

Abstract
Background
Congenital hemophilia A is a recessive inherited hemorrhagic disorder. According to the activity of functional coagulation factors, the severity of hemophilia A is divided into three levels: mild, moderate and severe. The first bleeding episode in severe and moderate congenital hemophilia A occurs mostly in early childhood and mainly involves soft tissue and joint bleeds. At present, there are limited reports on severe congenital hemophilia A with low factor XII (FXII) activity during the neonatal period.
Case presentation
A 13-day-old neonate was admitted to the hospital with hematoma near the joints of both upper arms. Coagulation tests showed he had low activity of factor VIII (FVIII) and FXII. He was diagnosed with congenital hemophilia A and treated with human coagulation factor VIII (recombinant FVIII). Although the hematoma became smaller, FVIII activity was only increased to a certain extent and FXII activity decreased gradually. Unfortunately, the child responded poorly to recombinant human coagulation factor VIII and his guardian rejected prophylactic inhibitors and genetic testing and refused further treatment. Three months later, the child developed intracranial hemorrhage (ICH) due to low FVIII activity.
Conclusions
In hemophilia A, the presence of FVIII inhibitors, drug concentration and testing are three important aspects that must be considered when FVIII activity does not reach the desired level. Early positive disease treatment and prophylaxis can decrease the frequency of bleeding and improve quality of life. We recommend that pregnant women with a family history of hemophilia A undergo early prenatal and neonatal genetic testing.
Keywords: Congenital hemophilia A, Factor VIII, Factor XII, Inhibitor, Neonate
Background
Congenital hemophilia A is an inherited hemorrhagic disorder caused by the x-linked chromosome factor VIII (FVIII) and it occurs in 0.01% of newborns. Thirty percent of these patients have spontaneous mutations in FVIII and do not have a family history of hemophilia A [1, 2]. In an observational cohort study consisting of 679 patients with severe or moderate hemophilia A, the researchers found that the first bleeding episode in hemophilia A occurred at the median age of 0.82 years in severe disease and 1.47 years in moderate disease [3]. Acute joint hemorrhage is a common symptom. Patients often suffer from repeated bleeding and chronic joint injury [4]. In addition, in 1–4% of patients, intracranial hemorrhage could be the first symptom [5]. When the FVIII activity is < 1%, repeated episodes of spontaneous bleeding occur in approximately 50–60% of patients [6]. Primary prevention in patients with severe hemophilia A can reduce the progression of arthropathy [7].
It is found that the lower the FVIII activity in the body, the more frequent bleeding episodes [5]. The severity of hemophilia is classified according to the activity of functional coagulation factors, with 5–40% being mild, 1–5% being moderate and < 1% being severe [8, 9]. Currently, the main prevention and treatment method is to use plasma-derived or recombinant FVIII products [9]. In addition, bleeding episodes could also be treated with activated prothrombin complex concentrate (aPCC) or recombinant activating factor VII (rFVIIa) [10]. Here, we report the case of a very early onset of severe neonatal congenital hemophilia A and perform a brief literature review on its mechanism.
Case presentation
We encountered a 13-day-old neonatal patient with congenital hemophilia A with low FXII activity. At first, he was admitted for hematoma near the joints of both upper arms. Blood test results showed that activated partial thromboplastin time (APTT) was prolonged without extended prothrombin time (PT) and the activity of factors VIII, IX, XI and XII was low, especially the activity of FVIII (0.7%) and factor XII (15.3%). The newborn had a family history of hemophilia A, with an uncle diagnosed with hemophilia A. The neonate was diagnosed with congenital hemophilia A. After admission, he was treated with human coagulation factor VIII with standard dosage according to the guidelines for the management of hemophilia [11, 12]. In general, each unit of FVIII/kg per 8–12 h infused intravenously raises plasma FVIII levels by approximately 2% in the absence of an inhibitor [11, 12]. After 5 days of treatment, the APTT returned to normal and the hematomas became smaller. But the FVIII activity of this patient did not reach the desired level, remaining below 20%, and FXII gradually decreased (Fig. 1). We believed that the plasma factor peak level response was inadequate and that the duration of administration needed to be longer. However, despite the continued risk of bleeding, the family members stopped treatment and refused further prophylactic inhibitors and genetic testing due to financial and other reasons.
Fig. 1.

After treatment with human coagulation factor VIII for 5 days, FVIII activity was not significantly increased and FXII gradually decreased
Prophylaxis prevents bleeding and joint destruction and must be initiated 2–3 times per week [11]. Unfortunately, 3 months after all of the treatments were discontinued, he developed convulsions and brain CT scanning revealed intracranial hemorrhage (Fig. 2).
Fig. 2.
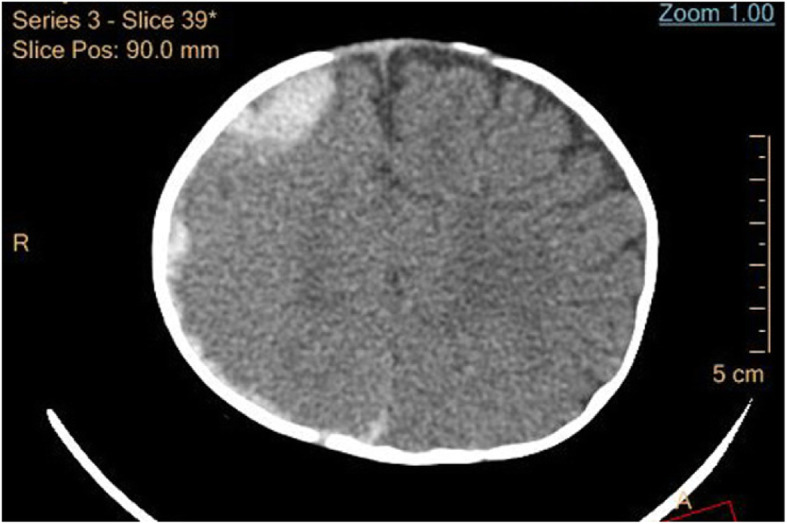
Three months after initial presentation, brain CT scanning showed intracranial hemorrhage
Discussion and conclusions
Reports of neonatal patients with low FVIII:C and FXII:C are relatively rare. In this case, the neonate’s FVIII activity did not reach the desired level with treatment, and his FXII activity gradually decreased. This case presents some of the challenges of treating patients with hemophilia A. Our patient had severe hemophilia A with FXII deficiency and may have had chromosome and gene mutations. To address these challenges, this paper reviews some new knowledge about congenital hemophilia A and FXII. Currently, three important aspects, namely, the presence of FVIII inhibitors, drug concentration and genetic testing, must be considered when poor treatment response occurs in hemophilia A.
The newborn had low FVIII activity. However, after treatment with human coagulation factor VIII, the activity of FVIII did not increase significantly, as inhibition of FVIII may have occurred. The presence of inhibitors is a common adverse effect of human coagulation factor VIII therapy in patients with hemophilia [13, 14]. Generally, 30 % of patients with severe hemophilia A may develop inhibitors during the first 20 days of exposure to recombinant coagulation factor VIII [15]. The presence of inhibitors may be an immune response to foreign proteins in patients with severe hemophilia A [16]. The inhibitor development depends on the proper activation of antigen-presenting cells (APCs) that encounter FVIII in the periphery, which is a T-cell-dependent process [9]. The total risk of developing inhibitors over a lifetime is 25–40% for severe hemophilia and 5–15% for moderate/mild hemophilia A [16]. However, inhibitors can be produced without previous treatment with human coagulation factor VIII or with only a small amount of blood components [17]. Besides, high doses of recombinant FVIII therapy and surgery may increase the risk of inhibitor development in patients with non-severe hemophilia A [18]. In general, the emergence of inhibitors increases the risk of progressive and disabling joints disease. The sensitive inhibitor screening or the Nijmegen modification method of Bethesda should be used for screening [14, 19, 20].
Despite the potential for the production of inhibitors, the benefits of recombinant coagulation factor VIII still seem to outweigh the risks [21], and better treatment with recombinant FVIII product is possible. Studies have shown that patients treated with factor VIII containing von Willebrand factor (VWF) have a lower incidence of inhibitor production than patients treated with recombinant FVIII product [17]. This may be because the von Willebrand factor obscures the inhibitor epitope in the concentrate, resulting in a longer half-life of the product [14]. Another study suggested that when patients with hemophilia A had inhibitors, clinicians could initiate an immune tolerance induction (ITI) protocol to reduce levels of the inhibitor [14, 22]. A randomized trial showed that ITI eliminated anti-FVIII alloantibodies in about two-thirds of patients [23].
Elimination of inhibitors is important because some asymptomatic patients remain at risk of severe bleeding or life-threatening conditions until the inhibitors are eliminated. Prednisolone has been reported to achieve a complete immunosuppressive response (CR) in some patients [14]. Recombinant FVIII concentrates that produce fewer inhibitors are under study, and a new treatment option for hemophilia patients with inhibitors is the bispecific monoclonal antibody emicizumab [5].
Research findings have shown that plasma-derived or full-length recombinant FVIII have a half-life of between 6 and 25 h [24]. A regimen of lower doses of prophylaxis given more frequently may be an effective option to decrease the frequency of bleeding, joint disease and intracranial hemorrhage. Recombinant FVIII, which effectively prevent spontaneous bleeding, must be injected intravenously three times a week or every other day to maintain FVIII levels ≥1% in patients with severe hemophilia A [25, 26]. Weight-adjusted clearance (CL) of FVIII is related to age and weight. From infancy to adulthood, CL decreases with age and/or weight, and the terminal half-life increases accordingly [27]. Extended plasma half-life FVIII products can produce higher FVIII plasma levels and reduce the number of intravenous injections, which increase the possibility of a more active lifestyle [28]. Currently, half-life extension technology for Fc-fusion proteins or modification with polyethylene glycol (PEG) can prolong the plasma half-life of FVIII, as with efmoroctocog alfa and BAX 855 [26, 29]. Coagulation FVIII produced by Fc fusion technology has few adverse effects because the components of the fusion peptide are plasma proteins, causing fewer allergic reactions [25].
Accurate testing of FVIII is critical for guiding clinical treatment. The test results for coagulation factors are greatly affected by laboratory test methods, and the reason for variation between laboratories is not the bias of instrument calibration, but the differences in reagents, instruments used and test design. The following sampling protocol is recommended for more effective detection of inhibitors: FVIII samples were taken at pre-dose, 15 min, 30 min and at 3, 6, 9, 24, 28 and 32 h post-dose administration to obtain more test information [14]. Results of experiments have shown that test designs for three samples produce more stable results than designs that test only one or two of the diluents. It is recommended that at least three different sample diluents be used in each FVIII:C (FVIII activity) assay with a commutable lyophilised FVIII:C calibrator, which results in a limited reduction of the inter-laboratory variation [30]. However, there are no comparable data to reliably predict an individual patient’s FVIII:C level to guide clinical treatment. Current studies have shown that adults and adolescents need less FVIII/kg than young children to maintain serum drug concentrations. Personalized drug delivery is therefore more suitable in clinical practice [24].
Activated factor XII (Hageman factor, FXII) can trigger the internal coagulation pathway [31], which is measured by APTT [32]. Hageman factor deficiency is usually an autosomal recessive disorder but can be autosomal dominant. Matsushita et al. reported a female patient with hemophilia with an FXII deficiency who had an extremely inactivated normal X chromosome [33]. The exact prevalence of Hageman factor deficiency is not known because patients are normally asymptomatic. Hageman factor deficiency is usually detected by chance in coagulation assay results that isolated prolonged APTT or unexplained coagulation disease [32, 34].
FXII plays an important role in the coagulation system. FXII respectively drives the contact system to initiate coagulation and inflammation through the intrinsic coagulation pathway and the bradykinin-producing kallikrein-kinin system [35]. Humans and animals with low FXII activity have a normal hemostatic ability, but animal models show that FXII is involved in the thrombotic process [36]. FXII was associated with thromboembolic complications, but it was only rarely associated with severe hemorrhagic disease [32].
FXII activity is generally lower in Asians. Deficiency of FXII is an autosomal recessive disorder, but few cases have been reported. Alleles in homozygotes or complex heterozygotes are associated with very low FXII activity (< 1%) compared to unaffected individuals [34]. The autosomal recessive genetic diseases can be prevented by avoiding intermarriage, which requires counselling and education [32]. The average FXII activity depends on the race of the person. One study showed that 95% of healthy Chinese subjects had FXII activity between 47 and 160.25%, and identified some mutations associated with low FXII activity [34].
In addition, results of the coagulation assays are highly age-dependent and must be used to ensure the correct evaluation of coagulation assays in children, especially in the first year of life [37]. For newborns, coagulation factors are already low and gradually increase to adult levels after 6 months [38–40]. On the whole, the low FXII:C in hemophilia A is related to race and age, which is determined by chromosomal and genetic testing.
Unfortunately, the newborn described in this report had a family history of hemophilia A, and the child’s mother did not receive a prenatal genetic diagnosis, nor did she agree to have the child tested for inhibitors and genetic mutations as soon as possible after birth. In one study, Chen et al. developed a noninvasive prenatal diagnosis (NIPD) method for Hemophilia A by sequencing a small target region [41]. A genetic diagnosis can help couples at risk of hemophilia reduce their anxiety about childbirth. The obstetrician must discuss the birth plan with the expectant mother [5]. Hemophilia A is diagnosed by informational gene tracking and/or measurement of fetal FVIII: C level [42]. Determination of a woman’s genetic and phenotypic status before pregnancy is optimal so that she can understand her options and the requirements for a safe delivery [43–45].
In conclusion, this paper presents the case of a newborn with severe neonatal congenital hemophilia A with FXII deficiency. This case highlights the importance of FVIII inhibitors, serum recombinant FVIII concentration and testing in hemophilia A. Prophylaxis treatment is an effective option for decreasing the frequency of bleeding and improving quality of life. We suggest that expectant mothers identified as congenital hemophilia A gene carriers test their fetuses for the FVIII or FXII activity levels and that newborns undergo genetic testing as soon as possible after birth to assess for the risk of the disease.
Acknowledgements
Not applicable.
Abbreviations
- FVIII
Factor VIII
- ICH
Intracranial hemorrhage
- aPCC
Activated Prothrombin complex concentrate
- rFVIIa
Recombinant activating factor VII
- FVIII:C
FVIII activity
- FXII
Factor XII
- FXII:C
FXII activity
- VWF
Von Willebrand factor
- CR
Complete remission
- CL
Weight-adjusted clearance
- PEG
Polyethylene glycol
- APTT
Activated partial thromboplastin time
- PT
Prothrombin time
- FXIIa
Free factor XIIa
- NIPD
Noninvasive prenatal diagnosis
Authors’ contributions
(I) Conception and design: Haiyan Feng; (II) Provision of study materials: All of the authors; (III) Manuscript writing: All of the authors; (IV) All of the authors read and approved the final manuscript.
Funding
This work was supported by the High-level Hospital Construction Research Project of Maoming People’s Hospital.
Availability of data and materials
All of the data presented in this article can be found in our hospital.
Declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The neonate’s guardians consented to publication of this case.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Baoyu Lei and Chuang Liang contributed equally to this work.*Correspondence should be addressed to Haiyan Feng (haiyanfeng90@163.com).
References
- 1.Mannucci PM, Tuddenham EG. The hemophilias--from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773–1779. doi: 10.1056/NEJM200106073442307. [DOI] [PubMed] [Google Scholar]
- 2.Simpson ML, Desai V, Maro GS, Yan S. Comparing Factor Use and Bleed Rates in U.S. Hemophilia A Patients Receiving Prophylaxis with 3 Different Long-Acting Recombinant Factor VIII Products. J Manag Care Spec Pharm. 2020;26:504–12. [DOI] [PMC free article] [PubMed]
- 3.Clausen N, Petrini P, Claeyssens-Donadel S, Gouw SC, Liesner R, PedNet et al. Similar bleeding phenotype in young children with haemophilia a or B: a cohort study. Haemophilia. 2014;20(6):747–755. doi: 10.1111/hae.12470. [DOI] [PubMed] [Google Scholar]
- 4.VandenDriessche T, Chuah MK. Hemophilia gene therapy: ready for prime time? Hum Gene Ther. 2017;28(11):1013–1023. doi: 10.1089/hum.2017.116. [DOI] [PubMed] [Google Scholar]
- 5.Kurnik K, Bidlingmaier C, Olivieri M. How do I counsel parents of a newly diagnosed boy with Haemophilia a? Hamostaseologie. 2020;40(1):88–96. doi: 10.1055/s-0039-3402805. [DOI] [PubMed] [Google Scholar]
- 6.Frampton JE. Efmoroctocog alfa: a review in Haemophilia a. Drugs. 2016;76(13):1281–1291. doi: 10.1007/s40265-016-0622-z. [DOI] [PubMed] [Google Scholar]
- 7.Nagae C, Yamashita A, Ashikaga T, Mori M, Akita M, Kitsukawa K, Yamazaki S, Yoshikawa K, Yamamoto H, Taki M. A cohort study of the usefulness of primary prophylaxis in patients with severe haemophilia a. Int J Hematol. 2016;104(2):208–215. doi: 10.1007/s12185-016-2005-3. [DOI] [PubMed] [Google Scholar]
- 8.Blanchette VS, Key NS, Ljung LR, Manco-Johnson MJ, van den Berg HM, Srivastava A, the Subcommittee on Factor VIII, Factor IX and Rare Coagulation Disorders Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12(11):1935–1939. doi: 10.1111/jth.12672. [DOI] [PubMed] [Google Scholar]
- 9.Lovgren KM, Sondergaard H, Skov S, Wiinberg B. Non-genetic risk factors in haemophilia a inhibitor management - the danger theory and the use of animal models. Haemophilia. 2016;22(5):657–666. doi: 10.1111/hae.13075. [DOI] [PubMed] [Google Scholar]
- 10.van Velzen AS, Eckhardt CL, Streefkerk N, Peters M, Hart DP, Hamulyak K, et al. The incidence and treatment of bleeding episodes in non-severe haemophilia a patients with inhibitors. Thromb Haemost. 2016;115(03):543–550. doi: 10.1160/th15-03-0212. [DOI] [PubMed] [Google Scholar]
- 11.Srivastava A, Brewer AK, Mauser-Bunschoten EP, Key NS, Kitchen S, Llinas A, Ludlam CA, Mahlangu JN, Mulder K, Poon MC, Street A, Treatment Guidelines Working Group The World Federation Of Hemophilia Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–47. doi: 10.1111/j.1365-2516.2012.02909.x. [DOI] [PubMed] [Google Scholar]
- 12.Thrombosis, Hemostasis Group CSoH Chinese Medical Association/Hemophilia Treatment Center Collaborative Network of C. [Chinese guidelines on the treatment of hemophilia (version 2020)] Zhonghua Xue Ye Xue Za Zhi. 2020;41:265–271. doi: 10.3760/cma.j.issn.0253-2727.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Messori A, Peyvandi F, Mengato D, Mannucci PM. Incidence of low-titre factor VIII inhibitors in patients with haemophilia a: meta-analysis of observational studies. Haemophilia. 2017;23(2):e87–e92. doi: 10.1111/hae.13193. [DOI] [PubMed] [Google Scholar]
- 14.Hay CRM, Brown S, Collins PW, Keeling DM, Liesner R. The diagnosis and management of factor VIII and IX inhibitors: a guideline from the United Kingdom Haemophilia Centre doctors organisation. Br J Haematol. 2006;133(6):591–605. doi: 10.1111/j.1365-2141.2006.06087.x. [DOI] [PubMed] [Google Scholar]
- 15.Klamroth R. A new era of treatment for patients with haemophilia a? Hamostaseologie. 2017;37(3):216–218. doi: 10.5482/HAMO-16-07-0028. [DOI] [PubMed] [Google Scholar]
- 16.Tieu P, Chan A, Matino D. Molecular mechanisms of inhibitor development in hemophilia. Mediterr J Hematol Infect Dis. 2020;12(1):e2020001. doi: 10.4084/mjhid.2020.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peyvandi F, Mannucci PM, Garagiola I, El-Beshlawy A, Elalfy M, Ramanan V, et al. A randomized trial of factor VIII and neutralizing antibodies in hemophilia A. N Engl J Med. 2016;374(21):2054–2064. doi: 10.1056/NEJMoa1516437. [DOI] [PubMed] [Google Scholar]
- 18.van Velzen AS, Eckhardt CL, Peters M, Leebeek FWG, Escuriola-Ettingshausen C, Hermans C, et al. Intensity of factor VIII treatment and the development of inhibitors in non-severe hemophilia A patients: results of the INSIGHT case-control study. J Thromb Haemost. 2017;15(7):1422–1429. doi: 10.1111/jth.13711. [DOI] [PubMed] [Google Scholar]
- 19.White GC, 2nd, Rosendaal F, Aledort LM, Lusher JM, Rothschild C, Ingerslev J, et al. Definitions in hemophilia. Recommendation of the scientific subcommittee on factor VIII and factor IX of the scientific and standardization committee of the international society on thrombosis and Haemostasis. Thromb Haemost. 2001;85:560. doi: 10.1055/s-0037-1615621. [DOI] [PubMed] [Google Scholar]
- 20.Pinto P, Shetty S, Lacroix-Desmazes S, Bayry J, Kaveri S, Ghosh K. Antibody profile in Indian severe haemophilia a patients with and without FVIII inhibitors. Immunol Lett. 2016;169:93–97. doi: 10.1016/j.imlet.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Lusher JM, Arkin S, Abildgaard CF, Schwartz RS. Recombinant factor VIII for the treatment of previously untreated patients with hemophilia A. safety, efficacy, and development of inhibitors. Kogenate previously untreated patient study group. N Engl J Med. 1993;328(7):453–459. doi: 10.1056/NEJM199302183280701. [DOI] [PubMed] [Google Scholar]
- 22.DiMichele DM, Hoots WK, Pipe SW, Rivard GE, Santagostino E. International workshop on immune tolerance induction: consensus recommendations. Haemophilia. 2007;13(Suppl 1):1–22. doi: 10.1111/j.1365-2516.2007.01497.x. [DOI] [PubMed] [Google Scholar]
- 23.Hay CRM, DiMichele DM. International Immune Tolerance S. The principal results of the International Immune Tolerance Study: a randomized dose comparison. Blood. 2012;119(6):1335–1344. doi: 10.1182/blood-2011-08-369132. [DOI] [PubMed] [Google Scholar]
- 24.Collins PW, Fischer K, Morfini M, Blanchette VS, Bjorkman S, International prophylaxis study group pharmacokinetics expert working G Implications of coagulation factor VIII and IX pharmacokinetics in the prophylactic treatment of haemophilia. Haemophilia. 2011;17(1):2–10. doi: 10.1111/j.1365-2516.2010.02370.x. [DOI] [PubMed] [Google Scholar]
- 25.Mancuso ME, Mannucci PM. Fc-fusion technology and recombinant FVIII and FIX in the management of the hemophilias. Drug Des Devel Ther. 2014;8:365–71. [DOI] [PMC free article] [PubMed]
- 26.Tiede A. Half-life extended factor VIII for the treatment of hemophilia A. J Thromb Haemost. 2015;13:S176–SS79. doi: 10.1111/jth.12929. [DOI] [PubMed] [Google Scholar]
- 27.Bjorkman S, Blanchette VS, Fischer K, Oh M, Spotts G, Schroth P, et al. Comparative pharmacokinetics of plasma- and albumin-free recombinant factor VIII in children and adults: the influence of blood sampling schedule on observed age-related differences and implications for dose tailoring. J Thromb Haemost. 2010;8(4):730–736. doi: 10.1111/j.1538-7836.2010.03757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mannucci PM. Benefits and limitations of extended plasma half-life factor VIII products in hemophilia A. Expert Opin Investig Drugs. 2020;29(3):1–7. doi: 10.1080/13543784.2020.1723547. [DOI] [PubMed] [Google Scholar]
- 29.Mahlangu J, Young G, Hermans C, Blanchette V, Berntorp E, Santagostino E. Defining extended half-life rFVIII-A critical review of the evidence. Haemophilia. 2018;24(3):348–358. doi: 10.1111/hae.13438. [DOI] [PubMed] [Google Scholar]
- 30.van den Besselaar AM, Haas FJ, Kuypers AW. Harmonisation of factor VIII:C assay results: study within the framework of the Dutch project 'Calibration 2000′. Br J Haematol. 2006;132(1):75–79. doi: 10.1111/j.1365-2141.2005.05829.x. [DOI] [PubMed] [Google Scholar]
- 31.Vorlova S, Koch M, Manthey HD, Cochain C, Busch M, Chaudhari SM, Stegner D, Yepes M, Lorenz K, Nolte MW, Nieswandt B, Zernecke A. Coagulation factor XII induces pro-inflammatory cytokine responses in macrophages and promotes atherosclerosis in mice. Thromb Haemost. 2017;117(1):176–187. doi: 10.1160/TH16-06-0466. [DOI] [PubMed] [Google Scholar]
- 32.Chaudhry LA, El-Sadek WYM, Chaudhry GA, Al-Atawi FE. Factor XII (Hageman factor) deficiency: a rare harbinger of life threatening complications. Pan Afr Med J. 2019;33:39. doi: 10.11604/pamj.2019.33.39.18117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsushita T, Takamatsu J, Kagami K, Takahashi I, Sugiura I, Hamaguchi M, Kamiya T, Saito H. A female hemophilia a combined with hereditary coagulation factor XII deficiency: a case report. Am J Hematol. 1992;39(2):137–141. doi: 10.1002/ajh.2830390212. [DOI] [PubMed] [Google Scholar]
- 34.Han Y, Zhu T, Jiao L, Hua B, Cai H, Zhao Y. Normal range and genetic analysis of coagulation factor XII in the general Chinese population. Thromb Res. 2015;136(2):440–444. doi: 10.1016/j.thromres.2015.06.012. [DOI] [PubMed] [Google Scholar]
- 35.Maas C, Renne T. Coagulation factor XII in thrombosis and inflammation. Blood. 2018;131(17):1903–1909. doi: 10.1182/blood-2017-04-569111. [DOI] [PubMed] [Google Scholar]
- 36.Nickel KF, Long AT, Fuchs TA, Butler LM, Renne T. Factor XII as a therapeutic target in thromboembolic and inflammatory diseases. Arterioscler Thromb Vasc Biol. 2017;37(1):13–20. doi: 10.1161/ATVBAHA.116.308595. [DOI] [PubMed] [Google Scholar]
- 37.Liu J, Dai Y, Yuan E, Li Y, Wang Q, Wang L, Su Y. Paediatric reference intervals for common coagulation assays in Chinese children as performed on the STA-R coagulation analyzer. Int J Lab Hematol. 2019;41(5):697–701. doi: 10.1111/ijlh.13098. [DOI] [PubMed] [Google Scholar]
- 38.Andrew M, Paes B, Milner R, Johnston M, Mitchell L, Tollefsen DM, Powers P. Development of the human coagulation system in the full-term infant. Blood. 1987;70(1):165–172. doi: 10.1182/blood.V70.1.165.165. [DOI] [PubMed] [Google Scholar]
- 39.Andrew M, Paes B, Milner R, Johnston M, Mitchell L, Tollefsen DM, Castle V, Powers P. Development of the human coagulation system in the healthy premature infant. Blood. 1988;72(5):1651–1657. doi: 10.1182/blood.V72.5.1651.1651. [DOI] [PubMed] [Google Scholar]
- 40.Toulon P, Berruyer M, Brionne-François M, Grand F, Lasne D, Telion C, Arcizet J, Giacomello R, de Pooter N. Age dependency for coagulation parameters in paediatric populations. Results of a multicentre study aimed at defining the age-specific reference ranges. Thromb Haemost. 2016;116(1):9–16. doi: 10.1160/TH15-12-0964. [DOI] [PubMed] [Google Scholar]
- 41.Chen C, Sun J, Yang Y, Jiang L, Guo F, Zhu Y, et al. Noninvasive prenatal diagnosis of hemophilia A by a haplotype-based approach using cell-free fetal DNA. Biotechniques. 2020. 10.2144/btn-019-0113. [DOI] [PubMed]
- 42.Sasanakul W, Chuansumrit A, Ajjimakorn S, Krasaesub S, Sirachainan N, Chotsupakarn S, Kadegasem P, Rurgkhum S. Cost-effectiveness in establishing hemophilia carrier detection and prenatal diagnosis services in a developing country with limited health resources. Southeast Asian J Trop Med Public Health. 2003;34(4):891–898. [PubMed] [Google Scholar]
- 43.Street AM, Ljung R, Lavery SA. Management of carriers and babies with haemophilia. Haemophilia. 2008;14(Suppl 3):181–187. doi: 10.1111/j.1365-2516.2008.01721.x. [DOI] [PubMed] [Google Scholar]
- 44.Lavery S. Preimplantation genetic diagnosis of haemophilia. Br J Haematol. 2009;144(3):303–307. doi: 10.1111/j.1365-2141.2008.07391.x. [DOI] [PubMed] [Google Scholar]
- 45.Tizzano EF, Barcelo MJ, Baena M, Cornet M, Vencesla A, Mateo J, et al. Rapid identification of female haemophilia a carriers with deletions in the factor VIII gene by quantitative real-time PCR analysis. Thromb Haemost. 2005;94(3):661–664. doi: 10.1160/TH05-03-0144. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All of the data presented in this article can be found in our hospital.