

Abstract
Purpose
Filamin A (FLNa) cross-links actin filaments into dynamic orthogonal networks and interacts with binding proteins of diverse cellular functions that are implicated in cell growth and motility regulation. Here, we tested the hypothesis that FLNa plays a role in cancer proliferation and metastasis via the regulation of epidermal growth factor receptor (EGFR) function.
Methods
Ectopic expression and knockdown of FLNa in human melanoma cell lines was performed to investigate changes in cellular proliferation, migration and invasion in vitro and tumor growth in a xenograft model in the mouse. The role of FLNa in EGFR expression and signaling was evaluated by Western blot. Immunohistochemistry was performed on histological sections of human melanoma tumors to determine whether an association existed between FLNa and overall survival.
Results
The depletion of FLNa significantly reduced the proliferation, migration and invasion of two melanoma cell lines in vitro and was associated with smaller tumors in a xenograft model in vivo. EGF-induced phosphorylation of EGFR and activation of the Raf-MEK-ERK cascade was negatively affected by the silencing of FLNa both in vitro and in vivo. Cancer patients with low melanoma tumor FLNa expression have improved survival benefit.
Conclusion
These data indicate that enhanced tumorigenesis occurs through increase in EGF-induced EGFR activation in FLNa-expressing melanoma cells and that high FLNa levels are predictors of negative outcome for patients with melanoma tumors.
Keywords: Malignant melanoma, FLNa, EGFR, Metastasis, Proliferation
Introduction
Melanoma is an aggressive cutaneous malignancy that can spread to other organs of the body, with more than 160,000 new cases and 48,000 deaths per year in the world (Eggermont et al. 2014). The initiation and progression of melanoma is a complex cellular process mediated by genetic mutations or epigenetic factors affecting key molecules (Hodis et al. 2012; Kunz 2014), result in the transformation of normal melanocyte stem cells into populations of cancer cells with inherited properties of self-renewal and differentiation.
Filamin A (FLNa) belongs to the filamin family, which consists of three homologous proteins (FLNa, FLNb and FLNc), and FLNa is the first actin filament cross-linking protein identified in non-muscle cells (Savoy and Ghosh 2013). It forms a homo-dimer and cross-links cortical actin filaments into a dynamic three-dimensional structure (Djinovic-Carugo and Carugo 2010). FLNa is known to scaffold over 90 binding partners, including plasma membrane receptors to regulate cellular functions (Nakamura et al. 2011) and processes such as cell shape, cellular motion, division and intracellular transport (Wickstead and Gull 2011; Razinia et al. 2012). Abnormal expression of FLNa has been found in cancer progression and a wide range of other diseases (Wang et al. 2007; Kyndt et al. 2007), which are caused by impaired interactions between FLNa and binding partners (Nakamura et al. 2009; Zhou et al. 2010).
Epidermal growth factor receptor (EGFR) is the first receptor identified in a family of receptors known as the type I receptor tyrosine kinases or ErbB receptors (Hynes and MacDonald 2009). EGFR is the major contributor of a complex signaling cascade that modulates growth, signaling, differentiation, adhesion, migration and survival of cells, and its deregulation is implicated in oncogenesis (Yewale et al. 2013). Homo- or hetero-dimerization of ligand-activated EGFR results in receptor tyrosine kinase autophosphorylation and channeling of mitogenic signals predominantly via the Ras-extracellular signal-regulated kinase (ERK), Akt and c-Jun NH2-terminal kinase (JNK) pathways (Hynes and MacDonald 2009; Baselga and Albanell 2002). Regulation of the pleiotropic responses of EGFR involves activation of EGFR and endocytosis of the ligand-activated receptors (Grovdal et al. 2004; Barriere et al. 2007). Oncogenesis is not only associated with persistent activation and impaired endocytic down-regulation of EGFR (Roepstorff et al. 2008; Grandal and Madshus 2008), but it also involves a complex network of signaling molecules that interact with FLNa (Nallapalli et al. 2012; Ravid et al. 2008). Despite these advances, the mechanistic link between FLNa and the signaling events associated with malignancy remains elusive.
In this study, we measured the changes in proliferation, migration and invasion of two human melanoma cell lines of distinct origins after alteration in FLNa expression. The proliferative properties of FLNa were then explored in a xenotransplantation melanoma tumor model in the mouse. In a second series of experiments, the contribution of FLNa in ligand-mediated activation of EGFR and its downstream signaling pathway was investigated both in vitro and in vivo. Finally, immunohistochemical analysis was performed on sections of malignant melanoma tumors to determine whether an association existed between FLNa expression and overall survival of these cancer patients.
Materials and methods
Cell culture
The use of human melanoma cell lines M2 (spontaneously lost FLNa expression), M2A7 (derivative of M2 cells with stable FLNa expression) and UACC647 (high expression of FLNa) was reported earlier (Zhu et al. 2007). M2 cells were grown in minimal essential medium (MEM) (Life Technologies Gibco, Grand Island, NY, USA) with 10 % (v/v) fetal bovine serum (FBS) (Thermo Scientific HyClone, Waltham, MA, USA) and penicillin (100 units/ml), streptomycin (100 μg/ml) (North China Pharmaceutical Co., Shijiazhuang, Hebei, China) at 37 °C in 5 % CO2. M2A7 cells were maintained in the same medium as M2 cells except for the additional supplementation of 500 μg/ml G418 (Amersco, Inc., Solon, OH, USA). UACC647 cells were grown in RPMI 1640 medium (Life Technologies Gibco) with 10 % (v/v) FBS and antibiotics.
FLNa knockdown experiments
UACC647 melanoma cells were stably transfected either with a plasmid expressing shRNA against FLNa (pSIF1-FLNa shRNA) or control shRNA using FuGENE HD transfection reagent (Promega, Madison, WI, USA) and selected with puromycin (3 μg/mL), resulting in UACC647(KD) and UACC647(ctrl) cell lines. The knockdown efficiency of FLNa was measured by Western blot assay.
Cell proliferation assay and Clonogenic assay
MTT assay was used to measure the role of FLNa in cell growth and viability in vitro. Cells were seeded in 96-well plates (5 × 103/well), starved for 4 h and then were left untreated or treated with various concentrations of EGF (4, 20, 100 nM) for 44 h. MTT (Sigma-Aldrich, Saint Louis, MO, USA) in PBS (5 mg/mL) was added (20 μL/well) and plates were incubated at 37 °C for 4 h, after which culture media was removed, DMSO was added and the absorbance was measured at 570 nm. In each MTT assay, cells were kept under the same culture conditions, plated at the same cell density and treated with EGF for the same periods of time.
For colony assay, M2 and M2A7 cells or UACC647(ctrl) and UACC647(KD) cells, were trypsinized, counted and seeded at density 300 or 200 cells/35 mm dish, and incubated in medium containing 1 % FBS in the presence of EGF or left untreated. Two weeks later, cell colonies were stained with hematoxylin and counted.
To evaluate the anchorage-independent cell growth, cells were mixed with medium containing 0.7 % agar to result in a final agar concentration of 0.35 %. Then 1 ml samples of this cell suspension (300 cells/well) were immediately plated in six-well plates coated with 0.6 % agar in regular medium (2 ml per well) and cultured in medium containing 1 % FBS in the presence of EGF or left untreated. After 2 weeks, the numbers of colonies were counted.
In vitro scratch motility assay
Scratch assay was carried out as described (Fiori et al. 2009). In brief, melanoma cells were seeded in 24-well plates at a concentration of 2 × 105 per well and allowed to form a confluent monolayer for 24 h. Cells were serum-starved for 4 h, and the monolayer was then scratched with a pipette tip and washed with serum-free medium (SFM) to remove floating cells. Following addition of vehicle or 20 nM epidermal growth factor (EGF; Miltenyi Biotec, Auburn, CA, USA), cells were photographed at the same field every 2 h until closure of the scratch.
In vitro invasion assay
This invasion assay was performed according to a published protocol (Jiang et al. 2013) with some modifications. Serum-starved cells (1 × 105 cells in 200 μL of serum-free MEM) were placed in the top chamber of transwell invasion chambers (8.0 μm polycarbonate membrane inserts; Corning, Lowell, MA, USA). The lower chamber was filled with 600 μL MEM supplemented with 10 % FBS. After a 24-h incubation period in a humidified atmosphere of 5 % CO2 at 37 °C, noninvasive cells were removed from the upper surface of the transwell membrane with a cotton swab, and invasive cells on the lower membrane surface were fixed with methanol, stained with H&E, photographed and counted under a microscope at 200× magnification in five fields. These experiments were repeated twice.
Immunoprecipitation
Cells were lysed in immune precipitation buffer as described previously (He et al. 2006). The clarified lysates (500 μg) were incubated with 2 μg of mouse anti-EGFR antibodies and non-immune IgG (Santa Cruz, Dallas, Texas, USA) overnight at 4 °C followed by the addition of 20 μl of protein G-agarose (Millipore, Temecula, CA, USA) for 2 h at 4 °C. Total cell extracts and immunoprecipitated complexes were subjected to Western blotting.
Western blotting
Serum-starved cells were left untreated or treated with EGF (20 nM) for 5, 10 and 30 min, after which cells were placed immediately on ice, washed twice in cold PBS and lysed with cold lysis buffer as described previously (Zhu et al. 2007). After determining the protein concentration using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA), equal amount of proteins were separated on 8 % SDS-PAGE and transferred onto polyvinylidene difluoride membranes (Millipore, Billerica, MA, USA). The membranes were blocked in 5 % nonfat milk or 3 % bovine serum albumin, incubated with various primary antibodies, followed by incubation with HRP-conjugated secondary antibodies and visualization with enhanced chemiluminescence (ECL) detection reagents (Beyotime, Haimen, Jiangsu, China). Signals were quantitated by densitometry using the ImageJ software (National Institutes of Health, Bethesda, MD, USA). Primary antibody against phosphotyrosine (clone 4G10) was obtained from Millipore (Temecula, CA, USA); antibody against phospho-ERK1/2 was from Cell Signaling Technology (Danvers, MA, USA); antibodies against ERK1/2 and β-actin were from Santa Cruz Biotechnology, Inc. (Dallas, Texas, USA); antibodies against FLNa were purchased from Millipore (Cat. number: MAB1680) and Santa Cruz (Cat. number: sc-17749), while anti-EGFR antibodies were from Cell Signaling Technology (Cat. number: D38B1) and Millipore (Cat. number: 05–101).
In vivo xenograft model for tumor growth
Five-week-old female BALB/c nude mice were purchased from Vital River Laboratory Animal Technology (Beijing, China) and used in accordance with institutional guidelines under approved protocols. UACC647(ctrl) and UACC647(KD) cells were inoculated subcutaneously within the right thigh region (2 × 106 cells per mouse) to generate tumors (n = 6 mice for each cell line). Tumor size was measured with calipers twice weekly, and tumor volume estimated according to the formula: Volume = 1/2 × length × width2.
Mice were killed 4 weeks after tumor inoculation. Tumor tissue was weighted and then cut into two fragments: one-half was snap-frozen in liquid nitrogen for protein analysis, the other half was fixed in 10 % formalin and subsequently dehydrated and blocked in paraffin. Lysates from frozen tissues were separated on SDS-PAGE, and immunoblot analyses were carried out. Immunostaining was performed on 5-μm formalin-fixed, paraffin-embedded tissue sections using an immunoperoxidase method with mouse anti-FLNa monoclonal antibody (1:200; Santa Cruz) and rabbit anti-Ki-67 monoclonal antibody (Sunbiote, Shanghai, China). Antigen retrieval were performed with EDTA (pH 9, Autoclave at 120 °C for 2 min) and 10 mM citrate buffer (pH 6.0, Autoclave at 120 °C for 2 min), respectively.
Two pathologists blinded to the experiments assessed the extent and intensity of immunostaining. Semi-quantitative scoring for FLNa was performed using the H-score method (Flanagan et al. 2008). For H-score assessment, the staining intensity was scored as 0–3 (0 = none, 1 = weak, 2 = moderate, and 3 = strong), and the proportion of the tumor staining for that intensity was recorded from 0 to 100. A final H-score was given as the sum of the percent staining multiplied by an ordinal value corresponding to the intensity level: H-score = (% of cells stained at intensity category 1 × 1) + (% of cells stained at intensity category 2 × 2) + (% of cells stained at intensity category 3 × 3). With four intensity levels, the final resulting score ranges from 0 (no staining in the tumor) to 300 (diffuse intense staining of the tumor). For better interpretation, categories “low” and “high” expression levels were used based on the median value of H-score algorithm. The percentage of nuclear Ki-67 immunoreactivity was calculated by determining the average expression of positive Ki-67 signal in 500 nuclei.
Immunohistochemical (IHC) staining of tissue
Thirty cases of malignant melanoma tissues were obtained from the surgical pathology files at Bethune International Peace Hospital with the approval from the Institutional Review Board. FLNa (1:200; Santa Cruz) and Ki-67 (Sunbiote) immunostaining was performed using a procedure similar to that indicated in the experiments in nude mice.
Statistical analysis
For all experiments, raw data were analyzed and graphed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA), and appropriate statistical tests were chosen according to the data analyzed using GraphPad Prism 5 or SPSS version 13.0 (SPSS Software, Armonk, NY, USA). Unless otherwise stated, all data are reported as mean ± SEM; * and ** indicate P < 0.05 and P < 0.01, respectively.
Results
FLNa promotes the proliferation, migration and invasion in human melanoma cell lines
Previous studies have illustrated that FLNa interacts with many proteins related to cancer progression (Kim et al. 2010; Leung et al. 2010), a condition that involves acquisition of growth, motility and invasive properties. The effects of FLNa on proliferation, migration and invasion were evaluated in two human melanoma cell lines of distinct origins: (a) the FLNa-deficient M2 melanoma cells and M2A7 cells, a subclone with stable reintroduction of human FLNa to levels that approximate endogenous FLNa concentration (Fiori et al. 2009), and (b) UACC647 melanoma cells. Stable knockdown of FLNa gene was carried out in UACC647 cells to create the UACC647(KD) cells. These cells were found to have a 70 % reduction in FLNa protein levels as compared either with UACC647 cells transfected with control shRNA (ctrl) or mock-transfected cells (Fig. 1a, b).
Fig. 1.
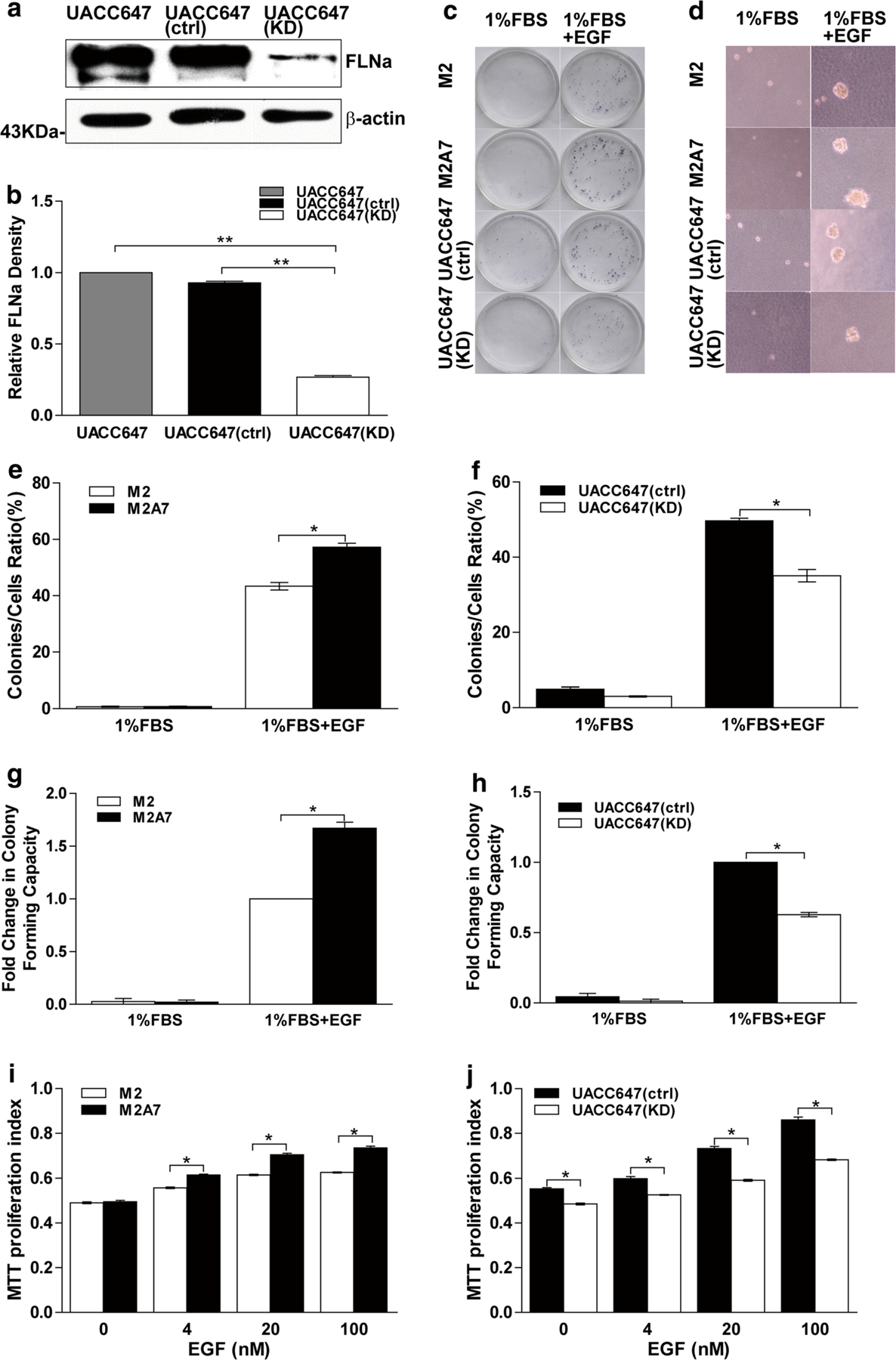
FLNa-deficient cells have impaired cell proliferation capacity. a Human melanoma UACC647 cells were mock-treated or stably transfected with control shRNA (ctrl) or FLNa shRNA (KD) plasmid and selected by puromycin. Cell lysates were prepared and immunoblotted for FLNa. Membrane was reprobed for β-actin, which was used as loading control. b Bars represent ratio of FLNa signal normalized to β-actin. c Colony assays were performed with M2 and M2A7 or UACC647(ctrl) and UACC647(KD) cells in the medium containing 1 % FBS in the absence or presence of EGF (20 nM). Images of representative colonies were showed. e, f The ratio of colony formation was measured. d Anchorage-independent growth assays were performed with M2 and M2A7 or UACC647(ctrl) and UACC647(KD) cells in the medium containing 1 % FBS in the absence or presence of EGF (20 nM). Images of representative colonies formed in soft agar were showed. g Fold changes in colony-forming capacity in soft agar in M2 and M2A7 cells. h Fold changes in colony-forming capacity in soft agar in UACC647(ctrl) and UACC647(KD) cells. Cells were serum-starved for 4 h and then incubated with EGF (0, 4, 20, 100 nM) for 44 h followed by MTT assay. i M2 and M2A7 cells; j UACC647(ctrl) and UACC647(KD) cells. Bars represent mean ± SEM of three experiments, each assayed in quintuple. *P < 0.05
The role of FLNa in basal and EGF-induced cell growth and viability was then evaluated using the MTT assay. M2A7 cells exhibited a higher rate of viability than M2 cells when stimulated with 4–100 nM EGF, but not in basal conditions (Fig. 1i). Depletion of FLNa in UACC647(KD) cells was accompanied by significant reduction in basal and EGF-stimulated growth as compared with UACC647(ctrl) cells (Fig. 1j). Subsequently, to investigate whether alteration of FLNa affects the growth potential of malignant melanoma cells, we performed the colony formation assay at low cell density (Fig. 1c). Interestingly, our results indicated that reduction of FLNa expression inhibited the ability of melanoma cells to form colonies in medium containing 1 % FBS in the presence of EGF. The colony formation ratios in FLNa-deficient M2 cells (43.39 ± 1.33 %) and UACC647(KD) cells (35.08 ± 1.66 %) were significantly lower than that in FLNa-sufficient M2A7 cells (57.22 ± 1.44 %) and UACC647(ctrl) cells (49.67 ± 0.72 %) (P < 0.05, respectively) (Fig. 1e, f). Finally, the soft agar assay was used as a measure of anchorage-independent growth, a defining characteristic of transformed cells (Fig. 1d). We found that the capacity for clone formation of FLNa-sufficient M2A7 cells and UACC647(ctrl) cells in soft agar were higher than that in FLNa-deficient M2 cells and UACC647(KD) cells (P < 0.05, respectively) (Fig. 1g, h).
Using wound-healing assays, it was determined that addition of EGF to FLNa-expressing M2A7 melanoma cells stimulated wound closure within 12 h. Ectopic expression of FLNa was associated with efficient closure of the wound, which closed faster in the presence of EGF (Fig. 1a, b). Consistent with these results, the rate of basal and EGF-induced wound closure were markedly higher in UACC647(ctrl) cells as compared to the FLNa-depleted UACC647(KD) cells, with 50 % closure of the wound at 9 and 15 h, respectively (Fig. 1c, d). Thus, reduction in FLNa expression was correlated with impaired motility and EGF-induced migration in melanoma cells.
Another critical step in cancer metastasis is the detachment of tumor cells from the primary sites and invasion of the surrounding tissues (Kim et al. 2011). M2A7 cells had a 1.9-fold higher invasive potential in Matrigel invasion assays than M2 cells (P < 0.01) (Fig. 2i). Similarly, there was a 1.8-fold increase in invasive potential of UACC647(ctrl) cells as compared to UACC647(KD) cells (P < 0.01) (Fig. 2j). Taken together, these results indicate that FLNa has essential roles in cellular migration and proliferation potential of melanoma cells, especially in the presence of EGF.
Fig. 2.
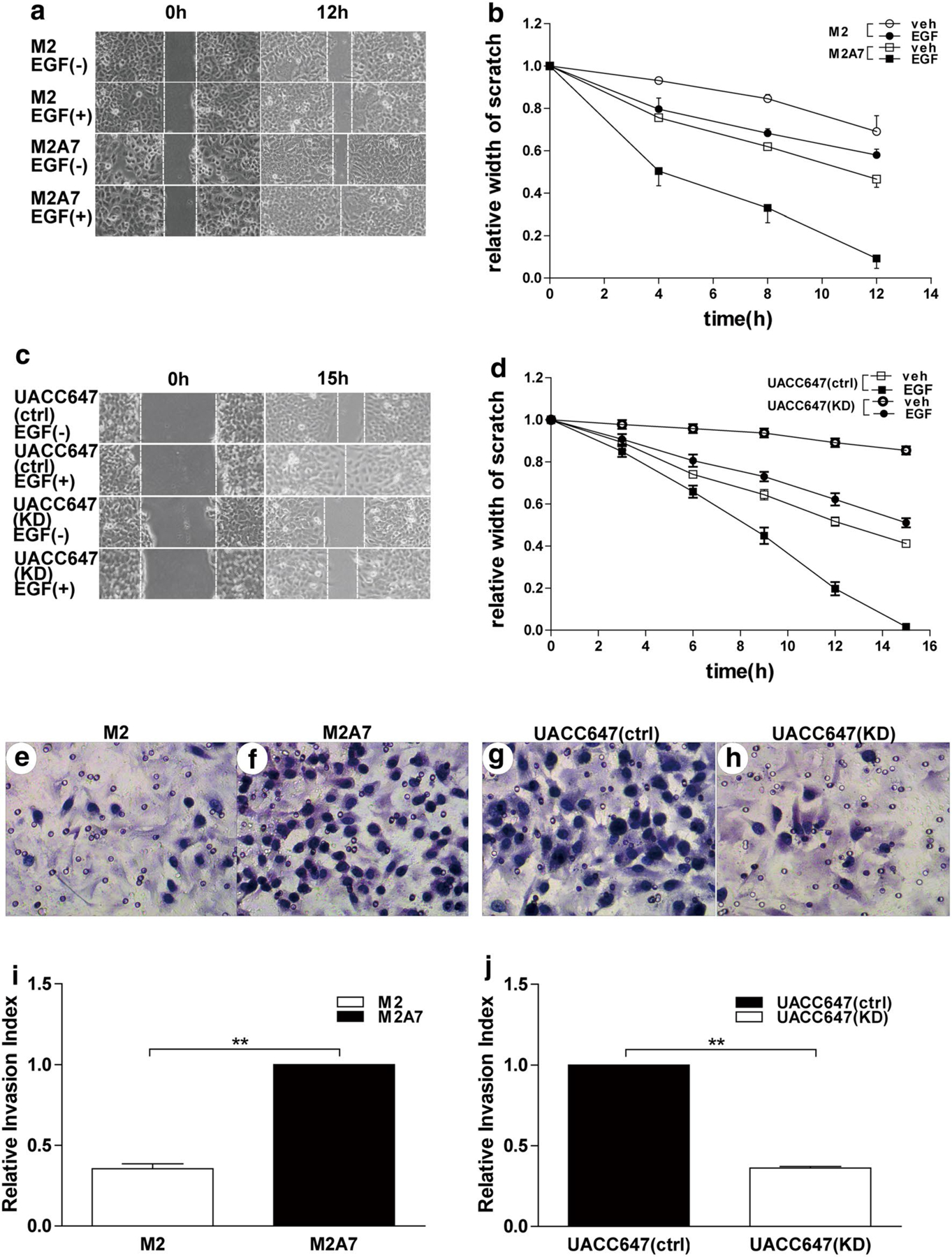
Depletion in FLNa reduces cell migration and invasion. Scratch assays were performed with human melanoma M2 and M2A7 cells (a) or UACC647(ctrl) and UACC647(KD) cells (c) in the absence or presence of EGF (20 nM) for the indicated times. (b) and (d) The relative scratch width was measured over time and plotted. Transwell migration chambers were used to determine invasive capacity of M2 and M2A7 cells (e, f) as well as UACC647(ctrl) and UACC647(KD) cells (g, h). After 24 h of invasion through matrigel, cells were stained with hematoxylin, images were taken (e–h) and cells were counted (i, j). The data are representative of three independent experiments and are shown as mean ± SEM. **P < 0.01
Role of FLNa in ligand-induced EGFR phosphorylation in human melanoma cells
Activation of EGFR results in recruitment and phosphorylation of several intracellular substrates, which leads to uncontrolled cell proliferation, migration, invasion, adhesion, survival and cellular repair (Mendelsohn and Baselga 2003). We hypothesized that the ability of FLNa to enhance motility and growth of human melanoma cells stems from the regulation of EGFR phosphorylation and activation of downstream proteins in the Raf-MEK–ERK cascade. Immunoblot analysis using antibodies raised against phosphotyrosine (clone 4G10) [to detect active, phosphorylated form of EGFR (pY-EGFR)] and phosphorylated ERK (pERK1/2) illustrated a rapid increase in pY-EGFR and pERK1/2 levels in response to EGF stimulation in both M2 and M2A7 cells, reaching a maximum at 5 min and declining thereafter (Fig. 3a). As anticipated, the lack of FLNa rendered M2 cells somewhat unresponsive to EGF-induced phosphorylation of EGFR, even though these cells expressed noticeably higher levels of total EGFR as compared with M2A7 cells (Fig. 3a, top and second panels). As a result, the pY-EGFR to total EGFR ratio was significantly higher in EGF-stimulated M2A7 cells (Fig. 3b). Similar results were observed when anti-EGFR immunoprecipitates were subjected to Western blot analysis using 4G10 (Fig. 3d). As anticipated, pERK1/2 levels were found to be significantly higher in M2A7 cells as compared to M2 cells upon EGF stimulation (Fig. 3c). These experiments were repeated using UACC647(ctrl) and UACC647(KD) cells, and the results indicated that knockdown of FLNa was accompanied by sharp reduction in EGF responsiveness when looking at pY-EGFR and pERK1/2 levels (Fig. 3e, top and third panels). The phosphorylated to total EGFR protein ratio was 70 % lower in UACC647(KD) cells as compared with UACC647(ctrl) cells after a 10-min stimulation with EGF (Fig. 3f). Immunoprecipitation experiments were carried out to further confirm the differential tyrosine phosphorylation of EGFR in UACC647(ctrl) cells vs. UACC647(KD) cells (Fig. 3h). The depletion of FLNa in UACC647(KD) cells was accompanied by a significant 1.4-fold reduction in EGF-dependent increase in pERK1/2 levels when compared to UACC647(ctrl) cells (Fig. 3g). It would appear therefore that FLNa enhances EGF-induced phosphorylation of EGFR and activation of the Raf-MEK-ERK cascade.
Fig. 3.
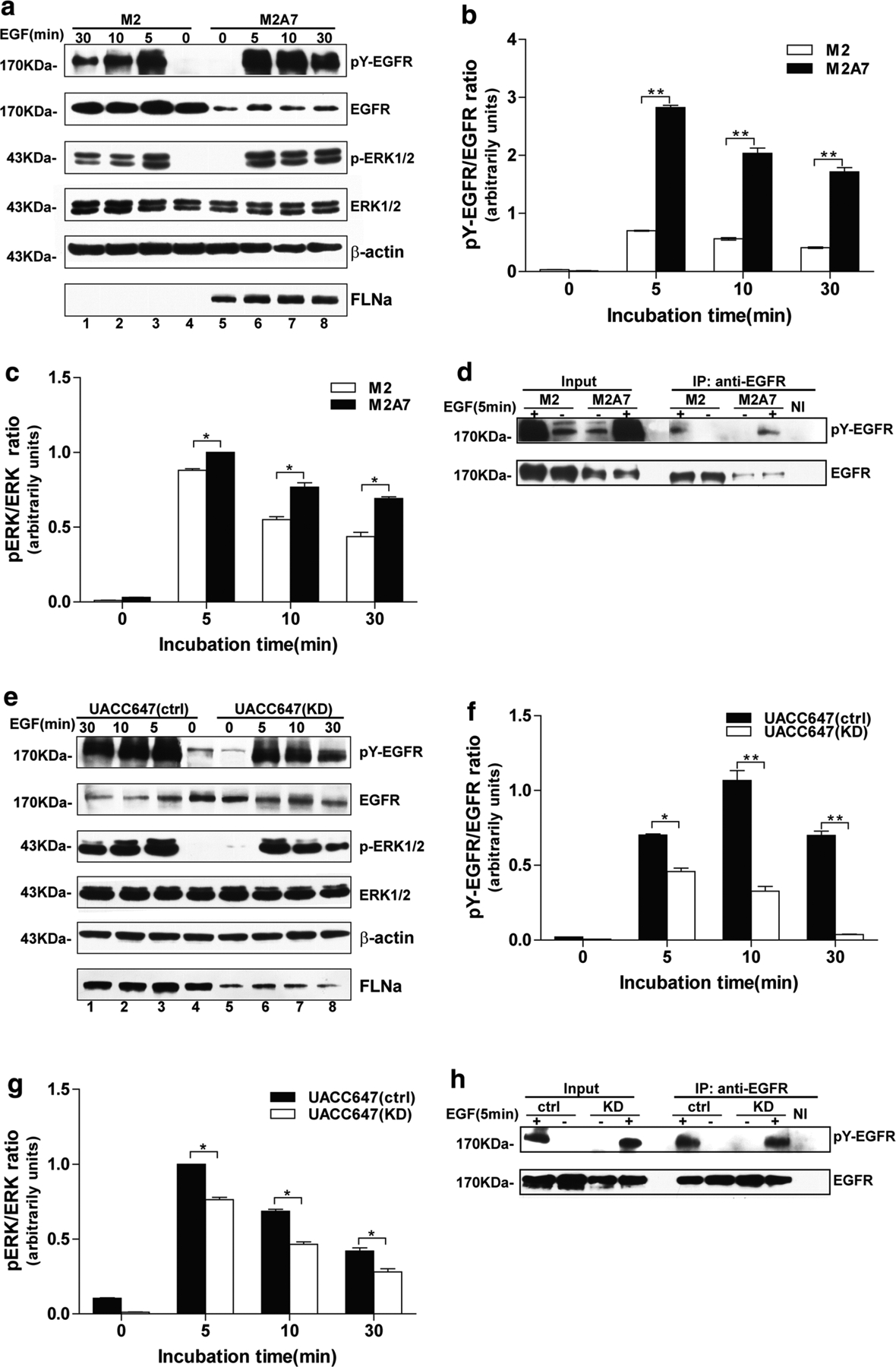
Role of FLNa in ligand-induced EGFR phosphorylation in human melanoma cell lines. a Human melanoma M2 and M2A7 cells were serum-starved for 4 h and then left alone or incubated with EGF (20 nM) for 5, 10, or 30 min. Western blot analysis was performed with a portion of cell lysates using antibodies against FLNa, phosphotyrosine (4G10 clone), EGFR, phosphorylated and total ERK1/2, or β-actin shown here as a loading control. Densitometric analysis of EGFR (second panel, lane 4) and ERK (fourth panel, lane 4) in unstimulated M2 cells was arbitrarily given the value of 1.0. Ratios of pY-EGFR to total EGFR (b) and pERK to total ERK (c) were calculated. d Lysates from M2 and M2A7 cells were immunoprecipitated (IP) with anti-EGFR antibody and analyzed by Western blot with antibodies against phosphotyrosine (4G10 clone) and EGFR. NI, non-immune IgG. e Serum-starved UACC647 (ctrl) and UACC647 (KD) cells were left alone or incubated with EGF (20 nM) for 5, 10, or 30 min. Western blot analysis was performed as in (a). Densitometric analysis of EGFR (second panel, lane 4) and ERK (fourth panel, lane 4) in unstimulated UACC647 (ctrl) cells was arbitrarily given the value of 1.0. Ratios of pY-EGFR to total EGFR (f) and pERK to total ERK (g) were calculated. h Lysates from UACC647(ctrl) and UACC647(KD) cells were immunoprecipitated (IP) with anti-EGFR antibody and analyzed by Western blot with antibodies against 4G10 and EGFR. NI, non-immune IgG. All data shown are representative of four independent experiments, each performed in duplicate dishes. *P < 0.05; **P < 0.01
FLNa increases human melanoma cell proliferation in nude mice xenograft model
The in vivo proliferative properties of FLNa were tested after subcutaneous implantation of UACC647(ctrl) and UACC647(KD) cells in nude mice. These cells were used because of their ability to form solid tumors in nude mice and their high proliferative potential. The tumor size was measured over a 4-week period after which animals were killed, and tumor masses were weighted and processed for further analysis. A significant reduction in melanoma tumor growth was observed in FLNa-deficient UACC647(KD) cells as compared to UACC647(ctrl) cells (Fig. 4a, b). However, there were no visualized distant sites to evaluate the changes of metastasis in FLNa altered mice.
Fig. 4.
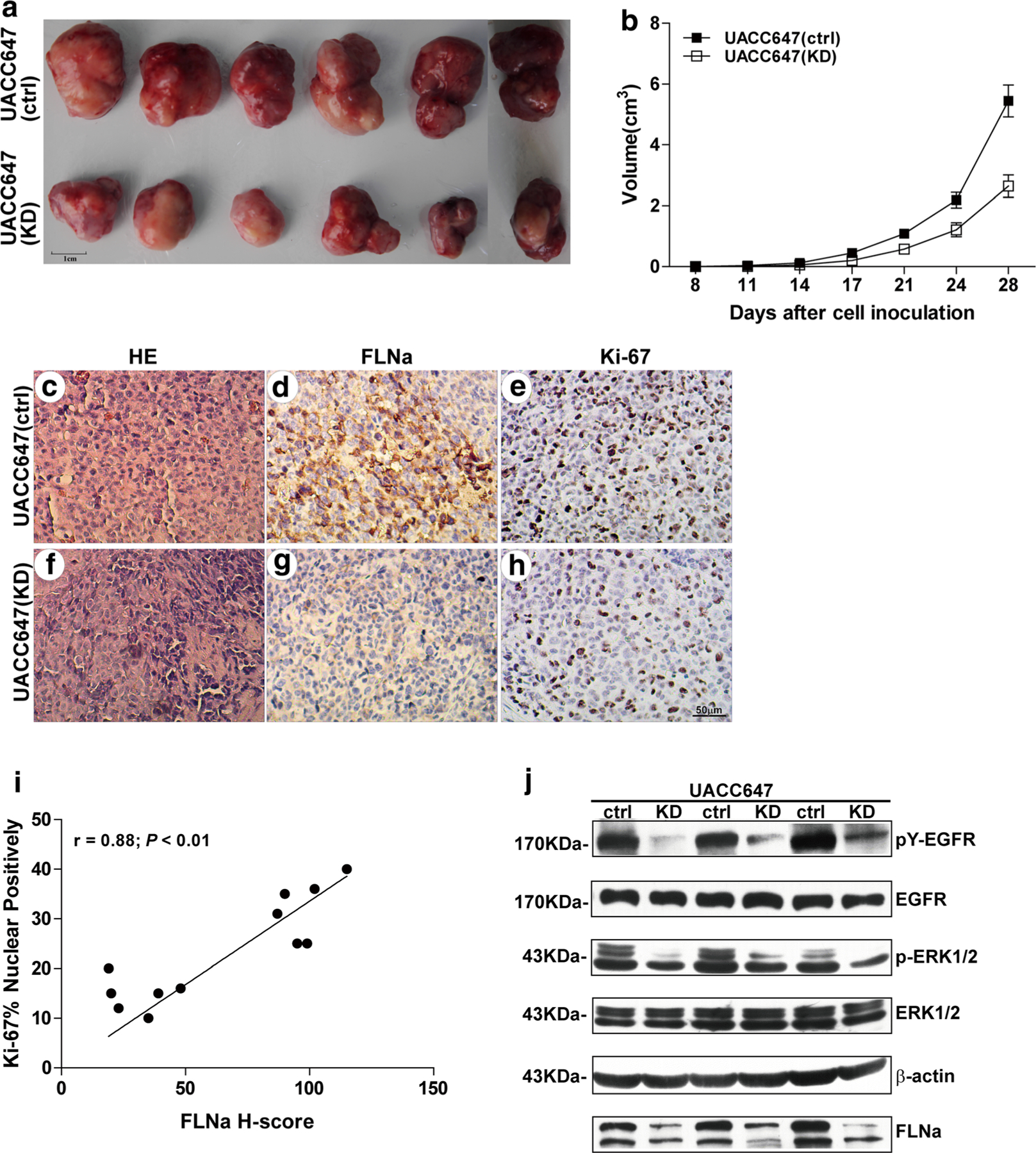
Impaired tumor growth by FLNa knockdown in tumor-bearing mice. Tumor-bearing mice were generated by subcutaneous injection of UACC647(ctrl) and UACC647(KD) melanoma cells. Mice were killed 28 days after inoculation. a Representative photographs of tumors after surgical resection. Bar 1 cm. b Tumor growth in mice injected with UACC647(ctrl) and UACC647(KD) cells. Tumor volume in each group was expressed as mean ± SEM. (N = 6). IHC staining with FLNa, Ki-67 and H&E staining of tumor tissue sections from mice injected with UACC647(ctrl) (c–e) and UACC647(KD) (f–h) cells. Bar 50 μm. i Correlation between the H-score for FLNa and Ki-67 (r = 0.88; P < 0.01; y = 7.62 + 0.24x; r2 = 0.77). j Western blot analysis was performed with a portion of tumor lysates using antibodies against FLNa, phosphotyrosine (4G10 clone), EGFR, phosphorylated and total ERK1/2, or β-actin shown here as a loading control
Immunohistochemical analysis illustrated that the diffuse and strong nuclear staining of Ki-67 (Fig. 4e) correlated with strong FLNa cytoplasmic signal in tumors from UACC647 (ctrl) cells (Fig. 4d). In contrast, tumor tissues from UACC647(KD) cells exhibited weak staining of Ki-67 (Fig. 4h) and low levels of FLNa staining (Fig. 4g). The H-score for FLNa was 64.33 ± 10.59, and a significant positive correlation was detected between the immunoreactivity for FLNa and Ki-67 (r = 0.88; P < 0.01; Fig. 4i). Thus, knockdown of FLNa correlated with reduction in melanoma growth and the proportion of FLNa-positive cells in these tumor xenografts.
Western blot analysis revealed a significant attenuation in pY-EGFR and pERK1/2 levels in UACC647(KD) cells by 80 and 46 %, respectively (Fig. 4j). These data support the notion that knockdown of FLNa decreases proliferation and melanoma tumor growth both in vitro and in vivo.
Reduction of FLNa predicts better survival in malignant melanoma
Our results suggest that reduction in FLNa expression alters the motility and growth of melanoma cells. IHC staining of FLNa (Fig. 5b, e) and Ki-67 (Fig. 5c, f) on a panel of paraffin-embedded malignant melanoma tissues was performed and was found to validate these cellular and biochemical findings. Based on the scoring by two independent pathologists, the H-score value (mean ± SEM) for FLNa in 30 cases was 69.77 ± 5.17. There were significant positive correlations between the immunoreactivity for FLNa and Ki-67 (r = 0.60; P < 0.01; Fig. 5g).
Fig. 5.
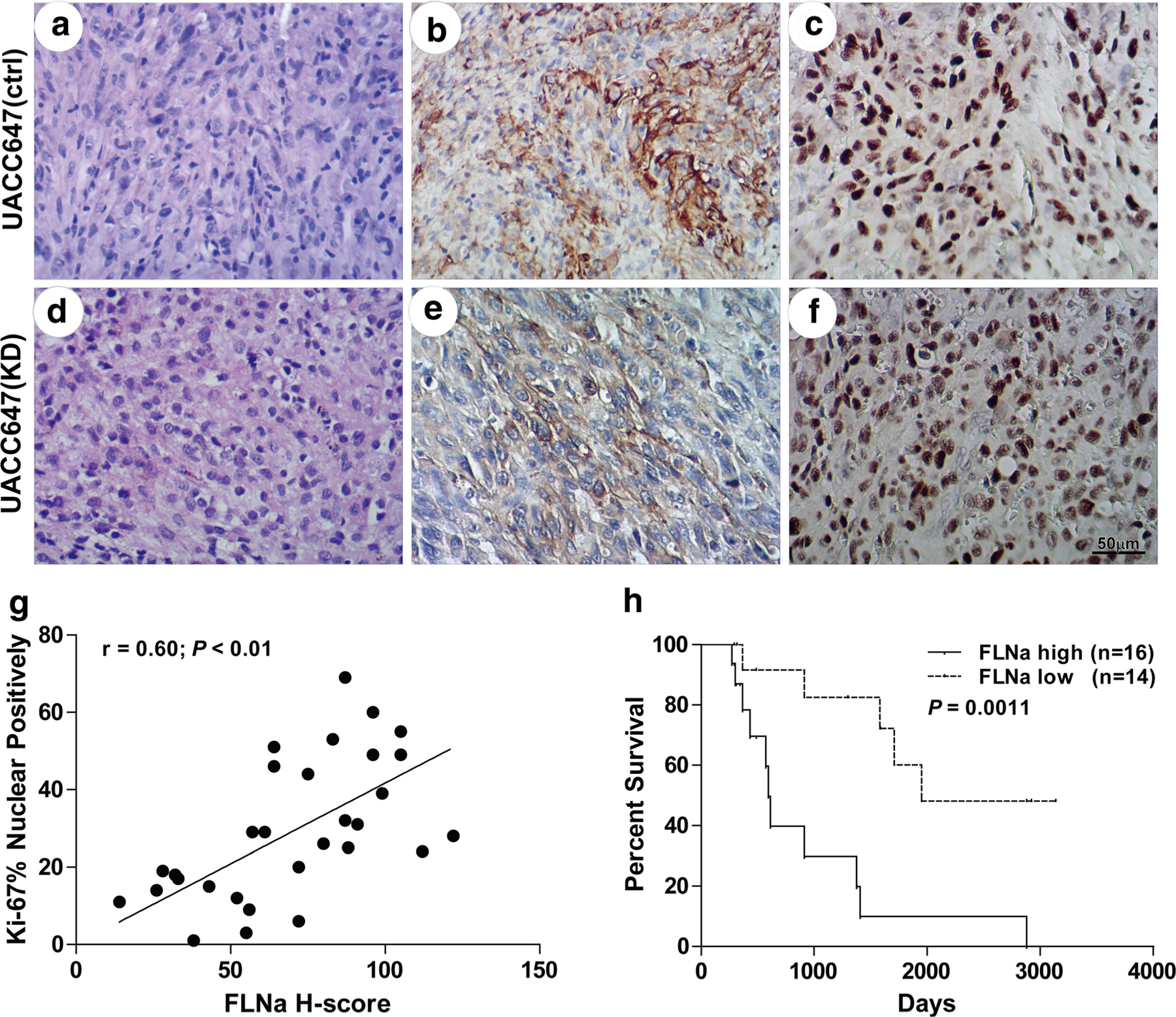
Correlation of FLNa expression status with malignancy and patient survival. H&E and immunohistochemical staining for FLNa and Ki-67 was performed with paraffin-embedded malignant melanoma tissues from patients with high H-values of FLNa (a-c) and low H-values (d–f). Bar 50 μm. g Correlation between the H-score for FLNa and Ki-67 (r = 0.60; P < 0.01; y = 2.65 + 0.38x; r2 = 0.36). h Kaplan–Meier survival analysis of patients with malignant melanoma tumors. A significant difference was found between the overall survival of patients with melanoma tumors that have low and high FLNa levels (Hazard Ratio 6.232, 95 % CI of ratio 2.073 to 18.73, P value by Log–rank 0.0011)
The association between FLNa immunoreactivity and clinical characteristics is summarized in Table 1. There was an inverse correlation between FLNa immunoreactivity and over survival (r = −0.45; P = 0.013). Gather the cases based on the median value of H-score, patients with high FLNa expression in their tumors had a markedly lower survival (median survival was 594 days) than those with low FLNa content (median survival was 1,949 days; P = 0.0011; Fig. 5h). However, in this study, there was no significant relationship between FLNa immunoreactivity and patient age, sex and recurrence or metastases.
Table 1.
The relationship between FLNa expression and clinical characteristics of patients with melanoma tumors
| n | FLNa H-score | p | r | |
|---|---|---|---|---|
| Number of patients | 30 | N/A | N/A | N/A |
| Age (years) | N/A | N/A | 0.746 | −0.06 |
| Gender | 0.532 | |||
| Male | 15 | 66.47 ± 6.25 | ||
| Female | 15 | 73.07 ± 8.36 | ||
| Recurrence/metastases | 0.071 | |||
| Yes | 20 | 76.35 ± 4.89 | ||
| No | 10 | 56.60 ± 11.33 | ||
| Vital status | 0.030 | |||
| Alive | 14 | 58.00 ± 7.64 | ||
| Deceased | 16 | 80.06 ± 6.11 | ||
| Over survival (days) | N/A | N/A | 0.013 | −0.45 |
| Ki-67 % positivity | N/A | N/A | <0.01 | 0.6 |
Discussion
FLNa binds to actin and participates in cytoskeletal rear-rangement. By virtue of its scaffolding function, FLNa also interacts with more than 90 functionally diverse cellular proteins, many of which participate in signaling complexes needed for oncogenesis and metastasis (Zhou et al. 2010; Razinia et al. 2012; Nakamura et al. 2011). The aims of this study were to determine the role of FLNa in proliferation, migration and invasion of melanoma cells in vitro and melanoma development in vivo.
The knockdown of FLNa was associated with a decrease in metastasis and proliferation of human melanoma cells in vitro. These studies also illustrated that EGF-induced EGFR activation up-regulates cell motility and growth, in agreement with an earlier report (Lemmon and Schlessinger 2010). Like FLNa, EGFR is an actin-binding protein. Upon EGF binding, EGFR undergoes dimerization and autophosphorylation, thereby triggering activation of downstream signaling cascades, including the Ras-Raf-MAP kinase pathway (Avraham and Yarden 2011), with the latter serving as a converging point in all known biological functions acquired during the multistep development of human tumors (Hanahan and Weinberg 2011). It is unclear whether varying levels of EGFR phosphorylation has any impact on expression and/or ability of FLNa to interact with binding partners.
EGF-induced activation of EGFR is a dynamic process, which includes autophosphorylation of the cell surface receptor on tyrosine residues and subsequent internalization, ubiquitination and degradation of the internalized EGFR (Citri and Yarden 2006; Yarden and Pines 2012; Grandal and Madshus 2008). Dysregulation of the endocytic and/or trafficking pathway can result in tumorigenesis. In the presence of FLNa, ligand-bound, tyrosine-phosphorylated EGFR is internalized and sorted into the degradation pathway (Fiori et al. 2009). Although we were unable to demonstrate direct interaction between FLNa and EGFR, our results support the notion that FLNa enhances proliferation, migration and invasion in melanoma cells in vitro through increase in EGFR activity and activation of downstream signaling.
The Ras/Raf/MEK/ERK signaling cascade which has been shown to contribute to melanoma tumorigenesis by increasing cell proliferation and tumor metastasis, and by inhibiting apoptosis is a hallmark of cutaneous malignant melanoma. Ras and B-Raf was found to be mutated in approximately 15–20 and 40–60 % of human malignant melanoma, respectively (Fernandez-Flores 2012). And the activated ERK was found in almost all late-stage melanoma cell lines and in tumor tissues, which plays a pivotal role in melanoma cell proliferation by controlling the G1-phase to S-phase transition, increasing transcription and stability of c-Jun and inhibition differentiation (McCubrey et al. 2007; Neuzillet et al. 2014). Activation of this pathway not only increase proliferation of melanoma cells but also promote their metastatic potential. A critical step of metastatic is degradation of the extracellular matrix to allow extravasation and migration of the metastatic cells. It has been demonstrated that the Ras/Raf/MEK/ERK signaling cascade is active in melanoma and is the dominant pathway to regulate the expression and activity of matrix metalloproteinases (MMPs) which are involved in matrix remodeling(Kim and Choi 2010). Thus, the possible molecular mechanism of FLNa promotes proliferation and metastasis in melanoma is through up-regulate the activity of Ras/Raf/MEK/ERK pathway during EGF stimulated.
And FLNa were also direct required for normal cell migration, although in some cases defects in migration were not evident following loss of a single FLN isoform. The FLNa-deficient cells fail to polarize and move because they have highly unstable surfaces that continuously expand and contract circumferential blebs (Flevaris et al. 2007). But overexpression of FLNa could prevent migration by sequestering signaling molecules from their normal position and altering F-actin and focal adhesion turnover to enhance adhesion.
Since FLNa interacts with many proteins related to cancer metastasis, it displays a dual role in cell migration, spreading and invasion. Appropriate FLNa levels are essential in tumor cells keep motility, invasive, resist mechanical stress and attachment to extracellular matrix (ECM) to form metastases. In another side, Baldassarre et al. showed that reduction of FLNa in fibrosarcoma increased matrix metalloprotease 2 (MMP-2) activity and enhanced the ability of cell to remodel ECM. And FLNa also act as a negative regulator of integrin activation that is essential in cell migrate (Kim and McCulloch 2011). The apparent discrepancies among the researches indicate that the role of FLNa in metastases dependent on the type of tumor cells, concurrent expression of other relevant genes in the tumor cells, their interactions with the extracellular environment and the proteolysis of FLNa.
Various mechanisms about the complexity of molecules in tumorigenesis have been described, some of them probably showing this phenomenon due to clonal heterogeneity of the tumors (Grunewald et al. 2011). It means a functional and/or phenol-typical differentiation in various subsets of tumor cells. Melanoma is not always a genetic-related malignancy but many times an epigenetic one. Because of the genetic heterogeneity, the regulators affected by variations of miRNAs or methylation were often specific to the cell types and may have different functions in the primary and distal tumors (Bullock et al. 2012; Cock-Rada and Weitzman 2013). Evidence suggested that the Ras/Raf/MEK/ERK pathway is required for epithelial to mesenchymal transition (EMT) and contributes to the maintenance of an undifferentiated/mesenchymal state in tumor cells (Maurer et al. 2011). It cooperates with other pathways to up-regulate expression of EMT-related genes, including mesenchymal genes and transcription repressors of epithelial genes (Cano et al. 2010). Hence, the effects of FLNa performed on several compartments via the Ras/Raf/MEK/ERK pathway perhaps resulted the promotion in melanoma proliferation and metastases in this study.
In vivo experiments in nude mice illustrated that the knockdown of FLNa in UACC647 cells led to lower tumor growth and that the volume and weight of tumors were correlated with the phosphorylation status of EGFR and ERK. But, there were no visualized metastases to be detected in both groups. Possibly, the reason is that 4 weeks is too short to generate metastases after subcutaneous inoculation of tumor cells. A recent study has reported the reduction of K-RAS-induced tumor growth in vivo and decrease in fibroblast proliferation in vitro through down-regulation of activated ERK and AKT after knockout of the FLNa gene (Nallapalli et al. 2012). One potential explanation for these results is that FLNa is required for efficient spatiotemporal activation of effectors in the RAS pathway; another possibility is that FLNa is involved in regulating the dynamics of the actin cytoskeleton and that the impact of FLNa deficiency on tumor growth reflects a more general role in cellular structure and function; and, lastly, perhaps a more intriguing possibility, is that cleavage of FLNa in the hinge region enables its translocation to the nucleus to regulate gene transcription (Loy et al. 2003).
A number of studies have identified the impact of FLNa status in cancer prognosis. For example, the levels of FLNa are correlated with the pathological grade in gliomas (Alper et al. 2009) and in prostate cancer, the cytoplasmic localization of full-length FLNa correlates with metastasis (Bedolla et al. 2009). Here, our data illustrated a poor survival index for patients with melanoma tumors that have high FLNa levels. The association between FLNa and Ki-67 staining provides clinical evidence for the need to investigate further the interaction of FLNa with EGFR at the molecular level.
In conclusion, this study provides new insight into the biology of FLNa in vitro and in vivo. The molecular mechanism(s) by which FLNa affects ligand-induced activation of EGFR remains unclear, but may involve the participation of, yet to be identified, interaction partner(s).
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81071846, to T.N.Z); Natural Science Foundation of Hebei Province of China (No. H2013505059, to T.N.Z); Department of Science and Technology of Hebei Province of China (No. 12396107D, to R.J.Z); Wu Jieping Foundation (No. 320.6750.12604, to T.N.Z) and, in part, by the Intramural Research Program of National Institutes of Health, National Institute on Aging.
Abbreviations
- FLNa
Filamin A
- EGFR
Epidermal growth factor receptor
- ERK
Ras-extracellular signal-regulated kinase
- JNK
c-Jun NH2-terminal kinase
- MEM
Minimal essential medium
- FBS
Fetal bovine serum
- SFM
Serum-free medium
- EGF
Epidermal growth factor
- ECL
Enhanced chemiluminescence
- pY-EGFR
Phosphorylated form of EGFR
- pERK1/2
Phosphorylated ERK1/2
- IHC
Immunohistochemical
- MMPs
Matrix metalloproteinases
- ECM
Extracellular matrix
Footnotes
Conflict of interest We, all authors declare that there is no any financial arrangements that could be related to the manuscript and confirm that we do not have any disclosure to make at submission. None of the authors has any potential financial conflict of interest related to this manuscript.
Contributor Information
Kai Zhang, Department of Oncology, Hebei Medical University, Shijiazhuang 050017, Hebei, China.
Tienian Zhu, Key Laboratory of Immune Mechanism and Intervention, on Serious Disease in Hebei Province, Department of Immunology, Hebei Medical University, Shijiazhuang 050017, Hebei, China; Department of Medical Oncology, Bethune International Peace, Hospital, Shijiazhuang 050082, Hebei, China.
Dongmei Gao, Department of Medical Oncology, Bethune International Peace, Hospital, Shijiazhuang 050082, Hebei, China.
Yimei Zhang, Department of Medical Oncology, Bethune International Peace, Hospital, Shijiazhuang 050082, Hebei, China.
Qinglan Zhao, Department of Medical Oncology, Bethune International Peace, Hospital, Shijiazhuang 050082, Hebei, China.
Shuang Liu, Department of Pathology, Bethune International Peace Hospital, Shijiazhuang 050082, Hebei, China.
Tongyi Su, Department of Medical Oncology, Bethune International Peace, Hospital, Shijiazhuang 050082, Hebei, China.
Michel Bernier, Translational Gerontology Branch, National Institute on Aging, National Institutes of Health, Baltimore, Maryland 21224, USA.
Ruijing Zhao, Key Laboratory of Immune Mechanism and Intervention, on Serious Disease in Hebei Province, Department of Immunology, Hebei Medical University, Shijiazhuang 050017, Hebei, China.
References
- Alper O, Stetler-Stevenson WG, Harris LN, Leitner WW, Ozdemirli M, Hartmann D, Raffeld M, Abu-Asab M, Byers S, Zhuang Z, Oldfield EH, Tong Y, Bergmann-Leitner E, Criss WE, Nagasaki K, Mok SC, Cramer DW, Karaveli FS, Goldbach-Mansky R, Leo P, Stromberg K, Weil RJ (2009) Novel anti-filamin-A antibody detects a secreted variant of filamin-A in plasma from patients with breast carcinoma and high-grade astrocytoma. Cancer Sci 100(9):1748–1756. doi: 10.1111/j.1349-7006.2009.01244.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham R, Yarden Y (2011) Feedback regulation of EGFR signaling: decision making by early and delayed loops. Nat Rev Mol Cell Biol 12(2):104–117. doi: 10.1038/nrm3048 [DOI] [PubMed] [Google Scholar]
- Barriere H, Nemes C, Du K, Lukacs GL (2007) Plasticity of poly-ubiquitin recognition as lysosomal targeting signals by the endosomal sorting machinery. Mol Biol Cell 18(10):3952–3965. doi: 10.1091/mbc.E07-07-0678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Albanell J (2002) Epithelial growth factor receptor interacting agents. Hematol/Oncol Clin N Am 16(5):1041–1063 [DOI] [PubMed] [Google Scholar]
- Bedolla RG, Wang Y, Asuncion A, Chamie K, Siddiqui S, Mudryj MM, Prihoda TJ, Siddiqui J, Chinnaiyan AM, Mehra R, de Vere White RW, Ghosh PM (2009) Nuclear versus cytoplasmic localization of filamin A in prostate cancer: immunohistochemical correlation with metastases. Clin Cancer Res 15(3):788–796. doi: 10.1158/1078-0432.CCR-08-1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullock MD, Sayan AE, Packham GK, Mirnezami AH (2012) Micro-RNAs: critical regulators of epithelial to mesenchymal (EMT) and mesenchymal to epithelial transition (MET) in cancer progression. Biol Cell 104(1):3–12. doi: 10.1111/boc.201100115 [DOI] [PubMed] [Google Scholar]
- Cano CE, Motoo Y, Iovanna JL (2010) Epithelial-to-mesenchymal transition in pancreatic adenocarcinoma. Sci World J 10:1947–1957. doi: 10.1100/tsw.2010.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Yarden Y (2006) EGF–ERBB signaling: towards the systems level. Nat Rev Mol Cell Biol 7(7):505–516. doi: 10.1038/nrm1962 [DOI] [PubMed] [Google Scholar]
- Cock-Rada A, Weitzman JB (2013) The methylation landscape of tumour metastasis. Biol Cell 105(2):73–90. doi: 10.1111/boc.201200029 [DOI] [PubMed] [Google Scholar]
- Djinovic-Carugo K, Carugo O (2010) Structural portrait of filamin interaction mechanisms. Cur Protein Pept Sci 11(7):639–650 [DOI] [PubMed] [Google Scholar]
- Eggermont AM, Spatz A, Robert C (2014) Cutaneous melanoma. Lancet 383(9919):816–827. doi: 10.1016/S0140-6736(13)60802-8 [DOI] [PubMed] [Google Scholar]
- Fernandez-Flores A (2012) Prognostic factors for melanoma progression and metastasis: from Hematoxylin–Eosin to genetics. Rom J Morphol Embryol 53(3):449–459 [PubMed] [Google Scholar]
- Fiori JL, Zhu TN, O’Connell MP, Hoek KS, Indig FE, Frank BP, Morris C, Kole S, Hasskamp J, Elias G, Weeraratna AT, Bernier M (2009) Filamin A modulates kinase activation and intracellular trafficking of epidermal growth factor receptors in human melanoma cells. Endocrinology 150(6):2551–2560. doi: 10.1210/en.2008-1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan MB, Dabbs DJ, Brufsky AM, Beriwal S, Bhargava R (2008) Histopathologic variables predict Oncotype DX recurrence score. Mod Pathol 21(10):1255–1261. doi: 10.1038/modpathol.2008.54 [DOI] [PubMed] [Google Scholar]
- Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X (2007) A molecular switch that controls cell spreading and retraction. J Cell Biol 179(3):553–565. doi: 10.1083/jcb.200703185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandal MV, Madshus IH (2008) Epidermal growth factor receptor and cancer: control of oncogenic signaling by endocytosis. J Cell Mol Med 12(5A):1527–1534. doi: 10.1111/j.1582-4934.2008.00298.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grovdal LM, Stang E, Sorkin A, Madshus IH (2004) Direct interaction of Cbl with pTyr 1045 of the EGF receptor (EGFR) is required to sort the EGFR to lysosomes for degradation. Exp Cell Res 300(2):388–395. doi: 10.1016/j.yexcr.2004.07.003 [DOI] [PubMed] [Google Scholar]
- Grunewald TG, Herbst SM, Heinze J, Burdach S (2011) Understanding tumor heterogeneity as functional compartments—superor-ganisms revisited. J Trans Med 9:79. doi: 10.1186/1479-5876-9-79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674. doi: 10.1016/j.cell.2011.02.013 [DOI] [PubMed] [Google Scholar]
- He HJ, Zhu TN, Xie Y, Fan J, Kole S, Saxena S, Bernier M (2006) Pyrrolidine dithiocarbamate inhibits interleukin-6 signaling through impaired STAT3 activation and association with transcriptional coactivators in hepatocytes. J Biol Chem 281(42):31369–31379. doi: 10.1074/jbc.M603762200 [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, Dicara D, Ramos AH, Lawrence MS, Cibulskis K, Sivachenko A, Voet D, Saksena G, Stransky N, Onofrio RC, Winckler W, Ardlie K, Wagle N, Wargo J, Chong K, Morton DL, Stemke-Hale K, Chen G, Noble M, Meyerson M, Ladbury JE, Davies MA, Gershenwald JE, Wagner SN, Hoon DS, Schadendorf D, Lander ES, Gabriel SB, Getz G, Garraway LA, Chin L (2012) A landscape of driver mutations in melanoma. Cell 150(2):251–263. doi: 10.1016/j.cell.2012.06.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes NE, MacDonald G (2009) ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol 21(2):177–184. doi: 10.1016/j.ceb.2008.12.010 [DOI] [PubMed] [Google Scholar]
- Jiang X, Yue J, Lu H, Campbell N, Yang Q, Lan S, Haffty BG, Yuan C, Shen Z (2013) Inhibition of filamin-A reduces cancer metastatic potential. Int J Biol Sci 9(1):67–77. doi: 10.7150/ijbs.5577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EK, Choi EJ (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802(4):396–405. doi: 10.1016/j.bbadis.2009.12.009 [DOI] [PubMed] [Google Scholar]
- Kim H, McCulloch CA (2011) Filamin A mediates interactions between cytoskeletal proteins that control cell adhesion. FEBS Lett 585(1):18–22. doi: 10.1016/j.febslet.2010.11.033 [DOI] [PubMed] [Google Scholar]
- Kim H, Nakamura F, Lee W, Hong C, Perez-Sala D, McCulloch CA (2010) Regulation of cell adhesion to collagen via beta1 integrins is dependent on interactions of filamin A with vimentin and protein kinase C epsilon. Exp Cell Res 316(11):1829–1844. doi: 10.1016/j.yexcr.2010.02.007 [DOI] [PubMed] [Google Scholar]
- Kim Y, Stolarska MA, Othmer HG (2011) The role of the microenvironment in tumor growth and invasion. Prog Biophys Mol Biol 106(2):353–379. doi: 10.1016/j.pbiomolbio.2011.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunz M (2014) Oncogenes in melanoma: an update. Eur J Cell Biol 93(1–2):1–10. doi: 10.1016/j.ejcb.2013.12.002 [DOI] [PubMed] [Google Scholar]
- Kyndt F, Gueffet JP, Probst V, Jaafar P, Legendre A, Le Bouffant F, Toquet C, Roy E, McGregor L, Lynch SA, Newbury-Ecob R, Tran V, Young I, Trochu JN, Le Marec H, Schott JJ (2007) Mutations in the gene encoding filamin A as a cause for familial cardiac valvular dystrophy. Circulation 115(1):40–49. doi: 10.1161/CIRCULATIONAHA.106.622621 [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Schlessinger J (2010) Cell signaling by receptor tyrosine kinases. Cell 141(7):1117–1134. doi: 10.1016/j.cell.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung R, Wang Y, Cuddy K, Sun C, Magalhaes J, Grynpas M, Glogauer M (2010) Filamin A regulates monocyte migration through Rho small GTPases during osteoclastogenesis. J Bone Min Res 25(5):1077–1091. doi: 10.1359/jbmr.091114 [DOI] [PubMed] [Google Scholar]
- Loy CJ, Sim KS, Yong EL (2003) Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc Natl Acad Sci USA 100(8):4562–4567. doi: 10.1073/pnas.0736237100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer G, Tarkowski B, Baccarini M (2011) Raf kinases in cancer-roles and therapeutic opportunities. Oncogene 30(32):3477–3488. doi: 10.1038/onc.2011.160 [DOI] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA (2007) Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta 1773(8):1263–1284. doi: 10.1016/j.bbamcr.2006.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn J, Baselga J (2003) Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J Clin Oncol 21(14):2787–2799. doi: 10.1200/JCO.2003.01.504 [DOI] [PubMed] [Google Scholar]
- Nakamura F, Heikkinen O, Pentikainen OT, Osborn TM, Kasza KE, Weitz DA, Kupiainen O, Permi P, Kilpelainen I, Ylanne J, Hartwig JH, Stossel TP (2009) Molecular basis of filamin A-FilGAP interaction and its impairment in congenital disorders associated with filamin A mutations. PLoS ONE 4(3):e4928. doi: 10.1371/journal.pone.0004928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Stossel TP, Hartwig JH (2011) The filamins: organizers of cell structure and function. Cell Adh Migr 5(2):160–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nallapalli RK, Ibrahim MX, Zhou AX, Bandaru S, Sunkara SN, Redfors B, Pazooki D, Zhang Y, Boren J, Cao Y, Bergo MO, Akyurek LM (2012) Targeting filamin A reduces K-RAS-induced lung adenocarcinomas and endothelial response to tumor growth in mice. Mol Cancer 11:50. doi: 10.1186/1476-4598-11-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuzillet C, Tijeras-Raballand A, de Mestier L, Cros J, Faivre S, Raymond E (2014) MEK in cancer and cancer therapy. Pharmacol Ther 141(2):160–171. doi: 10.1016/j.pharmthera.2013.10.001 [DOI] [PubMed] [Google Scholar]
- Ravid D, Chuderland D, Landsman L, Lavie Y, Reich R, Liscovitch M (2008) Filamin A is a novel caveolin-1-dependent target in IGFI-stimulated cancer cell migration. Exp Cell Res 314(15):2762–2773. doi: 10.1016/j.yexcr.2008.06.004 [DOI] [PubMed] [Google Scholar]
- Razinia Z, Makela T, Ylanne J, Calderwood DA (2012) Filamins in mechanosensing and signaling. Ann Rev Biophys 41:227–246. doi: 10.1146/annurev-biophys-050511-102252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roepstorff K, Grovdal L, Grandal M, Lerdrup M, van Deurs B (2008) Endocytic downregulation of ErbB receptors: mechanisms and relevance in cancer. Histochem Cell Biol 129(5):563–578. doi: 10.1007/s00418-008-0401-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoy RM, Ghosh PM (2013) The dual role of filamin A in cancer: can’t live with (too much of) it, can’t live without it. Endocr-Related Cancer 20(6):R341–R356. doi: 10.1530/ERC-13-0364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kreisberg JI, Bedolla RG, Mikhailova M, de Vere White RW, Ghosh PM (2007) A 90 kDa fragment of filamin A promotes Casodex-induced growth inhibition in Casodex-resistant androgen receptor positive C4–2 prostate cancer cells. Oncogene 26(41):6061–6070. doi: 10.1038/sj.onc.1210435 [DOI] [PubMed] [Google Scholar]
- Wickstead B, Gull K (2011) The evolution of the cytoskeleton. J Cell Biol 194(4):513–525. doi: 10.1083/jcb.201102065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden Y, Pines G (2012) The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer 12(8):553–563. doi: 10.1038/nrc3309 [DOI] [PubMed] [Google Scholar]
- Yewale C, Baradia D, Vhora I, Patil S, Misra A (2013) Epidermal growth factor receptor targeting in cancer: a review of trends and strategies. Biomaterials 34(34):8690–8707. doi: 10.1016/j.biomaterials.2013.07.100 [DOI] [PubMed] [Google Scholar]
- Zhou AX, Hartwig JH, Akyurek LM (2010) Filamins in cell signaling, transcription and organ development. Trends Cell Biol 20(2):113–123. doi: 10.1016/j.tcb.2009.12.001 [DOI] [PubMed] [Google Scholar]
- Zhu TN, He HJ, Kole S, D’Souza T, Agarwal R, Morin PJ, Bernier M (2007) Filamin A-mediated down-regulation of the exchange factor Ras-GRF1 correlates with decreased matrix metalloproteinase-9 expression in human melanoma cells. J Biol Chem 282(20):14816–14826. doi: 10.1074/jbc.M611430200 [DOI] [PubMed] [Google Scholar]