

Abstract
Development of new chemical entities is costly, time-consuming, and has a low success rate. Accurate prediction of pharmacokinetic properties is critical to progress compounds with favorable drug-like characteristics in lead optimization. Of particular importance is the prediction of hepatic clearance, which determines drug exposure and contributes to projection of dose, half-life, and bioavailability. The most commonly employed methodology to predict hepatic clearance is termed in vitro to in vivo extrapolation (IVIVE) that involves measuring drug metabolism in vitro, scaling-up this in vitro intrinsic clearance to a prediction of in vivo intrinsic clearance by reconciling the enzymatic content between the incubation and an average human liver, and applying a model of hepatic disposition to account for limitations of protein binding and blood flow to predict in vivo clearance. This manuscript reviews common in vitro techniques used to predict hepatic clearance as well as current challenges and recent theoretical advancements in IVIVE.
Graphical Abstract
1. INTRODUCTION
Accurate prediction of human pharmacokinetic properties of new chemical entities (NCEs) is essential in the drug discovery process. Due to the time-consuming and costly nature of developing a drug,1 and because very few can be examined directly in humans, it is of interest early on in the drug discovery process to exclude compounds that may display unfavorable pharmacokinetic or ADME (absorption, distribution, metabolism, excretion) properties. Of particular importance is the prediction of human hepatic clearance, which largely determines the exposure of drug in the body, influencing both the efficacy and safety of an NCE. Hepatic clearance also contributes to projection of dose, half-life, and bioavailability and greatly aids in prioritization of compounds with desired drug-like properties for in vivo studies, such as decreased systemic clearance, adequate oral bioavailability, and half-life to permit once-a-day oral dosing. To predict the in vivo hepatic clearance of NCEs, in vitro metabolic stability studies are routinely performed, and if resulting data can be accurately extrapolated, significant benefit can be gained in the development of a new candidate drug. Thus, drug metabolism is considered the leading issue to address in lead optimization efforts and often finds itself as a tier 1 screen for newly synthesized compounds. Here, we discuss common in vitro techniques used to predict hepatic clearance of NCEs as well as review recent advancements and current challenges in the in vitro to in vivo extrapolation (IVIVE) of hepatic clearance.
2. IN VITRO TO IN VIVO EXTRAPOLATION
The universally accepted and most utilized method of predicting in vivo hepatic clearance (CLH) from in vitro measures of drug metabolism is a process known as IVIVE (Figure 1). The three steps of IVIVE are (1) experimentally measuring an in vitro intrinsic clearance (CLint), (2) calculating an in vivo CLint, and (3) applying a model of hepatic disposition to predict CLH. In the first step, drug metabolism measurements are conducted in human liver tissue, isolated cells (hepatocytes), or subcellular fractions such as microsomes, and what is being measured in vitro is the CLint, or the intrinsic ability of the liver to remove drug in the absence of the limitations of organ blood flow and protein binding. To achieve this, the rate of unbound drug elimination is measured (kinc,u), and with consideration of the volume of the incubation (Vinc), an in vitro CLint can be determined.2 Step two involves reconciling enzymatic or cell content differences between the in vitro incubation and the in vivo average human liver, resulting in a prediction of in vivo CLint. Finally, CLH is predicted by applying a model of hepatic disposition, such as the well-stirred model,3,4 which accounts for liver blood flow, the free fraction of drug in the blood, as well as the predicted in vivo CLint.
Figure 1.
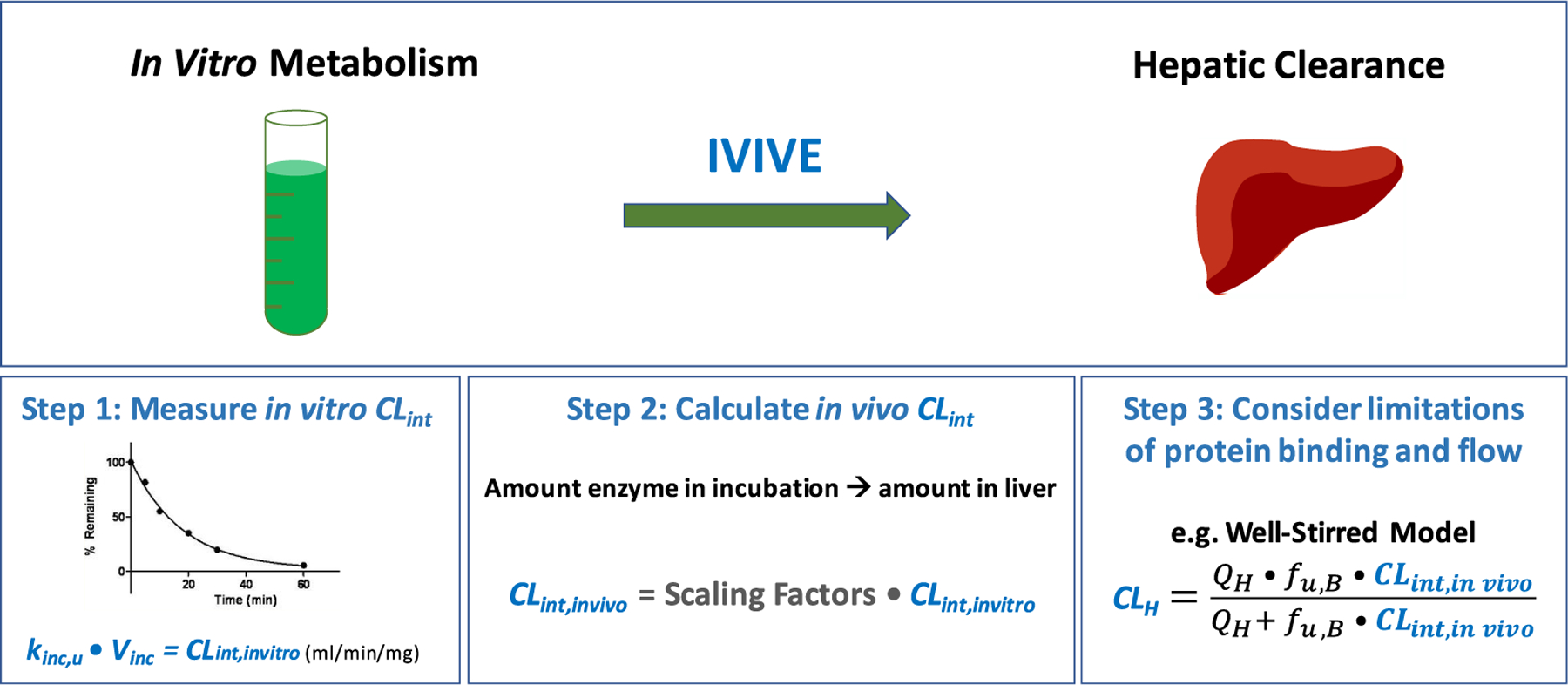
In vitro to in vivo extrapolation (IVIVE). Abbreviations: CLH, hepatic clearance; CLint, intrinsic clearance; CLint,invitro, in vitro intrinsic clearance; CLint,invivo, in vivo intrinsic clearance; fu,B, fraction unbound in blood; IVIVE, in vitro to in vivo extrapolation; kinc,u, unbound rate of incubational drug loss; QH, hepatic blood flow; Vinc, volume of in vitro incubation.
2.1. Experimental Tools to Study Drug Metabolism.
There are a number of different model systems that can be used to study drug metabolism, as outlined in Figure 2. An in vivo pharmacokinetic study (for example, in a preclinical species) most resembles the true in vivo scenario, and the complexity of experimental systems decreases as alternate techniques are utilized, such as isolated perfused liver studies, stability assays (in liver slices, hepatocytes, cell fractions, or recombinant enzymes), or an in silico prediction of drug metabolism. However, tremendous benefit can be gained by utilizing less-complex systems, such as decreased cost, increased utility, increased throughput, and using more ethically acceptable methodologies. Table 1 further details useful information for the in vitro systems (i.e., liver slices, hepatocytes, microsomes, cytosol, and recombinant enzymes) with respect to which enzymes are contained in each system, cofactors required, the presence of transporters, storage and throughput considerations, advantages, disadvantages, as well as the range of ADME assays that could be performed with each system.
Figure 2.
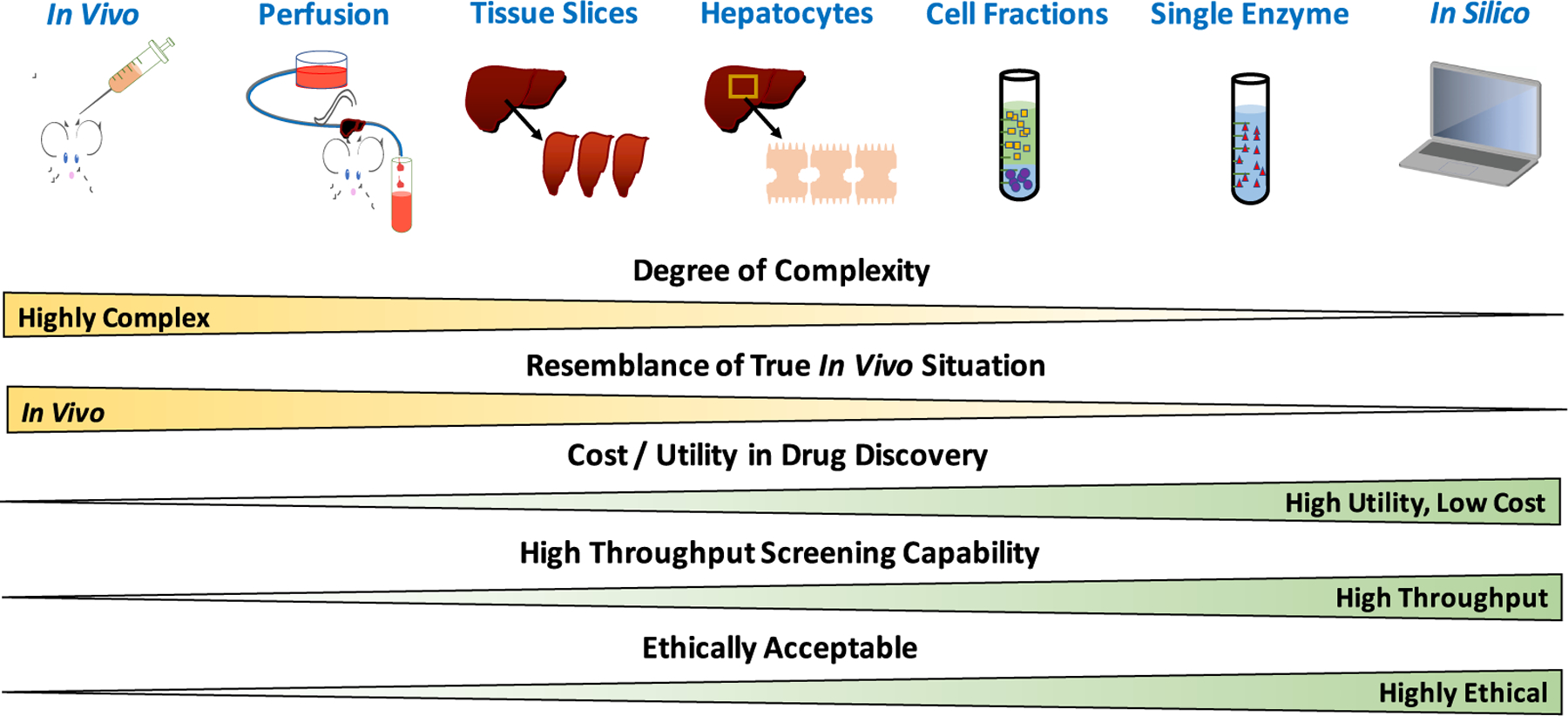
Experimental methodologies employed to study drug metabolism.
Table 1.
Overview of In Vitro Tools Integral in Lead Optimization Effortsa
| liver slices | hepatocytes | microsomes | cytosol | recombinant | |
|---|---|---|---|---|---|
| enzymes | all enzymes | all enzymes | CYPs | NAT | singularly expressed CYP isoform |
| FMO | GST | ||||
| UGT | SULT | ||||
| membrane proteins | AO soluble proteins | ||||
| cofactors | none | none | NADPH (CYP/FMO) | PAPS (SULT) | NADPH (CYP) |
| UDPGA (UGT) | GSH (GST) acetyl-CoA (NAT) | ||||
| transporters | all transporters | all transporters | none | none | none |
| storage | fresh | fresh/cryopreserved | −80 °C freezer | −80 °C freezer | −80 °C freezer |
| advantages | tissue heterogeneity | transporters/enzymes (+ phase II) | robust, low cost | non-P450 metabolism | specific metabolic pathway |
| drug distribution | pooled (5–100 donors) | withstands freeze/thaw pooled (150 donors) | pooled (150 donors) | easy to purchase | |
| disadvantages | difficult to prepare | cell viability | cofactors needed | cofactors needed | difficult to prepare |
| difficult to scale-up | difficult to scale-up | ||||
| throughput | very low | moderate | high | high | high |
| ADME assays | seldom used | metabolic stability | metabolic stability | metabolic stability | reaction phenotyping |
| toxicity | CL prediction | CL prediction | metabolite ID | metabolism of stable compounds | |
| metabolic profiling | metabolite ID | metabolite ID | |||
| drug interaction | drug interaction | reaction phenotyping | |||
| reaction phenotyping sandwich culture | reaction phenotyping |
Abbreviations: AO, aldehyde oxidase; CL, clearance; CYPs, cytochrome P450s; FMO, flavin-containing monooxygenase; GSH, glutathione; GST, glutathione S-transferase; NADPH, nicotinamide adenine dinucleotide phosphate; NAT, N-acetyl transferase; PAPS, 3′-phosphoadenosine-5′-phosphosulfate; SULT, sulfotransferase; UDPGA, uridine diphosphate glucuronic acid; UGT; UDP-glucuronosyltransferase.
The most commonly used experimental tools in lead optimization efforts are hepatocyte and microsomal stability assays, and Figure 3 outlines how each are isolated from liver tissue. Hepatocytes (liver cells) can be isolated from an intact liver via a two-step collagenase digestion of liver tissue.5–8 At that point, hepatocytes can be immediately utilized in a suspension assay or plated as primary cell cultures. Often metabolic stability assays are conducted using hepatocyte suspensions, whereas enzyme induction studies require culturing of plated hepatocytes. Alternatively, the isolated hepatocytes can be cryopreserved7,8 for future use in ADME or toxicity assays. Microsomes are prepared by homogenization of the liver and a process of consecutive centrifugation steps commonly termed “differential centrifugation”.9,10 First, the liver homogenate is centrifuged at a low speed (9000g) to separate the pellet (cell debris) from the supernatant (commonly referred to as the S9 fraction), which contains all the soluble and membrane-bound hepatic proteins. A high-speed centrifugation step (100 000g) can then be performed using the S9 fraction to isolate the supernatant, which contains the soluble cytosolic proteins, from the pellet, which contains the endoplasmic reticulum membrane-bound proteins such as the cytochrome P450s (CYPs). Thus, microsomes are artificial vesicles of hepatic endoplasmic reticulum that contain the CYP enzymes that form as a result of this differential centrifugation process. Microsomes can be stored at −80 °C long-term and withstand multiple freeze–thaw cycles while still retaining enzymatic activity11 and can be thawed at the convenience of the investigator for use in a range of ADME assays (Table 1).
Figure 3.
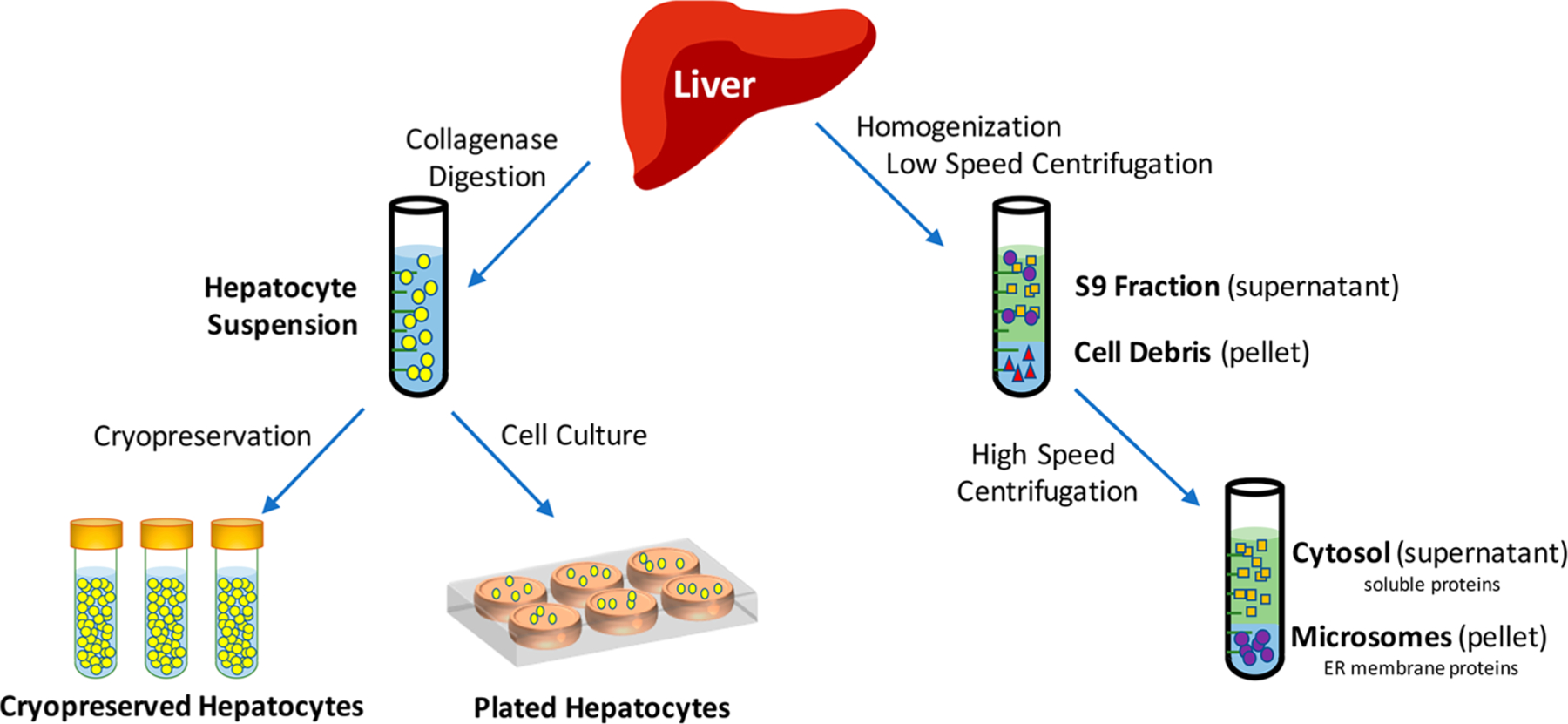
Isolation of hepatocytes and microsomes from hepatic tissue.
Based on the robustness, ease-of-use, and low cost, microsomal incubations are most often utilized as tier 1 screens in lead optimization efforts. Microsomes contain the membrane-bound phase I enzymes such as the CYPs and flavin-containing monooxygenase (FMO), both of which are primarily oxidative and require the addition of the cofactor NADPH (nicotinamide adenine dinucleotide phosphate).12 Microsomes also contain the membrane-bound UDP-glucuronosyltransferases (UGTs), which catalyze the phase II conjugation of glucuronic acid to xenobiotics, typically at hydroxyl, carboxyl, carbonyl, or amino functional groups.13 The reaction requires addition of the cofactor uridine diphosphate glucuronic acid (UDPGA) as well as a pore-forming agent such as alamethicin, as the catalytic active site of UGTs is located within the lumen of the endoplasmic reticulum.14 It has also been reported by a number of investigators that cytosolic contamination of microsomal fractions can occur as a result of the preparation process, resulting in appreciable nonmicrosomal metabolism by enzymes such as aldehyde oxidase.15–17 In fact, many investigators choose to perform tier 1 stability assays using the S9 fraction (supplemented with NADPH) to capture both the microsomal Phase I CYP-mediated metabolism as well as contributions of Phase I and II cytosolic enzymes such as esterases, aldehyde oxidase, xanthine oxidase, glutathione S-transferase, and sulfotransferase.18
Microsomes are versatile, as they can be used for a range of ADME assays such as metabolic profiling or reaction phenotyping,19,20 drug–drug interaction studies,21,22 estimation of drug metabolism and clearance predictions,2,23,24 and detection of reactive metabolites.25 Due to the ease-of-use of microsomes, many of these ADME assays are amenable to high-throughput formats, allowing for weekly screens of hundreds of compounds.21–23 Pooled microsomes are readily available with up to 150 donors to overcome issues related to interindividual differences in activity or expression of metabolic enzymes. Additionally, microsomes can be prepared from any organ (i.e., liver, intestine, lung, kidney, heart, etc.) to allow for evaluation of potential extrahepatic metabolism.26,27 However, there are some key assumptions when utilizing human liver microsomes for IVIVE of CLH, most notably that CYPs are primarily responsible for clearance with no involvement of xenobiotic transporters. It is also assumed that the liver is the major site of metabolism with minor contribution of extrahepatic metabolism, and that the in vitro compound concentration is representative of the in vivo concentrations (i.e., that enzyme saturation is not occurring).
Hepatocytes are considered the gold standard in lead optimization efforts, as they are intact liver cells containing all the major Phase I and II enzymes with cofactors at physiologically relevant concentrations. In addition, membrane transport mechanisms (including xenobiotic transporters) and intracellular compartments are maintained; thus, it is possible to measure the permeability, transport, and metabolism of a test compound in a hepatocyte stability incubation. Hepatocytes can also be specially cultured between two layers of gelled collagen to capture hepatobiliary elimination.28,29 Thus, hepatocytes are a versatile in vitro system that can be utilized to conduct a range of ADME and toxicity assays, including clearance prediction,30,31 transporter-mediated uptake,31,32 drug–drug interaction potential,33 enzyme and xenobiotic transporter induction,34–36 biliary clearance or toxicity,28,29,37 and hepatotoxicity.38–40 Cryopreserved hepatocytes are readily available from multiple vendors and can be procured as pooled lots of up to 100 donors. In comparison to microsomes, hepatocytes are more expensive and typically considered moderate-throughput as additional care is needed in the thawing of hepatocytes and throughout the assay to ensure adequate cell viability. This results in a more labor-intensive assay that poses additional challenges to successfully automate and often finds itself as a tier 2 screen in many lead optimization paradigms.
2.2. IVIVE Step 1: Measure In Vitro Intrinsic Clearance.
With firm understanding of the experimental tools available to study drug metabolism, we now highlight each individual step of IVIVE in further detail to cover the theory, process, and limitations of this approach. The first step of IVIVE involves measurement of in vitro CLint, typically in hepatocyte or microsomal incubations. A chemical reaction can be considered analogous to drug elimination when metabolism is the major route of elimination, however, intrinsic clearance is not a parameter typically used in chemistry. The rate of a chemical reaction (v) is typically characterized by the Michaelis–Menten relationship:
| (1) |
where [S] indicates the drug concentration, Vmax is the maximum rate of decrease in drug concentration (units of concentration/time), and Km is the drug concentration corresponding to half of Vmax (units of concentration), which results in a reaction rate or velocity in the units of inverse time. To determine these kinetic parameters, a wide range of substrate concentrations is required (to capture both above and below the Km), metabolite formation should be determined under linear conditions with respect to time and protein concentration (thus requiring a metabolite standard for analytical quantification), and one must ensure less than 20% of parent drug depletion has occurred in each incubation.41 Figure 4A displays a typical plot of Michaelis–Menten kinetics, visually highlighting the labor-intensive nature of such determinations.
Figure 4.
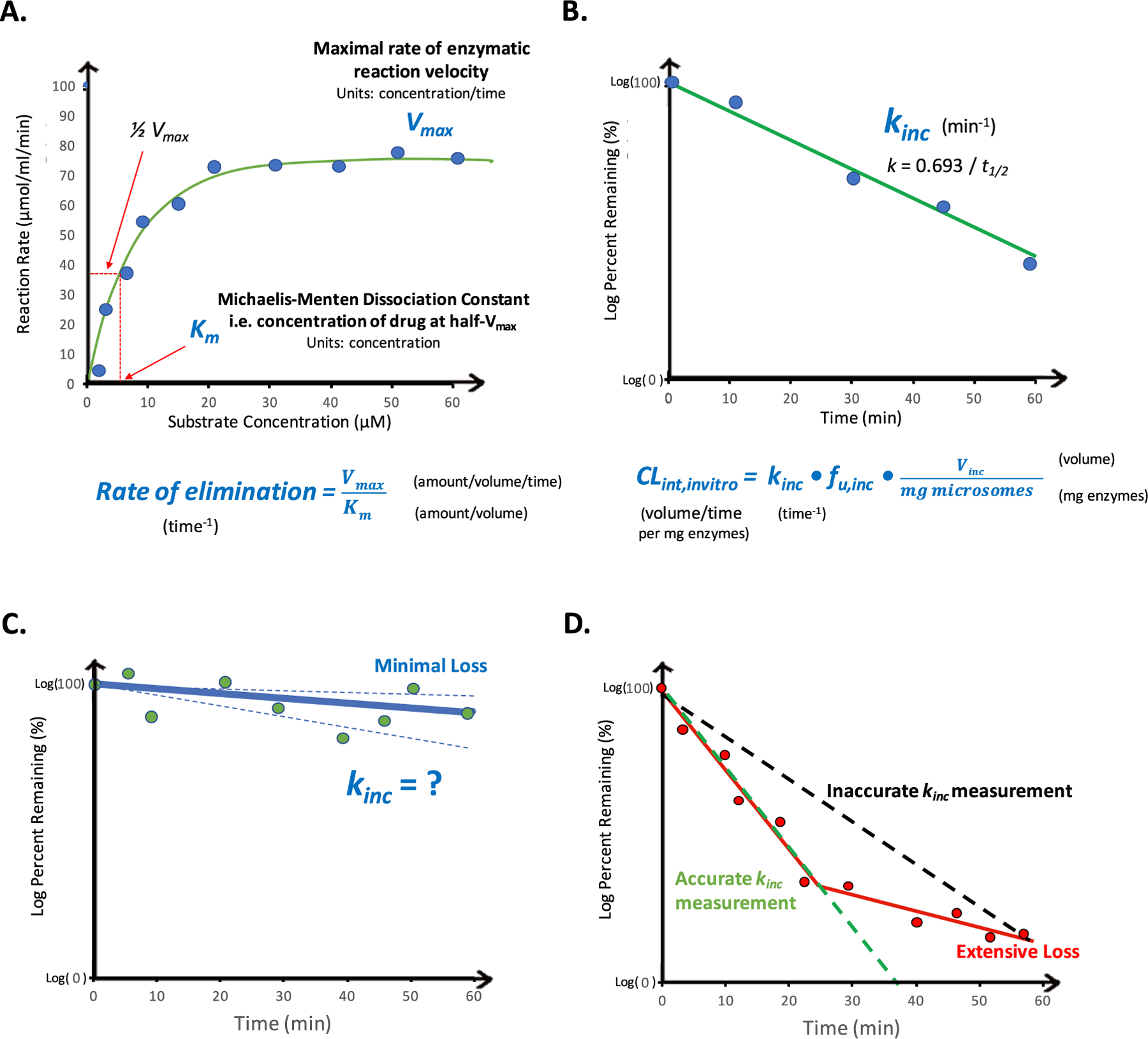
In vitro determinations of intrinsic clearance and high throughput assay considerations. Abbreviations: CLint,invitro, in vitro intrinsic clearance; fu,inc, fraction unbound in the incubation; kinc, rate of incubational drug loss, Km, Michaelis–Menten dissociation constant; Vinc, volume of in vitro incubation; Vmax, maximal rate of enzymatic reaction velocity.
Under conditions where drug concentrations are much less than the Km value (i.e., [S] ≪ Km), the Michaelis–Menten relationship can be simplified and the rate of drug loss (kinc) can be determined under such linear conditions.2 Thus, this “substrate-depletion” or “in vitro half-life” approach is most often utilized in lead optimization efforts (Figure 4B). Depletion of drug is measured at low concentrations, typically 0.1 or 1 μM, under the assumption that this is much less than the Km value. Measurement of rate of depletion is conducted using the log–linear portion of the curve, resulting in a rate constant with units of inverse time.
To calculate an intrinsic clearance (with units volume/time) from the resulting measured rate of elimination (units of inverse time) from either of the above-discussed methodologies, it is necessary to introduce a volume term into the relationship. This is achieved by multiplying the unbound rate constant of elimination by the in vitro incubational volume (Vinc), which occurs when normalizing for enzymatic content:2,42,43
| (2) |
where fu,inc is the fraction unbound in the incubation. Although this aspect was acknowledged in the original publication by Obach et al. that introduced the “in vitro half-life” approach,2 the implications of utilizing a fixed-volume in clearance predictions has not been widely recognized by the field, and this aspect will be discussed in further detail in a subsequent section.
The substrate-depletion approach is commonly utilized in high-throughput screens and some experimental considerations related to the measurement of very low turnover or very high turnover compounds poses additional challenges. Low clearance compounds are becoming increasingly common in drug discovery efforts, due to effective design strategies to overcome metabolic liability in combination with increased assay throughput that can facilitate the rapid establishment of data-rich structure–stability relationships.44,45 For very low turnover compounds, the loss of compound in the assay should be sufficient to confidently measure kinc to reliably distinguish substrate depletion from bioanalytical or assay variability (Figure 4C). Inadequate attempted solutions include prolonging the incubation time, as significant decreases in enzymatic activity can occur,46 or increasing the incubational enzymatic content, as issues related to incubational binding can be introduced47 because fu,inc is rarely measured for NCEs in tier 1 assays. It should be noted that multiple groups have attempted to elucidate and assess predictive relationships between physicochemical properties and fu,inc for both microsomal and hepatocyte incubations,48–54 and we emphasize the importance of considering incubational binding in all CLint determinations. Instead, “very low” clearance cutoffs should be employed rather than reporting clearance predictions from such results. For example, it has been recommended that when less than 10–20% turnover occurs by the end of an incubation, a clearance cutoff should be reported rather than an exact value.46,55,56 This issue can potentially be overcome by alternatively measuring metabolite formation. However, in the lead optimization stage, there is a lack of authentic metabolite standards, and it is not practical in most workflows to anticipate and detect major metabolites for newly synthesized molecules. Of late, a number of in vitro systems and methodologies have been developed to more accurately measure low turnover compounds, which will be discussed in a subsequent section together with other notable attempts to improve IVIVE by the field. Extensive loss of drug in the in vitro assays also poses challenges, as only the log–linear portion of the curve should be utilized for kinc measurement. As depicted in Figure 4D, inclusion of all time points may result in an underprediction of rate of drug loss, thereby potentially resulting in underprediction of in vivo clearance. This aspect is often overlooked in high-throughput screens and is of particular concern in determining rate of drug loss by utilizing assays that only sample a single end-of-incubation time point plus the initial time zero.
2.3. IVIVE Step 2: Calculate In Vivo Intrinsic Clearance.
The second step of IVIVE involves estimating in vivo CLint from measurements of in vitro CLint. This is achieved by reconciling the enzymatic or cell content difference between the incubation and an average whole liver with use of physiologically-based scaling factors:
| (3) |
These scaling factors first consider microsomal protein or hepatocellularity per gram of liver and then account for liver weight per kg of body weight. Typically used values for human microsomal protein content range from 32 to 48.8 mg microsomal protein per gram of liver57–60 and values for human hepatocellularity range from 99 to 139 million hepatocytes per gram liver.57,59,61 The typically utilized value of human liver weight per kg body weight ranges from 21.4 to 25.7 g liver/kg body weight.62,63 Thus, an in vitro CLint can be scaled up to a prediction of in vivo CLint.
2.4. IVIVE Step 3: Apply a Hepatic Disposition Model to Predict Hepatic Clearance.
To predict total hepatic clearance, the physiologic limitations of hepatic blood flow (QH) and fraction of unbound drug in the blood (fu,B) must be considered by utilizing a hepatic disposition model. To describe hepatic drug elimination without being able to measure intraorgan drug concentrations, pharmacokineticists based clearance concepts on chemical engineering reactor models for which only entering and exiting reactant amounts are known but no measurements within the reactor are possible.64 Common assumptions of liver models are that (1) only unbound drug can cross membranes and occupy enzyme active sites, (2) no diffusional barriers exist (i.e., passive membrane passage is much larger than metabolic CLint), and (3) hepatic enzymes are homogeneously distributed throughout the liver. Thus, in vitro CLint measures, in vitro fu,B determinations, and physiologic values of QH (20.7 mL/min/kg62) can be utilized to predict clearance using a hepatic disposition model.
Figure 5 depicts the most common hepatic disposition models utilized for clearance predictions, including the well-stirred model, the parallel tube model, and the dispersion model, as well as the mathematical relationships that relate entering drug concentration (Cin), exiting drug concentration (Cout), and QH to total clearance CLH for each model. The simplest model is the well-stirred model:
| (4) |
The well-stirred model assumes that drug is homogeneously distributed throughout the liver (Figure 5A). Although this well-stirred representation of the liver is far from capturing the complex physiologic aspects of the liver, the simple well-stirred relationship depicted in eq 4 is very useful.
Figure 5.
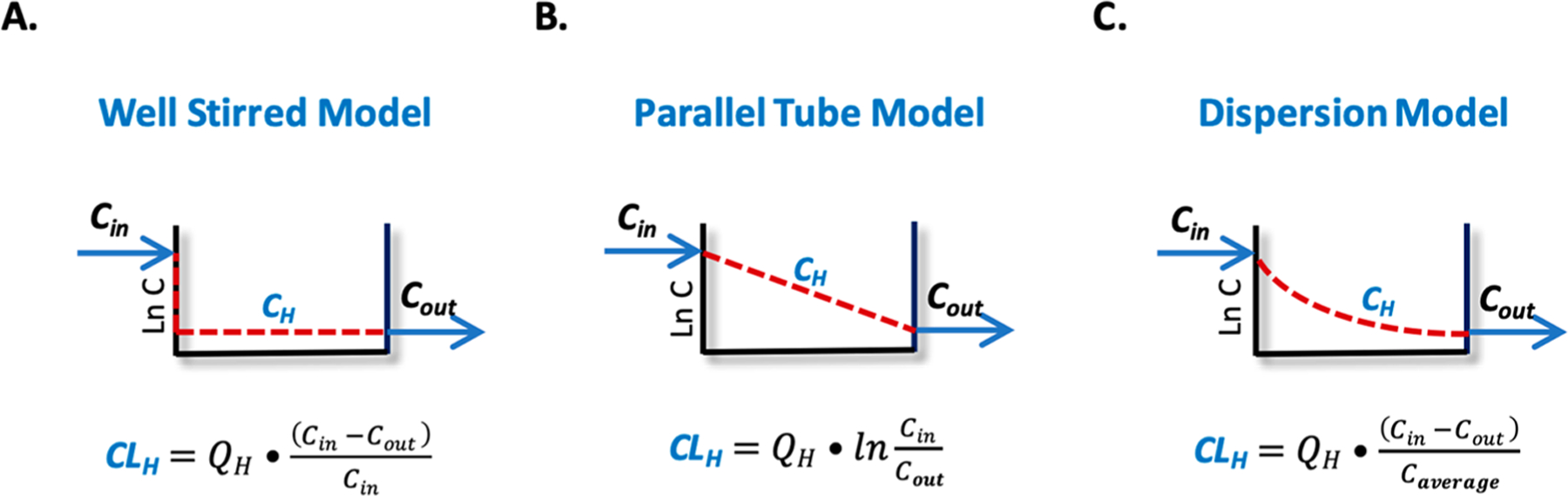
Hepatic disposition models. Abbreviations: CH, hepatic drug concentration; Cin, entering drug concentration; CLH, hepatic clearance; Cout, exiting drug concentration; QH, hepatic blood flow.
The parallel tube model assumes incremental metabolism where drug concentrations decrease by a first order process throughout the liver. The well-stirred model and the parallel tube model are the two boundary conditions, and there are an infinite number of dispersion models between these two boundary models that are characterized by different dispersion numbers (DN) that can range from zero (parallel tube mode) to infinity (well-stirred model). A representative dispersion model is depicted in Figure 5C. From examination of each model in Figure 5, one can see that based on the same Cin and Cout the concentration profile of drug in each model differs significantly, resulting in different hepatic drug exposures (area under the curve) and different average driving force concentrations (CH) responsible for hepatic drug elimination between the models.
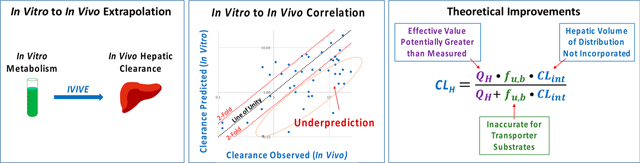
3. IVIVE UNDERPREDICTS IN VIVO HEPATIC CLEARANCE
Although measuring drug metabolism in microsomes or hepatocytes is widely used throughout the industry to predict hepatic clearance, in vitro measures of drug metabolism significantly and systematically underpredict in vivo hepatic clearance.65–67 It had been reported in 2009 by Chiba et al. that the underprediction of CLH is approximately 3- to 6-fold in human hepatocytes and approximately 9-fold in human microsomes.65 More recently, Wood et al.66 reported that the human hepatocyte underprediction of CLint was 4.2-fold and human microsomes was 2.8-fold, with similar findings in rat hepatocytes (4.7-fold) and rat microsomes (2.3-fold). Bowman and Benet67 evaluated 11 IVIVE data sets, showing that human hepatocytes underpredicted 1.4- to 21.7-fold and human microsomes underpredicted 1.5- to 7.9-fold, although these reported underpredictions are sometimes associated with CLH and are sometimes associated with CLint depending on the comparisons being made in the original publications. More recently, we have pointed out that it is more appropriate to evaluate IVIVE success with respect to total CLH rather than CLint because potential errors in CLint for high extraction ratio (ER) compounds may not translate to significant error when CLH is calculated.42 Further, back-calculating an in vivo CLint from total CLH measurements requires the assumption that the in vivo CLH measurement, the experimentally determined fu,B measurement, and value of QH are accurate, and thus any resulting errors in IVIVE are primarily attributed to issues with determining CLint. To date, these assumptions have been considered reasonable by the field; however, we have recently pointed out potential errors in these assumptions.42 To clarify, we are not implying that accurate determination of CLint is unimportant for high ER drugs, as in vivo CLint determines unbound drug exposure for hepatically cleared drugs regardless of ER,68 we are simply highlighting the additional potential errors that are associated with each parameter that determines total observed CLH.
The greatest challenge with IVIVE underprediction is that the degree of underprediction can vary greatly from drug-to-drug, and the field does not yet understand why. Attempts to explain this issue by the field have been unsuccessful to date. Explanations of lack of IVIVE have most commonly been attributed to (1) extrinsic factors such as the loss of enzymatic activity due to suboptimal storage or preparation of human liver tissues or due to the presence of metabolic inhibitors present during the isolation process, (2) the inability of in vitro incubations to recapitulate hepatic architecture, (3) nonspecific or protein binding that is not fully accounted for in clearance prediction calculations, (4) a neglected contribution of extrahepatic clearance or other clearance mechanisms, or (5) the potential differences between the donors of liver tissue and the young healthy volunteers in which clinical clearance determinations are conducted.65,69 A number of groups have attempted to simply mitigate the unexplainable underprediction issue by employing a regression-based “fudge” factor to their data,69–72 and such approaches are common in lead optimization as a practical approach to predict clearance (or rank-order compounds by CLint) despite the unpredictability of IVIVE. Such approaches are commonly referred to as IVIVC, or in vitro to in vivo correlation. For instance in a simplified example, if it is observed that in vitro data underpredicts in vivo clearance by 2- to 6-fold for a series of compounds, investigators may choose to apply a 4-fold scaling factor to other compounds in this series to get in vitro predictions into the ballpark of in vivo values. However, this is a temporary solution that does not address the underlying reasons for underprediction, demonstrating the clear need for a mechanistic understanding of the reasons for underprediction of hepatic clearance.
Throughout the field, many groups both academic and within industry have attempted to understand, explain and mitigate IVIVE underpredictions spanning more than two decades. Many notable efforts to improve IVIVE predictability have addressed issues with nonspecific or protein binding,24,47,70,73–76 considered differences in drug ionization in extracellular and intracellular liver regions,77–79 conducted hepatocyte uptake experiments for hepatic or renal transporter substrates,31,32,80 developed experimental methodologies to account for biliary clearance,28,29 introduced the Extended Clearance Model that integrates metabolism with membrane passage intrinsic clearances such as hepatic uptake, biliary excretion, and sinusoidal efflux,81 incorporated the fraction unbound in the liver or liver-to-plasma partition coefficient of unbound drug (Kpuu) for transporter substrates,82–85 incorporated intestinal absorption, first-pass elimination and other extrahepatic metabolic contributions,26,27,86 developed experimental methodologies such as the relay method to extend hepatocyte incubations to 20+ hours and coculture techniques with additional cell types to prolong hepatocyte function in long-term cultures to more accurately measure very low turnover compounds,87–89 and have investigated the potential for albumin-facilitated uptake by adding serum into hepatocyte incubations.90–92 We emphasize that each one of these aspects are crucial to consider when attempting to accurately predict in vivo clearance, and that the referenced studies are merely representative examples of the extensive efforts by the field to improve IVIVE success in drug discovery.
More recently, our laboratory has thoroughly evaluated the current state of IVIVE and confirmed the interlab variability93 and clearance-dependent underprediction trends94 observed by the field.65,66 We have also found that poor IVIVE for metabolized drugs is not due to transporter involvement.67 With respect to observations of albumin-facilitated uptake, we have proposed that the albumin effect is due to a protein binding shift due to increased affinity for transporters.95 Further, we have identified a CYP3A4 underprediction anomaly where microsomes result in markedly higher CLint values and IVIVE success than hepatocytes, but the same trend was not observed for other CYP isoforms,96 suggesting the potential for enzyme-transporter interplay with the efflux transporter P-glycoprotein present in hepatocyte membranes that may prevent drug access to CYP3A4 as a result of their overlapping substrate specificities,97 and these results have been further confirmed with a larger number of substrates more recently by the field.98
4. ADVANCING IVIVE THEORY
As outlined above, for decades IVIVE underprediction has challenged pharmaceutical scientists both in industry and academia, with extensive efforts directed primarily toward improving experimental outcomes for certain categories of “problem” or hard-to-predict drugs. Although countless mechanistic studies have been conducted representing tremendous efforts across the field, there have only been incremental advances in IVIVE success, and there is still no consensus on the reasons behind the shortcomings of IVIVE for all drugs. Efforts have not yet been able to identify the types of compounds for which IVIVE can be trusted to quantitatively predict clearance. Therefore, in lead optimization IVIVE is regarded as a practical approach to rank-order NCEs based on metabolic stability, but uncertainties remain surrounding the quantitative utilization of IVIVE-based clearance predictions, for instance, in human dose projections. It is surprising that IVIVE is not successful for those drugs that are exclusively eliminated by metabolism (with no involvement of xenobiotic transporters). Why is it that measures of in vitro drug metabolism measured in actual liver tissue cannot provide adequate predictions of in vivo hepatic elimination? We hypothesize that perhaps the theoretical basis of current IVIVE practices is flawed, and thus significant efforts of our laboratory have been toward advancing IVIVE theory.
4.1. Implicit Well-Stirred Model Assumptions.
We have taken a measured approach to critically evaluate both in vitro and in vivo assumptions in basic clearance concepts to elucidate the potential reasons for IVIVE underprediction. First, we have recognized by derivation that the extended clearance model,99 Kpuu,100 and organ ER101 have all inherently assumed the well-stirred model. Thus, when xenobiotic transporters are involved in drug disposition, there is no advantage in the utilization of alternate models of hepatic disposition (that are considered more physiologic).99 This is particularly relevant for physiologically based pharmacokinetic (PBPK) modeling approaches, as the dispersion model appears to be universally utilized to model hepatic clearance throughout the literature. Further, the basis of Kpuu on the well-stirred model indicates that nuances of intracellular drug distribution are not considered.100 Therefore, using Kpuu to improve clearance predictions cannot capture the differences in average drug concentration driving metabolic elimination from the concentrations at the basolateral or apical hepatocyte membranes that drive efflux and biliary elimination, respectively, and thus may provide limited benefits. The recognition that clearance calculations based on ER have inherently assumed the well-stirred model101 indicates that all clearance calculations are model-dependent when drug concentrations entering and exiting an organ at steady-state are utilized. We have further critically analyzed all such published experimental data that use ER to calculate clearance in isolated perfused rat liver studies, concluding that all in situ and in vitro data can be described by the well-stirred model.102
4.2. The Lower Boundary of IVIVE.
We have also derived IVIVE from first-principles,42 noting that the lower boundary condition for IVIVE predictions to have the potential to be valid is
| (5) |
That is, for all drugs regardless of their ER, the product of fu,B and CLint will always be larger than observed CLH, holds for all models of hepatic disposition, and this relationship is the prerequisite for IVIVE predictions to be accurate. Evaluation of a large IVIVE database66 and notable IVIVE studies24,84 revealed that approximately two-thirds of the available published IVIVE data violate the lower boundary condition of the predictive relationship. Until recently, the field has primarily attributed that error to the underprediction of CLint; however, there are a number of assumptions that must also be accurate related to measurements of CLH and determinations of fu,B for that assessment to be true.
Many investigators believe that the reason in vitro rates often fail to predict in vivo rates can be due to a variety of assay-centric reasons, such as the ability of enzymes to perform once isolated, the limited architecture of the microsomes and hepatocyte environment, or issues during isolation including the presence of agents that may be inhibitors of metabolic enzymes. This could be true; however, our analysis of the published data suggests that this is not the reason for the observed poor predictability. Obach24 initially investigated 29 drugs, all based on the same experimental methodology using human hepatic microsomes, and found that 31% of the drugs resulted in accurate clearance predictions within 2-fold. The compilation of Wood et al.66 for 83 drugs in human microsomes from many different investigators results in 42% within 2-fold. We find it hard to believe that for 69% of the Obach24 study drugs there were assay issues, but for 31% there were not because the same procedure was followed for all 29 drugs (and the same point can be made for the Wood et al.66 IVIVE database). Alternatively, based on our analysis,42 the drugs exhibiting poor predictability predominantly violated eq 5, while those drugs giving accurate predictability did not violate eq 5, suggesting that poor predictability is drug-specific and not a function of assay-centric reasons.
4.3. Valid Experimental Determinations.
4.3.1. Total Hepatic Clearance.
Most published IVIVE investigations evaluate error between in vitro CLint and in vivo CLint. The in vivo CLint is back-calculated from total CLH plasma measurements under the assumptions that each of the individual parameters that determine total clearance are correct. Thus, any resulting errors in IVIVE are primarily attributed to issues with in vitro determination of CLint rather than the other factors discussed here. Some of these assumptions may be reasonable, however, because IVIVE has continued to challenge the field, we suggest it is more appropriate to compare total CLH values and recognize the potential contribution of error in each term. Again, we are not suggesting that accurate CLint determination is not critical, because it is in vivo CLint that determines unbound drug exposure (that drives pharmacodynamic outcomes) for all hepatically cleared drugs, regardless of ER.69 We are just pointing out that there may be additional potential errors associated with each parameter that determines total observed CLH, thus may introduce additional errors in back-calculations of in vivo CLint. We recognize that investigators are aware there may be errors inherent in the experimental determinations of each parameter, for instance due to difficulties in measuring binding parameters for highly bound drugs or due to intrasubject variability. However, here, we further discuss the potential theoretical errors associated with determination of each parameter.
Measurement of in vivo clearance values are typically considered to be accurate, however, it must be determined following intravenous dosing or with an accurate estimation of bioavailability (F) following oral dosing. One must also consider interindividual variability, potential for saturation of absorption or metabolism, as well as adequate sampling of the terminal phase (to minimize any errors introduced with extrapolation) and of the absorption phase for high clearance compounds (to accurately capture initial concentrations). Many of these aspects are given due consideration in clinical trial design, however, clearance determinations in vivo are typically conducted in plasma. This value is converted to a blood clearance based on a measurement of a blood-to-plasma partitioning ratio , and thus it is crucial that such experimental measurements are accurate:
| (6) |
In the absence of experimental data, investigators often make the assumption that the value is equal to 0.55 for acidic compounds and equal to 1 for basic and neutral compounds.
4.3.2. Fraction Unbound in the Blood.
The ratio is also required to convert measured values of fraction unbound in the plasma (fu,P) to fu,B:
| (7) |
Plasma is experimentally and analytically advantageous for in vitro experimentation; thus, determinations of fu,P are routinely conducted and are converted to fu,B based on a separate experimental determination of . But what has not been widely recognized as a potential source of IVIVE error is that in order for the relationship in eq 7 to be true, the free drug theory must hold, which assumes that the free drug concentrations in the blood cell must be equal to that in the plasma. In other words, it is assumed that xenobiotic transporters expressed in the red blood cell are not involved in drug distribution,42 and this was not an unreasonable assumption at the time this equation was developed, as it was prior to the recognition that transporters were relevant to drug disposition. Xenobiotic transporters have been identified within erythrocyte membranes,103–105 could potentially have a large impact on the observed unpredictability of IVIVE, and is a fruitful area of future research.
4.3.3. Hepatic Blood Flow Value.
The QH value utilized in clearance predictions is based on physiologic determinations of the total blood flow rate entering and exiting the liver. Based on recently published simulations, we have suggested that perhaps blood flow in contact with the metabolic enzymes within the liver may be greater than the actual blood flow into the liver.42 In all models of hepatic disposition (Figure 5), CLH cannot exceed QH, as drug cannot be eliminated until it is presented to the elimination organ. However, a clearance-dependent underprediction has been observed throughout the field (where the IVIVE underprediction becomes larger with increasing clearance values),65,66,94 suggesting that such an error could potentially be observed if the commonly used value of QH was an underprediction. Simulations revealed that the widely used QH value of approximately 20 mL/min/kg underpredicts effective blood flow by about 2.5-fold.42 At present, this is only a hypothesis that requires experimental validation. However, with recent advancements in hepatic imaging capabilities, it may be possible to improve our understanding of hepatic physiology and potentially revise the relevant QH value that should be utilized in clearance predictions.
4.3.4. In Vitro CLint Determinations: Chemistry versus Pharmacokinetics.
We have speculated that a significant source of error in the determination of CLint in basic IVIVE methodologies is that a “chemistry” approach is utilized to predict a “pharmacokinetic” parameter.42 The term “chemistry” is utilized to describe the in vitro scenario in which the incubational volume is fixed, whereas the term “pharmacokinetics” refers to the in vivo scenario where volume of distribution can be different for each drug due to each drug’s unique physicochemical properties. The major differences between these fields with respect to IVIVE are in the definition of Vmax and the pharmacokinetic volume of distribution that can vary from drug to drug, which is not considered in chemistry where the relevant reaction rates are measured in fixed volumes.
As outlined in detail above, IVIVE is based on principles of Michaelis–Menten kinetics that describe the rate of a chemical (or biochemical) reaction (eq 1) based on reactant concentrations. Under the linear conditions in which the substrate concentration is much less than the Km of the reaction, the relationship is simplified and the slope of the depletion of parent drug can be used to approximate the eq 1 relationship. The results of such determinations provide the rate of drug loss in units of time−1 and are conducted in a fixed incubation volume. But, the desired outcome of IVIVE is to predict a drug clearance in units of volume/time.
In contrast to chemistry, in pharmacokinetics, all derivations are based on mass balance considerations (i.e., amounts rather than concentrations), thus in pharmacokinetics the units of Vmax are in terms of an amount change in contrast to the chemistry-based Vmax that has always been expressed as a concentration change. This results in the ratio of Vmax/Km in pharmacokinetics as a clearance parameter with the units of volume/time (because Vmax has the units of amount/time and Km has the units of amount/volume). However, pharmacokineticists have not derived the classic Michaelis–Menten relationship based on amounts to obtain a Vmax parameter that has units of amount/time. Rather they just take the chemistry Michaelis–Menten derivation and then change the units of Vmax for convenience based on no theoretical rationale.
A second potential pharmacokinetic versus chemistry difference relates to volume of distribution. From the incubation, the in vitro CLin is implicitly calculated by multiplying the rate constant for elimination (units time−1) by the volume of the incubational fluid (Vinc) as outlined in eq 2.42 This detail (and its implications) have not been widely recognized because the volume term is introduced by dividing the measured kinc,u (determined in IVIVE Step 1) by the concentration of enzymes in the incubation (which is half of the enzyme reconciliation that occurs in IVIVE Step 2). eqs 2 and 3 have been combined here as eqs 8a and 8b to further illustrate how the investigator-selected Vinc is incorporated into IVIVE predictions:
| (8a) |
| (8b) |
where the first two terms on the right-hand side of the equality in eq 8a are how in vitro CLint is currently calculated by the field by normalizing kinc,u for in vitro enzymatic/cellular content, and rearrangement of this relationship (eq 8b) highlights how Vinc is introduced into the IVIVE relationship.
Pharmacokinetics is a field founded on mass-balance considerations; thus, measurements of systemic drug concentrations are effectively converted to amounts by incorporating a volume of distribution that does not have physiological relevance and can vary by drug. It is a theoretical volume in which a drug must distribute to relate the observed systemic concentrations to the amount of drug present in the body. It is recognized that rate of loss is dependent on both clearance and volume of distribution, and thus changes in either parameter (as a result of drug–drug interactions, disease state, or pharmacogenomic variance of metabolizing enzymes and transporters) can have an impact on observed drug half-life.106 Current IVIVE approaches are conducted in a fixed-volume incubation and do not account for the pharmacokinetic volume of distribution that can vary for each drug, and drug distribution is not currently recapitulated in traditional metabolic stability incubations.
Figure 6A depicts current IVIVE models that have considered the liver to be a simplified, homogeneous system. Drug enters and exits the liver with QH, and the difference between entering and exiting concentrations are attributed to CLH (and the value of CLH can be modeled using any of the relationships in Figure 5). However, physiologically the liver is a heterogeneous organ comprised of both aqueous and lipophilic regions into which drugs can distribute. Figure 6B depicts the liver as a two-compartmental model comprised of a hepatocyte water and a lipophilic (nonhepatocyte water) compartment. Drugs primarily cleared by metabolism are typically lipophilic,107,108 and it is expected that each drug will partition differently into the lipophilic components of the liver (including the hepatocyte membrane) depending on its unique physicochemical properties. Due to the potential for drug distribution within the liver itself, it is highly unlikely that the volume of distribution of drug in the whole liver at steady state (Vss,H) is equal to the volume of distribution of drug in the hepatocyte water (Vhep) in contact with the drug metabolizing enzymes (Figure 6A–B), and we suggest that the difference of these two volumes of distribution result in the 60–80% of drugs where present IVIVE methods underpredict the in vivo measured clearance.42 We maintain that examination of this potential volume of distribution difference should be a major issue of investigation, as has been recently examined by Riccardi et al.84
Figure 6.
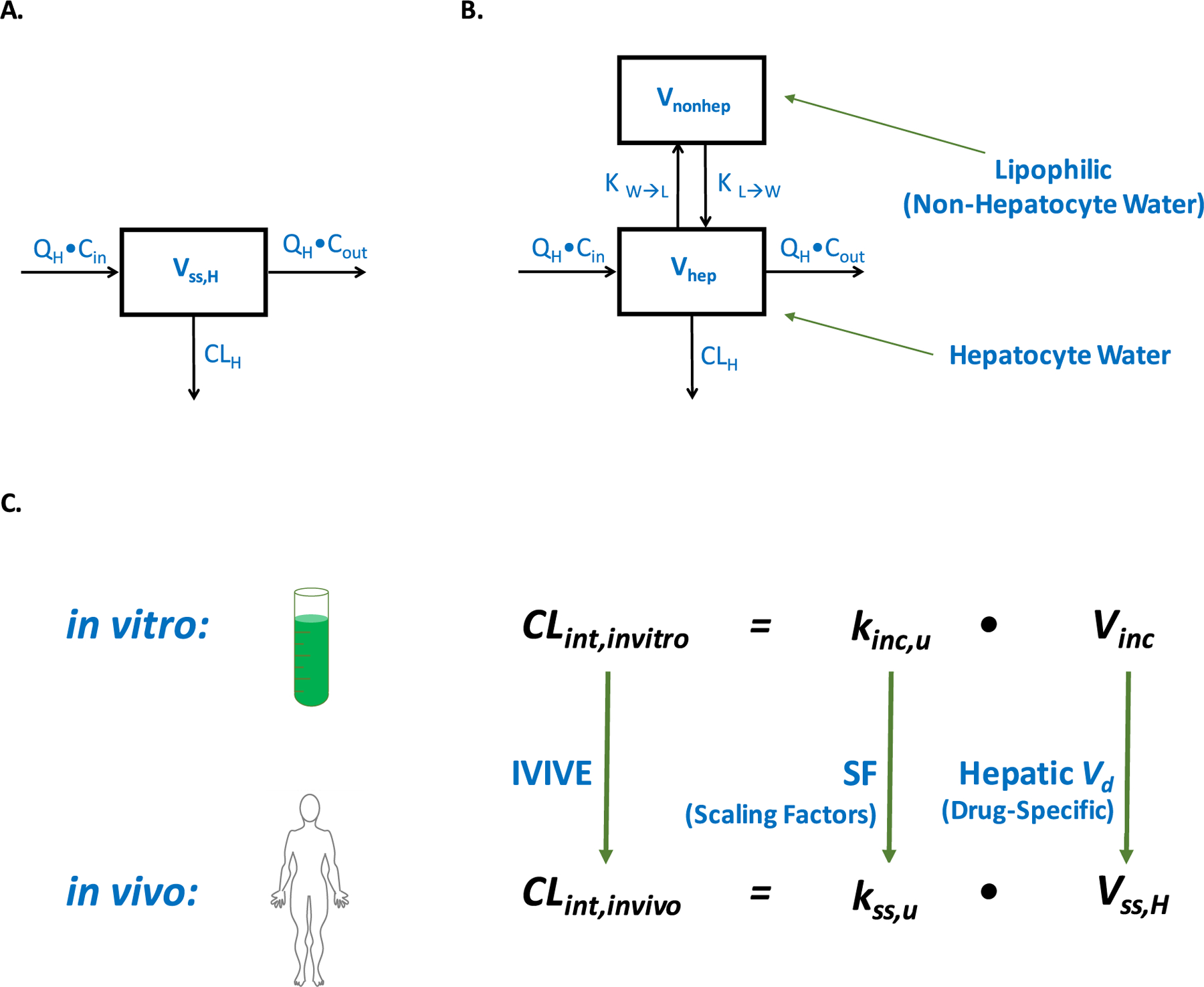
Hepatic volume of distribution and IVIVE. Abbreviations: CLH, hepatic clearance; Cin, entering drug concentration; CLint,invitro, in vitro intrinsic clearance; CLint,invivo, in vitro intrinsic clearance; Cout, exiting drug concentration; IVIVE, in vitro to in vivo extrapolation; kinc,u, unbound rate of incubational drug loss; KL→W, partition coefficient from the lipophilic to aqueous hepatic compartments; kss,u, steady-state in vivo rate of unbound drug loss; KW→L, partition coefficient from the aqueous to lipophilic hepatic compartments; QH, hepatic blood flow; SF, physiologically based scaling factors; Vd, volume of distribution; Vhep, volume of distribution of drug in the hepatocyte water at steady state; Vinc, volume of in vitro incubation; Vnonhep, volume of distribution of drug in the nonhepatocyte water (lipophilic regions) at steady state; Vss,H, volume of distribution of drug in the whole liver at steady state.
By inaccurately assuming the liver is a one-compartment homogeneous system, the field has overlooked the potential of drug to distribute out of the hepatocyte water away from the drug metabolizing enzymes. Thus, if one assumes that Vss,H = Vhep, which is what the field has been unknowingly doing, one is not accurately determining the concentration of drug exposed to drug metabolizing enzymes in vivo. Because this difference in volume of distribution is a function of drug distribution within the liver and the physiological characteristics of the liver itself, it is hypothesized that this difference will undoubtedly vary from drug to drug. Therefore, a universal biological scaling factor alone is not appropriate for IVIVE, which many in the field presently believe will succeed (Figure 6C). Theoretical and experimental aspects related to estimating appropriate drug specific correction factors for marketed drugs (to extrapolate to NCEs) and incorporation into IVIVE practices for improved clearance predictions should, in our opinion, be an area of active research in drug metabolism.
5. CONCLUSIONS
In vitro metabolic stability is critically important in lead-optimization for prediction of in vivo clearance, and there are a number of experimental systems that could be leveraged for clearance predictions. Microsomal stability is particularly amenable to high-throughput screening for early stages of drug discovery due to the relatively low cost and ease-of-use of microsomal fractions. However, it is critical to anticipate the most likely in vivo clearance mechanism to select the appropriate in vitro tool for clearance determinations. Although IVIVE approaches are very useful in rank-ordering the metabolic stability of NCEs, IVIVE methods tend to underpredict clearance for reasons that have not yet been fully elucidated, despite significant experimental efforts by the field. Improved methodologies are continuously emerging;109–111 however, the theoretical basis of the IVIVE process currently employed requires recognition of its inherent assumptions and limitations. There are inherent assumptions with determination of in vivo CLH and fu,B, and it is possible that the currently utilized value of QH is underpredicted. It is likely that the major limitation of IVIVE is that a chemistry-based determination of rate of drug loss (conducted in a fixed incubation volume) is being utilized to predict an in vivo pharmacokinetic clearance parameter in which drug can distribute into hepatic tissues where metabolizing enzymes are not expressed. Thus, it is possible the inexplicable IVIVE underprediction issue challenging the field is due to the fact that current approaches do not account for the pharmacokinetic volume of distribution that can vary for each drug, and drug distribution is not currently recapitulated in traditional metabolic stability incubations nor considered in clearance calculations.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Jason S. Halladay for the inspiration of Figure 2. This work was supported in part by a Mary Anne Koda-Kimble Seed award for innovation. J.K.S. was supported in part by an American Foundation for Pharmaceutical Education Pre-Doctoral Fellowship, NIGMS Grant R25 GM56847 and a Louis Zeh Fellowship. L.Z.B. is a member of the UCSF Liver Center supported by NIH Grant P30 DK026743.
ABBREVIATIONS USED
blood-to-plasma partitioning ratio
- CH
average hepatic drug concentration
- Cin
entering drug concentration
- CLH
hepatic clearance
- CLint
intrinsic clearance
- CLint,invitro
in vitro intrinsic clearance
- CLint,invivo
in vivo intrinsic clearance
- Cout
exiting drug concentration
- CYP
cytochrome P450
- DN
dispersion number
- ER
extraction ratio
- F
bioavailability
- FMO
flavin-containing monooxygenase
- fu,B
fraction unbound of drug in blood
- fu,inc
fraction unbound of drug in the in vitro incubation
- fu,p
fraction unbound of drug in plasma
- IVIVC
in vitro to in vivo correlation
- IVIVE
in vitro to in vivo extrapolation
- kinc
rate of drug elimination in the in vitro incubation
- kinc,u
rate of unbound drug elimination in the in vitro incubation
- KL→W
partition coefficient from the lipophilic to aqueous hepatic compartments
- Km
Michaelis–Menten dissociation constant
- Kpuu
liver-to-plasma partition coefficient
- kss,u
steady-state in vivo rate of unbound drug loss
- KW→L
partition coefficient from the aqueous to lipophilic hepatic compartments
- NCEs
new chemical entities
- QH
hepatic blood flow
- SF
physiologically based scaling factors
- UGT
UDP-glucuronosyltransferase
- Vd
volume of distribution
- Vhep
volume of distribution of drug in the hepatocyte water at steady state
- Vinc
volume of the in vitro incubation
- Vmax
maximal rate of enzymatic reaction velocity
- Vnonhep
volume of distribution of drug in the nonhepatocyte water (lipophilic regions) at steady state
- Vss,H
volume of distribution of drug in whole liver at steady state
Biographies
Dr. Jasleen K. Sodhi received her undergraduate degree from the University of California Berkeley and then spent 9 years in the pharmaceutical industry, primarily at Genentech in the Drug Metabolism and Pharmacokinetics department, where she ran the suite of in vitro ADME assays and experimentally investigated IVIVE disconnects. More recently, Jasleen received her Ph.D. from the University of California San Francisco under the mentorship of Dr. Leslie Benet, where she also focused on improving the IVIVE of hepatic clearance and understanding complex drug–drug interactions but from a theoretical perspective. Jasleen now leads the Drug Metabolism and Pharmacokinetics efforts at Plexxikon, Inc.
Dr. Leslie Z. Benet, Professor and former Chairman (1978–1998) of Bioengineering and Therapeutic Sciences, University of California San Francisco (UCSF), received his A.B., B.S., and M.S. from the University of Michigan, Ph.D. from UCSF, and has nine honorary doctorates. Dr. Benet was the first President of the American Association of Pharmaceutical Sciences. In 1987, he was elected to membership in the National Academy of Medicine of the US National Academy of Sciences. He previously served as the Treasurer of the International Society for the Study of Xenobiotics (ISSX), Chair of the Drug Metabolism Gordon Conference, and in 2015 received the ISSX North American Achievement Award. Dr. Benet has published over 600 scientific articles and book chapters, holds 12 patents, and has edited 7 books. His peer reviewed publications have been cited more than 29 000 times.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c01930
The authors declare no competing financial interest.
Contributor Information
Jasleen K. Sodhi, Department of Bioengineering and Therapeutic Sciences, Schools of Pharmacy and Medicine, University of California San Francisco, San Francisco, California 94143, United States
Leslie Z. Benet, Department of Bioengineering and Therapeutic Sciences, Schools of Pharmacy and Medicine, University of California San Francisco, San Francisco, California 94143, United States;.
REFERENCES
- (1).DiMasi JA; Grabowski HG; Hansen RW Innovation in the pharmaceutical industry: New estimates of R&D costs. J. Health Econ 2016, 47, 20–33. [DOI] [PubMed] [Google Scholar]
- (2).Obach RS; Baxter JG; Liston TE; Silber BM; Jones BC; MacIntyre F; Rance DJ; Wastall P The prediction of human pharmacokinetic parameters from preclinical and in vitro metabolism data. J. Pharmacol. Exp. Ther 1997, 283, 46–58. [PubMed] [Google Scholar]
- (3).Rowland M; Benet LZ; Graham GG Clearance concepts in pharmacokinetics. J. Pharmacokinet. Biopharm 1973, 1, 123–135. [DOI] [PubMed] [Google Scholar]
- (4).Wilkinson GR; Shand DG Commentary: a physiologic approach to hepatic drug clearance. Clin. Pharmacol. Ther 1975, 18, 377–390. [DOI] [PubMed] [Google Scholar]
- (5).Strom SC; Jirtle RL; Jones RS; Novicki DL; Rosenberg MR; Novonty A; Irons G; McLain JR; Michalopoulos G Isolation, culture, and transplantation of human hepatocytes. J. Natl. Cancer Inst 1982, 68 (50), 771–778. [PubMed] [Google Scholar]
- (6).Lee SML; Schelcher C; Demmel M; Hauner M; Thasler WE Isolation of human hepatocytes by a two-step collagenase perfusion procedure. J. Vis. Exp 2013, 79, 50615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mitry RR; Hughes RD; Dhawan A Progress in human hepatocytes: isolation, culture and cryopreservation. Semin. Cell Dev. Biol 2002, 13 (6), 463–467. [DOI] [PubMed] [Google Scholar]
- (8).Li AP Human hepatocytes: isolation, cryopreservation and applications in drug development. Chem.-Biol. Interact 2007, 168 (1), 16–29. [DOI] [PubMed] [Google Scholar]
- (9).Claude A Fractionation of mammalian liver cells by differential centrifugation: II. Experimental procedures and results. J. Exp. Med 1946, 84 (1), 61–69. [PubMed] [Google Scholar]
- (10).von Jagow R; Kampffmeyer H; Kinese M The preparation of microsomes. Naunyn-Schmiedeberg’s Arch. Pharmacol 1965, 251, 73–87. [DOI] [PubMed] [Google Scholar]
- (11).Pearce RE; McIntyre CJ; Madan A; Sanzgiri U; Draper AJ; Bullock PL; Cook DC; Burton LA; Latham J; Nevins C; Parkinson A Effects of freezing, thawing and storing human liver microsomes on cytochrome P450 activity. Arch. Biochem. Biophys 1996, 331 (2), 145–169. [DOI] [PubMed] [Google Scholar]
- (12).Cashman JR Some distinctions between flavin-containing and cytochrome P450 monooxygenases. Biochem. Biophys. Res. Commun 2005, 338 (1), 599–604. [DOI] [PubMed] [Google Scholar]
- (13).Miners JO; Mackenzie PI Drug glucuronidation in humans. Pharmacol. Ther 1991, 51, 347–369. [DOI] [PubMed] [Google Scholar]
- (14).Fisher MB; Campanale K; Ackermann BL; Vandenbranden M; Wrighton SA In vitro glucuronidation using human liver microsomes and the pore-forming peptide alamethicin. Drug Metab. Dispos 2000, 28 (5), 560–566. [PubMed] [Google Scholar]
- (15).Xie J; Saburulla NF; Chen S; Wong SY; Yap ZP; Zhang LH; Lau AJ Evaluation of carbazeran 4-oxidation and O6-benzylguanine 8-oxidation as catalytic markers of human aldehyde oxidase: impact of cytosolic contamination of liver microsomes. Drug Metab. Dispos 2019, 47 (1), 26–37. [DOI] [PubMed] [Google Scholar]
- (16).Crouch RD; Morrison RD; Byers FW; Lindsley CW; Emmitte KA; Daniels JS Evaluating the disposition of a mixed aldehyde oxidase/cytochrome P450 substrate in rats with attenuated P450 activity. Drug Metab. Dispos 2016, 44 (8), 1296–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Wilkinson DJ; Southall RL; Li M; Wright LM; Corfield LJ; Heeley TA; Bratby B; Mannu R; Johnson SL; Shaw V; Friett HL; Blakeburn LA; Kendrick JS; Otteneder MB Minipig and human metabolism of aldehyde oxidase substrates: in vitro-in vivo comparisons. AAPS J. 2017, 19 (4), 1163–1174. [DOI] [PubMed] [Google Scholar]
- (18).Richardson SJ; Bai A; Kulkarni AA; Moghaddam MF Efficiency in drug discovery: liver S9 fraction assay as a screen for metabolic stability. Drug Metab. Lett 2016, 10 (2), 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang H; Davis CD; Sinz MW; Rodrigues AD Cytochrome P450 reaction-phenotyping: an industrial perspective. Expert Opin. Drug Metab. Toxicol 2007, 3 (5), 667–687. [DOI] [PubMed] [Google Scholar]
- (20).Watt AP; Mortishire-Smith RJ; Gerhard U; Thomas SR Metabolite identification in drug discovery. Curr. Opin. Drug Discovery Devel 2003, 6 (1), 57–65. [PubMed] [Google Scholar]
- (21).Halladay JS; Delarosa EM; Tran D; Wang L; Wong S; Khojasteh SC High-throughput, 384-well, LC-MS/MS CYP inhibition assay using automation, cassette-analysis technique, and streamlined data analysis. Drug Metab. Lett 2011, 5 (3), 220–230. [PubMed] [Google Scholar]
- (22).Mukadam S; Tay S; Tran D; Wang L; Delarosa EM; Khojasteh SC; Halladay JS; Kenny JR Evaluation of time-dependent cytochrome P450 inhibition in a high-throughput, automated assay: introducing a novel area under the curve shift approach. Drug Metab. Lett 2012, 6 (1), 43–53. [DOI] [PubMed] [Google Scholar]
- (23).Halladay JS; Wong S; Jaffer SM; Sinhababu AK; Khojasteh-Bakht CS Metabolic stability screen for drug discovery using cassette analysis and column switching. Drug Metab. Lett 2007, 1 (1), 67–72. [DOI] [PubMed] [Google Scholar]
- (24).Obach RS Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos 1999, 27 (11), 1350–1359. [PubMed] [Google Scholar]
- (25).Garle MJ; Fry JR Detection of reactive metabolites in vitro. Toxicology 1989, 54 (1), 101–110. [DOI] [PubMed] [Google Scholar]
- (26).Igari Y; Sugiyama Y; Sawada Y; Iga T; Hanano M In vitro and in vivo assessment of hepatic and extrahepatic metabolism of diazepam in the rat. J. Pharm. Sci 1984, 73 (6), 826–828. [DOI] [PubMed] [Google Scholar]
- (27).Howe JL; Back DJ; Colbert J Extrahepatic metabolism of zidovudine. Br. J. Clin. Pharmacol 1992, 33 (2), 190–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dunn JC; Yarmush ML; Koebe HG; Tompkins RG Hepatocyte function and extracellular matrix geometry: long-term culture in a sandwich configuration. FASEB J. 1989, 3 (2), 174–177. [DOI] [PubMed] [Google Scholar]
- (29).Swift B; Pfeifer ND; Brouwer KLR Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab. Rev 2010, 42 (3), 446–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Brown HS; Griffin M; Houston JB Evaluation of cryopreserved human hepatocytes as an alternative in vitro system to microsomes for the prediction of metabolic clearance. Drug Metab. Dispos 2007, 35 (2), 293–301. [DOI] [PubMed] [Google Scholar]
- (31).Soars MG; Grime K; Sproston JL; Webborn PJH; Riley RJ Use of hepatocytes to assess the contribution of hepatic uptake to clearance in vivo. Drug Metab. Dispos 2007, 35 (6), 859–865. [DOI] [PubMed] [Google Scholar]
- (32).Jigorel E; Houston JB Utility of drug depletion-time profiles in isolated hepatocytes for accessing hepatic uptake clearance: identifying rate-limiting steps and role of passive processes. Drug Metab. Dispos 2012, 40 (8), 1596–1602. [DOI] [PubMed] [Google Scholar]
- (33).Li AP; Jurima-Romet M Applications of primary human hepatocytes in the evaluation of pharmacokinetic drug-drug interactions: evaluation of model drugs terfenadine and rifampin. Cell Biol. Toxicol 1997, 13, 365–374. [DOI] [PubMed] [Google Scholar]
- (34).Li AP; Maurel P; Gomez-Lechon MJ; Cheng LC; Jurima-Romet M Preclinical evaluation of drug-drug interaction potential: present status of the application of primary human hepatocytes in the evaluation of cytochrome P450 induction. Chem.-Biol. Interact 1997, 107 (1–2), 5–16. [DOI] [PubMed] [Google Scholar]
- (35).Hengstler JG; Ringel M; Biefang K; Hammel S; Milbert U; Gerl M; Klebach M; Diener B; Platt KL; Böttger T; Steinberg P; Oesch F Cultures with cryopreserved hepatocytes: applicability for studies of enzyme induction. Chem.-Biol. Interact 2000, 125 (1), 51–73. [DOI] [PubMed] [Google Scholar]
- (36).Williamson B; Dooley KE; Zhang Y; Back DJ; Owen A Induction of influx and efflux transporters and cytochrome P450 3A4 in primary human hepatocytes by rifampin, rifabutin, and rifapentine. Antimicrob. Agents Chemother 2013, 57 (12), 6366–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Stieger B; Mahdi ZM Model systems for studying the role of canalicular efflux transporters in drug-induced cholestatic liver disease. J. Pharm. Sci 2017, 106 (9), 2295–2301. [DOI] [PubMed] [Google Scholar]
- (38).Paillard F; Finot F; Mouche I; Prenez A; Vericat JA Use of primary cultures of rat hepatocytes to predict toxicity in the early development of new chemical entities. Toxicology 1999, 13 (4–5), 693–700. [DOI] [PubMed] [Google Scholar]
- (39).Jover R; Ponsoda X; Castell JV; Gómez-Lechón MJ Evaluation of cytotoxicity of ten chemicals on human cultured hepatocytes: predictability of human toxicity and comparison with rodent cell culture systems. Toxicol. In Vitro 1992, 6 (1), 47–52. [DOI] [PubMed] [Google Scholar]
- (40).Zhang C; Ma S; Delarosa EM; Tay S; Sodhi J; Musinipally V; Chang P; Pai R; Halladay JS; Misner D; Kenny JR; Hop CECA; Khojasteh SC For a series of methylindole analogs, reactive metabolite formation is a poor predictor of intrinsic cytotoxicity in human hepatocytes. Toxicol. Res 2014, 3, 184–190. [Google Scholar]
- (41).Rané A; Wilkinson GR; Shand DG Prediction of hepatic extraction ratio from in vitro measurement of intrinsic clearance. J. Pharmacol. Exp. Ther 1977, 200, 420–424. [PubMed] [Google Scholar]
- (42).Benet LZ; Sodhi JK Investigating the theoretical basis for in vitro-in vivo extrapolation (IVIVE) in predicting drug metabolic clearance and proposing future experimental pathways. AAPS J. 2020, 22, 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Smith DA; Beaumont K; Maurer TS; Di L Clearance in drug design. J. Med. Chem 2019, 62, 2245–2255. [DOI] [PubMed] [Google Scholar]
- (44).Di L; Trapa P; Obach RS; Atkinson K; Bi YA; Wolford AC; Tan B; McDonald TS; Lai Y; Tremaine LM A novel relay method for determining low-clearance values. Drug Metab. Dispos 2012, 40 (9), 1860–1865. [DOI] [PubMed] [Google Scholar]
- (45).Hop CECA; Cole MJ; Davidson RE; Duignan DB; Federico J; Janiszewski JS; Jenkins K; Krueger S; Lebowitz R; Liston TE; Mitchell W; Snyder M; Steyn SJ; Soglia JR; Taylor C; Troutman MD; Umland J; West M; Whalen KM; Zelesky V; Zhao SX High throughput ADME screening: practical considerations, impact on the portfolio and enabler of in silico ADME models. Curr. Drug Metab 2008, 9 (9), 847–853. [DOI] [PubMed] [Google Scholar]
- (46).Jones HM; Houston JB Substrate depletion approach for determining in vitro metabolic clearance: time dependencies in hepatocyte and microsomal incubations. Drug Metab. Dispos 2004, 32 (9), 973–982. [DOI] [PubMed] [Google Scholar]
- (47).Obach RS Nonspecific binding to microsomes: impact on scale-up of in vitro intrinsic clearance to hepatic clearance as assessed through examination of warfarin, imipramine, and propranolol. Drug Metab. Dispos 1997, 25 (12), 1359–1369. [PubMed] [Google Scholar]
- (48).Austin RP; Barton P; Cockroft SL; Wenlock MC; Riley RJ The influence of nonspecific microsomal binding on apparent intrinsic clearance, and its prediction from physicochemical properties. Drug Metab. Dispos 2002, 30 (12), 1497–1503. [DOI] [PubMed] [Google Scholar]
- (49).Austin RP; Barton P; Mohmed S; Riley RJ The binding of drugs to hepatocytes and its relationship to physicochemical properties. Drug Metab. Dispos 2005, 33 (3), 419–425. [DOI] [PubMed] [Google Scholar]
- (50).Abraham MH; Austin RP The effect of ionized species on microsomal binding. Eur. J. Med. Chem 2012, 47 (1), 202–205. [DOI] [PubMed] [Google Scholar]
- (51).Gao H; Steyn SJ; Chang G; Lin J Assessment of in silico models for fraction of unbound drug in human liver microsomes. Expert Opin. Drug Metab. Toxicol 2010, 6 (5), 533–542. [DOI] [PubMed] [Google Scholar]
- (52).Kilford PJ; Gertz M; Houston JB; Galetin A Hepatocellular binding of drugs: correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data. Drug Metab. Dispos 2008, 36 (7), 1194–1197. [DOI] [PubMed] [Google Scholar]
- (53).Nair PC; McKinnon RA; Miners JO Prediction of the nonspecific binding of drugs to hepatic microsomes. Drug Metab. Dispos 2016, 44 (11), 1794–1798. [DOI] [PubMed] [Google Scholar]
- (54).Chen S; Garcia LP; Bergström F; Nordell P; Grime K Intrinsic clearance assay incubational binding: a method comparison. Drug Metab. Dispos 2017, 45 (4), 342–345. [DOI] [PubMed] [Google Scholar]
- (55).Di L; Obach RS Addressing the challenges of low clearance in drug research. AAPS J. 2015, 17 (2), 352–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hutzler JM; Ring BJ; Anderson SR Low-turnover drug molecules: a current challenge for drug metabolism scientists. Drug Metab. Dispos 2015, 43 (12), 1917–1928. [DOI] [PubMed] [Google Scholar]
- (57).Barter ZE; Bayliss MK; Beaune PH; Boobis AR; Carlile DJ; Edwards RJ; Houston JB; Lake BG; Lipscomb JC; Pelkonen OR; Tucker GT; Rostami-Hodjegan A Scaling factors for the extrapolation of in vivo metabolic drug clearance from in vitro data: reaching a consensus on values of human microsomal protein and hepatocellularity per gram of liver. Curr. Drug Metab 2007, 8 (1), 33–45. [DOI] [PubMed] [Google Scholar]
- (58).Hakooz N; Ito K; Rawden H; Gill H; Lemmers L; Boobis AR; Edwards RJ; Carlile DJ; Lake BG; Houston JB Determination of a human hepatic microsomal scaling factor for predicting in vivo drug clearance. Pharm. Res 2006, 23 (3), 533–539. [DOI] [PubMed] [Google Scholar]
- (59).Wilson ZE; Rostami-Hodjegan A; Burn JL; Tooley A; Boyle J; Ellis SW; Tucker GT Inter-individual variability in levels of human microsomal protein and hepatocellularity per gram of liver. Br. J. Clin. Pharmacol 2003, 56 (3), 433–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Naritomi Y; Terashita S; Kimura S; Suzuki A; Kagayama A; Sugiyama Y Prediction of human hepatic clearance from in vivo animal experiments and in vitro metabolic studies with liver microsomes from animals and humans. Drug Metab. Dispos 2001, 29 (10), 1316–1324. [PubMed] [Google Scholar]
- (61).Sohlenius-Sternbeck AK Determination of the hepatocellularlity number for human, drug, rabbit, rat and mouse livers from protein concentration measurement. Toxicol. In Vitro 2006, 20 (8), 1582–1586. [DOI] [PubMed] [Google Scholar]
- (62).Davies B; Morris T Physiological parameters in laboratory animals and humans. Pharm. Res 1993, 10, 1093–1095. [DOI] [PubMed] [Google Scholar]
- (63).Pelkonen O; Turpeinen M In vitro-in vivo extrapolation of hepatic clearance: biological tools, scaling factors, model assumptions and correct concentrations. Xenobiotica 2007, 37 (10–11), 1066–1089. [DOI] [PubMed] [Google Scholar]
- (64).Folger HS Elements of chemical reaction engineering, 5th ed.; Prentice Hall: New York; 2016. [Google Scholar]
- (65).Chiba M; Ishii Y; Sugiyama Y Prediction of hepatic clearance in human from in vitro data for successful drug development. AAPS J. 2009, 11 (2), 262–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Wood FL; Houston JB; Hallifax D Clearance prediction methodology needs fundamental improvement: trends common to rat and human hepatocytes/microsomes and implications for experimental methodology. Drug Metab. Dispos 2017, 45 (11), 1178–1188. [DOI] [PubMed] [Google Scholar]
- (67).Bowman CM; Benet LZ Hepatic clearance predictions from in vitro-in vivo extrapolation and the Biopharmaceutics Drug Disposition Classification System. Drug Metab. Dispos 2016, 44 (11), 1731–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Benet LZ; Hoener B Changes in plasma protein binding have little clinical relevance. Clin. Pharmacol. Ther 2002, 71 (3), 115–121. [DOI] [PubMed] [Google Scholar]
- (69).Ito K; Houston JB Prediction of human drug clearance from in vitro and preclinical data using physiologically based empirical approaches. Pharm. Res 2005, 22, 103–112. [DOI] [PubMed] [Google Scholar]
- (70).Shibata Y; Takahashi H; Chiba M; Ishii Y Prediction of hepatic clearance and availability by cryopreserved human hepatocytes: an application of serum incubation method. Drug Metab. Dispos 2002, 30 (8), 892–896. [DOI] [PubMed] [Google Scholar]
- (71).Sohlenius-Sternbeck A-K; Jones C; Ferguson D; Middleton BH; Projean D; Floby E; Bylund J; Afzelius L Practical use of the regression offset approach for the prediction of in vivo intrinsic clearance from hepatocytes. Xenobiotica 2012, 42 (9), 841–853. [DOI] [PubMed] [Google Scholar]
- (72).Poulin P; Haddad S Toward a new paradigm for the efficient in vitro-in vivo extrapolation of metabolic clearance in humans from hepatocyte data. J. Pharm. Sci 2013, 102 (9), 3239–3251. [DOI] [PubMed] [Google Scholar]
- (73).Hallifax D; Houston JB Binding of drugs to hepatic microsomes: comment and assessment of current prediction methodology with recommendation for improvement. Drug Metab. Dispos 2006, 34 (4), 724–726. [DOI] [PubMed] [Google Scholar]
- (74).Riley RJ; McGinnity DF; Austin RP A unified model for predicting human hepatic, metabolic clearance from in vitro intrinsic clearance data in hepatocytes and microsomes. Drug Metab. Dispos 2005, 33 (9), 1304–1311. [DOI] [PubMed] [Google Scholar]
- (75).Austin RP; Barton P; Mohmed S; Riley RJ The binding of drugs to hepatocytes and its relationship to physicochemical properties. Drug Metab. Dispos 2005, 33 (3), 419–425. [DOI] [PubMed] [Google Scholar]
- (76).Blanchard N; Hewitt NJ; Silber P; Jones H; Coassolo P; Lavé T Prediction of hepatic clearance using cryopreserved human hepatocytes: a comparison of serum and serum-free incubations. J. Pharm. Pharmacol 2006, 58 (5), 633–641. [DOI] [PubMed] [Google Scholar]
- (77).Poulin P; Hop CECA; Ho Q; Halladay JS; Haddad S; Kenny JR Comparative assessment of in vitro-in vivo extrapolation method used for predicting hepatic metabolic clearance of drugs. J. Pharm. Sci 2012, 101 (11), 4308–4325. [DOI] [PubMed] [Google Scholar]
- (78).Berezhkovskiy LM The corrected traditional equations for calculation of hepatic clearance that account for the difference in drug ionization in extracellular and intracellular tissue water and the corresponding corrected PBPK equation. J. Pharm. Sci 2011, 100 (3), 1167–1183. [DOI] [PubMed] [Google Scholar]
- (79).Berezhkovskiy LM; Liu N; Halladay JS Consistency of the novel equation for determination of hepatic clearance and drug time course in liver that account for the difference in drug ionization in extracellular and intracellular tissue water. J. Pharm. Sci 2012, 101 (2), 516–518. [DOI] [PubMed] [Google Scholar]
- (80).Watanabe T; Maeda K; Kondo T; Nakayama H; Horita S; Kusuhara H; Sugiyama Y Prediction of hepatic and renal clearance of transporter substrates in rats using in vitro uptake experiments. Drug Metab. Dispos 2009, 37 (7), 1471–1479. [DOI] [PubMed] [Google Scholar]
- (81).Umehara K; Camenisch G Novel in vitro-in vivo extrapolation (IVIVE) method to predict hepatic organ clearance in rat. Pharm. Res 2012, 29 (2), 603–617. [DOI] [PubMed] [Google Scholar]
- (82).Poulin P Prediction of total hepatic clearance by combining metabolism, transporter, and permeability data in the in vitro-in vivo extrapolation methods: emphasis on an apparent fraction unbound in liver for drugs. J. Pharm. Sci 2013, 102 (7), 2085–2095. [DOI] [PubMed] [Google Scholar]
- (83).Riccardi K; Lin J; Li Z; Niosi M; Ryu S; Hua W; Atkinson K; Kosa RE; Litchfield J; Di L Novel method to predict in vivo liver-to-plasma Kpuu for OATP substrates using suspension hepatocytes. Drug Metab. Dispos 2017, 45 (5), 576–580. [DOI] [PubMed] [Google Scholar]
- (84).Riccardi KA; Tess DA; Lin J; Patel R; Ryu S; Atkinson K; Di L; Li R A novel unified approach to predict human hepatic clearance for both enzyme- and transporter-mediated mechanisms using suspended human hepatocytes. Drug Metab. Dispos 2019, 47 (5), 484–492. [DOI] [PubMed] [Google Scholar]
- (85).Izumi S; Nozaki Y; Komori T; Takenaka O; Maeda K; Kusuhara H; Sugiyama Y Comparison of the predictability of human hepatic clearance for organic anion transporting polypeptide substrate drugs between different in vitro-in vivo extrapolation approaches. J. Pharm. Sci 2017, 106 (9), 2678–2687. [DOI] [PubMed] [Google Scholar]
- (86).Cho H-J; Kim J-E; Kim D-D; Yoon I-S In vitro-in vivo extrapolation (IVIVE) for predicting human intestinal absorption and first-pass elimination of drugs: principles and applications. Drug Dev. Ind. Pharm 2014, 40 (8), 989–998. [DOI] [PubMed] [Google Scholar]
- (87).Di L; Trapa P; Obach RS; Atkinson K; Bi Y-A; Wolford AC; Tan B; McDonald TS; Lai Y; Tremaine LM A novel relay method for determining low-clearance values. Drug Metab. Dispos 2012, 40 (9), 1860–1865. [DOI] [PubMed] [Google Scholar]
- (88).Bonn B; Svanberg P; Janefeldt A; Hultman I; Grime K Determination of human hepatocyte intrinsic clearance for slowly metabolized compounds: comparison of a primary hepatocyte/stromal cell co-culture with plated primary hepatocytes and HepaRG. Drug Metab. Dispos 2016, 44 (4), 527–533. [DOI] [PubMed] [Google Scholar]
- (89).Hultman I; Vedin C; Abrahamsson A; Winiwarter S; Darnell M Use of Hμrel human coculture system for prediction of intrinsic clearance and metabolite formation for slowly metabolized compounds. Mol. Pharmaceutics 2016, 13 (8), 2796–2807. [DOI] [PubMed] [Google Scholar]
- (90).Bowman CM; Benet LZ An examination of protein binding and protein-facilitated uptake relating to in vitro-in vivo extrapolation. Eur. J. Pharm. Sci 2018, 123 (15), 502–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Bteich M; Poulin P; Haddad S The potential protein-mediated hepatic uptake: discussion on the molecular interactions between albumin and the hepatocyte cell surface and their implications for the in vitro-to-in vivo extrapolations of hepatic clearance of drugs. Expert Opin. Drug Metab. Toxicol 2019, 15 (8), 633–658. [DOI] [PubMed] [Google Scholar]
- (92).Kim S-J; Lee K-R; Miyauchi S; Sugiyama Y Extrapolation of in vivo hepatic clearance from in vitro uptake clearance by suspended human hepatocytes for anionic drugs with high binding to human albumin: improvement of the in vitro-to-in vivo extrapolation by considering the “albumin-mediated” hepatic uptake mechanism on the basis of the “facilitated-dissociation model. Drug Metab. Dispos 2019, 47 (2), 94–103. [DOI] [PubMed] [Google Scholar]
- (93).Bowman CM; Benet LZ Interlaboratory variability in human hepatocyte intrinsic clearance values and trends with physicochemical properties. Pharm. Res 2019, 36 (8), 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Bowman CM; Benet LZ In vitro-in vivo extrapolation and hepatic clearance-dependent underprediction. J. Pharm. Sci 2019, 108 (7), 2500–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Bowman CM; Benet LZ The presence of a transporter-induced protein binding shift: a new explanation for protein-facilitated uptake and improvement for in vitro-in vivo extrapolation. Drug Metab. Dispos 2019, 47 (4), 358–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Bowman CM; Benet LZ In vitro-in vivo inaccuracy: the CYP3A4 anomaly. Drug Metab. Dispos 2019, 47 (12), 1368–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Wacher VJ; Wu C-Y; Benet LZ Overlapping substrate specificities and tissue distribution of cytochrome P450 3A and P-glycoprotein: implications for drug delivery and activity in cancer chemotherapy. Mol. Carcinog 1995, 13 (3), 129–134. [DOI] [PubMed] [Google Scholar]
- (98).Williamson B; Harifinger S; McGinnity DF Evaluation of the disconnect between hepatocyte and microsome intrinsic clearance and in vitro in vivo extrapolation performance. Drug Metab. Dispos 2020, 48 (11), 1137–1146. [DOI] [PubMed] [Google Scholar]
- (99).Benet LZ; Bowman CM; Liu S; Sodhi JK The extended clearance concept following oral and intravenous dosing: theory and critical analyses. Pharm. Res 2018, 35 (12), 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Sodhi JK; Liu S; Benet LZ Challenging the relevance of unbound tissue-to-blood partition coefficient (Kpuu) on predictions of drug-drug interactions. Pharm. Res 2020, 37 (4), 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Benet LZ; Liu S; Wolfe AR The universally unrecognized assumption in predicting drug clearance and organ extraction ratio. Clin. Pharmacol. Ther 2018, 103 (3), 521–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Sodhi JK; Wang H-J; Benet LZ Are there any experimental perfusion data that preferentially support the dispersion and parallel-tube models over the well-stirred model of organ elimination? Drug Metab. Dispos 2020, 48 (7), 537–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Shi P; Liao M; Chuang B-C; Griffin R; Shi J; Hyer M; Fallon JK; Smith PC; Li C; Xia CQ Efflux transporter breast cancer resistance protein dominantly expresses on the membrane of red blood cells, hinders partitioning of its substrates into cells, and alters drug-drug interaction profiles. Xenobiotica 2018, 48 (11), 1173–1183. [DOI] [PubMed] [Google Scholar]
- (104).Várady G; Szabó E; Fehér Á; Németh A; Zámbó B; Pákáski M; Janka Z; Sarkadi B Alterations of membrane protein expression in red blood cells of Alzheimer’s disease patients. Alzheimers Dement 2015, 1 (3), 334–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Awasthi YC; Singh SV; Ahmad H; Wronski LW; Srivastava SK; LaBelle EF ATP dependent primary active transport of xenobiotic-glutathione conjugates by human erythrocyte membrane. Mol. Cell. Biochem 1989, 91 (1–2), 131–136. [DOI] [PubMed] [Google Scholar]
- (106).Rowland M; Tozer TN Clinical pharmacokinetics and pharmacodynamics: concepts and applications, 4th ed.; Lippincott Williams; Wilkens: Philadelphia, 2010. [Google Scholar]
- (107).Waring MJ Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5 (3), 235–248. [DOI] [PubMed] [Google Scholar]
- (108).Lewis DFV; Jacobs MN; Dickins M Compound lipophilicity for substrate binding to human P450s in drug metabolism. Drug Discovery Today 2004, 9 (12), 530–537. [DOI] [PubMed] [Google Scholar]
- (109).Chohan KK; Paine SW; Waters NJ Advancements in predictive in silico models for ADME. Curr. Chem. Biol 2008, 2 (3), 215–228. [Google Scholar]
- (110).Russell LE; Schleiff MA; Gonzalez E; Bart AG; Broccatelli F; Hartman JH; Humphreys WG; Lauschke VM; Martin I; Nwabufo C; Prasad B; Scott EE; Segall M; Takahashi R; Taub RE; Sodhi JK Advances in the study of drug metabolism - symposium report of the 12th meeting of the International Society for the Study of Xenobiotics (ISSX). Drug Metab. Rev 2020, 52 (3), 395–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Billington S; Shoner S; Lee S; Clark-Snustad K; Pennington M; Lewis D; Muzi M; Rene S; Lee J; Nguyen TB; Kumar V; Ishida K; Chen L; Chu X; Lai Y; Salphati L; Hop CECA; Xiao G; Liao M; Unadkat JD Positron emission tomography imaging of [11C]rosuvastatin hepatic concentrations and hepatobiliary transporter in humans in the absence and presence of cyclosporine A. Clin. Pharmacol. Ther 2019, 106 (5), 1056–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]