

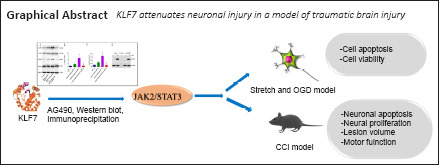
Key Words: apoptosis, hippocampus, JAK2/STAT3, Kruppel-like factor 7, neuroprotection, oxygen-glucose deprivation, stretch, traumatic brain injury
Abstract
Our previous study has shown that the transcription factor Krüppel-like factor 7 (KLF7) promotes peripheral nerve regeneration and motor function recovery after spinal cord injury. KLF7 also participates in traumatic brain injury, but its regulatory mechanisms remain poorly understood. In the present study, an HT22 cell model of traumatic brain injury was established by stretch injury and oxygen-glucose deprivation. These cells were then transfected with an adeno-associated virus carrying KLF7 (AAV-KLF7). The results revealed that, after stretch injury and oxygen-glucose deprivation, KLF7 greatly reduced apoptosis, activated caspase-3 and lactate dehydrogenase, downregulated the expression of the apoptotic markers B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax) and cleaved caspase-3, and increased the expression of βIII-tubulin and the antiapoptotic marker Bcl-2. Furthermore, KLF7 overexpression upregulated Janus kinase 2 (JAK2) and signal transducer and activator of transcription 3 (STAT3) phosphorylation in HT22 cells treated by stretch injury and oxygen-glucose deprivation. Immunoprecipitation assays revealed that KLF7 directly participated in the phosphorylation of STAT3. In addition, treatment with AG490, a selective inhibitor of JAK2/STAT3, weakened the protective effects of KLF7. A mouse controlled cortical impact model of traumatic brain injury was then established. At 30 minutes before modeling, AAV-KLF7 was injected into the ipsilateral lateral ventricle. The protein and mRNA levels of KLF7 in the hippocampus were increased at 1 day after injury and recovered to normal levels at 3 days after injury. KLF7 reduced ipsilateral hippocampal atrophy, decreased the injured cortex volume, downregulated Bax and cleaved caspase-3 expression, and increased the number of 5-bromo-2′-deoxyuridine-positive neurons and Bcl-2 protein expression. Moreover, KLF7 transfection greatly enhanced the phosphorylation of JAK2 and STAT3 in the ipsilateral hippocampus. These results suggest that KLF7 may protect hippocampal neurons after traumatic brain injury through activation of the JAK2/STAT3 signaling pathway. The study was approved by the Institutional Review Board of Mudanjiang Medical University, China (approval No. mdjyxy-2018-0012) on March 6, 2018.
Chinese Library Classification No. R453; R741; R363
Introduction
Traumatic brain injury (TBI) causes neurological functional deficits, and treatment for TBI remains limited. TBI leads to significant hippocampal atrophy accompanied by damaged verbal memory and cognition functions (Song et al., 2019; Rizk et al., 2021; Sun et al., 2021). Krüppel-like factors (KLFs) are transcription factors that regulate several important physiological and pathological processes, including proliferation, differentiation, and cell death (Puigdecanet et al., 2008; Tetreault et al., 2012; Xu et al., 2021). Across all 17 KLF family members, both KLF6 and KLF7 are necessary for optic axon regeneration; however, the ability of KLF7 to promote neurite growth is more than double that of KLF6 (Moore et al., 2009). KLF7 is broadly expressed in adult tissue, with a predominance in brain and spinal cord tissue (Laub et al., 2001). In TBI models, several KLF family members, such as KLF4, KLF11, and KLF6, play an important role in neuronal damage, hypoxia/reoxygenation injury, axonal growth, and motor neuron survival (Nagata et al., 2014; Cui et al., 2017; Li et al., 2017b). The Janus activated kinase (JAK)/signal transducer and activator of transcription (STAT) pathway has been reported to mediate these regulatory effects (Zhao et al., 2011b). The JAK/STAT pathway participates in the progress of various central nervous system diseases, including cerebral ischemia, TBI, and spinal cord injury (SCI) (Yamauchi et al., 2006; Zhao et al., 2011a, b). Additionally, a previous study has reported that KLF6 and STAT3 coregulate pro-regenerative genes (Wang et al., 2018). KLF6 and KLF7 act through the same transcriptional targets and DNA-binding domains (Veldman et al., 2007). Thus, KLF7 may also be associated with JAK2/STAT3 signaling because accumulating evidence indicates that KLF7 promotes neuronal survival and axonal regeneration after nerve injury (Li et al., 2017a, 2019b; Wang et al., 2017). The mechanisms of KLF7-mediated TBI pathogenesis, as well as the effects of its intervention, are worthy of investigation.
To create an in vitro TBI-like injury in cells, we induced stretch and oxygen-glucose deprivation (OGD) together, which closely replicates the events that occur in TBI (Torrente et al., 2014; Salvador et al., 2015, 2018). The in vivo TBI injury that we used was the induced controlled cortical impact (CCI), which is the most commonly used in vitro model of TBI in mice (Faden et al., 2005; Qu et al., 2020). We then investigated the function of KLF7 in stretch- and OGD-induced apoptosis of HT22 cells in vitro, and in TBI-induced hippocampal neuron damage in mice in vivo. We aimed to assess the possible benefits of KLF7 for rescuing hippocampal neuron injury and improving functional recovery, and explored the possible mechanisms responsible for any documented therapeutic benefits.
Materials and Methods
Cell culture and the in vitro model of neuronal cell injury in hippocampal cells
The hippocampal cell line HT22 (RRID:CVCL0321) was purchased from the Cell Bank at the Shanghai Institutes for Biological Sciences of the Chinese Academy of Sciences. Cell line authentication was performed by STR profiling before initiation of this study. Cells were cultured in Dulbecco’s minimal essential medium with 10% fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified incubator at 5% CO2(Sanyo, Hirakata, Japan). The medium was replaced every 2 days. Cells were seeded onto six-well plates (2 × 107 cells/well) for independent experiments.
Stretch combined with OGD has been demonstrated to successfully replicate the initial TBI event, and it has been recognized as an in vitro TBI model based on previous studies (Torrente et al., 2014; Salvador et al., 2015, 2018). As previously described (Salvador et al., 2015; Li et al., 2019a), a biaxial stretch injury model was performed. Briefly, HT22 cells were cultured on six-well plates coated with type I collagen (Cat# A1142801, Thermo Fisher Scientific, Waltham, MA, USA) until 80–90% confluence was reached, and they were then treated using the Cellular Injury Controller II (FlexCell International Corp., McKeesport, PA, USA). The delay was set to 50 ms, regulator pressure was set to 241 kPa, and peak pressure was set to 38 kPa. This level of pulse exposure corresponds to moderate stretch damage (Salvador et al., 2018). Cells were subjected to moderate damage every hour for 4 hours. Subsequently, cells were also deprived of glucose; in short, the complete medium (Dulbecco’s minimal essential medium containing 10% fetal bovine serum and 10 mM D-glucose) was replaced with Dulbecco’s minimal essential medium without D-glucose for 4 hours. Next, cells were kept in a hypoxic incubator for 4 hours and then reoxygenated for 20 hours prior to performing the following experiments (Salvador et al., 2015). Control HT22 cells were treated with same conditions but did not receive the stress pulse or OGD treatment.
AG490 pretreatment and adeno-associated virus carrying KLF7 transduction of HT22 cells
HT22 cells were inoculated onto six-well plates (2 × 107 cells/well). The control group consisted of untreated cells. The stretch + OGD + AAV-negative control (NC) group consisted of cells transduced with AAV-NC 24 hours after stretching and OGD. The stretch + OGD + adeno-associated virus carrying KLF7 (AAV-KLF7) group consisted of cells transduced with AAV-KLF7 24 hours after stretching and OGD. The stretch + OGD + AAV-KLF7 + AG490 group consisted of cells pre-treated with the JAK2/STAT3 inhibitor AG490 (1 μL in 50% ethanol, 5 mM; Cat#133550-30-8; Abcam, Cambridge, MA, USA) for 30 minutes, and then transduced with AAV-KLF7 24 hours after stretching and OGD. Finally, the stretch + OGD + AAV-NC + AG490 group consisted of cells pre-treated with AG490 for 30 minutes, and then transduced with AAV-NC 24 hours after stretching and OGD.
AAV transduction was performed 24 hours after stretching and OGD. Cells were infected with AAV-NC or AAV-KLF7 (both at 6.5 × 109 virus particles/mL; Vector Biolabs, Philadelphia, PA, USA) at a concentration of 150 μL/well (Figure 1A).
Figure 1.
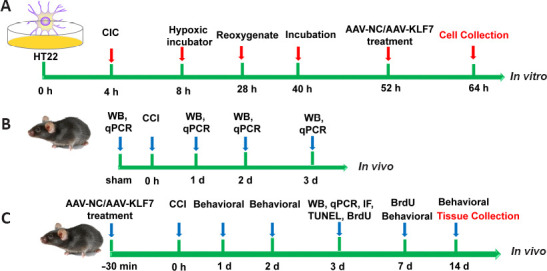
Cell (A) and animal (B, C) experiment time-course diagram.
AAV: Adeno-associated virus; BrdU: 5-bromo-2′-deoxyuridine; CCI: controlled cortical impact; CIC: cellular injury controller; IF: immunofluorescence; KLF7: Krüppel-like factor 7; NC: negative control; qPCR: quantitative real-time polymerase chain reaction; WB: western blot.
Animals
Seventy-two C57BL/6 mice (36 males and 36 females; 8 weeks old; weight 18–22 g) were purchased from the Experimental Animal Center of China Medical University (license No. SCXK (Liao) 2003-0009). All procedures strictly followed the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All experiments were designed and reported according to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines. The study was approved by the Institutional Review Board of Mudanjiang Medical University (approval No. mdjyxy-2018-0012) on March 6, 2018.
TBI and treatment
In the first animal experiment, changes in KLF7 expression were investigated in the mouse hippocampus following TBI. In this study, 12 adult mice were randomly divided into the following groups: sham or 1, 2, or 3 days following CCI injury (n = 3/group) (Figure 1B).
In the second animal experiment, all animals were randomly separated into the following three groups (n = 20/group): sham, TBI + AAV-NC (TBI treated with AAV-NC), and TBI + AAV-KLF7 groups (TBI treated with AAV-KLF7). CCI is a commonly used and highly regarded mechanical model of TBI, and the establishment of the CCI model was performed as described previously (Li et al., 2008; Gao et al., 2014; Wang et al., 2017). In brief, mice were anesthetized with an intra-peritoneal administration of 2,2,2-tribromoethanol (2.5%, 0.2 mL/20 g, Sigma-Aldrich, St. Louis, MO, USA) and placed in a stereotactic frame (Elekta Instruments, Tucker, GA, USA). The skull was exposed through a midline incision. A left hemisphere craniotomy of 4.5 mm diameter, 2.5 mm lateral to the midline, was performed between the lambda and bregma (Qu et al., 2020). A CCI injury with 1.0 mm impact depth (Impactor OneTM, MyNeuroLab, Leica Microsystems, Wetzlar, Germany) was then performed (3 mm diameter; round tip; dwell: 50 ms; speed: 3 m/s). The sham animals received the craniotomy only, without the impact.
For the TBI + AAV-NC and TBI + AAV-KLF7 groups, 10 μL of AAV-NC (6.5 × 109 viral particles/mL) and AAV-KLF7 (6.5 × 109 viral particles/mL), respectively, were injected into the ipsilateral lateral ventricle (from the bregma: lateral 1.5 mm; anteroposterior, –0.8 mm; depth 3.5 mm from the skull surface (Paxinos and Watson, 2007)) 30 minutes before impact on day 1 (Tao et al., 2018).
After surgery, the animals were placed on heating pads within recovery cages to recover from anesthesia. Three days after TBI, eight animals per group were sacrificed for tissue extraction for molecular analyses, including western blot, quantitative real-time polymerase chain reaction (qRT-PCR), immunohistochemistry, and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) or 5-bromo-2′-deoxyuridine (BrdU) staining. At 7 days after TBI, four animals per group were sacrificed for BrdU staining. The remaining eight animals per group were subjected to behavioral tests, and were then sacrificed for histopathological examinations at 14 days after TBI (Figure 1C).
Western blot analysis
For the in vitro experiments, cells were homogenized after treatment. In vivo, mice were anesthetized with 2,2,2-tribromoethanol (2.5%, 0.2 mL/20 g) and decapitated at 1, 2, or 3 days after injury, and the ipsilateral (right) hippocampus was immediately dissected and flash frozen on dry ice. In brief, proteins were separated via sodium dodecyl sulphate-polyacrylamide gel electrophoresis, followed by polyvinylidene difluoride membrane transfer (Millipore, Bedford, MA, USA). The membranes were then incubated with primary antibodies against KLF7 (Cat# PA5-101340; rabbit polyclonal; 1:500; Thermo Fisher Scientific), B-cell lymphoma 2 (Bcl-2; rabbit polyclonal; Cat# PA5-99356; 1:500; Thermo Fisher Scientific), Bcl-2-associated X protein (Bax; rabbit polyclonal; Cat# PA5-85918; 1:500; Thermo Fisher Scientific), cleaved caspase-3 (C-caspase3; Cat# ab2302;1:500, rabbit polyclonal; Abcam), phospho-JAK2 (p-JAK2; Tyr1007/1008; Cat# 44-426G; rabbit polyclonal; 1:500; Thermo Fisher Scientific), total-JAK2 (t-JAK2; Cat# 44-406G; rabbit polyclonal; 1:500; Thermo Fisher Scientific), phospho-STAT3 (p-STAT3; Ser727; Cat# PA5-17876; rabbit polyclonal; 1:500; Thermo Fisher Scientific), total-STAT3 (t-STAT3; Cat# 710077; rabbit polyclonal; 1:500; Thermo Fisher Scientific), or glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Cat# MA5-15738; rabbit polyclonal; 1:500; Thermo Fisher Scientific) overnight at 4°C. The next day, the samples were incubated with horseradish peroxidase-conjugated secondary antibody (Cat # 32260; goat anti-rabbit IgG; 1:5000; Thermo Fisher Scientific) for 1 hour at room temperature. The Enhanced Chemiluminescence Plus kit (GE Healthcare, Little Chalfont, UK) was used to visualize labeled proteins. The images were analyzed using ImageJ 1.42q software (National Institutes of Health, Bethesda, MD, USA). Optical densities (ODs) were calculated and normalized to GAPDH, the housekeeping control protein, and were imaged on a chemiluminometer (LAS-3000, Fujifilm, Tokyo, Japan).
qRT-PCR
HT22 cells and hippocampal tissue were homogenized using Trizol Reagent (Life Technologies, Carlsbad, CA, USA). Complementary DNA (cDNA) was generated according to the manufacturer’s instructions (TaKaRa, Dalian, China). The expression of KLF7 was normalized relative to the housekeeping gene β-actin. The KLF7 primers were forward: 5′-TTT CCT GGC AGT CAT CTG CAC-3′ and reverse: 5′-GGG TCT GTT TGT TTG TCA GTC TGT C-3′. The β-actin primers were forward: 5′-CCC ATC TAT GAG GGT TAC GC-3′ and reverse: 5′-TTT AAT GTC ACG CAC GAT TTC-3′. The cycling conditions were as follows: 95°C for 10 seconds, and 40 cycles of 95°C for 30 seconds, 60°C for 30 seconds, and 72°C for 40 seconds. Gene expression was quantified by the comparative CT (2–ΔΔCT) method (Fang et al., 2019).
Immunofluorescence
In vitro immunolabeling was performed based on a previous method (Li et al., 2020). Briefly, HT22 cells were labeled with immunofluorescent antibodies raised against KLF7 (rabbit polyclonal; Cat# PA5-101340; 1:200, Thermo Fisher Scientific) and βIII-tubulin (mouse polyclonal; Cat# T8328; 1:200; Sigma-Aldrich) overnight at 4°C. They were then incubated with the secondary antibodies fluorescein isothiocyanate-conjugated anti-rabbit IgG (Cat# F-2765; 1:200; Sigma-Aldrich) or Alexa 555-conjungated donkey anti-rabbit IgG (Cat# A32794; 1:200; Thermo Fisher Scientific) for 2 hours at room temperature, and with 4′,6-diamidino-2-phenylindole (1:200; Thermo Fisher Scientific) for 1 minute at room temperature. Images were taken using an Olympus IX71 fluorescence microscope (Olympus America Inc., Center Park, PA, USA). Fluorescent intensities were analyzed using MetaMorph software (Molecular Devices Inc., Downington, PA, USA).
Lactate dehydrogenase assay
An lactate dehydrogenase (LDH) cytotoxicity assay kit (Cat# 11644793001; Sigma-Aldrich) was used to measure LDH enzymatic activity. Briefly, HT22 cells from each group were mixed with 50-μL aliquots of the culture medium and reconstituted substrate and were then incubated in the dark at 37°C for 30 minutes. Subsequently, the reaction was stopped with a stop solution (1 M HCl, 50 μL). The color intensity of the reaction was proportional to the number of lysed cells. The OD reading at 490 nm (OD490 nm) was taken using a Synergy H4 microplate reader (BioTek Instruments, Winooski, VT, USA) and used for quantification. The percentage of cytotoxicity was then calculated by expressing the OD490 nm of the experimental cells as a percentage relative to the OD490 nm of untreated cells.
Caspase-3 activity assay
Caspase-3 activity was detected by using ELISA. A caspase-3 cellular activity assay kit (Cat# 235419; Sigma-Aldrich) was used to detect the activity of the pro-apoptotic protein caspase-3. Briefly, HT22 cells were lysed, and the cell supernatant (50 μL/well) was incubated with 5 μL of 4 mM DEVD-p-nitroaniline substrate and 50 μL reaction buffer for 2 hours at 37°C. A microplate reader (Bio-Rad, Sunnyvale, CA, USA) was used to determine the OD at 405 nm. The results were presented as the percentage of caspase-3-reactive ODs relative to the ODs of untreated controls.
Immunoprecipitation
Interactions between endogenous KLF7 and p-STAT3 were examined in both AAV-KLF7- and AAV-NC-transfected HT22 cells after stretching and OGD. After treatment, cells were harvested and homogenized in lysis buffer (2 mM ethylenediaminetetraacetic acid, 300 mM NaCl, 20 mM Tris-HCl, and 1% Nonidet P-40) including a cocktail of protease and phosphatase inhibitors. Next, 500 μg of protein was incubated with primary antibodies against KLF7 (1:50, mouse monoclonal; Cat# sc-101034; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA), p-STAT3 (1:50, rabbit polyclonal, Cat# PA5-17876, Thermo Fisher Scientific), or their respective isotype controls (Santa Cruz Biotechnology). After overnight incubation at 4°C, the samples were incubated with 50 μL of Protein A/G Plus Agarose (50% slurry, Cat# sc-2003, Santa Cruz Biotechnology) for 4 hours. Lysates were then immunoprecipitated, and non-binding proteins were denatured with sodium dodecyl sulfate loading buffer. The bound protein was then analyzed by western blot, as described in the western blot analysis section.
Propidium iodide staining
Cell viability was analyzed using propidium iodide (PI) staining. HT22 cells were incubated with Hoechst 33342 (5 μg/mL, Sigma-Aldrich) at 37°C for 15 minutes to label all cell nuclei. Next, to stain the apoptotic cells, 5 μg/mL PI (Sigma-Aldrich) was added for 10 minutes, followed by washing in phosphate-buffered saline for 5 minutes. Finally, the samples were fixed in 4% paraformaldehyde. Images were captured under a fluorescence microscope (Olympus America Inc.). Five to eight 200× fields were randomly selected, and ImageJ 1.42q software was used to quantify the percentage of PI-positive/Hoechst-positive cells, which was taken as cell viability.
Cell Counting Kit-8 assay
Cell viability was evaluated using the colorimetric Cell Counting Kit-8 (CCK-8) kit (Beyotime Biotechnology, Shanghai, China). Briefly, HT22 cells from each group were incubated for 2 hours at 37°C with 10 μL/well of CCK-8 solution, in 96-well plates. The OD was measured at 450 nm using a microplate reader (Bio-Rad). Cell viability was quantified proportional to the number of living cells. Cell viability (%) = (ODtreatment – ODblank) / (ODcontrol – ODblank) × 100.
TUNEL staining
TUNEL staining was used to detect apoptotic hippocampal cells. Briefly, 25 μm brain sections were harvested 3 days after TBI. Ipsilateral hippocampus sections were double-labeled with 4′,6-diamidino-2-phenylindole and the In Situ Cell Death Detection Kit, TMR Red (Roche Diagnostics, Pleasanton, CA, USA). The percentage of TUNEL-positive neurons in the ipsilateral hippocampal region was quantified using a Carl Zeiss LSM-780 confocal fluorescence microscope (Carl Zeiss Microscopy, Thornwood, NY, USA).
Immunofluorescence
Serial 25 μm cross-sections of ipsilateral hippocampal tissue were obtained and treated with 3% H2O2 to quench endogenous peroxidases. The sections were then blocked with phosphate-buffered saline with 5% bovine serum albumin (Sigma-Aldrich) at 37°C for 30 minutes, and incubated at 4°C overnight with rabbit polyclonal anti-KLF7 (Cat# PA5-101340; 1:200). Next, the sections were incubated with biotinylated anti-rabbit secondary antibody (Cat# 65-6140, 1:200; Thermo Fisher Scientific) for 1 hour at room temperature. A Vector Elite ABC kit (Cat# 32020; Thermo Fisher Scientific) was used to amplify the secondary antibody signal. Finally, 0.02% 3,3′-diaminobenzidine (Cat# TA-060-QHDX; Thermo Fisher Scientific) was used to visualize the antibody-biotin avidin-peroxidase complex. MetaMorph software was used to acquire images of the ipsilateral hippocampal tissue and analyze the fluorescent signal using integrated ODs.
To investigate neural proliferation, BrdU (50 mg/kg, intraperitoneal; Sigma-Aldrich) was injected 2 hours before perfusion (transcardially with 4.0% paraformaldehyde in 0.1 M phosphate buffer) to label the proliferating cells, which were analyzed at 3 and 7 days following TBI. The ipsilateral brain hemispheres were coronally sectioned at 25 μm. Sections were incubated with primary antibodies against BrdU (1:1000, Cat# ab1893, Abcam), doublecortin (DCX; Cat# 48-1200, 1:500, Invitrogen, Carlsbad, CA, USA), glial fibrillary acidic protein (GFAP; Cat# AB477035, 1:1000, Invitrogen), and NeuN (Cat# PA5-78499, 1:500, Invitrogen) at 4°C overnight. Alexa 555-conjungated donkey anti-rabbit IgG (Cat# A32794, 1:200, Thermo Fisher Scientific), fluorescein isothiocyanate-conjugated anti-rabbit IgG (Cat# F-2765, 1:200, Sigma-Aldrich), or Cy3-conjugated goat anti-rabbit IgG (Cat# AP187C, 1:200, Sigma-Aldrich) were added for 2 hours at room temperature, and sections were then incubated with 4′,6-diamidino-2-phenylindole (1:200) for 1 minute at room temperature. NeuN-, BrdU-, and DCX-positive cells were counted with a Carl Zeiss LSM-780 confocal fluorescence microscope using an unbiased stereological protocol in MetaMorph software (Völkening et al., 2017).
Histological assessments
As previously described (Qu et al., 2020), at 14 days post-TBI, brains were sectioned at 25 μm after perfusion with 4% paraformaldehyde. Sections were then stained with cresyl violet (Sigma-Aldrich). The contours of the bilateral cortices, hippocampi, and hemispheres were measured using an Olympus BX60 microscope (Olympus, Tokyo, Japan). The volume calculations followed Cavalieri’s principle (Coggeshall, 1992). The volume of spared tissue in the TBI brains was subtracted from the total sham brain volume to calculate tissue loss. Tissue loss was then divided by total tissue to calculate the percentage of the cortex volumetric tissue loss.
NeuroScore motor function analysis
A composite NeuroScore test was conducted to evaluate neurological motor function at 1, 2, 7, and 14 days post-TBI (Laurer et al., 2001). Mice walked on an open wire grid for 1 minute and were given a score from 4 (normal) to 0 (severely impaired) for indices including forelimb and hindlimb function and resistance to lateral pulsion (Liu et al., 2014). An experienced observer, blinded to study design and treatment, evaluated neurological motor function.
Rotarod test
The rotarod test was used to assess the coordination and sensorimotor function of mice (Liu et al., 2014; Qu et al., 2020). Mice walked on a rotating drum, and the time taken to fall from the drum was recorded. A rotational acceleration, from 1 to 18 r/min over 90 seconds, was performed to measure the latency to fall off the drum. Trials ended when mice either clung to or fell off the drum.
Statistical analysis
All values are expressed as the mean ± standard deviation (SD). SPSS software v.13.0 (SPSS, Chicago, IL, USA) was used. Power of analysis was performed to determine the sample size. One-way analysis of variance followed by Tukey’s post hoc test was used to compare three or more groups. The two-tailed Student’s t-test was applied to compare two groups. A P-value < 0.05 was considered statistically significant.
Results
Efficacy of AAV-KLF7 transfer to HT22 cells following stretching and OGD in vitro
To overexpress KLF7 in HT22 cells, we transduced cells with AAV-KLF7. Increased KLF7 expression was confirmed by western blot and qRT-PCR after stretching and OGD (Figure 2A–C). Compared with the control and stretch + OGD + AAV-NC groups, the protein and mRNA levels of KLF7 in the stretch + OGD + AAV-KLF7 group were increased (protein: F(2, 6) = 88.10, P < 0.001; mRNA: F(2, 6) = 59.75, P < 0.001).
Figure 2.

Efficacy of AAV-KLF7 gene transfer to HT22 cells following stretch and OGD in vitro.
(A) Representative western blot analysis of KLF7 expression in cultured HT22 cells at 1 day after stretch and OGD in vitro. (B) Quantitative results of the relative expression of KLF7 protein detected by western blot. The relative expression is expressed as the optical density ratio to GAPDH. (C) qRT-PCR analysis of KLF7 mRNA expression in the cultured HT22 cells at 1 day after stretch and OGD. (D) Immunofluorescence staining of KLF7 (green, stained by fluorescein isothiocyanate) and βIII-tubulin (red, stained by Alexa 555) with nuclear staining (DAPI; blue) in the three groups. The merged image shows that HT22 cells were co-labeled by KLF7, βIII-tubulin, and DAPI. The expression of KLF7 was increased after AAV-KLF7 infection compared with the other groups, and β-III-tubulin expression was also elevated in AAV-KLF7 cells compared with AAV-NC cells. Scale bars: 100 μm. (E, F) Quantitative results of the immunopositivity of KLF7 (E) and βIII-tubulin (F). Data are expressed as the mean ± SD. Each experiment was repeated three (A–C) or six (D–F) times. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; DAPI: 4′,6-diamidino-2-phenylindole; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; KLF7: Krüppel-like factor 7; NC: negative control; OGD: oxygen-glucose deprivation; qRT-PCR: quantitative real-time polymerase chain reaction.
Next, immunohistochemical staining was used to confirm the transduction efficiency of AAV-KLF7 (Figure 2D). Compared with the stretch + OGD + AAV-NC and control groups, KLF7 immunopositivity increased after AAV-KLF7 treatment (F(2, 15) = 264.0, P < 0.001; Figure 2E). The promoting effect of KLF7 overexpression on neurite length in HT22 cells was evaluated using βIII-tubulin immunolabeling. The neuronal marker βIII-tubulin, a type of microtubule, was considered an indicator of neurite growth because a previous study used immunostained βIII-tubulin to assess the effects of KLF7 on neurite outgrowth (Moore et al., 2009). Compared with the control group, βIII-tubulin immunopositivity was lower in cells treated with stretch and OGD, suggesting that stretch and OGD induce neurite injury. Compared with the stretch + OGD + AAV-NC group, the immunopositivity of βIII-tubulin was markedly elevated in AAV-KLF7-treated cells (F(2, 15) = 126.3, P < 0.001; Figure 2F). Therefore, AAV-KLF7 transduction significantly abrogated stretch- and OGD-induced neurite shortening.
KLF7 protects hippocampal neurons against stretch- and OGD-induced cell apoptosis in vitro
To assess the protective effects of KLF7 against stretch- and OGD-induced apoptosis in HT22 cells, PI-positive cells were counted as described previously (Qu et al., 2020) (Figure 3A–C). Consistent with a previous report (Faden et al., 2005), stretch and OGD induced apoptosis in HT22 cells, as indicated by a significantly increased proportion of PI-positive cells in the stretch + OGD + AAV-NC group compared with the control group. Furthermore, compared with the stretch + OGD + AAV-NC group, AAV-KLF7 transduction reduced the percentage of PI-positive cells (F(2, 15) = 75.71, P < 0.001; Figure 3D).
Figure 3.
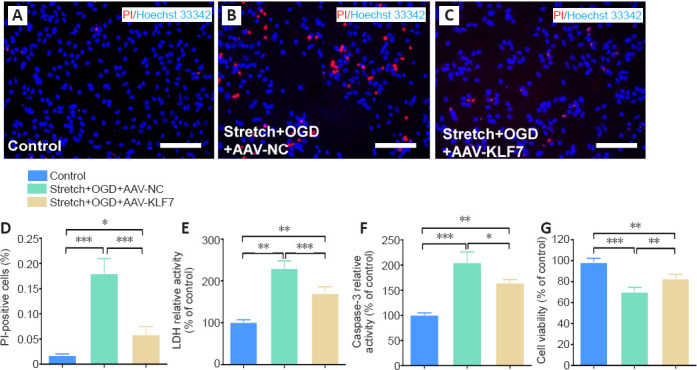
KLF7 protects hippocampal neurons against stretch- and OGD-induced cell apoptosis in vitro.
(A–C) HT22 cell apoptosis in the control (A), stretch + OGD + AAV-NC (B), and stretch + OGD + AAV-KLF7 (C) groups was measured by PI staining (red) with nuclear staining (Hoechst 33342; blue). AAV-KLF7 treatment reduced the percentage of PI-positive cells compared with the AAV-NC treatment group. Scale bars: 100 μm. (D) The percentage of PI-positive cells in each group. (E, F) LDH (E) and caspase-3 (F) activity was detected using LDH assay and Caspase-3 activity assay. (G) Cell viability was measured using CCK-8. Data are expressed as the mean ± SD. Each experiment was repeated three times. *P < 0.05, **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; CCK-8: Cell Counting Kit-8; DAPI: 4′,6-diamidino-2-phenylindole; ELISA: enzyme-linked immunosorbent assay; KLF7: Krüppel-like factor 7; LDH: lactate dehydrogenase; NC: negative control; OGD: oxygen-glucose deprivation; PI: propidium iodide.
Stretch and OGD also induced the activity of LDH and increased the apoptotic marker caspase-3 (Sîrbulescu and Zupanc, 2010); however, the increased activity of LDH and caspase-3 was reduced by AAV-KLF7 treatment (LDH: F(2, 15) = 62.45, P < 0.001; caspase-3: F(2, 15) = 51.14, P < 0.001; Figure 3E & F). Finally, the CCK-8 assay was used to examine cell viability, which remained higher in control cells compared with either the stretch + OGD + AAV-NC or stretch + OGD + AAV-KLF7 groups. Moreover, the stretch + OGD + AAV-KLF7 group had improved cell viability compared with the stretch + OGD + AAV-NC group (F(2, 15) = 43.06, P < 0.001; Figure 3G). These results indicate that AAV-KLF7 is neuroprotective in hippocampal neurons in response to stretch- and OGD-induced cell damage in vitro.
Involvement of the JAK2/STAT3 signaling pathway in the protective effects of KLF7
To investigate the underlying mechanisms by which KLF7 mediates its effects on stretch- and OGD-induced hippocampal neuronal injury, we explored how KLF7 regulates JAK2/STAT3 signaling. This signaling has been suggested to be a target of KLF6, which is a close relative of KLF7 (Nagata et al., 2014; Wang et al., 2018). Cells were pretreated with the JAK2/STAT3 inhibitor AG490 to explore whether the JAK2/STAT3 pathway mediated the effects of KLF7 in stretch- and OGD-induced HT22 injury.
Stretch and OGD significantly promoted the p-JAK2/t-JAK2 and p-STAT3/t-STAT3 ratios in the stretch + OGD + AAV-NC group compared with the control group (Figure 4A). Moreover, the p-JAK2/t-JAK2 and p-STAT3/t-STAT3 ratios were much higher in the stretch + OGD + AAV-KLF7 group compared with the stretch + OGD + AAV-NC group. However, the JAK2/STAT3 inhibitor AG490 significantly reduced the p-JAK2/t-JAK2 and p-STAT3/t-STAT3 ratios compared with the stretch + OGD + AAV-NC and stretch + OGD + AAV-KLF7 groups (p-JAK2/t-JAK2: F(3, 8) = 139.8, P < 0.001; p-STAT3/t-STAT3: F(3, 8) = 73.96, P < 0.001; Figure 4B & C). These data indicate the involvement of the JAK2/STAT3 signaling pathway in the KLF7-mediated regulation of an in vitro model of TBI in HT22 cells. In HT22 cells, direct interactions between KLF7 and p-STAT3 after stretch and OGD were further explored using co-immunoprecipitation assays (Figure 4D), which suggested that KLF7 may directly bind to p-STAT3 to activate STAT3 following TBI-like injury in vitro.
Figure 4.

Involvement of the JAK2/STAT3 signaling pathway in the neuroprotective effects of KLF7.
(A) Representative western blot of p-JAK2, t-JAK2, p-STAT3, and t-STAT3 expression in cultured HT22 cells at 1 day after stretch and OGD treatment. (B, C) Quantitative results of the relative optical densities of p-JAK2/t-JAK2 and p-STAT3/t-STAT3. (D) Co-immunoprecipitation analysis of the interactions between KLF7 and p-STAT3 in stretch- and OGD-damaged HT22 cells at 1 day, revealing a physical interaction between KLF7 and p-STAT3. Data are expressed as the mean ± SD. Each experiment was repeated three times. *P < 0.05, vs. control group; #P < 0.05, vs. stretch + OGD + AAV-NC group; &P < 0.05, vs. stretch + OGD + AAV-KLF7 group (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; IB: immunoblotting; IP: immunoprecipitation; KLF7: Krüppel-like factor 7; NC: negative control; OGD: oxygen-glucose deprivation; p-JAK2: phospho-Janus kinase 2; t-JAK2: total-Janus kinase 2; p-STAT3: phospho-signal transducer and activator of transcription 3; t-STAT3: total-signal transducer and activator of transcription 3.
AG490 reverses the protection of KLF7 against cell damage induced by stretch and OGD
To explore the regulatory effects of KLF7 on JAK2/STAT3 signaling, we investigated the effects of AG490 on cell damage in KLF7-overexpressing HT22 cells treated by stretch and OGD. Western blot results revealed that stretch- and PGD-induced apoptosis in HT22 cells was accompanied by inhibited anti-apoptotic Bcl-2 protein expression, as well as enhanced expression of the apoptotic protein Bax and C-caspase3 (stretch + OGD + AAV-NC group vs. control group). In contrast, Bcl-2 expression was markedly elevated in the stretch + OGD + AAV-KLF7 group compared with the stretch + OGD + AAV-NC group, while Bax and C-caspase3 expression was reduced. Furthermore, AG490 treatment decreased Bcl-2 expression and increased Bax and C-caspase3 expression (stretch + OGD + AAV-KLF7 + AG490 group vs. stretch + OGD + AAV-KLF7 group). There were no differences in Bcl-2, Bax, or C-caspase3 expression between the stretch + OGD + AAV-KLF7 + AG490 and stretch + OGD + AAV-NC + AG490 groups (Bcl-2: F(4, 10) = 43.34, P < 0.001; Bax: F(4, 10) = 173.6, P < 0.001; C-caspase3: F(4, 10) = 93.55, P < 0.001; Figure 5A–D). These data demonstrate that KLF7 reverses the changes in C-caspase3, Bcl-2, and Bax that are induced by stretch and OGD, and that these effects of KLF7 on apoptosis-related proteins are abrogated by the JAK2/STAT3 inhibitor AG490.
Figure 5.

The JAK2/STAT3 inhibitor AG490 reverses the protective effects of KLF7 on stretch-and OGD-induced cell apoptosis.
(A) Bcl-2, Bax, and C-caspase3 expression of cultured HT22 cells at 1 day in vitro. (B–D) Quantitative results of the relative expression of Bcl-2, Bax, and C-caspase3. The relative expression is expressed as the optical density ratio to GAPDH. (E) HT22 cell apoptosis measured by PI staining, with nuclear staining (Hoechst 33342; blue). AAV-KLF7 treatment reduced the expression of C-caspase-3 and Bax and elevated the expression of Bcl-2 compared with AAV-NC treatment. However, the effects of KLF7 on C-caspase-3, Bax, and Bcl-2 were abrogated by AG490 treatment. Scale bars: 100 μm. (F) Quantitative results of the percentage of PI-positive cells. (G) LDH activity was measured using ELISA. (H) Caspase-3 activity was detected using ELISA. (I) Cell viability was measured using CCK-8. Data are expressed as the mean ± SD. Each experiment was repeated three (A–D) or six (E–I) times. *P < 0.05, vs. control group; #P < 0.05, vs. stretch + OGD + AAV-NC group; &P < 0.05, vs. stretch + OGD + AAV-KLF7 group (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma 2; C-caspase3: cleaved caspase-3; CCK-8: Cell Counting Kit-8; DAPI: 4′,6-diamidino-2-phenylindole; ELISA: enzyme-linked immunosorbent assay; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; KLF7: Krüppel-like factor 7; LDH: lactate dehydrogenase; NC: negative control; OGD: oxygen–glucose deprivation; PI: propidium iodide.
Similarly, the percentage of PI-positive cells and LDH and caspase-3 activity were increased in the stretch + OGD + AAV-NC group compared with the control group, while cell viability was decreased. Compared with the stretch + OGD + AAV-NC group, the percentage of PI-positive cells and LDH and caspase-3 activity were decreased in the stretch + OGD + AAV-KLF7 group, while cell viability was increased. However, AG490 abrogated the protective effects of KLF7 on cell damage. The percentage of PI-positive cells and LDH and caspase-3 activity were increased in the stretch + OGD + AAV-KLF7 + AG490 group compared with the stretch + OGD + AAV-KLF7 group, while cell viability was decreased. There were no differences in the percentage of PI-positive cells, LDH or caspase-3 activity, or cell viability among the stretch + OGD + AAV-NC, stretch + OGD + AAV-KLF7 + AG490, and stretch + OGD + AAV-KLF7 + AG490 groups (PI-positive cells: F(4, 15) = 25.07, P < 0.001; LDH: F(4, 15) = 42.45, P < 0.001; caspase-3: F(4, 15) = 49.83, P < 0.001; cell viability: F(4, 15) = 32.64, P < 0.001; Figure 5E–I). These results indicate that AG490 can abrogate the protective effects of KLF7 on cell damage. Furthermore, they suggest that KLF7 protects against stretch- and OGD-induced cell damage in HT22 cells in vitro through activation of the JAK2/STAT3 pathway.
Changes in KLF7 expression in the mouse hippocampus following TBI
In the first animal experiment, KLF7 protein and mRNA expression in the hippocampus was evaluated using western blots and qRT-PCR (Figure 6A–C). Both KLF7 protein and mRNA levels in the hippocampus were increased at 1–2 days post-TBI, and normalized at about 3 days (protein: F(3, 8) = 75.12, P < 0.001; mRNA: F(3, 8) = 40.18, P < 0.001; Figure 6B & C). Immunohistochemical staining revealed that KLF7 was expressed in cortical and hippocampal neurons (NeuN-positive cells) in mice at 3 days after TBI. In contrast, no KLF7-expressing astrocytes (GFAP-positive cells) were observed (Figure 6D). These findings indicate that KLF7 is activated and mediates the pathological processes that occur after TBI.
Figure 6.
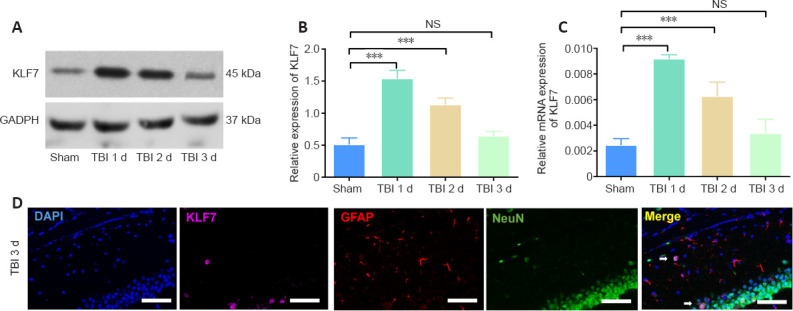
Changes in KLF7 in the mouse hippocampus following TBI.
(A) Western blot analysis showing the protein expression of KLF7 in the hippocampus after TBI. (B) Quantitative results of KLF7 protein expression, displayed as relative optical density normalized to GAPDH. (C) Quantitative results of KLF7 mRNA at different times by qRT-PCR. Data are expressed as the mean ± SD (n = 3). ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test). (D) To explore the expression of KLF7 in different cell types, double-labeling immunohistochemistry was performed for KLF7 (purple, stained by Cy3), GFAP (red, stained by Alexa 555), NeuN (green, stained by fluorescein isothiocyanate), and DAPI (blue). KLF7 was expressed in cortical and hippocampal neurons of mice 3 days after TBI. The arrow indicates KLF7 expression in a NeuN-positive cell. No GFAP-positive cells were observed to express KLF7. Scale bars: 200 μm. DAPI: 4′,6-Diamidino-2-phenylindole; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; GFAP: glial fibrillary acidic protein; KLF7: Krüppel-like factor 7; NS: not significant; qRT-PCR: quantitative real-time polymerase chain reaction; TBI: traumatic brain injury.
AAV-KLF7 significantly increases KLF7 expression in the mouse hippocampus following TBI
In the second animal experiment, KLF7 expression was evaluated using immunohistochemistry in hippocampal tissue collected 3 days after TBI (Figure 7A). Compared with the sham and TBI + AAV-NC groups, KLF7 expression was increased in the TBI + AAV-KLF7 group (F(2, 6) = 232.7, P < 0.001; Figure 7B). However, hippocampal KLF7 expression was elevated in the TBI + AAV-NC group compared with the sham group (F(2, 6) = 232.7, P < 0.001). These results indicate that AAV-KLF7 transduction significantly elevates KLF7 expression in the hippocampus following TBI.
Figure 7.
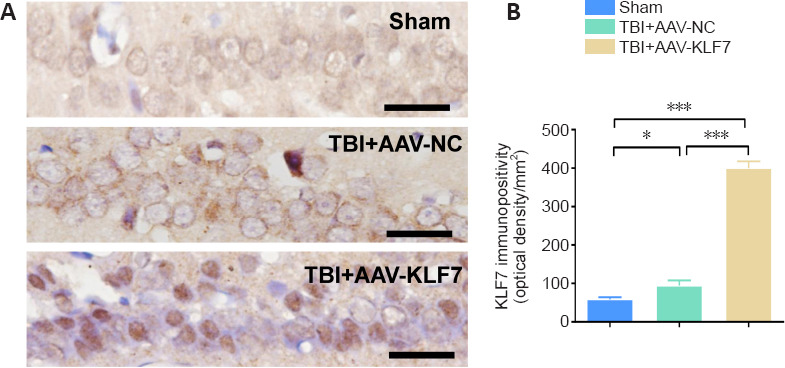
AAV-KLF7 increases KLF7 immunopositivity in the hippocampus at 3 days after TBI.
(A) Immunohistochemical staining for KLF7 in the hippocampus. KLF7 immunopositivity was increased in the TBI + AAV-KLF7 group compared with the other groups. Scale bars: 100 μm. (B) Quantitative results of KLF7 immunopositivity. Data are expressed as the mean ± SD (n = 3). ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; KLF7: Krüppel-like factor 7; NC: negative control; TBI: traumatic brain injury.
KLF7 protects hippocampal neurons from TBI-induced apoptosis
In the second animal experiment, western blot results for KLF7 protein expression in the hippocampus were similar to the immunohistochemical results in the three groups (F(2, 15) = 94.88, P < 0.001; Figure 8A & B). TBI-induced hippocampal apoptosis was characterized by reduced Bcl-2 expression and enhanced Bax and C-caspase3 expression in the TBI + AAV-NC group compared with the sham group. Moreover, Bcl-2 expression was markedly elevated and Bax and C-caspase3 expression was reduced in the TBI + AAV-KLF7 group compared with the TBI + AAV-NC group (Bcl-2: F(2, 6) = 50.67, P < 0.001; Bax: F(2, 6) = 68.10, P < 0.001; C-caspase3: F(2, 6) = 537.6, P < 0.001; Figure 8C–E).
Figure 8.

KLF7 protects hippocampal neurons against TBI-induced apoptosis at 3 days after surgery.
(A) Representative western blots of KLF7, Bcl-2, Bax, and C-caspase3 expression in the ipsilateral hippocampus at 3 days after TBI. (B–E) Quantitative results of the expression of KLF7 (B), Bcl-2 (C), Bax (D), and C-caspase3 (E). TBI decreased the expression of Bcl-2 and increased that of Bax and C-caspase3 compared with the sham group. Moreover, the expression of Bcl-2 was elevated in the TBI + AAV-KLF7 group compared with the TBI + AAV-NC group, while the expression of Bax and C-caspase3 was decreased. (F) Apoptotic cells in the ipsilateral hippocampus were stained by TUNEL (red) with nuclear staining (DAPI; blue). TBI increased the number of TUNEL-positive cells compared with the sham group, and the number of TUNEL-positive cells in the TBI + AAV-KLF7 group was lower than that in the TBI + AAV-NC group. The merged image shows that cells were double-labeled with TUNEL and DAPI. Scale bars: 100 μm. (G) Quantitative results of the TUNEL-positive cells in the ipsilateral hippocampus. Data are expressed as the mean ± SD (n = 3 in A–E, n = 6 in F–G). *P < 0.05, ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; Bax: Bcl-2-associated X protein; Bcl-2: B-cell lymphoma 2; C-caspase3: cleaved caspase-3; DAPI: 4′,6-diamidino-2-phenylindole; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; KLF7: Krüppel-like factor 7; NC: negative control; NS: not significant; TBI: traumatic brain injury; TUNEL: terminal deoxynucleotidyl transferase dUTP nick end labeling.
To evaluate hippocampal apoptosis, TUNEL staining was performed (Figure 8F). TUNEL-positive cells were increased in the TBI + AAV-KLF7 and TBI +AAV-NC groups compared with the sham group. In addition, TUNEL-positive cells in the TBI + AAV-KLF7 group were decreased compared with the TBI + AAV-NC group (F(2, 15) = 114.8, P < 0.001; Figure 8G). This finding indicates that KLF7 effectively ameliorates neuronal apoptosis in the hippocampus following TBI.
KLF7 activates the JAK2/STAT3 pathway in the hippocampus following TBI
To explore the mechanisms of the effects of KLF7 on TBI-induced injury, the p-STAT3/t-STAT3 and p-JAK2/t-JAK2 ratios in the hippocampus after AAV-KLF7 treatment were investigated using western blot in the second animal experiment (Figure 9A). Compared with the sham group, the p-STAT3/t-STAT3 and p-JAK2/t-JAK2 ratios were significantly increased in the TBI + AAV-NC group. Moreover, the p-STAT3/t-STAT3 and p-JAK2/t-JAK2 ratios were enhanced in the TBI + AAV-KLF7 group compared with the TBI + AAV-NC group (p-JAK2/t-JAK2: F(2, 6) = 37.57, P < 0.001; p-STAT3/t-STAT3: F(2, 6) = 116.5, P < 0.001; Figure 9B & C). These findings indicate that KLF7 activates the JAK2/STAT3 signaling pathway in the hippocampus following TBI.
Figure 9.
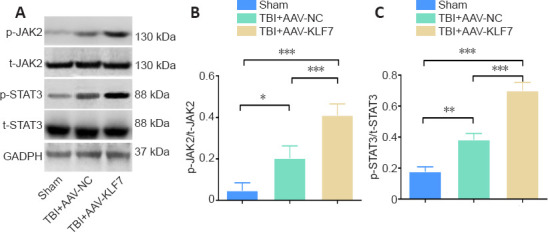
KLF7 activates the JAK2/STAT3 pathway at 3 days after TBI.
(A) Representative western blots of p-JAK2, t-JAK2, p-STAT3, and t-STAT3 expression in ipsilateral hippocampal tissue. (B, C) Quantitative results of the p-JAK2/t-JAK2 and p-STAT3/t-STAT3 ratios. Data are expressed as the mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; KLF7: Krüppel-like factor 7; NC: negative control; p-JAK2: phospho-Janus kinase 22; p-STAT3: phospho-signal transducer and activator of transcription 3; t-JAK2: total-Janus kinase; t-STAT3: total-signal transducer and activator of transcription 3; TBI: traumatic brain injury.
KLF7 increases neural proliferation in the mouse hippocampus following TBI
To explore the role of KLF7 in neural proliferation in a TBI model, double-labeling immunohistochemistry for BrdU (a proliferating neuronal marker (Falnikar et al., 2018)), GFAP (an astrocytic marker (Li et al., 2017a)), and DCX (a newly generated neuronal marker (Chen et al., 2015)) was performed in the second animal experiment (Figure 10A–C). We observed fewer BrdU-positive cells in the hippocampus of the TBI + AAV-NC group compared with the sham group. Compared with the TBI + AAV-NC group, BrdU-positive cells were increased in the TBI + AAV-KLF7 group at 3 and 7 days following TBI (3 days: F(2, 9) = 31.34, P < 0.001; 7 days: F(2, 9 = 22.12, P < 0.001; Figure 10A, B, D, & E). DCX-positive cells exhibited a similar pattern of change under the different treatment conditions (F(2, 9 = 135.6, P < 0.001; Figure 10C & F ). No BrdU-expressing astrocytes (GFAP-positive cells) were identified (Figure 10C). These data indicate that neural proliferation in the hippocampus can be inhibited by TBI, and that AAV-KLF7 treatment can rescue this impairment.
Figure 10.

KLF7 increases neural proliferation following TBI.
(A–C) Immunofluorescence staining of BrdU (proliferating neurons, red, stained by Alexa 555), DCX (newly generated neurons, red, stained by Alexa 555), and GFAP (astrocytes, green, stained by fluorescein isothiocyanate) with nuclear staining (DAPI; blue) in the ipsilateral hippocampus dentate gyrus (DG) in the three groups at 3 and 7 days following TBI. Compared with the TBI + AAV-NC group, BrdU-positive cells increased in the TBI + AAV-KLF7 group at 3 and 7 days following TBI. DCX-positive cells exhibited a similar pattern of change under the different treatment conditions. Scale bars: 100 μm in A, 200 μm in B and C. (D, E) Quantification of the number of BrdU-positive cells in the DG at 3 (D) and 7 (E) days following TBI. (F) Quantification of the density of DCX-positive cells at 3 days following TBI. Data are expressed as the mean ± SD (n = 4). *P < 0.05, ***P < 0.001 (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; BrdU: 5-bromo-2′-deoxyuridine; DAPI: 4′,6-diamidino-2-phenylindole; DCX: doublecortin; GFAP: glial fibrillary acidic protein; KLF7: Krüppel-like factor 7; NC: negative control; TBI: traumatic brain injury.
TBI-induced lesion volume in mice is reduced by KLF7
To evaluate whether AAV-KLF7 treatment reduced the lesion size following TBI, cresyl violet was used to stain brain sections, and cortical lesion volumes were measured in the second animal experiment. We did not observe any injury in the sham group (Figure 11A); however, there was a loss of cortical tissue in both the TBI + AAV-NC and TBI + AAV-KLF7 groups (Figure 11A). The percentage of cortex volumetric tissue loss was lower in the TBI + AAV-KLF7 group than in the TBI + AAV-NC group (t = 13.35, df = 4, P < 0.001; Figure 11B), indicating that AAV-KLF7 exerts neuroprotective effects after TBI.
Figure 11.
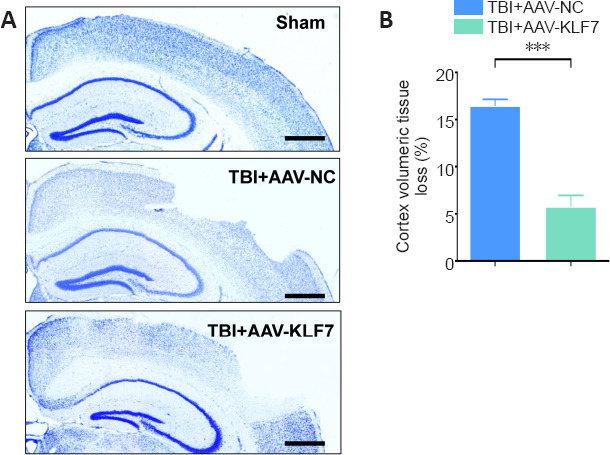
KLF7 protects mice from TBI-induced brain damage.
(A) Representative images of cresyl violet-stained sections, which were performed to evaluate cortex volumetric tissue loss at 14 days post-TBI. The percentage of cortex volumetric tissue loss was lower in the TBI + AAV-KLF7 group than in the TBI + AAV-NC group. Scale bars: 10 mm. (B) Quantification of the percentage of cortex volumetric tissue loss in the injured cortex among groups. Data are expressed as the mean ± SD (n = 3). ***P < 0.001 (two-tailed Student’s t-test). AAV: Adeno-associated virus; KLF7: Krüppel-like factor 7; NC: negative control; TBI: traumatic brain injury.
KLF7 improves mouse motor function after TBI
To evaluate motor function, the NeuroScore and rotarod tests were performed on consecutive days following TBI in the second animal experiment. NeuroScores were reduced on days 1, 2, 7, and 14 in the TBI + AAV-KLF7 and TBI + AAV-NC groups compared with the sham group. In addition, NeuroScores were increased in the TBI + AAV-KLF7 group compared with the TBI + AAV-NC group on days 7 and 14 (7 days: F(2, 15) = 75.55, P < 0.001; 14 days: F(2, 15) = 27.75, P < 0.001; Figure 12A).
Figure 12.
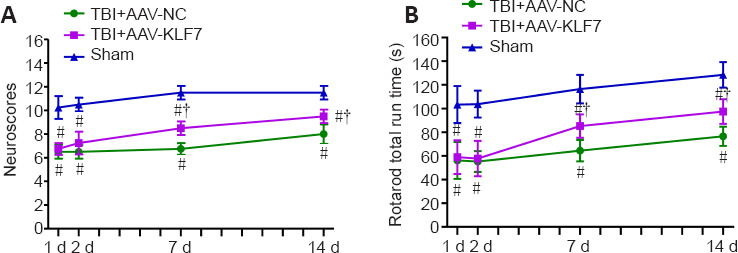
KLF7 significantly improves motor function after TBI.
(A) Composite NeuroScores. (B) Rotarod run time in the rotarod test. Data are expressed as the mean ± SD (n = 8). #P < 0.05, vs. sham group; †P < 0.05, vs. TBI + AAV-NC group (one-way analysis of variance followed by Tukey’s post hoc test). AAV: Adeno-associated virus; KLF7: Krüppel-like factor 7; NC: negative control; TBI: traumatic brain injury.
During the whole investigation period, there were significant deficits in rotarod performance in mice after TBI compared with the sham mice. AAV-KLF7 significantly improved rotarod performance on days 7 and 14 compared with the TBI + AAV-NC group (7 days: F(2, 15) = 27.87, P < 0.001; 14 days: F(2, 15) = 26.25, P < 0.001; Figure 12B). These results suggest that KLF7 has beneficial effects on motor function recovery.
Discussion
TBI-related death and disability are caused by the primary mechanical injury and the internal secondary injury, which is triggered by multiple physiological responses, including inflammatory responses, oxidative damage, and excitotoxicity (Liu et al., 2014). Because of the pathophysiological heterogeneity of TBIs, it is believed that a mechanism-based approach to TBI rehabilitation will help to yield novel targets and aid in the development of emerging therapies (Kochanek et al., 2015).
Previously, the role of the transcription factor KLF7 has been demonstrated as a promoter of axonal growth and regeneration in the spinal cord and peripheral nerves (Moore et al., 2011; Blackmore et al., 2012; Li et al., 2017a). Moreover, KLF7 is implicated in Schwann cell proliferation, survival, and myelination in sciatic nerve injury (Wang et al., 2017). In the present study, we used stretch- and OGD- or CCI-induced damage to model TBI in vitro or in vivo, respectively, and investigated whether KLF7 plays a similar role in cell and murine models of TBI.
Successful infection of the AAV-KLF7 construct into HT22 hippocampal cells demonstrated that KLF7 overexpression mitigated much of the stretch- and OGD-induced cell damage and cell damage markers, including decreased neurite shortening and improved expression of β-III-tubulin. KLF7 overexpression also led to the reduced expression of stretch- and OGD-induced apoptotic indicators, the upregulation of anti-apoptotic Bcl-2, and improved cell viability. Finally, our in vitro model of TBI revealed that KLF7 overexpression correlated with increased phosphorylation of JAK2/STAT3; this relationship is likely mediated through the direct binding of KLF7 to p-STAT3. Furthermore, inhibition of the JAK2/STAT3 signaling pathway restricted the protective effects of KLF7 in this cellular model (Figure 13).
Figure 13.
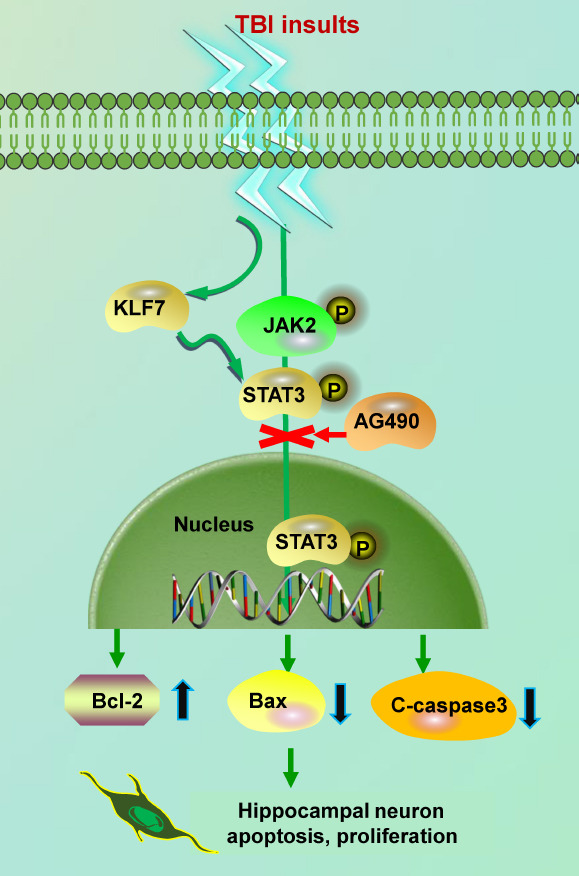
Schematic overview of KLF7 promoting JAK2/STAT3 signaling after TBI.
KLF7 promotes JAK2/STAT3 signaling, which stimulates downstream apoptosis and the proliferation of hippocampal neurons after TBI. C-caspase3: Cleaved caspase-3; JAK2: Janus kinase 2; KLF7: Krüppel-like factor 7; STAT3: signal transducer and activator of transcription 3; TBI: traumatic brain injury.
Among the KLF family, KLF7 and KLF6 share structural and functional features, such as comparable intron/exon organization, high homology of DNA binding domains, and similar transcriptional activation properties (Andreoli et al., 2010). The KLF7 relative KLF6 promotes axonal regeneration in injured corticospinal tract neurons. Specifically, co-expression of KLF6 and STAT3 leads to increased neurite growth in vitro (Wang et al., 2018). TBI-treated PC12 cells reportedly have increased carbonylation of β-tubulin (Zhang et al., 2016), which correlates with oxidative damage and inflammatory and neurodegenerative disease progression (Zheng and Bizzozero, 2010). Stretch injury is often used as an in vitro model for TBI, and OGD is a common ischemia model in vitro. The initial TBI event and the subsequent secondary injury (such as ischemia) can be replicated by a combination of these two methods (Salvador et al., 2015). In the current study, the stretch and OGD treatments simulated aspects of the damage caused by TBI in vitro. We observed stretch- and OGD-induced reductions in the cytoskeletal protein βIII-tubulin, which were reversed by KLF7 overexpression. This phenomenon indicates that KLF7 abrogates stretch- and OGD-induced neurite shortening and damage.
Our findings regarding apoptosis-related proteins following stretch and OGD were consistent with the literature. Moreover, KLF7 upregulation in a previous cell model of TBI injury has similarly demonstrated the decreased expression of C-caspase3 and Bax and increased expression of Bcl-2 (Lv et al., 2019).
We also confirmed a previously suspected interaction of KLF7 with the transcription factor STAT3 in the present study. The KLF7 relative KLF6 is predicted to harbor a functional STAT3 interaction domain in the promoter (Wang et al., 2018). Additionally, STAT3 is enriched in the promoter of KLF6-responsive genes, and co-occupancy has been detected in regulatory DNA in regeneration-relevant gene pathways (Cui et al., 2017). Using the JAK2/STAT3 inhibitor A490 and co-immunoprecipitation assay, we demonstrated that KLF7 directly interacted with STAT3 and that KLF7 upregulation coincided with JAK2/STAT3 phosphorylation. JAK2/STAT3 phosphorylation has previously been associated with the upregulation of anti-apoptotic proteins Bcl-2 and B-cell lymphoma-extra large (Bcl-xL) as well as cell survival in the pericontusional cortex (Zhao et al., 2011b). These results are in line with the characteristics observed in our KLF7-overexpressing cells. Moreover, similar to previous TBI studies, we revealed that inhibition of the JAK2/STAT3 pathway with the selective inhibitor A490 was sufficient to attenuate the neuroprotective effects of JAK2/STAT3 activation (Zhao et al., 2011b). Overall, our observations in hippocampal neurons subjected to stretch and OGD exhibited all of the expected neuronal damage, including increased apoptosis and reduced cell viability. Additionally, we observed that the cytoskeletal βIII-tubulin protein was preserved in neurites in proportion to the expression of KLF7, thus confirming its role in axonal regeneration and outgrowth, which has been well characterized in SCI.
Much of the lasting neurological damage of TBI occurs during the secondary phase, when the intricate cascade of biochemical signals converge to render classic neuronal damage such as inflammation, edema, and apoptosis. Ischemia is believed to be a major contributor to neuropathophysiology during the secondary phase of TBI (Morrison et al., 2011). The CCI model of TBI can induce hypoxia with ischemia through the induction of intracranial pressure elevations (Qu et al., 2020). Thus, in the current study, the CCI TBI model was applied because of its relatively easy procedure and focal injury capacity compared with other TBI models. By manually adjusting the parameters, we successfully created a model of moderate CCI (Qu et al., 2020).
Evidence from the sham control animals in this study demonstrated the successful induction of TBI damage, which included reductions in cell viability, the induction of apoptotic markers, and a decrease in anti-apoptotic Bcl-2. These results were on par with those observed in the in vitro model of TBI, thus indicating the successful establishment of a TBI model characterized by ischemia and secondary injury pathway activation. Additionally, we demonstrated for the first time that AAV-KLF7 can be successfully injected into the ipsilateral lateral ventricle to produce an overexpression of KLF7 protein and mRNA, which persisted up to 2 days post-TBI (peaking 1 day post-injury). Although this study confirmed the efficacy of AAV-KLF7 constructs for the successful overexpression of KLF7, previous SCI studies have reported that KLF7 can remain overexpressed for up to 14 days (Li et al., 2017a). This difference likely reflects differences in the injury response duration of relatively mild TBI compared with more severe SCI.
In vivo, KLF7 overexpression behaved similarly to its overexpression in HT22 cells; it mitigated increases in Bax and caspase-3 expression and promoted the anti-apoptotic factor Bcl-2. KLF7 overexpression in the TBI model additionally resulted in decreased apoptosis, as indicated by decreasing numbers of TUNEL-positive cells. This finding demonstrates a neuroprotective role of KLF7, as was corroborated by its anti-apoptotic effects in another cell model of hypoxic injury (Lv et al., 2019).
The cytokine receptor JAK2 and the transcription factor STAT3 are both upregulated in their phosphorylated state after TBI, along with STAT3-associated genes. In Schwann cells, activation of the JAK2/STAT3 pathway post-injury is related to the onset of re-myelination in the injury response. It also plays a role in modulating the expression of γ-aminobutyric acid type A receptor subunits after injury in a mouse model of TBI. Importantly, the JAK2/STAT3 pathway mediates the apoptotic injury response after TBI (Zhao et al., 2011b). This has been particularly demonstrated by the anti-apoptotic response that follows activation of the pathway with human recombinant erythropoietin therapy, and by the downregulation of that anti-apoptotic response after application of the JAK2/STAT3 inhibitor AG490. AG490 suppresses the activation of p-JAK2 and p-STAT3 without affecting the levels of non-phosphorylated JAK2 and STAT3 (Zhao et al., 2011b)
Similarly, activation of the pathway by AAV-KLF7, as evidenced by the increased abundance of p-JAK2 and p-STAT3, reduced TBI-induced apoptosis in the present study. Application of AG490 also abrogated KLF7-mediated activation of JAK2/STAT3, and consequently inhibited the KLF7-mediated anti-apoptotic protection. Together, these data highlight the important role that the JAK2/STAT3 pathway plays in the apoptosis cascade after TBI, and identify KLF7 as a novel activator. It should be noted that the AAV-NC injection elevated the relative density of phosphorylated JAK2/STAT3, despite non-significant elevations in KLF7 expression. This likely indicates some degree of JAK2/STAT3 response to the mechanics of AAV injection. However, because this group demonstrated no molecular or phenotypic similarities to the AAV-KLF7 group, no protective effects ascribed to JAK2/STAT3 activation were likely to be a factor. The interaction of KLF7 with JAK2/STAT3 resembles the activating relationship of KLF6, but more broadly strengthens the dynamic relationship of the KLF family with this signaling pathway during TBI. A limitation of the current study is that the in vitro TBI injury model that was used omitted the cellular microenvironment. Thus, further study is needed to determine the precise molecular mechanisms underlying JAK2/STAT3 signaling activated by KLF7 in vivo.
In the present study, we also revealed that KLF7 played a role in mitigating injury-related cell loss. This was demonstrated by the improved BrdU levels in KLF7-overexpressing animals and the improvement in cortex volumetric tissue loss that was observed after TBI. Previously, KLF7 has been proposed to promote Schwann cell survival and proliferation, and has been implicated in non-small-cell lung carcinoma proliferation (Wang et al., 2017). Unlike KLF7, KLF6 is a negative regulator of proliferation, although the contexts are almost always cancerous in nature (Andreoli et al., 2010). It is thus plausible that the regulatory role of KLF7 in proliferation may contribute to the preservation of cortical tissue, which would drastically improve outcomes.
Finally, in the current study, KLF7 overexpression was correlated with behavioral improvements; specifically of neurological function and sensorimotor capacity. KLF7-overexpressing animals had significant improvements in both categories at 7–14 days after TBI compared with NCs, although neither of the AAV-infected groups reached the levels of uninjured controls. In previous animal models of AAV-KLF7 in SCI, locomotor function was similarly preserved, supporting the functional therapeutic impact of KLF7 overexpression (Li et al., 2017a). However, further research into the molecular mechanisms underlying the effects of KLF7 knockdown on neurological function recovery is needed.
We demonstrated for the first time that the neuroprotective role of KLF7, which has been previously reported in SCI, extends to in vitro and in vivo models of mild TBI. KLF7 mitigated the TBI damage-related onset of pro-apoptotic signals and preserved cell viability and proliferation, likely underlying the preservation of cortical volume and functional improvements. Importantly, like its relative KLF6, KLF7 binds STAT3 to activate the JAK2/STAT3 signaling pathway, which has been previously implicated in the post-injury response. In this study, we have not explained the mechanisms of protective effect of KLF7 on cortical neurons and KLF7 reduced TBI-induced cortical lesion volume. Further work is needed to verify the protective ability of KLF7 overexpression across the spectrum of TBI severities, and to more clearly identify the JAK2/STAT3-associated targets that are regulated downstream of KLF7 activation.
Additional file: Open peer review report 1 (84.4KB, pdf) .
Footnotes
Conflicts of interest: The authors declare that they have no conflict of interest.
Financial support: This study was supported by the National Natural Science Foundation of China, No. 81870977 (to YW); the Natural Science Foundation of Heilongjiang of China, No. H2018068 (to WYL); the Basic Research Operating Expenses Program of Heilongjiang Provincial Universities of China, No. 2019-KYYWFMY-0018 (to WYL); the Graduate Innovative Research Projects of Mudanjiang Medical College of China, No. YJSCX-MY22 (to DM); Key projects of Education Department of Hebei Province of China, No. ZD2020178 (to XMF); and Hebei Key Laboratory of Nerve Injury and Repair of China (to XMF). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement: This study was approved by the Institutional Review Board of Mudanjiang Medical University (approval No. mdjyxy-2018-0012) on March 6, 2018.
Copyright license agreement: The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement: Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check: Checked twice by iThenticate.
Peer review: Externally peer reviewed.
Open peer reviewer: Hanin Abdel-Haq, Istituto Superiore di Sanità, Italy.
Funding: This study was supported by the National Natural Science Foundation of China, No. 81870977 (to YW); the Natural Science Foundation of Heilongjiang of China, No. H2018068 (to WYL); the Basic Research Operating Expenses Program of Heilongjiang Provincial Universities of China, No. 2019-KYYWFMY-0018 (to WYL); the Graduate Innovative Research Projects of Mudanjiang Medical College of China, No. YJSCX-MY22 (to DM); Key projects of Education Department of Hebei Province of China, No. ZD2020178 (to XMF); and Hebei Key Laboratory of Nerve Injury and Repair of China (to XMF).
P-Reviewer: Abdel-Haq H; C-Editor: Zhao M; S-Editors: Yu J, Li CH; L-Editors: Gardner B, Yu J, Song LP; T-Editor: Jia Y
References
- 1.Andreoli V, Gehrau RC, Bocco JL. Biology of Krüppel-like factor 6 transcriptional regulator in cell life and death. IUBMB Life. 2010;62:896–905. doi: 10.1002/iub.396. [DOI] [PubMed] [Google Scholar]
- 2.Blackmore MG, Wang Z, Lerch JK, Motti D, Zhang YP, Shields CB, Lee JK, Goldberg JL, Lemmon VP, Bixby JL. Krüppel-like Factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proc Natl Acad Sci U S A. 2012;109:7517–7522. doi: 10.1073/pnas.1120684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen L, Gao X, Zhao S, Hu W, Chen J. The small-molecule TrkB agonist 7, 8-dihydroxyflavone decreases hippocampal newborn neuron death after traumatic brain injury. J Neuropathol Exp Neurol. 2015;74:557–567. doi: 10.1097/NEN.0000000000000199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coggeshall RE. A consideration of neural counting methods. Trends Neurosci. 1992;15:9–13. doi: 10.1016/0166-2236(92)90339-a. [DOI] [PubMed] [Google Scholar]
- 5.Cui DM, Zeng T, Ren J, Wang K, Jin Y, Zhou L, Gao L. KLF4 knockdown attenuates TBI-induced neuronal damage through p53 and JAK-STAT3 signaling. CNS Neurosci Ther. 2017;23:106–118. doi: 10.1111/cns.12633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Faden AI, Movsesyan VA, Knoblach SM, Ahmed F, Cernak I. Neuroprotective effects of novel small peptides in vitro and after brain injury. Neuropharmacology. 2005;49:410–424. doi: 10.1016/j.neuropharm.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 7.Falnikar A, Stratton J, Lin R, Andrews CE, Tyburski A, Trovillion VA, Gottschalk C, Ghosh B, Iacovitti L, Elliott MB, Lepore AC. Differential response in novel stem cell niches of the brain after cervical spinal cord injury and traumatic brain injury. J Neurotrauma. 2018;35:2195–2207. doi: 10.1089/neu.2017.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang X, Zhang C, Yu Z, Li W, Huang Z, Zhang W. GDNF pretreatment overcomes Schwann cell phenotype mismatch to promote motor axon regeneration via sensory graft. Exp Neurol. 2019;318:258–266. doi: 10.1016/j.expneurol.2019.05.011. [DOI] [PubMed] [Google Scholar]
- 9.Gao S, Zheng Y, Cai Q, Deng Z, Yao W, Wang J, Wang X, Zhang P. Combination of acellular nerve graft and Schwann cells-like cells for rat sciatic nerve regeneration. Neural Plast. 2014;2014:139085. doi: 10.1155/2014/139085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kochanek PM, Jackson TC, Ferguson NM, Carlson SW, Simon DW, Brockman EC, Ji J, Bayır H, Poloyac SM, Wagner AK, Kline AE, Empey PE, Clark RS, Jackson EK, Dixon CE. Emerging therapies in traumatic brain injury. Semin Neurol. 2015;35:83–100. doi: 10.1055/s-0035-1544237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laub F, Aldabe R, Friedrich V, Jr, Ohnishi S, Yoshida T, Ramirez F. Developmental expression of mouse Krüppel-like transcription factor KLF7 suggests a potential role in neurogenesis. Dev Biol. 2001;233:305–318. doi: 10.1006/dbio.2001.0243. [DOI] [PubMed] [Google Scholar]
- 12.Laurer HL, Bareyre FM, Lee VM, Trojanowski JQ, Longhi L, Hoover R, Saatman KE, Raghupathi R, Hoshino S, Grady MS, McIntosh TK. Mild head injury increasing the brain’s vulnerability to a second concussive impact. J Neurosurg. 2001;95:859–870. doi: 10.3171/jns.2001.95.5.0859. [DOI] [PubMed] [Google Scholar]
- 13.Li D, Huang S, Zhu J, Hu T, Han Z, Zhang S, Zhao J, Chen F, Lei P. Exosomes from MiR-21-5p-increased neurons play a role in neuroprotection by suppressing Rab11a-mediated neuronal autophagy in vitro after traumatic brain injury. Med Sci Monit. 2019a;25:1871–1885. doi: 10.12659/MSM.915727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li WY, Jia H, Wang ZD, Zhai FG, Sun GD, Ma D, Liu GB, Li CM, Wang Y. Combinatory transplantation of mesenchymal stem cells with flavonoid small molecule in acellular nerve graft promotes sciatic nerve regeneration. J Tissue Eng. 2020;11:2041731420980136. doi: 10.1177/2041731420980136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li WY, Zhu GY, Yue WJ, Sun GD, Zhu XF, Wang Y. KLF7 overexpression in bone marrow stromal stem cells graft transplantation promotes sciatic nerve regeneration. J Neural Eng. 2019b;16:056011. doi: 10.1088/1741-2552/ab3188. [DOI] [PubMed] [Google Scholar]
- 16.Li WY, Wang Y, Zhai FG, Sun P, Cheng YX, Deng LX, Wang ZY. AAV-KLF7 promotes descending propriospinal neuron axonal plasticity after spinal cord injury. Neural Plast. 2017a;2017:1621629. doi: 10.1155/2017/1621629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Shi X, Li J, Zhang M, Yu B. Knockdown of KLF11 attenuates hypoxia/reoxygenation injury via JAK2/STAT3 signaling in H9c2. Apoptosis. 2017b;22:510–518. doi: 10.1007/s10495-016-1327-1. [DOI] [PubMed] [Google Scholar]
- 18.Li Z, Peng J, Wang G, Yang Q, Yu H, Guo Q, Wang A, Zhao B, Lu S. Effects of local release of hepatocyte growth factor on peripheral nerve regeneration in acellular nerve grafts. Exp Neurol. 2008;214:47–54. doi: 10.1016/j.expneurol.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Liu NK, Zhang YP, Zou J, Verhovshek T, Chen C, Lu QB, Walker CL, Shields CB, Xu XM. A semicircular controlled cortical impact produces long-term motor and cognitive dysfunction that correlates well with damage to both the sensorimotor cortex and hippocampus. Brain Res. 2014;1576:18–26. doi: 10.1016/j.brainres.2014.05.042. [DOI] [PubMed] [Google Scholar]
- 20.Lv JX, Zhou J, Tong RQ, Wang B, Chen XL, Zhuang YY, Xia F, Wei XD. Hypoxia induced miR 210 contributes to apoptosis of mouse spermatocyte GC 2 cells by targeting Kruppel like factor 7. Mol Med Rep. 2019;19:271–279. doi: 10.3892/mmr.2018.9644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moore DL, Apara A, Goldberg JL. Krüppel-like transcription factors in the nervous system: novel players in neurite outgrowth and axon regeneration. Mol Cell Neurosci. 2011;47:233–243. doi: 10.1016/j.mcn.2011.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Morrison B, 3rd, Elkin BS, Dollé JP, Yarmush ML. In vitro models of traumatic brain injury. Annu Rev Biomed Eng. 2011;13:91–126. doi: 10.1146/annurev-bioeng-071910-124706. [DOI] [PubMed] [Google Scholar]
- 24.Nagata K, Hama I, Kiryu-Seo S, Kiyama H. microRNA-124 is down regulated in nerve-injured motor neurons and it potentially targets mRNAs for KLF6 and STAT3. Neuroscience. 2014;256:426–432. doi: 10.1016/j.neuroscience.2013.10.055. [DOI] [PubMed] [Google Scholar]
- 25.Paxinos G, Watson C. 6th ed. Academic Press/Elsevier; 2007. The Rat Brain in Stereotaxic Coordinates. [Google Scholar]
- 26.Puigdecanet E, Espinet B, Lozano JJ, Sumoy L, Bellosillo B, Arenillas L, Alvarez-Larrán A, Solé F, Serrano S, Besses C, Florensa L. Gene expression profiling distinguishes JAK2V617F-negative from JAK2V617F-positive patients in essential thrombocythemia. Leukemia. 2008;22:1368–1376. doi: 10.1038/leu.2008.112. [DOI] [PubMed] [Google Scholar]
- 27.Qu W, Liu NK, Wu X, Wang Y, Xia Y, Sun Y, Lai Y, Li R, Shekhar A, Xu XM. Disrupting nNOS-PSD95 interaction improves neurological and cognitive recoveries after traumatic brain injury. Cereb Cortex. 2020;30:3859–3871. doi: 10.1093/cercor/bhaa002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rizk M, Vu J, Zhang Z. Impact of pediatric traumatic brain injury on hippocampal neurogenesis. Neural Regen Res. 2021;16:926–933. doi: 10.4103/1673-5374.297057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salvador E, Burek M, Förster CY. Stretch and/or oxygen glucose deprivation (OGD) in an in vitro traumatic brain injury (TBI) model induces calcium alteration and inflammatory cascade. Front Cell Neurosci. 2015;9:323. doi: 10.3389/fncel.2015.00323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salvador E, Burek M, Förster CY. An in vitro model of traumatic brain injury. Methods Mol Biol. 2018;1717:219–227. doi: 10.1007/978-1-4939-7526-6_17. [DOI] [PubMed] [Google Scholar]
- 31.Sîrbulescu RF, Zupanc GK. Inhibition of caspase-3-mediated apoptosis improves spinal cord repair in a regeneration-competent vertebrate system. Neuroscience. 2010;171:599–612. doi: 10.1016/j.neuroscience.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Song Y, Li T, Liu Z, Xu Z, Zhang Z, Chi L, Liu Y. Inhibition of Drp1 after traumatic brain injury provides brain protection and improves behavioral performance in rats. Chem Biol Interact. 2019;304:173–185. doi: 10.1016/j.cbi.2019.03.013. [DOI] [PubMed] [Google Scholar]
- 33.Sun TJ, Liu SJ, Xie FK, Huang XF, Zhang J, Jiang XH, Feng H, Yu AY. Role and hotspots of stem cell-derived exosome in the repair of traumatic brain injury. Zhongguo Zuzhi Gongcheng Yanjiu. 2021;25:123–127. [Google Scholar]
- 34.Tao J, Shen C, Sun Y, Chen W, Yan G. Neuroprotective effects of pinocembrin on ischemia/reperfusion-induced brain injury by inhibiting autophagy. Biomed Pharmacother. 2018;106:1003–1010. doi: 10.1016/j.biopha.2018.07.026. [DOI] [PubMed] [Google Scholar]
- 35.Tetreault MP, Alrabaa R, McGeehan M, Katz JP. Krüppel-like factor 5 protects against murine colitis and activates JAK-STAT signaling in vivo. PLoS One. 2012;7:e38338. doi: 10.1371/journal.pone.0038338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Torrente D, Avila MF, Cabezas R, Morales L, Gonzalez J, Samudio I, Barreto GE. Paracrine factors of human mesenchymal stem cells increase wound closure and reduce reactive oxygen species production in a traumatic brain injury in vitro model. Hum Exp Toxicol. 2014;33:673–684. doi: 10.1177/0960327113509659. [DOI] [PubMed] [Google Scholar]
- 37.Veldman MB, Bemben MA, Thompson RC, Goldman D. Gene expression analysis of zebrafish retinal ganglion cells during optic nerve regeneration identifies KLF6a and KLF7a as important regulators of axon regeneration. Dev Biol. 2007;312:596–612. doi: 10.1016/j.ydbio.2007.09.019. [DOI] [PubMed] [Google Scholar]
- 38.Völkening B, Schönig K, Kronenberg G, Bartsch D, Weber T. Deletion of psychiatric risk gene Cacna1c impairs hippocampal neurogenesis in cell-autonomous fashion. Glia. 2017;65:817–827. doi: 10.1002/glia.23128. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, Li WY, Jia H, Zhai FG, Qu WR, Cheng YX, Liu YC, Deng LX, Guo SF, Jin ZS. KLF7-transfected Schwann cell graft transplantation promotes sciatic nerve regeneration. Neuroscience. 2017;340:319–332. doi: 10.1016/j.neuroscience.2016.10.069. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Mehra V, Simpson MT, Maunze B, Chakraborty A, Holan L, Eastwood E, Blackmore MG, Venkatesh I. KLF6 and STAT3 co-occupy regulatory DNA and functionally synergize to promote axon growth in CNS neurons. Sci Rep. 2018;8:12565. doi: 10.1038/s41598-018-31101-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu JH, Qin XZ, Zhang HN, Ma YX, Qi SB, Zhang HC, Ma JJ, Fu XY, Xie JL, Saijilafu Deletion of Krüppel-like factor-4 promotes axonal regeneration in mammals. Neural Regen Res. 2021;16:166–171. doi: 10.4103/1673-5374.286978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamauchi K, Osuka K, Takayasu M, Usuda N, Nakazawa A, Nakahara N, Yoshida M, Aoshima C, Hara M, Yoshida J. Activation of JAK/STAT signalling in neurons following spinal cord injury in mice. J Neurochem. 2006;96:1060–1070. doi: 10.1111/j.1471-4159.2005.03559.x. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Q, Zhang M, Huang X, Liu X, Li W. Inhibition of cytoskeletal protein carbonylation may protect against oxidative damage in traumatic brain injury. Exp Ther Med. 2016;12:4107–4112. doi: 10.3892/etm.2016.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao J, Li G, Zhang Y, Su X, Hang C. The potential role of JAK2/STAT3 pathway on the anti-apoptotic effect of recombinant human erythropoietin (rhEPO) after experimental traumatic brain injury of rats. Cytokine. 2011a;56:343–350. doi: 10.1016/j.cyto.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 45.Zhao JB, Zhang Y, Li GZ, Su XF, Hang CH. Activation of JAK2/STAT pathway in cerebral cortex after experimental traumatic brain injury of rats. Neurosci Lett. 2011b;498:147–152. doi: 10.1016/j.neulet.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 46.Zheng J, Bizzozero OA. Accumulation of protein carbonyls within cerebellar astrocytes in murine experimental autoimmune encephalomyelitis. J Neurosci Res. 2010;88:3376–3385. doi: 10.1002/jnr.22488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.