

Abstract
It has become increasingly clear that the terms used to define memory T cell subsets no longer accurately reflect our understanding of memory T cell biology. Here, we discuss the limitations of our current terminology and propose a new approach for defining memory T cell subsets.
Memory T Cell Subsets: A Complex History
Memory T cells provide protective immunity against pathogens and also drive numerous inflammatory diseases. The development of more effective vaccination strategies as well as targeted therapies for inflammatory diseases will require delineating the mechanisms regulating the development, maintenance, and function of memory T cells. Over the last 30 years, there has been tremendous progress in defining the unique biology of memory T cells compared with naïve T cells in vivo. Landmark experiments on T cell trafficking in sheep revealed that memory T cells exhibit distinct trafficking patterns compared to naïve T cells [1]. Specifically, all T cells isolated from afferent lymphatics were found to exhibit a memory phenotype, demonstrating that memory T cells possess the ability to traffic from blood to nonlymphoid tissue (NLT) during homeostasis, allowing for broader immunosurveillance of the host [1]. Subsequent influential experiments revealed that memory T cells were not homogeneous, but rather, composed of phenotypically and functionally distinct subsets [2]. Blood-derived memory CD4+ and CD8+ T cells from humans could be separated into two subsets based on the expression of the chemokine receptor CCR7. Furthermore, re-stimulation of these two subsets ex vivo resulted in different cytokine expression profiles with CCR7+ cells preferentially producing interleukin (IL)-2, and CCR7− cells preferentially producing effector cytokines, such as IFN-γ or IL-4 and IL-5. Finally, CCR7− cells expressed receptors known to promote homing to NLTs while CCR7+ cells expressed CD62L (l-selectin). Given these observations of phenotypic and functional heterogeneity, CCR7− cells were named effector memory T (Tem) cells and CCR7+ cells as central memory T (Tcm) cells [2]. A model was proposed in which these two memory T cell subsets performed distinct roles during immunosurveillance in vivo; namely, that Tem cells circulated between the blood and NLT, poised to rapidly respond to recurrent antigen encounter. In contrast, Tcm cells preferentially localized within secondary lymphoid organs (SLOs), undergoing rapid proliferation and producing secondary effector T cells that would subsequently traffic into inflamed NLTs to augment the initial response provided by Tem cells [2]. The Tem–Tcm model inspired and dominated the memory T cell field for many years, in part, because it merged multiple different aspects of T cell biology, including phenotype, function, and trafficking into one elegant schema. However, the study defining Tem and Tcm cells was performed on blood-derived memory T cells without experimental evidence to support the proposed model in vivo.
As additional insights into memory T cell trafficking were made, multiple observations were inconsistent with the Tem–Tcm model. For instance, the concept that Tem cells are the predominant memory T cell subset surveying NLTs was called into question on two major fronts. First, the phenotype and biology of memory T cells isolated from various NLTs seemed distinct from Tem cells isolated from blood and spleen [3]. Second, two studies in mice, including one from our group, demonstrated that CCR7 expression on CD4+ and CD8+ T cells is required for efficient T cell egress from NLTs [4,5]. Consequently, the concept that CCR7− Tem cells trafficked through NLT and were the first-line responders to antigen re-exposure was incompatible with these experimental findings. The first discrepancy was clarified by the discovery that a distinct population of memory T cells establishes long-term residence in NLT [3]. These tissue-resident memory T (Trm) cells are a transcriptionally and functionally distinct population of memory T cells that serve as frontline responders. While the addition of Trm cells to the model of memory T cell biology has been an important advancement in our understanding, whether a distinct memory T cell subset could provide more global immunosurveillance by trafficking through NLT remained an open question. Our group subsequently showed that there is a unique population of memory CD4+ T cells in mice that are capable of trafficking from the skin to the draining lymph node in a CCR7-dependent manner [6]. We termed these cells recirculating memory T (Trcm) cells and demonstrated that they exhibit a distinct phenotype from other memory CD4+ T cell populations with intermediate expression of the lymph-node-homing molecules CD62L and CCR7 [6]. In support of these findings, others demonstrated that the chemokine receptor CX3CR1 defines three distinct circulating memory CD8+ T cell subsets in mice [7]. Tcm cells were found to be CX3CR1− while Tem cells were CX3CR1hi. In addition, there was a CX3CR1int population that represented ~15% of blood memory CD8+ T cells. CX3CR1int cells were the predominant circulating (nonresident) memory CD8+ T cells within NLT during homeostasis. The authors termed this CX3CR1int population as peripheral memory T (Tpm) cells [7]. Of note, CX3CR1hi Tem cells were not found in LNs or NLT, suggesting that Tem cells remain intravascular and might function to provide intravascular immunity. Notably, few memory CD4+ T cells in mice express CX3CR1, preventing this marker from being used to identify distinct subsets of circulating memory CD4+ T cells. Studies of human memory T cells within NLT have revealed heterogeneity within nonresident, CCR7-expressing cells, leading to various additional descriptive terms, such as tissue migratory memory T (TMC) cells and migratory memory T (TMM) cells [8,9]. Consequently, while we have gained insight into the phenotype and biology of memory T cells surveying NLT in different contexts, there is a lack of consensus in defining these populations.
In addition to our modifications of NLT-surveying memory T cells, recent studies have also challenged our understanding of Tcm cell function in vivo. Two independent groups using viral infection models in mice demonstrated that CD8+ Tcm cells are capable of rapidly trafficking to inflamed NLT, challenging the idea that Tcm cells exclusively respond within SLOs [10,11]. In one study, Tcm cells showed superior trafficking to NLT than Tem cells did, due in part, to enhanced synthesis of core 2 O-glycans, which yield functional ligands for E and P selectins [10]. These observations have enhanced our understanding of memory T cell biology, but they have rendered the current nomenclature of memory T cell subsets unhelpful and confusing. While defining a memory T cell subset is challenging, it is clear that memory T cells exhibit heterogeneous biology and applying terminology to describe this heterogeneity is helpful. For instance, while the presence of memory T cells within NLT has been known for many years, the recognition that the majority of these T cells are durably parked in NLT – and appropriately termed Trm cells – has added clarity and allowed for thoughtful investigation into the biology of tissue-resident memory. A universal approach is needed to describe memory T cell subsets in humans and mice, which allows for granular descriptions, but also exhibits flexibility in incorporating new findings.
Redefining Memory T Cell Subsets
We can consider multiple different approaches to define memory T cell subsets, including phenotype, transcriptional/epigenetic signatures, function, and trafficking patterns. While arguments can be made for different approaches, we suggest separating these different features into distinct axes for clarity. Specifically, given the benefits of defining memory T cell subsets based on trafficking patterns, we argue that trafficking should be a separate, defining axis. We propose maintaining the term Trm cells to describe memory T cells in dis-equilibrium with the circulation, while simplifying the terms used for nonresident memory T cells. We suggest describing all nonresident memory T cells as circulating memory T cells and using the designation Tcm as the counterpart to Trm. An alternative approach would be to define nonresident memory T cells as recirculating memory T cells with the designation Trcm. While the former proposal may lead to initial confusion with central memory T cells, we feel Trcm is a more cumbersome abbreviation and could also lead to confusion with ‘r’ standing for resident. Thus, redefining Tcm to mean circulating memory T cells is a simpler approach (Figure 1). Along with trafficking, additional axes that include phenotype, transcriptional/epigenetic signatures, and function can be superimposed on the Trm–Tcm subset system. As certain features/axes gain robustness as the best modifiers, these might become more universally adopted. While we believe the Trm–Tcm delineation is helpful, it is important to acknowledge that it is not absolute. Specifically, there is growing evidence that CD8+ and CD4+ Trm cells within NLT can egress via afferent lymphatics and become circulating memory T cells, while retaining phenotypic and transcriptional features of Trm cells, as well as exhibiting a predilection to traffic back to the NLT of origin (Figure 1, see dotted arrows) [12,13]. Nevertheless, we posit that having trafficking as a separate, defining axis, resolves the inaccuracies of our current nomenclature system. Furthermore, our proposed system allows for the integration of new concepts, such as the recent findings on transcriptionally distinct subsets of CD8+ Trm cells [14,15]. While our approach has limitations, and other modifications or alternative proposals may yield a better framework, we hope this Forum will spark ongoing discussions and further debates to develop a new standardized and optimal system for defining memory T cell subsets.
Figure 1. Distinct Feature Axes for Redefining Memory T Cell Subsets.
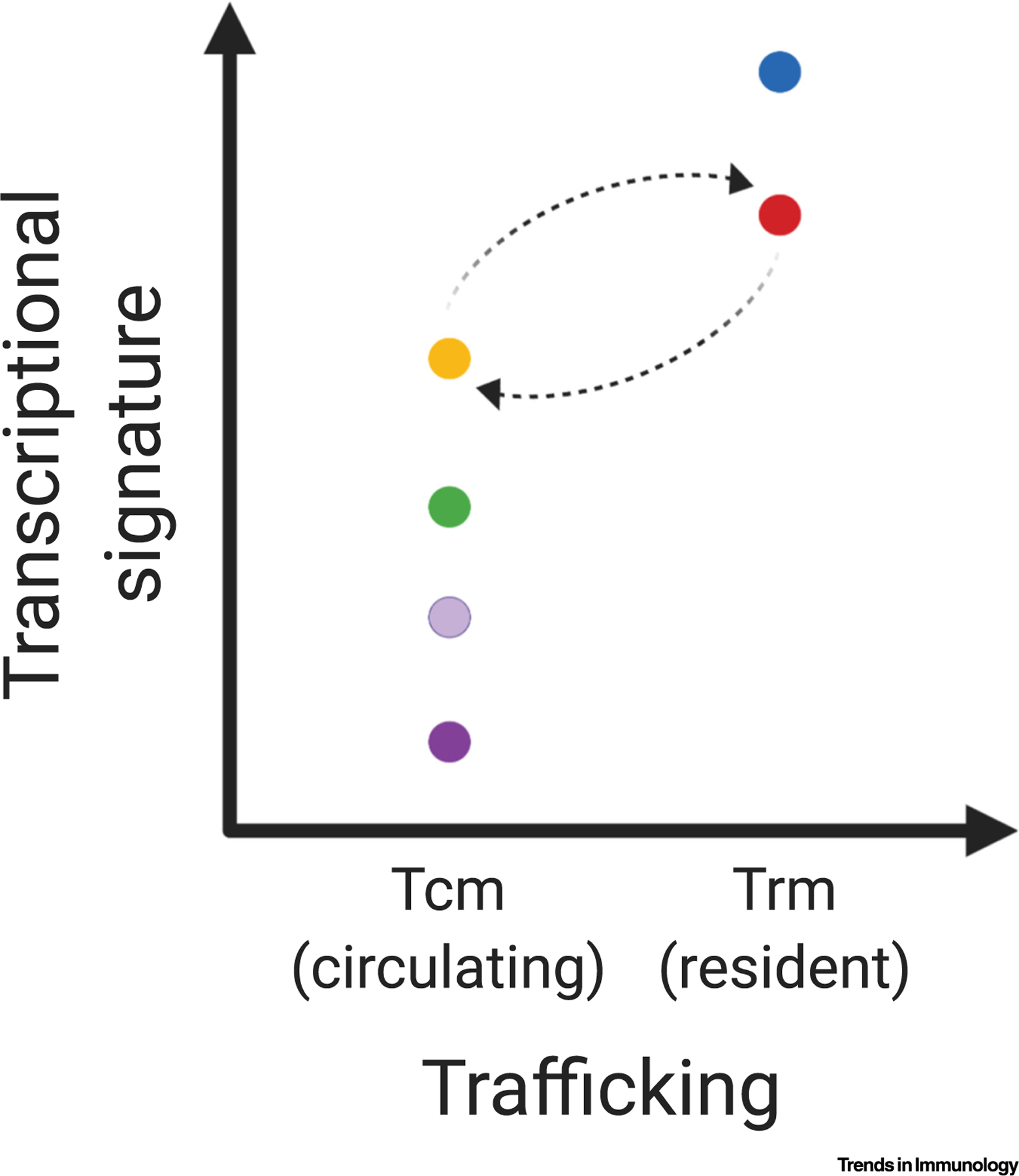
Graphical representation of the proposed model for redefining memory T cell subsets. A binary x axis indicates trafficking patterns, including circulating memory T (Tcm) cells and resident memory T (Trm) cells. Additional features are represented on distinct axes. For example, a continuous y axis can be used for distinct transcriptional signatures. Dotted arrows represent the ability of Trm cells to egress via lymphatics to join the Tcm pool in the circulation, as well as Tcm cells to subsequently re-establish residency in certain contexts. This figure was created using BioRender (https://biorender.com/).
Acknowledgments
This work was supported by grants from the National Institutes of Health R01 AI040618, R01 CA204028, the Training Program in Pulmonary Immunology and Allergy T32 HL116275 (A.D.L.), and Mentored Clinical Scientist Research Career Development Award K08 HL140173 (R.A.R).
References
- 1.Mackay CR et al. (1990) Naive and memory T cells show distinct pathways of lymphocyte recirculation. J. Exp. Med 171, 801–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sallusto F et al. (1999) Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 [DOI] [PubMed] [Google Scholar]
- 3.Schenkel JM and Masopust D (2014) Tissue-resident memory T cells. Immunity 41, 886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bromley SK et al. (2005) Chemokine receptor CCR7 guides T cell exit from peripheral tissues and entry into afferent lymphatics. Nat. Immunol 6, 895–901 [DOI] [PubMed] [Google Scholar]
- 5.Debes GF et al. (2005) Chemokine receptor CCR7 required for T lymphocyte exit from peripheral tissues. Nat. Immunol 6, 889–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bromley SK et al. (2013) Recirculating memory T cells are a unique subset of CD4+ T cells with a distinct phenotype and migratory pattern. J. Immunol 190, 970–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gerlach C et al. (2016) The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity 45, 1270–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe R et al. (2015) Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med 7, 279ra39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vieira Braga FA et al. (2019) A cellular census of human lungs identifies novel cell states in health and in asthma. Nat. Med 25, 1153–1163 [DOI] [PubMed] [Google Scholar]
- 10.Osborn JF et al. (2017) Enzymatic synthesis of core 2 O-glycans governs the tissue-trafficking potential of memory CD8+ T cells. Sci. Immunol 2, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steinert EM et al. (2015) Quantifying memory CD8 T cells reveals regionalization of immunosurveillance. Cell 161, 737–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klicznik MM et al. (2019) Human CD4+CD103+ cutaneous resident memory T cells are found in the circulation of healthy individuals. Sci. Immunol 4, eaav8995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fonseca R et al. (2020) Developmental plasticity allows outside-in immune responses by resident memory T cells. Nat. Immunol 21, 412–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Milner JJ et al. (2020) Heterogenous populations of tissue-resident CD8+ T cells are generated in response to infection and malignancy. Immunity 52, 808–824 e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurd NS et al. (2020) Early precursors and molecular determinants of tissue- resident memory CD8+ T lymphocytes revealed by single-cell RNA sequencing. Sci. Immunol 3, 16–19 [DOI] [PMC free article] [PubMed] [Google Scholar]