

Abstract
The incidence of cardiovascular disease (CVD) is higher in cancer survivors than in the general population. Several cancer treatments are recognized as risk factors for CVD, but specific therapies are unavailable. Many cancer treatments activate shared signaling events, which reprogram myeloid cells (MCs) towards persistent senescence-associated secretory phenotype (SASP) and consequently CVD, but the exact mechanisms remain unclear. This study aimed to provide mechanistic insights and potential treatments by investigating how chemo-radiation can induce persistent SASP. We generated ERK5 S496A knock-in mice and determined SASP in myeloid cells (MCs) by evaluating their efferocytotic ability, antioxidation-related molecule expression, telomere length, and inflammatory gene expression. Candidate SASP inducers were identified by high-throughput screening, using the ERK5 transcriptional activity reporter cell system. Various chemotherapy agents and ionizing radiation (IR) up-regulated p90RSK-mediated ERK5 S496 phosphorylation. Doxorubicin and IR caused metabolic changes with nicotinamide adenine dinucleotide depletion and ensuing mitochondrial stunning (reversible mitochondria dysfunction without showing any cell death under ATP depletion) via p90RSK-ERK5 modulation and poly (ADP-ribose) polymerase (PARP) activation, which formed a nucleus-mitochondria positive feedback loop. This feedback loop reprogramed MCs to induce a sustained SASP state, and ultimately primed MCs to be more sensitive to reactive oxygen species. This priming was also detected in circulating monocytes from cancer patients after IR. When PARP activity was transiently inhibited at the time of IR, mitochondrial stunning, priming, macrophage infiltration, and coronary atherosclerosis were all eradicated. The p90RSK-ERK5 module plays a crucial role in SASP-mediated mitochondrial stunning via regulating PARP activation. Our data show for the first time that the nucleus-mitochondria positive feedback loop formed by p90RSK-ERK5 S496 phosphorylation-mediated PARP activation plays a crucial role of persistent SASP state, and also provide preclinical evidence supporting that transient inhibition of PARP activation only at the time of radiation therapy can prevent future CVD in cancer survivors.
Keywords: Mitochondrial stunning, Atherosclerosis, Senescence-associated secretory phenotype (SASP), Efferocytosis, Antioxidants, Telomere length, p90RSK, Ionizing radiation, ERK5, Poly (ADP-Ribose) polymerase
Graphical abstract
Highlights
-
•
High-throughput screening detects chemo-radiation as p90RSK-ERK5 modulator, which induces senescence-associated secretory phenotype (SASP).
-
•
Chemo-radiation instigates mitochondrial (mt) stunning (reversible mt dysfunction without showing any cell death under ATP depletion).
-
•
Mt stunning is provoked by p90RSK-ERK5 modulation and PARP activation, which forms a nucleus-mitochondria positive feedback loop.
-
•
This feedback loop reprogramed myeloid cells (MCs) to induce a sustained SASP state, and ultimately prime MCs to be more sensitive to reactive oxygen species.
-
•
When PARP activity is transiently inhibited at the time of IR, mitochondrial stunning, priming, macrophage infiltration, and coronary atherosclerosis are all eradicated.
1. Introduction
The incidence of cardiovascular (CV) disease (CVD) is higher in cancer survivors than in the general population. Several cancer treatments are recognized as risk factors for CVD, especially coronary artery disease and stroke [[1], [2], [3], [4]]. CV comorbidities and toxicities are the leading causes of morbidity and mortality among cancer survivors [[5], [6], [7]]; indeed CVD is the leading causes of death in breast and colorectal cancer survivors 5–10 years after their cancer diagnosis [[8], [9], [10]]. Even without exposure to cardiotoxic treatments, cancer survivors frequently manifest signs of CVD, including lower left ventricular mass and more cardiac dysfunction than do their healthy siblings [3]. In addition, all cancer survivors, whether exposed to cardiotoxic treatments or not, had a greater mean body mass index, and the higher serum levels of insulin, c-reactive protein and non-high-density lipoprotein cholesterol, thereby putting them at a higher risk of CVD. Importantly, it has been noted that one of the common phenotypes observed in these patients is premature aging [3,11,12]. Chronological age (an indirect indicator of physiological age) is an independent predictor of CVD [13].
The use of ionizing radiation (IR) and anthracyclines has been associated, directly and indirectly, with premature aging and subsequent CVD [14,15]. The incidence of major coronary events increased linearly with the mean dose to the heart by 7.4% per gray (Gy), with no apparent threshold and an overall mean of 4.9 Gy to the whole heart. Coronary events emerged within the first 5 years after radiation therapy (RT) and continued to increase into the third decade [16]. Modern RT techniques have allowed for significant decreases in the radiation dose to the heart, but it may still receive doses of 1–5 Gy (and even higher in the case of thoracic malignancies) [17]. Anthracyclines are the classical cardiotoxins, causing acute and subacute congestive heart failure within weeks after treatment, and like RT, they are associated with senescence [18,19]. Vascular diseases, including myocardial infarction, ischemia, and stroke, have also been observed in survivors of breast cancer and hematologic malignancies 5–10 years after the completion of anthracycline-based treatments [20,21], and MRI studies have shown accelerated vascular aging after anthracycline exposure [22]. These observations indicate broader impact of anthracyclines on senescence of the CV system.
Senescence can be a reflection not only of aging, but also of chronic stress, which alters cell metabolism, and reprograms cells to acquire the senescence-associated secretory phenotype (SASP). Its relevance for CVD is in part due to the pro-inflammatory milieu it generates [[23], [24], [25]]. In individuals with human immunodeficiency virus infection treated with a combination of antiretroviral therapy, we found that the 4 components of SASP—1) telomere (TL) shortening-mediated DNA damage and subsequent induction of p53, p16, and p21; 2) mitochondrial reactive oxygen species (mtROS) induction; 3) inflammation; and 4) impairment of efferocytosis—were regulated by p90RSK-mediated ERK5 S496 phosphorylation in myeloid cells (MCs). This resulted in priming of MCs and instigated atherosclerosis [26]. Therefore, the key role of p90RSK-mediated ERK5 S496 phosphorylation in SASP-mediated MCs priming and atherosclerotic plaque formation has been demonstrated in our previous studies [26,27]. Of note, p90RSK-mediated ERK5 S496 phosphorylation inhibited ERK5 transcriptional activity, but showed no effect on ERK5 kinase activity. Therefore, p90RSK-mediated ERK5 S496 phosphorylation has a very different functional role from other poly (ADP-ribose) polymerase (PARP)-related MAP kinase including ERK1/2, JNK1/2, and p38 [27,28]. SASP cells can secrete several cocktail of pro-inflammatory cytokines (e.g. TNF-α, IL6, and IL8), chemokines, growth factors, pro-angiogenic factors, and ROS [29,30]. It is well known that cytokines stimuli increase inflammation, but these effects are temporarily. In contrast, SASP maintains and shows long-term effects to the cell, which is uniquely different from the temporary effects shown in non-senescent inflammatory cells [31,32]. In fact, Coppé et al. have suggested that a larger proportion of the SASP of senescent fibroblasts is irreversible once established [33]. However, it remains unclear why SASP is persistent and irreversible if we don't apply any further intervention to it. The importance of nuclear PARP activation in mitochondrial dysfunction and mtROS production has been suggested extensively [[34], [35], [36]]. Furthermore, telomere DNA damage-induced PARP activation is also reported [37]. Recently, Qian et al. have reported that mtROS caused rapid and preferential telomere DNA damage, but not gross nuclear DNA damage [38,39]. These previous data brought us an idea that the establishment of nucleus-mitochondria positive feedback loop induced by chemo-radiation may play a key role for inducing persistent and irreversible SASP. To the best of our knowledge, we could not find any paper even review, except for ours, describing any form of nucleus-mitochondria positive feedback loop to explain the persistent SASP and irreversible if we do not apply any further intervention to it status. Chemo-radiation can induce persistent SASP [[40], [41], [42]], but how SASP and subsequent MCs priming are induced by the cancer treatments has been incompletely elucidated. Furthermore, although we established the contribution of mtROS production to SASP in our previous study [26], how chemo-radiation can change mitochondrial function and reprogram MCs to SASP is not clear. Our goal of this study was to establish that p90RSK-mediated ERK5 S496 phosphorylation is the shared signaling event activated by various cancer treatments and to determine the molecular mechanism of the p90RSK-ERK5 modulation and how this modulation reprograms MCs to SASP and priming. Furthermore, since mtROS production induced by p90RSK-ERK5 modulation plays a significant role in SASP induction [26], we also investigated mitochondrial respiratory function by concurrently measuring oxygen consumption rate (OCR) and extracellular proton flux. Our study reveals a novel role of nucleus-mitochondria positive feedback loop formed by p90RSK-ERK5 S496 phosphorylation-mediated PARP activation in reprogramming of MCs toward persistent SASP status. Furthermore, we found that low dose of ionizing radiation (IR) and doxorubicin (DOX) induced very unique form of mt dysfunction, which showed high mtROS production even under severe ATP depletion without showing any immediate cell apoptosis or death. Moreover, this ATP depletion was reversible, and recovered by treating MC with p90RSK or PARP inhibitor, even 24 h after IR. We denoted this mitochondrial dysfunction with reversible ATP depletion as mitochondrial stunning.
2. Methods
See Supplementary material online for extended experimental procedures.
3. Results
3.1. IR and Chemo-radiation Inhibited ERK5 Transcriptional Activity in Macrophages and Human Peripheral Blood Mononuclear Cells, and p90RSK-ERK5 Chemical Modulators Detected by High Throughput Screening (HTS)
When mouse bone marrow-derived macrophages (BMDMs) were irradiated by IR, both p90RSK activity and ERK5 S496 phosphorylation (which inhibits ERK5 transcriptional activity) [27] were increased in a dose- and time-dependent manner; however, IR had no effect on ERK5 TEY motif phosphorylation (Fig. 1A, and quantification data in Figs. S1A and B). We transfected BMDMs with an ERK5 transcriptional activity reporter and stimulated ERK5 using constitutively active MEK5 (CA-MEK5), with or without IR, to demonstrate that IR inhibits ERK5 transcriptional activity but that this inhibition is overcome by FMK-MEA (the pharmacologic inhibitor of p90RSK) or dominant-negative p90RSK (DN-RSK-MTg; Fig. 1B and C). Similarly, in human peripheral mononuclear cells, IR increased p90RSK activation and ERK5 S496 phosphorylation, but the p90RSK and ERK5 S496 phosphorylation were blunted by FMK-MEA (Fig. 1D and E).
Fig. 1.

IR and DOX sensitized macrophages to oxidative stress and induced SASP, which are inhibited by inhibiting p90RSK activity and activating NRF2.
(A) BMDMs were analyzed by immunoblotting at various time points after IR (2 Gy) or with varying doses for 10 min. Western blot analyses were performed using specific antibodies indicated on the right. GM-CSF stimulation (20 ng/ml for 30 min) was used as the positive control. Representative images from 3 independent experiments are shown (quantification in Figs. S1A and B). (B, C) BMDMs were transfected with pBind-ERK5 and pG5-luc plasmids and pcDNA3.1 plasmid or CA-MEK5 for 30 h. They were then pre-treated with FMK-MEA (10 μM) or vehicle (DMSO, 0.1%) for 1 h and exposed to IR (2 Gy) (B). Alternatively, BMDMs from WT and DN-p90rsk-MTg mice were treated with IR, and after 6 h, ERK5 transcriptional activity was measured as described in Methods (C). Mean ± SD (n = 3). (D) Human monocytes were pre-treated with FMK-MEA (10 μM) or vehicle for 30 min and then exposed to IR (2 Gy). Cells were collected at the indicated times after IR, and total p90RSK, p90RSK phosphorylation, ERK5 S496 phosphorylation, ERK5 TEY motif phosphorylation, and total ERK5 were detected by Western blotting. (E) Quantification of IR-induced p90RSK S380 phosphorylation (left), ERK5 S496 phosphorylation (middle), and ERK5 TEY motif phosphorylation (right) is shown after normalization by total protein levels. The data represent the mean ± SD (n = 3). Blue line: DMSO control pre-treatment, red line: FMK-MEA pre-treatment. (F) Percentage of apoptotic BMDMs after 24 h treatment with IR or DOX is shown at the indicated doses. Cells were stained with annexin V and analyzed by flow cytometry. Data are expressed as mean ± SD (n = 3) from at least 3 independent experiments. G) MTT assays were performed to measure cell viability after 24 h of IR and DOX treatment at the indicated doses. Results are expressed as % compared to time 0. Mean ± SD, (n = 3). (H) BMDMs were exposed to IR (2 Gy), DOX (1 μM), or vehicle and after 24 h, cells were incubated with H2O2 (200 μM) for 0–30 min. Western blotting was performed with the indicated antibodies. Representative images from 3 independent experiments are shown. (I) BMDMs were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h and irradiated by IR. After 0–24 h, Western blotting was performed with the indicated antibodies. Representative images from 3 independent experiments are shown. (J, K) BMDMs were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h and then exposed to IR. (J), Alternatively, BMDMs from WT and DN-p90rsk-MTg mice were exposed to IR. After 24 h, TL lengths were determined by measuring the fluorescent telomeric signal intensity of the fluorescein-conjugated PNA probe as described in Methods. Results are presented as the relative TL length (%) (mean ± SD). The TL length of the human T-cell leukemia cell line (1301) cells was set to 100% (n = 3). (L) BMDMs were transfected with the NF-κB luciferase reporter and the constitutively expressing Renilla luciferase vector for 16 h. Cells were pre-treated with FMK-MEA (10 μM) or BI-D1870 (5 μM) for 1 h and then exposed to IR or left untreated. After 12 h, NF-κB transcriptional activity was measured as described in Methods. Mean ± SD, (n = 3). (M) BMDMs were isolated from WT and DN-p90rsk-MTg mice and exposed to IR or left untreated. After 24 h, cells were incubated with the IncuCyte pHrodo-labeled apoptosis detection probe, and pHrodo-positive cells quantified. Mean ± SD, (n = 3). (N) BMDMs were pre-treated with NRF2A (CAS 1362661) or vehicle for 6 h and exposed to IR or non-IR. After 18 h, BMDMs were incubated with H2O2 (200 μM) for 10 min. Western blotting was performed using specific antibodies indicated on the right. Representative images from 3 independent experiments are shown. **P < 0.01.
To determine whether chemotherapy drugs also regulate the p90RSK-ERK5 module similar to IR, we generated a HeLa cell line that stably expressed the pG5-Luc and pBIND vector containing ERK5. Cells were treated for 18 h with compounds in the MicroSource SPECTRUM Collection, and luciferase activity was assayed as we reported previously [43] and also described in the methods. Outcomes (Pubchem AID 1508626, External ID, ERK5 transcriptional activity-HTS) were then used to select 4 compounds (doxorubicin [DOX]), ifosfamide, paclitaxel, and methotrexate) that showed 50% or less signal compared with controls (vehicle-treated cells). All compounds significantly increased p90RSK activity and ERK5 S496 phosphorylation, but not ERK5 TEY motif phosphorylation, in a dose- and time-dependent manner. We also confirmed that these 4 compounds inhibited CA-MEK5-induced ERK5 transcriptional activity by activating p90RSK (Fig. S1C-D, and S3 and 4). It is noteworthy that each drug concentration used was within the therapeutic range of the plasma concentration in humans (see Methods).
Fig. 4.

IR inhibited both OXPHOS and glycolysis via the p90RSK and ERK5 S496 phosphorylation.
(A-D, F–I) BMDMs isolated from WT mice were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h and exposed to IR (A-D). Alternatively, BMDMs from WT and ERK5 S496A KI mice were exposed to IR (F–I). These cells were then seeded on Seahorse plates. After 24 h, OXPHOS and glycolysis parameters were measured. During extracellular flux analysis, cells were sequentially treated with (A, F) Oligomycin (OM), carbonyl cyanide 4-(tri 4-(trifluoromethoxy) phenylhydrazone (FCCP), and rotenone plus antimycin A (ROT/AA) and used to assess OXPHOS parameters based on oxygen consumption rates. (B, H) The basal respiration, mitochondrial ATP production, maximal respiration, and spare respiratory capacity were calculated and plotted as oxygen consumption rates in pmoles/minutes. Mean ± SD, (n = 3). (C, G) Glucose (GLUC), OM, and 2-deoxyglucose (2-DG) were used to determine glycolysis parameters from extracellular acidification rates. (D, I) Glycolysis, glycolytic reserve, glycolytic capacity, and non-glycolytic acidification were calculated and plotted as the extracellular acidification rate in mpH/minutes. Mean ± SD, (n = 3). (E, J) BMDMs from WT mice were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h and then exposed to IR (E); alternatively, BMDMs from WT and ERK5 S496A KI mice were exposed to IR (J). **P < 0.01 (K, L) BMDMs from WT mice were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h and then exposed to IR, and after 24 h of IR, the expressions of M1- (K) and M2 (L) -like markers mRNA levels were detected by the fold inductions relative to the expression in unexposed BMDMs. Mean ± SD, (n = 3).
3.2. IR and DOX primed macrophages to oxidative stress by decreasing antioxidant expression and driving senescence
Since accelerated vascular aging after IR and anthracycline exposure has been reported [22,44], first, we assessed apoptosis in macrophages treated with IR and DOX. Low doses of IR (2 Gy) and DOX (1 μM) did not increase the number of annexin V-positive cells, but these doses affected cell metabolic viability, as determined by the 3-(4,5-dimethylthiazole-2-yl)-2.5-diphenyl tetrazolium bromide (MTT) assay (Fig. 1F and G) [45,46]. 2 Gy (IR) and 1 μM (DOX) are within the range of the treatment dose [[47], [48], [49]], and perturbed cell metabolism without causing catastrophic apoptosis or necrosis, therefore we used these doses in subsequent experiments unless specified otherwise. Treatment of BMDMs with IR or DOX for 24 h significantly enhanced the p90RSK and ERK5 S496 phosphorylation by H2O2 (200 μM) (Fig. 1H). We reported earlier that anti-HIV drugs induced priming of monocytes and macrophages to a secondary insult of ROS, which is a hallmark of SASP [26]. Our data here support the theory that IR and DOX also induce SASP.
To gain insights into the mechanism of this priming effect, we tested the following 2 potential mechanisms reported previously [26]: (a) decreased expression of antioxidants and (b) TL shortening. p90RSK activation inhibits NRF2 transcriptional activity and anti-oxidant expression via ERK5 S496 phosphorylation [26]. We found that IR (Fig. 1I) and DOX (Figs. S1E and F) significantly decreased the expression of antioxidant molecules such as thioredoxin 1 (Trx1) and heme oxygenase 1 (HO-1) and that this decrease was completely reversed by the p90RSK-specific inhibitor FMK-MEA. Without changing NRF2 expression (both 60 and 110 kDa isoforms), IR and DOX inhibited NRF2 transcriptional activity detected by anti-oxidant response element (ARE) reporter luciferase activity. This inhibition was reversed by FMK-MEA or BI-D1870 (another p90RSK-specific inhibitor) or by overexpressing dominant-negative p90RSK (DN-RSK) (Figs. S5A–I). IR (Fig. 1I–K and S4D) and DOX (Fig. S1E, F and S4H) also increased the expression of senescence markers (p16, p21, and p53) and increased TL erosion, all of which were inhibited by FMK-MEA. These results indicate that p90RSK activation not only suppresses the expression of antioxidant molecules, but also induces TL dysfunction and senescence in macrophages, both promoting chronic priming to ROS. We also showed the increase of various cytokines and chemokines secretion after IR for 24 h, which were inhibited by FMK-MEA (Fig. S2A).
ERK5 and NRF2 regulate macrophage efferocytosis via upregulating the expression of opsonins such as Gas6 [50] and inflammatory genes [26]. In BMDMs, we found that Gas6 expression was inhibited but that TNFα expression was increased by IR and DOX in a p90RSK activation-dependent manner (Fig. 1I and S1E, F). DOX and IR also increased NF-κB activation while reducing efferocytosis, and these effects were reversed in BMDMs pre-treated with a p90RSK-specific inhibitor (Fig. 1L and M and Figs. S5E, F, and I). To determine the role of NRF2 in the IR- and DOX-induced reduction of antioxidant expression and TL length, we used a cell-penetrating, NRF2-KEAP1-binding inhibitory peptide, CAS 1362661 (NRF2A), that can specifically activate NRF2 transcriptional activity [51]. The IR- and DOX-mediated reduction of NRF2 transcriptional activity was completely prevented in BMDMs pre-treated with NRF2A (Figs. S5J and K). We also confirmed that pre-treatment with IR, followed by H2O2 stimulation, inhibited the Trx1, HO-1, and Gas6 expression while increasing the p21, p16, p53, and TNFα expression (Fig. 1N) and TL shortening (Fig. S5K) and enhancing p90RSK activation and ERK5 S496 phosphorylation (Fig. 1N). All of these events were profoundly diminished by NRF2A (Fig. 1N and Fig. S5K), indicating that NRF2 transcriptional activity plays a crucial role in IR-induced SASP and subsequent priming of macrophages to ROS.
3.3. Radiation treatment primed peripheral monocytes to ROS in cancer patients
To determine whether radiation primes monocytes in cancer patients, we enrolled 15 patients (Fig. 2A). Blood was collected at the time of enrollment and 2–3 weeks and 3 months after the initial radiation treatment (Fig. 2B), and mononuclear cells were isolated. We then measured the levels of total and phosphorylated p90RSK in CD14+ cells with or without H2O2 stimulation by flow cytometry. No significant changes were found in the expression and p90RSK activity levels in the absence of H2O2 stimulation (Fig. 2C and E). However, when the cells were treated with H2O2 (200 μM) for 10 min, we observed time-dependent increases in p90RSK phosphorylation without changes in protein expression (Fig. 2D and E). These results indicate that radiation treatment causes chronic sensitization (i.e. priming) of monocytes and macrophages towards ROS. They further indicate that by adding a certain stressor to cancer patients’ peripheral mononuclear cells in vitro, we were able to unmask underlying molecular predisposition that may relate to increased adverse cardiovascular outcomes among cancer survivors.
Fig. 2.

Priming of peripheral monocytes for oxidative stress after radiation.
(A) Patient profiles. Abbreviations: NSCLC; non-small cell lung cancer, SCLC; small cell lung cancer. (B) Timing of blood sampling after radiation therapy (RT). (C-E) CD14+ peripheral blood monocytes were collected from patients before radiation treatment (pre-RT), 2–3 weeks after RT, and 3 months after RT, and p90RSK activity was determined with and without H2O2 (200 μM for 10 min) stimulation by flow cytometry as described in Methods. (C) The basal (i.e. no H2O2 stimulation). p90RSK activity is shown as the level of phospho-p90RSK over the total p90RSK. (D) The p90RSK activity after H2O2 stimulation at the 3 time points were measured, and the ratio between these values and the basal values at each time point for each patient is plotted. Note that RT caused priming of these cells to H2O2. (E) p90RSK levels in CD14+ -positive cells. No significant differences were detected. Mean ± SD, *P < 0.05, **P < 0.01.
3.4. ERK5 S496A mutation inhibited the IR- and DOX-Induced SASP
We generated ERK5 S496A knock-in (KI) mice by mutating 2 bases of the serine 496 codon (AGT) in the Erk5 (Mapk7) gene of C57Bl/6 mice to an alanine codon (GCT) through homologous recombination using the CRISPR/Cas9 technique. We isolated BMDMs from wild-type and ERK5 S496A KI mice, assayed for IR- and DOX-induced ERK5 S496 phosphorylation and found that this phosphorylation, but not p90RSK activation, was completely abolished in BMDMs from ERK5 S496A KI mice (Fig. 3A). The reduced ERK5 and NRF2 transcriptional activity induced by IR and DOX was also reversed in BMDMs from the KI mice, supporting the key role of ERK5 S496 phosphorylation in regulating ERK5 and NRF2 transcriptional activity (Fig. 3B and C). We also found that IR led to reduced protein expression of Gas6, HO-1, and Trx1, and the accumulation of TNFα, p16, and p21. However, these effects were not seen in BMDMs from ERK5 S496A KI mice (Fig. 3D). Similarly, the IR- and DOX-induced TL shortening (Fig. 3E) and NF-κB activation (Fig. 3F) observed in BMDMs from WT mice were significantly attenuated in cells from ERK5 S496A KI mice. Lastly, BMDMs isolated from ERK5 S496A KI mice exhibited significantly improved efferocytosis (Fig. 3G). Overall, these data demonstrate the critical role of ERK5 S496 phosphorylation in regulating the chemo-radiation-induced SASP.
Fig. 3.

Role of ERK5 S496 phosphorylation in IR- and DOX-induced reduction of ERK5 transcriptional activity and subsequent SASP.
(A) BMDMs isolated from WT and ERK5S496A KI mice were treated with IR (2 Gy). After the indicated times, Western blotting was performed using the antibodies against the indicated proteins. Representative images from 3 independent experiments are shown. (B) BMDMs from WT and ERK5S496A KI mice were transfected with pBind vector containing ERK5, pG5-luc plasmids, as well as pcDNA3.1 plasmid (control) or pcDNA3.1 containing a constitutively active MEK5 (CA-MEK5) for 30 h and then treated with IR (left) or DOX (right). After 6 h, ERK5 transcriptional activity was measured as described in Methods. Mean ± SD, (n = 3). (C) BMDMs from WT and ERK5S496A KI mice were transfected with the ARE luciferase reporter and the constitutively expressing Renilla luciferase vector for 16 h. Cells were treated with IR or DOX, and 6 h later, NRF2 transcriptional activity from 3 independent experiments was measured as described in Methods. (D) BMDMs from WT and ERK5S496A KI mice were exposed to IR or non-IR. After 24 h, Western blotting was performed using antibodies against the indicated proteins. (E) BMDMs from WT and ERK5S496A KI mice were treated with IR or DOX. After 24 h, TL lengths were determined as described in Methods and presented relative (%) to the TL length of human T-cell leukemia (1301) cells, which was set to 100%. Mean ± SD, (n = 3). (F) BMDMs from WT and ERK5S496A KI mice were transfected with the NF-κB luciferase reporter and the constitutively expressing Renilla luciferase vector for 16 h and then exposed to IR or left untreated. After 12 h, NF-κB transcriptional activity was measured as described in Methods. Mean ± SD, (n = 3). (G) BMDMs from WT and ERK5S496A KI mice were exposed to IR (left) or DOX (right). After 24 h, pHrodo-positive cells were quantified. Mean ± SD, (n = 3), **P < 0.01.
3.5. IR and DOX abolished both glycolysis and oxidative phosphorylation in mouse macrophages by activating p90RSK and ERK5 S496 phosphorylation
It is well known that mitochondria are directly or indirectly targeted by a variety of drugs [52]. Indeed, we found that low doses of IR and DOX inhibit cell metabolism as assessed by the MTT assay, which measures the activity of succinate dehydrogenase, and enzyme complex in the mitochondrial inner membrane (Fig. 1G). We, thus, investigated whether IR and DOX affect mitochondrial metabolism using an extracellular flux analysis. We determined oxygen consumption rate (OCR) in response to oligomycin, carbonyl cyanide 4-(tri 4-(trifluoromethoxy) phenylhydrazone, and rotenone + antimycin A treatments, and detected changes in the extracellular acidification rate as a marker for glycolysis in response to glucose, oligomycin, and 2-deoxyglucose injection as described [53]. We found that while IR (24 h after 2 Gy exposure) and DOX (1 μM for 24 h) completely inhibited both glycolysis and oxidative phosphorylation (OXPHOS) in BMDMs, these inhibitory effects were blunted in cells pre-treated with FMK-MEA for 1 h (Fig. 4A–D and Figs. S6A–D). We confirmed the effect of FMK-MEA using an ATP synthesis bioluminescence assay (Fig. 4E). We also evaluated the role of ERK5 S496 phosphorylation in IR-induced mitochondrial dysfunction in BMDMs isolated from ERK5 S496A KI mice. IR reduced glycolysis and OXPHOS in BMDMs isolated from control mice, but this inhibition was significantly reversed in BMDMs from ERK5 S496A KI mice (Fig. 4F–I). Both IR and DOX reduced the ATP level in control cells but not in BMDMs from ERK5 S496A KI mice (Fig. 4J).
We also determined M1 and M2-like cell polarization after IR and DOX by detecting 1) the increase of the cytokines and chemokines expression including IL-1β, TNF-α, and iNOS as markers of M1-like phenotype, and 2) no significant changes of Arg-1, Ym1, and Fizz1 as markers of M2-like phenotype. The pre-treatment of FMK-MEA significantly inhibited both IR and Dox-induced M1-like phenotype induction (Fig. 4K, L and Figs. S2B and C). We also found that various cytokines and chemokines expression were increased by low dose of IR, which are inhibited by p90RSK inhibitor (Fig. S2A). These data suggest that low dose of IR and DOX induced M1-like phenotype in BMDMs in a p90RSK activation dependent manner.
3.6. p90RSK-mediated ERK5 S496 phosphorylation provoked MtROS production and PARP activation, resulting in positive feedback loop between the nucleus and mitochondria after chemo-radiation
Various cancer treatments, including IR and chemotherapies, increased both acutely (10 min) and chronically (12 h later) mtROS production in BMDMs. This effect was significantly inhibited by mitoTEMPOL, a preferential inhibitor of mtROS generation (Fig. 5A). Further, mitoTEMPOL completely inhibited NF-κB activation and attenuated the IR-induced reduction of NRF2 transcriptional activity (Fig. 5B and C), suggesting that mitochondria are a common target of various cancer treatments in macrophages. Using MitoSOX Red, we detected mtROS and found that p90RSK activation was involved in the IR- and DOX-induced chronic mtROS production in BMDMs (Fig. 5D–G), but FMK-MEA had no effect on the IR-induced acute mtROS production, especially at 30 min after IR (Fig. 5E). Similarly, FMK-MEA inhibited only the chronic mtROS production in BMDMs from ERK5 S496A KI mice (Fig. 5F). These data suggest that p90RSK-mediated ERK5 S496 phosphorylation plays a critical role in regulating chemo-radiation-induced mtROS production, especially at the chronic phase.
Fig. 5.

Only late phase of mtROS production after chemo-radiation was dependent on.
(A) BMDMs from WT mice were incubated with various cancer drugs or TNFα with or without MitoTEMPOL (10 μM) as indicated. MitoSox Red was added, and mtROS levels were detected as described in Methods. Cells exposed to IR were assayed 10 min or 12 h later. Mean ± SD, (n = 3). (B, C) WT BMDMs were transfected with the NF-κB (B) or ARE (C) luciferase reporter and the constitutively expressing Renilla luciferase vector for 16 h. Cells were then pretreated with MitoTEMPOL (10 μM) for 1 h and treated with or without IR. After 12 h, NF-κB (B) or ARE (C) transcriptional activity was measured (see Methods). Mean ± SD, (n = 3). (D) BMDMs from WT and DN-p90rsk-MTg mice were exposed to IR or non-IR and 24 h later, mtROS was measured as described in A, Mean ± SD, (n = 3). (E-G) BMDMs from WT mice were pretreated with vehicle or FMK-MEA (10 μM) for 1 h (E, G) or BMDMs from WT or ERK5 S496A KI mice (F) were exposed to IR or treated with DOX, and mtROS levels detected. Mean ± SD, (n = 3).
Poly (ADP-ribose) polymerase (PARP), a DNA damage response (DDR)-related molecule that is localized in the nucleus, can induce cytosolic nicotinamide adenine dinucleotide (NAD+) depletion, leading to changes in mitochondrial metabolism and mtROS production [54,55]. Recently, the key role of PARP activation in inhibiting both OXPHOS and glycolysis and depleting NAD+, NADH, and ATP after DNA damage was reported [56]. The crucial role of mtROS-induced TL damage in the DDR has been reported [39]. Since p90RSK-mediated ERK5 S496 phosphorylation plays a decisive role in mtROS levels and TL shortening after 12 h of IR and DOX treatment, we examined the role of p90RSK-mediated ERK5 S496 phosphorylation in PARP activation. IR and DOX treatment for 24 h increased PARP activation in BMDMs, but this increase was inhibited by FMK-MEA or in cells isolated from ERK5 S496A KI mice (Fig. 6A and B, and quantification in Figs. S6E–G). IR- and DOX-induced NAD+ depletion was also inhibited by FMK-MEA and in BMDMs isolated from ERK5 S496A KI mice (Fig. 6C and D). Taken together, these data support that p90RSK-mediated ERK5 S496 phosphorylation induces PARP activation, which then plays a crucial role in cellular NAD+ depletion.
Fig. 6.

Roles of p90RSK-ERK5 S496 phosphorylation in chemo-radiation-induced PARP activation, and complex II activity in mtROS production.
(A, B) BMDMs from WT mice were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h and then exposed to IR (left) or treated with DOX (right) (A). Alternatively, BMDMs from WT and ERK5 S496A KI mice were exposed to IR (left) or treated with DOX (right) (B). After 24 h, levels of PARylation was determined by Western blotting using the indicated antibodies Representative images from 3 independent experiments are shown (quantification in Figs. S5E and F). (C, D) NAD+ levels were measured using the NAD kit (Cell Biolabs) in BMDMs that were treated as described in A. Mean ± SD, (n = 3). (E) A scheme of complexes I-III of the electron transport chain, which plays a key role in mtROS production. Electrons (e−) from NADH and FADH2 are passed to oxygen and generates ROS. CI, CII, CIII: complex I-III, CoQ: coenzyme Q. (F) WT BMDMs were pre-treated with atpenin A5 (20 nM) or vehicle (DMSO, 0.1%) for 1 h and then treated with IR (left) or DOX (right) as in G. After 24 h, mtROS levels were measured as described in A. **P < 0.01 (G) Succinate dehydrogenase (SDH) activity was measured after 24 h of IR at the indicated doses. Results are expressed as % compared to time 0. Mean ± SD, (n = 3). (H) BMDMs from WT mice were pre-treated with FMK-MEA (10 μM) or vehicle for 1 h and then exposed to IR, and succinate levels were measured after 24 h of IR (2 Gy). Mean ± SD, (n = 3).
Although IR and DOX reduced ATP levels, persistent mtROS production was increased in BMDMs (Fig. 5). Since electrons from succinate can enter the electron transport chain through FADH2 in complex II [57], we predicted that mtROS is a product of increased complex II electron flux resulting from depletion of NAD+ and NADH induced by PARP activation (Fig. 6E).
When cells BMDMs were treated with the complex II-specific Q-site inhibitor atpenin A5 (AA5) [58] the IR- and DOX-induced mtROS production was completely abolished, suggesting that complex II sourced electrons contribute to mtROS production (Fig. 6F). Fig. 6G show that IR (2 Gy) inhibited only 25% of succinate dehydrogenase activity, but this level of inhibition was not sufficient for affecting mtROS production even when ATP is depleted (Figs. 4E, and Fig. 5G). Furthermore, we also found that IR increased succinate levels, which were inhibited by p90RSK inhibitor (Fig. 6H), also suggesting the involvement of complex II activity in mtROS production (Fig. 6E). Finally, PARP inhibition abolished DOX-induced mtROS production (Fig. S6G), and the inhibition of mtROS production by mitoTEMPOL prevented IR- and DOX-induced p90RSK and ERK5 S496 phosphorylation (Figs. S6H and I). These results indicate a positive feedback loop between the nucleus and mitochondria in which PARP-mediated mtROS production and mtROS-induced p90RSK-ERK5 S496 phosphorylation play key roles.
3.7. PARP and p90RSK activation has a crucial role in establishing mitochondrial stunning
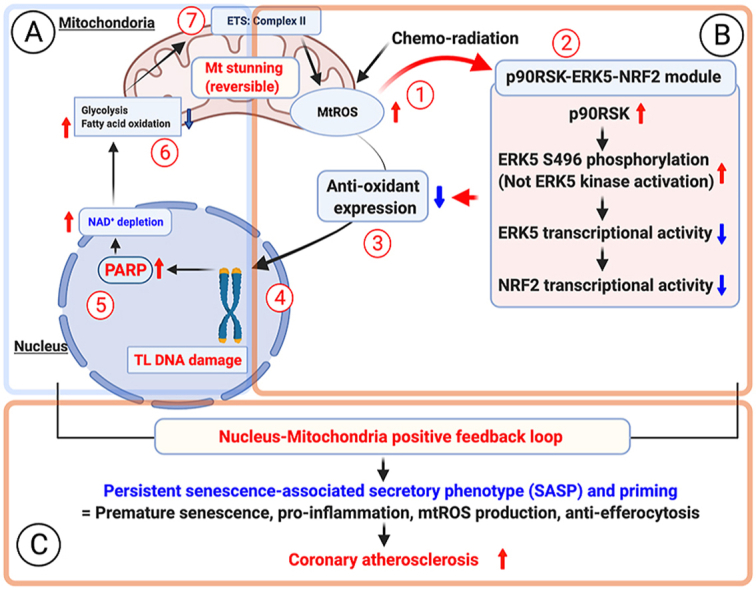
Since the ATP level was remarkably depleted by IR and DOX, we examined cell viability after IR by the trypan blue exclusion assay and found that cells were viable for at least 48 h after IR (Fig. 7A–C). We also detected the release of lactate dehydrogenase (LDH) into culture medium after IR, and confirmed that IR (2 Gy) did not show any cell death at least 24 h after IR, which was completely different from those after high dose of IR (50 Gy) (Fig. 7B, right). These data suggested that low dose of IR-induced ATP depletion did not cause immediate cell death or apoptosis (Fig. 1, Fig. 7B, C) in BMDMs, which was unique compared to the cases of ATP depletion induced by other stimuli, which has been reported previously [35,59]. Next, we investigated whether the effects of NAD+ and ATP depletion are reversible in irradiated cells (Fig. 7A). One day (1R) after IR, both NAD+ and ATP levels were decreased, but this reduction was preventable when cells were pre-treated by olaparib or FMK-MEA (R0: Fig. 7D–G). The IR-induced depletion of NAD+ and ATP recovered partially in 3 days (D3) without any treatment. Treating cells with olaparib or FMK-MEA, even 24 h after IR, increases the reduced NAD+ and ATP levels to that of the non-IR and vehicle-treated cells (R1). Addition of these drugs 48 h after IR was less effective, especially for recovery of ATP levels (R2; Fig. 7A and D-G). These data suggest that mitochondria ATP deletion can be reversible without showing any immediate cell death or apoptosis, which we denominated as mitochondrial stunning. Mitochondria can be stunned for up to 48 h after IR and that p90RSK and PARP activation plays a critical role in IR-induced mitochondrial stunning. As we discussed above, PARP activation induces mtROS production [55], which plays a key role in p90RSK-ERK5-NRF2 module and subsequent priming (Fig. 1 and Figs. S6H and I). We also found that transient treatment with the PARP inhibitor at the time of IR significantly inhibited PARP activation, γH2AX levels, and priming in response to H2O2 in CD11b-positive cells in vivo (Fig. 7H–K). Taken together, these data suggest that mtROS production initiated by chemo-radiation forms a nucleus-mitochondria positive feedback loop by mtROS-induced p90RSK-ERK5-NRF2 modultion and PARP activation, which is triggered by mitochondrial stunning (Figure 7L).
Fig. 7.

Roles of p90RSK and PARP activation in mitochondrial stunning, cell viability, DDR, and IR-induced priming.(A) A schema of how to detect mitochondrial stunning (recovery) after IR with PARP and p90RSK inhibitors. R0: On day 0 (D0), BMDMs from WT mice were pre-treated with the PARP inhibitor, olaparib (10 μM) or FMK-MEA (10 μM) for 1 h and then exposed to IR (2 Gy). One day (D1) after IR, NAD+ and ATP levels were measured. R1: BMDMs were exposed to IR and 1 day later, were treated with olaparib or FMK-MEA as in R0. After 2 days of IR (D2), NAD+ and ATP levels were measured. R2: WT BMDMs were exposed to IR and after 2 days, treated with olaparib or FMK-MEA as in R0. On day 3 (D3), NAD+ and ATP levels were measured. (B, C) BMDMs were treated with olaparib or FMK-MEA as described in A, and cell viability was measured by the Trypan blue-exclusion assay (B, left and C) and cell death by detecting LDH release into the culture medium. Mean ± SD, (n = 3). (D–G) BMDMs were treated with olaparib or FMK-MEA as in A, and NAD+ and ATP levels were measured as described in A. Mean ± SD, (n = 3). (H–K) Olaparib inhibited IR-induced monocyte priming. C57Bl/6 wild-type mice were treated with olaparib (10 mg/kg/day ip) from day (D) 0 to D2 and exposed to IR (2 Gy) on D1. On D5, PBMCs were isolated (top), and Western blotting was performed using the indicated antibodies (H). (I, J) The graphs represent PARylation and γH2AX densitometry data from 3 independent gels, one of which is shown in H (I: PARylation, J: γH2AX). (K) On D5, PBMCs were isolated as described in H (top), and p90RSK activity was measured in CD11b-positive cells before and after 10 min stimulation with H2O2 (200 μM). (L) A schematic showing the role of nucleus-mitochondria positive feedback loop mediated by the p90RSK activation-ERK5 S496 phosphorylation module and PARP activation in regulating mitochondrial stunning, priming, and consequent atherosclerosis. *P < 0.05, and **P < 0.01. . (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3.8. Transient inhibition of PARP, only at the time of IR, prevented IR-Induced monocyte priming, atherosclerosis, and macrophage infiltration
To obtain pre-clinical data on the combined effects of PARP inhibitors and radiation on future CVD, we modified the mouse coronary atherosclerosis model [60] by combining it with the transverse aortic coarctation (TAC) model in LDLR−/- mice that are fed a high-fat diet and treated with or without IR (Fig. 8A). We found coronary atherosclerosis (Fig. 8B, second from the top) with high macrophage accumulation (Fig. S7A) in the plaque in 7 of 18 mice in the IR + TAC group, and 3 of 11 mice in the IR + TAC with olaparib treated group after a total of 10 Gy (5 Gy administered twice for a total dose of 10Gy) to the neck and thorax region. In contrast, no plaques were found in the non-IR + TAC group, as reported [60]. Of note, we observed no difference among these three groups in body weights and cholesterol levels (Figs. S7D and E). As shown in Fig. 8C, we found diffuse LAD stenosis with atherosclerotic lesions in the IR group, which has been reported in human coronary arteries after radiation [61]. The LAD stenosis was significantly increased by IR, and transient olaparib treatment significantly inhibited this compared to the IR + vehicle group (Fig. 8D, top), demonstrating the crucial role of PARP activation in IR-induced coronary atherosclerosis.
Fig. 8.

Role of PARP activation in coronary atherosclerosis, vascular wall thickening, macrophage infiltration, as well as subsequent cardiac dysfunction and myocardial infarction.(A) A schematic showing the timing of each procedure for evaluating IR-induced cardiovascular pathologies. LDLR−/- mice were fed an HFD for ≈2 weeks and exposed to localized IR (5 Gy) in the neck and thoracic area twice. Mice were fed a normal chow diet (ND) for ≈2 weeks until their body weight had recovered, as we described previously [100], and HFD feeding was restarted for ≈2 weeks. Thoracic aorta coarctation (TAC) was then performed and ≈4 weeks later, we performed echocardiography and euthanized the mice for sample collection. (B) Representative images of the left anterior descending artery (LAD) stained with hematoxylin and eosin 4 weeks after TAC at different distances from the aortic valve (AV) to the tip of the heart (Apex), depicting varying degrees of atherosclerosis and vessel wall thickness between the IR and non-IR groups. (C) Stenosis grades, as described in Methods, at different distances from AV (section #1 [AV] to #11 [apex]). Mean ± SD, (n = 11). (D) Quantification of stenosis grades (top, 11 sections examined for each group), the normalized and averaged ratio of vascular wall thickness to the vessel diameter of LAD (middle, Non-IR, n = 9 (3 male (M)/6 female (F); IR + vehicle, n = 11 (3 M/8F); IR + olaparib, n = 11 (5 M/6F)) and fractional shortening (FS) (bottom, Non-IR, n = 21 (4 M/17F); IR + vehicle, n = 17 (6 M/11F); IR + Olaparib, n = 11 (5 M/6F)) 4 weeks after TAC-surgery as indicated in A. Mean ± SD (Top) Repeated measures 1-way ANOVA, Turkey's multiple comparison test with individual variances (grades/section) were used for each comparison. (Middle and bottom) ordinary 1-way ANOVA. (E) Representative immunofluorescence staining of the coronary artery for ECs (CD31 [green]), macrophages (Mac3 [red]), and nuclei (DAPI [blue]). The number of anti-Mac3 positive cells in the perivascular region were normalized by the area of perivascular region (x10 [4] μm [2]). Mean ± SD; non-IR, n = 9 (3 M/6F); IR + vehicle, n = 16 (7 M/9F); IR + olaparib, n = 11 (5 M/6F). The area between two dotted lines is the perivascular region. Statistical significance was assessed by 1-way ANOVA and multiple comparisons were made by Bonferroni's t-test. Scale bar = 10 μm. (F) The numbers of anti-Mac3 positive cells in the perivascular region of LAD without atherosclerotic plaques were normalized by the area of perivascular region (x10 [4] μm [2]). Mean ± SD; non-IR, n = 9 (3 M/6F); IR + vehicle, n = 10 (3 M/7F); IR + olaparib, n = 8 (2 M/6F). *P < 0.05, **P < 0.01. Macrophage infiltration into perivascular area was significantly increased by IR + TAC, which was completely inhibited by transient olaparib treatment. *P < 0.05, **.. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
We also determined the mean LAD wall thickness from 11 whole heart sections equally spaced (400 μm) from the proximal (level 1) to the distal tip (level 11) of the heart. LDA without atherosclerotic plaque throughout the entire length were analyzed (Fig. 8D, middle). A significant increase was noted in the IR + TAC group compared to the non-IR + TAC group, but olaparib did not affect the LAD wall thickness. Although no effects of IR were observed in interstitial fibrosis after TAC (Fig. S7B), we found a significant increase in perivascular fibrosis after IR. Olaparib, however, did not inhibit the IR-induced perivascular and vascular fibrosis (Fig. S7C). This may explain the slight but not significant decrease in the left ventricular (LV) mass after olaparib treatment (Fig. 9A and B).
Fig. 9.

Transient PARP inhibition improved IR (localized 5 Gy twice)-induced cardiac dysfunction after TAC.(A) Echocardiographic parameters after IR and TAC. LV: left ventricular, LVAW; s: LV end-systolic anterior wall thickness, LVAW; d: LV end-diastolic anterior wall thickness, LVPW; s: LV end-systolic posterior wall thickness, LVPW; d: LV end-diastolic posterior wall thickness. (B) LV mass (left), LVAW; d (middle), and LVPW; d (right) before and after TAC from the non-IR after TAC, IR after TAC, and IR + olaparib treatment after TAC groups. The data are mean ± SD, n = 6–21, **P < 0.01 and *P < 0.05. (C) Representative M-mode echocardiograph from the non-IR after TAC, IR after TAC, and IR + olaparib treatment after TAC groups. (D) Representative B-mode images of a mouse heart in the parasternal long axis with infarct (IR after TAC group) or without infarct (non-IR after TAC group), showing the thin ventricular wall and akinesis in the apex area. (E) Representative Masson trichrome stained heart from the IR + vehicle treatment group after TAC.
Cardiac function was assessed by echocardiography, and we observed cardiac hypertrophy about 4 weeks after TAC in both the non-IR and IR groups; this hypertrophy (LV mass) was exacerbated by IR (Fig. 9). The IR group exhibited reduction in fractional shortening (FS), by comparison to the non-IR group. Transient olaparib treatment prevented this (Fig. 8D, bottom). In addition, localized cardiac dysfunction and infarction were found in 4 of the 18 IR + TAC group and 1 of 11 in the IR + TAC olaparib-treated group. No akinesis or thinning of the ventricular wall was noted in the non-IR + TAC group. We confirmed the infarcted area by Masson's trichrome staining in mice with significant coronary atherosclerosis (Fig. 9D and E). These data suggest that IR exacerbates coronary atherosclerosis-mediated ischemia and localized cardiac dysfunction in the murine model, an effect that is attenuated by PARP inhibition.
Next, we stained heart sections with Mac3 and CD31 and found a significant increase in Mac3-positive cells in perivascular lesions in the IR + TAC group compared with the TAC only group.; Mac3+ cells infiltration was significantly inhibited in the olaparib-treated IR + TAC group (Fig. 8E). Most importantly, IR increased numbers of Mac3-positive cells in the perivascular regions of both atherosclerotic and non-atherosclerotic coronaries (Fig. 8E and F). Although olaparib did not avert IR-induced increases in vascular wall thickness and perivascular fibrosis, it improved cardiac function, suggesting that macrophage infiltration may play a role in IR-induced cardiac dysfunction.
4. Discussion
Chemo-radiation including DOX, ifosfamide, paclitaxel, mitoxantrone, and IR significantly increased p90RSK activity in monocytes and macrophages both in vitro and in vivo. p90RSK activation decreases NRF2-ARE transcriptional activity by increasing ERK5 S496 phosphorylation, leading to SASP. Interestingly, we found p90RSK-mediated ERK5 S496 phosphorylation caused mitochondrial stunning, which was associated with increased mtROS production even after ATP depletion and primed myeloid cells to a secondary insult of ROS. This priming effect was also detected in cancer patients even 1–2 months after the completion of radiation therapy. The epigenetic changes play a role in reprogramming, which in turn activates MCs and subsequently elicits atherogenesis [[62], [63], [64]]. Several studies support the link between mitochondria-mediated metabolites production and epigenetic reprograming that induces trained innate immunity, which is a similar concept to our priming to ROS in MCs [64,65]. However, the exact mechanism for priming including the role of epigenetic changes and the manner by which the state of priming is maintained are unclear. IR-induced epigenetic changes have been extensively studied, but the results are conflicting [66]. Therefore, the concept that priming is induced and maintained solely by epigenetic changes becomes questionable. In this current study, we found that IR forms a feedback loop involving phosphorylation of p90RSK and ERK5 and PARP activation which causes sustained mtROS production and subsequently priming of MCs. Indeed, transient PARP inhibition at the time of IR obstructed priming (Fig. 7K and L). Our data indicate that not only epigenetic changes but also the establishment of the feedback loop between the p90RSK-ERK5 module and PARP activation is crucial for priming MCs after IR.
As we discussed in the introduction, there are a plenty of studies to define the role and regulatory mechanisms of how nuclear PARP activation induces mitochondrial dysfunction (Part A in Fig. 10). However, to understand how SASP status becomes persistent, we investigated whether mtROS-induced telomere DNA damage can form the nucleus-mitochondria feedback loop, because telomere DNA damage induced by mtROS has been reported [39]. Therefore, in our current study, we focused on trying to determining how chemo-radiation-induced mtROS instigates telomere DNA damage and subsequent PARP activation and mitochondrial dysfunction (From part B to A in Fig. 10). Previously, we have reported the role of p90RSK-ERK5 S496-NRF2 pathway in the combined antiretroviral therapy (cART)-induced telomere shortening [26]. However, to the best of our knowledge, there is no data on how mitochondrial dysfunction can be regulated by this pathway and how p90RSK-mediated ERK5 S496 phosphorylation can form a positive feedback loop and induce persistent SASP. In this study, we found a crucial role for p90RSK-ERK5 S496 phosphorylation and subsequent attenuation of NRF2 transcriptional activity in chemo-radiation-induced telomere DNA damage and subsequent PARP activation, leading to mitochondrial dysfunction (From Part B to A in Fig. 10), inducing a positive feedback loop between the nucleus and the mitochondria by provoking persistent mtROS production. In this study, we found the crucial role of p90RSK-ERK5 S496 phosphorylation and subsequent attenuation of NRF2 transcriptional activity in chemo-radiation-induced telomere DNA damage and subsequent PARP activation, leading to mitochondrial dysfunction and persistent mtROS production (From Part B to A in Fig. 10), and form a nucleus-mitochondria positive feedback loop.
Fig. 10.

A new model of nucleus-mitochondria positive feedback loop formed by p90RSK-ERK5-NRF2 module-mediated PARP activation, and subsequent persistent SASP induction and coronary atherosclerosis formation after chemo-radiation.
(1) Chemo-radiation initiates mtROS production before p90RSK-ERK5 S496 phosphorylation, because initial phase of mtROS production induced by low dose of IR and DOX could not inhibit by p90RSK inhibitor and ERK5 S496A mutant (Fig. 5E and F).
(2) Next, chemo-radiation increased p90RSK-mediated ERK5 S496 phosphorylation, which subsequently inhibited ERK5 and NRF2 transcriptional activity, because mtROS specific inhibiotor (MitoTEMPOL) significantly inhibited low dose of both IR and DOX-induced p90RSK activity and ERK5 S496 phosphorylation. However, no change of ERK5 kinase activation detected by ERK5 TEY motif phosphorylation was detected (Supplementary Figs. 6H and I).
(3) The impairment of NRF2 transcriptional activity decreases anti-oxidant expressions including HO1 and Trx1 (Fig. 1H and N), which plays a crucial role of instigating persistent SASP status (i. senescence, ii. Inflammation, iii. mtROS production, iv. Attenuation of efferocytosis) (Fig. 1H-N), and primes MCs for enhancing the secondary insult of extracellular ROS-induced p90RSK-ERK5 S496 phosphorylation (Fig. 1H, I, and N).
(4) p90RSK-mediated ERK5 S496 phosphorylation and subsequent downregulation of NRF2 transcriptional activity and anti-oxidant expression is necessary for low dose of IR and DOX-induced telomere shortening (Fig. 1J and K, Fig. 3E, and Supplementary Figs. 5D, H, and K).
(5) Telomere DNA damage-induced PARP activation has been reported [37].
(6) PARP activation-mediated mitochondrial damage and cell death have been extensively studied [[34], [35], [36]]. For example, the involvement of the reduction of MKP1 induced by PARylated ATF-4 in ERK1/2/JNK/p38 activation-mediated mitochondrial dysfunction and cell death has been reported [55]. The implication of PARP activation on significant inhibition of both glycolysis and OXPHOS has been reported, and these conditions lead to severe and irreversible mitochondrial damage and cell death [35,56,59]. However, we found that low dose of IR and DOX did not induce any immediate cell death (Fig. 7B and C), and the depletion of ATP and NAD+ was recovered by PARP and p90RSK inhibitors (Fig. 7D–G), suggesting the reversibility of mitochondrial dysfunction. We referred this unique reversible form of mitochondrial dysfunction with severe ATP depletion as “mitochondrial (mt) stunning”.
(7) Another unique aspect of this mitochondrial stunning is its metabolically active status even under the depleted ATP condition. We found persistent mtROS production and the increase of succinate level after low dose of IR (Fig. 5A, E, F and G), and the induction of late phase of mtROS (but not early phase of mtROS production) (Fig. 5E and F) and succinate were p90RSK dependent (Fig. 6H). We also observed the necessity of complex II activity for mitochondrial stunning-induced mtROS production (Fig. 6F). Significant and persistent mtROS production without killing the cells would be important for chronic inflammation and unceasing SASP status, which are noted even long after the completion of cancer therapy as late effects.
As we showed in Fig. 5E and F, both the inhibition of p90RSK and the mutant of ERK5 S496A cannot inhibit the early phase (within 30 min of stimulation) of mtROS production induced by IR and DOX, but can inhibit the mtROS production in later phase after 6 h of IR and DOX treatment. MitoTEMPOL completely inhibited IR and DOX-induced p90RSK activation and ERK5 S496 phosphorylation as shown in the Supplementary Figs. 6H and J. These findings suggest that p90RSK-ERK5 S496 phosphorylation is mtROS dependent (Fig. 10 #2). Taken together, although late phase of mtROS production after chemo-radiation is p90RSK-ERK5 S496 phosphorylation dependent, the early phase of mtROS production is independent on p90RSK-ERK5 S496 phosphorylation, indicating that mtROS production is one of the initial steps for forming the nucleus-mitochondria positive feedback loop (Fig. 10 #1).
We found that p90RSK-ERK5 S496 phosphorylation is an upstream event of PARP activation (Fig. 6A and B, and Fig. 10 #5), and subsequent mitochondrial dysfunction (Figs. 4 and 10 #6) and late phase of mtROS production (Fig. 5E and F, and Fig. 10 #7). Since p90RSK-ERK5 S496 phosphorylation is not a downstream event of PARP activation as reported in other MAP kinase family [55,67], our finding is quite novel. One of the unique aspects of chemo-radiation-induced mtROS production is persistency, and we found the mtROS production was sustained over 24 h after low dose of IR and DOX treatment (Fig. 5G), which is very different from TNFα-induced mtROS production as we showed in Fig. 5A. This low dose of IR and DOX-induced persistent mtROS production forms a nucleus-mitochondria positive feedback loop as shown in Fig. 10 (#1–7).
Surprisingly, we detected mtROS production even under the ATP-depleted condition after chemo-radiation at the dose that resulted in no apoptosis and cell death but a slight decrease in succinate dehydrogenase (SDH) activity (Fig. 6G). This is a quite unique form of mitochondrial dysfunction compared to other stimuli, which have been reported previously. For example, Virág et al. showed that PARP activation induced cell necrosis, but no data on OXPHOS and glycolysis were included [35]. Andrabi et al. reported that N-methyl-N-nitroso-N′-nitroguanidine (MNNG) inhibits both OXPHOS and glycolysis via PARP activation in mouse cortical neurons, but again MNNG caused over 55% cell death after 24 h of treatment by detecting propidium iodide and Hoechst staining [59]. Although we did observe similar OXPHOS and glycolysis reduction after low dose of IR and DOX treatment, we did not find any increase of apoptotic cells detecting by annexin V staining (Fig. 1F) and cell death detecting by trypan blue staining and the release of LDH even after 24 h of low dose of IR and DOX (Fig. 7A–C). This is a very surprising finding, because low dose of IR and DOX completely reduced ATP levels by inhibiting both OXPHOS and glycolysis, but we could not find any increase of cell apoptosis and death. Furthermore, we did find a significant increase of mtROS production even after IR and DOX completely depleted NAD+ and ATP level. To the best of our knowledge, no previous work determined the high mtROS production under ongoing both ATP and NAD+ depleted condition (not after reperfusion as Choiuchani et al. have reported [68]). Since complex II activity is NAD+ independent and complex II is able to produce mtROS production, we investigated the potential role of complex II activity in mtROS production, and showed that the complex II specific inhibitor of Atepenin A5 significantly inhibited IR and DOX-induced mtROS production (Fig. 6F). These data suggest that complex II activity is necessary for mtROS production. Furthermore, we also found that IR increased succinate levels, which were inhibited by p90RSK inhibition (Fig. 6H). Thus, macrophages treated by low dose of IR and DOX are metabolically active, and the p90RSK-dependent increase of succinate level after IR and DOX can also explain why mtROS production was increased even when SDH activity is partially decreased (Fig. 6G). It is also important to note that succinyl coenzyme A (CoA) synthetase can generate GTP during the reaction from succinyl-CoA to succinate [69], which can be used as an alternative source for ATP in some reaction [70]. We will determine how succinate is increased by chemo-radiation in a p90RSK dependent manner in our future studies.
Another unique aspect of chemo-radiation-induced mitochondria dysfunction is its reversibility. As shown in Fig. 7A–G, the depletion of ATP and NAD+ was sustained, but treating cells with PARP or p90RSK inhibitor, even 24 h after IR, increased the reduced NAD+ and ATP levels to that of non-IR and vehicle-treated cells, suggesting that ATP and NAD+ depletion after IR is reversible. To the best of our knowledge, we could not find any papers that described the reversibility of ATP and NAD+ depletion after a significant decrease of both OXPHOS and glycolysis, and most of the papers showed cell death or severe mitochondrial damage [35,56,59]. Based on the unique aspects of mitochondrial dysfunction as we discussed above, we called this as “mitochondria stunning”. Mitochondrial stunning is a state of mitochondrial ATP depletion without showing any immediate cell death or apoptosis, and mitochondrial ATP depletion condition is reversible. Also, mitochondrial stunning is metabolically active with high persistent mtROS production. p90RSK-mediated ERK5 S496 phosphorylation is necessary for IR- and DOX-induced PARP activation, and all of these events in the nucleus-mitochondria positive feedback loop (#1-#7 in Fig. 10) contribute to inducing mitochondrilal stunning. Of note, during mitochondrial stunning, which could last for up to 48 h, mtROS production was increased by sustained complex II flux, possibly due to the increased p90RSK-mediated succinate level (Fig. 6H). These data also suggest that macrophage exposed to low dose of IR and DOX are not dying, and they are metabolically very active, which also supports the very unique aspects of the status of mitochondrial stunning.
Recently, it was proposed that PARP inhibitors can induce radiosensitization and that tumor cells are preferentially sensitized to RT by PARP inhibitors in repair-deficient hypoxic cells [71]. Of note, a biologically active dose of PARP inhibitors caused minimal toxicity in clinical trials; several clinical trials are evaluating the combination of RT and PARP inhibitors as radiosensitizers [71], but not as atherosclerosis inhibitors, for preventing the late effects of RT that result in CVD. In our study, we found that the combination of RT and PARP inhibition significantly decreased IR-induced coronary atherosclerosis formation, macrophage infiltration, and cardiac dysfunction in our TAC + IR model. Although the long-term inhibition of PARP can inhibit atherosclerosis formation in animal studies [72], it has not been recommended for the clinical use as an anti-atherosclerosis drug because of a potentially harmful side effect of long-term PARP inhibition on tumorigenesis by increasing chromosomal instability [73]. Our data provide pre-clinical evidence that the transient PARP inhibition restricted to the time of RT not only induces radiosensitization in tumor cells but also inhibits cardiovascular damage after RT without causing potential tumorigenesis induced by long-term PARP inhibition.
In this study, we found a significant macrophage infiltration into perivascular regions after IR, regardless of coronary plaque formation; this macrophage infiltration was inhibited by the PARP inhibitor. It is becoming evident that cardiac remodeling is, at least in part, controlled by a complex cross-talk between cardiomyocytes and macrophages [74]. For example, inhibition of monocyte and monocyte-derived macrophage recruitment improves cardiac dysfunction after myocardial infarction [75]. On the other hand, macrophages could cause conduction abnormalities by secreting IL-1β which destabilizes the electrical activity of cardiomyocytes, potentially promoting ventricular arrhythmia [76]. Thus, macrophage infiltration could play a role in instigating cardiac dysfunction after IR, which could be prevented by PARP activation. Monocytes and macrophages are cells of the innate immune system and are also relevant to aortic stiffness associated with atherosclerotic plaque formation. Although resident macrophages can be derived from a unique lineage [77], recent evidence indicates that circulating monocytes can traffic into and out of tissues without becoming macrophages [78]. Therefore, circulating monocytes may play a significant role in regulating arterial stiffness and atherogenesis. Indeed, decreased arterial stiffness in angiotensin II-infused macrophage colony stimulating factor-deficient mice, which are deficient in both monocytes and macrophages, was reported [79]. Although RT targets localized cancer, it can have systemic effects [[80], [81], [82], [83]]. The current study shows that localized radiation exposure of cancer patients leads to priming of circulating monocytes to H2O2, even 1–2 months after the completion of RT, suggesting that radiation exerts a systemic effect, at least through circulating monocytes. Since we previously reported that p90RSK activation in MCs has a crucial role in atherosclerosis development, the presence of readily activatable p90RSK in monocytes may predispose cancer survivors to CVD.
Based on prior publications [[84], [85], [86], [87], [88], [89], [90], [91], [92]], and the new data presented in this study, we have formulated a conceptual model as the followings. Long established risk factors such as smoking, hypertension, and diabetes, “prime” the system towards a pro-inflammatory state, which is further exacerbated by various cancer treatments, especially by ionizing radiation (IR) [[84], [85], [86], [87], [88], [89], [90], [91], [92]]. Inflammation is a critical driver of atherosclerosis and an independent risk factor for myocardial infarction, heart failure and cardiovascular death [93]. At the central hub of this chronic inflammation are the monocyte/myeloid cells (MC), which play a key role in healing processes. It is the abnormal activation of MC through various processes, including IR, that progresses through a series of steps that lead to microvascular damage and radiation-induced CVD (RICVD) (Fig. S8): (1) MC activation and reprogramming, which promotes cellular infiltration and extravasation into areas of IR-induced perivascular injury. Reprogrammed MCs at the site of IR elicit priming of circulating MC, probably in the spleen or bone marrow, and accelerate MC recruitment even long after IR [94,95]; (2) Infiltrated MCs interact with cardiomyocytes, and induce cardiac dysfunction and arrhythmias [96] (3) Activation of endothelial cells (EC) stimulates EC inflammation and damage induced by cytokines secreted from MC [97]; (4) EC activation provokes microvascular dysfunction, which promotes and predicts future CV events and coronary atherosclerosis (5) [98,99]. Each step proposed here definitely needs further investigation in the context of chemo-radiation-induced cardiovascular disease, but our novel animal model would be helpful to define these steps.
We did not see a significant decrease of ERK5 transactivation induced by chemo-radiation without CA-MEK5 transfection in myeloid cells (Fig. 1B). There are two possibilities to explain this. The basal level of ERK5 transactivation was very low in myeloid cells when we used our mammalian one-hybrid assay system with Gal4-ERK5 and the Gal4-responsive luciferase reporter pG5-Luc constructs. Therefore, it may be difficult to detect more down-regulating effects of ERK5 S496 phosphorylation on ERK5 transactivation at the basal level in myeloid cells because of the low basal level of ERK5 transactivation and sensitivity of the ERK5 transactivation assay system in myeloid cells. Of note, since we did see the decrease of ERK5 transactivation induced by various chemotherapy reagents in Hela cells with HTS analysis without CA-MEK5 transfection, the low basal level of ERK5 transactivation might be cell type-specific.
However, it is also possible that ERK5 S496 phosphorylation can regulate NRF2 transactivation, independent of ERK5 transactivation. We also found that NRF2 SUMOylation was regulated by ERK5 S496 phosphorylation, which was independent of ERK5 transcriptional activity (data not shown). Therefore, NRF2 transcriptional activity may be regulated by ERK5 S496 phosphorylation-mediated NRF2 SUMOylation without affecting ERK5 transactivation. We will investigate this possibility in our future study.
In conclusion, this study uncovered that various cancer treatments instigate p90RSK-mediated ERK5 S496 phosphorylation as the shared signaling event in circulating monocytes, and this p90RSK-ERK5 module plays key roles in priming MCs for ROS and establishing the nucleus-mitochondrial feedback loop through telomere DNA damage-mediated PARP activation, mitochondrial stunning, and mtROS production. The activation of this nucleus-mitochondria feedback loop reprograms MCs toward SASP while keeping the cells’ physiologic states in reduced antioxidant expression, heightened telomere DNA damage, and increased sensitivity to ROS. We also showed that the inhibition of PARP effectively suppressed the IR-induced mitochondrial stunning, priming, and coronary atherosclerosis. Based on these results, we propose that the PARP inhibitor is a promising candidate that inhibits the IR- and anti-cancer drug-induced pathologic events in MCs. Thus, PARP inhibitors are not only radio-sensitizers but also effective agents that could reduce CV events after radiation.
As we explained above, Part A (except the aspect of mt stunning) has been well studied [[34], [35], [36]], and we have reported the role of p90RSK-ERK5 S496 phosphorylation-NRF2 module in regulating combined antiretrovirals therapy-induced telomere DNA damage [26] (Part B). However, to the best of our knowledge, how part B regulates part A remains largely unknown. In this study, we are successfully closing the gap between part A and B by finding the key role of p90RSK-ERK5 S496 phosphorylation-NRF2 module in chemo-radiation-induced PARP activation and mitochondrial dysfunction and establish the novel concept of nucleus-mitochondria positive feedback loop. More importantly, we discover the crucial role of reversible mitochondrial stunning without causing immediate cell death for explaining persistent mtROS production in MC after low dose of IR and DOX treatment. This persistent mtROS production is also essential for maintaining nucleus-mitochondria positive feedback loop and consequently long-lasting SASP status as described in Part C.
In part C, we showed that each component of nucleus-mitochondria positive feedback loop formed by part A and B contributes to prolonged SASP status as i) telomere DNA damage to senescence, ii) ERK5 S496 phosphorylation-NRF2 and PARP activation to inflammation and ROS [26,101], iii) mt stunning to mtROS, and iv) p90RSK-ERK5 S496 phosphorylation to efferocytosis [26]. The reduction of anti-oxidant expression contributes to the priming effects of MC in response to the secondary ROS insult. Although the contribution of SASP in atherosclerosis has been suggested [102,103], to the best of our knowledge, nobody has tested whether the transient inhibition of PARP activation at the time of IR only attenuates SASP induction and priming of MC after IR, and suppresses IR-induced coronary atherosclerosis. Our data provide a novel pre-clinical evidence that the transient PARP inhibition restricted to the time of RT not only induces radiosensitization in tumor cells but also inhibits cardiovascular damage after RT.
Author contributions
S.K., A.Z., M.I., and K.A.K. performed the experiments, interpreted the data, and wrote the manuscript. Y.J.G. performed the experiments and analyzed the data. S.H.L., C.C., S.A.M., and S.K. planned the clinical studies, supervised the data acquisition, and interpreted the data. D.A. and H.G. evaluated study participants and contributed to the interpretation of the data. A.G. supervised data management and supported data analyses. T.N.T maintained mouse colony. K.L.S., J.B., S.W.Y., Y.H.S., J.H., E.S.K., M.L.E., J.P.C., G.S., S.B.M., Z.S.P.,Y.Y., A.D., and P.S.B. contributed to the interpretation of the data. K.F. analyzed and interpreted the imaging data and edited the manuscript. E.M. designed and generated ERK5 S496A knock-in mice. S.K., NT.L., D.J.H., and J.A. planned and generated the study design, obtained funding, interpreted data, and wrote the manuscript.
Declaration of competing interest
S.H.L is an Advisory Board member of AstraZeneca, Beyond Spring Pharmaceuticals, STCube Pharmaceuticals. The other authors report no conflicts.
Acknowledgements
We thank Scientific Publications, Research Medical Library at The University of Texas MD Anderson Cancer Center for editing and Carolyn J. Giancursio for her technical assistance. We thank the study coordinators and supporting staff at MD Anderson for enrolling the study participants and managing data and specimen collection. This clinical study would not have been possible without the generous contribution of time by the study participants.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2021.102132.
Contributor Information
Sivareddy Kotla, Email: skotla@mdanderson.org.
Jun-ichi Abe, Email: jabe@mdanderson.org.
Data availability
The individual data will be shared or reasonable request to the corresponding authors.
Funding
This work was partially supported by grants from the National Institutes of Health (NIH) to Drs. Abe (AI-156921), Maggirwar (HL-123346), Schifitto (HL-123346), Cooke (HL-149303), Le (HL-134740 and HL-149303), National Aeronautics and Space Administration (NASA) to Dr. Caroline Chung (80NSSC18K1639 P00007), and from Cancer Prevention and Research Institute of Texas (CPRIT) to Drs. Abe and Schadler (RP190256). This research performed in the Flow Cytometry and Cellular Imaging Facility (NC) is supported in part by the National Institutes of Health through MD Anderson's Support Grant CA016672.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Monsuez J.J., Belin C., Bouchaud O. Microvascular function in aging among women living with HIV. Curr. HIV AIDS Rep. 2016;13(6):392–398. doi: 10.1007/s11904-016-0339-3. [DOI] [PubMed] [Google Scholar]
- 2.Kaplan-Lewis E., Aberg J.A., Lee M. Atherosclerotic cardiovascular disease and anti-retroviral therapy. Curr. HIV AIDS Rep. 2016;13(5):297–308. doi: 10.1007/s11904-016-0331-y. [DOI] [PubMed] [Google Scholar]
- 3.Lipshultz S.E., Landy D.C., Lopez-Mitnik G., Lipsitz S.R., Hinkle A.S., Constine L.S., French C.A., Rovitelli A.M., Proukou C., Adams M.J., Miller T.L. Cardiovascular status of childhood cancer survivors exposed and unexposed to cardiotoxic therapy. J. Clin. Oncol. 2012;30(10):1050–1057. doi: 10.1200/JCO.2010.33.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chow E.J., Chen Y., Hudson M.M., Feijen E.A.M., Kremer L.C., Border W.L., Green D.M., Meacham L.R., Mulrooney D.A., Ness K.K., Oeffinger K.C., Ronckers C.M., Sklar C.A., Stovall M., van der Pal H.J., van Dijk I., van Leeuwen F.E., Weathers R.E., Robison L.L., Armstrong G.T., Yasui Y. Prediction of ischemic heart disease and stroke in survivors of childhood cancer. J. Clin. Oncol. 2018;36(1):44–52. doi: 10.1200/JCO.2017.74.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hull M.C., Morris C.G., Pepine C.J., Mendenhall N.P. Valvular dysfunction and carotid, subclavian, and coronary artery disease in survivors of hodgkin lymphoma treated with radiation therapy. J. Am. Med. Assoc. 2003;290(21):2831–2837. doi: 10.1001/jama.290.21.2831. [DOI] [PubMed] [Google Scholar]
- 6.Moslehi J. The cardiovascular perils of cancer survivorship. N. Engl. J. Med. 2013;368(11):1055–1056. doi: 10.1056/NEJMe1215300. [DOI] [PubMed] [Google Scholar]
- 7.Oeffinger K.C., Mertens A.C., Sklar C.A., Kawashima T., Hudson M.M., Meadows A.T., Friedman D.L., Marina N., Hobbie W., Kadan-Lottick N.S., Schwartz C.L., Leisenring W., Robison L.L., Childhood Cancer Survivor S. Chronic health conditions in adult survivors of childhood cancer. N. Engl. J. Med. 2006;355(15):1572–1582. doi: 10.1056/NEJMsa060185. [DOI] [PubMed] [Google Scholar]
- 8.Patnaik J.L., Byers T., DiGuiseppi C., Dabelea D., Denberg T.D. Cardiovascular disease competes with breast cancer as the leading cause of death for older females diagnosed with breast cancer: a retrospective cohort study. Breast Cancer Res. 2011;13(3):R64. doi: 10.1186/bcr2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Erning F.N., van Steenbergen L.N., Lemmens V.E., Rutten H.J., Martijn H., van Spronsen D.J., Janssen-Heijnen M.L. Conditional survival for long-term colorectal cancer survivors in The Netherlands: who do best? Eur. J. Canc. 2014;50(10):1731–1739. doi: 10.1016/j.ejca.2014.04.009. [DOI] [PubMed] [Google Scholar]
- 10.Ness K.K., Krull K.R., Jones K.E., Mulrooney D.A., Armstrong G.T., Green D.M., Chemaitilly W., Smith W.A., Wilson C.L., Sklar C.A., Shelton K., Srivastava D.K., Ali S., Robison L.L., Hudson M.M. Physiologic frailty as a sign of accelerated aging among adult survivors of childhood cancer: a report from the St Jude Lifetime cohort study. J. Clin. Oncol. 2013;31(36):4496–4503. doi: 10.1200/JCO.2013.52.2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanoff H.K., Deal A.M., Krishnamurthy J., Torrice C., Dillon P., Sorrentino J., Ibrahim J.G., Jolly T.A., Williams G., Carey L.A., Drobish A., Gordon B.B., Alston S., Hurria A., Kleinhans K., Rudolph K.L., Sharpless N.E., Muss H.B. Effect of cytotoxic chemotherapy on markers of molecular age in patients with breast cancer. J. Natl. Cancer Inst. 2014;106(4):dju057. doi: 10.1093/jnci/dju057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hurria A., Jones L., Muss H.B. Cancer treatment as an accelerated aging process: assessment, biomarkers, and interventions. Am Soc Clin Oncol Educ Book. 2016;35:e516–e522. doi: 10.1200/EDBK_156160. [DOI] [PubMed] [Google Scholar]
- 13.Katsuumi G., Shimizu I., Yoshida Y., Minamino T. Vascular senescence in cardiovascular and metabolic diseases. Front Cardiovasc Med. 2018;5:18. doi: 10.3389/fcvm.2018.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ishida T., Ishida M., Tashiro S., Yoshizumi M., Kihara Y. Role of DNA damage in cardiovascular disease. Circ. J. 2014;78(1):42–50. doi: 10.1253/circj.cj-13-1194. [DOI] [PubMed] [Google Scholar]
- 15.Cappetta D., Rossi F., Piegari E., Quaini F., Berrino L., Urbanek K., De Angelis A. Doxorubicin targets multiple players: a new view of an old problem. Pharmacol. Res. 2018;127:4–14. doi: 10.1016/j.phrs.2017.03.016. [DOI] [PubMed] [Google Scholar]
- 16.Darby S.C., Ewertz M., Hall P. Ischemic heart disease after breast cancer radiotherapy. N. Engl. J. Med. 2013;368(26):2527. doi: 10.1056/NEJMc1304601. [DOI] [PubMed] [Google Scholar]
- 17.Wang X., Palaskas N.L., Yusuf S.W., Abe J.I., Lopez-Mattei J., Banchs J., Gladish G.W., Lee P., Liao Z., Deswal A., Lin S.H. Incidence and onset of severe cardiac events after radiotherapy for esophageal cancer. J. Thorac. Oncol. 2020 doi: 10.1016/j.jtho.2020.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindsay S., Kohn H.I., Dakin R.L., Jew J. Aortic arteriosclerosis in the dog after localized aortic x-irradiation. Circ. Res. 1962;10:51–60. doi: 10.1161/01.res.10.1.51. [DOI] [PubMed] [Google Scholar]
- 19.Curigliano G., Cardinale D., Suter T., Plataniotis G., de Azambuja E., Sandri M.T., Criscitiello C., Goldhirsch A., Cipolla C., Roila F., Group E.G.W. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO Clinical Practice Guidelines. Ann. Oncol. 2012;23(Suppl 7):vii155–v166. doi: 10.1093/annonc/mds293. [DOI] [PubMed] [Google Scholar]
- 20.Carver J.R., Shapiro C.L., Ng A., Jacobs L., Schwartz C., Virgo K.S., Hagerty K.L., Somerfield M.R., Vaughn D.J., Panel A.C.S.E. American Society of Clinical Oncology clinical evidence review on the ongoing care of adult cancer survivors: cardiac and pulmonary late effects. J. Clin. Oncol. 2007;25(25):3991–4008. doi: 10.1200/JCO.2007.10.9777. [DOI] [PubMed] [Google Scholar]
- 21.Drafts B.C., Twomley K.M., D'Agostino R., Jr., Lawrence J., Avis N., Ellis L.R., Thohan V., Jordan J., Melin S.A., Torti F.M., Little W.C., Hamilton C.A., Hundley W.G. Low to moderate dose anthracycline-based chemotherapy is associated with early noninvasive imaging evidence of subclinical cardiovascular disease. JACC Cardiovasc Imaging. 2013;6(8):877–885. doi: 10.1016/j.jcmg.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chaosuwannakit N., D'Agostino R., Jr., Hamilton C.A., Lane K.S., Ntim W.O., Lawrence J., Melin S.A., Ellis L.R., Torti F.M., Little W.C., Hundley W.G. Aortic stiffness increases upon receipt of anthracycline chemotherapy. J. Clin. Oncol. 2010;28(1):166–172. doi: 10.1200/JCO.2009.23.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J.C., Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012;111(2):245–259. doi: 10.1161/CIRCRESAHA.111.261388. [DOI] [PubMed] [Google Scholar]
- 24.Childs B.G., Baker D.J., Wijshake T., Conover C.A., Campisi J., van Deursen J.M. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354(6311):472–477. doi: 10.1126/science.aaf6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaitiekus D., Muckiene G., Vaitiekiene A., Maciuliene D., Vaiciuliene D., Ambrazeviciute G., Sereikaite L., Verikas D., Jurkevicius R., Juozaityte E. Impact of arterial hypertension on doxorubicin-based chemotherapy-induced subclinical cardiac damage in breast cancer patients. Cardiovasc. Toxicol. 2020;20(3):321–327. doi: 10.1007/s12012-019-09556-3. [DOI] [PubMed] [Google Scholar]
- 26.Singh M.V., Kotla S., Le N.T., Ae Ko K., Heo K.S., Wang Y., Fujii Y., Thi Vu H., McBeath E., Thomas T.N., Jin Gi Y., Tao Y., Medina J.L., Taunton J., Carson N., Dogra V., Doyley M.M., Tyrell A., Lu W., Qiu X., Stirpe N.E., Gates K.J., Hurley C., Fujiwara K., Maggirwar S.B., Schifitto G., Abe J.I. Senescent phenotype induced by p90RSK-NRF2 signaling sensitizes monocytes and macrophages to oxidative stress in HIV-positive individuals. Circulation. 2019;139(9):1199–1216. doi: 10.1161/CIRCULATIONAHA.118.036232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Le N.T., Heo K.S., Takei Y., Lee H., Woo C.H., Chang E., McClain C., Hurley C., Wang X., Li F., Xu H., Morrell C., Sullivan M.A., Cohen M.S., Serafimova I.M., Taunton J., Fujiwara K., Abe J. A crucial role for p90RSK-mediated reduction of ERK5 transcriptional activity in endothelial dysfunction and atherosclerosis. Circulation. 2013;127(4):486–499. doi: 10.1161/CIRCULATIONAHA.112.116988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heo K.S., Berk B., Abe J.I. Disturbed flow-induced endothelial pro-atherogenic signaling via regulating post-translational modifications and epigenetic events. Antioxidants Redox Signal. 2015;25(7):435–450. doi: 10.1089/ars.2015.6556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coppe J.P., Patil C.K., Rodier F., Sun Y., Munoz D.P., Goldstein J., Nelson P.S., Desprez P.Y., Campisi J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Faget D.V., Ren Q., Stewart S.A. Unmasking senescence: context-dependent effects of SASP in cancer. Nat. Rev. Canc. 2019;19(8):439–453. doi: 10.1038/s41568-019-0156-2. [DOI] [PubMed] [Google Scholar]
- 31.He S., Sharpless N.E. Senescence in health and disease. Cell. 2017;169(6):1000–1011. doi: 10.1016/j.cell.2017.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Acosta J.C., O'Loghlen A., Banito A., Guijarro M.V., Augert A., Raguz S., Fumagalli M., Da Costa M., Brown C., Popov N., Takatsu Y., Melamed J., d'Adda di Fagagna F., Bernard D., Hernando E., Gil J. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell. 2008;133(6):1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- 33.Coppe J.P., Rodier F., Patil C.K., Freund A., Desprez P.Y., Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011;286(42):36396–36403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bai P., Canto C., Oudart H., Brunyanszki A., Cen Y., Thomas C., Yamamoto H., Huber A., Kiss B., Houtkooper R.H., Schoonjans K., Schreiber V., Sauve A.A., Menissier-de Murcia J., Auwerx J. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metabol. 2011;13(4):461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Virag L., Salzman A.L., Szabo C. Poly(ADP-ribose) synthetase activation mediates mitochondrial injury during oxidant-induced cell death. J. Immunol. 1998;161(7):3753–3759. [PubMed] [Google Scholar]
- 36.Bai P., Nagy L., Fodor T., Liaudet L., Pacher P. Poly(ADP-ribose) polymerases as modulators of mitochondrial activity. Trends Endocrinol. Metabol. 2015;26(2):75–83. doi: 10.1016/j.tem.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 37.Gomez M., Wu J., Schreiber V., Dunlap J., Dantzer F., Wang Y., Liu Y. PARP1 Is a TRF2-associated poly(ADP-ribose)polymerase and protects eroded telomeres. Mol. Biol. Cell. 2006;17(4):1686–1696. doi: 10.1091/mbc.E05-07-0672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar N., Qian W., Van Houten B. Sick mitochondria cause telomere damage: implications for disease. Mol Cell Oncol. 2020;7(1):1678362. doi: 10.1080/23723556.2019.1678362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian W., Kumar N., Roginskaya V., Fouquerel E., Opresko P.L., Shiva S., Watkins S.C., Kolodieznyi D., Bruchez M.P., Van Houten B. Chemoptogenetic damage to mitochondria causes rapid telomere dysfunction. Proc. Natl. Acad. Sci. U. S. A. 2019;116(37):18435–18444. doi: 10.1073/pnas.1910574116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang S., Liu X., Bawa-Khalfe T., Lu L.S., Lyu Y.L., Liu L.F., Yeh E.T. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat. Med. 2012;18(11):1639–1642. doi: 10.1038/nm.2919. [DOI] [PubMed] [Google Scholar]
- 41.Davalos A.R., Coppe J.P., Campisi J., Desprez P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Canc. Metastasis Rev. 2010;29(2):273–283. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Coppe J.P., Desprez P.Y., Krtolica A., Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Le N.T., Takei Y., Izawa-Ishizawa Y., Heo K.S., Lee H., Smrcka A.V., Miller B.L., Ko K.A., Ture S., Morrell C., Fujiwara K., Akaike M., Abe J. Identification of activators of ERK5 transcriptional activity by high-throughput screening and the role of endothelial ERK5 in vasoprotective effects induced by statins and antimalarial agents. J. Immunol. 2014;193(7):3803–3815. doi: 10.4049/jimmunol.1400571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lowe D., Raj K. Premature aging induced by radiation exhibits pro-atherosclerotic effects mediated by epigenetic activation of CD44 expression. Aging Cell. 2014;13(5):900–910. doi: 10.1111/acel.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dong L.F., Low P., Dyason J.C., Wang X.F., Prochazka L., Witting P.K., Freeman R., Swettenham E., Valis K., Liu J., Zobalova R., Turanek J., Spitz D.R., Domann F.E., Scheffler I.E., Ralph S.J., Neuzil J. Alpha-tocopheryl succinate induces apoptosis by targeting ubiquinone-binding sites in mitochondrial respiratory complex II. Oncogene. 2008;27(31):4324–4335. doi: 10.1038/onc.2008.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang L., Zhang X., Cui G., Chan J.Y., Wang L., Li C., Shan L., Xu C., Zhang Q., Wang Y., Di L., Lee S.M. A novel agent exerts antitumor activity in breast cancer cells by targeting mitochondrial complex II. Oncotarget. 2016;7(22):32054–32064. doi: 10.18632/oncotarget.8410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Maraldo M.V., Giusti F., Vogelius I.R., Lundemann M., van der Kaaij M.A., Ramadan S., Meulemans B., Henry-Amar M., Aleman B.M., Raemaekers J., Meijnders P., Moser E.C., Kluin-Nelemans H.C., Feugier P., Casasnovas O., Fortpied C., Specht L., European Organisation for R., Treatment of Cancer Lymphoma G. Cardiovascular disease after treatment for Hodgkin's lymphoma: an analysis of nine collaborative EORTC-LYSA trials. Lancet Haematol. 2015;2(11):e492–502. doi: 10.1016/S2352-3026(15)00153-2. [DOI] [PubMed] [Google Scholar]
- 48.Drost L., Yee C., Lam H., Zhang L., Wronski M., McCann C., Lee J., Vesprini D., Leung E., Chow E. A systematic review of heart dose in breast radiotherapy. Clin. Breast Canc. 2018;18(5):e819–e824. doi: 10.1016/j.clbc.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 49.Wihlm J., Limacher J.M., Leveque D., Duclos B., Dufour P., Bergerat J.P., Methlin G. [Pharmacokinetic profile of high-dose doxorubicin administered during a 6 h intravenous infusion in breast cancer patients] Bull. Cancer. 1997;84(6):603–608. [PubMed] [Google Scholar]
- 50.Heo K.S., Cushman H.J., Akaike M., Woo C.H., Wang X., Qiu X., Fujiwara K., Abe J. ERK5 activation in macrophages promotes efferocytosis and inhibits atherosclerosis. Circulation. 2014;130(2):180–191. doi: 10.1161/CIRCULATIONAHA.113.005991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Steel R., Cowan J., Payerne E., O'Connell M.A., Searcey M. Anti-inflammatory effect of a cell-penetrating peptide targeting the nrf2/keap1 interaction. ACS Med. Chem. Lett. 2012;3(5):407–410. doi: 10.1021/ml300041g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu D., Wang X., Sun H. The role of mitochondria in cellular toxicity as a potential drug target. Cell Biol. Toxicol. 2018;34(2):87–91. doi: 10.1007/s10565-018-9425-1. [DOI] [PubMed] [Google Scholar]
- 53.Van den Bossche J., Baardman J., Otto N.A., van der Velden S., Neele A.E., van den Berg S.M., Luque-Martin R., Chen H.J., Boshuizen M.C., Ahmed M., Hoeksema M.A., de Vos A.F., de Winther M.P. Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 2016;17(3):684–696. doi: 10.1016/j.celrep.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 54.Schriewer J.M., Peek C.B., Bass J., Schumacker P.T. ROS-mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia-reperfusion. J Am Heart Assoc. 2013;2(2) doi: 10.1161/JAHA.113.000159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hocsak E., Szabo V., Kalman N., Antus C., Cseh A., Sumegi K., Eros K., Hegedus Z., Gallyas F., Jr., Sumegi B., Racz B. PARP inhibition protects mitochondria and reduces ROS production via PARP-1-ATF4-MKP-1-MAPK retrograde pathway. Free Radic. Biol. Med. 2017;108:770–784. doi: 10.1016/j.freeradbiomed.2017.04.018. [DOI] [PubMed] [Google Scholar]
- 56.Murata M.M., Kong X., Moncada E., Chen Y., Imamura H., Wang P., Berns M.W., Yokomori K., Digman M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell. 2019;30(20):2584–2597. doi: 10.1091/mbc.E18-10-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gerbitz K.D., Gempel K., Brdiczka D. Mitochondria and diabetes. Genetic, biochemical, and clinical implications of the cellular energy circuit. Diabetes. 1996;45(2):113–126. doi: 10.2337/diab.45.2.113. [DOI] [PubMed] [Google Scholar]
- 58.Wojtovich A.P., Brookes P.S. The complex II inhibitor atpenin A5 protects against cardiac ischemia-reperfusion injury via activation of mitochondrial KATP channels. Basic Res. Cardiol. 2009;104(2):121–129. doi: 10.1007/s00395-009-0001-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Andrabi S.A., Umanah G.K., Chang C., Stevens D.A., Karuppagounder S.S., Gagne J.P., Poirier G.G., Dawson V.L., Dawson T.M. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc. Natl. Acad. Sci. U. S. A. 2014;111(28):10209–10214. doi: 10.1073/pnas.1405158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Marino A., Zhang Y., Rubinelli L., Riemma M.A., Ip J.E., Di Lorenzo A. Pressure overload leads to coronary plaque formation, progression, and myocardial events in ApoE-/- mice. JCI Insight. 2019;4(9) doi: 10.1172/jci.insight.128220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yusuf S.W., Sami S., Daher I.N. Radiation-induced heart disease: a clinical update. Cardiol. Res. Pract. 2011;2011:317659. doi: 10.4061/2011/317659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seijkens T., Hoeksema M.A., Beckers L., Smeets E., Meiler S., Levels J., Tjwa M., de Winther M.P., Lutgens E. Hypercholesterolemia-induced priming of hematopoietic stem and progenitor cells aggravates atherosclerosis. Faseb. J. 2014;28(5):2202–2213. doi: 10.1096/fj.13-243105. [DOI] [PubMed] [Google Scholar]
- 63.van Kampen E., Jaminon A., van Berkel T.J., Van Eck M. Diet-induced (epigenetic) changes in bone marrow augment atherosclerosis. J. Leukoc. Biol. 2014;96(5):833–841. doi: 10.1189/jlb.1A0114-017R. [DOI] [PubMed] [Google Scholar]
- 64.Christ A., Bekkering S., Latz E., Riksen N.P. Long-term activation of the innate immune system in atherosclerosis. Semin. Immunol. 2016;28(4):384–393. doi: 10.1016/j.smim.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 65.Bekkering S., Joosten L.A., van der Meer J.W., Netea M.G., Riksen N.P. Trained innate immunity and atherosclerosis. Curr. Opin. Lipidol. 2013;24(6):487–492. doi: 10.1097/MOL.0000000000000023. [DOI] [PubMed] [Google Scholar]
- 66.Miousse I.R., Kutanzi K.R., Koturbash I. Effects of ionizing radiation on DNA methylation: from experimental biology to clinical applications. Int. J. Radiat. Biol. 2017;93(5):457–469. doi: 10.1080/09553002.2017.1287454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Veres B., Radnai B., Gallyas F., Jr., Varbiro G., Berente Z., Osz E., Sumegi B. Regulation of kinase cascades and transcription factors by a poly(ADP-ribose) polymerase-1 inhibitor, 4-hydroxyquinazoline, in lipopolysaccharide-induced inflammation in mice. J. Pharmacol. Exp. Therapeut. 2004;310(1):247–255. doi: 10.1124/jpet.104.065151. [DOI] [PubMed] [Google Scholar]
- 68.Chouchani E.T., Pell V.R., Gaude E., Aksentijevic D., Sundier S.Y., Robb E.L., Logan A., Nadtochiy S.M., Ord E.N.J., Smith A.C., Eyassu F., Shirley R., Hu C.H., Dare A.J., James A.M., Rogatti S., Hartley R.C., Eaton S., Costa A.S.H., Brookes P.S., Davidson S.M., Duchen M.R., Saeb-Parsy K., Shattock M.J., Robinson A.J., Work L.M., Frezza C., Krieg T., Murphy M.P. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515(7527):431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sanadi D.R., Gibson D.M., Ayengar P., Jacob M. Alpha-ketoglutaric dehydrogenase. V. Guanosine diphosphate in coupled phosphorylation. J. Biol. Chem. 1956;218(1):505–520. [PubMed] [Google Scholar]
- 70.Rogne P., Rosselin M., Grundstrom C., Hedberg C., Sauer U.H., Wolf-Watz M. Molecular mechanism of ATP versus GTP selectivity of adenylate kinase. Proc. Natl. Acad. Sci. U. S. A. 2018;115(12):3012–3017. doi: 10.1073/pnas.1721508115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chalmers A.J., Lakshman M., Chan N., Bristow R.G. Poly(ADP-ribose) polymerase inhibition as a model for synthetic lethality in developing radiation oncology targets. Semin. Radiat. Oncol. 2010;20(4):274–281. doi: 10.1016/j.semradonc.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 72.Pacher P., Szabo C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: the therapeutic potential of PARP inhibitors. Cardiovasc. Drug Rev. 2007;25(3):235–260. doi: 10.1111/j.1527-3466.2007.00018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Patel A.G., Sarkaria J.N., Kaufmann S.H. Nonhomologous end joining drives poly(ADP-ribose) polymerase (PARP) inhibitor lethality in homologous recombination-deficient cells. Proc. Natl. Acad. Sci. U. S. A. 2011;108(8):3406–3411. doi: 10.1073/pnas.1013715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gomez I., Duval V., Silvestre J.S. Cardiomyocytes and macrophages discourse on the method to govern cardiac repair. Front Cardiovasc Med. 2018;5:134. doi: 10.3389/fcvm.2018.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lavine K.J., Epelman S., Uchida K., Weber K.J., Nichols C.G., Schilling J.D., Ornitz D.M., Randolph G.J., Mann D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. U. S. A. 2014;111(45):16029–16034. doi: 10.1073/pnas.1406508111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Monnerat G., Alarcon M.L., Vasconcellos L.R., Hochman-Mendez C., Brasil G., Bassani R.A., Casis O., Malan D., Travassos L.H., Sepulveda M., Burgos J.I., Vila-Petroff M., Dutra F.F., Bozza M.T., Paiva C.N., Carvalho A.B., Bonomo A., Fleischmann B.K., de Carvalho A.C.C., Medei E. Macrophage-dependent IL-1beta production induces cardiac arrhythmias in diabetic mice. Nat. Commun. 2016;7:13344. doi: 10.1038/ncomms13344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hashimoto D., Chow A., Noizat C., Teo P., Beasley M.B., Leboeuf M., Becker C.D., See P., Price J., Lucas D., Greter M., Mortha A., Boyer S.W., Forsberg E.C., Tanaka M., van Rooijen N., Garcia-Sastre A., Stanley E.R., Ginhoux F., Frenette P.S., Merad M. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804. doi: 10.1016/j.immuni.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jakubzick C., Gautier E.L., Gibbings S.L., Sojka D.K., Schlitzer A., Johnson T.E., Ivanov S., Duan Q., Bala S., Condon T., van Rooijen N., Grainger J.R., Belkaid Y., Ma'ayan A., Riches D.W., Yokoyama W.M., Ginhoux F., Henson P.M., Randolph G.J. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39(3):599–610. doi: 10.1016/j.immuni.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.De Ciuceis C., Amiri F., Brassard P., Endemann D.H., Touyz R.M., Schiffrin E.L. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler. Thromb. Vasc. Biol. 2005;25(10):2106–2113. doi: 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- 80.Formenti S.C., Demaria S. Local control by radiotherapy: is that all there is? Breast Cancer Res. 2008;10(6):215. doi: 10.1186/bcr2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Formenti S.C., Demaria S. Systemic effects of local radiotherapy. Lancet Oncol. 2009;10(7):718–726. doi: 10.1016/S1470-2045(09)70082-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nesslinger N.J., Sahota R.A., Stone B., Johnson K., Chima N., King C., Rasmussen D., Bishop D., Rennie P.S., Gleave M., Blood P., Pai H., Ludgate C., Nelson B.H. Standard treatments induce antigen-specific immune responses in prostate cancer. Clin. Canc. Res. 2007;13(5):1493–1502. doi: 10.1158/1078-0432.CCR-06-1772. [DOI] [PubMed] [Google Scholar]
- 83.Rerkpattanapipat P., D'Agostino R.B., Jr., Link K.M., Shahar E., Lima J.A., Bluemke D.A., Sinha S., Herrington D.M., Hundley W.G. Location of arterial stiffening differs in those with impaired fasting glucose versus diabetes: implications for left ventricular hypertrophy from the Multi-Ethnic Study of Atherosclerosis. Diabetes. 2009;58(4):946–953. doi: 10.2337/db08-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Halle M., Hall P., Tornvall P. Cardiovascular disease associated with radiotherapy: activation of nuclear factor kappa-B. J. Intern. Med. 2011;269(5):469–477. doi: 10.1111/j.1365-2796.2011.02353.x. [DOI] [PubMed] [Google Scholar]
- 85.Madan R., Benson R., Sharma D.N., Julka P.K., Rath G.K. Radiation induced heart disease: pathogenesis, management and review literature. J. Egypt. Natl. Canc. Inst. 2015;27(4):187–193. doi: 10.1016/j.jnci.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 86.Taunk N.K., Haffty B.G., Kostis J.B., Goyal S. Radiation-induced heart disease: pathologic abnormalities and putative mechanisms. Front Oncol. 2015;5:39. doi: 10.3389/fonc.2015.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Halle M., Gabrielsen A., Paulsson-Berne G., Gahm C., Agardh H.E., Farnebo F., Tornvall P. Sustained inflammation due to nuclear factor-kappa B activation in irradiated human arteries. J. Am. Coll. Cardiol. 2010;55(12):1227–1236. doi: 10.1016/j.jacc.2009.10.047. [DOI] [PubMed] [Google Scholar]
- 88.Stoecklein V.M., Osuka A., Ishikawa S., Lederer M.R., Wanke-Jellinek L., Lederer J.A. Radiation exposure induces inflammasome pathway activation in immune cells. J. Immunol. 2015;194(3):1178–1189. doi: 10.4049/jimmunol.1303051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rana S., Espinosa-Diez C., Ruhl R., Chatterjee N., Hudson C., Fraile-Bethencourt E., Agarwal A., Khou S., Thomas C.R., Jr., Anand S. Differential regulation of microRNA-15a by radiation affects angiogenesis and tumor growth via modulation of acid sphingomyelinase. Sci. Rep. 2020;10(1):5581. doi: 10.1038/s41598-020-62621-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chatterjee S., Pietrofesa R.A., Park K., Tao J.Q., Carabe-Fernandez A., Berman A.T., Koumenis C., Sielecki T., Christofidou-Solomidou M. LGM2605 reduces Space radiation-induced NLRP3 inflammasome activation and damage in in vitro lung vascular networks. Int. J. Mol. Sci. 2019;20(1) doi: 10.3390/ijms20010176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang S., Che J., Chu Q., Zhang P. The role of NLRP3 inflammasome in radiation-induced cardiovascular injury. Front Cell Dev Biol. 2020;8:140. doi: 10.3389/fcell.2020.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wu Q., Allouch A., Martins I., Modjtahedi N., Deutsch E., Perfettini J.L. Macrophage biology plays a central role during ionizing radiation-elicited tumor response. Biomed. J. 2017;40(4):200–211. doi: 10.1016/j.bj.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Libby P., Ridker P.M., Hansson G.K. Inflammation in atherosclerosis: from pathophysiology to practice. J. Am. Coll. Cardiol. 2009;54(23):2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koelwyn G.J., Newman A.A.C., Afonso M.S., van Solingen C., Corr E.M., Brown E.J., Albers K.B., Yamaguchi N., Narke D., Schlegel M., Sharma M., Shanley L.C., Barrett T.J., Rahman K., Mezzano V., Fisher E.A., Park D.S., Newman J.D., Quail D.F., Nelson E.R., Caan B.J., Jones L.W., Moore K.J. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat. Med. 2020 doi: 10.1038/s41591-020-0964-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Milanovic M., Yu Y., Schmitt C.A. The senescence-stemness alliance - a cancer-hijacked regeneration principle. Trends Cell Biol. 2018;28(12):1049–1061. doi: 10.1016/j.tcb.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 96.Chen M., Li X., Wang S., Yu L., Tang J., Zhou S. The role of cardiac macrophage and cytokines on ventricular arrhythmias. Front. Physiol. 2020;11:1113. doi: 10.3389/fphys.2020.01113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gistera A., Hansson G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017;13(6):368–380. doi: 10.1038/nrneph.2017.51. [DOI] [PubMed] [Google Scholar]
- 98.Kinlay S., Libby P., Ganz P. Endothelial function and coronary artery disease. Curr. Opin. Lipidol. 2001;12(4):383–389. doi: 10.1097/00041433-200108000-00003. [DOI] [PubMed] [Google Scholar]
- 99.Libby P., Sukhova G., Lee R.T., Galis Z.S. Cytokines regulate vascular functions related to stability of the atherosclerotic plaque. J. Cardiovasc. Pharmacol. 1995;25(Suppl 2):S9–S12. doi: 10.1097/00005344-199500252-00003. [DOI] [PubMed] [Google Scholar]
- 100.Ko K.A., Wang Y., Kotla S., Fujii Y., Vu H.T., Venkatesulu B.P., Thomas T.N., Medina J.L., Gi Y.J., Hada M., Grande-Allen J., Patel Z.S., Milgrom S.A., Krishnan S., Fujiwara K., Abe J.I. Developing a reliable mouse model for cancer therapy-induced cardiovascular toxicity in cancer patients and survivors. Front Cardiovasc Med. 2018;5:26. doi: 10.3389/fcvm.2018.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Albadawi H., Crawford R.S., Atkins M.D., Watkins M.T. Role of poly(ADP-ribose) polymerase during vascular reconstruction. Vascular. 2006;14(6):362–365. doi: 10.2310/6670.2006.00061. [DOI] [PubMed] [Google Scholar]
- 102.Rea I.M., Gibson D.S., McGilligan V., McNerlan S.E., Alexander H.D., Ross O.A. Age and age-related diseases: role of inflammation triggers and cytokines. Front. Immunol. 2018;9:586. doi: 10.3389/fimmu.2018.00586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dominic A., Banerjee P., Hamilton D.J., Le N.T., Abe J.I. Time-dependent replicative senescence vs. disturbed flow-induced pre-mature aging in atherosclerosis. Redox Biol. 2020;37:101614. doi: 10.1016/j.redox.2020.101614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The individual data will be shared or reasonable request to the corresponding authors.