

SUMMARY
To maintain secretory pathway fidelity, misfolded proteins are commonly retained in the endoplasmic reticulum (ER) and selected for ER-associated degradation (ERAD). Soluble misfolded proteins use ER chaperones for retention, but the machinery that restricts aberrant membrane proteins to the ER is unclear. In fact, some misfolded membrane proteins escape the ER and traffic to the lysosome/vacuole. To this end, we describe a model substrate, SZ*, that contains an ER export signal but is also targeted for ERAD. We observe decreased ER retention when chaperone-dependent SZ* ubiquitination is compromised. In addition, appending a linear tetra-ubiquitin motif onto SZ* overrides ER export. By screening known ubiquitin-binding proteins, we then positively correlate SZ* retention with Ubx2 binding. Deletion of Ubx2 also inhibits the retention of another misfolded membrane protein. Our results indicate that polyubiquitination is sufficient to retain misfolded membrane proteins in the ER prior to ERAD.
Graphical Abstract
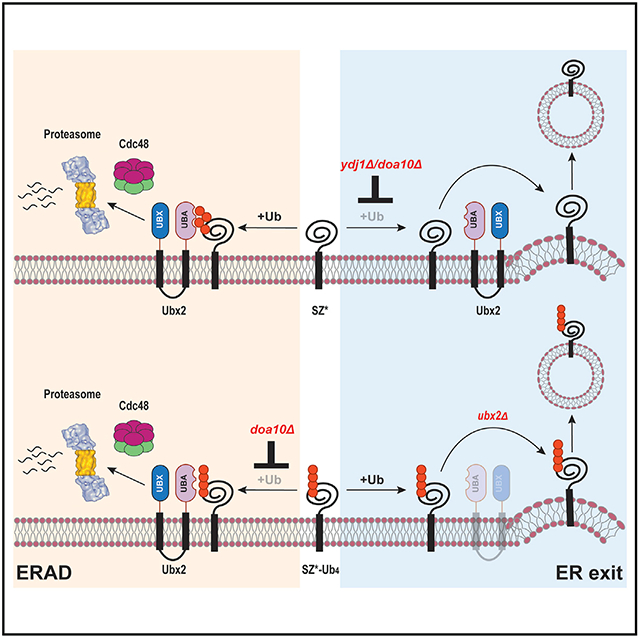
In brief
Sun et al. characterize how misfolded membrane proteins are delivered for either ERAD or post-ER degradation in the secretory pathway. By using a model substrate that can access both pathways, they show that substrate retention requires chaperone-dependent substrate ubiquitination and interaction with a conserved ER membrane protein, Ubx2.
INTRODUCTION
In eukaryotes, approximately one-third of the proteome enters the secretory pathway at the endoplasmic reticulum (ER). After proteins are synthesized, they are translocated into the ER, fold under the guidance of resident chaperones, are post-translationally modified, and may assemble into multi-subunit complexes (Balchin et al., 2016; Braakman and Hebert, 2013; Xu and Ng, 2015). During these events, ER quality-control systems monitor folding efficacy and usually ensure that only folded and assembled proteins leave the ER in coat protein complex II (CO-PII) vesicles for delivery to the Golgi (Barlowe and Helenius, 2016; Gomez-Navarro and Miller, 2016). Despite the investment of cellular resources dedicated to ER protein quality control, misfolding can occur quite frequently due to genetic mutations, translational errors, and environmental stresses, of which each can lead to myriad human diseases (Guerriero and Brodsky, 2012).
Proteins terminally misfolded or assembled inefficiently in the ER are degraded by ER-associated degradation (ERAD) (McCracken and Brodsky, 1996). ERAD substrates are first recognized by ER lumenal (Bole et al., 1986; Gardner et al., 1993; Knittler et al., 1995; Nishikawa et al., 2001; Plemper et al., 1997) and cytosolic (Han et al., 2007; Nakatsukasa et al., 2008) chaperones as well as chaperone-like lectins (Bhamidipati et al., 2005; Hosokawa et al., 2006; Kim et al., 2005; Szathmary et al., 2005). Depending on the site of a folding lesion, ERAD substrates in yeast are ubiquitinated by distinct E3 ubiquitin ligases by one of three branches of the ERAD pathway, as follows: ERAD-C, ERAD-M, and ERAD-L (Carvalho et al., 2006; Denic et al., 2006; Vashist and Ng, 2004). Concomitant with ubiquitination, misfolded substrates are retrotranslocated from the ER to the cytoplasm in an ATP-dependent manner by the Cdc48/p97 complex (Jarosch et al., 2002; Rabinovich et al., 2002; Ye et al., 2001). Finally, substrates are delivered to the 26S proteasome, often with the assistance of ubiquitin-binding protein shuttles (Medicherla et al., 2004).
To protect against the transport of misfolded proteins to later compartments in the secretory pathway, non-native proteins are largely excluded from COPII vesicles and are instead retained in the ER for ERAD. Several models have been proposed to explain how misfolded proteins are retained. Chaperone-mediated retention was first shown for misfolded soluble proteins in the ER, such as thyroglobulin, a vesicular stomatitis virus G protein mutant, and C-terminally truncated fragments of influenza hemagglutinin (de Silva et al., 1990; Ellgaard and Helenius, 2003; Muresan and Arvan, 1998; Zhang et al., 1997). In contrast, other ERAD-L substrates enter COPII vesicles and are liberated from the ER when a strong export motif is appended, suggesting passive ER retention or, less likely, that ER chaperones are limiting (Kincaid and Cooper, 2007). In this scenario, misfolded proteins fail to exit the ER because export signals are absent or are unable to present a signal to COPII adapters (Barlowe and Miller, 2013). Alternatively, some misfolded proteins may be degraded so rapidly that there is no opportunity for COPII engagement. In this case, inhibiting ERAD may spare substrates and favor export, particularly when combined with small molecule folding “correctors” (Grove et al., 2009; Mu et al., 2008; Wang et al., 2011). However, these varied ER retention mechanisms are not mutually exclusive. For example, structural changes may recruit chaperones for active retention while simultaneously obscuring COPII recognition. Nevertheless, misfolded proteins with folding lesions located in distinct domains from ER export signals may still be recruited to COPII vesicles. How the ER retains these misfolded proteins–especially misfolded membrane proteins–with remote export signals and excludes them from Golgi-targeted COPII vesicles is unclear.
We now report on the competition between ERAD and COPII-mediated export of a model substrate, SZ*, a single-spanning membrane protein that contains both an ERAD-C degron and a functional ER export signal (Sun and Brodsky, 2018). We show that inhibition of post-ubiquitination steps in the ERAD pathway, e.g., proteasome-dependent degradation, fails to augment SZ* export from the ER. Instead, compromised SZ* ubiquitination by inhibiting chaperone-dependent selection or E3-dependent ubiquitination enhances ER export both in vivo and in vitro. To test if substrate ubiquitination actively mediates ER retention, we appended a linear ubiquitin moiety to SZ* and observed enhanced retention. After screening ER-associated ubiquitin receptors, Ubx2 was found to facilitate retention through its ubiquitin-binding domain (UBD). A negative correlation was then uncovered between ER export and Ubx2 binding for both SZ* as well as another substrate. Thus, in contrast to the quality control of soluble secreted proteins, these data indicate that potentially toxic membrane proteins are unable to - access later compartments in the secretory pathway because–once ubiquitinated–they are actively retained in the ER with the assistance of a ubiquitin-binding protein.
RESULTS
An Hsp40 selects a misfolded membrane protein for ERAD
To investigate how the decision between retaining or secreting a misfolded membrane protein is made, we considered several previously characterized substrates (Figure S1; Kincaid and Cooper, 2007; Reggiori and Pelham, 2002; Sun and Brodsky, 2018; Wang and Ng, 2010). The substrates fall into five classes, with all except one targeted for either post-ER degradation or ERAD. The exception is SZ*, which is exported inefficiently from the ER because it also contains an ERAD-C degron (Sun and Brodsky, 2018). In addition, SZ* lacks an ER lumenal domain, so ERAD-L–and thus retention by lumenal chaper-ones–is minimized. We also previously showed that SZ* that escapes ERAD in COPII vesicles is targeted for ESCRT and vacuole-dependent degradation. Based on the ability of SZ* to toggle between the ERAD and post-ER degradation pathways, we explored how the fate of SZ* is regulated.
Consistent with the presence of a misfolded domain facing the cytosol, SZ* degradation requires the Hsp70 Ssa1 (Sun and Brodsky, 2018). The cytosolic, ER-tethered Hsp40 cochaperone Ydj1 activates the ATPase activity of Ssa1 (Caplan et al., 1992) and facilitates the degradation of other ERAD-C substrates (Han et al., 2007; Nakatsukasa et al., 2008; Zhang et al., 2001). Therefore, we asked if Ydj1 is also required for SZ* degradation. As predicted, SZ* turnover was slowed in yeast lacking Ydj1 (Figure 1A). Next, SZ* was immunoprecipitated from cells maintained at 26°C or shifted to 37°C, a temperature at which ERAD (versus SZ* trafficking) is enhanced (Sun and Brodsky, 2018). A ~2-fold increase in the SZ*-Ydj1 interaction at 37°C was measured (Figures 1B and C). In contrast, Ydj1 was dispensable for the degradation of Wsc1*, which is degraded in the vacuole (Wang and Ng, 2010; Figure S2A). We then asked if another ER-associated Hsp40 homolog, Hlj1, facilitates SZ* degradation, but turnover was identical between wild-type and hlj1Δ cells (Figure S2B). These data suggest that Ydj1 recognizes SZ* and targets the substrate for ERAD. Interestingly, this role of Ydj1 contrasts with the action of an ER-resident Hsp40, ERdj3, during the selection of a secreted protein for ERAD. In this case, ERdj3 escorts misfolded/aggregation-prone proteins to the extracellular space, thus preventing ERAD (Genereux et al., 2015).
Figure 1. The cytosolic Hsp40 Ydj1 is required for the ERAD of SZ*.

(A) The stability of SZ* from wild-type and ydj1Δ yeast expressing SZ* and the indicated vector in a CEN plasmid was determined in a cycloheximide chase. The QAA mutant lacks the Hsp70-interacting HPD motif. G6PD in this and other figures controls for equal loading. Data are means ± SE of 3–6 independent experiments; black asterisks apply to the +vector (ydj1Δ) versus +Ydj1 (ydj1Δ) comparison, and red asterisks apply to the +QAA mutant (ydj1Δ) versus +Ydj1 (ydj1Δ) comparison. **p < 0.005, ***p < 0.0005.
(B) HA-tagged SZ* was immunoprecipitated from wild-type yeast containing a vector control or ydj1Δ yeast expressing SZ* and Ydj1 or the QAA mutant. Cultures were shifted to 37°C or remained at 26°C for 1 h prior to harvest. A total of 1% of the input was analyzed to show equal loading. –LYS, indicates a control experiment performed in the absence of cell lysate. The asterisk denotes a contaminating species.
(C) This graph depicts relative Ydj1 binding to SZ* in yeast incubated at 26°C or 37°C, as quantified from the data in part (B). Data are the means ± SE of 3 independent experiments; *p < 0.05; NS, not significant.
We next measured the degree of SZ* ERAD as Ydj1 levels increase. Consistent with a receptor-mediated phenomenon, proteasome-dependent degradation rose with elevated Ydj1 levels (Figure 2A). Moreover, a negative correlation between Ydj1 levels and ER exit was observed. This was determined by assessing the accumulation of vacuole-generated GFP liberated from an SZ*-GFP fusion protein (Figures 2B and 2C). In the vacuole, GFP is clipped from fusion proteins but is resistant to vacuolar proteases (Li et al., 1999). As expected, neither SZ*-GFP stability nor GFP cleavage was altered in a strain deleted for the gene encoding Hlj1 (Figure S2C, left). Similarly, the results of a pulse-chase assay indicated that GFP cleavage from newly synthesized SZ*-GFP was accelerated when Ydj1 was absent (Figure 2D). These data show that Ydj1 limits SZ* vacuolar sorting by targeting it for ERAD.
Figure 2. Ydj1 facilitates the ER retention of SZ*.

(A) Proteasome-dependent degradation of SZ* was measured in pdr5Δ yeast expressing both SZ* and Ydj1 versus a vector control in the presence or absence of 100 μM MG132. Data are the means ± SE of 6–9 independent experiments; black asterisks apply to the MG132 (Ydj1 OE) versus DMSO (Ydj1 OE) comparison, and red asterisks apply to the MG132 (Ydj1 OE) versus MG132 comparison. **p < 0.005, ***p < 0.0005.
(B) A schematic of SZ*-GFP sorting into the vacuole by the MVB pathway. N and C indicate SZ*-GFP topology with the N terminus facing the ER lumen (not shown), but the protein is then delivered to the vacuole in MVBs.
(C) GFP cleavage in yeast expressing variable amounts of Ydj1 was determined by immunoblotting. –, denotes ydj1Δ; +, denotes YDJ1 (wild-type); ++, denotes wild-type yeast overexpressing Ydj1. Numbers indicate average values of normalized GFP cleavage levels from 3 independent experiments.
(D) GFP cleavage was measured by pulse-chase in wild-type and ydj1Δ yeast. Cleavage was determined by normalizing free GFP to full-length SZ*-GFP at 0 min. Data are the means ± SE of 3 independent experiments; *p < 0.05.
(E) A schematic of the in vitro budding assay to measure ER exit by COPII vesicles.
(F) In vitro COPII budding efficiency of SZ*-GFP in the presence or absence of Ydj1, Doa10, or Ubx2, respectively, was measured. V, 50% of total budded vesicles; M, 2.5% unbudded microsomes used in the reaction; +COPII/NTP and −COPII/NTP, indicate experiments performed in the presence or absence of the purified COPII proteins and energy, respectively.
(G) A graph depicting in vitro budding efficiency of SZ*-GFP and the positive control Erv46, as quantified from part (F). Data are the means ± SE of 4 independent experiments; **p < 0.005; NS, not significant.
To more directly assess how Ydj1 regulates ER exit, we used an in vitro assay that recapitulates COPII- and energy-dependent vesicle budding from ER-enriched membranes (Figure 2E). In brief, microsomes containing SZ* were isolated from wild-type and ydj1Δ cells, which was followed by incubation in the presence or absence of highly enriched COPII components (i.e., Sar1, Sec23/Sec24, and Sec13/Sec31) and ATP/GTP. In the presence of these reagents and an energy-regenerating system, freed COPII vesicles are resolved from membranes by differential centrifugation (Barlowe et al., 1994). By using this confirmatory assay, we first noted that COPII budding of SZ* from wild-type microsomes was inefficient (Figure 2F and G), even though a low but measurable amount of SZ* is targeted for vacuolar degradation in yeast (Sun and Brodsky, 2018). This phenomenon likely reflects the absence of accessory factors that support the recognition and budding of specific cargo proteins in vivo (Gimeno et al., 1995; Kodera et al., 2011; Supek et al., 2002). Yet, the absence of Ydj1, which is normally ER associated, significantly increased SZ* incorporation into COPII vesicles, consistent with the enhanced GFP cleavage observed in the ydj1Δ strain (Figure 2C). Unlike SZ*, the amount of incorporated Erv46 in this experiment and others (see below) was unchanged when budding from wild-type and ydj1Δ microsomes was compared (Figures 2F and 2G).
Substrate ubiquitination facilitates ER retention
Because cytosolic molecular chaperones are involved in substrate recognition as well as the recruitment of ubiquitin ligases (McClellan et al., 2005; Nakatsukasa et al., 2008), we next investigated if Ydj1 binding is required for SZ* ubiquitination. SZ* was expressed in wild-type and ydj1Δ yeast along with myc-tagged ubiquitin, and after substrate capture, SZ* ubiquitination was measured. As expected, heterogeneous high-molecular-weight polyubiquitinated SZ* was apparent in wild-type yeast, which was enhanced when cells were shifted to 37°C (Figure S2D). This result is consistent with previous data demonstrating increased ERAD targeting after heat shock (Sun and Brodsky, 2018). Notably, SZ* polyubiquitination was negligible when Ydj1 was absent, irrespective of the temperature. In turn, the presence of Ydj1 had little if any effect on Wsc1* ubiquitination (Figure S2E).
The dual functions of Ydj1, i.e., substrate binding (Figure 1B) and facilitating substrate ubiquitination (see Figure S2D, above), presented an obstacle to understand how each phenomenon contributes to retention, so we sought to uncouple these events. Notably, Ydj1 stimulates the ATPase activity of Ssa1 through the HPD motif in the J domain (Tsai and Douglas, 1996). Given the role of Ssa1 in recruiting ubiquitin ligases for ERAD substrate ubiquitination (Nakatsukasa et al., 2008), we hypothesize that the HPD motif would also be required for SZ* ubiquitination. Indeed, a negligible amount of polyubiquitinated SZ* was seen in ydj1Δ yeast expressing an HPD mutant (QAA) (Figure S2F). In line with defective substrate ubiquitination, SZ* degradation was slowed in cells expressing the mutant (Figure 1A). In contrast, the QAA mutation had no effect on Ydj1-substrate interaction (Figures 1B and 1C). Therefore, the ability of the QAA mutant to compromise substrate ubiquitination while remaining substrate binding proficient made it possible to distinguish possible dual effects of Ydj1 during ER retention.
To test if ubiquitination regulates ER retention, we examined the steady-state GFP cleavage of SZ* in ydj1Δ yeast expressing wild-type Ydj1 or the QAA Ydj1 mutant. Higher levels of GFP cleavage were observed in ydj1Δ yeast expressing the QAA mutant (or containing a vector control) than ydj1Δ yeast expressing wild-type Ydj1 (Figure S2C, right), suggesting that ubiquitination plays a critical role in retention. Of note, the QAA mutant also exhibited a somewhat decreased ability to maintain substrate solubility, which could also negatively affect ER exit (Figure S2G). Consequently, the amount of SZ* exiting the ER in yeast expressing the QAA mutant probably underrepresents the effect of substrate ubiquitination on ER retention.
To confirm that ubiquitination underlies retention, we instead examined SZ* biogenesis in yeast deleted for the Doa10 ubiquitin ligase, which is also required for the ERAD of this substrate (Sun and Brodsky, 2018). The absence of Doa10, as anticipated, led to decreased SZ* ubiquitination (Figure 3A), but in contrast to the absence of Ydj1, the loss of Doa10 had no effect on SZ* solubility, most likely because the cytosolic chaperones remained unaffected in doa10Δ yeast (Figure 3B). To more definitively test if ubiquitination regulates the retention of SZ* in the ER, we next asked if deleting Doa10 augmented ER exit. Therefore, we measured the level of cleaved GFP from SZ*-GFP at steady-state along with the amount of protein that built up in the vacuole. Western blot analysis detected both a greater level of accumulated GFP, which was cleaved from SZ*-GFP (Figure 3C), and enhanced levels of vacuole-resident GFP in doa10Δ and ydj1Δ yeast (Figures 3D and 3E). Newly synthesized SZ* was also more rapidly targeted to the vacuole in the absence of Doa10, as measured by the generation of cleaved GFP in a pulse-chase assay (Figures 3F and 3G). In accordance with these data, a budding assay showed substantially more SZ* incorporation into COPII vesicles when Doa10 was absent (see data corresponding to “doa10Δ” in Figures 2F and 2G). Together, these data link the acquisition of a ubiquitin chain to the retention of a misfolded membrane protein in the ER.
Figure 3. Disrupting SZ* ubiquitination facilitates ER exit.

(A) SZ* ubiquitination in wild-type and doa10Δ yeast was examined by immunoprecipitation of SZ* under denaturing conditions. The arrowhead to the left is the location at which SZ* migrates, and a molecular mass ladder (×103 Dalton) is shown to the right. Total SZ* (HA) is also depicted. Data are the means ± SE of 3 independent experiments; ***p < 0.0005.
(B) Microsomes from wild-type or doa10Δ yeast expressing SZ* were treated with 1% DDM. Protein in the supernatant (S) and pellet (P) fractions was analyzed after centrifugation and immunoblotting. Sec61 was blotted as a control. Data are the means ± SE of 3 independent experiments; NS, non-significant (p > 0.05).
(C) GFP cleavage was determined by immunoblotting in the presence or absence of Doa10. Numbers indicate average values of normalized free GFP from 5 independent experiments.
(D) Live-cell fluorescence and differential interference contrast (DIC) imaging of SZ*-GFP in wild-type, ydj1Δ, and doa10Δ yeast. CMAC marks the yeast vacuole.
(E) Quantification of the GFP signal in the vacuole from (D). Greater than 100 cells in each strain were counted; **p < 0.005, ***p < 0.0005; a.u., arbitrary units.
(F) GFP cleavage from SZ*-GFP was measured by pulse-chase in wild-type and doa10Δ yeast. Cleavage was determined as in Figure 2D.
(G) A graph depicting GFP cleavage rate of SZ*-GFP as quantified from (F). Data are the means ± SE of 3–4 independent experiments; *p < 0.05. Please note that DOA10 (wild-type) in this figure (F) is identical to YDJ1 (wild-type) in Figure 2D.
An appended ubiquitin moiety overrides ER export
We next asked if ubiquitin was sufficient to retain SZ* in the ER. Therefore, a linear tetra-ubiquitin moiety was appended onto the C terminus of SZ* (SZ*-GFP-Ub4; Figure 4A). Although the K48 linkage commonly signals proteasome degradation (Ravid and Hochstrasser, 2008; Varshavsky, 2017), linear tetra-ubiquitin also supported the degradation of a cytosolic protein in a Cdc48- and proteasome-dependent manner (Zhao and Ulrich, 2010). Next, to avoid further modification on available Lys side chains in the linear tetra-ubiquitin moiety–which might then mask an ER exit signal–three Lys residues (K29, K48, and K63) on each ubiquitin were mutated to Arg. As expected, polyubiquitinated SZ*-GFP-Ub4 was barely detected (Figures S3A-S3C) compared to the signal when SZ*-GFP was examined (Figures S3D and S3E). We next assessed the level of cleaved GFP in the vacuole at steady state in yeast expressing SZ*-GFP-Ub4 and found that the tetra-ubiquitin appendage reduced the levels of vacuole-resident GFP (Figure 4B and C).
Figure 4. Tetra-ubiquitin attenuates ER exit.

(A) Predicted topologies of SZ*-GFP and SZ*-Ub4. N and C indicate the topology with respect to the ER.
(B) Live-cell fluorescence and DIC imaging of SZ*-Ub4 in wild-type and doa10Δ yeast and SZ*-GFP in wild-type yeast. CMAC marks the yeast vacuole.
(C) Quantification of GFP intensity in the vacuole from (B). Greater than 100 cells in each strain were counted; **p < 0.005, ***p < 0.0005.
(D) Live-cell fluorescence imaging of wild-type yeast expressing the integrated fluorescent marker mCherry-Scs2-tm (ER-cherry), Sec13-RFP, or Sec24-mCherry, and containing the SZ*-Ub4 expression plasmid.
(E) Live-cell fluorescence imaging of wild-type or sec12-4 yeast expressing SZ*-Ub4. Cultures were shifted to 37°C for 30 min prior to imaging.
(F) GFP cleavage was measured by pulse-chase in wild-type and doa10Δ yeast. Cleavage was determined by normalizing free GFP to full-length SZ*-Ub4 at 0 min. Data are the means ± SE of 3 independent experiments; *p < 0.05. Quantification of SZ*-GFP in wild-type and doa10Δ yeast is also shown.
(G) In vitro COPII budding efficiency of SZ*-Ub4 in the presence or absence of Doa10 or Ubx2, respectively, was measured. V, 50% of total budded vesicles; M, 2.5% unbudded microsomes used in the reaction; +COPII/NTP and −COPII/NTP, indicate experiments performed in the presence or absence of the purified COPII proteins and energy, respectively.
(H) Graph depicts in vitro budding efficiency of SZ*-GFP as quantified from (G). Data are the means ± SE of 4 independent experiments; ***p < 0.0005.
Interestingly, SZ*-GFP-Ub4 appeared to reside in puncta (Figure 4B). To characterize the nature of these puncta, we first confirmed that SZ*-GFP-Ub4 was retained in the ER. As shown in Figure 4D, the SZ*-GFP-Ub4 puncta co-localized with an ER marker, mCherry-Scs2-tm. The puncta were reminiscent of ER-associated compartments (ERACs), which form when the cystic fibrosis transmembrane conductance regulator (CFTR) or select Ste6 mutants are expressed in yeast (Fu and Sztul, 2003; Huyer et al., 2004; Kakoi et al., 2013). Although ERACs are characterized by their co-localization with COPII components, SZ*-GFP-Ub4 failed to co-localize with Sec13 or Sec24 (Figure 4D). In addition, because CFTR-generated ERACs require COPII function (Fu and Sztul, 2003), SZ*-GFP-Ub4 puncta formation was next examined when COPII transport was compromised in a sec12-4 strain (Nakano et al., 1988). When Sec12 was inactivated upon temperature shift, SZ*-GFP-Ub4 was still found in puncta (Figure 4E). Importantly, even though there was minimal vacuolar targeting of SZ*-GFP-Ub4, residual vacuole transport/GFP cleavage still required both vacuolar protease activity (pep4Δ) and ESCRT (vps36Δ) (Figure S3F). Combined with the data presented above, our results suggest the existence of an ER retention mechanism that acts upstream of COPII capture (also see below). Moreover, retained substrates appear to be sequestered away from ER exit sites (ERESs).
At most, ~10% of SZ*-GFP is polyubiquitinated (Figure S3D; by comparing a polyubiquitin signal in the hemagglutinin (HA) blot, not shown, to the HA signal, shown below). Instead, the C-terminal non-cleavable tetra-ubiquitin motif represents most of the overall ubiquitination signal. Not surprisingly, then, the loss of Doa10 has little if any effect on SZ*-GFP-Ub4 ubiquitination (Figures S3B and S3C). Therefore, in contrast to the impact of deleting Doa10 on SZ*-GFP exit (see above and Figure 3), we reasoned that SZ*-GFP-Ub4 would still be retained in a doa10Δ strain. As hypothesized, there was no affect on vacuole targeting of either steady-state (Figures 4B and 4C) or newly synthesized (Figure 4F) SZ*-GFP-Ub4 in doa10Δ yeast. Moreover, the loss of Doa10 increased the incorporation of SZ* (Figure 2F) but not SZ*-GFP-Ub4 (Figures 4G and 4H) into COPII vesicles. Collectively, these results indicate that membrane protein ubiquitination can override an ER exit signal for a substrate subject to ER and post-ER quality-control pathways.
Given the effect of appending a linear tetra-ubiquitin motif on misfolded membrane protein retention, we next examined if appending tetra-ubiquitin also retained a native protein in the ER. To address this question, we fused tetra-ubiquitin to the C terminus of a GFP-tagged yeast ABC transporter, Yor1 (Yor1-GFP) (Figure S4A), which confers oligomycin resistance. In contrast to the ER-localized puncta formed by SZ*-GFP-Ub4 (Figure 4D), the resulting Yor1-GFP-Ub4 fusion protein was routed to the vacuole in wild-type and doa10Δ yeast (Figure S4B). Next, we asked if ubiquitination was sufficient for retention if a degron was also present. Therefore, we added a cytosolic degron, Pca1(1-392) (Adle et al., 2009), to the N terminus of Yor1-GFP-Ub4, thus creating a Pca1(1-392)-Yor1-GFP-Ub4 construct (Figure S4A). In wild-type yeast, we found that Pca1(1-392)-Yor1-GFP and Pca1(1-392)-Yor1-GFP-Ub4 were rapidly degraded based on fluorescence imaging and immunoblotting (Figures S4B and S4E). However, the loss of Doa10 rescued the ER export of Pca1(1-392)-Yor1-GFP and led to plasma membrane localization and oligomycin resistance (Figures S4B, S4C, S4D, and S4E; Adle et al., 2009). In contrast, attaching the tetra-ubiquitin moiety led to the formation of ER puncta (see Pca1(1-392)-Yor1-GFP-Ub4 in Figures S4B and S4C), similar to what we observed when SZ*-GFP-Ub4 was examined (Figure 4D). Although the loss of Doa10 rescued the growth of Pca1(1-392)-Yor1-GFP-expressing yeast on oligomycin, doa10Δ yeast expressing Pca1(1-392)-Yor1-GFP-Ub4 remained oligomycin sensitive (Figure S4D). These data indicate that a linear tetra-ubiquitin appendage can retain a second membrane protein, but only one with folding lesions, in the ER.
Ubx2 facilitates ER retention
UBD-containing proteins regulate a variety of cellular processes (Husnjak and Dikic, 2012). To test which protein might orchestrate SZ* retention, we selected yeast lacking one of seven ER-associated proteins that contain UBA, CUE, or NZF domains. We then examined GFP cleavage from SZ*-GFP under steady-state conditions. Even though most strains exhibited similar or lower cleavage levels, the exception was the ubx2Δ strain, in which cleavage was ~50% higher (Figure 5A).
Figure 5. The loss of Ubx2 enhances SZ* ER exit.

(A) GFP cleavage in wild-type and the indicated UBD-containing mutant strains was determined by immunoblotting. Data are the means ± SE of 3 independent experiments; *p < 0.05.
(B) Live-cell fluorescence imaging of SZ*-GFP in wild-type and ubx2Δ yeast. CMAC marks the yeast vacuole.
(C) Quantification of GFP intensity in the vacuole of SZ*-GFP in (B). More than 100 cells in each strain were counted; ***p < 0.0005.
(D) GFP cleavage was measured by pulse-chase in wild-type and ubx2Δ yeast. Cleavage was determined as in Figure 2D. Data are the means ± SE of 3–9 independent experiments; *p < 0.05; **p < 0.005. UBX2 (wild-type) in this figure is identical to YDJ1 (wild-type) in Figure 2D.
To test if Ubx2 retains a ubiquitinated, misfolded membrane protein in the ER, we next measured GFP liberated from SZ*-GFP under steady-state (Figures 5B and 5C) and pulse-chase (Figure 5D) conditions. In both analyses, the loss of Ubx2 enhanced the vacuolar targeting of SZ*. Consistent with this result, SZ* was incorporated more efficiently into COPII vesicles when microsomes lacked this ER-integrated membrane protein (see above and Figures 2F and 2G). Furthermore, in contrast to the rescue of ER export when Ydj1 or Doa10 were absent–which blocked substrate ubiquitination (Figure S2D; Figure 3A)–deleting Ubx2 enhanced SZ* ubiquitination (Figures S5A and S5B). Magnified ubiquitination and a build-up of SZ* in the ER may arise due to the role of Ubx2 as a Cdc48 receptor (Neuber et al., 2005; Schuberth and Buchberger, 2005), which facilitates ERAD, as well as due to its putative role as an ER retention factor for SZ*.
We next examined the effect of mutating Ubx2 on SZ*-GFP-Ub4 transport. Consistent with its function as a ubiquitin receptor, SZ*-GFP-Ub4 exited the ER more efficiently when Ubx2 was absent (Figures S6A-S6D). Consistent with these in vivo data, the export of SZ*-GFP-Ub4 from the ER increased when COPII budding was measured using microsomes prepared from ubx2Δ yeast (Figures 4G and 4H; Figures S6E and S6F). To provide further support for the role of Ubx2 as an ER gate-keeper, we asked if ER retention requires an interaction between Ubx2 and SZ* or between Ubx2 and ubiquitin. SZ* was immunoprecipitated from wild-type and doa10Δ yeast, and as shown above (Figure 3A), SZ* ubiquitination was reduced in a doa10Δ strain. More pertinent, the interaction between SZ* and Ubx2 was reduced in this strain (Figures 6A and 6B). There was also a dramatic increase (~10-fold) in Ubx2-SZ*-GFP-Ub4 binding relative to the Ubx2-SZ*-GFP complex (Figures 6A and 6B). As expected, the interaction between SZ*-GFP-Ub4 and Ubx2 was Doa10 independent. In addition, we observed a negative correlation between Ubx2- SZ* binding and ER export efficiency (Figure 6C), consistent with Ubx2 acting in a receptor-mediated pathway.
Figure 6. The interaction between SZ* and Ubx2 is enhanced by an appended tetra-ubiquitin moiety.

(A) HA-tagged SZ*-GFP and SZ*-Ub4 were immunoprecipitated from both wild-type and doa10Δ yeast expressing the indicated substrates or containing a vector control. Ubx2 contains a triple FLAG tag. A total of 1% of the input was analyzed to show equal loading.
(B) A bar graph depicts relative Ubx2 binding by SZ*-GFP and SZ*-Ub4 in the indicated yeast strains quantified from (A). Data are the means ± SE of 4 independent experiments; *p < 0.05 and **p < 0.005.
(C) Relative Ubx2 binding was determined by native immunoprecipitation from (A), and percent cleaved GFP at 120 min was determined by pulse-chase from Figure 4F. Data are the means of 4 independent experiments for relative Ubx2 binding and 3–4 independent experiments for percent GFP cleavage.
(D) Schematic of Ubx2 and truncated variants; FL, 1–585 amino acid (aa); ΔUBA, 71–585 aa; ΔUBX, 1–410 aa; ΔUBAΔUBX, 71–410 aa.
(E) SZ*-GFP-Ub4 was immunoprecipitated from ubx2Δ yeast expressing the Ubx2 variants or containing a vector control. After SDS-PAGE, the indicated antibodies were used to detect SZ*-GFP-Ub4 (anti-HA) and the Ubx2 variants. A total of 1% of the input was analyzed to show equal loading.
(F) A bar graph depicts SZ*-Ub4 binding quantified from (E). Data are the means ± SE of 3 independent experiments; *p < 0.05, **p < 0.005.
(G) Live-cell fluorescence imaging of SZ*-GFP in ubx2Δ yeast containing a vector or the indicated Ubx2 variants. CMAC marks the yeast vacuole.
(H) Quantification of GFP intensity in the vacuole in (G). Greater than 100 cells in each strain were counted; **p < 0.005, ***p < 0.0005.
To confirm that SZ*-GFP-Ub4 retention requires direct interaction with Ubx2, we disrupted the association by mutating key residues in the linear tetra-ubiquitin motif. Based on other studies (Dikic et al., 2009; Hicke et al., 2005), three conserved hydrophobic residues were mutated to polar residues (L8E, I44E, and V70D) (Keren-Kaplan et al., 2013), leading to the creation of a SZ*-GFP-Ub4* construct. As expected, significantly reduced binding was observed between Ubx2 and SZ*-GFP-Ub4* relative to SZ*-GFP- Ub4 (Figure S7A). In fact, SZ*-GFP- Ub4* binding to Ubx2 was comparable to the level observed between Ubx2 and SZ*-GFP. Next, we investigated if defects in SZ*-GFP- Ub4* and Ubx2 binding restored ER export and vacuolar trafficking. Indeed, when the biogenesis of the SZ*-GFP- Ub4* protein was examined, ER export and vacuolar trafficking were enhanced, as shown in a GFP cleavage assay (Figure S7B) and by live-cell fluorescence imaging (Figures S7C and S7D). As predicted, SZ*-GFP-Ub4* shares the same vacuolar targeting pathway (ESCRT-mediated multi-vesicular body [MVB] pathway) as SZ*-GFP (Figure S7B). Interestingly, the ER puncta pattern of SZ*-GFP-Ub4 also disappeared when the L8E/I44E/V70D mutations were introduced into the expression vector that contained K29R, K48R, and K63R (Figure S7C). This result suggests that the puncta depend on the interaction between ubiquitin and Ubx2. More generally, these data indicate that a ubiquitin-Ubx2 axis oversees the ER retention of SZ*-GFP-Ub4.
Ubx2 contains both a UBA domain and a UBX domain, which are responsible for ubiquitin binding and Cdc48 recruitment, respectively (Neuber et al., 2005; Schuberth and Buchberger, 2005; Schuberth et al., 2004). To identify the domain that binds ubiquitinated SZ*, we used a series of mutants lacking UBA, UBX, or both domains (Figure 6D; Wang and Lee, 2012). Loss of UBA on its own or in combination with the UBX domain abolished the SZ*-GFP-Ub4 complex (Figures 6E and 6F). Although deleting the UBX domain decreased SZ*-GFP-Ub4 binding, SZ* still associated with modified Ubx2, and this level was greater than Ubx2 that lacked the UBA. It is possible that reduced binding in the UBX mutant relative to the wild-type protein reflects secondary effects of domain-domain interactions or the contributions of additional factors that support Ubx2-ubiquitin binding. Regardless and consistent with these results, the loss of the UBA domain enhanced vacuolar targeting (Figures 6G and 6H).
Based on our proposition that ubiquitinated SZ* builds up in the ER in the absence of Ubx2 (see above), we asked if SZ* accumulation in the ER facilitated ER exit, regardless of its ubiquitination state. To this end, we inhibited proteasome activity with MG132, and the extent of SZ* stabilization was comparable to the loss of Ydj1, Doa10, or Ubx2 (compare Figure 2 and Figures S8A and S8C; data summary in Figure S8D; Sun and Brodsky, 2018). Proteasome inhibition also enhanced SZ* ubiquitination to a similar degree as the loss of Ubx2 (Figures S5A-5D). Nevertheless, GFP cleavage and thus ER exit were unchanged (Figures S8B, S8C, and S8D). These data confirm that it is the ubiquitin-Ubx2 axis, rather than SZ* abundance, that establishes SZ* retention.
Ubx2 facilitates the ER retention of another model substrate
To confirm that Ubx2 and substrate ubiquitination can orchestrate the retention of a misfolded membrane protein in the ER, we developed a third model substrate. In this case, we instead constructed a protein bearing a folding lesion in the ER lumen. Therefore, the substrate would contain both an ERAD-L degron and an ER exit signal. To this end, we designed a substrate based on ED-Wsc1-L63R (Wang and Ng, 2010), which contains both (1) the CPY N-terminal domain that functions as an ERAD-L degron and (2) an ER export signal from Wsc1. Previous work indicated that the turnover of ED-Wsc1-L63R required both Cue1 and Pep4, suggesting that both ERAD and the vacuole contribute to its degradation (Wang and Ng, 2010). We then appended tetra-ubiquitin to the C terminus of ED-Wsc1-L63R, thereby creating “ED-Ub4” (Figure 7A). Like SZ*-GFP-Ub4, the attachment of tetra-ubiquitin to ED-Wsc1-L63R sequestered the substrate in ER puncta (Figure 7B). Although some ED-Ub4 could be routed for vacuolar transport (Figure S3F), the loss of Ubx2 significantly enhanced vacuolar targeting of ED-Ub4 (Figures 7B, 7C, and 7D). Consistent with the function of Ubx2 in the ER retention of ED-Ub4, the deletion of UBX2 also rescues the vacuolar targeting of Pca1(1-392)-Yor1-GFP-Ub4 (Figures S4F and S4G).
Figure 7. Ubx2 facilitates ER retention of an additional misfolded protein.

(A) Schematic of SZ*-GFP, SZ*-Ub4, and ED-Ub4. N and C indicate the relative topology with respect to the ER.
(B) Live-cell fluorescence imaging of ED-Ub4 in wild-type and ubx2Δ yeast. CMAC marks the yeast vacuole.
(C) Quantification of GFP intensity in the vacuole in (B). Greater than 100 cells in each strain were counted; ***p < 0.0005.
(D) GFP cleavage was measured by pulse-chase in wild-type and ubx2Δ yeast. Cleavage was determined as in Figure 2D. Data are the means ± SE of 3 independent experiments; *p < 0.05.
DISCUSSION
Proteins traversing the secretory pathway are subject to multiple checkpoints, but those failing to achieve their native states during or soon after synthesis are retained in the ER (Anelli and Sitia, 2008; Arvan et al., 2002; MacGurn et al., 2012; Phillips et al., 2020; Sun and Brodsky, 2019). Here, we delineate a previously uncharacterized mechanism that retains misfolded membrane proteins in the ER. We find that substrate ubiquitination–catalyzed by a chaperone-ubiquitin ligase complex–is sufficient to prevent misfolded membrane protein trafficking. In contrast to some ERAD-L substrates (Kroeger et al., 2009; Mu et al., 2008; Wang et al., 2011; White et al., 1999), compromising a post-ubiquitination step during ERAD (i.e., proteasomal degradation) failed to liberate an ERAD-C substrate from the ER. Moreover, the presence of a C-terminally appended tetra-ubiquitin tag was sufficient for retention. We propose that retention of ubiquitinated membrane proteins–marked as a result of compromised folding–prevents proteotoxicity in later secretory pathway compartments. This is especially important when substrates contain strong ER export signals (Kawaguchi et al., 2010; Kincaid and Cooper, 2007). We also found that the retained substrates failed to enter COPII-marked ERES. These data are consistent with studies noting that ER-resident chaperones BiP, calnexin, and PDI, which interact with premature or misfolded proteins, are depleted from ERES (Anelli and Sitia, 2008; Barlowe and Helenius, 2016). For soluble proteins, it is generally thought that the binding of ER molecular chaperones precludes COPII entry (de Silva et al., 1990; Ellgaard and Helenius, 2003; Muresan and Arvan, 1998; Zhang et al., 1997), but the mechanism underlying this phenomenon for membrane proteins has been unclear.
We also show that Ubx2 is required for retention and propose that this ER-resident ubiquitin-binding protein is positioned at a quality-control site distinct from ERES. The presence of quality-control sites in the mammalian ER has long been appreciated (Shenkman and Lederkremer, 2019). Recent data also suggest that ERAD-active zones are distinct from ERES (Albert et al., 2020). These results suggest the ER is organized to distinguish machineries required for quality control and exit. Consistent with this view, Ubx2 associates with several ERAD-requiring factors, including Cdc48 and ubiquitin ligases (Neuber et al., 2005; Schuberth and Buchberger, 2005; Schuberth et al., 2004), and the unfolded protein response is induced in cells lacking Ubx2 (Jonikas et al., 2009). Our demonstration of the critical role played by Ubx2 in overseeing the retention of misfolded membrane proteins–but not a wild-type protein–was based on several results. First, deleting Ubx2 or mutating key residues in tetra-ubiquitin required for UBD recognition enhanced ER export and vacuolar trafficking of four substrates (SZ*-GFP, SZ*-GFP-Ub4, Pca1(1-392)-Yor1-GFP-Ub4, and ED-Ub4). Second, mutating these residues reversed the ER puncta phenotype, so the substrate again exhibited a diffuse pattern in the ER. Chaperones also likely contribute to this ER retention network. Together, our findings support a model whereby the ER employs Ubx2 to actively retain misfolded membrane proteins prior to the final steps of ERAD, i.e., retrotranslocation and degradation.
Our work was made possible by the use of a model substrate that accesses both ERAD and post-ER degradation pathways (Sun and Brodsky, 2018), by the characterization of additional substrates that exhibit this feature, and by appending a short ubiquitin moiety at the C terminus. In contrast, larger polyubiquitin chains may mask ER exit motifs and/or enlarge a substrate so it is unable to enter COPII vesicles. This would confound efforts to resolve the relationship between ubiquitin conjugation and ER exit. Instead, appending a mutated tetra-ubiquitin chain overrides these limitations. In addition, the relatively short half-lives of ERAD substrates have traditionally made it difficult to detect an independent ER retention step. However, tetra-ubiquitin extends substrate half-life (Zhao and Ulrich, 2010), thus allowing us to focus on retention. Furthermore, because the ubiquitin ligase complex that forms linear ubiquitin chains is absent in yeast, the yeast ERAD machinery may inefficiently decode linear ubiquitin chains, thus slowing ERAD but contributing to more robust ER retention (Kirisako et al., 2006; Rahighi et al., 2009; Tokunaga et al., 2009). Yet, this might also be a limitation in our Ubx2-substrate binding studies because Ubx2 likely evolved to bind isopeptide ubiquitin linkages containing K48 and K63. Unfortunately, tools to readily append defined isopeptide-containing ubiquitin motifs to proteins are currently lacking. We also note that the two tetra-ubiquitin-containing substrates assayed in this study trafficked to the vacuole through the MVB pathway, albeit at low levels (Figure S3F). The ability to access this pathway likely arose from both the ER exit signal in SZ* along with the fact that linear and K63-linked ubiquitin chains, which facilitate MVB association, exhibit similar topologies (Erpapazoglou et al., 2012; Lauwers et al., 2009; Stringer and Piper, 2011). It is also important to comment on the fact that SZ*-GFP-Ub4 remained detergent soluble, ruling out a potential contribution of ER-phagy (Cui et al., 2019; Grumati et al., 2018), and SZ* does not appear to be clipped prior to ERAD (data not shown), which facilitates ERAD (Knopf et al., 2020). And, finally, because SZ* accesses both ERAD and post-ER degradation pathways, degron-like sequences required for ERAD and ER exit by the diacidic motif (Barlowe, 2003) in the cytosolic domain and in the Wsc1 transmembrane domain (Karsten et al., 2004; Rayner and Pelham, 1997; Roberts et al., 1992; Singh and Mittal, 2016; Wang and Ng, 2010) were accessible.
Misfolded proteins with ER exit signals are subject to a dynamic interplay between ERAD and ER export. In mammalian cells, inhibiting ERAD can liberate some substrates from the ER, a phenomenon co-opted to correct misfolded, disease-causing proteins (Guerriero and Brodsky, 2012; Ong and Kelly, 2011; Qi et al., 2017). Yet, not every step during ERAD is available for correction. Our work suggests that inhibiting steps prior to substrate ubiquitination is most efficacious. Consistent with this view, stabilizing the disease-causing F508del CFTR protein with proteasome inhibitors failed to rescue ER export and led to the accumulation of ubiquitinated, insoluble aggresomes (Johnston et al., 1998). In contrast, calnexin overexpression, which blunts F508del CFTR ubiquitination, increased plasma membrane residence (Okiyoneda et al., 2004). Similarly, inhibiting F508del CFTR ubiquitination by genetic manipulations or with small molecules potentiates the effects of a drug that aids F508del CFTR folding (Chung et al., 2016; Grove et al., 2011). However, for proteins that are subjected to complex ER quality-control decisions, reducing the function of quality-control factors can fail to restore ER export due to compensation (Pagant et al., 2007). A similar phenomenon was likely evident when proteasome inhibitors were investigated (Kroeger et al., 2009; Mu et al., 2008).
Our studies provide yet another indication that Ubx2 regulates proteostasis. In addition to ERAD, Ubx2 maintains lipid droplet homeostasis and mitochondrial protein quality control. In mitochondria, Ubx2 recruits Cdc48 to remove arrested precursor proteins from the translocase in the outer membrane (Mårtensson et al., 2019). Similar to its yeast homolog, human UBXD8 recruits p97 to support the retrotranslocation of several substrates, such as class I major histocompatibility complex heavy chain (Mueller et al., 2008), lipidated ApoB-100 (Suzuki et al., 2012), and Insig-1 (Lee et al., 2008). Mammalian UBXD8 can also restore defects in both ERAD and lipid droplet homeostasis in ubx2Δ yeast (Wang and Lee, 2012), suggesting that the function of Ubx2 in these pathways–as well as in the retention of misfolded membrane proteins (this study)–is conserved.
Finally, our work suggests the existence of a quality-control center in the ER that precludes the access of misfolded membrane proteins to ERES. Given the challenge of removing transmembrane helices from the ER membrane, it is likely that retrotranslocation is the rate-limiting step in the degradation of these proteins (Guerriero et al., 2017). Therefore, Ubx2 may position membrane proteins destined for retrotranslocation at a Cdc48-active site (Neuber et al., 2005; Schuberth and Buchberger, 2005), which is distinct from ERES. Still, it is possible that polyubiquitination yields an enlarged substrate that fails to enter COPII vesicles in yeast because this organism lacks components to enlarge secretory vesicles (Raote and Malhotra, 2019). Consequently, the retention of ubiquitinated substrates may result from active retention by ER-associated ubiquitin-binding proteins, such as Ubx2, and by passive retention. Future studies will test these hypotheses.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for materials can be directed to the Lead Contact, Dr. Jeffrey L. Brodsky, University of Pittsburgh (jbrodsky@pitt.edu).
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact without restriction.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast was grown at 26°C on rich or selective media as described previously (Adams et al., 1997). A complete list of Saccharomyces cerevisiae strains employed in this study is presented in Key resources table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-HA Peroxidase High Affinity (3F10, rat) | Roche | 12013819001 |
| Anti-HA (12CA5, mouse) | Roche | 11583816001 |
| Anti-glucose-6-phosphate-dehydrogenase (Rabbit) | Sigma-Aldrich | A9521 |
| Anti-GFP (3E6, mouse) | Invitrogen | A-11120 |
| Anti-GFP (mouse) | Roche | 11814460001 |
| Anti-Ubiquitin (P4D1, mouse) | Santa Cruz Biotechnology | SC-8017 |
| Anti-FLAG (DYKDDDDK tag antibody 2368, rabbit) | Cell Signaling Technology | 2368S |
| Anti-rabbit IgG, HRP-linked antibody (Goat) | Cell Signaling Technology | 7074S |
| Anti-mouse IgG, HRP-linked antibody (Horse) | Cell Signaling Technology | 7076S |
| Anti-Ydj1 (Rabbit) | Avrom Caplan Lab | Caplan and Douglas, 1991 |
| Anti-Erv46 (Rabbit) | Charles Barlowe Lab | N/A |
| Anti-Sec61 (Rabbit) | This Lab | N/A |
| Anti-Ubx2 (Rabbit) | Thomas Becker Lab | N/A |
| Bacterial and virus strains | ||
| CBB205 | Sar1-GST | Barlowe et al., 1994 |
| E.coli (BL21) | NEB | C2530H |
| Chemicals, peptides, and recombinant proteins | ||
| ProSignal Pco ECL Reagent | Genesee Scientific | 20-300B |
| ProSignal Femto ECL Reagent | Genesee Scientific | 20-302B |
| Cycloheximide | Sigma-Aldrich | C7698 |
| Oligomycin | Sigma-Aldrich | O4876-5MG |
| MG132 | Millipore | 474790 |
| GTP | Sigma-Aldrich | G8877 |
| ATP | Sigma-Aldrich | A2383 |
| Creatine phosphate | Roche | 10621722001 |
| Creatine Phosphokinase | Sigma-Aldrich | C3755 |
| Phenylmethanesulfonyl fluoride (PMSF) | Sigma-Aldrich | P7626 |
| Leupeptin | Sigma-Aldrich | L9783 |
| Pepstatin A | Sigma-Aldrich | P5318 |
| N-ethylmaleimide (NEM) | Sigma-Aldrich | E1271 |
| Thrombin from human plasma | Sigma-Aldrich | T6884 |
| Isopropyl β-D-thiogalactoside (IPTG) | Sigma-Aldrich | 16758 |
| CellTracker Blue CMAC Dye | Invitrogen | C2110 |
| 4’,6’-Diamidino-2-phenylinadole (DAPI) | Sigma-Aldrich | D8417 |
| Triton X-100 | Sigma-Aldrich | X100 |
| Dodecyl-beta-D-maltoside (DDM) | Calbiochem | D310 |
| Critical commercial assays | ||
| Pierce BCA Protein Assay Kit | Thermo Fisher | 23225 |
| EasyTag EXPRESS 35S Protein Labeling Mix | PerkinElmer Life Sciences | NEG772014MC |
| Deposited data | ||
| Raw data files | This study | N/A |
| Experimental models: Organisms/strains | ||
| BY4742 | MATα, his3Δ1, leu2Δ0, lys2Δ0 ura3Δ3 | Winzeler et al., 1999 |
| pdr5Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 pdr5Δ::KanMX | Winzeler et al., 1999 |
| pdr5Δpep4Δ | MATα, met15Δ0, his3Δ1, leu2Δ0, ura3Δ3 pdr5Δ::KanMX, pep4Δ::KanMX | Winzeler et al., 1999 |
| ydj1Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 ydj1Δ::KanMX | Winzeler et al., 1999 |
| vps36Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 vps36Δ::KanMX | Winzeler et al., 1999 |
| hrd1Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 hrd1Δ::KanMX | Winzeler et al., 1999 |
| doa10Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 doa10Δ::KanMX | Winzeler et al., 1999 |
| ubx1Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 ubx1Δ::KanMX | Winzeler et al., 1999 |
| ubx2Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 ubx2Δ::KanMX | Winzeler et al., 1999 |
| ubx4Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 ubx4Δ::KanMX | Winzeler et al., 1999 |
| cuelΔ | MATα, his3Δ1, leu2Δ0, ura3Δ3 cue1Δ::KanMX | Winzeler et al., 1999 |
| npl4Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 npl4Δ::KanMX | Winzeler et al., 1999 |
| rad23Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 rad23Δ::KanMX | Winzeler et al., 1999 |
| dsk2Δ | MATα, his3Δ1, leu2Δ0, ura3Δ3 dsk2Δ::KanMX | Winzeler et al., 1999 |
| cdc48-2 | Backcrossed 3X to BY4742 | Moir et al., 1982 |
| ySZ083 | UBX2-3FLAG::HIS BY4742 | This study |
| ySZ084 | doa10Δ::KanMX UBX2-3FLAG::HIS BY4742 | This study |
| ySZ087 | ubx2Δ::KanMX PUBX2-UBX2::LEU BY4742 | This study |
| ySZ088 | ubx2Δ::KanMX PUBX2-UBX2(ΔUBA)::LEU BY4742 | This study |
| ySZ089 | ubx2Δ::KanMX PUBX2-UBX2(ΔUBX)::LEU BY4742 | This study |
| ySZ090 | ubx2Δ::KanMX PUBX2-UBX2(ΔUBA ΔUBX)::LEU BY4742 | This study |
| Sec13-RFP | Allison O’Donnell lab | |
| CBY120 | Sec13/Sec31 purification | Charles Barlowe lab |
| CBY1285 | Sec23/Sec24 purification | Charles Barlowe lab |
| sec12-4 | MATa, SUC2, mal mel gal2 CUP1, sec12-4 | Novick et al., 1980 |
| Oligonucleotides | ||
| OSZ08 | GTACTCTAGAATGTTTTTCAACAGACTAAG | Sun and Brodsky, 2018 |
| OSZ14 | GCCTTTAGACATGGAGCCTGAGCCACCTCCT | This study |
| OSZ15 | AGGAGGTGGCTCAGGCTCCATGTCTAAAGGC | This study |
| OSZ16 | GTCAGGATCCCTTTGTACAATTCGTCCATTC | This study |
| RE113 | GCACTGGATGAAGAAGATGAAGAAGATGAGGAAAATGAAGAACAAGGGGGAGGCGGGGGTGGA | Surma et al., 2013 |
| RE114 | CTCTTTGTACGCGTTTGTCGTTTTTAACGATATGCTATTTTAGAATTCGAGCTCGTTTAAAC | Surma et al., 2013 |
| OSZ17 | AGCTTCCTCACTTGGATTCTTAGCTGCTTGGTATTTTAAGGCGCATTTTC | This study |
| OSZ18 | GAAAATGCGCCTTAAAATACCAAGCAGCTAAGAATCCAAGTGAGGAAGCT | This study |
| OSZ21 | GTCATCTAGAACTAGTATGAAAGCATTCACCAGT | This study |
| OSZ22 | GTCAGGATCCCAGCTTTGTACAATTCGTCCAT | This study |
| oSZ23 | GCTTTGTACAATTCGTCCATTCCGCGGCCGCGTTTGTATAGTTCAT | This study |
| OSZ24 | GAACTTAGTTTCGACGGATT | This study |
| OSZ25 | ATGAACTATACAAACGCGGCCGCGGAATGGACGAATTGTACAAAGC | This study |
| OSZ26 | GTGACATAACTAATTACATGACTCGAGTTAGCGGCCGCAAGCTTGCATGCCGGTAGAGGTGTG | This study |
| Recombinant DNA | ||
| pSZ01 | CEN URA3 P-TEF SZ* | Sun and Brodsky, 2018 |
| pSZ03 | CEN URA3 P-TEF SZ*-GFP | Sun and Brodsky, 2018 |
| pSW148 | CEN LEU2 P-GAS Wsc1* | Wang and Ng, 2010 |
| pSW144 | CEN LEU2 P-PRC1 ED-Wsc1-L63R | Wang and Ng, 2010 |
| BPM390 | CEN LEU2 P-YDJ1 Ydj1 | Elizabeth Craig lab |
| pTY40 | Sar1-GST | Barlowe et al., 1994 |
| pSZ10 | CEN URA3 P-TEF SZ*-GFP-Ub4 | This study |
| pSZ11 | CEN LEU2 P-YDJ1 Ydj1 (HPD/QAA) | This study |
| pSZ12 | CEN URA3 P-TEF SZ*-GFP-Ub4* | This study |
| pSZ13 | CEN LEU2 P-TEF SZ*-GFP | This study |
| pRS315 | CEN LEU2 | ATCC |
| pKN31 | 2μ HIS3 Pcup1-mycUb-Tcyc1 | Nakatsukasa et al., 2008 |
| pSZ14 | CEN URA3 P-TEF ED-Ub4 | This study |
| pSZ15 | CEN LEU2 P-GPD Yor1-GFP-Ub4 | This study |
| pSZ16 | CEN LEU2 P-GPD Pca1(1-392)-Yor1-GFP-Ub4 | This study |
| pFA6a-6xGly-3xFLAG-HIS3MX6 | 6xGly-3xFLAG-HIS3MX6 | Lee et al., 2013 |
| 4625 | LEU2 Pubx2 Ubx2(1-585)-Cyc1term | Wang and Lee, 2012 |
| 4628 | LEU2 Pubx2 Ubx2(71-585)-Cyc1term | Wang and Lee, 2012 |
| 4626 | LEU2 Pubx2 Ubx2(1-410)-Cyc1term | Wang and Lee, 2012 |
| 4623 | LEU2 Pubx2 Ubx2(71-410)-Cyc1term | Wang and Lee, 2012 |
| SFNB2443 | LEU2 pRS305-SEC24-2xmCherry | Susan Ferro-Novick Lab |
| 105 | CEN LEU2 PGPD-Yor1-GFP | Adle et al., 2009 |
| 106 | CEN LEU2 PGPD-Pca1(1-392)-Yor1-GFP | Adle et al., 2009 |
| 1677 | CEN LEU2 Ppol30 Pol30-Ub-Ub-Ub-Ub | Zhao and Ulrich, 2010 |
| Software and algorithms | ||
| ImageJ 1.48V | NIH | https://imagej.net/ |
| FIJI (2.0.0-rc-69/1.52i) | NIH | https://imagej.net/software/fiji |
| GraphPad Prism (7.0c) | GraphPad Software | https://www.graphpad.com/ |
| Other | ||
| Anti-HA Affinity Matrix | Roche | 11815016001 |
| Protein A-Sepharose CL-4B | GE Healthcare | GE17-0780-01 |
| DEAE Sepharose Fast Flow | GE Healthcare | 17070910 |
| Q Sepharose XL | GE Healthcare | 10252711 |
| Ni-NTA agarose | QIAGEN | 30230 |
| Glutathione-Agarose | Millipore | G4510 |
| Amicon Ultra-15 Centrifugal Filter Unit | Millipore | UFC903024 |
METHOD DETAILS
Yeast strains and plasmid construction
Plasmids and primers used in this study are listed in the STAR Methods. Tagging of Ubx2 with FLAG tag was performed by standard PCR-based homologous recombination (Longtine et al., 1998). In brief, the Ubx2-6xGly-3xFLAG fragment was first PCR amplified using primers RE113 and RE114 and wild-type genomic DNA as a template (Surma et al., 2013). The fragment was then transformed into wild-type and doa10Δ BY4742 yeast, and positive clones were then screened by western blotting, thus generating the ySZ083, and ysZ084 yeast strains, respectively. Plasmids 4623, 2625, 4626 and 4628 harboring full-length UBX2 and its truncation variants were inserted into the genome of ubx2Δ yeast, generating yeast ySZ087, ySZ088, ysZ089, and y090, respectively.
To construct SZ*-GFP-Ub4, we first mutated the BamHI site within the SZ*-GFP coding region by site-directed mutagenesis, and amplified the modified SZ*-GFP fragment using primers OSZ08 and OSZ16. The amplified SZ*-GFP fragment was then digested with XbaI and BamHI. The Ub4 fragment was generated and collected by cutting plasmid 1677 with BamHI and HindIII. Both SZ*-GFP and the Ub4 fragments were then inserted into p416TEF plasmid digested with XbaI and HindIII, thus generating plasmid pSZ10. Plasmid pSZ11 that expresses Ydj1-HPD/QAA was generated by site-directed mutagenesis of BPM390 using primers OSZ17 and OSZ18. To create the pSZ12, SZ*-GFP-Ub4* construct, we first ordered the Ub4* sequence from IDT with the L8E, I44E, and V70D mutations (on top of the K29R, K48R, and K63R mutations that are localized in SZ*-GFP-Ub4 construct) with BamHI and XhoI sites on the N- and C-terminal, respectively. A subcloning step (BamHI-HindIII) was done to replace original Ub4from SZ*-GFP-Ub4 with Ub4*, thus making the SZ*-GFP-Ub4* construct. For plasmid pSZ13, XbaI and XhoI enzymes were used to cut plasmid pSZ03 to release SZ*-GFP fragment, which was then ligated with 415TEF plasmid treated with same enzymes. In order to generate plasmid pSZ14, which harbors ED-Ub4, the ED-Wsc1-L63R fragment was first amplified using primers OSZ21 and OSZ22 and pSW144 as a template. The vector containing the Ub4 fragment was made by cutting SZ*-GFP fragment from pSZ10 using XbaI and BamHI. After being digested by the same enzymes, the ED-Wsc1-L63R fragment was inserted into the vector with Ub4, generating pSZ14.
Yor1-GFP-Ub4 (pSZ15) and Pca1(1-392)-Yor1-GFP-Ub4 (pSZ16) were constructed by Gibson assembly. In brief, a vector fragment was generated by digesting plasmid 105 with XbaI and XhoI enzymes. Next, the Yor1-GFP and Pca1(1-392)-Yor1-GFP fragments were PCR amplified using primers OSZ23 and OSZ24 with plasmid 105 and 106 as a template, respectively. The Ub4 fragment was then PCR amplified using primers OSZ25 and OSZ26 with plasmid pSZ10 as the template. Finally, the vector, Yor1-GFP, and the Ub4 fragments were ligated by Gibson assembly, thus making Yor1-GFP-Ub4. In contrast, the vector, Pca1(1-392)-Yor1-GFP, and the Ub4 fragments were ligated by Gibson assembly to make Pca1(1-392)-Yor1-GFP-Ub4. The DNA sequences of all constructs were confirmed by Genewiz.
Clycloheximide chase assays
To determine protein stability, cycloheximide chase assays were performed as described previously (Sun and Brodsky, 2018). In brief, the indicated yeast strains expressing the desired substrates were grown in synthetic complete (SC) medium lacking specific amino acids and containing glucose to log phase (A600~0.8) at 26°C. Cycloheximide was added to the culture media to a final concentration of 150 μg/ml. A 1 mL aliquot of culture was taken immediately after adding cycloheximide for the zero time point. At each indicated time point, 1 mL aliquots were taken and snap-frozen immediately in liquid nitrogen. Protein extraction was conducted using the TCA precipitation method as previously described (Zhang et al., 2002). Samples were then resolved on 10% SDS-PAGE and subject to western blot analysis using the indicated antibodies or antisera.
Pulse chase assays
To determine the rate of GFP cleavage from GFP-fusion proteins, pulse chase assays was performed as described previously with minor modifications (Tansey, 2007). In brief, yeast expressing distinct substrates were grown in SC medium lacking uracil (-ura) and methionine (-met) but containing glucose to early log phase (A600 ~0.5) at 26°C, and newly synthesized proteins were pulse labeled with 35S-cys/met for 20 min at a final concentration of 28 μCi/OD cells. The yeast cells were then washed twice with culture medium and resuspended in 4 mL chase media (culture medium supplemented with 5 mg/ml methionine and 5 mg/ml cysteine). A 1.2 mL aliquot of the culture was immediately taken as the zero time point. At each indicated time point, 1.2 mL aliquots were removed, and cells were pelleted by centrifugation followed by snap freezing in liquid nitrogen. Cell lysis was conducted by agitation with glass beads, and the indicated GFP tagged proteins were immunoprecipitated with a 2 h incubation with anti-GFP antibody (Invitrogen) followed by overnight incubation with protein A Sepharose beads (GE healthcare). After washing, the indicated substrates were eluted and subject to 12.5% SDS-PAGE. Gels were subsequently dried and exposed to a storage phosphor screen (FUJI film), and substrates were detected by phosphorimager analysis on a Amersham Typhoon imager (GE healthcare).
Detection of steady state levels of cleaved GFP
Plasmids expressing the indicated substrates were transformed into distinct yeast strains, and 7 days post-transformation the yeast were grown to log phase (A600 ~1.0), and 1 OD of cells was harvested. Total protein extraction was conducted using the TCA precipitation method (see above). Proteins were then resolved by 10% SDS-PAGE and subject to western blot analysis.
Isolation of ER-enriched microsomes from yeast
Yeast expressing the indicated substrates were grown in SC medium (-ura) containing glucose at 26°C until the A600 was ~1.5. Cells were collected by centrifugation and ER microsomes were purified using the large-scale technique described previously (Nakatsukasa et al., 2008). In brief, cells were first resuspended in Buffer A (100mM Tris-HCl, pH9.4, and 10mM DTT) for 15 min at room temperature and then collected and resuspended in lyticase buffer (10mM Tris-HCl, pH7.4, 0.75% yeast extract, 1.5% peptone, 0.5% glucose, and 0.7M sorbitol) plus 20 units of lyticase for 1 h at 26°C to digest the cell wall. The treated cells were overlaid onto an equal volume of Cushion 1 (20mM HEPES, pH 7.4, 0.8M sucrose, and 1.5% Ficoll 400) followed by centrifugation. Next, the pellet were resuspended in cold lysis buffer (20mM HEPES, pH 7.4, 2mM EDTA, 50mM KOAC, 0.1M sorbitol, 1mM of freshly added DTT) plus protease inhibitors (1mM PMSF, 1.5 μg/ml pepstatin A, and 3 μg/ml leupeptin) and lysed with a motor driven Potter-Elvehjem homogenizer. The lysed cells were then overlaid onto an equal volume of Cushion 2 (20mM HEPES, pH 7.4, 1M sucrose, 50mM KOAC, 1mM of freshly added DTT) and centrifuged. The ER microsomes in the pellet was washed with Buffer 88 (20mM HEPES, pH 6.8, 5mM MgOAC, 150mM KOAC, and 250mM sorbitol) and re-centrifuged. The final pellet (ER microsomes) was resuspended in Buffer 88 to a final concentration of 10mg/ml as determined by measuring the A280 in 1% SDS.
Detergent solubility assays
The solubility of the indicated proteins was measured in 1% dodecylmaltoside (DDM) as described previously (Sun and Brodsky, 2018). In brief, ER microsomes from distinct yeast backgrounds expressing the indicated substrates were prepared as described above and incubated on ice for 30 min in Buffer 88 with protease inhibitors in the presence of 1% DDM (EMD Millipore). The detergent soluble fraction was separated from the insoluble fraction by centrifugation at 18,000 g for 30 min at 4°C. Protein pellets from both the soluble (after TCA precipitation; see above) and insoluble fractions were resuspended in the same volume of TCA sample buffer and resolved by 10% SDS-PAGE followed by immunoblotting with anti-HA. Sec61 was used as an internal control for microsomal membrane solubilization and protein extraction. Quantification was performed using ImageJ version 1.48V.
Live cell fluorescence microscopy
Plasmids expressing the indicated substrates were transformed into distinct yeast strains, and 7 d post-transformation the cells were grown to log phase (A600 ~0.8), harvested, and incubated with CMAC in the dark for 15 min. The treated cells were then imaged by fluorescent microscopy using FITC, DAPI and DIC channels on an Nikon ECLIPSE Ti2 epifluorescence microscope.
In vivo ubiquitination assays
To investigate the roles of select E3 ubiquitin ligases on the ubiquitination of select substrates, a 30 mL culture of the indicated yeast strains co-expressing myc-tagged ubiquitin were grown at 26°C until the A600 was ~0.8. Copper sulfate was then added with to the culture medium at a final concentration of 100 μM to induce the expression of myc-tagged ubiquitin for 1 h before the cells were harvested. An equal number of cells were then resuspended in denaturing lysis buffer (50mM Tris-HCl pH 7.0, 150mM NaCl, 1% Triton X-100, 0.2% SDS) supplemented with protease inhibitors and 1mM NEM. Lysis of yeast cells was conducted by agitation with glass beads, and indicated substrates were immunoprecipitated from these samples by overnight incubation with HA-conjugated beads at 4°C to precipitate the HA-tagged substrates. After washing, substrates were eluted and subject to 10% SDS-PAGE. Samples were next processed for western blotting as described previously (Sun and Brodsky, 2018), except that the nitrocellulose membrane was immersed in a boiling water bath for 1 h prior to blocking to better expose antibody epitopes on polyubiquitin chains. Polyubiquitin and the precipitated substrate were visualized using anti-ubiquitin (P4D1) anti-HA (3F10) antibodies, respectively.
Native immunoprecipitation assays
Yeast cells (ySZ084 and ySZ083) expressing SZ*-GFP or SZ*-GFP-Ub4 were grown in SC (-ura) medium containing glucose to A600~1.2. Approximately 30 OD cells of ySZ084 and 60 OD cells of ySZ083 (Ubx2-FLAG steady-state level in ySZ084 is about two fold of that in ySZ083) expressing indicated substrates were harvested and resuspended in native lysis buffer (50mM Tris-HCl, pH 7.0, 150mM NaCl, 1% Triton X-100) supplemented with protease inhibitors. Cell lysate was prepared using glass beads beating method followed by centrifugation at 18,000 g for 15min to isolate soluble membrane fractions. The clear membrane fraction was incubated with protein A beads for 2 hours at 4°C as pre-clearance step. The supernatant was next transferred to new micro-centrifuge tube with HA antibody (12CA5 Roche) for 4 hours incubation at 4°C. Following that, protein A beads was added into the reaction for another 2 hours incubation at 4°C. Three times washing with native lysis buffer were applied to protein A beads, and substrates were eluted with 1X SDS sample buffer (250mM Tris-HCl pH 6.8, 4% SDS, 30% glycerol, 500mM DTT, and 0.25% bromo-phenol blue). Finally, samples were resolved by 10% SDS-PAGE and analyzed by immunoblotting.
COPII protein purification
Sar1 was purified from the pTY40 plasmid (Barlowe et al., 1994) in an E. coli BL21-based CBB205 strain (NEB). Cells were first grown to saturation in 25 mL of LB medium with ampicillin as a seed culture, which was then inoculated to 3 l of medium. When the cells reached an OD600 of ~0.5, the expression of Sar1-GST was induced by adding 2 mL of 1M IPTG (Sigma-Aldrich). The culture was incubated at 37°C for 4 h, the ells were harvested by centrifugation, and frozen at −80°C. To lyse cells, the frozen pellet was first resuspended in 30 mL TBS buffer (50mM Tris-HCl pH7.5, and 150mM NaCl) supplemented with protease inhibitors, and then lysed 3 times at 1000 psi with an Avestin Emulsiflex C3 homogenizer. Triton X-100 was next added to the cell lysate at a final concentration of 0.5%, which was then rotated at 4°C for 15 min. After centrifugation at 14,000 rpm for 15 min in an SS34 rotor, the supernatant was added to a bed volume of 3 mL of glutathione Sepharose beads (Millipore) and incubated at 4°C for 1 h. The beads were then washed with 15 mL TBST (TBS with 1% Triton X-100), and 70 μL of 1 M Tris-HCl, pH 9.4, and 400 μL of a 0.1M CaCl2 stock were added together with 500 U of thrombin to the beads in 7 mL TBS to remove the GST tag via a 1 h incubation at 25°C. The flow through was collected and dialyzed against Buffer 88 overnight at 4°C. The final dialyzed Sar1 faction was aliquoted and analyzed by 10% SDS-PAGE followed by Coomassie blue staining.
Sec23/Sec24 was purified from yeast strain CBY1285 (Barlowe et al., 1994). A 400 mL seed culture in SC (-ura-leu) medium containing glucose was grown overnight. Yeast were inoculated at an initial A600 of ~0.05 in eight, 1.5 l cultures and grown at 23°C until an A600 of ~1.5 was reached. The cells were then harvested by centrifugation and frozen at −80°C. Next, the frozen pellet was resuspended in 100 mL Buffer A (50mM HEPES, pH 7.0, 50 mM KOAC, 2 mM EDTA, 0.25 M sorbitol) supplemented with 2 mM 2-mercaptoethanol and protease inhibitors, and then snap frozen in liquid nitrogen in small droplets. The frozen droplets were then lysed using a stainless steel blender filled half full with liquid nitrogen and ground for 1 min 10 times with a 1min rest between each round. After thawing the frozen yeast powder on ice, 100 mL of Buffer B (50 mM HEPES, pH 7.0, 1 M KOAC, 2 mM EDTA, 0.25 M sorbitol) supplemented with 2 mM 2-mercaptoethanol and protease inhibitors was added to the lysate, which was homogenized with 5 strokes using a motor-driven Potter-Elvehjem homogenizer. A clarified supernatant was collected after two rounds of centrifugation at 14,000 rpm for 10 min in an SS34 rotor. The top layer of lipid was removed by pouring supernatant through four layers of cheesecloth. The clear lysate was then loaded onto a 35 mL DEAE column (GE healthcare), which was then washed with 500 mL Buffer C (50 mM HEPES, pH 7.0, 500 mM KOAC, 0.25 M sorbitol), and protein was eluted with 50ml Buffer D (50 mM HEPES, pH 7.0, 0.75 M KOAC, 0.01% Triton X-100) supplemented with protease inhibitors which was next loaded onto a 10ml Ni-NTA column. Prior to elution with NTA buffer C (50 mM HEPES, pH 7.0, 0.15 M KOAC, 200 mM imidazole), the resin was washed with 25 mL of NTA Buffer A (50mM HEPES, pH 7.0, 0.5M KOAC) and 50ml of NTA Buffer B (50 mM HEPES, pH 7.0, 0.15 M KOAC, 15 mM imidazole). Elution fractions of 1ml were manually collected and analyzed by SDS-PAGE to determine the Sec23/Sec24 containing fractions, which were then combined and went through desalting column (PD-10, GE healthcare). The final enriched proteins were eluted with elution buffer (50 mM HEPES, pH 7.0, 1 mM MgOAC, 0.3 M KOAC) and analyzed by SDS-PAGE followed by Coomassie blue staining.
Sec13/Sec31 was purified from CBY120 (Barlowe et al., 1994) yeast, which were grown in eight 1.51 of SC (-ura) media containing glucose and harvested and lysed as described above except that a cell suspension was made using 50 mL Buffer 88 supplemented with protease inhibitors. A clarified supernatant was obtained as described above, and was loaded onto a 10 mL Ni-NTA column for nickel column, which was washed with 25 mL NTA Buffer A and 50 mL NTA Buffer B, and then protein was eluted with 20 mL NTA Buffer C. Elution fractions of 1ml were manually collected and analyzed by SDS-PAGE followed by Coomassie blue staining to determine the Sec13/Sec31 containing fractions, which were then combined and loaded onto Q Sepharose XL (GE Healthcare). As above, a wash was done with 75% Q BufferA+24% Q Buffer B, and 70% Q Buffer A +30% Q buffer B, and elution was achieved with 20ml 25% Q buffer A+75% Q buffer B in 1ml fractions. Peak fractions, determined by SDS-PAGE followed by Coomassie blue staining, were isolated after a 100% Q Buffer B wash. The peak fractions were then concentrated and sanp frozen in liquid nitrogen. Prior to storage at −80°C, protein aggregates were removed by centrifugation at 18,000 g for 20 min.
In vitro budding assays
To determine the in vitro COPII budding efficiency of the indicated substrates, an in vitro budding assay was performed as described previously (Barlowe et al., 1994; Shibuya et al., 2015). In brief, both ER microsomes from yeast cells containing vectors for the desired substrates and COPII components (Sar1, Sec23-Sec24, and Sec13-Sec31) were prepared as described above. A 100 μL reaction was then set-up by incubating 5 μg of ER microsomes in Buffer 88 supplemented with GTP (0.1mM) and an ATP regeneration system (1 mM ATP, 40 μM creatine phosphate, 200 μg/ml creatine phosphokinase) in the presence or absence of purified COPII proteins (Sar1:5 μg/ml; Sec23/Sec24, 4 μg/ml; Sec13/Sec31, 40 μg/ml) at 25°C for 20 min followed by a 5 min incubation on ice. After centrifugation at 17,000 g for 5 min, unbudded membranes (in the pellet) were separated from budded vesicles (in the supernatant), which were then collected by ultracentrifugation at 195,500 g for 30 min. Unbudded membranes and budded vesicle pellet fractions were solubilized with 60 μL and 12 μL of SDS-sample buffer, respectively. A total of 6 μL (50% of the total) of the budded fractions and 1.5 μL (2.5% of the total) of the unbudded membranes were resolved on 10% SDS-PAGE followed by immunoblotting for HA and Erv46.
Oligomycin resistance assay
To measure the oligomycin resistant phenotype of yeast expressing various substrates, wild-type or doa10Δ yeast containing a vector or the Yor1-GFP, Yor1-GFP-Ub4, Pca1(1-392)-Yor1-GFP, and Pca1(1-392)-Yor1-GFP-Ub4 were grown overnight to stationary phase. Six 5-fold dilutions were made and transferred to YPEG solid medium supplemented with 2.5 μg/ml oligomycin (dissolved in DMSO). The plates were incubated at 26°C for 5 days and imaged 5 using a Bio-Rad imaging station.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data represent the means ± SE unless otherwise stated. The number of independent experiments performed for each experiment is indicated in the figure legends. Band intensities from western blotting and pulse chase experiments were quantified using ImageJ version 1.48V and normalized to the loading control or to total protein. GFP fluorescence intensities were quantified using FIJI (2.0.0-rc-69/1.52i). Statistical analysis in this work was determined by Prism 7 using either unpaired t- tests, ordinary one-way ANOVA with Holm-Sidak’s multiple comparison tests, or ordinary two-way ANOVA with a Dunnett’s multiple comparisons test. Significance is defined by a p < 0.05. However, three variations of significance were also presented in this study, which were used to show variations in the level of significance for all data; * denotes p < 0.05, ** p < 0.005, and *** p < 0.0005.
Supplementary Material
Highlights.
Ubiquitination is sufficient to retain misfolded membrane proteins in the ER for ERAD
Ubx2 is as a ubiquitin-dependent ER retention factor for misfolded membrane proteins
A tetra-ubiquitin fusion sequesters a misfolded membrane protein from ER exit sites
Inhibiting the Ubx2-ubiquitin axis allows for ER exit of misfolded membrane proteins
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants GM131732 and DK079307 to J.L.B. and DK124659 to C.J.G. We thank Drs. Charles Barlowe, Thomas Becker, Miguel Betegon, David Fushman, Elizabeth Craig, Robert Ernst, Susan Ferro-Novick, Jaekwon Lee, Thibault Mayor, Davis Ng, Allyson O’Donnell, Helle Ulrich, and Chao-Wen Wang for reagents and valuable discussions.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109717.
REFERENCES
- Adams A, Gottschling D, Kaiser C, and Stearns T (1997). Media and stock preservation. Methods in Yeast Genetics (Cold Spring Harbor Laboratory Pres; ), pp. 145–160. [Google Scholar]
- Adle DJ, Wei W, Smith N, Bies JJ, and Lee J (2009). Cadmium-mediated rescue from ER-associated degradation induces expression of its exporter. Proc. Natl. Acad. Sci. USA 106, 10189–10194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert S, Wietrzynski W, Lee CW, Schaffer M, Beck F, Schuller JM, Salomé PA, Plitzko JM, Baumeister W, and Engel BD (2020). Direct visualization of degradation microcompartments at the ER membrane. Proc. Natl. Acad. Sci. USA 117, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anelli T, and Sitia R (2008). Protein quality control in the early secretory pathway. EMBO J. 27, 315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvan P, Zhao X, Ramos-Castaneda J, and Chang A (2002). Secretory pathway quality control operating in Golgi, plasmalemmal, and endosomal systems. Traffic 3, 771–780. [DOI] [PubMed] [Google Scholar]
- Balchin D, Hayer-Hartl M, and Hartl FU (2016). In vivo aspects of protein folding and quality control. Science 353, aac4354. [DOI] [PubMed] [Google Scholar]
- Barlowe C (2003). Signals for COPII-dependent export from the ER: what’s the ticket out? Trends Cell Biol. 13, 295–300. [DOI] [PubMed] [Google Scholar]
- Barlowe C, and Helenius A (2016). Cargo Capture and Bulk Flow in the Early Secretory Pathway. Annu. Rev. Cell Dev. Biol 32, 197–222. [DOI] [PubMed] [Google Scholar]
- Barlowe CK, and Miller EA (2013). Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 193, 383–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, and Schekman R (1994). COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77, 895–907. [DOI] [PubMed] [Google Scholar]
- Bhamidipati A, Denic V, Quan EM, and Weissman JS (2005). Exploration of the topological requirements of ERAD identifies Yos9p as a lectin sensor of misfolded glycoproteins in the ER lumen. Mol. Cell 19, 741–751. [DOI] [PubMed] [Google Scholar]
- Bole DG, Hendershot LM, and Kearney JF (1986). Posttranslational association of immunoglobulin heavy chain binding protein with nascent heavy chains in nonsecreting and secreting hybridomas. J. Cell Biol 102, 1558–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braakman I, and Hebert DN (2013). Protein folding in theendoplasmic reticulum. Cold Spring Harb. Perspect. Biol 5, a013201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AJ, and Douglas MG (1991). Characterization of YDJ1: a yeast homologue of the bacterial dnaJ protein. J. Cell Biol 114, 609–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan AJ, Cyr DM, and Douglas MG (1992). YDJ1p facilitates polypeptide translocation across different intracellular membranes by a conserved mechanism. Cell 71, 1143–1155. [DOI] [PubMed] [Google Scholar]
- Carvalho P, Goder V, and Rapoport TA (2006). Distinct ubiquitin-ligase complexes define convergent pathways for the degradation of ER proteins. Cell 126, 361–373. [DOI] [PubMed] [Google Scholar]
- Chung WJ, Goeckeler-Fried JL, Havasi V, Chiang A, Rowe SM, Plyler ZE, Hong JS, Mazur M, Piazza GA, Keeton AB, et al. (2016). Increasing the Endoplasmic Reticulum Pool of the F508del Allele of the Cystic Fibrosis Transmembrane Conductance Regulator Leads to Greater Folding Correction by Small Molecule Therapeutics. PLoS One 11, e0163615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Parashar S, Zahoor M, Needham PG, Mari M, Zhu M, Chen S, Ho HC, Reggiori F, Farhan H, et al. (2019). A COPII subunit acts with an autophagy receptorto target endoplasmic reticulum for degradation. Science 365, 53–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva AM, Balch WE, and Helenius A (1990). Quality control in the endoplasmic reticulum: folding and misfolding of vesicular stomatitis virus G protein in cells and in vitro. J. Cell Biol 111, 857–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denic V, Quan EM, and Weissman JS (2006). A luminal surveillance complex that selects misfolded glycoproteins for ER-associated degradation. Cell 126, 349–359. [DOI] [PubMed] [Google Scholar]
- Dikic I, Wakatsuki S, and Walters KJ (2009). Ubiquitin-binding domains - from structures to functions. Nat. Rev. Mol. Cell Biol 10, 659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, and Helenius A (2003). Quality control in the endoplasmic reticulum. Nat. Rev. Mol. Cell Biol 4, 181–191. [DOI] [PubMed] [Google Scholar]
- Erpapazoglou Z, Dhaoui M, Pantazopoulou M, Giordano F, Mari M, Léon S, Raposo G, Reggiori F, and Haguenauer-Tsapis R (2012). A dual role for K63-linked ubiquitin chains in multivesicular body biogenesis and cargo sorting. Mol. Biol. Cell 23, 2170–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu L, and Sztul E (2003). Traffic-independent function of the Sar1p/COPII machinery in proteasomal sorting of the cystic fibrosis transmembrane conductance regulator. J. Cell Biol 160, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner AM, Aviel S, and Argon Y (1993). Rapid degradation of an unassembled immunoglobulin light chain is mediated by a serine protease and occurs in a pre-Golgi compartment. J. Biol. Chem 268, 25940–25947. [PubMed] [Google Scholar]
- Genereux JC, Qu S, Zhou M, Ryno LM, Wang S, Shoulders MD, Kaufman RJ, Lasmézas CI, Kelly JW, and Wiseman RL (2015). Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis. EMBO J. 34, 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimeno RE, Espenshade P, and Kaiser CA (1995). SED4 encodes a yeast endoplasmic reticulum protein that binds Sec16p and participates in vesicle formation. J. Cell Biol 131, 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Navarro N, and Miller E (2016). Protein sorting at the ER-Golgi interface. J. Cell Biol 215, 769–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove DE, Rosser MF, Ren HY, Naren AP, and Cyr DM (2009). Mechanisms for rescue of correctable folding defects in CFTRDelta F508. Mol. Biol. Cell 20, 4059–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grove DE, Fan CY, Ren HY, and Cyr DM (2011). The endoplasmic reticulum-associated Hsp40 DNAJB12 and Hsc70 cooperate to facilitate RMA1 E3-dependent degradation of nascent CFTRDeltaF508. Mol. Biol. Cell 22, 301–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grumati P, Dikic I, and Stolz A (2018). ER-phagy at a glance. J. Cell Sci 131, jcs217364. [DOI] [PubMed] [Google Scholar]
- Guerriero CJ, and Brodsky JL (2012). The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev 92, 537–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerriero CJ, Reutter KR, Augustine AA, Preston GM, Weiberth KF, Mackie TD, Cleveland-Rubeor HC, Bethel NP, Callenberg KM, Nakatsukasa K, et al. (2017). Transmembrane helix hydrophobicity is an energetic barrier during the retrotranslocation of integral membrane ERAD substrates. Mol. Biol. Cell 28, 2076–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Liu Y, and Chang A (2007). Cytoplasmic Hsp70 promotes ubiquitination for endoplasmic reticulum-associated degradation of a misfolded mutant of the yeast plasma membrane ATPase, PMA1. J. Biol. Chem 282, 26140–26149. [DOI] [PubMed] [Google Scholar]
- Hicke L, Schubert HL, and Hill CP (2005). Ubiquitin-binding domains. Nat. Rev. Mol. Cell Biol 6, 610–621. [DOI] [PubMed] [Google Scholar]
- Hosokawa N, Wada I, Natsuka Y, and Nagata K (2006). EDEM accelerates ERAD by preventing aberrant dimer formation of misfolded alpha1-anti-trypsin. Genes Cells 11, 465–476. [DOI] [PubMed] [Google Scholar]
- Husnjak K, and Dikic I (2012). Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu. Rev. Biochem 81, 291–322. [DOI] [PubMed] [Google Scholar]
- Huyer G, Longsworth GL, Mason DL, Mallampalli MP, McCaffery JM, Wright RL, and Michaelis S (2004). A striking quality control subcompartment in Saccharomyces cerevisiae: the endoplasmic reticulum-associated compartment. Mol. Biol. Cell 15, 908–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosch E, Taxis C, Volkwein C, Bordallo J, Finley D, Wolf DH, and Sommer T (2002). Protein dislocation from the ER requires polyubiquitination and the AAA-ATPase Cdc48. Nat. Cell Biol 4, 134–139. [DOI] [PubMed] [Google Scholar]
- Johnston JA, Ward CL, and Kopito RR (1998). Aggresomes: a cellular response to misfolded proteins. J. Cell Biol 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, and Schuldiner M (2009). Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323, 1693–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakoi S, Yorimitsu T, and Sato K (2013). COPII machinery cooperates with ER-localized Hsp40 to sequester misfolded membrane proteins into ER-associated compartments. Mol. Biol. Cell 24, 633–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsten V, Hegde RS, Sinai AP, Yang M, and Joiner KA (2004). Transmembrane domain modulates sorting of membrane proteins in Toxoplasma gondii. J. Biol. Chem 279, 26052–26057. [DOI] [PubMed] [Google Scholar]
- Kawaguchi S, Hsu CL, and Ng DTW (2010). Interplay of substrate retention and export signals in endoplasmic reticulum quality control. PLoS One 5, e15532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren-Kaplan T, Attali I, Estrin M, Kuo LS, Farkash E, Jerabek-Willemsen M, Blutraich N, Artzi S, Peri A, Freed EO, et al. (2013). Structure-based in silico identification of ubiquitin-binding domains provides insights into the ALIX-V:ubiquitin complex and retrovirus budding. EMBO J. 32, 538–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Spear ED, and Ng DT (2005). Yos9p detects and targets misfolded glycoproteins for ER-associated degradation. Mol. Cell 19, 753–764. [DOI] [PubMed] [Google Scholar]
- Kincaid MM, and Cooper AA (2007). Misfolded proteins traffic from the endoplasmic reticulum (ER) due to ER export signals. Mol. Biol. Cell 18, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, Sano S, Tokunaga F, Tanaka K, and Iwai K (2006). A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 25, 4877–4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knittler MR, Dirks S, and Haas IG (1995). Molecular chaperones involved in protein degradation in the endoplasmic reticulum: quantitative interaction of the heat shock cognate protein BiP with partially folded immunoglobulin light chains that are degraded in the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 92, 1764–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knopf JD, Landscheidt N, Pegg CL, Schulz BL, Kühnle N, Chao CW, Huck S, and Lemberg MK (2020). Intramembrane protease RHBDL4 cleaves oligosaccharyltransferase subunits to target them for ER-associated degradation. J. Cell Sci 133, jcs243790. [DOI] [PubMed] [Google Scholar]
- Kodera C, Yorimitsu T, Nakano A, and Sato K (2011). Sed4p stimulates Sar1p GTP hydrolysis and promotes limited coat disassembly. Traffic 12, 591–599. [DOI] [PubMed] [Google Scholar]
- Kroeger H, Miranda E, MacLeod I, Pérez J, Crowther DC, Marciniak SJ, and Lomas DA (2009). Endoplasmic reticulum-associated degradation (ERAD) and autophagy cooperate to degrade polymerogenic mutant serpins. J. Biol. Chem 284, 22793–22802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauwers E, Jacob C, and André B (2009). K63-linked ubiquitin chains as a specific signal for protein sorting into the multivesicular body pathway. J. Cell Biol 185, 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JN, Zhang X, Feramisco JD, Gong Y, and Ye J (2008). Unsaturated fatty acids inhibit proteasomal degradation of Insig-1 at a postubiquitination step. J. Biol. Chem 283, 33772–33783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lim WA, and Thorn KS (2013). Improved blue, green, and red fluorescent protein tagging vectors for S. cerevisiae. PLoS One 8, e67902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kane T, Tipper C, Spatrick P, and Jenness DD (1999). Yeast mutants affecting possible quality control of plasma membrane proteins. Mol. Cell. Biol 19, 3588–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- MacGurn JA, Hsu PC, and Emr SD (2012). Ubiquitin and membrane protein turnover: from cradle to grave. Annu. Rev. Biochem 81, 231–259. [DOI] [PubMed] [Google Scholar]
- Mårtensson CU, Priesnitz C, Song J, Ellenrieder L, Doan KN, Boos F, Floerchinger A, Zufall N, Oeljeklaus S, Warscheid B, and Becker T (2019). Mitochondrial protein translocation-associated degradation. Nature 569, 679–683. [DOI] [PubMed] [Google Scholar]
- McClellan AJ, Scott MD, and Frydman J (2005). Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 121, 739–748. [DOI] [PubMed] [Google Scholar]
- McCracken AA, and Brodsky JL (1996). Assembly of ER-associated protein degradation in vitro: dependence on cytosol, calnexin, and ATP. J. Cell Biol 132, 291–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medicherla B, Kostova Z, Schaefer A, and Wolf DH (2004). A genomic screen identifies Dsk2p and Rad23p as essential components of ER-associated degradation. EMBO Rep. 5, 692–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moir D, Stewart SE, Osmond BC, and Botstein D (1982). Cold-sensitive cell-division-cycle mutants of yeast: isolation, properties, and pseudoreversion studies. Genetics 100, 547–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR 3rd, Segatori L, and Kelly JW (2008). Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 134, 769–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller B, Klemm EJ, Spooner E, Claessen JH, and Ploegh HL (2008). SEL1L nucleates a protein complex required for dislocation of misfolded glycoproteins. Proc. Natl. Acad. Sci. USA 105, 12325–12330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muresan Z, and Arvan P (1998). Enhanced binding to the molecular chaperone BiP slows thyroglobulin export from the endoplasmic reticulum. Mol. Endocrinol 12, 458–467. [DOI] [PubMed] [Google Scholar]
- Nakano A, Brada D, and Schekman R (1988). A membrane glycoprotein, Sec12p, required for protein transport from the endoplasmic reticulum to the Golgi apparatus in yeast. J. Cell Biol 107, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatsukasa K, Huyer G, Michaelis S, and Brodsky JL (2008). Dissecting the ER-associated degradation of a misfolded polytopic membrane protein. Cell 132, 101–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuber O, Jarosch E, Volkwein C, Walter J, and Sommer T (2005). Ubx2 links the Cdc48 complexto ER-associated protein degradation. Nat. Cell Biol 7, 993–998. [DOI] [PubMed] [Google Scholar]
- Nishikawa SI, Fewell SW, Kato Y, Brodsky JL, and Endo T (2001). Molecular chaperones in the yeast endoplasmic reticulum maintain the solubility of proteins for retrotranslocation and degradation. J. Cell Biol 153, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P, Field C, and Schekman R (1980). Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21, 205–215. [DOI] [PubMed] [Google Scholar]
- Okiyoneda T, Harada K, Takeya M, Yamahira K, Wada I, Shuto T, Suico MA, Hashimoto Y, and Kai H (2004). Delta F508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol. Biol. Cell 15, 563–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong DS, and Kelly JW (2011). Chemical and/or biological therapeutic strategies to ameliorate protein misfolding diseases. Curr. Opin. Cell Biol 23, 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagant S, Kung L, Dorrington M, Lee MCS, and Miller EA (2007). Inhibiting endoplasmic reticulum (ER)-associated degradation of misfolded Yor1p does not permit ER export despite the presence of a diacidic sorting signal. Mol. Biol. Cell 18, 3398–3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips BP, Gomez-Navarro N, and Miller EA (2020). Protein quality control in the endoplasmic reticulum. Curr. Opin. Cell Biol 65, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemper RK, Böhmler S, Bordallo J, Sommer T, and Wolf DH (1997). Mutant analysis links the translocon and BiP to retrograde protein transport for ER degradation. Nature 388, 891–895. [DOI] [PubMed] [Google Scholar]
- Qi L, Tsai B, and Arvan P (2017). New Insights into the Physiological Role of Endoplasmic Reticulum-Associated Degradation. Trends Cell Biol. 27, 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich E, Kerem A, Fröhlich KU, Diamant N, and Bar-Nun S (2002). AAA-ATPase p97/Cdc48p, a cytosolic chaperone required for endoplasmic reticulum-associated protein degradation. Mol. Cell. Biol 22, 626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, et al. (2009). Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell 136, 1098–1109. [DOI] [PubMed] [Google Scholar]
- Raote I, and Malhotra V (2019). Protein transport by vesicles and tunnels. J. Cell Biol 218, 737–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T, and Hochstrasser M (2008). Diversity of degradation signals in the ubiquitin-proteasome system. Nat. Rev. Mol. Cell Biol 9, 679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner JC, and Pelham HR (1997). Transmembrane domain-dependent sorting of proteins to the ER and plasma membrane in yeast. EMBO J. 16, 1832–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reggiori F, and Pelham HR (2002). A transmembrane ubiquitin ligase required to sort membrane proteins into multivesicular bodies. Nat. Cell Biol 4, 117–123. [DOI] [PubMed] [Google Scholar]
- Roberts CJ, Nothwehr SF, and Stevens TH (1992). Membrane protein sorting in the yeast secretory pathway: evidence that the vacuole may be the default compartment. J. Cell Biol 119, 69–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuberth C, and Buchberger A (2005). Membrane-bound Ubx2 recruits Cdc48 to ubiquitin ligases and their substrates to ensure efficient ER-associated protein degradation. Nat. Cell Biol 7, 999–1006. [DOI] [PubMed] [Google Scholar]
- Schuberth C, Richly H, Rumpf S, and Buchberger A (2004). Shp1 and Ubx2 are adaptors of Cdc48 involved in ubiquitin-dependent protein degradation. EMBO Rep. 5, 818–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenkman M, and Lederkremer GZ (2019). Compartmentalization and Selective Tagging for Disposal of Misfolded Glycoproteins. Trends Biochem. Sci 44, 827–836. [DOI] [PubMed] [Google Scholar]
- Shibuya A, Margulis N, Christiano R, Walther TC, and Barlowe C (2015). The Erv41-Erv46 complex serves as a retrograde receptor to retrieve escaped ER proteins. J. Cell Biol 208, 197–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, and Mittal A (2016).Transmembrane Domain Lengths Serve as Signatures of Organismal Complexity and Viral Transport Mechanisms. Sci. Rep 6, 22352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stringer DK, and Piper RC (2011). A single ubiquitin is sufficient for cargo protein entry into MVBs in the absence of ESCRT ubiquitination. J. Cell Biol 192, 229–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, and Brodsky JL (2018). The degradation pathway of a model misfolded protein is determined by aggregation propensity. Mol. Biol. Cell 29, 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, and Brodsky JL (2019). Protein quality control in the secretory pathway. J. Cell Biol 218, 3171–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Madden DT, Hamamoto S, Orci L, and Schekman R (2002). Sec16p potentiates the action of COPII proteins to bud transport vesicles. J. Cell Biol 158, 1029–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surma MA, Klose C, Peng D, Shales M, Mrejen C, Stefanko A, Braberg H, Gordon DE, Vorkel D, Ejsing CS, et al. (2013). A lipid E-MAP identifies Ubx2 as a critical regulatorof lipid saturation and lipid bilayer stress. Mol. Cell 51, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Otsuka T, Ohsaki Y, Cheng J, Taniguchi T, Hashimoto H, Taniguchi H, and Fujimoto T (2012). Derlin-1 and UBXD8 are engaged in dislocation and degradation of lipidated ApoB-100 at lipid droplets. Mol. Biol. Cell 23, 800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szathmary R, Bielmann R, Nita-Lazar M, Burda P, and Jakob CA (2005). Yos9 protein is essential for degradation of misfolded glycoproteins and may function as lectin in ERAD. Mol. Cell 19, 765–775. [DOI] [PubMed] [Google Scholar]
- Tansey WP (2007). Pulse-chase assay for measuring protein stability in yeast. CSH Protoc. 2007, pdb.prot4641. [DOI] [PubMed] [Google Scholar]
- Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, et al. (2009). Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell Biol 11, 123–132. [DOI] [PubMed] [Google Scholar]
- Tsai J, and Douglas MG (1996). A conserved HPD sequence of the J-domain is necessary for YDJ1 stimulation of Hsp70 ATPase activity at a site distinct from substrate binding. J. Biol. Chem 271, 9347–9354. [DOI] [PubMed] [Google Scholar]
- Varshavsky A (2017). The Ubiquitin System, Autophagy, and Regulated Protein Degradation. Annu. Rev. Biochem 86, 123–128. [DOI] [PubMed] [Google Scholar]
- Vashist S, and Ng DT (2004). Misfolded proteins are sorted by a sequential checkpoint mechanism of ER quality control. J. Cell Biol 165, 41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CW, and Lee SC (2012). The ubiquitin-like (UBX)-domain-containing protein Ubx2/Ubxd8 regulates lipid droplet homeostasis. J. Cell Sci 125, 2930–2939. [DOI] [PubMed] [Google Scholar]
- Wang S, and Ng DT (2010). Evasion of endoplasmic reticulum surveillance makes Wsc1p an obligate substrate of Golgi quality control. Mol. Biol. Cell 21, 1153–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Song W, Brancati G, and Segatori L (2011). Inhibition of endoplasmic reticulum-associated degradation rescues native folding in loss of function protein misfolding diseases. J. Biol. Chem 286, 43454–43464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AL, Guerra B, Wang J, and Lanford RE (1999). Presecretory degradation of apolipoprotein [a] is mediated by the proteasome pathway. J. Lipid Res 40, 275–286. [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, Bangham R, Benito R, Boeke JD, Bussey H, et al. (1999). Functional characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science 285, 901–906. [DOI] [PubMed] [Google Scholar]
- Xu C, and Ng DT (2015). Glycosylation-directed quality control of protein folding. Nat. Rev. Mol. Cell Biol 16, 742–752. [DOI] [PubMed] [Google Scholar]
- Ye Y, Meyer HH, and Rapoport TA (2001). The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656. [DOI] [PubMed] [Google Scholar]
- Zhang JX, Braakman I, Matlack KE, and Helenius A (1997). Quality control in the secretory pathway: the role of calreticulin, calnexin and BiP in the retention of glycoproteins with C-terminal truncations. Mol. Biol. Cell 8, 1943–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nijbroek G, Sullivan ML, McCracken AA, Watkins SC, Michaelis S, and Brodsky JL (2001). Hsp70 molecular chaperone facilitates endoplasmic reticulum-associated protein degradation of cystic fibrosis transmembrane conductance regulator in yeast. Mol. Biol. Cell 12, 1303–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Michaelis S, and Brodsky JL (2002). CFTR expression and ER-associated degradation in yeast. Methods Mol. Med 70, 257–265. [DOI] [PubMed] [Google Scholar]
- Zhao S, and Ulrich HD (2010). Distinct consequences of posttranslational modification by linear versus K63-linked polyubiquitin chains. Proc. Natl. Acad. Sci. USA 107, 7704–7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request.
This paper does not report original code.
Additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.