytes. Isoprotenerol may have masked such effect of Tofacitinib. Nonetheless, our findings are in agreement with the idea that JAK2 inhibition in white adipocytes may be beneficial in obese conditions.
In conclusion, our data support the notion that OSM negatively regulates thermogenesis in subcutaneous WAT via gp130-STAT3 signaling, thus making it an attractive target for the treatment of obesity.
Author's contribution
P.P.v.K and S.W. designed and performed experiments, analyzed data, and wrote the manuscript. T.S.O., M.Bo., and M.Bl. performed experiments. D.K. designed experiments, analyzed data, and wrote the manuscript. All authors reviewed and commented on the manuscript.
Acknowledgments
This work was supported by a grant from the Swiss National Science Foundation (#310030-179344 to DK), a grant from the Wolfermann-Nägeli Stiftung, and a grant from the Hartmann-Müller Stiftung, University of Zurich (#2292) (both to SW). Human studies were supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) through CRC 1052, project number 209933838, subproject B1 to MBl. We are grateful to Werner Muller, Faculty of Biology, Medicine and Health, University of Manchester, U.K., for providing gp130Δadipo mice.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2021.101341.
Conflict of interest
None declared.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Cannon B., Nedergaard J. Brown adipose tissue: function and physiological significance. Physiological Reviews. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 2.Kajimura S., Spiegelman B.M., Seale P. Brown and beige fat: physiological roles beyond heat generation. Cell Metabolism. 2015;22:546–559. doi: 10.1016/j.cmet.2015.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cypess A.M., Lehman S., Williams G., Tal I., Rodman D., Goldfine A.B. Identification and importance of brown adipose tissue in adult humans. New England Journal of Medicine. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Marken Lichtenbelt W.D., Vanhommerig J.W., Smulders N.M., Drossaerts J.M., Kemerink G.J., Bouvy N.D. Cold-activated brown adipose tissue in healthy men. New England Journal of Medicine. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 5.Virtanen K.A., Lidell M.E., Orava J., Heglind M., Westergren R., Niemi T. Functional brown adipose tissue in healthy adults. New England Journal of Medicine. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 6.Bartelt A., Heeren J. Adipose tissue browning and metabolic health. Nature Reviews Endocrinology. 2014;10:24–36. doi: 10.1038/nrendo.2013.204. [DOI] [PubMed] [Google Scholar]
- 7.Giordano A., Frontini A., Cinti S. Convertible visceral fat as a therapeutic target to curb obesity. Nature Reviews Drug Discovery. 2016;15:405–424. doi: 10.1038/nrd.2016.31. [DOI] [PubMed] [Google Scholar]
- 8.Donath M.Y., Shoelson S.E. Type 2 diabetes as an inflammatory disease. Nature Reviews Immunology. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 9.Ma D., Wang Y., Zhou G., Wang Y., Li X. Review: the roles and mechanisms of glycoprotein 130 cytokines in the regulation of adipocyte biological function. Inflammation. 2019;42:790–798. doi: 10.1007/s10753-019-00959-6. [DOI] [PubMed] [Google Scholar]
- 10.Taga T., Kishimoto T. Gp130 and the interleukin-6 family of cytokines. Annual Review of Immunology. 1997;15:797–819. doi: 10.1146/annurev.immunol.15.1.797. [DOI] [PubMed] [Google Scholar]
- 11.White U.A., Stewart W.C., Stephens J.M. Gp130 cytokines exert differential patterns of crosstalk in adipocytes both in vitro and in vivo. Obesity. 2011;19:903–910. doi: 10.1038/oby.2010.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wueest S., Item F., Lucchini F.C., Challa T.D., Muller W., Bluher M. Mesenteric fat lipolysis mediates obesity-associated hepatic steatosis and insulin resistance. Diabetes. 2016;65:140–148. doi: 10.2337/db15-0941. [DOI] [PubMed] [Google Scholar]
- 13.Wueest S., Laesser C.I., Boni-Schnetzler M., Item F., Lucchini F.C., Borsigova M. IL-6-Type cytokine signaling in adipocytes induces intestinal GLP-1 secretion. Diabetes. 2018;67:36–45. doi: 10.2337/db17-0637. [DOI] [PubMed] [Google Scholar]
- 14.Babaei R., Schuster M., Meln I., Lerch S., Ghandour R.A., Pisani D.F. Jak-TGFbeta cross-talk links transient adipose tissue inflammation to beige adipogenesis. Science Signaling. 2018;11 doi: 10.1126/scisignal.aai7838. [DOI] [PubMed] [Google Scholar]
- 15.Banks A.S., McAllister F.E., Camporez J.P., Zushin P.J., Jurczak M.J., Laznik-Bogoslavski D. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature. 2015;517:391–395. doi: 10.1038/nature13887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moisan A., Lee Y.K., Zhang J.D., Hudak C.S., Meyer C.A., Prummer M. White-to-brown metabolic conversion of human adipocytes by JAK inhibition. Nature Cell Biology. 2015;17:57–67. doi: 10.1038/ncb3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Y., Li R., Meng Y., Li S., Donelan W., Zhao Y. Irisin stimulates browning of white adipocytes through mitogen-activated protein kinase p38 MAP kinase and ERK MAP kinase signaling. Diabetes. 2014;63:514–525. doi: 10.2337/db13-1106. [DOI] [PubMed] [Google Scholar]
- 18.Knudsen J.G., Murholm M., Carey A.L., Bienso R.S., Basse A.L., Allen T.L. Role of IL-6 in exercise training- and cold-induced UCP1 expression in subcutaneous white adipose tissue. PloS One. 2014;9 doi: 10.1371/journal.pone.0084910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petruzzelli M., Schweiger M., Schreiber R., Campos-Olivas R., Tsoli M., Allen J. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metabolism. 2014;20:433–447. doi: 10.1016/j.cmet.2014.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Sanchez-Infantes D., White U.A., Elks C.M., Morrison R.F., Gimble J.M., Considine R.V. Oncostatin m is produced in adipose tissue and is regulated in conditions of obesity and type 2 diabetes. Journal of Clinical Endocrinology & Metabolism. 2014;99:E217–E225. doi: 10.1210/jc.2013-3555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lam Y.Y., Ha C.W., Campbell C.R., Mitchell A.J., Dinudom A., Oscarsson J. Increased gut permeability and microbiota change associate with mesenteric fat inflammation and metabolic dysfunction in diet-induced obese mice. PloS One. 2012;7 doi: 10.1371/journal.pone.0034233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rolle-Kampczyk U., Gebauer S., Haange S.B., Schubert K., Kern M., Moulla Y. Accumulation of distinct persistent organic pollutants is associated with adipose tissue inflammation. The Science of the Total Environment. 2020;748:142458. doi: 10.1016/j.scitotenv.2020.142458. [DOI] [PubMed] [Google Scholar]
- 23.Betz U.A., Bloch W., van den Broek M., Yoshida K., Taga T., Kishimoto T. Postnatally induced inactivation of gp130 in mice results in neurological, cardiac, hematopoietic, immunological, hepatic, and pulmonary defects. Journal of Experimental Medicine. 1998;188:1955–1965. doi: 10.1084/jem.188.10.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kovsan J., Osnis A., Maissel A., Mazor L., Tarnovscki T., Hollander L. Depot-specific adipocyte cell lines reveal differential drug-induced responses of white adipocytes-relevance for partial lipodystrophy. American Journal of Physiology. Endocrinology and Metabolism. 2009;296:E315–E322. doi: 10.1152/ajpendo.90486.2008. [DOI] [PubMed] [Google Scholar]
- 25.Rapold R.A., Wueest S., Knoepfel A., Schoenle E.J., Konrad D. Fas activates lipolysis in a Ca2+-CaMKII-dependent manner in 3T3-L1 adipocytes. The Journal of Lipid Research. 2013;54:63–70. doi: 10.1194/jlr.M028035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wueest S., Mueller R., Bluher M., Item F., Chin A.S., Wiedemann M.S. Fas (CD95) expression in myeloid cells promotes obesity-induced muscle insulin resistance. EMBO Molecular Medicine. 2014;6:43–56. doi: 10.1002/emmm.201302962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu Y., Wang Y., Liu J., Cao W., Li L., Du H. Adipose tissue-derived autotaxin causes cardiomyopathy in obese mice. Journal of Molecular Endocrinology. 2019;63:113–121. doi: 10.1530/JME-18-0242. [DOI] [PubMed] [Google Scholar]
- 28.Odermatt T.S., Dedual M.A., Borsigova M., Wueest S., Konrad D. Adipocyte-specific gp130 signalling mediates exercise-induced weight reduction. International Journal of Obesity. 2020;44:707–714. doi: 10.1038/s41366-019-0444-7. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez-Infantes D., Cereijo R., Peyrou M., Piquer-Garcia I., Stephens J.M., Villarroya F. Oncostatin m impairs brown adipose tissue thermogenic function and the browning of subcutaneous white adipose tissue. Obesity. 2017;25:85–93. doi: 10.1002/oby.21679. [DOI] [PubMed] [Google Scholar]
- 30.Dodd G.T., Decherf S., Loh K., Simonds S.E., Wiede F., Balland E. Leptin and insulin act on POMC neurons to promote the browning of white fat. Cell. 2015;160:88–104. doi: 10.1016/j.cell.2014.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plum L., Rother E., Munzberg H., Wunderlich F.T., Morgan D.A., Hampel B. Enhanced leptin-stimulated Pi3k activation in the CNS promotes white adipose tissue transdifferentiation. Cell Metabolism. 2007;6:431–445. doi: 10.1016/j.cmet.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 32.Rodriguez A., Becerril S., Mendez-Gimenez L., Ramirez B., Sainz N., Catalan V. Leptin administration activates irisin-induced myogenesis via nitric oxide-dependent mechanisms, but reduces its effect on subcutaneous fat browning in mice. International Journal of Obesity. 2015;39:397–407. doi: 10.1038/ijo.2014.166. [DOI] [PubMed] [Google Scholar]
- 33.Federico L., Ren H., Mueller P.A., Wu T., Liu S., Popovic J. Autotaxin and its product lysophosphatidic acid suppress brown adipose differentiation and promote diet-induced obesity in mice. Molecular Endocrinology. 2012;26:786–797. doi: 10.1210/me.2011-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun S., Wang R., Song J., Guan M., Li N., Zhang X. Blocking gp130 signaling suppresses autotaxin expression in adipocytes and improves insulin sensitivity in diet-induced obesity. The Journal of Lipid Research. 2017;58:2102–2113. doi: 10.1194/jlr.M075655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elks C.M., Zhao P., Grant R.W., Hang H., Bailey J.L., Burk D.H. Loss of oncostatin M signaling in adipocytes induces insulin resistance and adipose tissue inflammation in vivo. Journal of Biological Chemistry. 2016;291:17066–17076. doi: 10.1074/jbc.M116.739110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyaoka Y., Tanaka M., Naiki T., Miyajima A. Oncostatin M inhibits adipogenesis through the RAS/ERK and STAT5 signaling pathways. Journal of Biological Chemistry. 2006;281:37913–37920. doi: 10.1074/jbc.M606089200. [DOI] [PubMed] [Google Scholar]
- 37.Song H.Y., Kim M.R., Lee M.J., Jeon E.S., Bae Y.C., Jung J.S. Oncostatin M decreases adiponectin expression and induces dedifferentiation of adipocytes by JAK3- and MEK-dependent pathways. The International Journal of Biochemistry & Cell Biology. 2007;39:439–449. doi: 10.1016/j.biocel.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 38.Rui L. Brown and beige adipose tissues in health and disease. Comparative Physiology. 2017;7:1281–1306. doi: 10.1002/cphy.c170001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komori T., Tanaka M., Senba E., Miyajima A., Morikawa Y. Deficiency of oncostatin M receptor beta (OSMRbeta) exacerbates high-fat diet-induced obesity and related metabolic disorders in mice. Journal of Biological Chemistry. 2014;289:13821–13837. doi: 10.1074/jbc.M113.542399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pandit R., Beerens S., Adan R.A.H. Role of leptin in energy expenditure: the hypothalamic perspective. American Journal of Physiology - Regulatory, Integrative and Comparative Physiology. 2017;312:R938–R947. doi: 10.1152/ajpregu.00045.2016. [DOI] [PubMed] [Google Scholar]
- 41.Porte D., Jr., Baskin D.G., Schwartz M.W. Insulin signaling in the central nervous system: a critical role in metabolic homeostasis and disease from C. elegans to humans. Diabetes. 2005;54:1264–1276. doi: 10.2337/diabetes.54.5.1264. [DOI] [PubMed] [Google Scholar]
- 42.Cornelissen C., Luscher-Firzlaff J., Baron J.M., Luscher B. Signaling by IL-31 and functional consequences. European Journal of Cell Biology. 2012;91:552–566. doi: 10.1016/j.ejcb.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Dillon S.R., Sprecher C., Hammond A., Bilsborough J., Rosenfeld-Franklin M., Presnell S.R. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nature Immunology. 2004;5:752–760. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- 44.Wunderlich C.M., Hovelmeyer N., Wunderlich F.T. Mechanisms of chronic JAK-STAT3-SOCS3 signaling in obesity. JAK-STAT. 2013;2 doi: 10.4161/jkst.23878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zvonic S., Baugh J.E., Jr., Arbour-Reily P., Mynatt R.L., Stephens J.M. Cross-talk among gp130 cytokines in adipocytes. Journal of Biological Chemistry. 2005;280:33856–33863. doi: 10.1074/jbc.M508020200. [DOI] [PubMed] [Google Scholar]
- 46.Komori T., Tanaka M., Furuta H., Akamizu T., Miyajima A., Morikawa Y. Oncostatin M is a potential agent for the treatment of obesity and related metabolic disorders: a study in mice. Diabetologia. 2015;58:1868–1876. doi: 10.1007/s00125-015-3613-9. [DOI] [PubMed] [Google Scholar]
- 47.Qurania K.R., Ikeda K., Wardhana D.A., Barinda A.J., Nugroho D.B., Kuribayashi Y. Systemic inhibition of Janus kinase induces browning of white adipose tissue and ameliorates obesity-related metabolic disorders. Biochemical and Biophysical Research Communications. 2018;502:123–128. doi: 10.1016/j.bbrc.2018.05.131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.