

Abstract
The mammalian SWI/SNF complex, or BAF complex, has a conserved and direct role in antagonizing Polycomb-mediated repression. Yet, BAF also promotes repression by Polycomb in stem cells and cancer. How BAF both antagonizes and promotes Polycomb-mediated repression remains unknown. Here, we utilize targeted protein degradation to dissect the BAF-Polycomb axis in mouse embryonic stem cells on short timescales. We report that rapid BAF depletion redistributes Polycomb repressive complexes PRC1 and PRC2 from highly occupied domains, like Hox clusters, to weakly occupied sites normally opposed by BAF. Polycomb redistribution from highly repressed domains results in their decompaction, gain of active epigenomic features and transcriptional derepression. Surprisingly, through dose-dependent degradation of PRC1 and PRC2, we identify a conventional role for BAF in Polycomb-mediated repression, in addition to global Polycomb redistribution. These findings provide new mechanistic insight into the highly dynamic state of the Polycomb-Trithorax axis.
Chromatin regulation establishes and maintains the precise gene expression states that determine cellular identity and prevent human pathologies1. The balance between different states primarily involves the antagonism between activating (Trithorax-group) and repressive (Polycomb-group) proteins2. Opposition between these two classes of chromatin regulator was first demonstrated genetically during Drosophila development3. For example, deletion of Polycomb-group genes results in Hox gene derepression, which gives rise to homeotic transformations, whereas Trithorax-group mutations dominantly suppress these phenotypes4,5. Similarly, loss of function mutations in Trithorax-group genes also produces homeotic transformations, but these are instead due to insufficient Hox gene expression6. Following these pioneering studies, the genes encoding members of these groups have been shown to be mutated in many human diseases. This is especially true for the BAF (mammalian switch/sucrose non-fermentable or mSWI/SNF) complex, a Trithorax-group homolog, which is frequently mutated in cancers, neurodevelopmental disorders and intellectual disabilities7,9.
BAF complexes are combinatorially assembled chromatin remodeling enzymes of ~15 subunits that hydrolyze adenosine triphosphate (ATP) to mobilize nucleosomes and generate accessible DNA10. In mammalian cells, BAF directly evicts Polycomb repressive complex 1 (PRC1)11,12, leading to transcriptional derepression13. Thus, the ability of Trithorax-group proteins to antagonize Polycomb-mediated repression is conserved from Drosophila to mammals. Polycomb repressive complexes PRC1 and PRC2 direct histone H2A ubiquitination (H2AK119ub1) and trimethylation of histone H3 at lysine 27 (H3K27me3), respectively, spatially constraining the genome in support of transcriptional repression2. BAF-mediated Polycomb antagonism is essential during development and is thought to underlie the tumor suppressive role in human cancers14. Conversely, BAF’s potent ability to antagonize Polycomb-mediated repression is co-opted in synovial sarcoma, where fusion of SSX onto the SS18 subunit retargets BAF, opposing Polycomb-mediated repression to drive tumor growth7,15.
Despite extensive evidence that BAF has a dominant role in Polycomb opposition, BAF is required for Polycomb-mediated repression in embryonic stem cells (ESCs), during lineage commitment and in sub-types of rare atypical teratoid rhabdoid tumors (ATRT) characterized by Hox gene derepression16–18. ATRTs are highly malignant tumors that are typically seen in children younger than 3 years. These tumors are characterized by inactivation of either SMARCB1 (BAF47) or SMARCA4 (BRG1), the core ATPase subunit, and rarely contain mutations in other genes19,20. Currently, the mechanism by which BAF simultaneously supports active and repressed states remains unclear.
A major limitation to resolving this question has been the loss of function approaches that lack sufficient temporal resolution to distinguish primary from secondary effects. Chromatin regulators tend to be stable for days following conventional depletion methods and are subject to numerous feedback mechanisms. This is especially problematic when studying chromatin remodelers like BAF, which regulate accessibility for many DNA-based processes and have numerous ascribed roles. To overcome these limitations, we implemented a chemical genetic approach to enable rapid degradation of essential BAF, PRC1 and PRC2 subunits in mouse ESCs (mESCs). We demonstrate that targeted degradation of the BAF ATPase subunit immediately derepresses genes that are highly occupied by Polycomb, such as Hox genes and developmental regulators. We show that BAF depletion results in the genome-wide redistribution of PRC1 and PRC2 from highly occupied domains like Hox clusters to sites that are normally opposed by BAF, independent of transcription, resulting in their physical decompaction. Our mechanistic study reconciles the dual role for BAF in opposition and maintenance of Polycomb-mediated repression, highlighting the dynamic nature of the Polycomb–Trithorax axis with implications for human disease.
Results
Brg1 degradation with auxin is rapid and near complete.
To temporally resolve the effects of BAF inactivation, we developed an ESC line where the sole ATPase subunit Brg1 (also known as Smarca4) can be rapidly degraded. Both endogenous alleles of Brg1 were tagged with the minimal 44 amino acid auxin inducible degradation tag (AID*) using CRISPR-Cas9 (CRISPR, clustered regularly interspaced short palindromic repeats) with homology-dependent repair21,22 (Extended Data Fig. 1a,b). The F-box protein osTIR1 was inducibly expressed in these cells, forming a hybrid SCF ubiquitin ligase complex that targets Brg1 for degradation by the proteasome when the small molecule auxin is added (Fig. 1a). Tagging Brg1 with AID* did not affect protein abundance, such that for all experiments Brg1 levels were equivalent to WT before adding auxin (Extended Data Fig. 1c). Additionally, the cells divided at the same rate and were indistinguishable from the parent mESC line. Consistent with other studies, auxin treatment in the absence of osTIR1 was innocuous to cell viability and growth23. Yet, when auxin was added to cells expressing osTIR1, Brg1 was rapidly degraded, with a protein half-life of ~30 min and maximal, near-complete, degradation by 2 h (Fig. 1b,c). This degron strategy resulted in much faster loss of function than genetic deletion (~2 h versus three days, Extended Data Fig. 1d) and induced changes to colony morphology at the 24-h time point (Extended Data Fig. 1d,e), so we conducted all subsequent experiments at short time points (≤8 h), shorter than one cell cycle. Thus, the Brg1 degron is a tractable and robust strategy to resolve the direct effects of BAF inactivation on the timescale of minutes to hours.
Fig. 1|. Brg1 degradation results in the derepression of many highly Polycomb-bound genes.
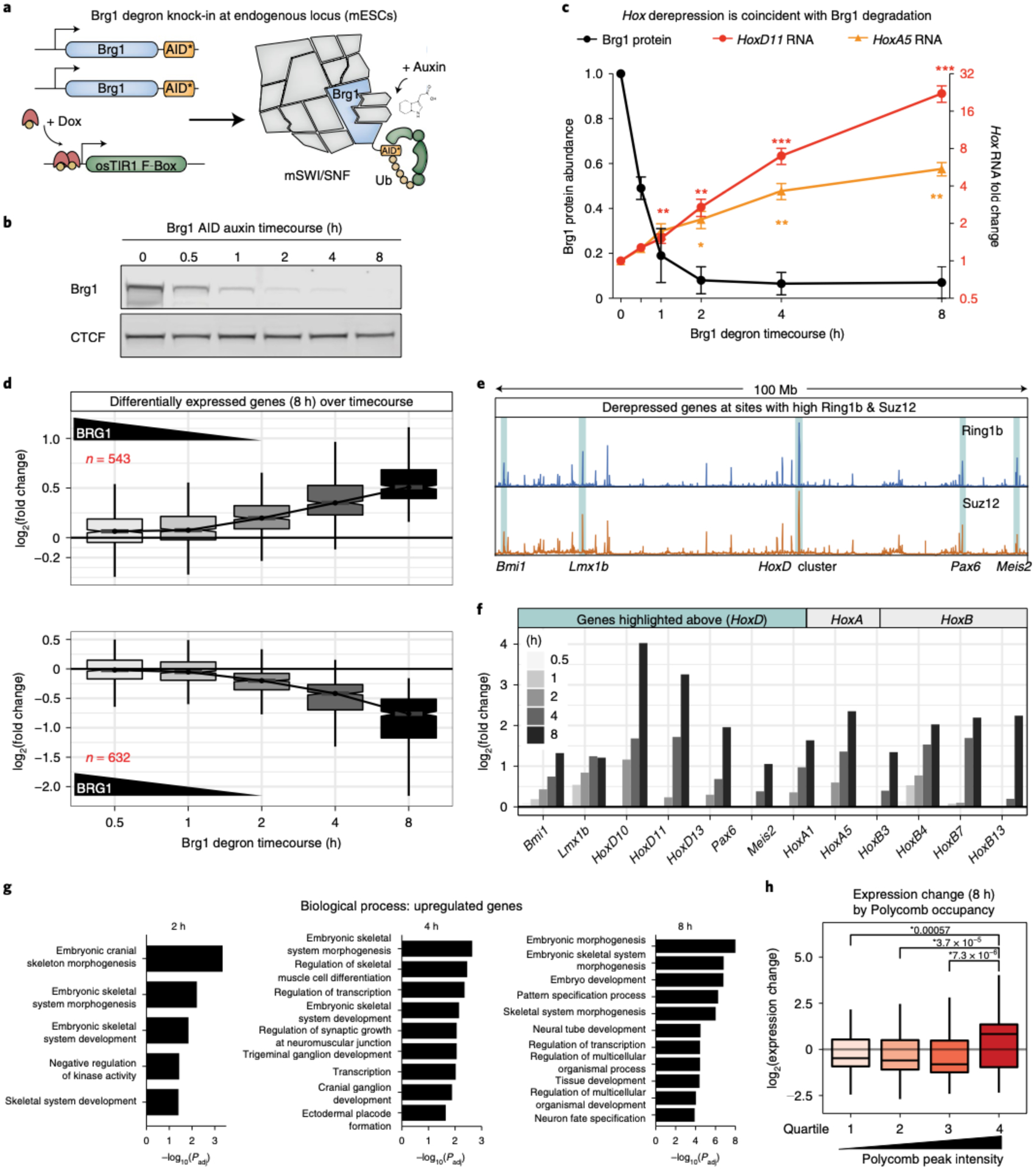
a, Schematic depicting the Brg1-AID strategy in mESCs. Both alleles of Brg1 are endogenously tagged at the C terminus with the minimal AID* tag. osTIR1 is induced with doxycycline before degrading Brg1 with auxin. b, Representative western blot showing Brg1-AID degradation kinetics and efficiency, with CTCF as the loading control. c, Quantitation of Brg1 degradation kinetics (left axis, maximal by 2h with T1/2 = ~30min) and induction of HoxD11 and HoxA5 transcription before Brg1 is maximally degraded (right axis, fold change by ddCt). Error bars represent mean ± s.e.m. from n = 3 and 4 biological replicates, respectively. Significance was calculated by individual two-sided Student’s t-test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). d, Fold change in expression level (log2) for genes that are differentially expressed at 8 h (FDR-corrected P < 0.05) across all time points (increased n = 543 (top), decreased n = 632 (bottom)) with Brg1 degradation kinetics overlaid. e, Browser tracks showing Ring1b and Suz12 ChIP-seq signal (0 h) at 100 Mb of 181-Mb chromosome 2. Regions with high Polycomb occupancy that become derepressed by Brg1 degradation are highlighted. f,g Timecourse of log2 fold changes (f) for the genes listed in g and genes from HoxA and B clusters that are derepressed by Brg1 degradation. h, Gene set enrichment analysis for differentially upregulated genes at 2, 4 and 8 h time points. Genes with high Ring1b and Suz12 occupancy at the TSS (±2 kb) become derepressed by Brg1 degradation (by quartile of peak intensity; two-sided Wilcoxon rank sum P values are shown, n = 4 biological replicates). Boxplot bounds depict quartile 1, median and quartile 3, with whiskers at 1.5× interquartile range (d,h). All experiments were conducted in serum/leukemia inhibitory factor (LIF).
Brg1 degradation derepresses highly Polycomb-bound genes.
A previous study reported derepression of Hox genes following three days of Brg1 conditional knockout18. This time point is unable to capture primary effects and undoubtedly combines secondary changes driven by a smaller number of primary events (Extended Data Fig. 2d). We first leveraged the fast Brg1 depletion kinetics to test whether this effect is causal. Indeed, we observed a time-dependent increase in HoxA5 and HoxD11 transcription that was directly coincident with Brg1 degradation, visible as early as 0.5 h, with both genes becoming significantly derepressed by the time that Brg1 was maximally degraded and expression continued to increase for 8 h (P < 0.05; Fig. 1c). By contrast, Hox gene derepression was not caused by auxin-induced degradation of Pbrm1, a defining member of the polybromo-associated BAF (pBAF) complex, controlling for artifacts of the experimental approach (Extended Data Fig. 2b) and also demonstrating that this repressive role is specific to the canonical BAF complex.
To comprehensively define the transcriptional kinetics, we next conducted a RNA-seq timecourse at 0.5, 1, 2, 4 and 8 h after degrading Brg1 with auxin. The number of differentially expressed genes increased at each successive time point with n = 543 upregulated and n = 632 downregulated genes at 8 h (false discovery rate (FDR)-corrected P < 0.05; Extended Data Fig. 2a). Consistent with the trend for HoxA5 and HoxD11, the genome-wide response was also directly coincident with Brg1 degradation (Fig. 1d). For genes upregulated at 8 h, median log2 fold changes were higher as early as 0.5 h (~50% Brg1 levels) and increased at each successive time point. Downregulated genes exhibited a minor time lag relative to upregulated genes, as expected, considering the time needed for transcript decay and completion of elongation, yet a reduction in transcription was clearly seen by 2 h and transcription of these genes continued to decrease at each successive time point (Fig. 1d, bottom). Most transcriptional changes occurred before Brg1 was even completely degraded, at timescales of ~0.5–2 h. Considering that the protein maturation time and half-life of protein/RNA is generally much longer than this, we conclude that these changes are the direct result of rapid Brg1 degradation and not a secondary effect.
We were intrigued to find that, in addition to Hox, many strongly derepressed genes colocalized with the strongest PRC1 and PRC2 peaks on the chromosome (Fig. 1e). Previous studies have shown that these sites form extra long-range chromosomal interactions that require PRC1, for example between HoxD and Bmi1, Pax6, Meis2 and Lmx1b24. HoxA, B and D genes were among the most strongly derepressed and showed a time-dependent increase in transcription by ~2 h (Fig. 1f), consistent with the results in Fig. 1c,d. Bmi1 (PCGF4) was also derepressed quickly upon Brg1 degradation and is a reliable BAF repressed gene that has been used as a reporter in a high-throughput inhibition screen25. Yet, strong ectopic overexpression did not cause Hox gene derepression (Extended Data Fig. 2e), providing further support that derepression within Polycomb domains is directly caused by rapid Brg1 depletion.
In ESCs, the Polycomb system represses developmental regulators, including many transcription factors, to prevent differentiation26. In light of our finding that many Polycomb-bound genes are derepressed by Brg1 depletion, we conducted gene set enrichment analysis (Fig. 1g). As expected, we find that many patterning, differentiation and developmental categories are enriched, even at the 2 h time point (Extended Data Fig. 2c shows downregulated genes). Considering that many genes with high Polycomb occupancy become derepressed, we sought to determine if this is a general trend that can be predicted by Polycomb occupancy. At 8 h, where 1,175 genes are differentially expressed, weakly Polycomb-bound genes (average Ring1b and Suz12 intensity ±2 kb from the transcription start site (TSS), quartiles 1–3) become repressed, whereas genes with the highest PRC1 and PRC2 levels (quartile 4) tend to become derepressed by Brg1 degradation (Fig. 1h, P < 0.05 between quartile 4 and 1–3). Thus, rapid Brg1 depletion has opposing effects on Polycomb-mediated gene regulation, with derepression of highly and repression of lowly Polycomb-occupied genes.
PRC1 and PRC2 are quickly redistributed by Brg1 degradation.
We observed quick transcriptional derepression within Polycomb domains, so we next sought to determine the effect of Brg1 degradation on PRC1&2 occupancy genome-wide by conducting chromatin immunoprecipitation sequencing (ChIP-seq) for Ring1b and Suz12 (core subunits of the PRC1 and PRC2 complexes) (Extended Data Fig. 3a). Consistent with the established role for BAF in evicting Polycomb, we found that more Ring1b and Suz12 peaks increase than decrease following 8 h of Brg1 degradation (for Ring1b, 931 increased and 641 decreased, where 14.9% of 10,569 peaks were differentially bound; for Suz12, 457 increased and 200 decreased, where 7.4% of 8,853 peaks were differentially bound, FDR-corrected P < 0.1). Differential peaks with low Ring1b and Suz12 occupancy (as assessed by normalized peak counts) are mostly increased, and >90% are bound by BAF, whereas highly occupied peaks mostly decrease following Brg1 degradation (Fig. 2a). This general trend can be seen in representative genome-browser snapshots of increased (Cyp2s1 and Ptk2b) and decreased (HoxA/D clusters) sites (Fig. 2b and Extended Data Fig. 3b,c). Collectively, these results demonstrate that the loss of PRC2 at Hox clusters, previously seen after 72 h in the Brg1 conditional deletion18, occurs coincident with Brg1 removal, and show that not only is PRC2 lost, but PRC1 is also depleted. In fact, Brg1 degradation-induced changes to PRC1 and PRC2 are highly correlated across all peaks (R = 0.79, P < 2.2 × 10−16) and display a high degree of overlap between peaks (Fig. 2c,d). Importantly, these changes are independent of global changes to the core proteins and the histone modifications placed by these complexes, even at much longer time points (Extended Data Fig. 3e).
Fig. 2 |. PRC1 and PRC2 are quickly redistributed following Brg1 degradation.
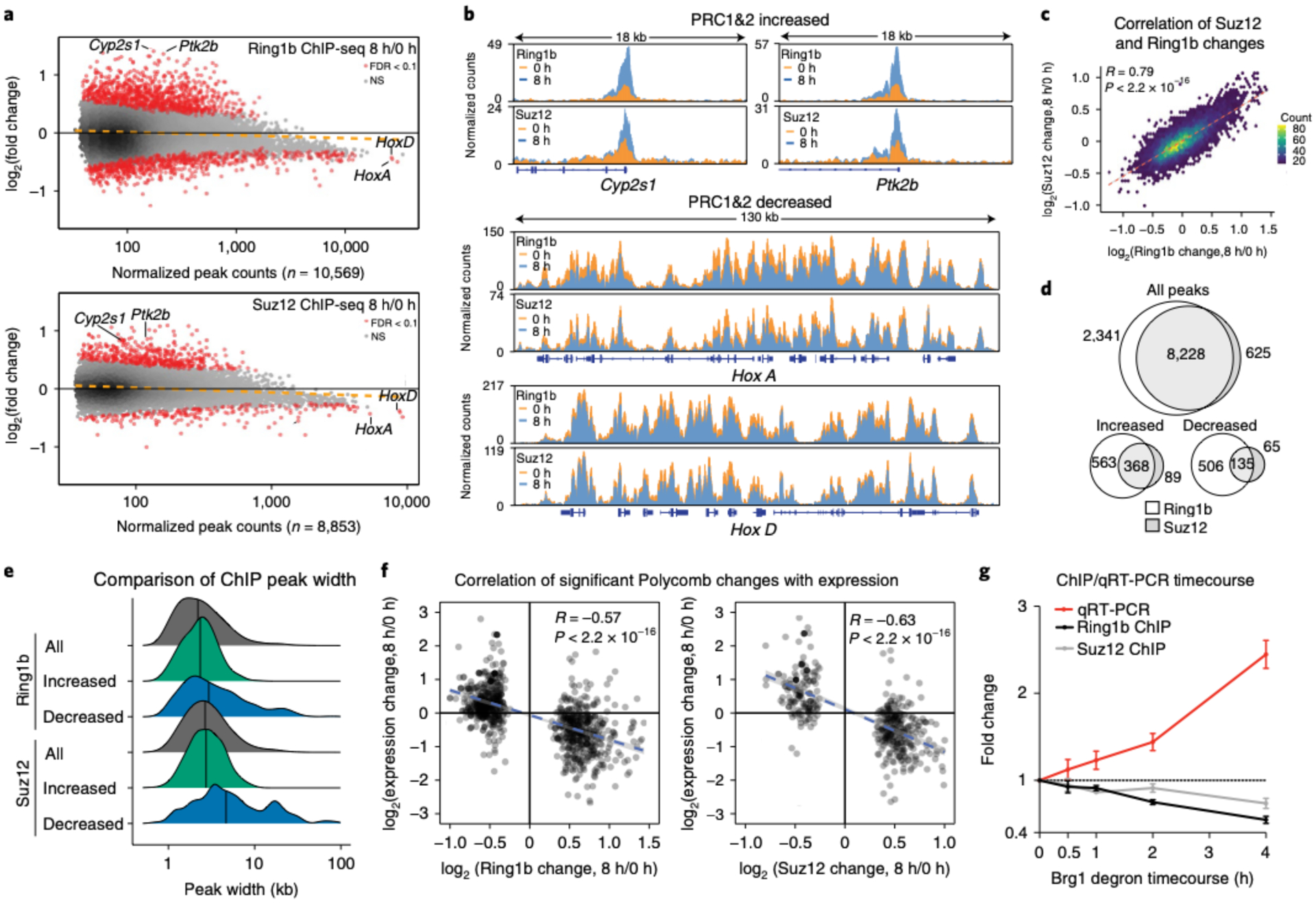
a, MA (log-intensity ratios (M-values) versus log-intensity averages (A-values)) plot showing genome-wide changes to Ring1b and Suz12 ChIP-seq peaks (n = 10,569 and n = 8,853, respectively) at 8-h Brg1 degradation, with differentially bound peaks in red (FDR-corrected P < 0.1), from four biological replicates. For Ring1b, there are n = 930 increased and n = 640 decreased peaks, and for Suz12 there are n = 457 increased and n = 198 decreased peaks. NS, not significant. b. Representative genome-browser tracks for peaks labeled in a that increase (Cyp2s1 and Ptk2b) and decrease (HoxA and HoxD clusters), normalized by read depth. c, Correlation plot between Ring1b and Suz12 peak changes as a hexagonal heatmap of two dimensional bin counts. Correlation and P values were obtained from Pearson’s product moment correlation. d, Peak overlap for all, increased and decreased peaks for Ring1b and Suz12. e, Peak widths for all, increased and decreased peak changes for Ring1b and Suz12. f, Correlation of significantly changed Ring1b and Suz12 peaks that are ±2 kb from gene TSSs and gene expression changes at 8-h Brg1 degradation. g, ChIP-qPCR and qRT-PCR timecourse at the Bmi1 locus showing coincident PRC1/2 loss and derepression. Data are presented as mean ± standard error of the mean from three biological replicates. Correlation and P values were obtained from Pearson’s product moment correlation. All experiments were conducted in serum/LIF.
Chromatin state enrichment analysis27 revealed that regions occupied by PRC1&2 are highly enriched for bivalent chromatin, with slight enrichment for the fully repressed state at regions where PRC1&2 are decreased following Brg1 degradation (Extended Data Fig. 3g). We wondered whether the high normalized peak counts seen at decreased sites were due to enrichment for broader domains as a general principle, as seen at the Hox clusters (Fig. 2e). We found that the increased peaks have a very similar distribution to all PRC1 and PRC2 peaks (Ring1b median for all peaks = 2.2 kb, increased = 2.3 kb and Suz12 median for all peaks = 2.9 kb, increased = 2.7 kb), but decreased peaks initially contain broader Polycomb domains (Ring1b median decreased = 2.9 kb, Suz12 median decreased = 4.7 kb). We previously showed that the basal occupancy level of PRC1 and PRC2 is predictive of the direction of differential gene expression (Fig. 1h). We next sought to determine how global Polycomb changes influence transcription at the same 8-h time point. We found that, for all genes, there is a modest but highly significant negative correlation between changes to Polycomb occupancy and transcription (Extended Data Fig. 3f; Ring1b, R = −0.31, P < 2.2 × 10−16; Suz12, R = −0.33, P < 2.2 × 10−16). Yet, considering only differential peaks, we observed an even stronger negative correlation (R = −0.57, P < 2.2 × 10−16 and R = −0.63, P < 2.2 × 10−16 for Ring1b and Suz12, respectively; Fig. 2f). These results suggest that Polycomb redistribution leads to transcriptional derepression. To define the temporal dynamics, we conducted ChIP quantitative PCR (qPCR) and qPCR with reverse transcription (qRT-PCR) at Bmi1, a canonical BAF and Polycomb repressed gene. Here, Polycomb loss and transcriptional derepression are coincident, with subtle Ring1b and Suz12 loss as early as 0.5 h where Brg1 levels are ~50%, which continues to decrease in a time-dependent manner, consistent with Polycomb loss leading to derepression (Fig. 2g and Extended Data Figs. 3d (longer time courses) and 3h). Altogether, these results suggest that BAF activity is constantly required to evict Polycomb from lowly occupied bivalent sites to accumulate at highly occupied broad domains, such as Hox clusters.
Brg1 degradation and Polycomb loss decompact Hox clusters.
Polycomb complexes constrain and physically compact chromatin, which is thought to facilitate repression28–30, leading us to ask if the partial PRC1 and PRC2 loss from sites with high Polycomb occupancy is sufficient to spatially decompact them. To test this, we conducted optical reconstruction of chromatin architecture (ORCA) experiments28,31,32 at HoxA and HoxD clusters, which lose Polycomb and become transcriptionally derepressed by Brg1 degradation. ORCA enables reconstruction of chromatin trajectories (100–700 kb) by tiling short regions (2–10 kb) with unique barcodes and measuring their nanoscale three-dimensional (3D) positions (Fig. 3a,b). We tiled 290 kb at HoxA and 234 kb at HoxD in 5-kb steps (that is, with different barcode for each 5-kb step) that completely cover the Polycomb repressed domains and extend into the flanking regions (Fig. 3c,d).
Fig. 3 |. Brg1 degradation and Polycomb loss result in decompaction of HoxA and HoxD regions.
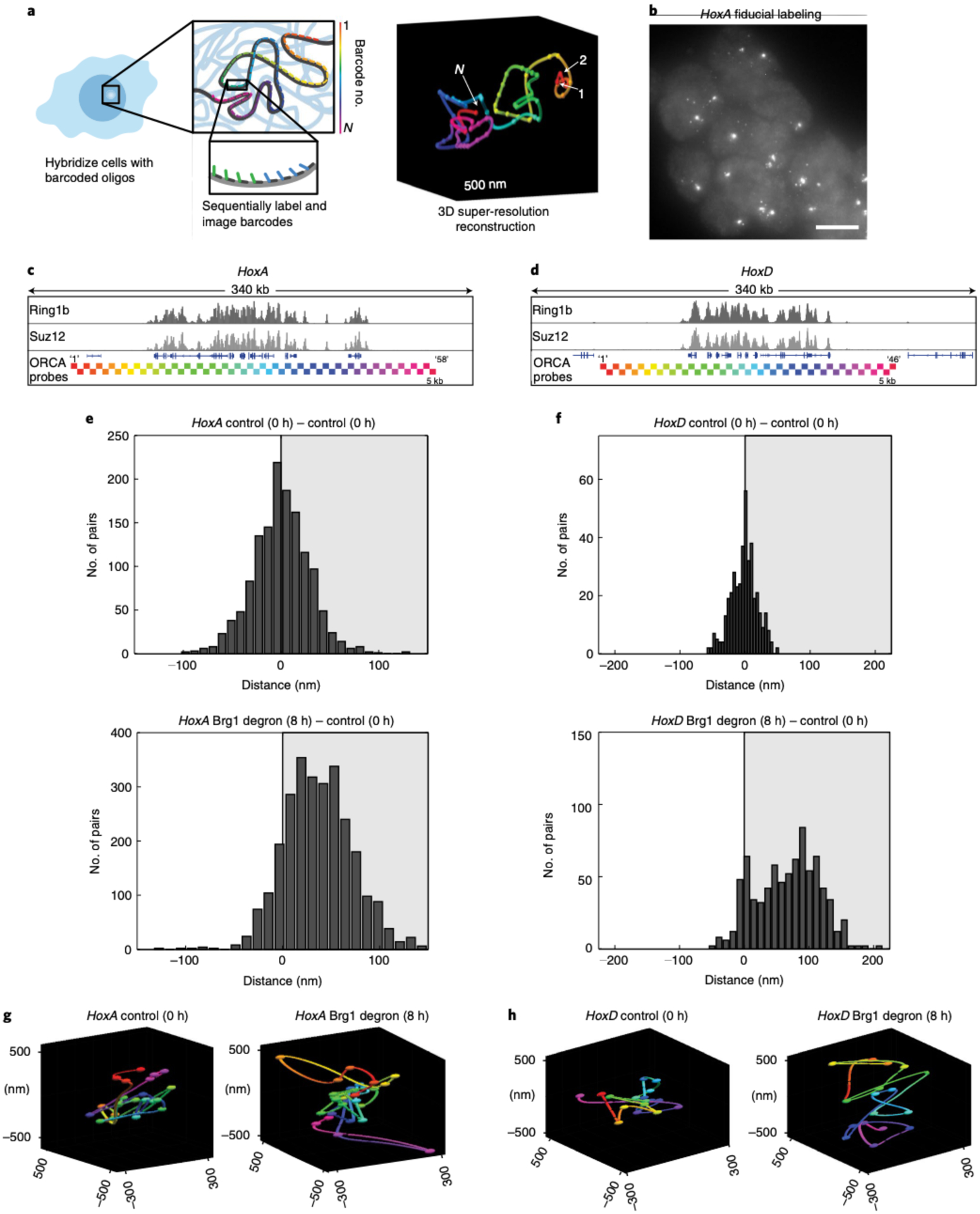
a, Schematic of the ORCA method, where sequential imaging and labeling of fluorescent barcodes (numbers 1 to N) are artificially color-coded by barcode number, enabling reconstruction of the 3D DNA path of a DNA region. b, mESCs labeled with the HoxA probe. Each spot corresponds to one allele, and shown here is the fiducial staining, which labels the entire 290-kb HoxA region. The experiment was repeated at least three times with similar results. Scale bar, 10 μm. c,d, Genomic HoxA (c) and HoxD (d) regions labeled by ORCA. The bottom tracks illustrate the positions of probes, color-coded by barcode number, as in a. e,f, Histograms plotting the difference in median distance for all barcode pairs, where the median was calculated over all cells, between AID-treated and control cells, for HoxA (e) and HoxD (f). g,h, Representative polymers in the control and Brg1-depleted cells for HoxA (g) and HoxD (h). All experiments were conducted in serum/LIF.
Applying ORCA in untreated cells and calculating the contact frequency over all cells revealed similar sub-TAD (TAD, topologically associating domain) domain architecture and a high correlation with published Hi-C data in mESCs33 (Extended Data Fig. 4a,b). We then leveraged the unique ability of ORCA to measure 3D nanoscale distances and quantified the median inter-barcode distance at HoxA and HoxD following Brg1 degradation. Consistent with redistribution of PRC1 and PRC2 from Hox detected by ChIP, we found higher inter-barcode distances for both HoxA and HoxD when Brg1 is degraded (8 h) (Fig. 3e,f and Extended Data Fig. 4c–e), which is also apparent in the reconstructed polymer traces from single cells (Fig. 3g,h). Thus, the redistribution of Polycomb away from Hox clusters, despite being incomplete, is sufficient to spatially decompact them.
PRC redistribution results in opposite epigenomic changes.
Transcriptional regulation involves a dynamic interplay between activating and repressive forces. We next sought to determine how the genome-wide redistribution of Polycomb-mediated repression impacts other chromatin features associated with active transcription. We conducted ChIP-seq for H3K4me3 and H3K27ac following 8 h of Brg1 degradation. H3K4me3 is a histone modification associated with poised and transcriptionally active genes that is deposited by the COMPASS complex, which has homologous subunits in Drosophila that are Trithorax-group members, like the BAF complex34. H3K27ac is primarily deposited at active genes and enhancers by the CBP/p300 complex, which binds to BAF35 and antagonizes the mutually exclusive H3K27me3 modification that is placed by PRC236. Interestingly, we found that at 8 h of Brg1 degradation, both H3K27ac and H3K4me3 were negatively correlated with changes to Ring1b (R = −0.45, P < 2.2 × 10−16 and R = −0.5, P < 2.2 × 10−16) and Suz12 (R = −0.37, P < 2.2 × 10−16 and R = −0.44, P < 2.2 × 10−16) genome-wide (Fig. 4a,b), such that where PRC1 and PRC2 increase, active marks are decreased (Fig. 4c), and where PRC1 and PRC2 decrease, active marks increase after Brg1 degradation (Fig. 4d) (Extended Data Fig. 5a,b). These data suggest that the global Polycomb redistribution results in opposite epigenomic changes associated with active transcription.
Fig. 4 |. Genome-wide Polycomb redistribution changes chromatin state from repressed to active.
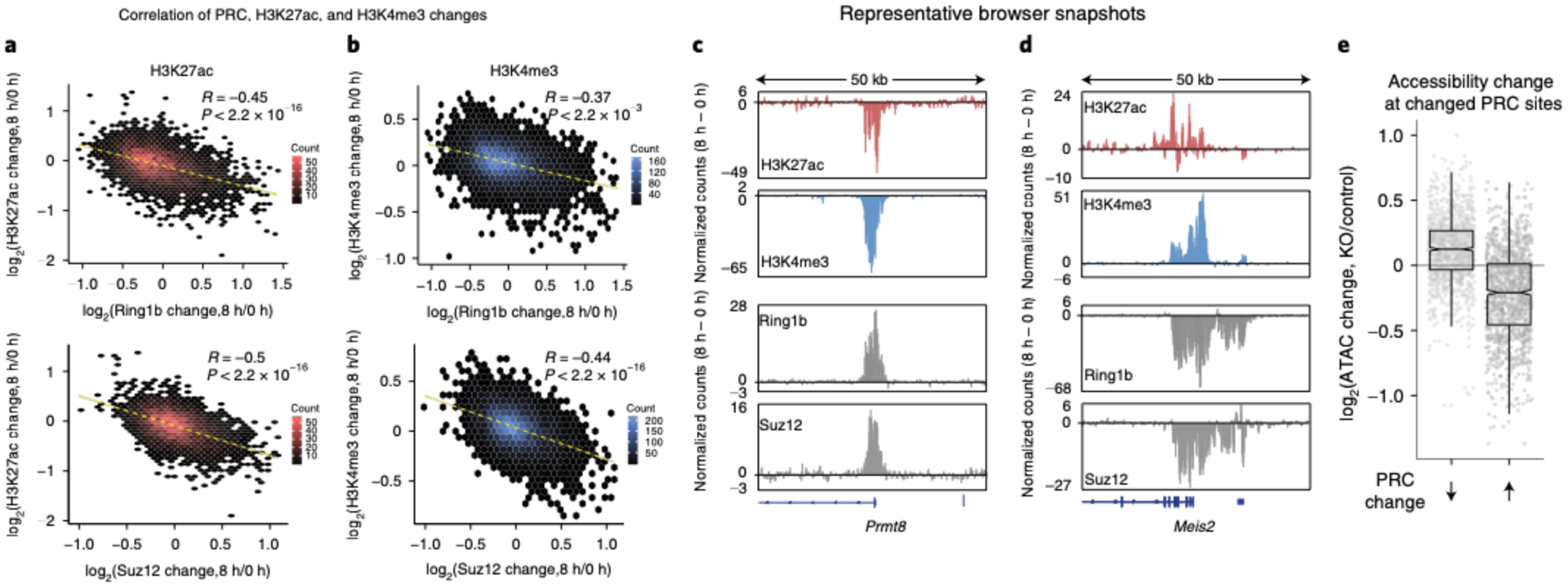
a,b, Correlation plot of fold change (log2) to H3K27ac (a) H3K4me3 (b) and Ring1b or Suz12 at 8 h of Brg1 degradation as a hexagonal heatmap of two-dimensional bin counts. Correlation and P values were obtained from Pearson’s product moment correlation. c, Representative browser snapshots showing the Prmt8 locus that gains Ring1b and Suz12 and loses both H3K27ac and H3K4me3 as the difference in normalized counts (8 h to 0 h degradation). d, Browser snapshot at the Meis2 locus, which loses Ring1b and Suz12 and gains both H3K27ac and H3K4me3. e, Boxplot showing the change in accessibility to Tn5 transposition in Brgfl/fl deleted cells (TAM) over control (EtOH) at peaks that have decreased or increased Ring1b or Suz12 signal after 8 h of Brg1 degradation. (Assay for transposase-accessible chromatin sequencing (ATAC-seq) data are from ref.44, n = 2 biological replicates). Boxplot bounds depict quartile 1, median and quartile 3, with whiskers at 1.5× interquartile range. All experiments were conducted in serum/LIF.
We next sought to understand what drives the global redistribution of Polycomb from highly occupied sites. One possibility is that BAF inhibits transcription through nucleosome positioning37, which then selectively drives Polycomb loss due to physical disruption from the transcription machinery or by competitive removal through preferential binding of PRC2 to RNA38. We tested this idea by blocking transcription initiation with Triptolide, ±auxin, for 8 h39 (Extended Data Fig. 6a,b). We chose this time point for consistency with our other ChIP-seq datasets and to minimize toxicity40. We then correlated the changes to Ring1b and Suz12 caused by Brg1 degradation ± complete inhibition of transcription (auxin + triptolide/triptolide to auxin/DMSO). This revealed that Polycomb is still redistributed, despite global transcription inhibition for both Ring1b (R = 0.68, P < 2.2 × 10−16) and Suz12 (R = 0.65, P < 2.2 × 10−16) (Extended Data Fig. 6c). Importantly, this correlation is strengthened even further for peaks that are significantly changed by Brg1 degradation (R = 0.86, P < 2.2 × 10−16 and R = 0.83, P < 2.2 × 10−16 for Ring1b and Suz12, respectively; Extended Data Fig. 6d). By contrast, the effect of triptolide versus auxin alone shows zero or a very weak correlation (R = 0.015 for Ring1b and R = 0.25 for Suz12; Extended Data Fig. 6e). Thus, Polycomb redistribution is independent of transcription, consistent with our observation that Polycomb is lost across large 100+ kb domains, and not just at genes that become derepressed.
In general, BAF and PRC1 and PRC2 overlap extensively genome-wide, but sites that are highly occupied for either appear mutually antagonistic (Extended Data Fig. 7a). Our Polycomb ChIP-seq results in the Brg1 degron showed that PRC1 and PRC2 accumulate at sites normally opposed by BAF. To see if the opposite is true, that is, does PRC1 produce resistance to BAF binding as suggested by in vitro experiments41,42, we used ESCs in which we could conditionally delete Ring1bfl/fl using a tamoxifen-inducible actin-CreER in a Ring1a−/− background, to completely ablate PRC1 activity43. After tamoxifen treatment, we examined BAF binding using the BAF155 core subunit (Extended Data Fig. 7a,b). In contrast to expectations, we found that while some BAF155 peaks do change, the vast majority (>95%) are unchanged following PRC1 deletion, and only 1% of differential peaks are within PRC1 domains (Extended Data Fig. 7c–g). Therefore, the BAF–Polycomb axis appears mostly unidirectional, such that BAF antagonizes Polycomb at weakly bound sites but is not excluded from PRC1 domains.
To investigate this further, we looked at Brg1-dependent accessibility changes by the assay for transposase-accessible chromatin sequencing44 at sites where Polycomb changes upon loss of Brg1. Accessibility changes reflect SWI/SNF chromatin remodeling activity, because the readout is the endpoint of the catalytic cycle (that is, nucleosome dynamics). We reasoned that if BAF is required to generate accessibility for the DNA-binding subunits that target Polycomb complexes, then accessibility changes upon Brg1 loss should mimic Polycomb changes. In sharp contrast to this, we found that sites where Polycomb is lost become more accessible, and sites that gain Polycomb become less accessible (Fig. 4e and Extended Data Fig. 5c,d). Yet, Polycomb does not repress accessibility to Tn5 transposition in mESCs45,46, which suggests that Brg1 is required to repress accessibility at these sites. These results are most consistent with a model where BAF facilitates Polycomb-mediated repression by limiting accessibility at highly PRC-bound sites in addition to distant eviction, which liberates PRC1 and PRC2 for accumulation elsewhere.
BAF promotes repression directly and by PRC redistribution.
If the BAF-driven Polycomb redistribution and subsequent decompaction alone is sufficient to drive derepression, we reasoned that this should result in insufficient Polycomb to both accumulate and maintain distal repression. To test this, we wanted a system where we could rapidly degrade PRC1&2 in a dose-dependent way and measure the transcriptional response relative to Brg1 depletion at the exact time point. For this goal, we implemented the dTAG targeted degradation approach, which enables dose-dependent, specific and efficient protein degradation47. We tagged endogenous alleles of essential subunits of PRC1 (Ring1b) and PRC2 (EED) with the FKBPF36V tag, which enables targeted degradation in the presence of dTAG13, a heterobifunctional analog of rapamycin. Tagging these proteins did not noticeably affect protein abundance, cell viability or growth (Extended Data Fig. 8a–d).
Treating ESCs with a 10-fold serial dilution of dTAG13 ligand resulted in dose-dependent, near-complete degradation of Ring1b/EED to 75, 50, 12 and 3% of WT levels at the 8 h time point (Fig. 5a; maximal degradation is achieved by 2 h, similar to the Brg1 degron). We next conducted RNA-seq from these four Polycomb doses at the 8 h time point to enable comparison with the Brg1 degron. Surprisingly, we found that reducing PRC1&2 to 75% and 50% has very modest transcriptional effects. Yet, depleting PRC1&2 to 12% and 3% results in many more derepressed genes (n = 179 and n = 970, respectively, FDR-corrected P < 0.05; Fig. 5b). Consistent with the statistical cutoff, the magnitude of log2 fold changes also displays modest dosage sensitivity, until the higher levels of depletion (Fig. 5c).
Fig. 5 |. BAF promotes repression directly and by genome-wide Polycomb redistribution.
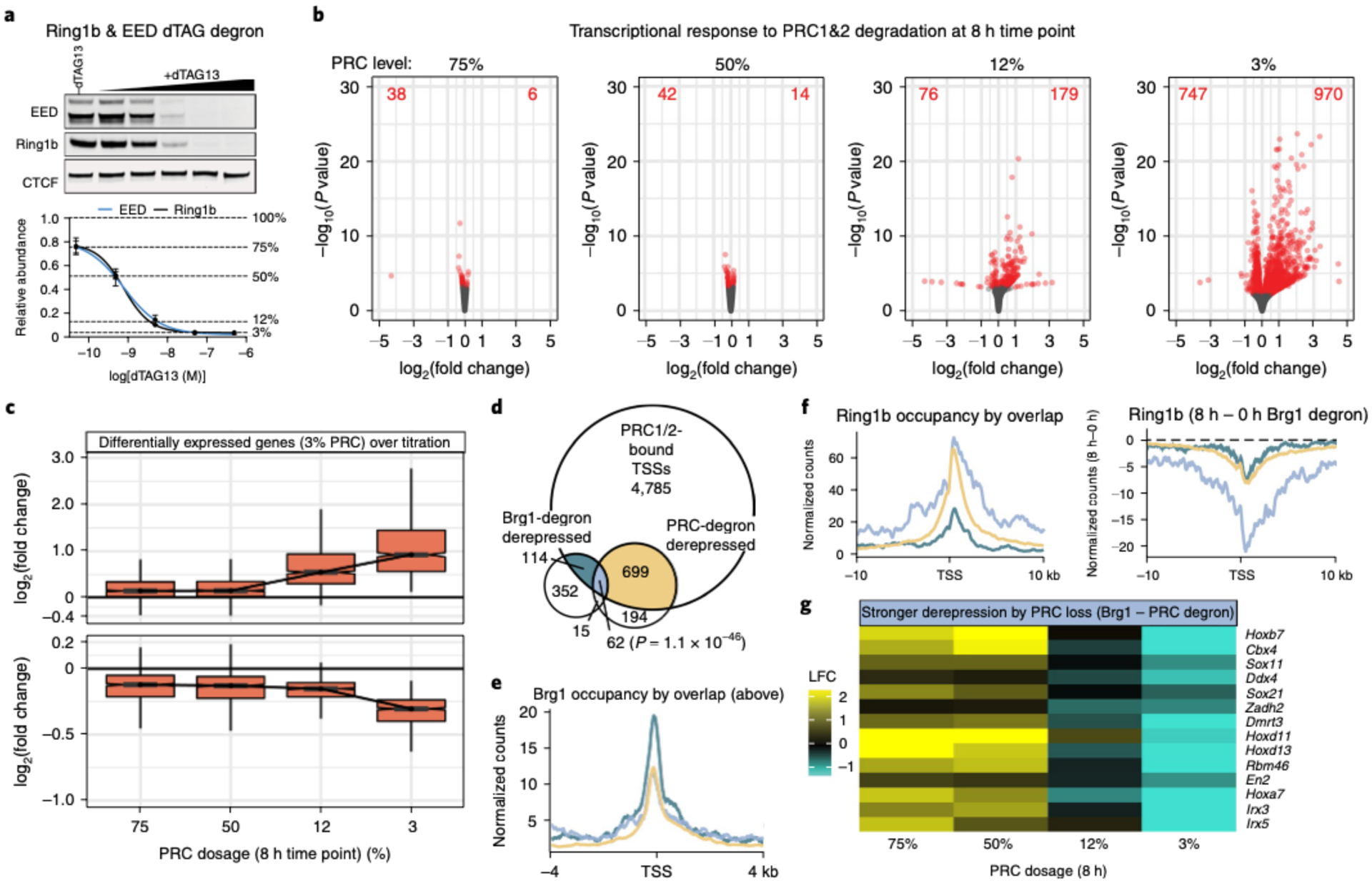
a. Representative western blot (top) and quantification (bottom) of Ring1b (PRC1) and EED (PRC2) degron lines across 10-fold dilution treatment with dTAG13 proteolysis-targeting chimera (PROTAC) for 8 h. Data are presented as mean ± s.e.m. from four cell lines. b, Transcriptional response to degrading PRC1/2 to 75, 50, 12 and 3% of WT levels for 8 h. The volcano plots depict gene expression changes across four PRC doses, with differentially expressed genes colored red (FDR-corrected P < 0.05) from n = 4 cell lines. c, Fold change in expression level (log2) for genes that are differentially expressed at 3% PRC dosage (FDR-corrected P < 0.05) across all doses (increased n = 970 (top), decreased n = 747 (bottom)). The boxplot bounds depict quartile 1, median and quartile 3, with whiskers at 1.5× interquartile range. d, Plot showing significant overlap between genes that are bound by PRC1/2 (Ring1b and Suz12 ± 2kb from TSS), Brg1-degron derepressed (8 h) and PRC1/2-degron derepressed (3% dosage, 8 h). The significance of the overlap of PRC-bound, Brg1 and PRC upregulated genes was calculated by Fisher’s exact test (P = 1.1 × 10−46). e, Brg1 occupancy is substantially higher over PRC1/2 bound genes solely derepressed in the Brg1 degron compared to genes derepressed by PRC or PRC and Brg1 degradation. Colors are as in d. f, Left: Polycomb domains are higher and broader over genes derepressed by both Brg1 and PRC degron than genes derepressed by Brg1 degron alone. Right: broad domains over genes derepressed by both Brg1 and PRC degron show substantially more redistribution in the Brg1 degron. Colors are as in d. g, Heatmap depicting the log2 fold change (LFC) in expression between the Brg1 degron and each PRC degron dose for select genes more strongly derepressed by PRC than Brg1 degradation. All experiments were conducted in serum/LIF.
We next compared the effects of the Brg1 and PRC1&2 degron at 8 h. Our ChIP-seq analysis identified 5,660 genes with both Ring1b and Suz12 at their TSS (±2 kb). Of these genes, 699 are derepressed by maximum PRC1&2 depletion, 114 by Brg1 degradation and 62 are derepressed by both (Fig. 5d, Fisher’s exact test P = 1.1 × 10−46). This confirms that Brg1 degradation has both divergent and convergent (~35% of Brg1 derepressed genes that are bound by PRC) effects with PRC1&2 depletion. Considering that Brg1 depletion causes rapid transcriptional responses that cannot be explained by downstream feedback mechanisms, we focused on direct mechanisms to explain these results. We found that BAF and Polycomb occupancy overlap extensively, but PRC1 deletion has almost no effect on BAF binding (Extended Data Fig. 7), and that BAF is bound to the promoter of all differentially expressed genes, including those with high PRC1 and PRC2 (Extended Data Fig. 8e,f). So, we hypothesized that conventional BAF remodeling activity might contribute to repression, in addition to redistributing Polycomb (similar to the results in Fig. 4e). In support of this, Brg1 occupancy is substantially higher at Polycomb-bound genes that are derepressed by Brg1 (Polycomb-independent) than at genes derepressed by Brg1/PRC1&2 and PRC1&2 depletion (Fig. 5e and Extended Data Fig. 8g). Consistently, genes derepressed by Brg1 depletion alone, and both Brg1/PRC1&2 required BAF to repress accessibility at the promoter (Extended Data Fig. 8 h, P = 1.1 × 10−9).
We next investigated PRC occupancy at genes derepressed by Brg1 (Polycomb-independent), PRC1&2 alone and both Brg1 and PRC1&2. We found that genes derepressed only by Brg1 loss have substantially less PRC1 (Fig. 5f) and PRC2 (Extended Data Fig. 8i) compared to those also derepressed by loss of PRC1&2. Although Brg1 depletion induces Polycomb loss at all groups, the genes derepressed by both BAF and PRC1&2 depletion showed substantially greater reduction across a broader range (>20 kb). These genes are also modestly dosage-sensitive (Extended Data Fig. 8j), with a subset exhibiting much stronger derepression by PRC1&2 depletion than BAF, including a few Hox genes (Fig. 5e). It is likely that these genes require greater PRC1&2 depletion than the ~20–60% loss seen by redistribution (Fig. 2a,b,g and Extended Data Fig. 3d) because BAF also contributes modestly to their repression. Our results point to a mechanism of BAF-PRC opposition, in which BAF facilitates repression both by suppressing accessibility at TSSs and by globally redistributing Polycomb across the genome.
Increased PRC1 dosage inhibits derepression.
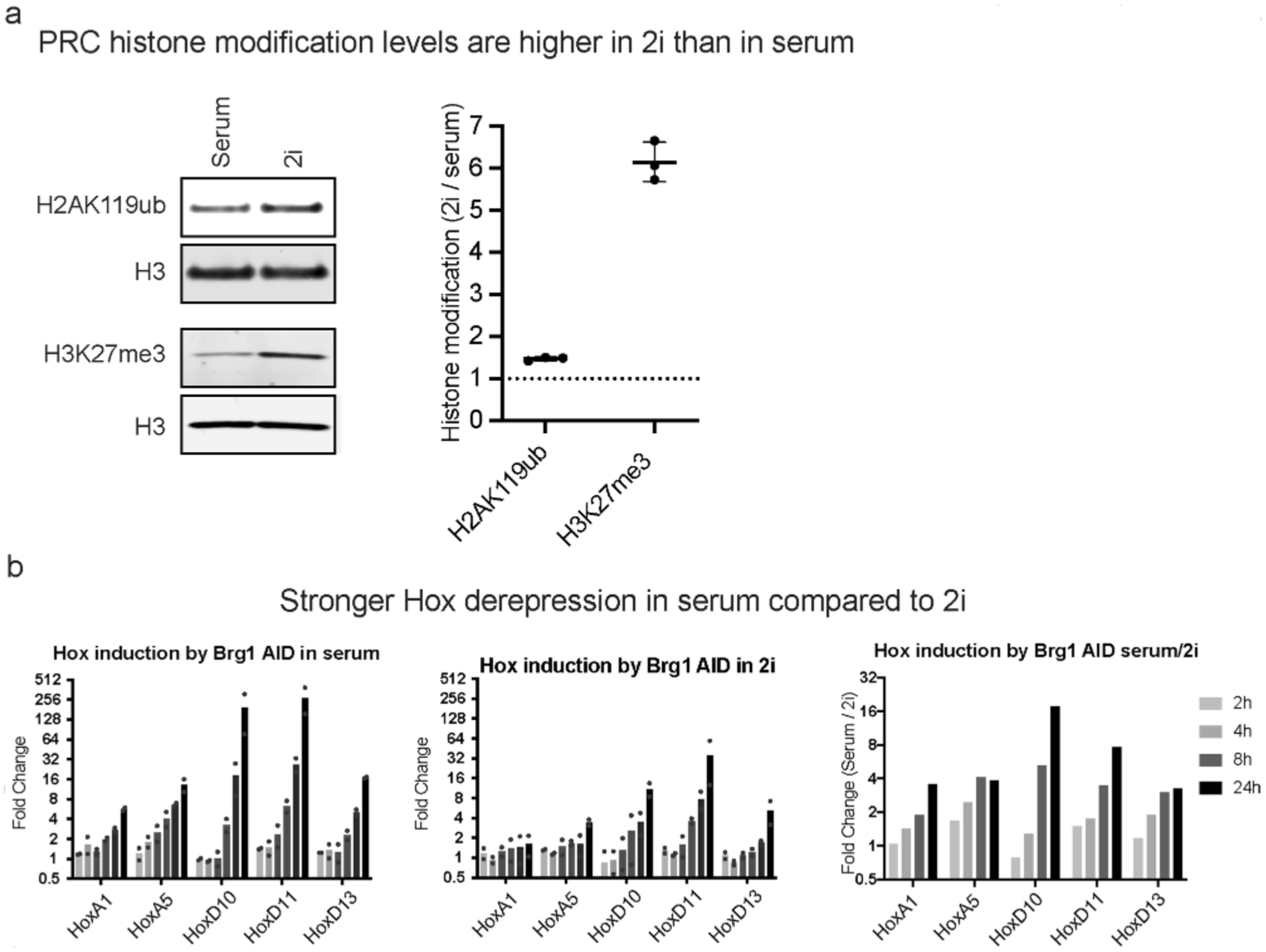
To evaluate the contribution of Polycomb redistribution, we reasoned that if we conduct the opposite of our PRC1&2 degron experiments and instead overexpress Polycomb, then the increased dosage should be sufficient to both redistribute and maintain repression of Hox genes. This is inherently challenging considering the complexity of the Polycomb system. Because it has not been feasible to increase the dosage of each subunit of multisubunit complexes, we sought to overexpress a minimal complex that has a dominant role in repression. To accomplish this, we overexpressed a minimal variant PRC1 complex containing Ring1b and Pcgf1, because it has a dominant role in depositing the H2AK119ub repressive mark48,50. Using this strategy, we obtained an ~2× increase in H2AK119ub1 levels over the empty vector control, despite ~10× RNA overexpression from the strong EF1a promoter (Fig. 6a). Next, we conducted a series of 8-h Brg1 degron experiments to test the effect of vPRC1 overexpression (vPRC1OE) on Brg1 degron-mediated derepression. In both vector and vPRC1OE cells, we obtained similar Brg1 degradation efficiencies (95 ± 3.5% and 91 ± 3.0% degraded, respectively). However, overexpressing variant PRC1 significantly inhibited the derepression caused by Brg1 degradation for 10/14 genes that were amenable to qRT-PCR (Fig. 6b, P < 0.05). To further explore the influence of PRC dosage on Brg1 degron-mediated derepression, we compared 2i versus serum conditions at a few genes. Consistent with other reports, 2i culture resulted in a net increase in H2AK119ub (~1.5×) and H3K27me3 (~6×) (Extended Data Fig. 9a)51,52. Consistent with our overexpression experiments, we found that Hox genes were derepressed less efficiently in 2i culturing conditions with increased PRC activity (~3× to 16×; Extended Data Fig. 9b). Thus, increased dosage of just a minimal two-subunit variant PRC1 complex or both PRC1&2 inhibits Brg1 degron-mediated derepression, consistent with our model that BAF frees Polycomb for passive accumulation across the genome.
Fig. 6 |. Increased PRC1 dosage inhibits Brg1 degron-mediated derepression.
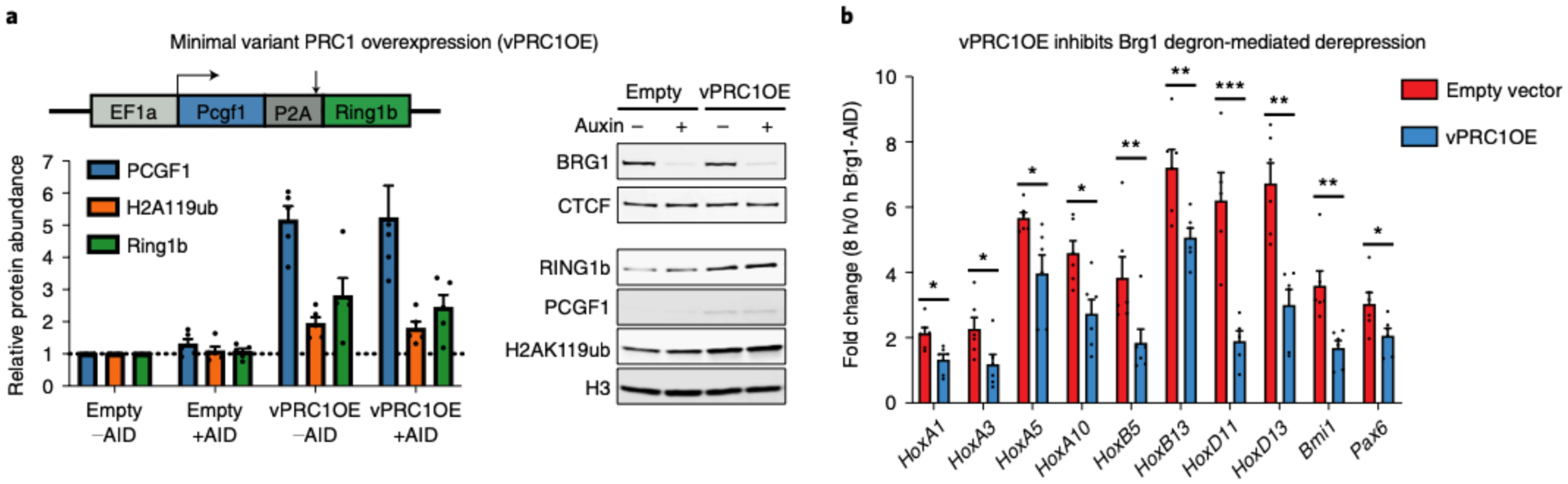
a, Left: western blot quantification of PCGF1, H2AK119ub and Ring1b relative to vector only −AID control cells. Right: representative western blot showing Brg1 degron and minimal vPRC1 overexpression. b, Fold change (8 h/0 h Brg1-AID) for cells stably expressing either empty vector or variant PRC1. n = 6 biological replicates from two transductions. Significance was calculated by individual Student’s t-test (*P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001). Error bars represent s.e.m.
Discussion
Here, we have used a targeted degradation approach to comprehensively dissect the BAF-Polycomb axis with high temporal precision in mESCs. Our studies demonstrate that BAF directly promotes Polycomb-mediated repression through conventional remodeling activity and by evicting PRC1 and PRC2, enabling accumulation at distal sites, such as the hox clusters (Fig. 7). This mechanism of repression reconciles previous observations that BAF is involved in both Polycomb antagonism53 and repression in stem cells and cancer16–18. Although it is not possible to rule out a direct role for BAF in Polycomb loading, our results are inconsistent with such a model, in that BAF limits accessibility where Polycomb is maintained instead of generating access. Additionally, direct chemically induced BAF recruitment only leads to Polycomb eviction, not recruitment11,13. Although the magnitude of PRC1 and PRC2 loss across these 100+ kb scale domains is incomplete, the level of depletion was sufficient to physically decompact HoxA and HoxD loci by ORCA. These results are consistent with previous findings that heterozygous Ring1b deletion decondenses Hox clusters29, but in this case BAF modulates Polycomb dosage at a distance.
Fig. 7 |. Model and summary of findings.
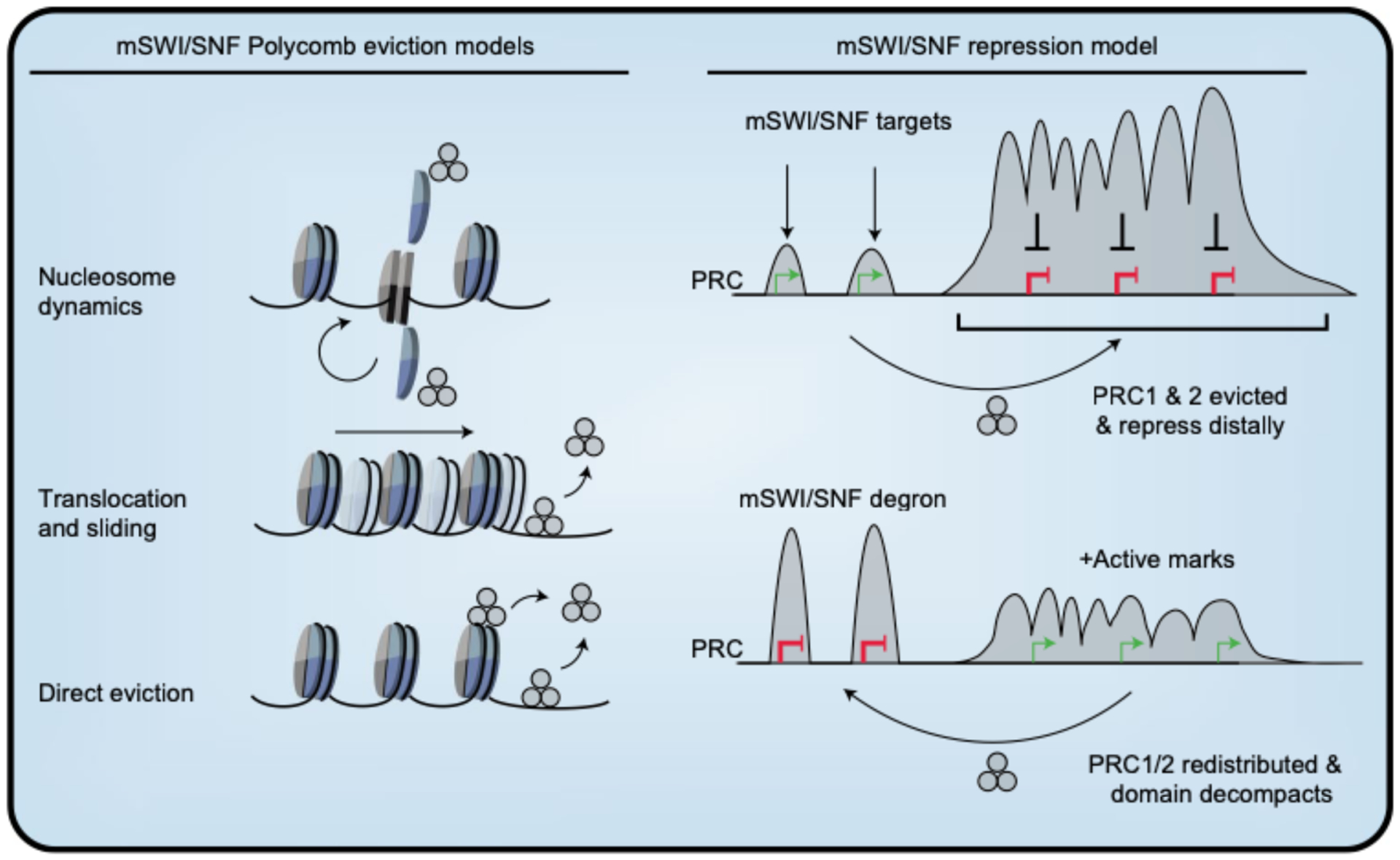
Left: mSWI/SNF Polycomb eviction models–nucleosome dynamics (top), DNA translocation and nucleosome sliding (middle) and direct ATP-dependent eviction (bottom). mSWI/SNF repression model: mSWI/SNF evicts Polycomb, through mechanisms described on the left, enabling accumulation at heavily occupied broad domains like Hox clusters, promoting distal repression. Within these repressed domains, BAF is weakly bound at gene promoters, supporting another level of repression. When mSWI/SNF is rapidly degraded, PRC complexes accumulate, causing redistribution away from heavily occupied sites. This leads to PRC redistribution, physical decompaction of the genome, accumulation of active marks and derepression.
Our observation that genes are derepressed within ~30 min of Brg1 degradation indicates that PRC1&2 began redistributing around the time that Brg1 dosage reached ~50%. This result was further supported by short timecourse ChIP experiments at the well-studied Bmi1 locus, showing coincident PRC1&2 depletion and transcriptional derepression. Considering the time needed for transcript accumulation, this reveals that PRC dynamics must be extremely rapid, consistent with chemically induced BAF recruitment studies, where PRC1 eviction was detected in less than 5 min, and PRC2 somewhat later11. Our studies support an understanding of BAF-Polycomb opposition that is far more dynamic than the textbook view of permanent epigenetic fixation.
The BAF–Polycomb axis is multifaceted. These combinatorially assembled complexes colocalize extensively across the genome11 and yet, where either is highly bound, the other appears to be excluded. It is tempting to conclude that PRC excludes BAF. However, we found that PRC1 deletion has a very minimal effect on BAF binding genome-wide, revealing that although BAF potently opposes Polycomb, Polycomb does not exclude BAF. Along these lines, our temporally resolved, dose-dependent Polycomb degradation approach has revealed that there is more to the repression equation than Polycomb redistribution. We found modest BAF binding to PRC1&2 repressed genes and that BAF is required to repress accessibility there. These genes also exhibited disproportionate PRC1 and PRC2 loss from broader domains, consistent with redistribution playing a role there as well. It is possible, then, that BAF cooperates with other proteins to facilitate repression, such as HDACs, BRD4S or the NuRD complex, which are known to interact with BAF54,55; however, future unbiased studies are needed to fully explore this possibility and define all contributors. In essence, these results highlight the power of chemical genetics approaches, which have enabled us to mechanistically dissect a causal role for BAF in Polycomb-mediated repression, independent of transcription, that is lacking in other studies17,18.
Polycomb-mediated repression has long been known to be dosage-sensitive, such that heterozygous genetic deletion alters the expression of Hox genes, giving rise to homeotic transformations4. Intriguingly, most PRC genes themselves do not appear to be as strongly dosage-sensitive in human disease56. An interesting conclusion from our work is that chromatin regulators that antagonize binding, like BAF and possibly others, titrate the effective dosage of all Polycomb complexes. This highlights the importance of gene dosage in regulating the BAF–PRC axis, where even modest overexpression of a single, minimal PRC1 complex is sufficient to inhibit transcriptional derepression caused by Brg1 depletion.
Previously, studies have shown that BAF evicts PRC1 complexes through a direct interaction with Brg112 and that ATPase activity is required for eviction11,12. Considering that there are extensive contacts between BAF subunits and the nucleosome core particle57, BAF probably evicts Polycomb by three non-mutually exclusive mechanisms: (1) nucleosome dynamics, (2) DNA translocation and sliding and (3) direct eviction by BAF (Fig. 7). While future structural studies are needed to precisely refine the eviction mechanism, there are structural and biochemical data supporting a DNA translocation model for another SWI/SNF subfamily remodeler, Mot1, which pries its substrate TATA-binding protein (TBP) off promoter DNA using ATP hydrolysis58. Our studies suggest that this type of antagonism is a determinant of Polycomb dynamics in ESCs, which have a hypermobile Polycomb fraction59. Thus, it is possible that this dynamic state is especially sensitive to modulation on chromatin, as BAF has been shown to facilitate Polycomb-mediated repression in pluripotency, during lineage commitment and in pediatric brain tumors, but not in more differentiated cells.
Although individual BAF subunits are frequently mutated in specific types of cancer, BAF is almost exclusively mutated in neuro-developmental disorders and not in other types of human disease60. The underlying mechanism for this specificity remains unknown. Our results suggest an explanation for BAF’s unique dual role in promoting Polycomb-mediated repression of genes involved in neurogenesis. A possible explanation for this specificity arises from the observation that the mRNA for certain posterior Hox genes such as Hoxd10-13 increase strongly upon BAF depletion. This would cause the embryo with a BAF mutation to enter neurogenesis with posterior hox genes expressed in developing neural tissue. Based on murine studies, this ‘hox confusion’ would lead to temporally disordered patterns of gene expression in neural progenitors and abnormal neural development61. Consistent with this, Brg1 knock-down in blastocysts was previously shown to result in aberrant Hox gene derepression62. Recently, Hobert and colleagues found that each class of neuron in Caenorhabditis elegans was found to be delineated by a unique hox code63, indicating that confusion of this code would have dramatic effects on neuronal sub-types and their circuitries. Although proper examination of this hypothesis would require complex genetic models, it provides a potential explanation for the surprising neural specificity of BAF subunit mutations.
Methods
Mouse ESC culture.
TC1(129) mouse ESCs were cultured in Knockout Dulbecco’s modified Eagles medium (Thermo Fisher, cat. no. 10829018) supplemented with 7.5% ES-qualified serum (Applied Stem Cell, cat. no. ASM-5017), 7.5% Knockout Serum replacement (Thermo Fisher, cat. no. 10828–028), 2 mM l-glutamine (Gibco, cat no. 35050061), 10 mM HEPES (Gibco, cat. no. 15630080), 100 U ml−1 penicillin/streptomycin (Gibco, cat. no. 151401222), 0.1 mM non-essential amino acids (Gibco, cat. no. 11140050), 0.1 mM β-mercaptoethanol (Gibco, cat. no. 21985023) and LIF. The ESCs were maintained on gamma-irradiated mouse embryonic fibroblast (MEF) feeders for passage or gelatin coated dishes for assays at 37°C with 5% CO2, seeded at ~3.6 × 104 cm−2 every 48 h, with daily media changes. 2i/LIF conditions were as previously described64. To degrade Brg1, osTIR1 was induced overnight with 1.0 μg ml−1 doxycycline, and medium containing 0.5 mM 3-indoleacetic acid (Sigma, cat. no. 12886) and doxycycline was added at the indicated time points. To degrade EED and Ring1b, dTAG13 was added for 8 h at 5 × 10−7 to 5 × 10−11 M. All lines tested negative for mycoplasma.
CRISPR-Cas9 genome editing.
Passage 11 TC1(129) mouse ESCs were thawed onto MEF-coated dishes and passaged once on gelatin-coated dishes before transfection. The cells (2 × 106) cells were nucleofected (Lonza #VVPH-1001, A-013 program) with 8 μg of homology-directed repair (HDR) template and 4 μg of PX459V2.0 (Addgene 62988) containing single-guide RNAs (below) or 2 μM RNP (IDT, cat. no. 1081058) for dTAG-Ring1b. HDR templates contained 0.5- or 1-kb homology arms flanking AID* or FKBPF36V degradation and epitope tags, which were inserted with a flexible (GGGGS)3 linker between endogenous protein and tag. Brg1 and EED were tagged at the C terminus, and Ring1b was tagged at the N terminus. Following transfection, 0.5 × 106 cells were seeded onto 6-cm dishes coated with DR4 MEFs and cultured for 24 h before puromycin selection (1.0 μg ml−1, 1.25 μg ml−1 or 1.5 μg ml−1) for 48 h or 1 × 103 to 1 × 105 cells without selection for ribonucleoprotein (RNP). Single colonies were manually picked, dissociated with trypsin and expanded on MEFs. Colonies containing homozygous insertions were confirmed by PCR and western blotting. Guide sequences are as follows: Brg1 sgRNA, TTGGCTGGGACGAGCGCCTC; EED sgRNA, TGATGCCAGCATTTGGCGAT; Ring1b sgRNA, TTTATTCCTAGAAATGTCTC.
Lentiviral preparation and delivery.
HEK293T cells were transfected with gene delivery constructs and packaging plasmids Md2G and psPAX2 using polyethylenimine (PEI). Two days post-transfection, the medium was collected, filtered and centrifuged at 50,000g for 2 h at 4°C. The concentrated viral pellet was resuspended in PBS and used directly for transduction or frozen at −80°C. Lentivirus encoding rtTA and TRE-osTIR1 was added to low-passage Brg1-AID* edited cells and selected with 1 μg mT−1 puromycin and 100 μg mL−1 hygromycin B.
RNA isolation and qRT-PCR.
Cells were dissociated with trypsin, quenched with medium, washed with PBS, and immediately resuspended in TRIsure (Bioline, cat. no. BIO-38033). Total RNA was isolated following the manufacturer’s guidelines, digested with DNasel (Thermo Fisher, cat. no. 18068015) and digestion reaction was cleaned up with acid-phenol:chloroform. cDNA was synthesized from 1 μg of RNA using a sensiFAST kit (Bioline, cat. no. BIO-65054). Primer sequences are provided in Supplementary Table 1 and were normalized to GAPDH using the ΔΔCt method.
RNA-seq and data analysis.
RNA sequencing libraries were made from 1 μg RNA (RNA integrity number > 9) using a SMARTer kit (Takara Bio, cat. no. 634874), which produces stranded libraries from ribosomal RNA-depleted total RNA. Libraries were amplified with 12 PCR cycles, quantified with Qubit, and the size distribution was determined by Bioanalyzer. Libraries were sequenced single-end with 76 cycles on an Illumina NextSeq or paired end with 150 cycles and the first three bases were trimmed with cutadapt65 before quantification. Transcript abundances were quantified by Kallisto66 using the ENSEMBL v96 transcriptomes. Transcript-level abundance estimates from Kallisto and gene-level count matrices were created using Tximport67. Differential expression analysis was conducted with DESeq2 version 1.22.2 (ref.68) using default parameters, after pre-filtering genes with low counts (rowSums > 10). Differential calls were made by requiring FDR-corrected P < 0.05. The log, fold changes were shrunk with ASHR69 for plotting volcano plots. Boxplot whiskers represent 1.5× interquartile range. Genome browser tracks were generated by aligning trimmed reads to the mm10 genome with HISAT2 (ref.70) with the appropriate RNA-strandedness option, and deepTools71 was used to generate read-depth-normalized bigwig files, scaled to the mouse genome size (RPGC), for viewing with IGV72. Gene ontology analysis was conducted with g:Profiler R client73. Gene overlap statistics were calculated using the GeneOverlap R package.
ChIP and ChIP-qPCR.
ChIP experiments were performed essentially as described in ref.13. Briefly, at the end of the time point 30 × 106 cells were fixed with 1% formaldehyde for 12 min, quenched, and sonicated with a Covaris focused ultrasonicator (~200–800 bp). Sonicated chromatin was split into separate tubes (~5 × 106 to 10 × 106 cell equivalent per immunoprecipitation) and incubated with 5 μg of primary antibody and 25 μl of Protein G Dynabeads (Life Technologies, 10009 D) overnight Beads were washed four times and DNA was isolated for qPCR or library preparation. The antibodies are listed in Supplementary Table 2. Primer sequences for qPCR experiments are provided in Supplementary Table 1.
ChIP-seq and data analysis.
ChIP-seq libraries were made using NEBnext Ultra II (NEB E7013) with ≤12 PCR cycles. Libraries were quantified with Qubit, and the size distribution was determined by Bioanalyzer. Libraries were sequenced single-end with 76 cycles on an Illumina NextSeq. Reads were aligned to the mm 10 genome with Bowtie 2 (ref.74) version 2.3.4.1 with the very sensitive option. Alignments were removed with the following criteria: quality score < 20, PCR duplicates, secondary alignments and supplemental alignments. Peaks were called with MACS2 using default parameters and requiring that q < 0.01. Peaks from control and treated datasets within ±1 kb were merged, filtered against the mouse blacklist75, and the numbers of reads overlapping these peak sets were compared for differential peak calling. Differential peak calls were determined using DESeq2 after pre-filtering peaks with low counts (rowSums > 400 for Ring1b and Suz12 with four biological replicates, and rowSums > 100 for histone marks with two biological replicates). Differential calls were made by requiring FDR-corrected P < 0.1 and data were plotted with ggplot2 (ref.76). For browser snapshots, replicate BAM files were merged and read-depth-normalized genome coverage files (bigwig) were generated with deepTools71, scaled to the mouse genome size (RPGC). For subtractions, normalized bigwig files were subtracted using deepTools71 (8 h–0 h) for visualization by heatmap, metagene plot or by browser (IGV).
RNA polymerase inhibition.
Transcription initiation was blocked by treating cells with 10 μM triptolide (Sellec, cat. no. S3604). To validate triptolide treatment, quantitative RT-PCR was performed by comparing derepressed genes in triptolide, triptolide and IAA, to IAA only cells, normalized to U6 snRNA using the ΔΔCt method.
ORCA imaging and data analysis.
The primary probes tiling the HoxA and HoxD DNA regions at 5-kb resolution (Supplementary Information) were designed as previously described32. Probes were amplified from the oligopool (CustomArray) and amplified according to the protocol described in refs.28,32.
In preparation for imaging, mESCs were plated on 40-mm glass coverslips (Bioptechs) coated with 0.1–0.2% gelatin, and fixed on the following day in 4% PFA in 1× PBS for 10 min. The hybridization and imaging were performed as previously described32. Briefly, for primary probe hybridization, cells were permeabilized for 10 min with 0.5% Triton-X in 1× PBS, then the DNA was denatured by treatment with 0.1 M HC1 for 5 min. Primary probes (2 μg) in hybridization solution were then added directly onto cells, placed on a heat block for at 90°C for 3 min and incubated overnight at 42°C in a humidified chamber. Before imaging, the samples were post-fixed for 1 h in 8% PFA + 2% glutaraldehyde in 1× PBS. The samples were then washed in 2× SSC and either imaged directly or stored for up to a week in 4°C before imaging. For imaging, samples were mounted into a Bioptechs flow chamber, and secondary probe hybridization, step-by-step imaging of individual barcodes and image processing were performed as in ref32.
Image analysis was performed as described in ref.32. For all analysis in Fig. 3. we excluded all barcodes that had a low labeling efficiency in either control or AID-treated condition (labeled in less than 10% of cells). This resulted in 52 barcodes for HoxA and 28 barcodes for HoxD. For comparison with Hi-C, mESC data from ref.33 were downloaded from Jukebox77,78 and matched to ORCA barcode coordinates by finding the closest genomic bins in Hi-C and removing bins corresponding to excluded barcodes. ORCA data were processed to calculate contact frequency across all cells (Extended Data Fig. 4a,b), where contact frequency was computed by calculating the fraction of cells where the probes were within a 200 nm distance. We use ‘cell’ to refer to all detected spots, with ~2 spots per cell corresponding to each allele. For calculation of median probe distance (to generate Fig. 4e,f) we calculated the median distance across all cells in each condition. For both HoxA and HoxD imaging, we obtained more cells in the control condition (1,238 for HoxA, 2,419 for HoxD) than in the AID-treated condition (587 for HoxA. 1,183 for HoxD). To control for sample size, we randomly split the cells in the control dataset into two, calculated the median of pairwise probe distances for each subset and computed the difference in pairwise distances between the two halves and between each control subset and Brg1 depleted cells (Extended Data Fig. 3d,e).
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this Article.
Extended Data
Extended Data Fig. 1|. Extended characterization of Brg1-AID degron, related to Fig. 1.
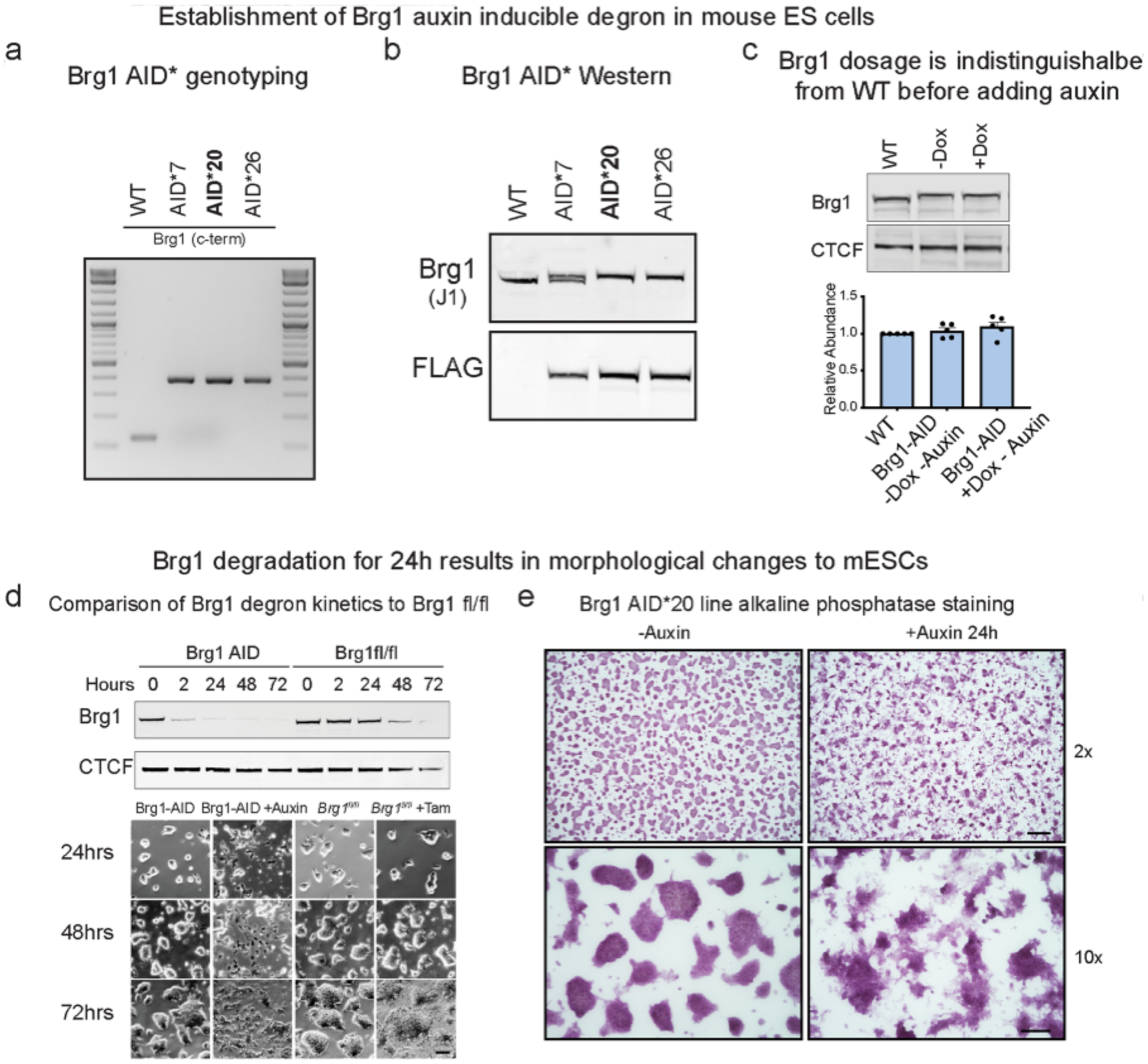
(a, Genotyping PCR for three homozygous knock in clones (AID*20 was used for most experiments in the manuscript). b, Western blot of whole-cell extracts from knock-in lines showing 8.8kDa shift in migration from c-terminal tag (G4S)3-AID*-G4S-3xFLAG. Line 20 and 26 have homozygous insertions in frame that were also confirmed by Sanger sequencing. c, Representative western blot and quantitation of Brg1 abundance in parent line compared to Brg1-AID* ± osTIR1 induction (-auxin). Error bars represent mean ± standard deviation from five biological replicates. d, Comparison of Brg1 depletion kinetics between AID and Cre-lox genetic deletion by western blot and changes to colony morphology that occur at the 24-hour time-point. Cells were only grown on the same dish for 72 h to illustrate timing for Brg1fl/fl colony morphology changes. Experiment was repeated at least 3 times with similar results. Scale bar = 50 μm. e, Alkaline phosphatase staining of Brg1 AID* 20 line on gelatin coated dishes, showing that changes to colony morphology occur ~24 h. Top scale bar = 200 μm, bottom scale bar = 50μm. Experiment was repeated at least 3 times with similar results.
Extended Data Fig. 2 |. Extended summary of RNA-seq results, related to Fig. 1.
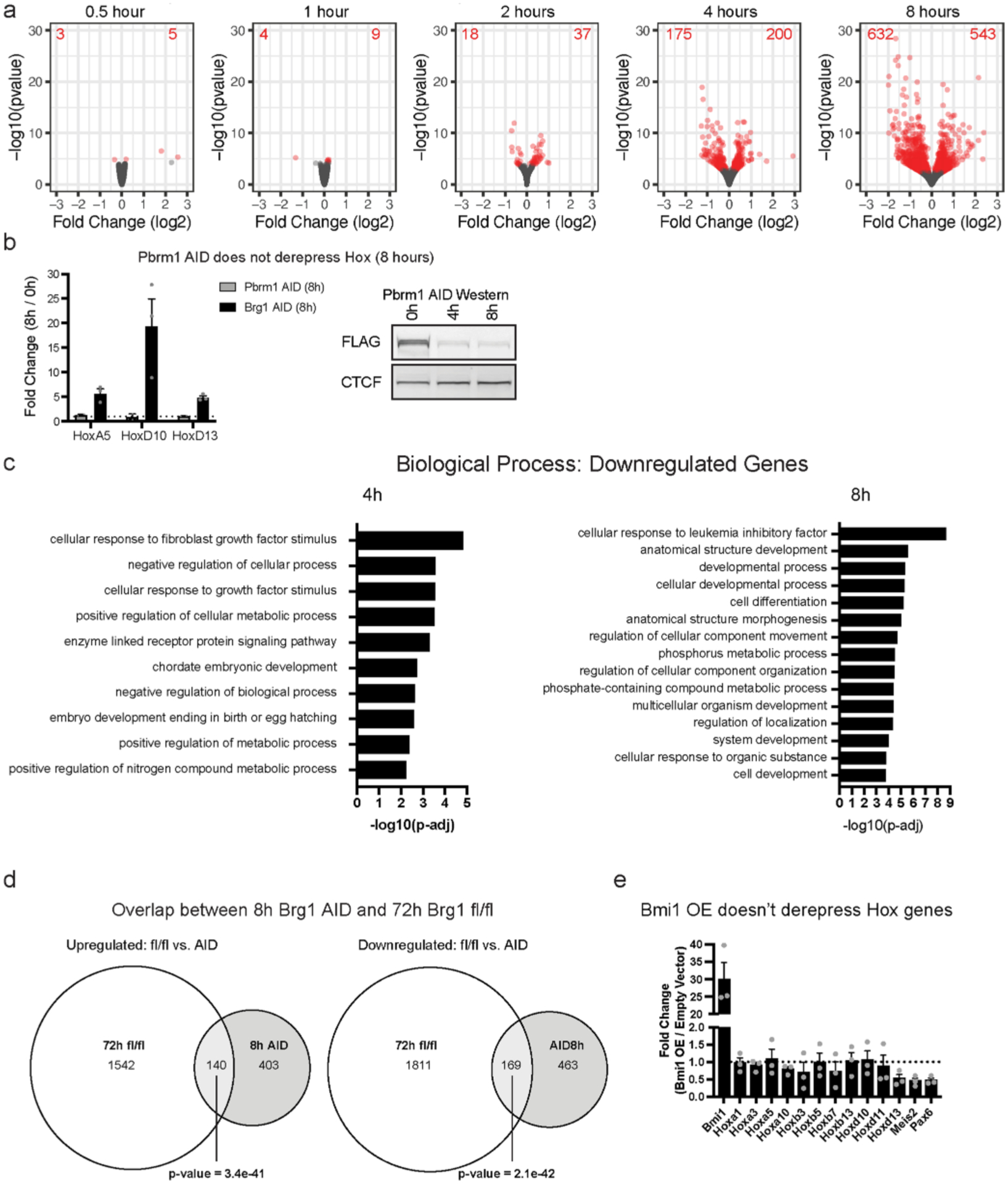
a, Volcano plots depicting gene expression changes at 0.5, 1, 2, 4, and 8 hours of Brg1 degradation. Differentially expressed genes are colored red (FDR-corrected P < 0.05) from four biological replicates for each time-point. b, Pbrm1 degradation with auxin, does not derepress Hox genes, revealing that pBAF is not required for their repression and controls for auxin, Tir1 expression, and proteasomal degradation in the Brg1 degron. Error bars are mean ± standard error for n = 3 replicates. c, Gene set enrichment analysis for differentially downregulated genes at 4 and 8-hour timepoints. d, Overlap of differentially expressed genes in 72 h Brg1 fl/fl from12 and 8 h Brg1 AID calculated by Fisher’s exact test (p-value = 1.1e-46). e, Strong lentiviral Bmi1 overexpression doesn’t derepress Hox genes or other genes derepressed by Brg1 degron. Error bars depict standard error of the mean from three biological replicates.
Extended Data Fig. 3 |. Brg1 degradation results in quick redistribution of PRC1&2 but doesn’t affect net dosage, related to Fig. 2.
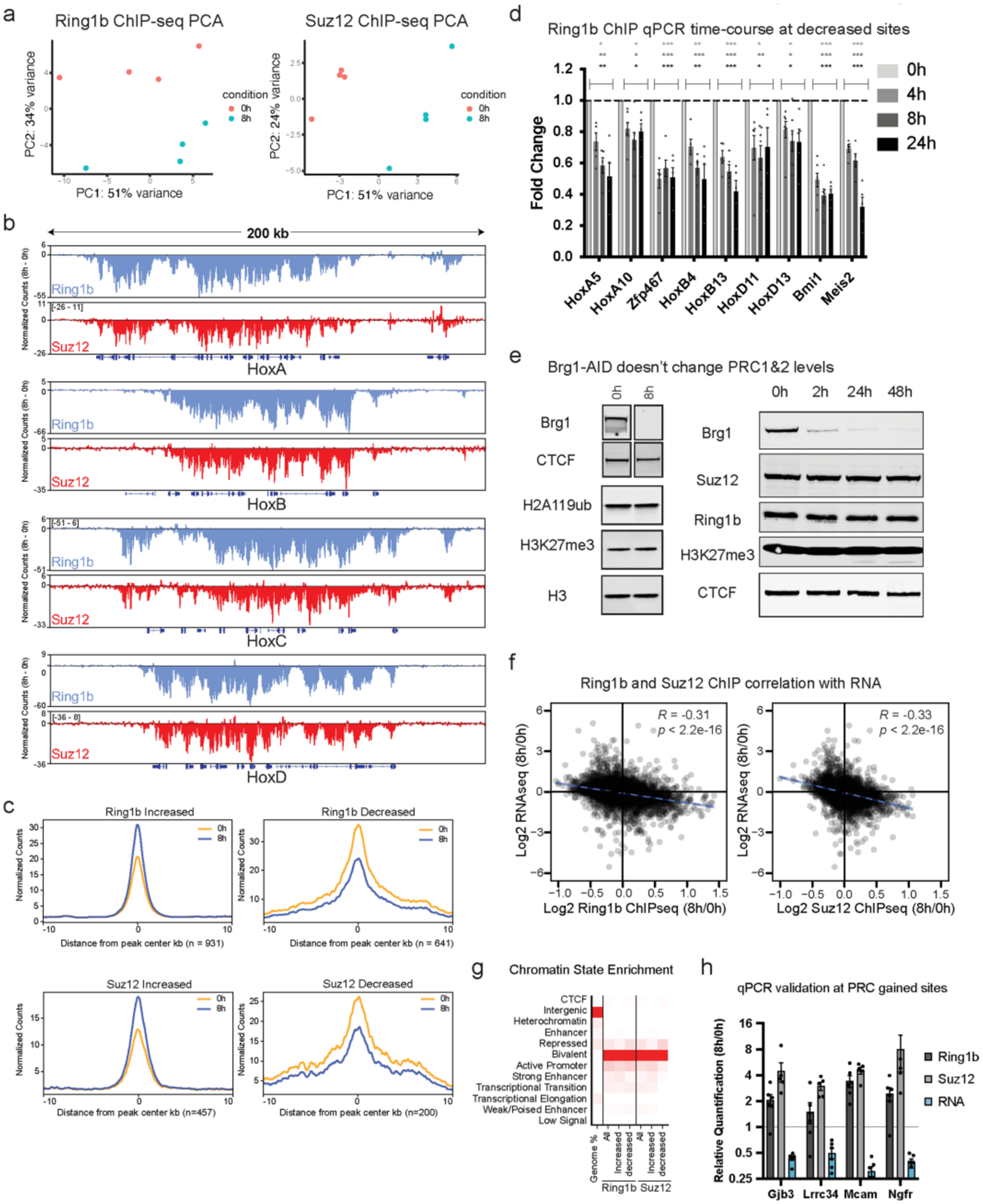
a, Principal component analysis of Ring1b and Suz12 ChIP-seq (n = 4 replicates) b, Browser snapshot of the difference in normalized counts (8 h – 0 h) for Ring1b and Suz12 at Hox A, B, C, and D depicting net loss across all four clusters. c, Average metagene plots of normalized counts (to read-depth) at sites that were significantly increased or decreased by DESeq2 analysis (FDR-corrected P < 0.1) and normalized to peak intensity, providing additional confirmation that normalization is correct. d, ChIP qPCR timecourse at 4, 8, and 24 h Brg1 degradation for Ring1b at sites that were significantly decreased by ChIP-seq. Error bars depict standard error of the mean from four biological replicates. (* = p < 0.05, ** = p < 0.005, *** = p < 0.0005; two-sided t test with Holm-Sidak multiple comparison correction). e, (left) Representative western blot showing Brg1 degradation efficiency and that H2AK119ub and H3K27me3 marks don’t change at 8 h time-point (ChIP-seq time-point for the complexes that deposit these marks), (right) Representative western showing Brg1 degradation efficiency over a longer timecourse and that Suz12, Ring1b, and H3K27me3 marks don’t noticeably change even at 48 h. Experiment was repeated at least 2 times with similar results (f) Correlation of all Ring1b and Suz12 peaks that are ± 2kb from gene transcription start sites and expression changes at 8 h Brg1 degradation. Correlation and p values were obtained from Pearson’s product moment correlation. g, chromatin state enrichment for all, increased, and decreased peak changes for Ring1b and Suz12. h, ChIP qPCR and qRT-PCR validation of genes that gain Ring1b and Suz12 and have reduced gene expression following 8 h Brg1 degradation. Error bars depict standard error of the mean from five biological replicates.
Extended Data Fig. 4 |. Comparison of ORCA to Hi-C, related to Fig. 3.
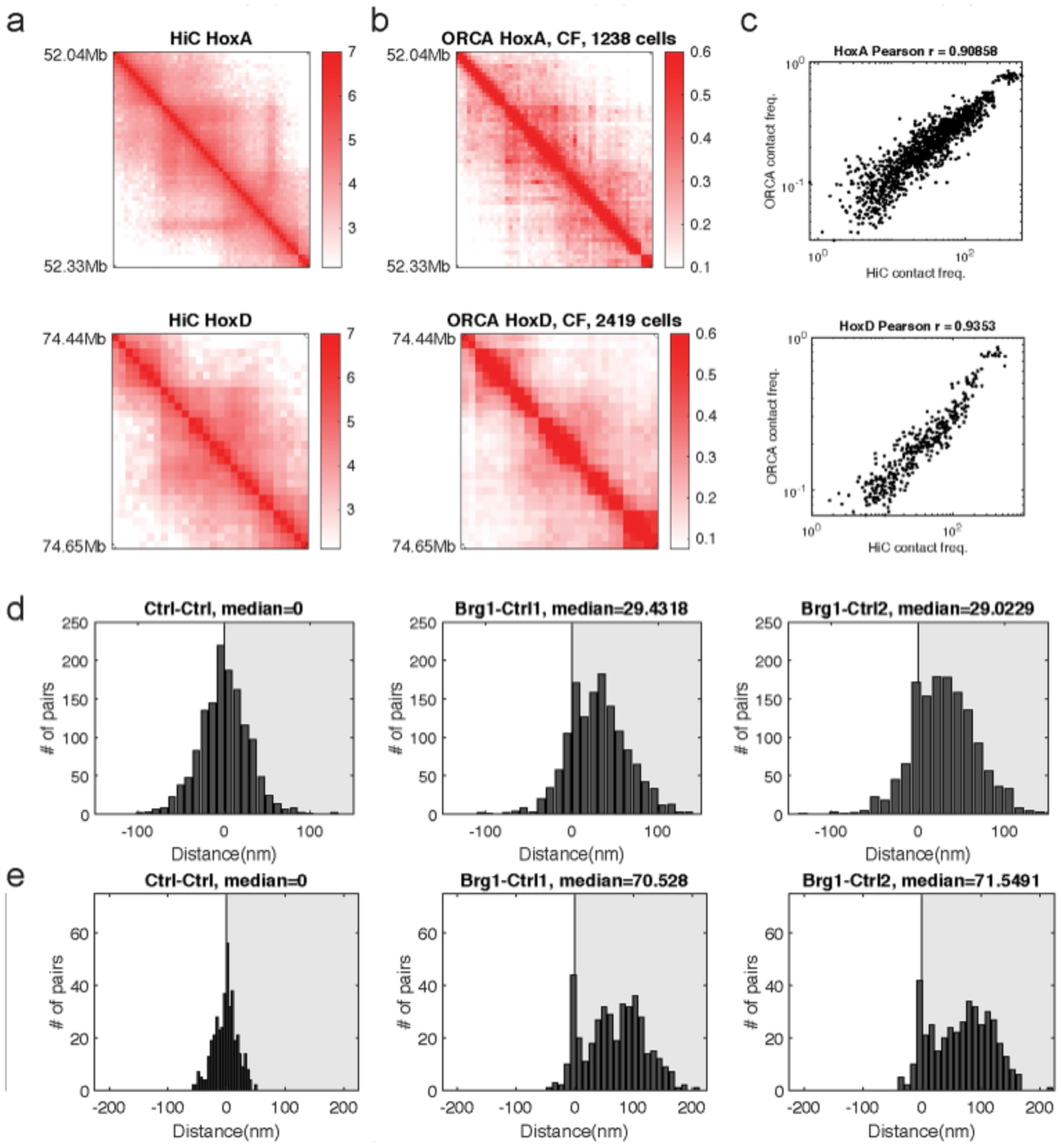
a, Average contact frequency as measured by high resolution Hi-C from serum grown cells33. b, Average contact frequency as measured by ORCA (200-nm threshold). c, Pearson’s correlation and Pearson’s r were computed using all unique pairwise combinations of X,Y barcodes measured. d, Histograms depicting the difference in median distance for barcode pair combinations at HoxA. e, Histograms depicting the difference in median distance for barcode pair combinations at HoxD.
Extended Data Fig. 5 |. Extended analysis related to Fig. 4.
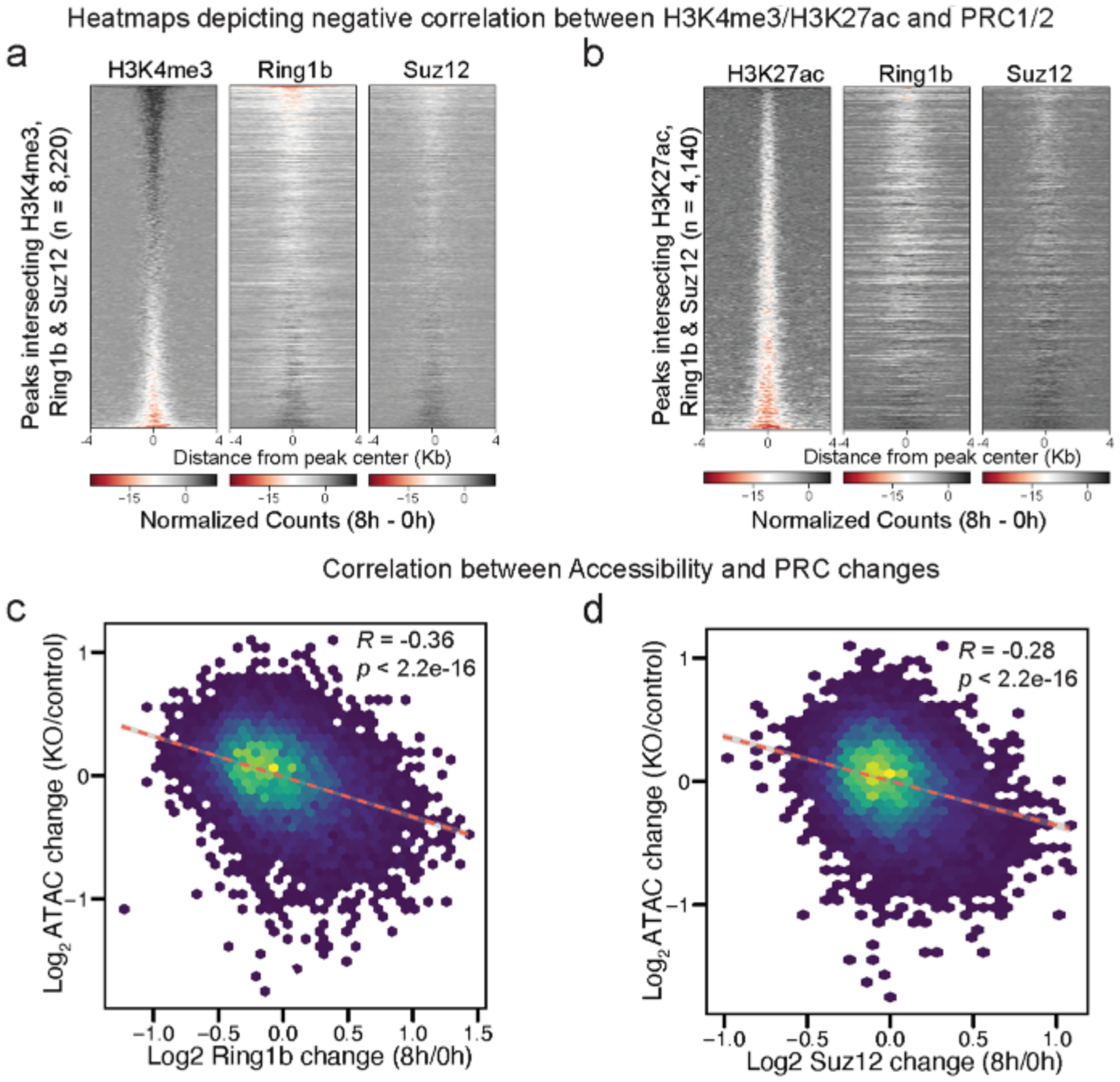
a, Heatmap depicting the negative correlation between H3K4me3 and Ring1b and Suz12 as the difference in normalized counts (8 h – 0 h) at peaks (8,220) that intersect H3K4me3, Ring1b, and Suz12 at merged peak center, sorted by decreasing H3K4me3 signal. b, Heatmap depicting the negative correlation between H3K27ac and PRC1&2 as the difference in normalized counts (8 h – 0 h) at peaks (4,140) that intersect H3K27ac, Ring1b, and Suz12 at merged peak center, sorted by decreasing H3K27ac signal. c,d, Scatterplots showing negative correlation between ATAC-seq changes (KO/control)44 and Ring1b (8 h/0 h) or Suz12 (8 h/0 h).
Extended Data Fig. 6 |. Transcription is not required for polycomb redistribution.
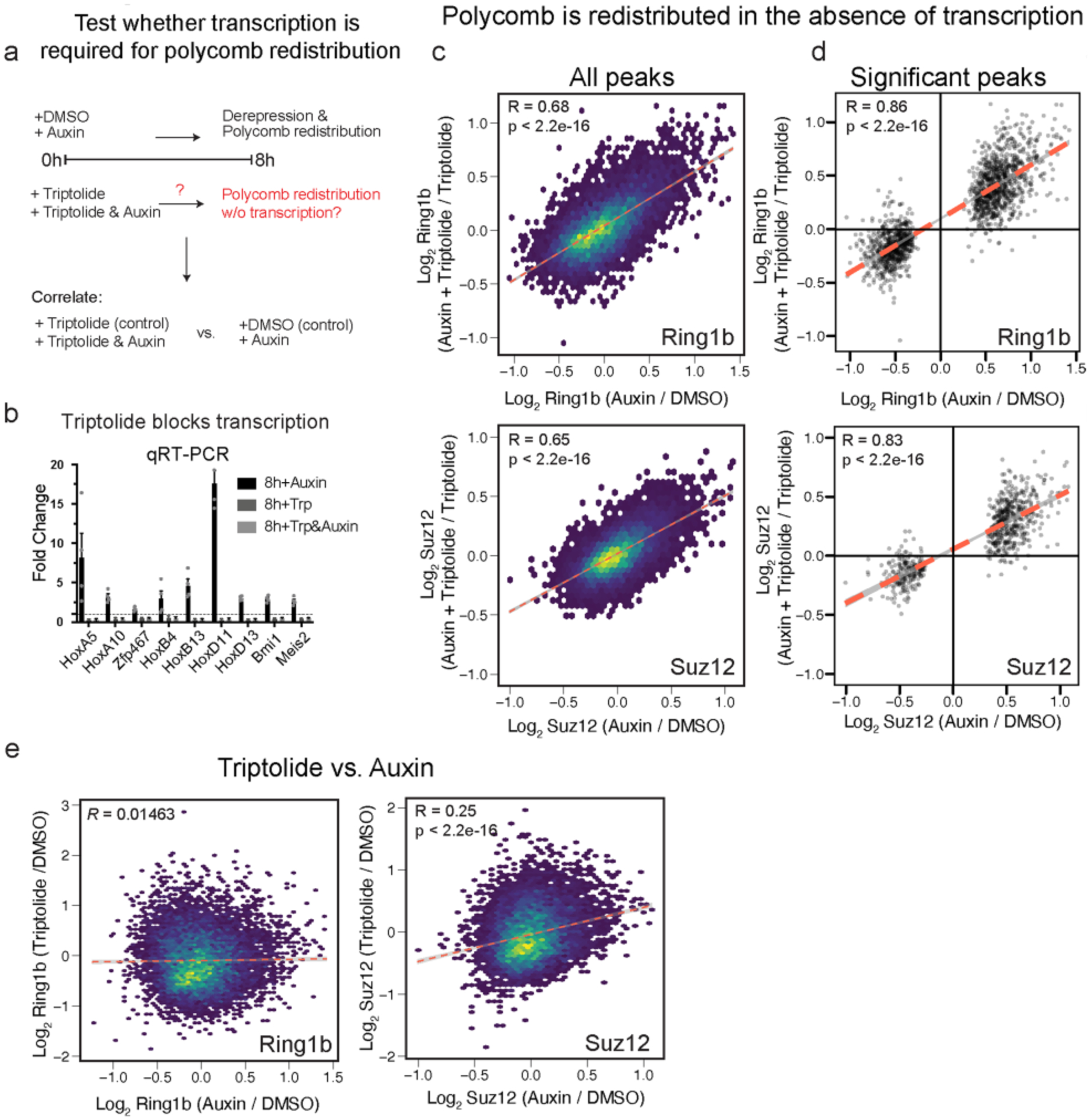
a, Experimental design to test whether polycomb redistribution requires transcription. b, qRT-PCR depicting fold change by ΔΔCt method with indicated treatments from n = 3 biological replicates. c,d, Scatterplot showing that Ring1b and Suz12 changes following Brg1 degradation are highly correlated despite global inhibition of transcription for all peaks c, and peaks that were significantly changed by Brg1 degradation alone (d) (FDR < 0.1). e, Scatterplot of changes to Ring1b and Suz12 upon treatment with auxin (auxin/control) or triptolide (triptolide/control) showing distinct effects from the two treatments. Correlation and p-values were obtained from Pearson’s product moment correlation for c-e, from n = 4 biological replicates at the 8 h timepoint.
Extended Data Fig. 7 |. PRC1 does not restrict BAF binding despite extensive overlap.
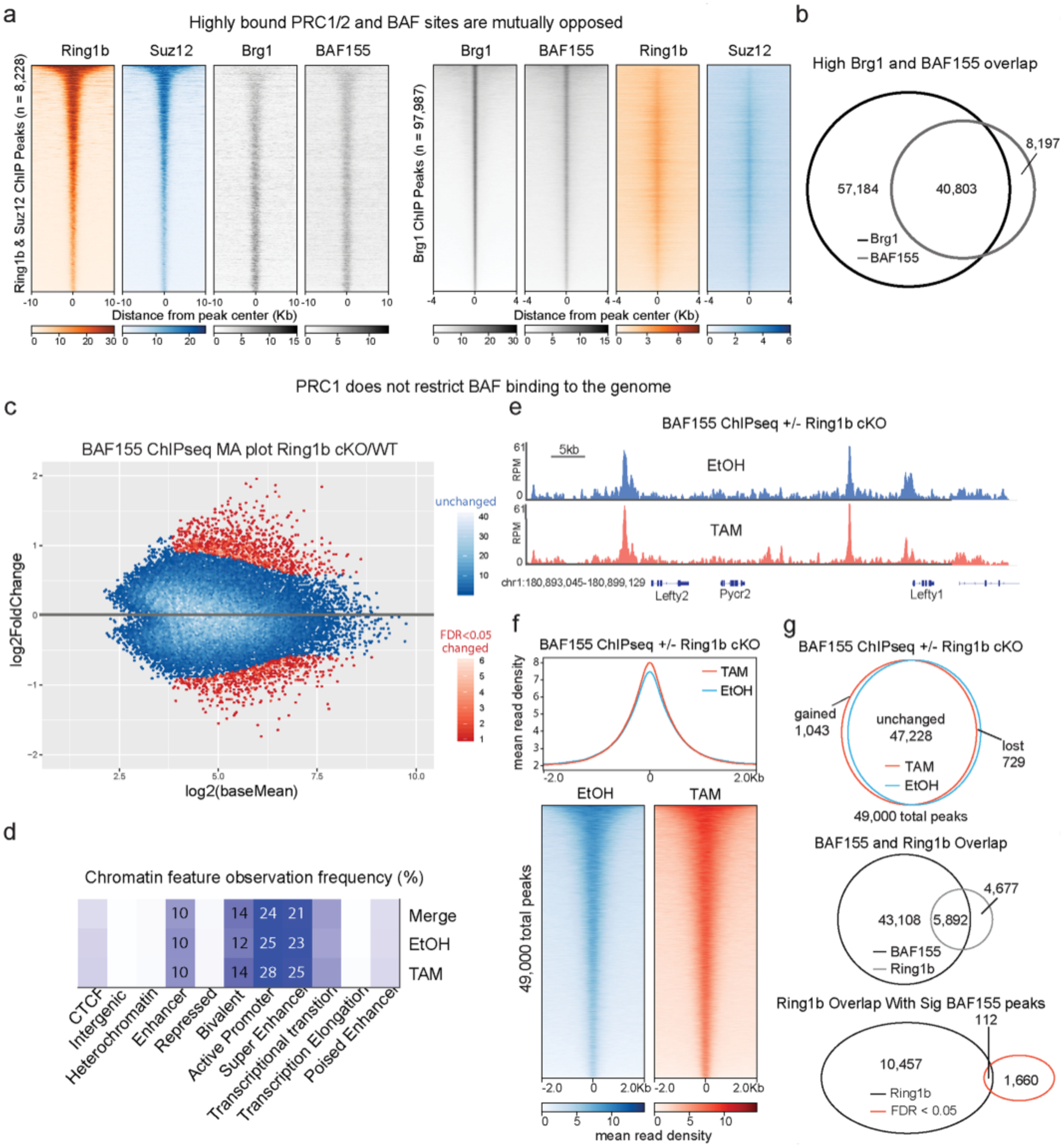
a, Heatmaps depicting Ring1b, Suz12, BAF155, and Brg1 ChIPseq signal from79 at peaks (8,228) that overlap Ring1b and Suz12, sorted by decreasing Ring1b signal (left) or Brg1 peaks (97,987) sorted by decreasing Brg1 signal (right). b, High overlap between Brg1 and BAF155 despite different number of peaks called due to antibody quality and read depth. c, BAF155 ChIP-seq in Ring1bfl/fl × Ring1a−/− × ActinCreER ESCs treated with Tamoxifen or EtOH for 72 h. MA plot (TAM/EtOH) highlights large number of unchanged peaks (>95% in blue) (n = 2). d, Analysis of the chromatin features that characterize BAF155 binding sites reveal that Ring1a/b double KO does not alter BAF complex binding, as the complex remains mainly bound to enhancers and bivalent promoters in both control and mutant ESCs. e, Representative genome tracks showing similar BAF155 levels across the Lefty1/2 locus in Tamoxifen and EtOH treated cells. f, Metagene plot and heatmaps showing that BAF155 ChIPseq signal is minimally affected by Ring1b deletion (g) Characterization of BAF155 peaks in EtOH and Tamoxifen conditions. Of the 49,000 detected BAF155 peaks in this dataset, less than 5% were significantly changed upon Ring1a/b double KO in ESCs and the small number of gained and decreased sites was similar. Despite 56% of Ring1b peaks overlapping with a BAF peak, only 1% of differentially bound peaks are within a PRC1 domain (bottom).
Extended Data Fig. 8 |. Characterization of EED/Ring1b degron and extended analysis related to Fig. 5.
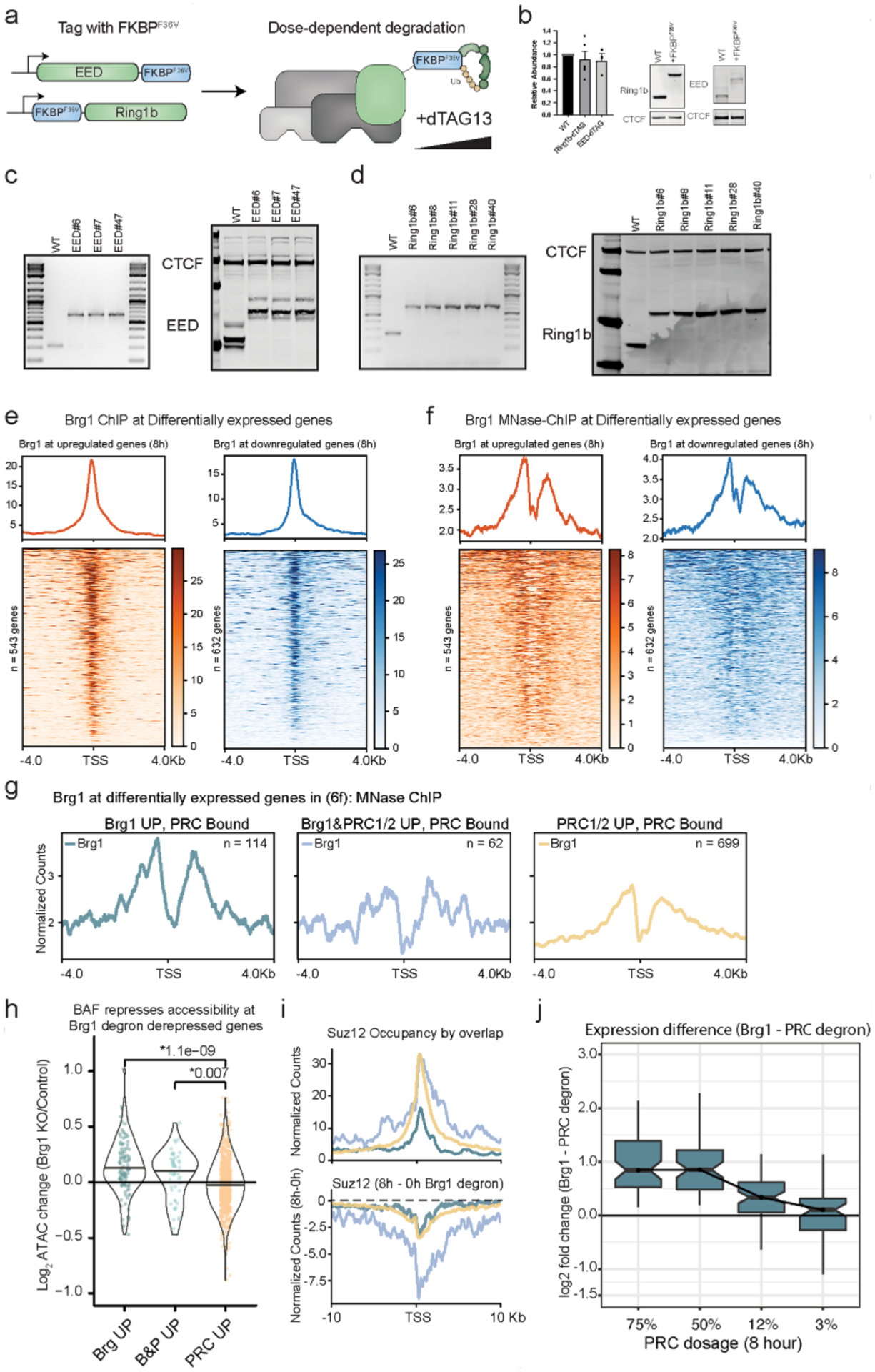
a, Schematic depicting EED and Ring1b dTAG targeted protein degradation strategy in mESCs, where EED is tagged at the C-terminus and Ring1b is tagged at the N-terminus with FKBPF36V to enable targeted degradation with dTAG13 PROTAC. b, Representative western blot and quantitation of EED and Ring1b abundance in parent line compared to lines tagged with FKBPF36V. Error bars depict mean ± standard error of the mean from three tagged EED lines and four tagged Ring1b lines. c, Genotyping PCR and western from whole cell extract showing 14.6kDa shift in migration from C-terminus (G4S)3-FKBPF36V-G4S-V5 tag on three different EED clones used for experiments. d, Genotyping PCR and western from whole cell extract with 14.2kDa shift in migration from N-terminus FKBPF36V-G4S-HA-(G4S)3. Experiment was repeated at least 2 times with similar results (e) Brg1 ChIPseq showing localization to the TSS of differentially expressed genes, ChIP from79. f, Brg1 MNase ChIPseq showing localization to the TSS of differentially expressed genes, ChIP from80. g, Brg1 MNase ChIPseq showing higher occupancy at PRC bound genes repressed by Brg1 (independent of polycomb). Compare to Fig. 5f. h, Brg1 represses accessibility at PRC bound genes derepressed by Brg1 degron and Brg1/PRC degron. Accessibility change (Brg1 KO/control) by overlap (Fig. 6f) for peaks ± 2kb from gene promoter, ATAC data from44 with two-sided t-test p-value above. (i,top) Higher and broader polycomb domains over genes derepressed by both Brg1 and PRC degron than genes derepressed by Brg1 degron alone, with Suz12 occupancy shown here, (i, bottom) Broad domains over genes derepressed by both Brg1 and PRC degron show substantially more redistribution in the Brg1 degron. Compare to Ring1b signal in Fig. 6. (j) Expression difference (Brg1 - PRC Log2 Fold Change) for genes derepressed by Brg1 and PRC degron, over dosage titration (Most dosage sensitive genes are shown in Fig. 6g), from n = 4 biological replicates. Boxplot bounds depict quartile 1, median, and quartile 3 with whiskers at 1.5× interquartile range.
Extended Data Fig. 9 |. Histone modifications deposited by PRC1 and PRC2 are higher in 2i growth conditions and results in weaker derepression, related to Figs. 2 and 6.
a, Western blot and relative quantification of H2AK119ub and H3K27me3 normalized to H3 in cells grown in Serum/LIF or 2i growth conditions from n = 3 biological replicates. b, Hox gene derepression measured by qRT-PCR for cells grown in serum, 2i, and the ratio of serum/2i from n = 2 biological replicates.
Supplementary Material
Acknowledgements
We thank members of the Crabtree laboratory for insightful comments and discussions over the course of the study. We are grateful to A. Krokhotin, S. Ramachandran (CU Anschutz) and V. Ramani (UCSF) for providing critical comments on the manuscript and for helpful discussions. This study was supported by NIH grants R01CA163915 (G.R.C.), R37NS046789 (G.R.C.) and DP2GM132935 (A.N.B.); the Howard Hughes Medical Institute (G.R.C.); a Swiss National Science Foundation (SNSF) postdoctoral fellowship (S.M.G.B.); the Walter V. and Idun Berry Postdoctoral fellowship program (C.M.W. and A.H.); and the Sir James Black postdoctoral fellowship (C.M.W. and S.M.G.B.).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41594-021-00604-7.
Competing interests
G.R.C. is founder and stockholder of Foghorn Therapeutics. The other authors declare no competing interests.
Extended data is available for this paper at https://doi.org/10.1038/s41594-021-00604-7.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41594-021-00604-7.
Data availability
All sequencing data have been deposited in the GEO (accession no. GSE145016). Source data are provided with this paper.
References
- 1.Flavahan WA, Gaskell E & Bernstein BE Epigenetic plasticity and the hallmarks of cancer. Science 357, eaal2380 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schuettengruber B, Bourbon HM, Di Croce L & Cavalli G Genome regulation by Polycomb and Trithorax; 70 years and counting. Cell 171, 34–57 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Kingston RE & Tamkun JW Transcriptional regulation by trithorax group proteins. Cold Spring Harb. Perspect. Biol 6, a019349 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kennison JA & Tamkun JW Dosage-dependent modifiers of Polycomb and antennapedia mutations in Drosophila. Proc. Natl Acad. Sci. USA 85, 8136–8140 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tamkun JW et al. brahma: a regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 68, 561–572 (1992). [DOI] [PubMed] [Google Scholar]
- 6.Yu BD, Hess JL, Horning SE, Brown GA & Korsmeyer SJ Altered Hox expression and segmental identity in Mll-mutant mice. Nature 378, 505–508 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Kadoch C & Crabtree GR Reversible disruption of mSWI/SNF (BAF) complexes by the SS18–SSX oncogenic fusion in synovial sarcoma. Cell 153, 71–85 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shain AH & Pollack JR The spectrum of SWI/SNF mutations, ubiquitous in human cancers. PLoS ONE 8, e55119 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ronan JL, Wu W & Crabtree GR From neural development to cognition: unexpected roles for chromatin. Nat. Rev. Genet 14. 347–359 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clapier CR, Iwasa J, Cairns BR & Peterson CL Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell Biol 18, 407–422 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kadoch C et al. Dynamics of BAF–Polycomb complex opposition on heterochromatin in normal and oncogenic states. Nat. Genet 49, 213–222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanton BZ et al. Smarca4 ATPase mutations disrupt direct eviction of PRC1 from chromatin. Nat. Genet 49, 282–288 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Braun SMG et al. Rapid and reversible epigenome editing by endogenous chromatin regulators. Nat. Commun 8, 560 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Valencia AM & Kadoch C Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat. Cell Biol 21, 152–161 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McBride MJ et al. The SS18–SSX fusion oncoprotein hijacks BAF complex targeting and function to drive synovial sarcoma. Cancer Cell 33, 1128–1141 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johann PD et al. Atypical teratoid/rhabdoid tumors are comprised of three epigenetic subgroups with distinct enhancer landscapes. Cancer Cell 29, 379–393 (2016). [DOI] [PubMed] [Google Scholar]
- 17.Alexander JM et al. Brg1 modulates enhancer activation in mesoderm lineage commitment. Development 142, 1418–1430 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho L et al. esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating Polycomb function. Nat. Cell Biol 13, 903–913 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakayama RT et al. SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat. Genet 49, 1613–1623 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masliah-Planchon J, Bièche I, Guinebretière J-M, Bourdeaut F & Delattre O SWI/SNF chromatin remodeling and human malignancies. Annu. Rev. Pathol 10, 145–171 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T & Kanemaki M An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods 6, 917–922 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Morawska M & Ulrich HD An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast 30, 341–351 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nora EP et al. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joshi O et al. Dynamic reorganization of extremely long range promoter promoter interactions between two states of pluripotency. Cell Stem Cell 17, 748–757 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Dykhuizen EC, Carmody LC, Tolliday N, Crabtree GR & Palmer MA Screening for inhibitors of an essential chromatin remodeler in mouse embryonic stem cells by monitoring transcriptional regulation. J. Biomol. Screen 17, 1221–1230 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Boyer LA et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Ernst J & Kellis M ChromHMM: automating chromatin-state discovery and characterization. Nat. Methods 9, 215–216 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mateo LJ, Sinnott-Armstrong N & Boettiger AN Tracing DNA paths and RNA profiles in cultured cells and tissues with ORCA. Nat. Protoc 16, 1647–1713 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eskeland R et al. Ring IB compacts chromatin structure and represses gene expression independent of histone ubiquitination. Mol. Cell 38, 452–464 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Francis NJ, Kingston RE & Woodcock CL Chromatin compaction by a Polycomb group protein complex. Science 306, 1574–1577 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Bintu B et al. Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mateo LJ et al. Visualizing DNA folding and RNA in embryos at single-cell resolution. Nature 568, 49–54 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bonev B et al. Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Piunti A & Shilatifard A Epigenetic balance of gene expression by Polycomb and COMPASS families. Science 352, aad9780 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Dallas PB et al. p300/CREB binding protein-related protein p270 is a component of mammalian SWI/SNF complexes. Mol. Cell Biol 18, 3596–3603 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tie F et al. CBP-mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136, 3131–3141 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hainer SJ et al. Suppression of pervasive noncoding transcription in embryonic stem cells by esBAF. Genes Dev. 29, 362–378 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang X et al. Molecular analysis of PRC2 recruitment to DNA in chromatin and its inhibition by RNA. Nat. Struct. Mol. Biol 24, 1028–1038 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bensaude O Inhibiting eukaryotic transcription: which compound to choose? How to evaluate its activity? Transcription 2, 103–108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Riising EM et al. Gene silencing triggers Polycomb repressive complex 2 recruitment to CpG islands genome wide. Mol. Cell 55, 347–360 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Francis NJ, Saurin AJ, Shao Z & Kingston RE Reconstitution of a functional core Polycomb repressive complex. Mol. Cell 8. 545–556 (2001). [DOI] [PubMed] [Google Scholar]
- 42.Shao Z et al. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 98, 37–46 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Endoh M et al. Polycomb group proteins Ring1A/B are functionally linked to the core transcriptional regulatory circuitry to maintain ES cell identity. Development 135, 1513–1524 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Miller EL et al. TOP2 synergizes with BAF chromatin remodeling for both resolution and formation of facultative heterochromatin. Nat. Struct. Mol. Biol 24. 344–352 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.King HW, Fursova NA, Blackledge NP & Klose RJ Polycomb repressive complex 1 shapes the nucleosome landscape but not accessibility at target genes. Genome Res. 28, 1494–1507 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hodges HC et al. Dominant-negative SMARCA4 mutants alter the accessibility landscape of tissue-unrestricted enhancers. Nat. Struct. Mol. Biol 25, 61–72 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nabet B et al. The dTAG system for immediate and target-specific protein degradation. Nat. Chem. Biol 14, 431–441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blackledge NP et al. PRC1 catalytic activity is central to Polycomb system function. Mol. Cell 77, 857–874 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tamburri S et al. Histone H2AK119 mono-ubiquitination is essential for Polycomb-mediated transcriptional repression. Mol. Cell 77, 840–856 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fursova NA et al. Synergy between variant PRC1 complexes defines Polycomb mediated gene repression. Mol. Cell 74, 1020–1036 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar B & Elsasser SJ Quantitative multiplexed ChIP reveals global alterations that shape promoter bivalency in ground state embryonic stem cells. Cell Rep. 28, 3274–3284 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Mierlo G et al. Integrative proteomic profiling reveals PRC2-dependent epigenetic crosstalk maintains ground-state pluripotency. Cell Stem Cell 24, 123–137 (2019). [DOI] [PubMed] [Google Scholar]
- 53.Bracken AP, Brien GL & Verrijzer CP Dangerous liaisons: interplay between SWI/SNF, NuRD and Polycomb in chromatin regulation and cancer. Genes Dev. 33, 936–959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yildirim O et al. Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147, 1498–1510 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Conrad RJ et al. The short isoform of BRD4 promotes HIV-1 latency by engaging repressive SWI/SNF chromatin-remodeling complexes. Mol. Cell 67, 1001–1012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karczewski KJ et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581, 434–443 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.He S et al. Structure of nucleosome-bound human BAF complex. Science 367, 875–881 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Wollmann P et al. Structure and mechanism of the Swi2/Snf2 remodeller Mot1 in complex with its substrate TBP. Nature 475, 403–407 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fonseca JP et al. In vivo Polycomb kinetics and mitotic chromatin binding distinguish stem cells from differentiated cells. Genes Dev. 26, 857–871 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kadoch C & Crabtree GR Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci. Adv 1, el500447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Philippidou P & Dasen JS Hox genes: choreographers in neural development, architects of circuit organization. Neuron 80, 12–34 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kidder BL, Palmer S & Knott JG SWI/SNF-Brg1 regulates self-renewal and occupies core pluripotency-related genes in embryonic stem cells. Stem Cells 27, 317–328 (2009). [DOI] [PubMed] [Google Scholar]
- 63.Reilly MB, Cros C, Varol E, Yemini E & Hobert O Unique homeobox codes delineate all the neuron classes of C. elegans. Nature 584, 595–601 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ying QL et al. The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martin M CUTADAPT removes adapter sequences from high-throughput sequencing reads. EMBnet J. 10.14806/ej.17.1.200 (2011). [DOI] [Google Scholar]
- 66.Bray NL, Pimentel H, Melsted P & Pachter L Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol 34, 525–527 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Soneson C, Love MI & Robinson MD Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4, 1521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Love MI, Huber W & Anders S Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Stephens M False discovery rates: a new deal. Biostatistics 18, 275–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kim D, Langmead B & Salzberg SL HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ramirez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson JT et al. Integrative genomics viewer. Nat. Biotechnol 29, 24–26 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Raudvere U et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Amemiya HM, Kundaje A & Boyle AP The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep 9, 9354 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wickham H ggplot2: Elegant Graphics for Data Analysis 2nd edn (Springer, 2016). [Google Scholar]
- 77.Durand NC et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robinson JT et al. Juicebox.js provides a cloud-based visualization system for Hi-C data. Cell Syst. 6, 256–258 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gatchalian J et al. A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun 9, 5139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Dieuleveult M Genome-wide nucleosome specificity and function of chromatin remodellers in ES cells. Nature 530, 113–116 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data have been deposited in the GEO (accession no. GSE145016). Source data are provided with this paper.