

Summary
We describe sample preparation and visualization of fluorescently tagged cellulose synthases in cellulose synthase complexes at the plasma membrane of Arabidopsis hypocotyl epidermal cells using live-cell imaging via spinning disk microscopy. We present a technique for sample mounting that may be suitable for imaging other samples. Additionally, we offer free, open-source solutions for image analysis and provide extensive troubleshooting suggestions.
For complete information on the use and execution of this protocol, please refer to McFarlane et al., 2021.
Subject areas: Cell Biology, Microscopy, Model Organisms, Plant sciences
Graphical abstract
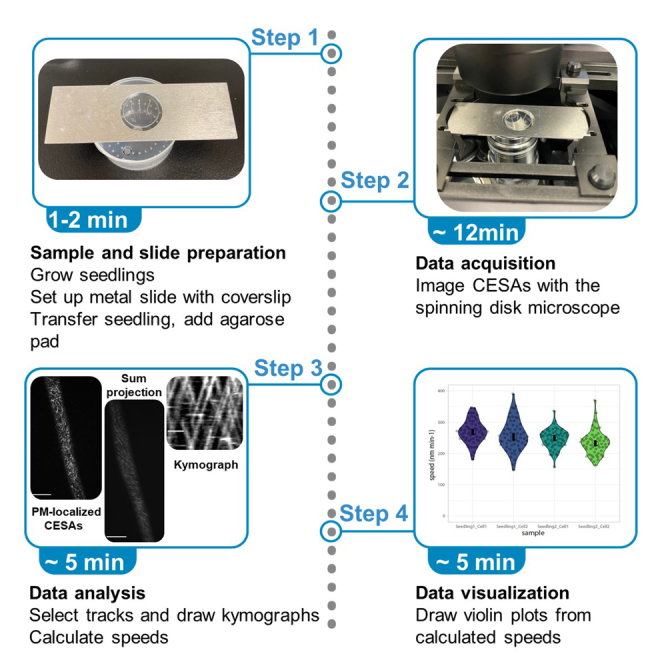
Highlights
-
•
Protocol to track cellulose synthesis in real time in living plant cells
-
•
Image analysis pipeline to quantify cellulose synthase complex speeds
-
•
Example dataset for analysis and example data display suggestions
We describe sample preparation and visualization of fluorescently tagged cellulose synthases in cellulose synthase complexes at the plasma membrane of Arabidopsis hypocotyl epidermal cells using live-cell imaging via spinning disk microscopy. We present a technique for sample mounting that may be suitable for imaging other samples. Additionally, we offer free, open-source solutions for image analysis and provide extensive troubleshooting suggestions.
Before you begin
The protocol below describes the specific steps for imaging fluorescently-tagged cellulose synthases (CESAs) in cellulose synthase complexes (CSCs) in Arabidopsis hypocotyl epidermal cells. However, we have also used a similar protocol in Arabidopsis leaf and root epidermal cells and similar steps should apply to fluorescently-tagged CESA visualization in other plant species. Additionally, this protocol for sample preparation and time-lapse imaging is also suitable for visualization of other plasma membrane-localized proteins, but data analysis might be different.
Preparation of ½ MS medium and plates
Timing: 2 h
-
1.
Dissolve 2.2 g Murashige & Skoog Medium (including vitamins) and 0.5 g MES in 800 mL ddH2O.
-
2.
Adjust pH to 5.8 with 1 M KOH.
-
3.
Add 8 g agar, fill up to 950 mL with ddH2O and autoclave for 20 min at 121°C.
-
4.
In a sterile hood, add 50 mL of sterile 20% sucrose solution (w/v in water) to a final concentration of 1% sucrose.
-
5.
Let the media cool to 50°C–60°C and pour approximately 20 square petri dishes.
-
6.
Let the plates cool completely and store them upside-down at 4°C until use, for up to 4 weeks.
-
7.
For preparation of ½ MS liquid medium omit the addition of agar and continue with steps as described. The ½ MS liquid medium can be stored at 4°C until use, for up to 4 weeks.
| Reagent | Final concentration | Amount |
|---|---|---|
| Murashige & Skoog Medium (including vitamins) | ½ concentration∗ | 2.2 g∗ |
| Sucrose | 1% | 50 mL of 20% (w/v) solution |
| MES | 2.35 mM | 0.5 g |
| Agar | 0.8% | 8 g |
| ddH2O | N/A | Fill up to 950 mL |
∗ Check manufacturer’s directions and use half as much MS media powder as directed to achieve ½ strength MS media.
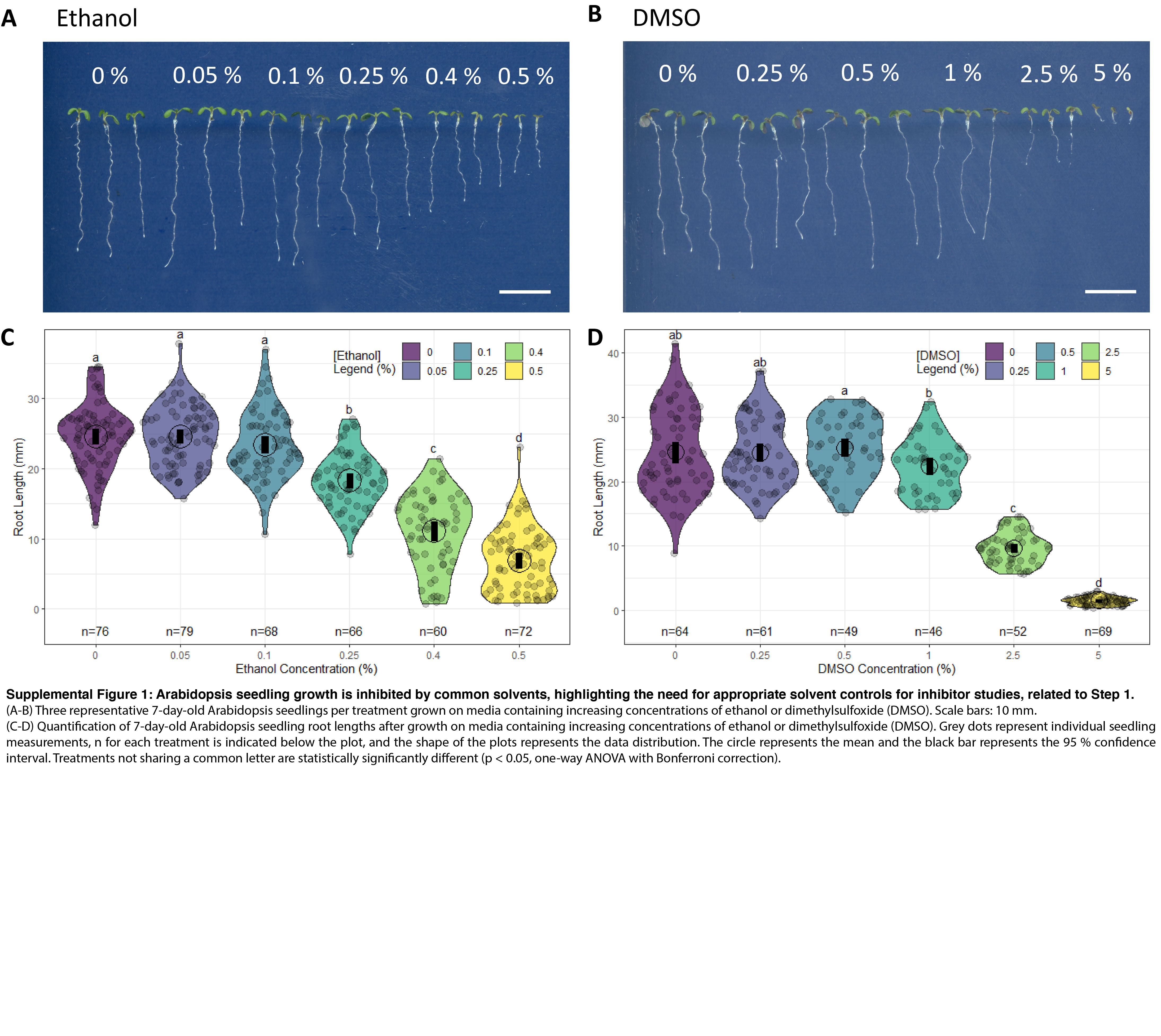
CRITICAL: If chemical inhibitors are used for long-term treatment, they should be dissolved in an appropriate solvent, filter sterilized and mixed into the cooled media after autoclaving, but before pouring into plates. Inhibitor plates should be used immediately, since some inhibitors can be degraded with time and/or light exposure. Appropriate solvent controls are essential; DMSO and ethanol can have significant effects on Arabidopsis seedling growth and subcellular dynamics (Figure S1). Therefore, dissolve the inhibitor at a concentration that minimizes the addition of the solvent in the final solution, e.g., isoxaben was prepared as a 20 μM solution in ethanol and 100 μL of the stock solution was added to a final volume of 1 L of ½ MS to have a final concentration of 2 nM isoxaben and 0.01% ethanol.
Plant growth
-
8.
In a sterile hood, sterilize seeds using a solution of 1.5% (final concentration, v/v) sodium hypochlorite and 0.1% (w/v) Triton X-100 solution for 15 min and then wash the seeds three times with sterile ddH2O.
-
9.
Using a sterile 1000 μL pipette tip, transfer seeds to ½ MS plates, leaving enough space above the seeds for hypocotyl growth and spacing seeds several mm apart (e.g., 2–3 rows of 15–20 seeds each per plate).
-
10.
Wrap the plates in aluminum foil and stratify for 3 days at 4°C.
-
11.
Expose the plates to light for 3 h (growth chamber conditions: 120 μmol m−2 s−1, 21°C), then wrap in a double layer of aluminum foil to block any light and incubate the plates vertically in the growth chamber in the dark for 3 days at 21°C.
Preparation of agarose pads
-
12.
Place several 15 mm round cover slips in a petri dish, on top of a piece of parafilm.
-
13.
Melt ½ MS with 0.8% agarose (for preparation refer to step 1) and pipet 80 μL onto each cover slip. Immediately place a second round cover slip on top to make a kind of “sandwich” and then wait for it to cool and solidify (a few minutes).
-
14.
Keep the agarose pads hydrated by placing a moistened tissue inside the petri dish.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals | ||
| Murashige & Skoog Medium (including vitamins) | Duchefa Biochemie | Cat# M0222 |
| Sucrose | Sigma-Aldrich | Cat# S9378 |
| Potassium hydroxide pellets | ChemSupply | Cat# PA161 |
| Agar | Sigma-Aldrich | Cat# A1296 |
| Agarose | Bioline | Cat# BIO-41025 |
| MES [2(n-morpholine) ethane sulphonic acid] | Sigma-Aldrich | Cat# M8250 |
| Isoxaben | Sigma-Aldrich | Cat# 36138 |
| Sodium hypochlorite solution, 12.5% | Thermo Fisher Scientific | Cat# AJA485 |
| Triton™ X-100, laboratory grade | Sigma-Aldrich | Cat# X100 |
| Silicone vacuum grease | Beckman Coulter | Cat# 335148 |
| Experimental models: Organisms/strains | ||
| Arabidopsis thaliana pCESA3:GFP-CESA3 | Desprez et al., 2007 | N/A |
| Arabidopsis thaliana pCESA6:YFP-CESA6 | Paredez et al., 2006 | N/A |
| Arabidopsis thaliana pCESA6:tdTomato-CESA6 | Sánchez-Rodríguez et al., 2012 | N/A |
| Software and algorithms | ||
| Fiji | Schindelin et al., 2012 | https://imagej.net/software/fiji/ |
| StackReg plugin | Thévenaz et al., 1998 | http://bigwww.epfl.ch/thevenaz/stackreg/ |
| PlotsOfDifference | Goedhart, 2019 | https://huygens.science.uva.nl/ PlotsOfDifferences/ |
| BoxPlotR | Spitzer et al., 2014 | http://shiny.chemgrid.org/ boxplotr/ |
| Other | ||
| Assistent® German coverslips for cell culture, round, 15 mm diameter | Neuvitro Corporation | Cat# GG-15 |
| Rectangular coverslips, #1.5 22 x 32 mm |
Thermo Scientific Gerhard Menzel |
Cat# 12322118 |
| Metal slides with round opening | Custom made (see Figure 1) | N/A |
| Kimwipes | Kimtech | Cat# WIPE4120 |
| Parafilm M (38m × 100 mm) | Bemis Company Inc | Cat#PM996 |
| Carl Zeiss™ Immersol™ Immersion Oil or other appropriate immersion media for the microscope objective | Fisher Scientific | Cat# 10539438 |
| Petri dish, square, clear, 120 x 120 x 17 mm, with vents | Greiner eHealth Technologies | Cat# 688102 |
| 6-Well plate | Greiner eHealth Technologies | Cat # 657160 |
| Growth chamber | e.g., Percival Scientific or Conviron | Model AR-36L2 or A1000 |
| Inverted microscope with a 100 x oil-immersion objective | Nikon | Eclipse Ti-E or Ti2-E with APOTIRF corrected N.A. 1.49, 100x objective |
| Spinning disk head | Yokogawa Electric Corporation | CSU-W1 or CSU-X1 |
| Focus maintenance system | Nikon | Perfect Focus System 4 |
| EM-CCD camera or sCMOS camera | Andor Technology or Photometrics | iXon Ultra 888 or Prime95B |
Figure 1.

Technical drawing of a custom-made metal microscopy slide
The drawing was created with FreeCAD (www.freecadweb.org).
Materials and equipment
Alternatives: Alternatively, Arabidopsis root, cotyledon or first true leaf epidermal cells can be imaged.
Fluorescently-tagged CESA lines are also currently available in Brachypodium distachyon (Liu et al., 2017) and Physcomitrium patens (Tran et al., 2018).
CESAs can be imaged using several varieties of spinning disk microscopes, including differences in microscope bases, objectives, spinning disk units, and cameras. Other imaging systems optimized for fast frame rate acquisition may also be suitable, but a spinning disk system is ideal for high-speed imaging.
Step-by-step method details
Sample and slide preparation
-
1.Prepare the metal slide for imaging (Figure 2A).
-
a.Attach a 22x32 mm coverslip on one side of the metal slide by adding a small amount of silicone vacuum grease on each side of the metal slide opening and pressing the coverslip firmly onto the slide without touching the center of the coverslip (Figures 2B and 2C).
-
b.Flip the metal slide over, so the coverslip is below the metal slide, and place it on a stand, such that the coverslip is not touching any surface (Figure 2D).
-
a.
-
2.Mount the etiolated seedling on the prepared metal slide and coverslip.
-
a.Add a minimal amount (∼10 μL) of sterile water or sterile liquid ½ MS medium in the middle of the coverslip without touching the glass (Figure 2D).
-
b.Carefully lift an etiolated seedling from the plate using a toothpick or fine forceps and transfer it to the coverslip. Place a total of one to three hypocotyls centered in the middle of the slide (Figure 2E).
-
c.Cover the etiolated seedling with an agarose pad.
-
i.Take the prepared round coverslips with agarose in the middle (Figure 2F) and separate the upper coverslip from the lower one; the agarose pad will remain attached to one of the round coverslips (like separating the cream filling from a popular type of chocolate sandwich cookie); discard the round coverslip without the agarose.
-
ii.Carefully place the agarose and coverslip, agarose side down, on top of the seedlings without moving or pressing the seedlings. The agarose pad will create a more stable environment for time-lapse imaging.
-
iii.Absorb the excess water or media with a tissue to reduce seedling drift or movement during imaging (Figures 2G and 2H).
-
i.
-
a.
Figure 2.

Sample preparation
(A) A metal slide with a round opening used for sample mounting.
(B) Vacuum grease is applied with a toothpick in two thin lines on each side of the round opening of the metal slide. Note that the metal slide is positioned on a stand to prevent contact with the surface.
(C) A rectangular coverslip is placed above the opening and pressed firmly to the metal slide on the sides.
(D) The metal slide is flipped over and a small amount of water or media is added to the coverslip.
(E) One to three etiolated seedlings are carefully transferred to the water/media droplets and placed in the middle of the coverslip.
(F) Preparation of an agarose pad. LEFT: Melted 0.8% agarose is added to a round coverslip. MIDDLE: A second round coverslip is carefully placed on top of the agarose. RIGHT: Prepared agarose pads are stored in a petri dish on parafilm.
(G) The upper round coverslip is removed from the agarose pad and the agarose pad with the attached round coverslip is carefully placed on top of the seedlings at an angle to avoid air bubbles. Excess water/media is absorbed by cautiously placing a corner of a tissue close to the agarose pad edge.
(H) The prepared sample is ready to be imaged.
(I) A thin layer of immersion oil is added to the 100x objective and the metal slide with the prepared sample is placed on top with the rectangular coverslip touching the oil.
CSC imaging with the spinning disk microscope
Etiolated hypocotyl epidermal cells of the seedlings are imaged using an inverted spinning disk confocal microscope (e.g., Nikon Eclipse Ti2-E with a CSU-W1 spinning disk head), a sCMOS camera (e.g., Photometrics Prime95B), and a 100 × oil-immersion objective (e.g., Nikon APOTIRF, MRD01991, NA 1.49, WD 0.12). Depending on the fluorophore attached to the CESA protein, use an excitation wavelength of 488 nm for GFP, 515 nm for YPF or 561 nm for RFP/tdTomato, and band emission filter 525/36 nm for GFP, 540/24 nm for YFP or 605/52 nm for RFP/tdTomato, or other appropriate excitation and emission parameters for the fluorophore.
-
3.
Put a drop of immersion oil on the objective and transfer the slide with the rectangular coverslip facing the microscope objective (Figure 2I).
-
4.
Using a low laser power (∼15% of 60 mW laser, power measured at fiber tip) and short exposure times (100–200 ms) to prevent photo-bleaching, find cells approximately 2 cells below the apical hook.
Note: Higher laser powers and longer exposure times increase the signal to noise ratio, but also increase photo-bleaching.
-
5.
Move to the plasma membrane focal plane, where there are active CSCs . Identify the plasma membrane focal plane by moving in towards the surface of the cell – the CSCs are small punctae, which will appear as clear and bright fluorescent signal. Troubleshooting 1 and 4. Avoid stressed or dead cells and cells with no fluorescent signal. Troubleshooting 2 and 3
-
6.
For time-lapse movies, activate the focus maintenance system (i.e., ‘Perfect Focus System’ for Nikon microscopes), then re-adjust the focus if necessary.
Note: The focus maintenance system stabilizes the selected focal plane and therefore reduces drift in the Z direction (‘Z-drift’). To minimize sample drift, water or media should be minimized when preparing the sample.
-
7.
Take a 10-min movie with 10 s time intervals with the exposure time of 300–800 ms, depending on the fluorescent marker (Methods video S1).
Note: It is very important that the imaging is performed in a temperature-stable environment as CESA movement/activity can vary in different temperatures (Fujita et al., 2011). The laser can increase sample temperature, it is therefore important to keep the imaging times to a minimum to avoid artefacts.
-
8.
Drift in the XY- and/or Z-direction is either caused by movement of the sample or extensive thermal drift. Significant drift can already be identified during live-cell imaging. In case of extensive drift, stop the acquisition and repeat the imaging with a new sample. Troubleshooting 5 and 6
Optional: Short-term inhibitor (e.g., isoxaben) treatment is possible before the sample preparation and imaging steps. For this, seedlings can be transferred to liquid ½ MS media with 1% sucrose (e.g., in a 6-well plate) with the appropriate inhibitor concentration and imaged immediately after the treatment.
TIFF stack of fluorescently-tagged CESAs in a hypocotyl cell at the plasma membrane focal plane. Scale is 9.0909 pixels = 1 μm. Time interval is 10 s per image. No data processing has been applied to these images so that readers may use this dataset for image analysis.
Image processing with Fiji
-
9.XY- and Z-drift can be easily detected by playing the movie or by generating a sum intensity projection of all images.
- a.
-
b.To generate the summed intensity image, go to ‘Image -> Stacks -> Z Project…’ and select all images and ‘Sum Slices’. Press OK. The output will be the movement of CESAs in the plasma membrane (and CESAs in Golgi stacks and other small intracellular compartments) over the time span of the imaging. Active CSCs should be observed as distinct linear tracks (Figure 3B).
-
10.If XY-drift occurred, the image stack can be processed with the StackReg plugin in Fiji (Thévenaz et al., 1998).
-
a.Go to ‘Plugins -> StackReg’ and select the ‘Rigid Body’ transformation correct for XY-drift; ‘Rigid Body’ allows for corrections of X- and Y- translations, as well as rotation, but will not resize the images (Figure 4).
-
b.Alternatively, the preinstalled Fiji plugin under ‘Plugins -> Registration -> Rigid Registration’ can be used to correct for XY-drift.
-
a.
-
11.Additional processing on the TIFF stack prior generating the image projection might be required to reduce background noise and enhance the signal intensity.
-
a.For background noise reduction select ‘Analyze -> Multi Kymograph -> Walking Average’ (3 frame average).
-
b.For the signal intensity enhancement select ‘Process -> Enhance contrast…’; the saturated pixels value should be around 0.4% and ‘Process all X slices’ should be selected to process all images in the stack.
-
c.For background subtraction, use ‘Process -> Subtract Background’ with a rolling ball radius of ∼50 px.
-
a.
Note: Selecting a bright and stable fluorophore with a high quantum yield and low photo-bleaching rate can reduce the need for image processing before the analysis.
Note: Laser power and exposure times should be optimized during live-cell imaging to yield images with high signal-to-noise ratio while reducing photo-bleaching and photo-damage.
-
12.
Adjusted stacks can be saved in the TIFF format and used for analysis.
Note: All images should be processed in an identical manner.
Note: Other analysis pipelines are also suitable, including using proprietary software such as IMARIS (Gutierrez et al., 2009; Vellosillo et al., 2015) or open-source software, such as ICY (Woodley et al., 2018).
Figure 3.

Visualization of cellulose synthase complexes (CSCs) by live-cell imaging
(A) Image of fluorescently-tagged CESAs in a hypocotyl cell at the plasma membrane focal plane. The insert is a close-up of the area inside the dashed rectangle and shows an example of an out-of-focus Golgi stack (white arrow), a SmaCC particle (yellow arrow) and several CSCs in a line (magenta arrow). Scale bar in the insert: 5 μm.
(B) A sum intensity projection of (A). CSCs can be visualized over a time span of 5–10 min as linear tracks at the plasma membrane.
Scale bars: 20 μm. See also Methods video S1.
Figure 4.

XY-drift correction of CSC time-lapse images using the StackReg plugin
(A–D) Time course at 0 (A), 1 (B), 2 (C) and 3 min (D) of imaging shows a slight XY-drift towards the upper right of the image.
(E) Sum intensity projection before applying StackReg for drift correction on a movie with XY-drift shows blurred CSC tracks.
(F–I) Time course at 0 (F), 1 (G), 2 (H) and 3 min (I) of the same movie as in (A–D) after XY-drift correction with StackReg.
(J) Sum intensity projection after running StackReg to correct for XY-drift shows successfully aligned images and clear CSC tracks.
Scale bars: 20 μm.
Expected outcomes
Fluorescently-tagged primary cell wall CESAs can be observed in bright, fast-moving Golgi stacks; in small, rapidly- and erratically-moving compartments inside the cell (i.e., below the plasma membrane focal plane), called small CESA compartments or microtubule-associated CESA compartments (SmaCCs/MASCs; Gutierrez et al., 2009; Crowell et al., 2009) and at the plasma membrane of Arabidopsis hypocotyls (Paredez et al., 2006) (Figure 3). CESA particles that are located at the plasma membrane can be observed as bright punctae that are moving in a linear manner over time and are presumed to be active CSCs, producing cellulose microfibrils (Figure 3B; Methods video S1).
Quantification and statistical analysis
CSC speeds are calculated from kymographs, which are graphical representations of individual CSCs in space over time (Figure 5). CSCs in a kymograph are observed as lines with a specific slope, from which speed can be calculated.
Figure 5.

CSC speed determination from kymographs in Fiji
(A) First image in the time-lapse movie.
(B) The sum intensity projection of all images in the time-lapse movie. Line segment in magenta is overlaying a chosen CSC track for speed analysis and was copied to the original time-lapse movie in (A).
(C) Kymograph generated from the magenta line segment in (A). The yellow line represents a single CSC trajectory chosen for speed measurement.
(D) Kymograph of a SmaCC particle, marked with white arrows. SmaCCs are usually bright and move erratically. Scale bars in A and B: 20 μ, scale bars in C and D: 2 μm. See also Supplemental Dataset 1.
Images often include fluorescently-tagged CESAs in SmaCCs/MASCs and Golgi stacks. SmaCCs are usually bright punctae with irregular movement (Figure 5D) and Golgi stacks are relatively large and bright objects with an irregular pattern of movement (Paredez et al., 2006), which can be easily observed in the kymograph and excluded.
It is also important to consider the definition of n for CSC speed calculation. The actual number of CSCs or CSC tracks analyzed is not considered as n, as many CSC tracks can be measured from a single cell (Figure 5). CSC tracks should be considered from several cells in different seedlings. Therefore, n is the number of cells in a certain number of seedlings, e.g., n=9 if CSC tracks are measured in 3 different cells of 3 different seedlings or n=6 if CSC tracks are measured in 3 different cells of 2 different seedlings. Different cells represent technical replicates and different seedlings represent biological replicates.
Kymograph analysis with Fiji
CSC speed data can be obtained with the ‘Multi Kymograph’ function in Fiji.
-
1.
Open the previously-processed TIFF stack of the time-lapse images (Figure 5A) and generate the sum intensity projection with ‘Image -> Stacks -> Z Project…’, select ‘all images’ and ‘Sum Slices’ (Figure 5B).
-
2.
In the sum intensity projection, where multiple CSC tracks should be visible, select the ‘segmented line’ tool and draw a line along one CSC track (Figures 3A and 5B).
-
3.
Go back to the previously opened TIFF stack and copy the drawn line to the stack with ‘Edit -> Selection -> Restore Selection’ to select the CSC track to follow through the whole imaging time (Figure 5A).
-
4.
To generate a kymograph, go to ‘Analyze -> Multi Kymograph -> Multi Kymograph’ and set the line width to 3 pixels (px). This kymograph is a two-dimensional representation of the time-lapse image, where the x-dimension is defined by the line drawn on the image stack, and the y-dimension is time. The kymograph usually shows the paths of several CSC tracks over time as diagonal lines (Figure 5C). Troubleshooting 7
-
5.The slope of those lines can be used to calculate CSC speeds.
-
a.Go to ‘Analyze -> Set measurements…’ and select the ‘Area’ and ‘Bounding Rectangle’ options.
-
b.With the ‘straight line’ tool, draw a line along one diagonal line on the kymograph (Figure 5C, yellow line) and press ‘M’ to measure the coordinates.
-
c.A ‘Results’ window with ‘width’ and ‘height’ values will automatically appear. Those values represent the X-distance (‘width’) and the Y-time (‘height’) in pixels by which the CSC particle moved.
-
a.
-
6.
With this data, CSC speed (i.e., the distance a particle has traveled over time) can be calculated. Each pixel in the y-dimension corresponds to one frame in the time-lapse, which is defined by the time interval between images. For the x-dimension of the kymograph, the pixel size is defined by the objective, camera, and any binning used. Scale information should be encoded in image metadata collected from a calibrated microscope but it can also be obtained using a micrometer calibration slide. In Fiji, scale information can be obtained by going to ‘Image -> Properties…’
-
7.
Example calculation: a time-series image was collected for 10 min with 10-s intervals and had a scale of 1 μm = 9.0909 px (i.e., 1 px = 110 nm). An example line drawn on the kymograph had a width of 23 px and a height of 54 px as measured by Fiji:
Convention in the field has been to present CSC speeds in nm min−1; however, nm s−1 are the most appropriate SI units; some authors call this measurement “velocity” but velocity is a vector quantity (speed plus direction), so speed is a more appropriate term.
-
8.
The data can be visualized as violin plots using statistical analysis software or tools available online, such as ‘PlotsOfDifferences - a web app for the quantitative comparison of unpaired data’ (Goedhart, 2019) (Figure 6) or ‘BoxPlotR: A web tool for generation of box plots’ (Spitzer et al., 2014).
Figure 6.

Data visualization of CSC speeds as violin plots generated with “PlotsOfDifferences”
Speed measurements from two different cells each from two different wild-type seedlings. Gray dots represent individual speed measurements (60 for each cell) and the shape of the plots represents the data distribution. The open circle represents the mean and the black bar represents the 95% confidence interval of the mean.
Limitations
The protocol requires stable fluorescently-labeled CESA lines, such as Arabidopsis GFP-CESA3, YFP-CESA6 or tdT-CESA6, transformed into wild-type or mutant backgrounds. All CESA marker lines reported to-date have the fluorescent protein fused to the N-terminus.
CESA imaging is temperature sensitive (Fujita et al., 2011). For example, Fujita et al., 2011 showed that transferring wild-type seedlings from 21°C to 29°C for 3 h increased the average CSC speed by 3.7-fold. Therefore, care must be taken to image the seedlings in a temperature-stable environment with as little temperature fluctuation as possible, e.g., performing the measurements in an air-conditioned laboratory.
Other factors can also affect CESA imaging. Exposing hypocotyls to light (during sample mounting) or to changes in gravity vector (i.e., mounting the sample on a horizontal stage) can also affect hypocotyl growth. Blue light (i.e., light used to excite GFP) can trigger microtubule reorientation in plants (Zandomeni and Schopfer, 1993), which can affect orientation of CSC trajectories in the plasma membrane (Paredez et al., 2006).
Osmotic and mechanical stress can induce CESA internalization from the plasma membrane and accumulation in SmaCCs (Gutierrez et al., 2009). Therefore, collecting images as quickly as possible and only mounting 1–3 seedlings at a time can reduce the effects of these abiotic factors on CSC speed in the plasma membrane and CESA distribution within the cell.
CESA movies with Z-drift or extensive XY-drift that cannot be corrected with the StackReg plugin in Fiji cannot be properly analyzed. We would advise against using these images for analyses for CESA speed or density. Troubleshooting 5 and 6
Troubleshooting
Problem 1
In step 4 of imaging CSCs, too few or too many plasma membrane-bound CSC can be observed due to imaging cells that are too young or too old (Figures 7A–7C).
Figure 7.

Selecting the correct cells for CSC imaging
(A) A low magnification stitched image of an etiolated seedling indicating the correct imaging region – epidermal cells in the upper hypocotyl (dashed box). The magenta arrow indicates cells of the apical hook that are too young to image and the white arrow indicates cells too far away from the apical hook that are too old for imaging. Scale bar: 200 μm.
(B) An image of a cell that is positioned too far away from the apical hook of the seedling (also indicated with a white arrow in A) and therefore has few CSCs in the plasma membrane.
(C) An image of cells that are positioned very close to the apical hook (also indicated with a magenta arrow in A) and are very young, therefore unsuitable for the analysis.
(D) An image of cells at the correct imaging region, positioned 2–3 cells below the apical hook of the etiolated seedling (also indicated with a dashed box in A).
(E) An example of stressed cells – very few plasma membrane-localized CSCs are present and CESA signal is much higher in Golgi stacks and SmaCCs.
(F) An example of a dead cell.
(G) An example of a seedling with no detected fluorescent CESA signal.
Scale bars B–G: 20 μm.
Potential solution
If there are too few or too many CSCs in the plasma membrane, the cells may be too young or too old. Cells that are too young/too close to the apical hook (Figures 7A and 7B) or too old/too far down the hypocotyl (Figures 7A and 7C) have unusually high or low CESA density in the plasma membrane, respectively and are unsuitable for imaging. Image cells that are approximately 2–3 cells away from the apical hook of the etiolated hypocotyl (Figures 7A and 7D; Bringmann et al., 2012).
Problem 2
In step 5 of imaging CSCs, few or no plasma membrane-bound CSCs can be observed due to stressed or dead cells (Figures 7E and 7F).
Potential solution
If there are many SmaCCs and few CSCs in the plasma membrane, the seedlings may be stressed due to over-handling or a long delay between mounting and imaging (Figure 7E). CESA localization and dynamics are sensitive to abiotic factors (Wang et al., 2016).Work very carefully when handing and mounting seedlings and image seedlings relatively quickly (within ∼30 min) after mounting.
Problem 3
In CSC imaging step 5, no plasma membrane-bound CSCs can be observed due to no fluorescent signal (Figure 7G).
Potential solution
If only vague outlines of the cells are visible and no fluorescently-tagged CESAs are visible in the plasma membrane or within the cell (Figure 7G), the signal may be segregating or silenced so there is little or no fluorescently-tagged CESA signal in the seedling; choose a new seedling for imaging.
Problem 4
In CSC imaging step 5, few or no plasma membrane-bound CSCs can be observed due to the wrong focal plane (Figure 8).
Figure 8.

Selecting the correct focal plane for CSC imaging
(A and B) Out of focus images taken above the plasma membrane (PM) focal plane. An image taken too close to the coverslip (A) is blurry and CSCs cannot be imaged. In the image taken just below image A (B) CSCs can be seen but are still out of focus.
(C) Image of CESAs at the correct PM focal plane. CESAs appear as bright punctae, are clear and can be easily imaged.
(D and E) Out of focus images taken below the PM focal plane. An image taken just below the PM focal plane (D) contains few CSCs, but includes many CESA-containing Golgi stacks (large bright dots). An image taken in the lower cell cortex below image D (E) contains almost exclusively CESA-containing Golgi stacks. Scale bars: 20 μm.
Potential solution
The focal plane may be incorrect and set above the cell (Figures 8A and 8B) or too deep into the cell (Figures 8D and 8E); adjust the focus so that the imaging plane is exactly at the plasma membrane (Figure 8C).
Problem 5
In CSC imaging step 8, XY-drift can occur (Figure 4).
Potential solution
Remove as much water as possible from the slide when mounting the sample by placing a corner of a tissue under the agarose pad to absorb water (Figure 2G). Wait for approximately 5 min after mounting the sample before starting image acquisition to allow the sample to settle into the agarose pad. If the XY-drift is moderate, the Fiji plugin ‘StackReg’ can be used for correction (Figures 4F–4J).
Problem 6
In CSC imaging step 8, Z-drift or XYZ-drift can occur (Figures 9A–9F).
Figure 9.

Example of an XYZ-drift and a resulting kymograph that is unsuitable for CSC speed analysis
(A) Sum projection of all images from a time-lapse image series with XYZ-drift without StackReg drift correction.
CSC tracks are blurry and not clearly distinguishable.
(B–E) Unsuccessful XYZ-drift correction with StackReg. Time course at 0 (B), 1 (C), 2 (D) and 3 min (E) after XY-drift correction with StackReg reveals substantial drift in the Z-direction. At the beginning of the movie, plasma membrane-bound CESAs are clear (B) but at the end of the movie the focal plane moves inside of the cell where large, bright CESA-containing Golgi stacks are more prevalent (E).
(F) Sum projection of all images from the StackReg-corrected time-lapse image series (B-E) with XYZ-drift. CSC tracks can be seen more clearly than before correction (A) but are still blurry and mixed between each other due to Z-drift. Scale bars A–F: 20 μm.
(G) An example of a kymograph that is unsuitable for analysis. CSC tracks are smeared and not clearly visible; therefore, a speed analysis cannot be performed. Scale bar: 2 μm.
Potential solution
Remove as much water as possible from the slide when mounting the sample, let the slide settle after mounting for ∼5 min. Use the focus maintenance system (e.g., Nikon ‘Perfect Focus System’) to reduce Z-drift (Figure 9F) and image hypocotyl cells that are closest to the cover slip. If the Z-drift is large, repeat the imaging on a different cell.
Problem 7
In data analysis step 4, linear tracks, which represent CSCs, may not be clearly visible either in the sum projection or the kymograph (Figure 9G). This makes it difficult or impossible to measure CSC speed.
Potential solution
Collect a new time-lapse image from a fresh sample, taking steps to minimize abiotic stress and XYZ-drift as described above.
Resource availability
Lead contact
The lead contact is Heather E. McFarlane (h.mcfarlane@utoronto.ca). Further information and requests for resources and reagents should be directed to and will be fulfilled by the technical contact, Jana Verbančič (jverbancic@student.unimelb.edu.au).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
Imaging was conducted using instruments supported by Canada Foundation for Innovation and the Ontario Research Fund grants (38721) to H.E.M. J.V. was supported by the University of Melbourne Research Scholarship and is affiliated with the International Max Planck Research School – Primary Metabolism and Plant Growth, and H.E.M. acknowledges funding from the Canada Research Chairs Program and a Natural Science and Engineering Council (NSERC) Discovery grant (2020-05959).
Author contributions
Conceptualization, J.V. and H.E.M.; resources, J.J.H. and H.E.M.; writing – original draft, J.V.; writing – reviewing and editing, J.V., J.J.H., and H.E.M.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xpro.2021.100863.
Contributor Information
Jana Verbančič, Email: jverbancic@student.unimelb.edu.au.
Heather E. McFarlane, Email: h.mcfarlane@utoronto.ca.
Supplemental information
(A and B) Three representative 7-day-old Arabidopsis seedlings per treatment grown on media containing increasing concentrations of ethanol or dimethylsulfoxide (DMSO). Scale bars: 10 mm.
(C and D) Quantification of 7-day-old Arabidopsis seedling root lengths after growth on media containing increasing concentrations of ethanol or dimethylsulfoxide (DMSO). Grey dots represent individual seedling measurements, n for each treatment is indicated below the plot, and the shape of the plots represents the data distribution. The circle represents the mean and the black bar represents the 95% confidence interval. Treatments not sharing a common letter are statistically significantly different (p < 0.05, one-way ANOVA with Bonferroni correction).
{kind=link}
Video of fluorescently-tagged CESAs in a hypocotyl cell at the plasma membrane focal plane. Time stamp is in minutes : seconds. Scale bar: 10 μm.
Data and code availability
This study did not generate unique datasets or code.
References
- Bringmann M., Li E., Sampathkumar A., Kocabek T., Hauser M.T., Perssona S. POM-POM2/CELLULOSE SYNTHASE INTERACTING1 is essential for the functional association of cellulose synthase and microtubules in Arabidopsis. Plant Cell. 2012;24:163–177. doi: 10.1105/tpc.111.093575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowell E.F., Bischoff V., Desprez T., Rolland A., Stierhof Y.D., Schumacher K., Gonneau M., Höfte H., Vernhettes S. Pausing of Golgi bodies on microtubules regulates secretion of cellulose synthase complexes in Arabidopsis. Plant Cell. 2009;21:1141–1154. doi: 10.1105/tpc.108.065334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desprez T., Juraniec M., Crowell E.F., Jouy H., Pochylova Z., Parcy F., Höfte H., Gonneau M., Vernhettes S. Organization of cellulose synthase complexes involved in primary cell wall synthesis in Arabidopsis thaliana. PNAS. 2007;104:15572–15577. doi: 10.1073/pnas.0706569104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita M., Himmelspach R., Hocart C.H., Williamson R.E., Mansfield S.D., Wasteneys G.O. Cortical microtubules optimize cell-wall crystallinity to drive unidirectional growth in Arabidopsis. Plant J. 2011;66:915–928. doi: 10.1111/j.1365-313X.2011.04552.x. [DOI] [PubMed] [Google Scholar]
- Goedhart J. PlotsOfDifferences – a web app for the quantitative comparison of unpaired data. bioRxiv. 2019 doi: 10.1101/578575. [DOI] [Google Scholar]
- Gutierrez R., Lindeboom J.J., Paredez A.R., Emons A.M.C., Ehrhardt D.W. Arabidopsis cortical microtubules position cellulose synthase delivery to the plasma membrane and interact with cellulose synthase trafficking compartments. Nat. Cell Biol. 2009;11:797–806. doi: 10.1038/ncb1886. [DOI] [PubMed] [Google Scholar]
- Liu D., Zehfroosh N., Hancock B.L., Hines K., Fang W., Kilfoil M., Learned-Miller E., Sanguinet K.A., Goldner L.S., Baskin T.I. Imaging cellulose synthase motility during primary cell wall synthesis in the grass Brachypodium distachyon. Sci. Rep. 2017;7:1–12. doi: 10.1038/s41598-017-14988-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarlane H.E., Mutwil-Anderwald D., Verbančič J., Picard K.L., Gookin T.E., Froehlich A., Trindade L.M., Alonso J.M., Assmann S.M., Persson S. A G protein-coupled receptor-like module regulates Cellulose Synthase secretion from the endomembrane system in Arabidopsis. Dev. Cell. 2021;56:14841497. doi: 10.1101/2020.10.20.345868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paredez A.R., Somerville C.R., Ehrhardt D.W. Visualization of cellulose synthase demonstrates functional association with microtubules. Science. 2006;312:1491–1495. doi: 10.1126/science.1126551. [DOI] [PubMed] [Google Scholar]
- Sánchez-Rodríguez C., Bauer S., Hématy K., Saxe F., Ibáñez A.B., Vodermaier V., Konlechner C., Sampathkumar A., Rüggeberg M., Aichinger E. CHITINASE-LIKE1/POM-POM1 and its homolog CTL2 are glucan interacting proteins important for cellulose biosynthesis in Arabidopsis. Plant Cell. 2012;24:589–607. doi: 10.1105/tpc.111.094672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. Fiji: an open-source platform for biological-image analysis. Nat. Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer M., Wildenhain J., Rappsilber J., Tyers M. BoxPlotR: a web tool for generation of box plots. Nat. Methods. 2014;11:121–122. doi: 10.1038/nmeth.2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thévenaz P., Ruttimann U.E., Unser M. A pyramid approach to subpixel registration based on intensity. IEEE Trans. Image Process. 1998;7:27–41. doi: 10.1109/83.650848. [DOI] [PubMed] [Google Scholar]
- Tran M.L., McCarthy T.W., Sun H., Wu S.Z., Norris J.H., Bezanilla M., Vidali L., Anderson C.T., Roberts A.W. Direct observation of the effects of cellulose synthesis inhibitors using live cell imaging of Cellulose Synthase (CESA) in Physcomitrella patens. Sci. Rep. 2018;8:1–9. doi: 10.1038/s41598-017-18994-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellosillo T., Yeats T., Sorek N. Analysis of in vivo cellulose biosynthesis in Arabidopsis cells by spinning disk confocal microscopy. Bio Protoc. 2015;5:e1617. doi: 10.21769/BioProtoc.1617. [DOI] [Google Scholar]
- Wang T., McFarlane H.E., Persson S. The impact of abiotic factors on cellulose synthesis. J. Exp. Bot. 2016;67:543–552. doi: 10.1093/jxb/erv488. [DOI] [PubMed] [Google Scholar]
- Woodley M., Mulvihill A., Fujita M., Wasteneys G.O. Exploring microtubule-dependent cellulose-synthase-complex movement with high precision particle tracking. Plants. 2018;7 doi: 10.3390/plants7030053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandomeni K., Schopfer P. Reorientation of microtubules at the outer epidermal wall of maize coleoptiles by phytochrome, blue-light photoreceptor, and auxin. Protoplasma. 1993;173:103112. doi: 10.1007/BF01378999. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TIFF stack of fluorescently-tagged CESAs in a hypocotyl cell at the plasma membrane focal plane. Scale is 9.0909 pixels = 1 μm. Time interval is 10 s per image. No data processing has been applied to these images so that readers may use this dataset for image analysis.
(A and B) Three representative 7-day-old Arabidopsis seedlings per treatment grown on media containing increasing concentrations of ethanol or dimethylsulfoxide (DMSO). Scale bars: 10 mm.
(C and D) Quantification of 7-day-old Arabidopsis seedling root lengths after growth on media containing increasing concentrations of ethanol or dimethylsulfoxide (DMSO). Grey dots represent individual seedling measurements, n for each treatment is indicated below the plot, and the shape of the plots represents the data distribution. The circle represents the mean and the black bar represents the 95% confidence interval. Treatments not sharing a common letter are statistically significantly different (p < 0.05, one-way ANOVA with Bonferroni correction).
Video of fluorescently-tagged CESAs in a hypocotyl cell at the plasma membrane focal plane. Time stamp is in minutes : seconds. Scale bar: 10 μm.
Data Availability Statement
This study did not generate unique datasets or code.