

Abstract
The pathology of fetal alcohol syndrome and the less severe fetal alcohol spectrum disorders includes brain dysmyelination. Recent studies have shed light on the molecular mechanisms underlying these white matter abnormalities. Rodent models of fetal alcohol syndrome and human studies have shown suppressed oligodendrocyte differentiation and apoptosis of oligodendrocyte precursor cells. Ethanol exposure led to reduced expression of myelin basic protein and delayed myelin basic protein expression in rat and mouse models of fetal alcohol syndrome and in human histopathological specimens. Several studies have reported increased expression of many chemokines in dysmyelinating disorders in central nervous system, including multiple sclerosis and fetal alcohol syndrome. Acute ethanol exposure reduced levels of the neuroprotective insulin-like growth factor-1 in fetal and maternal sheep and in human fetal brain tissues, while ethanol increased the expression of tumor necrosis factor α in mouse and human neurons. White matter lesions have been induced in the developing sheep brain by alcohol exposure in early gestation. Rat fetal alcohol syndrome models have shown reduced axon diameters, with thinner myelin sheaths, as well as reduced numbers of oligodendrocytes, which were also morphologically aberrant oligodendrocytes. Expressions of markers for mature myelination, including myelin basic protein, also were reduced. The accumulating knowledge concerning the mechanisms of ethanol-induced dysmyelination could lead to the development of strategies to prevent dysmyelination in children exposed to ethanol during fetal development. Future studies using fetal oligodendrocyte- and oligodendrocyte precursor cell-derived exosomes isolated from the mother’s blood may identify biomarkers for fetal alcohol syndrome and even implicate epigenetic changes in early development that affect oligodendrocyte precursor cell and oligodendrocyte function in adulthood. By combining various imaging modalities with molecular studies, it may be possible to determine which fetuses are at risk and to intervene therapeutically early in the pregnancy.
Key Words: alcohol, development, dysmyelination, ethanol, fetal alcohol syndrome, fetal brain, myelin basic protein, neurodegeneration, oligodendrocyte injury, oligodendrocyte precursor cells
Introduction
The pathophysiology of fetal alcohol syndrome (FAS) includes prominent dysmyelination in the brain due to abnormal development of oligodendrocytes (OLs). This article reviews recent studies on the effects of prenatal ethanol exposure on OL development. After describing the clinical and pathological features of FAS, including OL loss, we summarize the effects of alcohol exposure on markers of oligodendrocyte precursor cell (OPC) and OL mRNA and protein expression, and on the expression of specific cytokines and chemokines in the developing brain that may affect human fetal OL differentiation. We especially focus on tumor necrosis factor-α (TNF-α) and insulin-like growth factor-1 (IGF-1) because of their strong mutually antagonistic actions on cell survival in many tissues. Table 1 summarizes these data in human fetal brain. The discussion is extended to white matter defects seen in the less severe fetal alcohol spectrum disorders (FASDs). Table 2 summarizes recent information on the effects of EtOH exposure on OPC/OL markers in animal and human fetuses. Finally, we discuss the translational potential of the OL research on the mechanisms by which alcohol use during pregnancy might cause dysmyelination in fetal brain, summarized in Figure 1. In a concluding section on Future Directions, we pose the possibility that epigenetic changes in early development that can affect OPC and OL function in adulthood. OL- and OPC-derived exosomes could reveal histone modifications and miRNA profiles as diagnostic tools. Diffusion tensor imaging (DTI) and functional magnetic imaging (fMRI) could be used together with molecular studies to examine the correlation between OL injury and brain structural and functional connectivity in the same population. By providing greater insight into the mechanisms underlying prenatal alcohol exposure-related neurocognitive deficits, these methods may help to determine which fetuses are at risk and allow us to intervene therapeutically early in the pregnancy.
Table 1.
EtOH exposure and expression of OPC/OL markers and cytokines/chemokines in human fetal brain
| Down-regulation | Up-regulation | |
|---|---|---|
| OPC/OL markers | ||
| Oct4 | ↑ 5.1 | |
| Nanog | ↑ 2.1 | |
| NG2 | ↑ 1.3 | |
| A2B5 | ↑ 1.1 | |
| Nkx2.2 | ↓ 0.5 | |
| PDGFR | ↓ 1.9 | |
| Olig1 | ↓ 1.8 | |
| Olig2 | ↓ 1.9 | |
| MBP | ↓ 2.2 | |
| Neuronal markers | ||
| β-III Tubulin | ↓ 1.4 | |
| NeuN | ↓ 2.3 | |
| Cytokines / Chemokines | ||
| CXCL1/GRO | ↑ 3.8 | |
| IL-8 | ↑ 3.1 | |
| GCP2/CXCL6 | ↑ 5 | |
| ENA-78 | ↑ 5 | |
| MCP1 | ↑ 4.1 | |
| TNF-α | ↑ 7.3 | |
| IGF-1 | ↓ 4.9 | |
| Apoptosis | ||
| Active Caspase-3 | ↑ 2 |
Markers of OL lineage progression from immature to mature. After elective termination of pregnancy, levels of mRNAs and proteins for each stage of OL maturation were determined by qRT-PCR and FACS in fetal whole brain homogenates, and graphed as Fold Regulation. Values are normalized relative to EtOH-unexposed controls (n = 6 per group, GA = 18.6 to 21.3 weeks for Control group vs. GA = 19.3 to 21.4 weeks for the EtOH-exposed group). EtOH exposure increases cytotoxic cytokines/chemokines expression in male fetuses more than in females. Effects of maternal EtOH use on expression of cytokines/chemokines in fetal brain during OL lineage progression. Cytokine transcription was assayed by Real-Time qRT-PCR for: CXC ligands CXCL1/GRO, IL-8, GCP2/CXCL6, ENA-78, MCP1, TNF-α and IGF-1 mRNA using brain RNA and presented as fold regulation relative to unexposed controls) (Darbinian et al., 2021). A2B5: Cell surface ganglioside, marker of developing oligodendroglial progenitors and type 2 astrocyte precursor marker; CXC: cysteine-x-cysteine; CXCL1/GRO: growth regulated protein alpha/chemokine (C-X-C motif) ligand 1; ENA-78: epithelial-derived neutrophil-activating protein 78; FACS: fluorescence activated cell sorting; GCP2/CXCL6: granulocyte chemotactic protein 2/Chemokine (C-X-C motif) ligand 6; IGF-1: insulin-like growth factor-1; IL-8: interleukin 8/chemokine (C-X-C motif) ligand 8; MBP: myelin basic protein; MCP1: monocyte chemoattractant protein-1; Nanog: Tír na nÓg (Land of Youth) (Celtic land of youth) homeobox transcription factor; NeuN: neuronal nuclear monoclonal antibody; NG2: neural/glial antigen 2; Nkx2.2: nuclear transcription factor NK-2 homeobox locus 2; Oct4: octamer binding protein OCT4 (POU5F1) POU family transcription factor; Olig1: oligodendrocyte transcription factor 1; Olig2: oligodendrocyte transcription factor 2; OPC/OL: oligodendrocyte precursor cells/oligodendrocytes; PDGFR: platelet-derived growth factor receptor; qRT-PCR: quantitative reverse transcription polymerase chain reaction; TNF-α: tumor necrosis factor-α.
Table 2.
Effects of EtOH exposure on OPC/OL markers in animals and humans
| Reference | Organism | EtOH dose, mode/ timing of administration | Assay method/ parameter | Results |
|---|---|---|---|---|
| Sowell et al., 2002 | Human | FAS/FASD, Prenatal alcohol exposure four or more drinks per occasion at least once per week or 14 drinks or more per week 5–15 years old | MRI | Less white matter Less grey matter Volume reductions in striatal and thalamic regions bilaterally and in right prefrontal and left occipitoparietal cortices |
| Lebel et al., 2011 (33 studies on MRI) | Human | FAS/FASD 5–37 years old | MRI | Reduced brain volume Malformations of the corpus callosum Less cerebellum Less white matter Less grey matter Microcephally |
| Astley et al., 2009 | Human | FAS/PFAS SE/AE ND/AE Alcohol exposure through all 3 trimesters 8–15.9 years old | MRI | Disproportionately smaller frontal lobes Smaller caudate regions 6–15% smaller corpus callosum Smaller absolute volumes of white matter in the frontal lobes |
| Monnig et al., 2015 | Human | Heavy drinking 4–5 drinks/month 21–56 years old | MRI/DTI | Decreased white matter integrity |
| Pfefferbaum et al., 2014 | Human | Light drinking 0.7–4.1 kg alcohol/year; Heavy drinking 7.6–28.8 kg alcohol/year (> 5 kg of alcohol/year) 20–60 years old | MRI/DTI | Accelerating white matter damage Lower values indexing myelin integrity |
| Darbinian et al., 2021 | Human | Fetal brains Heavy Prenatal Alcohol exposure 57–1827.5 cumulative dose (6–320 drinks /month 1st and 2nd trimesters | FACS, ddPCR, qRT-PCR, ELISA | Accumulated OPC markers Reduced OL markers MBP reduction Delayed OL differentiation and decreased myelin production; OL death |
| Nardelli et al., 2011 | Human | FASD 6 to 17 years old | MRI | Reductions throughout the deep gray matter: Reductions of volume for the intracranial vault (7.6%), total white matter (8.6%), total cortical gray matter (7.8%), total deep gray matter (13.1%). All 6 deep gray matter reductions with the caudate (16%), globus pallidus (18%) |
| Rice and Gu, 2019 (review) | Human, mouse | FAS, AUD Heavy alcohol exposure | Microscopy | Deficits in white matter Less grey matter Brain shape abnormalities Myelin reduction Increased demyelination in different brain regions |
| Vangipuram and Lyman, 2012 | Human | In vitro FASD model, neural stem cells (fetal differentiating NSC) 20 or 100 mM EtOH | mRNA, Oligo GEArray for 263 genes | Changes in 22 genes in 5 pathways/cellular processes: axon guidance; hedgehog signaling; TGF-β signaling; cell adhesion molecules; and Wnt signaling Suppressed Wnt3a and Wnt5a, receptor complex proteins p-LRP6, LRP6 and DVL2, and cytoplasmic proteins Ser-p-GSK3β and β-catenin. Increased active Tyr-p-GSK3β |
| Newville et al., 2017 | Mouse | FASD mouse model 3.8 g/dL EtOH P2; 4.9 g/dL P3–P9 (128.2–160 mg/dL BAC) 7.1 g/dL P10-p15 (167.7 mg/dL BAC) | MRI/DTI | Oligodendrocyte loss, white matter injury reduction of proliferating OPC loss of OL (transient) |
| Ozer et al., 2000 | Mouse | Prenatal acohol exposure 12 g/kg GD6–GD17 | Immunohisochemistry | MBP reduction Reduction in brain myelination |
| Coutts et al., 2015 | Mouse | Primary mouse OLs: High EtOH 500 mM; 50 μM ACD 7 days | Microscopy; western blot assay | Reduced myelin in mature OLs White matter loss |
| Rice et al., 2019 | Mouse | 25% (v/v) EtOH 5 g/kg P28– P37 (300 mg/mL BAC) | Microscopy | Increased MBP+ myelin reduction; brain-region specific demyelination |
| Cantacorps et al., 2017 | Mouse | Prenatal and early neonatal alcohol 20% (v/v) (79.27–81.56 mg/dL BAC) | Behavioral studies; ELISA; western blot assay | Impaired cognitive functions; motor coordination; reduction in myelin proteins (myelin-associated glycoprotein, MBP, myelin proteolipid protein and myelin regulatory factor) in both the prefrontal cortex and hippocampus; myelin damage |
| Chiappelli et al., 1991 | Rat | Prenatal EtOH exposure 5% w/v at last 2 weeks of gestation | Immunostaining, ELISA, in vitro glial cultures | Delayed MBP expression during PD1–21 Delayed oligodendrocyte maturation |
| Miller, 2017 | Rat | 2.2%(v/v) EtOH G6–G7 4.5% G8–G10, 6.7% G11–G21 | EM at P5, P15, P30, or P90 | Early developmental events |
| Zoeller et al., 1994 | Rat | Prenatal EtOH, and EtOH on postnatal days 4–10 | Quantitative in situ hybridization, mRNA | Reduction in MBPs and MAG in adults from postnatal No reduction in MBP and MAG in the cerebellums of 15-day-old pups from prenatal exposure |
| Thomas et al., 2007 | Rat | 6.0 g/kg/d PD4–9 (3rd trimester) (79.29 mg/dL BAC) | Behavioral testing | Learning deficits in females |
| Watari et al., 2006 | Sheep | Early PAE: daily 90 min alcohol 1.5 g/kg i.v. daily 90 min at GD30-60, studied at GD127 (term = 147 d) | Immunohistology, Neuropathology Neuroimaging: Computer-assisted microscopy and quantitative morphometry | White matter injury The lesions in the temporal, parietal, and occipital white matter; WM reduction |
| Creeley et al., 2013 | Non-human primates (HNP) | 2.15 g/kg single dose at early or late third- trimester (300–400 mg/dL BAC) | Histopathology | WM reduction in fetal brain; reduced markers of OL progenitors (PDGFR) reduced MBP increased OL apoptosis G120: 12.7 times increase of dying WM cells |
Different ethanol exposure, route of administration, dose, and peak blood alcohol concentrations (BAC) are compared with the human studies. ACD: Acetaldehyde; AUDs: alcohol use disorders; DTI: diffusion tensor imaging; ELISA: enzyme-linked immunosorbent assay; EM: electron microscopy; FACS: fluorescent activated cell sorting; FAS/PFAS: Fetal Alcohol Syndrome or Partial Fetal Alcohol Syndrome; GD: gestation day; MAG: myelin associated glycoprotein; MBP: myelin basic protein; MRI: magnetic resonance imaging; ND/AE: neurobehavioral disorder/alcohol-exposed; OL: oligodendrocytes; PD: postnatal day; PDGFR: platelet-derived growth factor receptor; SE/AE: static encephalopathy/alcohol-exposed; WM: white matter.
Figure 1.
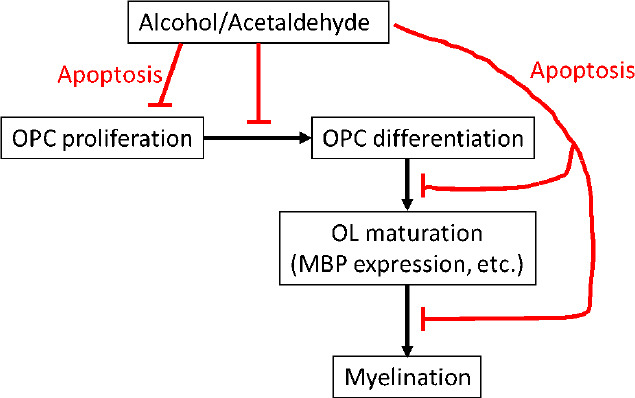
Mechanisms by which alcohol use during pregnancy might cause dysmyelination in fetal brain.
In brains of EtOH-exposed fetuses, studies of OL stage-specific biomarkers show a shift away from mature oligodendrocytes (OL) toward immature OL and OL precursor cells (OPC). But the absolute numbers of cells at each phase of OL development is reduced, and the proportions of OPCs and OLs showing caspase-3 activity is increased. Thus, EtOH itself, or its metabolite, acetaldehyde, can inhibit proliferation of OPC, as well as inhibit the differentiation of OPC to immature OL, or the maturation of immature OL to OL. Each of these effects may be due at least in part by increased apoptosis at each developmental stage. MBP: Myelin basic protein: OL: oligodendrocytes: OPC: oligodendrocyte precursor cells.
Search Strategy and Selection Criteria
Original studies on the effects of prenatal alcohol exposure on oligodendrocyte injury and dysmyelination cited in this review published from 1987 to 2021 were searched on the PubMed database using the following keywords: fetal alcohol syndrome, white matter injury, myelin basic protein, dysmyelination, neurodegeneration, oligodendrocyte injury, myelination.
Fetal Alcohol Syndrome and Oligodendrocyte Injury
Fetal exposure to ethanol (EtOH) during pregnancy is the leading cause of preventable cognitive impairment in the US (Ornoy and Ergaz, 2010; CDC, 2012; Mutch et al., 2016). The most severe and stereotyped combination of neurodevelopmental and somatic abnormalities due to prenatal exposure to EtOH is called “fetal alcohol syndrome,” but a broader array of congenital abnormalities including fetal alcohol syndrome (FAS), partial FAS, and alcohol-related neurodevelopmental disorders, in aggregate are called “fetal alcohol spectrum disorder” (FASD). These disorders frequently are not diagnosed (80%) or misdiagnosed (7%) (May et al., 2018). Past estimates of the prevalence of FAS in the US ranged from 0.2 to 7 per 1000 children, but newer research suggests that 3.1% to 9.9% of children have some form of FASD (Sampson et al., 1997; Andersen et al., 2012; Popova et al., 2017). In the US, 80,000 children are born with FASD each year, more than half undiagnosed. This was determined by studies performed in US schools, measuring children’s weight, height, and head circumference, and also evaluating the children for facial features of FAS, and other minor anomalies associated with FASD, using standard checklists. Neurodevelopmental performance was assessed by school psychologists using cognitive and behavioral testing to evaluate cognition, academic achievement, behavior, and adaptive skills (May et al., 2018). Because the EtOH exposure may occur before the mother is aware that she is pregnant, therapies based on knowledge of the molecular mechanisms and critical periods of EtOH exposure are needed. Direct fetal brain tissue examination is not feasible in human pregnancies, and non-invasive examination of the fetal brain has been limited to expensive and technically challenging in-utero functional and imaging studies. Thus, much of what we know has been derived from studies in animals and human cell lines, where dose and timing of EtOH exposure can be controlled.
In animal models, the pattern of brain injury depends on the developmental stage at EtOH exposure. Most research on effects of EtOH on the developing central nervous system (CNS) of experimental animals emphasizes reduced neurogenesis, abnormal neuronal differentiation and migration, induction of neuronal apoptosis (West, 1987, 1994; Luo and Miller, 1998; Ikonomidou et al., 2000; Goodlett and Horn, 2001; Guerri et al., 2001, 2009; Olney, 2002; Leigland et al., 2013; Donald et al., 2015). However, effects on glial cells have been reported (Guizzetti et al., 2014). MRI studies in children with FASD show that behavioral abnormalities correlate, not only with cortical thickening but also with disruption of white matter integrity (Sowell et al., 2008). The mechanisms underlying the white matter damage are not well understood, but because CNS myelin is formed by oligodendrocytes (OLs), a greater focus on these cells is warranted. OLs are the last cells generated during development. Myelination and expression of myelin basic protein (MBP) begin during the 2nd trimester (20 weeks gestational age (GA) in humans) and continue postnatally (Abraham et al., 2010; Nickel and Gu, 2018). OL precursor cells (OPCs) are produced much earlier (E16 in rats, GA 5.5 weeks in humans). Many human and animal studies have focused on late gestation effects of EtOH, including degeneration and death of neurons, synaptic loss, and activation of microglia and astrocytes (Gatford et al., 2007; Guizzetti et al., 2014; Saito et al., 2016). However, first trimester EtOH exposure is more common and thus, more likely to directly affect OPC differentiation and function (Darbinian et al., 2021). In addition, while the role of EtOH-induced neuroinflammation in FASD has been studied in mid to late gestation animal models (Kane et al., 2012, 2014; Kane and Drew, 2016; Newville et al., 2017), less is known regarding effects in humans, particularly in early gestation, including on OL development. Thus, investigation of the expression of cytokines and chemokines that regulate key signaling pathways in the human developing brain, including those that remain expressed in early postnatal life, is important. Male fetuses are more vulnerable than females to EtOH exposure (May et al., 2017; Darbinian et al., 2021), but human data regarding the mechanism of these sex differences, including on OL pathology, have been limited.
Effect of Ethanol Exposure on Markers of Oligodendrocyte Precursor Cell/Oligodendrocyte mRNA and Protein Expression
Despite their early development, some OPCs persist into adulthood, where they underlie late myelin formation and repair (El Waly et al., 2014). EtOH-induced apoptosis of OLs was shown in the fetal macaque brain (Creeley et al., 2013; Wang et al., 2020). EtOH exposure also led to a significantly weaker expression of MBP and delayed MBP expression in rats (Chiappelli et al., 1991; Zoeller et al., 1994). In a third trimester-equivalent mouse model of FASD, mice exposed to EtOH for two weeks during early postnatal development (postnatal day 3) and studied at postnatal day 16 showed a 58% decrease in the number of mature OLs and a 75% decrease in the number of proliferating OPCs within the corpus callosum (Newville et al., 2017). The EtOH-induced decreases in OL and OPC numbers were transient, although myelination deficits persisted into adulthood. A wide range of abnormalities of glial cells, including astrocytes, has been described in FASD (Guizzetti et al., 2014). Recent findings indicated that EtOH reduce late markers of OL maturation, and delay differentiation of OLs (Darbinian et al., 2021). It was suggested that EtOH exposure interferes with the expression of OL lineage-specific markers in human neural progenitor cells undergoing OL lineage progression, blocks the differentiation of OL lineage stem cells, and causes a downregulation of the late OPC marker, MBP. Apoptotic signaling was increased in both OPC and OL. Thus, two mechanisms were proposed to account for the effects of EtOH exposure in causing dysmyelination - a delay in maturation of OPC to OL, as well as a loss of mature OL, and apoptotic signaling may contribute to both of these (Darbinian et al., 2021).
Effects of Exposure to Ethanol on Expression of Specific Cytokines/Chemokines in Developing Brain during Oligodendrocyte Differentiation in Human Fetal Brain Tissues
Several extracellular signals, intracellular pathways, and transcription factors regulating OL differentiation and myelination have been studied in animal models of demyelination (Nave and Trapp, 2008; Young et al., 2013; Mitew et al., 2014; Jha et al., 2016; Rice and Gu, 2019). Some studies have reported increased expression of many chemokines in CNS demyelinating diseases including multiple sclerosis, or diseases with impaired myelination, including FAS. Knowledge regarding the molecular mechanisms causing the abnormalities is limited. Animal models of FAS have shown dysregulation of cytokine expression leading to apoptosis of OPCs and altered OL differentiation (Kirby et al., 2019). Whether this is true of human FAS is not known. However, a significant EtOH-induced upregulation has been demonstrated in the expression of several chemokines, including CXC ligands CXCL1/GRO, IL-8, GCP2/CXCL16, ENA-78 and MCP1, in developing brain. Recent findings of EtOH-induced suppression of OL maturation associated with altered expression of chemokines and fatty acids (Sowell et al, 2020) suggest that EtOH-induced delay in differentiation of OPCs can contribute to the failure of remyelination and repair in FAS (Darbinian et al., 2021). It is possible that secretion of these chemokines also may have pathological effects on other cells.
Tumor Necrosis Factor-α and Insulin-Like Growth Factor-1 in the Oligodendrocyte Pathology of Fetal Alcohol Syndrome
Acute EtOH exposure reduced levels of the neuroprotective cytokine insulin-like growth factor 1 (IGF-1) in fetal and maternal sheep (Gatford et al., 2007; Saito et al., 2016). On the other hand, EtOH increased the expression of tumor necrosis factor alpha (TNF-α) in microglial cultures, and conditioned medium from EtOH-treated microglia intensified the EtOH-induced apoptosis of neurons in primary hypothalamic cultures and in neonatal mice in vivo (Saito et al., 2016). Signaling pathways that may be involved in neuroimmune mechanisms contributing to FASD in the developing CNS or in OPCs and OLs in vitro, have been reviewed (Rice and Gu, 2019; Darbinian et al., 2021). Thus, part of the neurotoxic effect of EtOH may be mediated by release of TNF-α from EtOH-activated microglia. Despite considerable knowledge about the actions of hormones and cytokines in vitro, relatively little is known about their interactions in the brain. However, it is possible that much of the OL pathology of FASD reflects antagonistic effects between cytokines such as IGF-1 and TNF-α. Recent data on the effect of EtOH on OL development demonstrated that expression of the neuroprotective IGF-1 was reduced, while the neurotoxic TNF-α was increased (Table 1). Interestingly, the changes were greater in males than females (Darbinian et al., 2021), consistent with the difference in prevalence of FAS/D. These findings suggest that studying the mechanism of EtOH-induced OL toxicity in human fetuses may aid in the development of neuroprotective interventions for women who continue to use EtOH during pregnancy.
White Matter Defects in Clinical Fetal Alcohol Spectrum Disorder
Preclinical models of prenatal alcohol exposure have identified white matter damage. White matter lesions have been induced in the developing sheep brain by binge exposure to alcohol in early gestation (Watari et al., 2006). Some of the white matter damage associated with prenatal alcohol exposure appears to specifically target axons. Thus, rat models of fetal alcohol exposure have observed decreases in axon size, with thinner myelin sheaths. In addition to axonal damage, glial cells are vulnerable to the effects of prenatal alcohol exposure. Thus, oligodendrocytes from alcohol-exposed rats were morphologically aberrant, decreased in number, and showed lower expression levels of markers important for myelination, including myelin basic protein (Chiappelli et al., 1991). Similar inhibiting effect of prenatal alcohol exposure on OL markers was observed in human fetal brains (Darbinian et al., 2021). Children with FASD that combine cognitive, behavioral, and neurological impairments caused by prenatal alcohol exposure, need close examination, using a combination of diagnostic tools. Effects of prenatal alcohol exposure on cortical white matter were examined using diffusion tensor imaging (DTI) in 10-year-old children with FAS/FASD. Significantly lower fractional anisotropy was seen in four white matter regions, and higher mean diffusivity in seven regions in the FAS/FASD children compared to unexposed controls. DTI values were significantly associated with all three continuous measures of alcohol exposure (AA/day, AA/occasion and days/week) at cluster peaks. Low fractional anisotropy suggests low axon packing density and/or poor myelination. Mediation of behavioral deficits by white matter injury was most clearly observed in effects of alcohol exposure on processing speed and eyeblink conditioning. This information was consistent with numerous behavioral studies linking prenatal alcohol exposure to slower information processing in infancy and childhood (Table 2). Thus, the significant correlations that were observed between alcohol measures and DTI values indicated that the white matter damage found in several regions was dose-dependent. Multiple regression models indicated that cortical white matter impairment partially mediated adverse effects of prenatal alcohol exposure on information processing speed and eyeblink conditioning (Fan et al., 2016).
Translational Potential of the Oligodendrocyte Research
Despite considerable animal research, the mechanism of OL loss in the 1st and 2nd trimester human fetus, and the role this plays in FASD, is not fully understood. Retrospective studies are limited by the impossibility of accurately estimating EtOH exposure at each developmentally critical period, particularly because women often continue drinking before they are aware they are pregnant. Ongoing prospective studies such as the NIAAA Prenatal Alcohol and SIDS and Stillbirth Network allow more accurate exposure assessment but are confounded by the postnatal effects of ongoing maternal EtOH use. Those studies rely on behavioral assessments or imaging studies, and cannot address the mechanisms of teratogenesis. Recent research links EtOH exposure, quantified as accurately as clinically possible, directly with brain injury in the 1st and 2nd trimesters (Darbinian et al., 2021). The data identified therapeutic targets to reduce EtOH toxicity on brain development and its effects on molecular mechanisms in OL survival in FAS/D.
The OL research that links alcohol exposure directly to brain OL injury during early pregnancy will be vitally important to most pregnant women in the US. Among the most important molecular pathways to study in this regard are those that are involved in signaling by cytokines, including insulin, TNF-α, and IGF-1, because these pathways are strongly implicated in OL dysregulation. A critical gap in knowledge is how alcohol and cytokines interact in determining the OL differentiation and myelination in FASD. It is important to provide information about the mechanisms of EtOH-mediated OL injury during the early pregnancy, to better understand the environmental influences on fetal neurodevelopment, and how this is reflected by biomarkers of myelination dysregulation. Ultimately, the resulting knowledge could lead to the development of strategies to prevent myelination impairment and demyelination disorders in children exposed to EtOH during fetal development, and to better inform the pregnant women about the risks and potential therapies.
Future Directions
OLs damaged in FAS either fail to develop or undergo excessive apoptosis. Failure to repair the OLs hampers myelination and also leads to accumulation of neuronal damage. Outlines of major aspects of OL toxicity in FASD are presented in Figure 1. In developing therapeutic or preventive strategies for FASD, it will be important to target not only the OLs, but also the mechanisms of OL-neuron interaction. Studies of EtOH effects on brain development have been limited to animal or in vitro FAS models (Mooney and Varlinskaya, 2018). In animal models, except for non-human primates, the developmental stages equivalent to the human late-term fetus occur postnatally. Thus, recent animal studies show that third trimester-equivalent alcohol exposure leads to an acute decrease in OL lineage cell numbers, accompanied by white matter injury (Newville et al., 2017). Myelin loss and OL pathology in white matter also can lead to traumatic brain injury. Moreover, the role of sex in susceptibility of glial cells to the toxic effects of fetal alcohol exposure is as yet unexplored. Thus, sex differences in preclinical models of FASD have been reported previously, but the mechanisms are still unknown. In a rat model of FASD, prenatal alcohol exposure leads to increased fighting in males (Varlinskaya, 2014). This study demonstrated that prenatal exposure to EtOH on G15 increased social anxiety in early adolescent and adult females, but not in late adolescence, while males were previously shown to be vulnerable to an earlier EtOH exposure. The mechanisms of these effects also are still unknown. Sex differences in spatial learning deficits after neonatal alcohol exposure also have been reported (Goodlett and Peterson, 1995). Thus, male rats given the postnatal development 7–9 exposure had significant place learning deficits, which were as severe as with the full 6-day exposure. The hypothesis that glial cells are affected by alcohol selectively in males compared to females during brain development is worth further studies. Alcohol interferes with glial cell function in the developing brain, and this leads to neuronal deficits due to the critical importance of interactions between neurons and glial cells in the CNS. In order identify proper targets for therapeutic development, it will be necessary to further determine the complex biological disruptions induced in the fetal brain by alcohol exposure, and to identify the links between glial dysfunction and structural, functional and behavioral abnormalities. Because the incidence of EtOH use early in pregnancy is far greater than the number of children actually born with FAS/D, non-invasive methods to assess potential neuropathology will be very important. We have developed methods to study the contents of fetal OLs and OPCs by analyzing exosomes (OL-Es and OPC-Es) derived from these cells in the maternal blood (Darbinian et al., 2021), to target OL and OPC markers, inflammatory cytokines and chemokines, including TNF-α (Darbinian et al., 2021), markers for axonal signaling deficits, including Synaptophysin, BDNF (Goetzl et al., 2016, 2019), markers for microvascular density and vasculature in OL development (β-catenin), growth factor deficits (IGF-1, Darbinian et al., 2021), autophagy (ATG3, ATG7, LC3 (Girault et al., 2017)), facial markers for FAS, including markers for fetal eye development (BDNF, β-catenin, and PMP22), and maternal plasma fatty acid (Sowell et al., 2020). In addition, the possibility of epigenetic changes in early development that affect OPC and OL function in adulthood (Tiane et al., 2019) should be considered, and targeted using OL-Es and OPC-Es, including histone modification and miRNAs. Finally, it will be important to use DTI and fMRI, along with molecular studies, to examine the correlation between OL injury and brain structural and functional connectivity in the same population. This may provide greater insight into the mechanisms underlying prenatal alcohol exposure-related neurocognitive deficits. Such methods would help us to determine which fetuses are at risk and allow us to intervene therapeutically early in the pregnancy.
Additional file: Open peer review report 1 (81.2KB, pdf) .
Acknowledgments:
We thank members of the Shriners Hospitals Pediatric Research Center for their technical support.
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Financial support: This work was supported by: NIH grants R01NS97846, R01NS097846-02S1 and R01NS092876 awarded to MES; Shriners research grant SHC-85400 awarded to MES; and USA Pennsylvania State Department grant Project 10: 420491-04400-02 to ND.
Copyright license agreement: The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: C. Fernando Valenzuela, University of New Mexico Health Sciences Center, USA.
Funding: This work was supported by NIH grants R01NS97846, R01NS097846-02S1 and R01NS092876 awarded to MES; Shriners research grant SHC-85400 awarded to MES; and USA Pennsylvania State Department grant Project 10: 420491-04400-02 to ND.
P-Reviewer: Valenzuela CF; C-Editors: Zhao M, Liu WJ, Qiu Y; T-Editor: Jia Y
References
- 1.Abraham H, Vincze A, Jewgenow I, Veszpremi B, Kravjak A, Gomori E, Seress L. Myelination in the human hippocampal formation from midgestation to adulthood. Int J Dev Neurosci. 2010;28:401–410. doi: 10.1016/j.ijdevneu.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 2.Andersen AM, Andersen PK, Olsen J, Gronbaek M, Strandberg-Larsen K. Moderate alcohol intake during pregnancy and risk of fetal death. Int J Epidemiol. 2012;41:405–413. doi: 10.1093/ije/dyr189. [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention (CDC) Alcohol use and binge drinking among women of childbearing age -- United States 2006-2010. MMWR Morb Mortal Wkly Rep. 2012;61:534–538. [PubMed] [Google Scholar]
- 4.Chiappelli F, Taylor AN, Espinosa de los Monteros A, de Vellis J. Fetal alcohol delays the developmental expression of myelin basic protein and transferrin in rat primary oligodendrocyte cultures. Int J Dev Neurosci. 1991;9:67–75. doi: 10.1016/0736-5748(91)90074-v. [DOI] [PubMed] [Google Scholar]
- 5.Creeley CE, Dikranian KT, Johnson SA, Farber NB, Olney JW. Alcohol-induced apoptosis of oligodendrocytes in the fetal macaque brain. Acta Neuropathol Commun. 2013;1:23. doi: 10.1186/2051-5960-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Darbinian N, Darbinyan A, Merabova N, Bajwa A, Tatevosian G, Martirosyan D, Zhao H, Selzer ME, Goetzl L. Ethanol-mediated alterations in oligodendrocyte differentiation in the developing brain. Neurobiol Dis. 2021;148:105181. doi: 10.1016/j.nbd.2020.105181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Donald KA, Eastman E, Howells FM, Adnams C, Riley EP, Woods RP, Narr KL, Stein DJ. Neuroimaging effects of prenatal alcohol exposure on the developing human brain: a magnetic resonance imaging review. Acta Neuropsychiatr. 2015;27:251–269. doi: 10.1017/neu.2015.12. [DOI] [PubMed] [Google Scholar]
- 8.El Waly B, Macchi M, Cayre M, Durbec P. Oligodendrogenesis in the normal and pathological central nervous system. Front Neurosci. 2014;8:145. doi: 10.3389/fnins.2014.00145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gatford KL, Dalitz PA, Cock ML, Harding R, Owens JA. Acute ethanol exposure in pregnancy alters the insulin-like growth factor axis of fetal and maternal sheep. Am J Physiol Endocrinol Metab. 2007;292:E494–500. doi: 10.1152/ajpendo.00269.2006. [DOI] [PubMed] [Google Scholar]
- 10.Goodlett CR, Horn KH. Mechanisms of alcohol-induced damage to the developing nervous system. Alcohol Res Health. 2001;25:175–184. [PMC free article] [PubMed] [Google Scholar]
- 11.Guerri C, Pascual M, Renau-Piqueras J. Glia and fetal alcohol syndrome. Neurotoxicology. 2001;22:593–599. doi: 10.1016/s0161-813x(01)00037-7. [DOI] [PubMed] [Google Scholar]
- 12.Guerri C, Bazinet A, Riley EP. Foetal alcohol spectrum disorders and alterations in brain and behaviour. Alcohol Alcohol. 2009;44:108–114. doi: 10.1093/alcalc/agn105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guizzetti M, Zhang X, Goeke C, Gavin DP. Glia and neurodevelopment: focus on fetal alcohol spectrum disorders. Front Pediatr. 2014;2:123. doi: 10.3389/fped.2014.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Koch C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–1060. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 15.Jha MK, Lee WH, Suk K. Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem Pharmacol. 2016;103:1–16. doi: 10.1016/j.bcp.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Kane CJ, Drew PD. Inflammatory responses to alcohol in the CNS: nuclear receptors as potential therapeutics for alcohol-induced neuropathologies. J Leukoc Biol. 2016;100:951–959. doi: 10.1189/jlb.3MR0416-171R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kane CJ, Phelan KD, Drew PD. Neuroimmune mechanisms in fetal alcohol spectrum disorder. Dev Neurobiol. 2012;72:1302–1316. doi: 10.1002/dneu.22035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kane CJ, Phelan KD, Douglas JC, Wagoner G, Johnson JW, Xu J, Phelan PS, Drew PD. Effects of ethanol on immune response in the brain: region-specific changes in adolescent versus adult mice. Alcohol Clin Exp Res. 2014;38:384–391. doi: 10.1111/acer.12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirby L, Jin J, Cardona JG, Smith MD, Martin KA, Wang J, Strasburger H, Herbst L, Alexis M, Karnell J, Davidson T, Dutta R, Goverman J, Bergles D, Calabresi PA. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat Commun. 2019;10:3887. doi: 10.1038/s41467-019-11638-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leigland LA, Ford MM, Lerch JP, Kroenke CD. The influence of fetal ethanol exposure on subsequent development of the cerebral cortex as revealed by magnetic resonance imaging. Alcohol Clin Exp Res. 2013;37:924–932. doi: 10.1111/acer.12051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Luo J, Miller MW. Growth factor-mediated neural proliferation: target of ethanol toxicity. Brain Res Brain Res Rev. 1998;27:157–167. doi: 10.1016/s0165-0173(98)00009-5. [DOI] [PubMed] [Google Scholar]
- 22.May PA, Tabachnick B, Hasken JM, Marais AS, de Vries MM, Barnard R, Joubert B, Cloete M, Botha I, Kalberg WO, Buckley D, Burroughs ZR, Bezuidenhout H, Robinson LK, Manning MA, Adnams CM, Seedat S, Parry CDH, Hoyme HE. Who is most affected by prenatal alcohol exposure: Boys or girls? Drug Alcohol Depend. 2017;177:258–267. doi: 10.1016/j.drugalcdep.2017.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Mitew S, Hay CM, Peckham H, Xiao J, Koenning M, Emery B. Mechanisms regulating the development of oligodendrocytes and central nervous system myelin. Neuroscience. 2014;276:29–47. doi: 10.1016/j.neuroscience.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 24.Mutch RC, Jones HM, Bower C, Watkins RE. Fetal alcohol spectrum disorders: using knowledge, attitudes and practice of justice professionals to support their educational needs. J Popul Ther Clin Pharmacol. 2016;23:e77–89. [PubMed] [Google Scholar]
- 25.Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- 26.Newville J, Valenzuela CF, Li L, Jantzie LL, Cunningham LA. Acute oligodendrocyte loss with persistent white matter injury in a third trimester equivalent mouse model of fetal alcohol spectrum disorder. Glia. 2017;65:1317–1332. doi: 10.1002/glia.23164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nickel M, Gu C. Regulation of central nervous system myelination in higher brain functions. Neural Plast. 2018;2018:6436453. doi: 10.1155/2018/6436453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Olney JW. New insights and new issues in developmental neurotoxicology. Neurotoxicology. 2002;23:659–668. doi: 10.1016/S0161-813X(01)00092-4. [DOI] [PubMed] [Google Scholar]
- 29.Ornoy A, Ergaz Z. Alcohol abuse in pregnant women: effects on the fetus and newborn, mode of action and maternal treatment. Int J Environ Res Public Health. 2010;7:364–379. doi: 10.3390/ijerph7020364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Popova S, Lange S, Probst C, Gmel G, Rehm J. Estimation of national, regional, and global prevalence of alcohol use during pregnancy and fetal alcohol syndrome: a systematic review and meta-analysis. Lancet Glob Health. 2017;5:e290–299. doi: 10.1016/S2214-109X(17)30021-9. [DOI] [PubMed] [Google Scholar]
- 31.Rice J, Gu C. Function and mechanism of myelin regulation in alcohol abuse and alcoholism. Bioessays. 2019;41:e1800255. doi: 10.1002/bies.201800255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saito M, Chakraborty G, Hui M, Masiello K, Saito M. Ethanol-induced neurodegeneration and glial activation in the developing brain. Brain Sci. 2016;6:31. doi: 10.3390/brainsci6030031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sampson PD, Streissguth AP, Bookstein FL, Little RE, Clarren SK, Dehaene P, Hanson JW, Graham JM., Jr (1997) Incidence of fetal alcohol syndrome and prevalence of alcohol-related neurodevelopmental disorder. Teratology. 56:317–326. doi: 10.1002/(SICI)1096-9926(199711)56:5<317::AID-TERA5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 34.Sowell ER, Johnson A, Kan E, Lu LH, Van Horn JD, Toga AW, O’Connor MJ, Bookheimer SY. Mapping white matter integrity and neurobehavioral correlates in children with fetal alcohol spectrum disorders. J Neurosci. 2008;28:1313–1319. doi: 10.1523/JNEUROSCI.5067-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang X, Cuzon Carlson VC, Studholme C, Newman N, Ford MM, Grant KA, Kroenke CD. In utero MRI identifies consequences of early-gestation alcohol drinking on fetal brain development in rhesus macaques. Proc Natl Acad Sci U S A. 2020;117:10035–10044. doi: 10.1073/pnas.1919048117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.West JR. Fetal alcohol-induced brain damage and the problem of determining temporal vulnerability: a review. Alcohol Drug Res. 1987;7:423–441. [PubMed] [Google Scholar]
- 37.West JR. Recent findings on the mechanisms by which alcohol damages the developing nervous system. Alcohol Alcohol Suppl. 1994;2:395–399. [PubMed] [Google Scholar]
- 38.Young KM, Psachoulia K, Tripathi RB, Dunn SJ, Cossell L, Attwell D, Tohyama K, Richardson WD. Oligodendrocyte dynamics in the healthy adult CNS: evidence for myelin remodeling. Neuron. 2013;77:873–885. doi: 10.1016/j.neuron.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zoeller RT, Butnariu OV, Fletcher DL, Riley EP. Limited postnatal ethanol exposure permanently alters the expression of mRNAS encoding myelin basic protein and myelin-associated glycoprotein in cerebellum. Alcohol Clin Exp Res. 1994;18:909–916. doi: 10.1111/j.1530-0277.1994.tb00059.x. [DOI] [PubMed] [Google Scholar]
- 40.Zoeller RT, Butnariu OV, Fletcher DL, Riley EP. Limited postnatal ethanol exposure permanently alters the expression of mRNAS encoding myelin basic protein and myelin-associated glycoprotein in cerebellum. Alcohol Clin Exp Res. 1994;18:909–916. doi: 10.1111/j.1530-0277.1994.tb00059.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.