

Abstract
Peroxisome proliferator-activated receptor α (PPARα), a fatty acid oxidation regulator, inhibits alcohol-induced fatty liver (AFL). PPARα agonist WY-14,643 ameliorates AFL. Nicotine enhances AFL. In this study, we investigated whether PPARα activation also blocks nicotine-enhanced AFL. Mice were fed liquid diets containing ethanol in the presence or absence of nicotine, WY-14,643 was added to the above diets at 10 mg/L. The results showed that WY-14,643 blunted AFL and nicotine-enhanced AFL, which was paralleled with striking induction of PPARα target genes. However, serum ALT was dramatically increased by the ethanol/WY-14,643 feeding and was further increased by nicotine/ethanol/WY-14,643 feeding, which was confirmed by necro-inflammation and elevated oxidative stress. Interestingly, serum alcohol levels were dramatically decreased by WY-14,643. Ethanol is mainly metabolized by alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP2E1) and catalase. ADH and CYP2E1 were not increased by WY-14,643, but catalase was induced. What is more, injection of catalase inhibitor increased serum ethanol. Decreased serum alcohol, attenuated fatty liver, and enhanced liver injury were not induced by WY-14,643 in mice lacking PPARα. In conclusion, PPARα activation by WY-14,643 attenuates alcohol/nicotine-induced fatty liver but deteriorates ethanol/nicotine-induced liver injury; WY-14,643 enhances ethanol metabolism via induction of catalase.
Keywords: hydrogen peroxide, peroxisomal fatty acid oxidation, CYP2E1, fatty liver, inducible nitric oxide synthase (iNOS), nitrotyrosine (3-NT), nicotine
Graphic abstract
INTRODUCTION
Heavy alcohol drinking induces a spectrum of liver disorders including alcoholic fatty liver (AFL), steatohepatitis, fibrosis, cirrhosis, and even liver cancer, which are designated alcohol-related liver disease (ALD). Alcohol is mainly metabolized by alcohol dehydrogenase (ADH) to acetaldehyde in cytosol. In mitochondria, acetaldehyde is oxidized by aldehyde dehydrogenase (ALDH) to acetate. ADH-mediated ethanol oxidation consumes NAD+ and produces NADH. The increased ratio of NADH/NAD+ may inhibit fatty acid β-oxidation (FAO) leading to fat accumulation in liver, i.e., fatty liver (1). Chronic ethanol consumption leads to induction of cytochrome P450s especially CYP2E1 (2). CYP2E1 is a major enzyme in microsomal ethanol oxidation system (MEOS). Unlike ADH, CYP2E1 consumes NADPH to oxidize ethanol and produces reactive oxygen species (ROS). CYP2E1-mediated oxidative stress contributes to ALD (3–4). On the other hand, CYP2E1-derived ROS may up-regulate nuclear factor E2-related factor 2 (Nrf2), which in turn induces mouse CYP2A5 (CYP2A6 in human) (5). Indeed, ethanol induces CYP2A5 in a CYP2E1-dependent manner (6). CYP2A6 and CYP2A5 are major enzymes that metabolize nicotine (7–10). Nicotine can enhance AFL in mice, but it did not enhance alcohol-induced liver inflammation (11). Nicotine-enhanced AFL was not observed in CYP2A5 knockout (cyp2a5−/−) mice (12). Further study indicated that nicotine metabolism by CYP2A5 generates ROS, promotes alcohol-induced oxidative stress and lipid peroxidation, and inhibits hepatocyte proliferation (12,13).
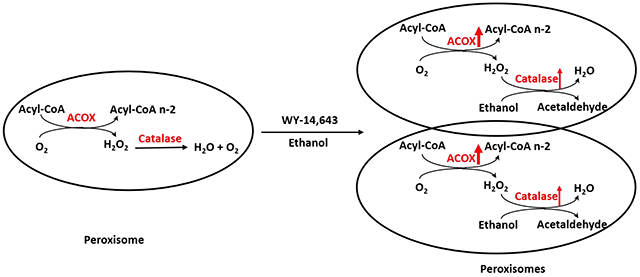
Peroxisome is one of cell organelles for fatty acid β-oxidation (FAO). Usually, very long chain fatty acids are oxidized in peroxisomes by acyl-CoA oxidase (ACOX), a rate-limiting enzyme (14). The resultant shorter chain fatty acids will be further oxidized in mitochondria. Unlike mitochondrial FAO, peroxisomal FAO produces hydrogen peroxide (H2O2), which is detoxified by peroxisomal catalase (15). Peroxisome proliferation is regulated by peroxisome proliferator-activated receptor α (PPARα), and many PPARα agonists are peroxisome proliferators (16–17). PPARα is a major regulator for lipid metabolism especially for FAO (14, 18). WY-14,643, a potent peroxisome proliferator and PPARα agonist, induced FAO-related enzymes such as ACOX and liver fatty acid binding protein (L-FABP) to prevent AFL (19). In the present study, we found that WY-14,643 also blunted nicotine-enhanced AFL.
Peroxisomes are also involved in ethanol metabolism via catalase, which is H2O2-dependent (20,21). Catalase oxidizes ethanol by consuming H2O2 (22,23). Peroxisomal catalase oxidation of alcohol can be stimulated by free fatty acids (FFA), because the peroxisomal FAO produces H2O2 (24–26). Peroxisomal catalase can be induced by peroxisome proliferators in rats because a peroxisome proliferator responsive element (PPRE) is identified in catalase promoter (27). Ciprofibrate, a peroxisome proliferator, can induce mRNA of catalase about 2-fold, but it can induce peroxisomal FAO activity up to 25-fold (28). The disproportionate induction of H2O2-generating ACOX and H2O2-scavenging catalase was proposed to be responsible for peroxisome-derived oxidative stress (29). It is unclear whether peroxisomal oxidative stress contributes to ALD.
In the present study, we examined (1) whether PPARα agonist WY-14,643 attenuates nicotine-enhance AFL and (2) whether WY-14,643 induces oxidative stress. We found that WY-14,643 attenuates fatty liver but enhanced oxidative liver injury induced by ethanol alone or in combination with nicotine. Interestingly, WY-14,643 escalated serum ethanol clearance by inducing catalase. The WY-14,643-enhanced ethanol metabolism was not observed in mice lacking PPARα (pparα−/−) but was observed in mice lacking L-FABP (L-fabp−/−). The attenuated fat accumulation in liver and enhanced liver injury induced by WY-14,643 in WT mice were not observed in pparα−/− mice and L-fabp−/− mice.
METHODS
Animals and Treatment
Colonies of C57BL/6J wild type mice (#000664) and pparα−/− mice (#008154, ref. 16) were purchased from Jackson Laboratory. L-fabp−/− mice were derived from L-fabp+/− mice (cryo recovered from Jackson Laboratory #022873, ref.30). All the mice were housed in temperature-controlled animal facilities with 12-hour light/dark cycles and were permitted consumption of tap water and Purina standard chow ad libitum. The mice received humane care, and experiments were carried out according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals. The animal studies were approved by Institution Committee of Animal Use and Care (IACUC).
Eight to ten weeks old female mice were selected for chronic ethanol feeding models. Female mice were selected because they reveal a propensity to greater ALD (31). The mice were fed the control liquid dextrose diet (Bio-Serv, Frenchtown, NJ, USA) for 3 days followed by the liquid ethanol diet (Bio-Serv) for 3 weeks. The concentration of ethanol in the liquid diet was gradually increased from 1.7% (v/v) to 2.5%, 3.4%, 4.2%, 5.1%, each concentration for 3 days, and finally 6% for the rest of time. The control group was continually fed control diet. Nicotine hydrogen tartrate salt (From Sigma) was mixed in the liquid ethanol diet at 30 mg free base/L. WY-14,643 (from Adipogen and Selleckchem, respectively) was mixed in the liquid diet at 10 mg/L. After a 6-h fasting, the mice were sacrificed following a body weight measurement. Blood was collected for serum biochemical assays. The livers were weighted, and liver index was calculated as liver weight/100g body weight. The liver aliquots from same lobes of different mice were put in neutral Formalin buffer for preparing paraffin sections for Hematoxylin & Eosin staining (H&E) and immunohistochemistry staining (IHC). The other liver tissue aliquots were stored at −80°C for future assays.
In binge model, the male mice were fed control liquid diets, with or without WY-14,643 (10 mg/L), for 3 weeks, followed by a single dose of ethanol at 5 g/kg by gavage. The mice were sacrificed after 8 hours of ethanol gavage. In acute-on-chronic models, male mice were fed diets containing 5% ethanol and 20 mg/L WY-14,643 for 2 weeks followed by a binge alcohol at 5 g/kg by gavage. Some mice received catalase inhibitor 3-amino-1,2,4-triazole (3-AT) by intra-peritoneal injection at 1 g/kg prior to the binge alcohol gavage. The mice were sacrificed after 8 hours of binge ethanol. In binge and acute-on-chronic models, male mice were selected because they displayed a lower mortality (32).
Activities of hepatic catalase, ADH, and CYP2E1
Liver homogenates were made using 0.15 M KCl. After 20 min of 9,000 x g centrifugation, the pellets were re-suspended to be used for measuring catalase activity by monitoring the decrease in absorbance at 240 nm in 50 mM potassium phosphate containing 20 mM H2O2 as described before (33). The supernatants were further centrifuged at 100,000 x g for 1 h. The resultant pellets were suspended for measuring CYP2E1 activity by PNP hydroxylation (33). The resultant supernatants were used for ADH assay using a commercially available assay kit.
Biochemical assays, Western Blotting analysis, and immunohistochemistry staining
Serum ALT, TG, FFA, alcohol and liver TG and FFA were measured using commercially available assay kits. For Western blotting, SDS-PAGE and chemiluminescence imaging was carried out as described before (7–10). Liver paraffin sections were used for IHC and liver homogenates were used for Western blotting. For IHC, a Broad Spectrum IHC Select® HRP/DAB kit (from EMD Millipore, Cat#: DAB150) was used. Antibodies against CYP2E1 and CYP2A5 are generous gifts from Drs Jerome Lasker (Hackensack Biomedical Research Institute, Hackensack, NJ) and Risto Juvonen (Department of Pharmacology and Toxicology, University of Kuopio, Kuopio, Finland). The other primary antibodies and all assay kits are listed in Table 1.
Table 1.
Antibodies, assay Kit, and other material sources
| Name | Cat# | Company |
|---|---|---|
| Anti-CYP4A11 | 11688-1-AP | Proteintech |
| Anti-Catalase | 21260-1-AP | Proteintech |
| Anti-ACOX1 | 10957-1-AP | Proteintech |
| Anti-ADH1C | 18897-1-AP | Proteintech |
| Anti-GAPDH | 60004-1-Ig | Proteintech |
| Anti-FABP1 (L-FABP) | 13626-1-AP | Proteintech |
| Anti-PPARα | 15540-1-AP | Proteintech |
| Anti-β-Tubulin | 66240-1-Ig | Proteintech |
| Anti-GCS | RB-1697-P1 | NeoMarkers |
| Anti-nitrotyrosine | sc-32757 | Santa Cruz Biotechnology |
| Anti-CYP reductase | Sc-25270 | Santa Cruz Biotechnology |
| Anti-XOD | 55156-1-AP | Proteintech |
| Anti-AOD | 19495-1-AP | Proteintech |
| Anti-SOD1 | Sc-8637 | Santa Cruz Biotechnology |
| Anti-FGF21 | Ab171941 | Abcam |
| Anti-iNOS | 610328 | BD Transduction Lab |
| Ethanol assay kit | mak076-1kt | Sigma |
| ADH assay kit | DADH-100 | BioAssay Systems |
| Free fatty acid assay kit | EFFA-100 | BioAssay Systems |
| Triglyceride assay kit | TR22421 | Thermo |
| ALT endpoint assay kit | 3460-08 | BIOO Research Corp. |
| FGF21 ELISA kit | RD291108200R | BioVendor |
| Broad Spectrum IHC Select® HRP/DAB kit | DAB150 | EMD Millipore |
Statistical analysis
Results are expressed as mean ± S.D. Statistical evaluation was carried out by using two-way analysis of variance (ANOVA) with subsequent the Student-Newman-Keuls post hoc test. P<0.05 was considered as statistical significance.
RESULTS
WY-14,643 inhibits hepatic TG accumulation induced by ethanol alone or in combination with nicotine.
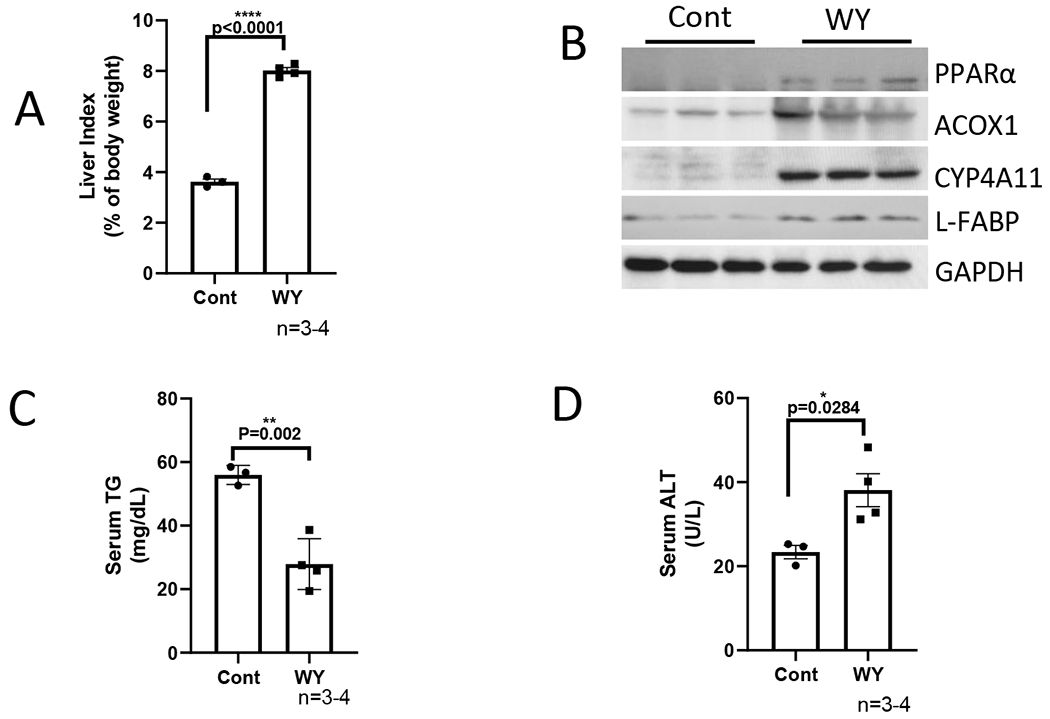
In previous studies that were carried out by other scientists, WY-14,643 was fed for 2 weeks at a dose of 0.1% either mixed in liquid diets (19) or solid chows (16, 17). Here, we added WY-14,643 in liquid diets at 10 mg/L (0.001%, w/v) and fed mice for 3 weeks. As shown in Fig. 1A, PPARα and its targets such as ACOX, CYP4A, L-FABP, and FGF21 were all induced dramatically by WY-14,643. Consistent with hepatic expression, serum FGF21 was increased by WY-14,643 (Fig. 1B). Liver contents of TG were increased by ethanol feeding and were further increased by ethanol/nicotine, both of them were blunted by WY-14,643 (Fig. 1C). Consistently, liver FFA contents were also decreased by WY-14,643 although they were not increased by ethanol or ethanol/nicotine (Fig. 1D). Serum TG (Fig. 1E) and serum FFA (Fig. 1F) were also decreased by WY-14,643. Please note that ethanol feeding increased serum FFA but not serum TG, which might be due to elevated lipolysis in adipose tissues after a 6-h fast. These results suggest that ethanol-induced fatty liver and nicotine-enhanced alcoholic fatty liver were ameliorated by WY-14,643, which was paralleled with induction of PPARα pathways.
Figure 1.
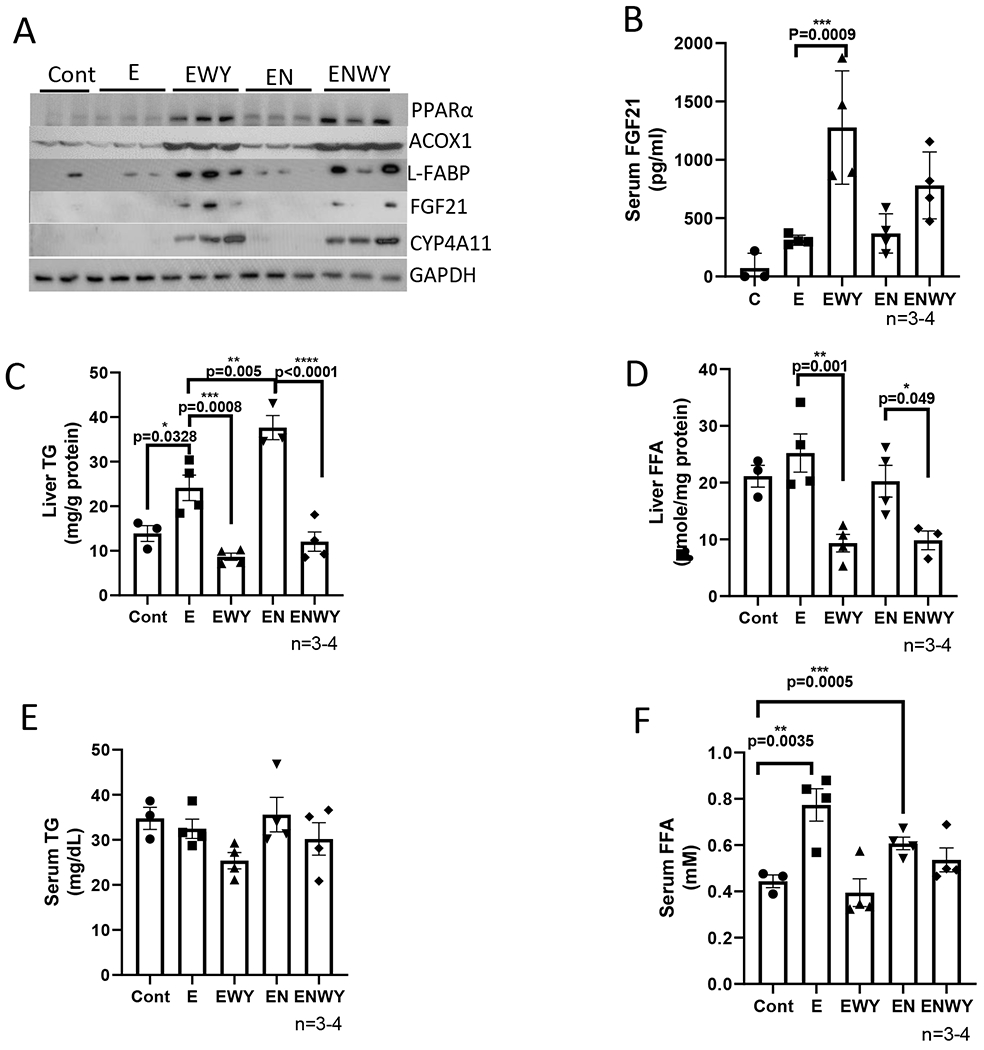
WY-14,643 mixed in ethanol diets induces PPARα pathway and inhibits hepatic TG accumulation induced by ethanol alone or in combination with nicotine. (A) Western blotting analyses for PPARα-regulated ACOX, L-FABP, FGF21, and CYP4A expression in liver. (B) Serum levels of FGF21. (C) Liver contents of TG. (D) liver contents of FFA. (E) Serum levels of TG. (F) Serum levels of FFA. Cont, Control group; E, Ethanol group; EWY, Ethanol/WY-14,643 group; EN, Ethanol/nicotine; ENWY, Ethanol/nicotine/WY-14,643.
WY-14,643 enhances liver injuries induced by ethanol alone or in combination with nicotine.
As a potent peroxisome proliferator, WY-14,643 can induce hepatomegaly (16). While liver index was increased identically by ethanol and ethanol/nicotine, respectively, the liver index was further increased by WY-14,643 in both groups in the same magnitude (Fig. 2A). Serum levels of ALT were increased dramatically in ethanol/WY-14,643 group and were further increased in ethanol/nicotine/WY-14,643 group (Fig. 2B). Consistently, in the liver sections with H&E staining, necroinflammation foci were observed in both groups with WY-14,643 (Fig. 2C). Liver contents of IL-1β were not changed significantly (Fig. 2D). However, liver TNFα was decreased by WY-14,643 (Fig. 2E). CYP2A5, a nicotine metabolism enzyme, was induced by ethanol feeding, but it was inhibited by WY-14,643, although CYP reductase was still induced by WY-14,643; aldehyde oxidase (AOD), an enzyme also involved in nicotine metabolism (34,35), was induced by WY-14,643 in the ethanol/nicotine group to a greater extent than in ethanol group. (Fig. 2F). Xanthine oxidase (XOD), a source of superoxide anion, was induced by WY-14,643 only in the ethanol/nicotine group but not in ethanol group, but superoxide dismutase 1 (SOD1) was not altered by WY-14,643 (Fig. 2F). The γ-glutamyl cysteine synthetase (γ-GCS), a glutathione (GSH) synthesis rate-limiting enzyme, was inhibited by WY-14,643, (Fig. 2F), but liver total glutathione (GSH) contents were increased by WY-14,643 (Fig. 2G). The inducible nitric oxide synthase (iNOS) was induced by WY-14,643 dramatically (Fig. 2F). Consistently, 3-nitrotyrosine (3-NT) adduct formation, which was induced by ethanol and ethanol/nicotine, was further induced by WY-14,643 (Fig. 2H). Similarly, ethanol- or ethanol/nicotine-induced formation of 4-hydroxynonenal (4-HNE), a lipid peroxidation end-product, was increased by WY-14,643 (Fig. 2I). These results suggest that WY-14,643 enhances oxidative liver injuries induced by ethanol alone or in combination with nicotine.
Figure 2.
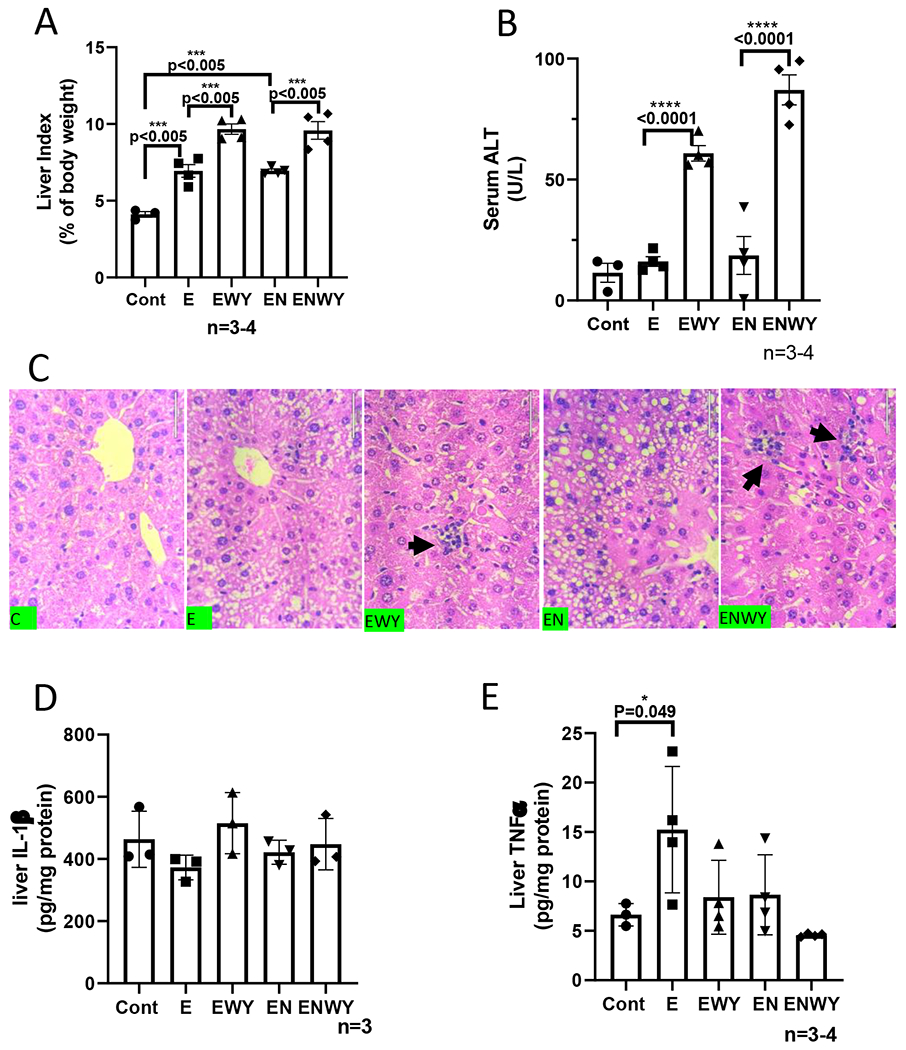
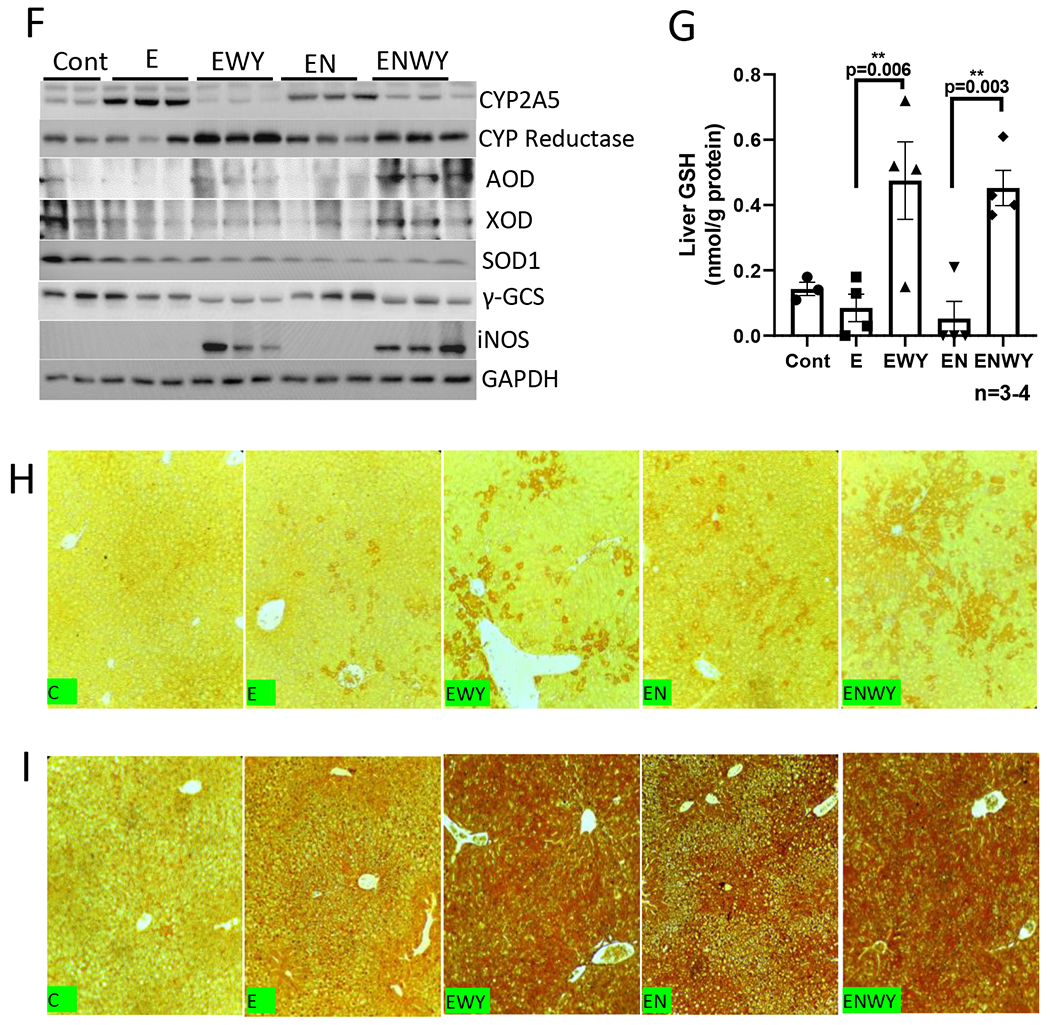
WY-14,643 enhances liver injuries induced by ethanol alone or in combination with nicotine. (A) Liver index. (B) Serum levels of ALT. (C) Liver sections with H&E staining. Black arrows indicating necroinflammation foci. (D) Liver contents of IL-1β. (E) Liver contents of TNFα. (F) Western blotting analyses for hepatic expression of CYP2A5, CYP reductase, AOD, XOD, SOD1, γ-GCS and iNOS, (G) Liver contents of total glutathione (GSH). (H) IHC for 3-NT adduct formation. (I) IHC for 4-HNE adduct formation.
WY-14,643 did not synergize with ethanol to induce hepatomegaly, because WY-14,643 also increased liver index in a similar magnitude in mice fed with control diet (Fig. 3A). WY-14,643 still induced PPARα, ACOX, CYP4A, and L-FABP (Fig. 3C) and had a hypolipidemic effect (Fig. 3D). However, WY-14,643 increased serum ALT less than 2 times in mice fed with control diet (Fig. 3B), but it induced serum ALT about 4-5 times in mice fed either ethanol or ethanol/nicotine (Fig. 2C). These results suggest that WY-14,643 synergizes with ethanol to induce liver injury.
Figure 3.
WY-14,643 mixed in control diets induces similar levels of hepatomegaly but less levels of serum ALT. (A) Liver index. (B) Western blotting analyses for PPARα-regulated ACOX, L-FABP, and CYP4A expression in liver. (C) Serum levels of TG. (D) Serum levels of ALT.
Enhancement of WY-14,643 on liver injuries induced by ethanol alone or in combination with nicotine is via a PPARα-L-FABP pathway.
To examine whether the enhancing effects of WY-14,643 on alcoholic liver injuries are regulated by PPARα or L-FABP, we fed pparα−/− mice and L-fabp−/− mice ethanol diets for 3 weeks in the presence or absence of WY-14,643 as above. As expected, ACOX and CYP4A were induced by WY-14,643 in L-fabp−/− mice but not in pparα−/− mice; L-FABP was absent in L-fabp−/− mice and was not induced by WY-14,643 in pparα−/− mice (Fig. 4A). Serum FGF21, which was almost undetectable in pparα−/− mice, was induced by WY-14,643 in L-fabp−/− mice but not in pparα−/− mice (Fig. 4B). Liver contents of TG and FFA and serum levels of TG and FFA were not decreased by WY-14,643 in pparα−/− mice or L-fabp−/− mice (Fig. 4C–4F), except for serum FFA was lowered by WY-14,643 in L-fabp−/− mice (Fig. 4F). Liver index in ethanol groups was higher in pparα−/− mice than in L-fabp−/− mice, but it was induced by WY-14,643 in L-fabp−/− mice but not in pparα−/− mice (Fig. 4G), suggesting that PPARα but not L-FABP is involved in hepatomegaly. Serum ALT was not increased by WY-14,643 in either L-fabp−/− mice or pparα−/− mice (Fig. 4H), suggesting that a PPARα-L-FABP pathway is involved in the WY-14,643 enhancing effects on alcoholic liver injury.
Figure 4.
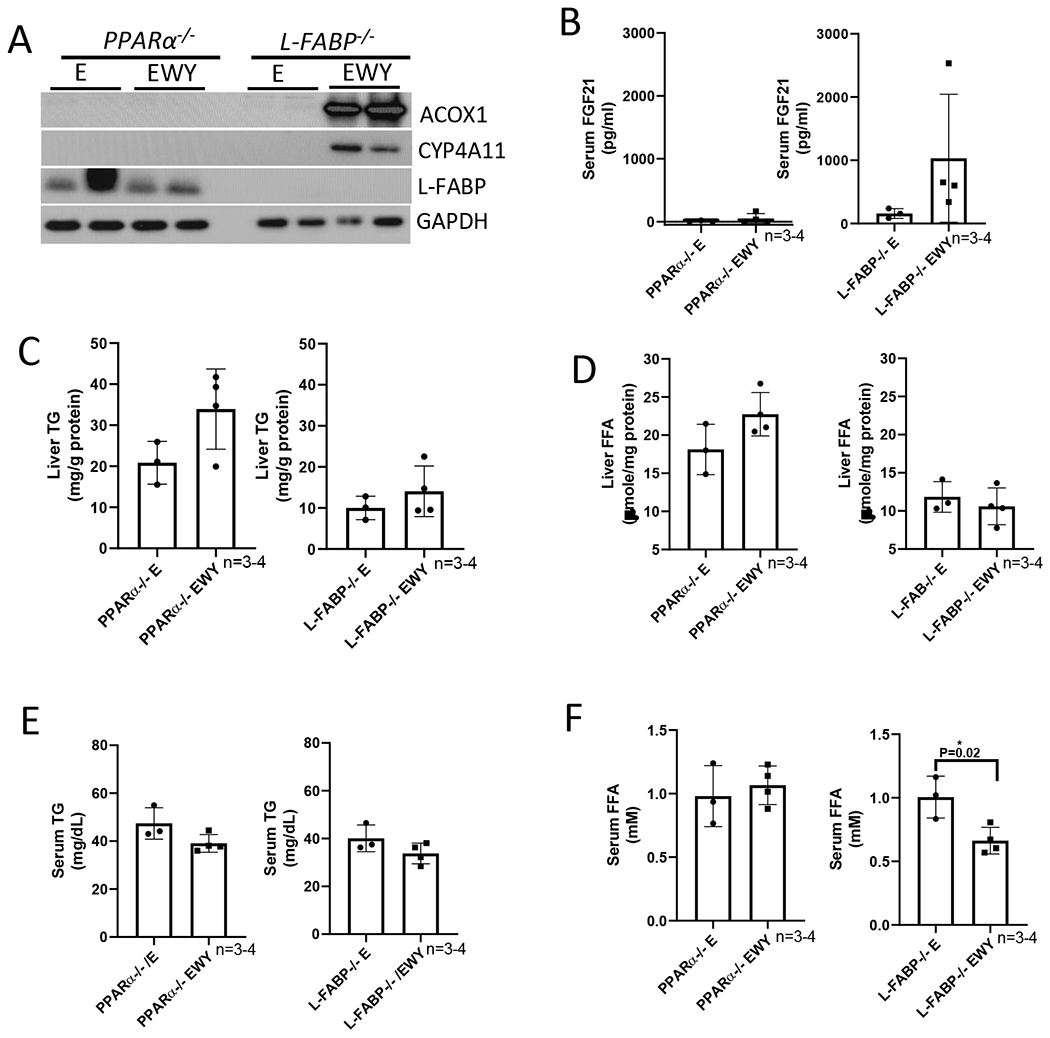
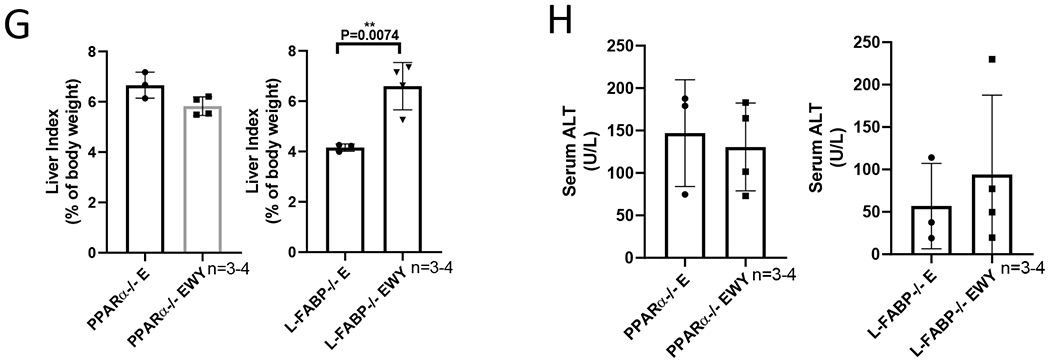
WY-14,643 feeding-induced changes are not observed in pparα−/− mice and L-fabp−/− mice. (A) Western blotting analyses for PPARα-regulated ACOX, L-FABP, and CYP4A expression in liver. (B) Serum levels of FGF21. (C) Liver contents of TG. (D) Liver contents of FFA. (E) Serum levels of TG. (F) Serum levels of FFA. (G) Liver index. (H) Serum levels of ALT. E, Ethanol diets; EWY, Ethanol diets plus WY-14,643.
WY-14,643 escalates ethanol clearance by inducing catalase.
Alcohol toxic effects are related to alcohol metabolism (1). To examine whether WY-14,643 has an influence on ethanol metabolism, serum ethanol was measured. As shown in Fig. 5A, serum alcohol was much lower in those mice fed with ethanol diets containing WY-14,643, whereas serum alcohol was comparable in mice fed with ethanol alone or in combination with nicotine. Major ethanol metabolism enzymes in liver including ADH, CYP2E1 and catalase were measured. Liver ADH expression was not significantly altered by WY-14,643, but CYP2E1 expression was decreased by WY-14,643 (Fig. 5B). Activities of hepatic ADH and CYP2E1 reflect the changes of protein expression (Fig. 5C–5D). In contrast, hepatic expression of catalase was increased by WY-14,643 (Fig. 5B) and activities of catalase were also higher in ethanol/nicotine/WY-14,643 group than in ethanol/nicotine group, although there was no significant induction in ethanol/WY-14,643 group compared with ethanol group (Fig. 5E). The induced catalase was also confirmed by IHC, which shows that the increased catalase was mainly expressed around the central veins (Fig. 5F). Interestingly, serum alcohol levels were not changed by WY-14,643 in pparα−/− mice, whereas they were dramatically decreased by WY-14,643 in L-fabp−/− mice (Fig. 6A). Consistently, catalase expression was induced by WY-14,643 in L-fabp−/− mice but not in pparα−/− mice, and CYP2E1 and ADH were not changed in either L-fabp−/− mice or pparα−/− mice (Fig. 6B). These results suggest that WY-escalates ethanol clearance by inducing catalase, which was PPARα-dependent.
Figure 5.
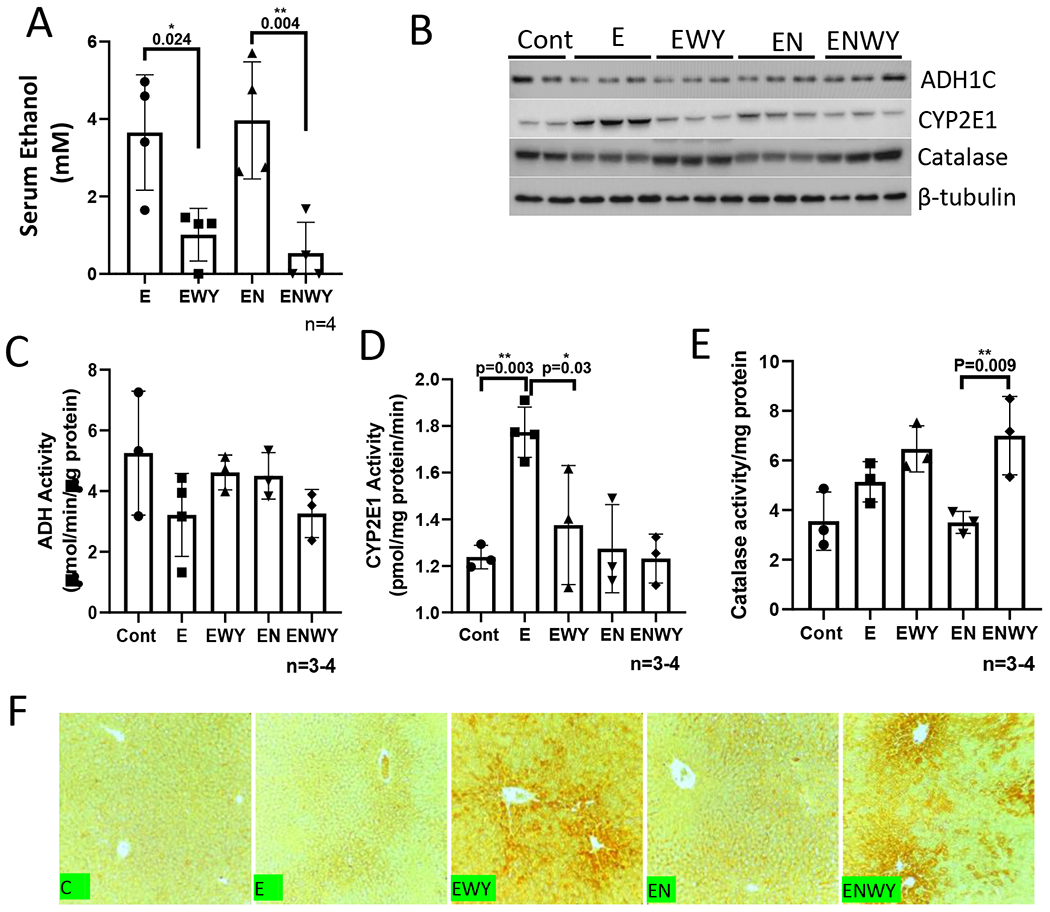
WY-14,643 decreases serum levels of alcohol and induces catalase. (A) Serum levels of ethanol. (B) Western blotting analysis for expression of CYP2E1, catalase, and ADH in liver. (C) Liver ADH activities. (D) Liver CYP2E1 activities. (E) Liver catalase activities. (F) IHC for catalase in liver sections.
Figure 6.
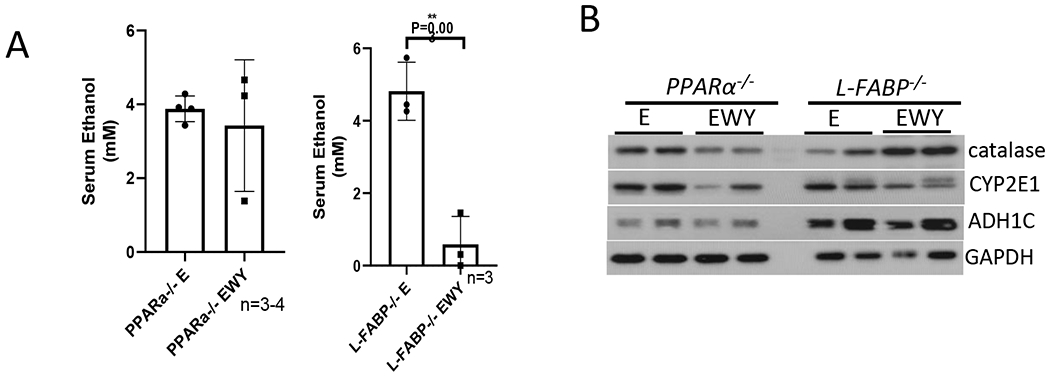
WY-14,643 diet-induced alcohol metabolism is observed in L-fabp−/− mice but not in pparα−/− mice. (A) Serum levels of ethanol. (B) Western blotting analysis for liver expression of ADH, CYP2E1, and catalase.
To further confirm the enhancing effect of WY-14,643 on blood ethanol clearance, a binge model was applied. After 3 weeks of consumption of control liquid diet with or without WY-14,643, the mice were administrated one single dose of ethanol at 5 g/kg by gavage. Like in the chronic model, WY-14,643 decreased serum alcohol levels in the binge model (Fig. 7A). Binge alcohol did not interrupt WY-14,643 induction of PPARα pathways as indicated by induction of ACOX and CYP4A (Fig. 7B). While catalase was induced by WY-14,643, CYP2E1 and ADH were not affected by WY-14,643 (Fig. 7B), suggesting that catalase induction is responsible of the enhanced alcohol clearance. WY-14,643-induced changes in liver TG and FFA, serum TG, FFA, liver index, and ALT were like what we observed in chronic feeding model (Fig. 7C–7H).
Figure 7.
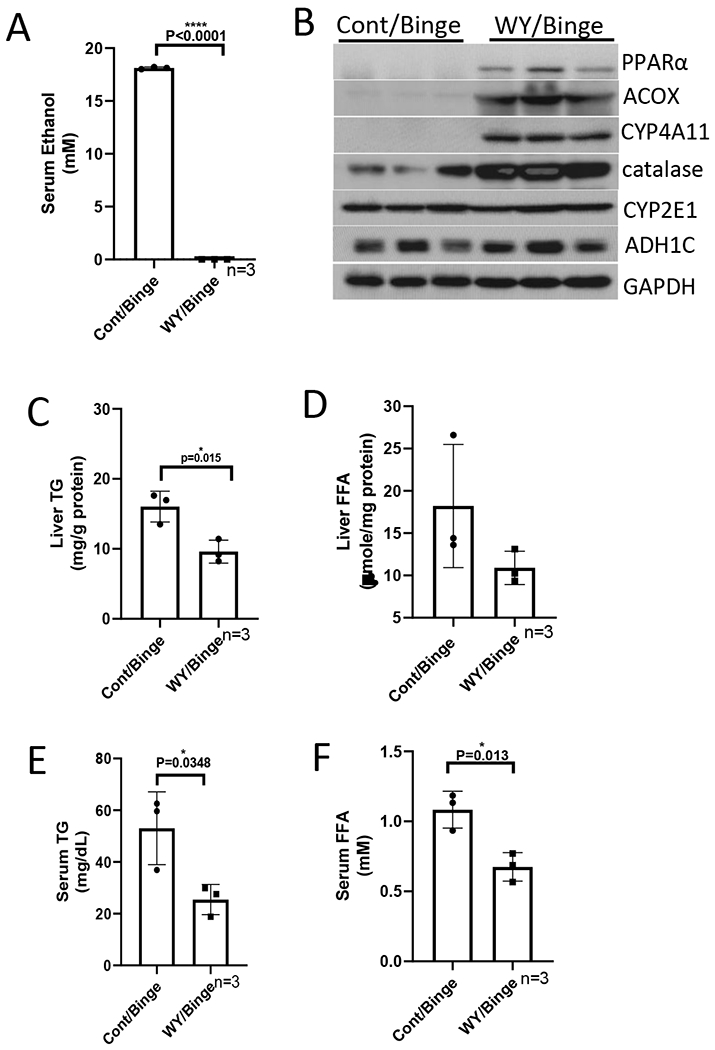
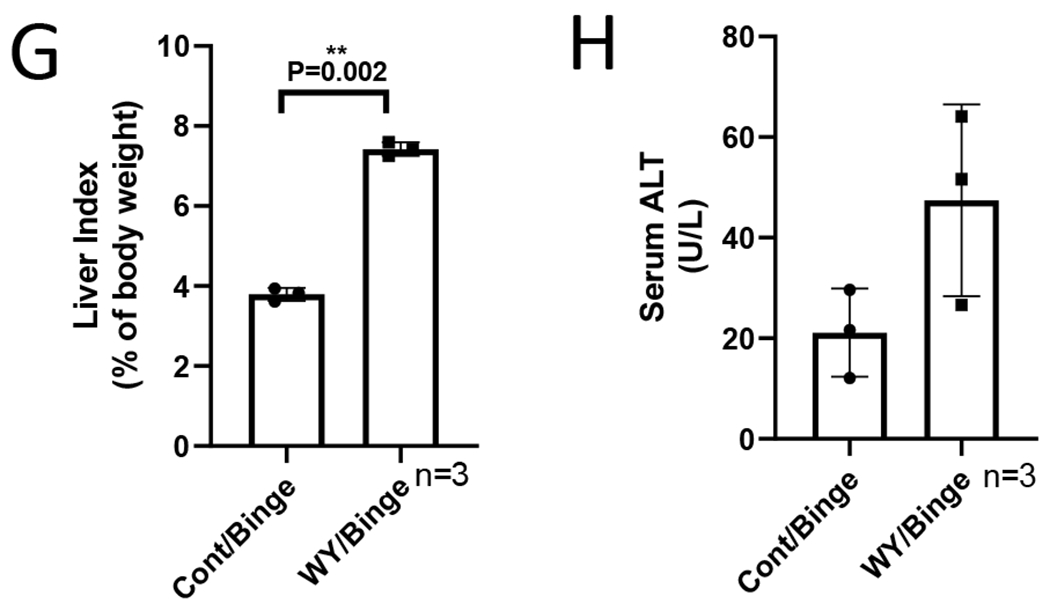
WY-14,643 feeding also escalates serum alcohol clearance in a binge alcohol model. (A) Serum levels of ethanol. (B) Western blotting analyses for PPARα-regulated ACOX, CYP4A, ADH, CYP2E1, and catalase expression in liver. (C) Liver contents of TG. (D) Liver contents of FFA. (E) Serum levels of TG. (F) Serum levels of FFA. (G) Liver index. (H) Serum levels of ALT.
Catalase inhibitor 3-AT elevates serum alcohol and ALT in an acute-on-chronic model
To further confirm the effect of catalase on the escalated ethanol clearance, catalase inhibitor 3-AT was applied. The mice were fed ethanol diet for 2 weeks followed by a binge alcohol gavage at 5 g/kg. The catalase inhibitor 3-AT was injected i.p. at 1 g/kg prior to binge alcohol administration. As shown in Figure 8, 3-AT increased serum ethanol levels (Fig. 8A), but it had no effect on liver index (Fig. 8B). Serum TG levels were reduced by 3-AT (Fig. 8C), but serum ALT was increased (Fig. 8D). These results suggest that 3-AT may inhibit ethanol clearance but increase liver injury.
Figure 8.
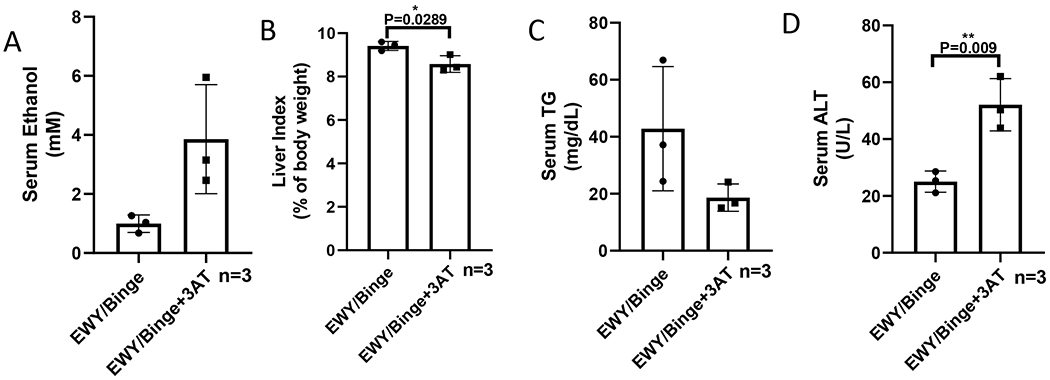
Catalase inhibitor 3-AT elevates serum alcohol and ALT in an acute-on-chronic model. (A) Serum levels of ethanol. (B) Liver index. (C) Serum levels of TG. (D) Serum levels of ALT.
DISCUSSION
It is already known that PPARα agonist WY-14,643 reversed abnormalities in hepatic lipid metabolism in ethanol-fed mice (19). We previously reported that nicotine enhances AFL (11). In this study, we confirmed that WY-14,643 suppressed AFL as well as nicotine-enhanced AFL. In addition, we have new findings: 1. WY-14,643 feeding enhanced liver injury induced by ethanol in the presence or absence of nicotine; 2. WY-14,643 administration escalated blood ethanol clearance through enhancing catalase-mediated ethanol metabolism; 3. WY-14,643 exerted all effects including hepatomegaly, attenuated fatty liver, enhanced ethanol clearance and liver injury via PPARα, but as a PPARα target gene, L-FABP did not affect WY-14,643-induced hepatomegaly and ethanol clearance.
PPARα is a major regulator for hepatic lipid metabolism (14,18). The pparα−/− mice develop more severe AFL in response to ethanol feeding (36). Previously we found that PPARα expression was upregulated by ethanol feeding in cyp2e1−/− mice but not in WT mice, and consistently, cyp2e1−/− mice developed much less AFL than WT mice (3). The basal level of PPARα expression was elevated in the cyp2a5−/− mice, but ethanol feeding did not upregulate PPARα in the cyp2a5−/− mice and development of AFL was enhanced in the cyp2a5−/− mice (37,38). Although WY-14,643 increased PPARα expression and its PPRE binding activity to ameliorate AFL, ethanol feeding did not alter PPARα expression including protein and mRNA but decreased the PPRE binding activity (19). These results suggest that ethanol feeding may induce AFL via decreasing PPARα transcriptive activity but not via decreasing PPARα expression.
As a PPARα agonist, WY-14,643 up-regulated PPARα target genes, which was confirmed by the striking induction of FAO related enzymes such as ACOX, CYP4A, and L-FABP. It was suggested that WY-14,643 ameliorates ethanol-induced fat accumulation in liver via up-regulation of these FAO enzymes (19). However, we found that in combination with ethanol, WY-14,643 also induced liver injury: serum ALT levels were dramatically increased by ethanol/WY-14,643, whereas WY-14,643 alone just slightly increased serum ALT although a similar magnitude of hepatomegaly was induced, suggesting that WY-14,643 synergizes with ethanol to induce liver injury. Nicotine only enhanced AFL but did not increase alcohol-induced serum ALT (11). However, WY-14,643 further increased serum ALT in ethanol/nicotine group to a greater extent than in ethanol group. It is well known that ethanol induces ROS generation (2). Nicotine metabolism also produces ROS (12). ACOX-mediated fatty acid oxidation produces H2O2 (14). These different sources of ROS combine together to induce oxidative liver injury as indicated by pathological observation of necro-inflammatory foci and positive staining of 3-NT and 4-HNE. Based on the ‘second-hit’ theory, fatty liver is vulnerable for second hits. However, we observed a minor fatty liver with a severe liver injury in mice fed with Lieber-DeCarli liquid diets containing ethanol and WY-14,643. Similar phenomenon was previously observed in CYP2E1 humanized transgenic mice fed with ethanol diets that developed a more severe liver injury with a less pronounced fatty liver (4).
WY-14,643 induced antioxidant catalase. WY-14,643-induced L-FABP and FGF21 also have antioxidant activities (39,40). Liver contents of total GSH were also increased by WY-14,643 although the GSH synthesis rate-limiting enzyme γ-GCS was inhibited. In contrast, ROS generator CYP2E1 was inhibited by WY-14,643. All WY-14,643-induced antioxidant effects offset oxidative stress. Nrf2-regulated CYP2A5 has an antioxidant activity (5,37,38, 41), but it was inhibited by WY-14,643. More importantly, nitric oxide (NO) producer iNOS was induced by WY-14,643 dramatically. NO may react with superoxide anion (O2•−) to form peroxynitrite (ONOO−), which may nitrate free and protein-associated tyrosine residues and form 3-NT (42). In consistent with iNOS induction, 3-NT formation was increased by WY-14,643. WY-14,643-enhanced liver injury seems to be specific for ethanol since methionine-choline deficient diet (MCD)-induced serum ALT was decreased by WY-14,643 (43).
An interesting finding in this study is that WY-14,643-induced ethanol clearance. Ethanol is mainly metabolized by three pathways: ADH, MEOS mainly CYP2E1, and catalase (44). After WY-14,643 feeding, ADH expression in liver was not changed, and CYP2E1 expression was decreased, only catalase was induced, albeit mildly. Furthermore, catalase inhibitor 3-AT elevated serum levels of ethanol. Therefore, catalase is, at least in part, responsible for the enhanced clearance of ethanol. As an antioxidant enzyme, catalase decomposes H2O2, which involves two steps: 1. A free catalase binds one molecule of H2O2 to form catalase-H2O2 complex; 2. The catalase-H2O2 complex reacts with a second H2O2 to decompose both H2O2 to 2 molecules of H2O and release one molecule of O2 (22). If the second molecule of H2O2 is replaced by ethanol, then the catalase-H2O2 complex will be decomposed to free catalase and H2O, and simultaneously ethanol is oxidized to acetaldehyde (22,23, 45). H2O2 production is called a primary oxidation system, and ethanol oxidation by H2O2 (catalyzed by catalase) is called coupled secondary oxidation. Therefore, catalase oxidizes ethanol by consuming H2O2. However, a solution of H2O2 cannot be catalyzed by catalase for coupled secondary ethanol oxidation, only “nascent” H2O2 gradually and continually released from a primary H2O2 generation system is effective for the coupled secondary ethanol oxidation (46). Ethanol oxidation by catalase mainly depends on the ratio of the H2O2 generation rate to catalase concentration (22). Fatty acids may stimulate ethanol oxidation by providing H2O2 generated by the peroxisomal FAO (ACOX is a rate limiting enzyme) for the catalase-catalyzed coupled secondary ethanol oxidation (26). Lieber-DeCarli ethanol diet and control diet are high fat diets, thus ACOX-initiated FAO in peroxisomes will increase H2O2 generation, which will increase the ratio of the H2O2 generation rate to catalase concentration and promote coupled secondary ethanol oxidation (Fig. 9B).
Figure 9.
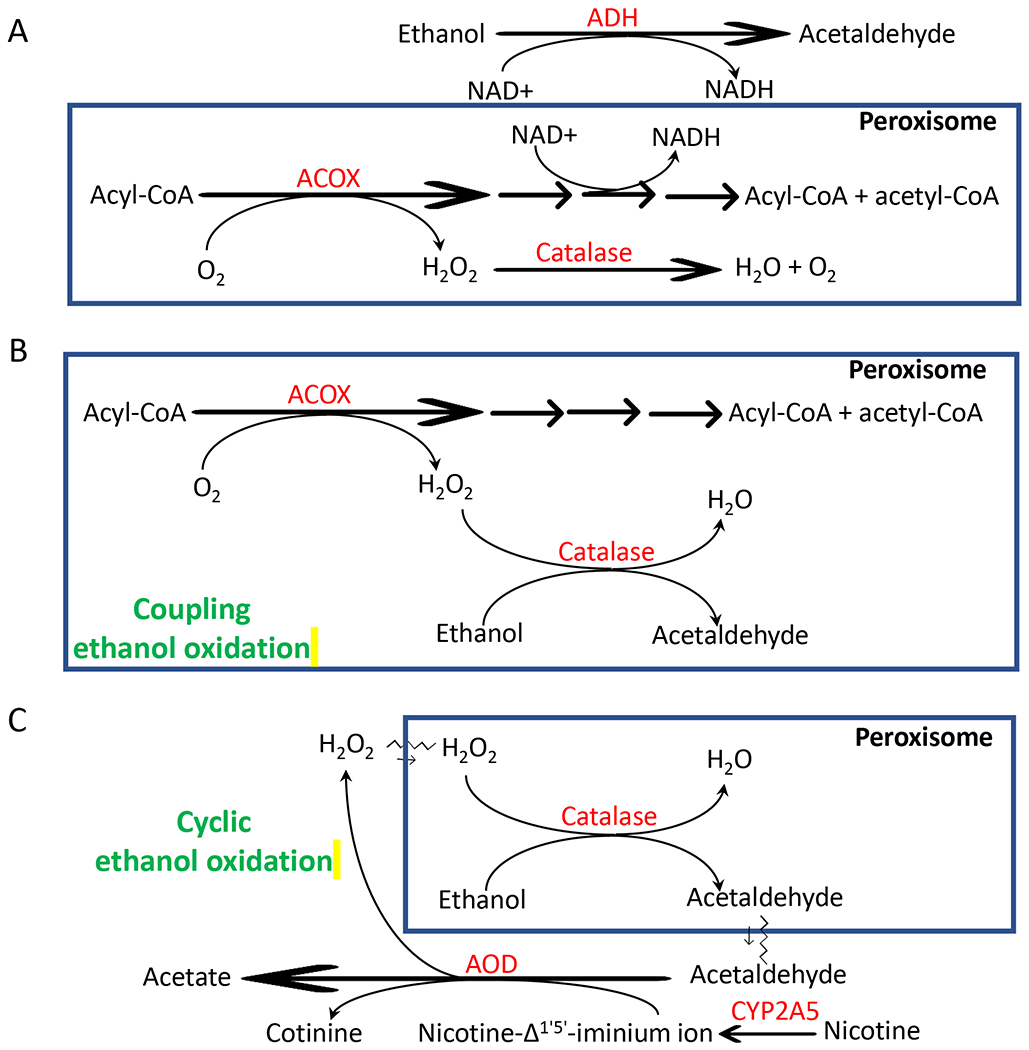
Scheme for peroxisomal oxidation of ethanol. For details, please see the texts in Discussion.
WY-14,643 induces hepatomegaly. ADH is a major ethanol metabolism enzyme. Unlike CYP2E1, ADH expression and activities were not decreased by WY-14,643 feeding. Thus, as the liver sizes are increased, total ADH activities in liver are increased. Is it possible that the escalated alcohol clearance due to the increased sizes of liver? Our data do not support this possibility: 1. ADH-mediated ethanol oxidation consumes NAD+ and produces NADH, which inhibits peroxisomal FAO and resultantly induces TG accumulation (31), but our data indicate that liver TG was decreased by WY-14,643; 2. The ADH-mediated inhibition of peroxisomal FAO should reduce the H2O2 generation rate and subsequently inhibit catalase-catalyzed coupled secondary oxidation of ethanol (47), but we observed a decreased serum alcohol level mediated by catalase. Further studies are needed to address this issue (Fig. 9A).
Most alcohol toxic effects are related to alcohol metabolism (1). When ethanol metabolism was escalated by WY-14,643, oxidative liver injury was also enhanced by WY-14,643. However, when catalase-mediated ethanol oxidation was inhibited by 3-AT, serum ALT was elevated. The 3-AT produces an irreversibly inhibited enzyme by reaction with catalase-H2O2 complex (48). Thus, 3-AT may inhibit H2O2 decomposition as well as ethanol oxidation. Therefore, 3-AT blunted ethanol metabolism and increased serum ethanol levels; 3-AT also induced liver injury and elevation of serum ALT via inhibiting catalase decomposition of H2O2. Further studies are needed to address the relationship between catalase-mediated ethanol metabolism and the development of liver injury.
As a peroxisome proliferator, WY-14,643 can induce peroxisome proliferation. Peroxisomes are responsible for about 20% oxygen consumption in liver (49). Besides ACOX, other oxidases like urate oxidase and D-amino acid oxidase in peroxisomes may also generate H2O2 for coupled secondary ethanol oxidation (50). Xanthine oxidase (XOD) is not located in peroxisomes, but it may oxidize ethanol metabolite acetaldehyde to acetate and generate H2O2, which was re-used for cyclic secondary ethanol oxidation in in vitro reaction systems (45, 46). Cytosolic aldehyde oxidase (AOD) can also oxidize acetaldehyde and the Km value of AOD for acetaldehyde is 30-fold lower than XOD for acetaldehyde (50). Nicotine-Δ1’,5’-iminium ion, an intermediate in the CYP2A5-mediated conversion of nicotine to cotinine, is also oxidized by AOD (34,35). CYP2A5 was decreased, but AOD was induced by WY-14,643. AOD-mediated conversion of nicotine-Δ1’,5’-iminium ion to cotinine also releases H2O2 (52), thus, nicotine oxidation via AOD may also provide H2O2 for “cyclic ethanol oxidation”. Under PPARα induction, nicotine may enhance alcoholic liver injury through oxidative stress resulted from AOD-mediated nicotine metabolism and catalase-mediated ethanol metabolism (Fig. 9C). XOD and AOD contribute to ethanol-induced lipid peroxidation in rodents (51,53). It will be interesting to address whether these cytosolic oxidases contribute to catalase oxidation of ethanol in in vivo whole organisms. In addition, WY-14,643 is not an only peroxisome proliferator. It is unclear whether other peroxisome proliferators like clofibrate also enhance ethanol metabolism and induce enhance alcoholic liver injury.
CONCLUSION
Alcohol consumption induced AFL, which is enhanced by nicotine. PPARα regulates lipid metabolism and PPARα agonist WY-14,643 blunts AFL and nicotine-enhanced AFL. On the other hand, WY-14,643 feeding also enhances catalase-mediated ethanol metabolism and alcoholic liver injury induced by ethanol in the presence or absence of nicotine. All the aforementioned effects of WY-14,643 including attenuated fatty liver, enhanced ethanol clearance and liver injury are through PPARα, but as a PPARα target gene, L-FABP did not affect WY-14,643-induced hepatomegaly and ethanol clearance.
Highlights:
WY-14,643 blunts alcoholic fatty liver and nicotine-enhanced alcoholic fatty liver.
WY-14,643 enhances alcoholic liver injury.
WY-14,643 escalates catalase-mediated serum alcohol clearance.
WY-14,643 effects on alcohol metabolism and liver injury are PPARα-dependent.
Acknowledgement
We thank Dr. Risto Juvonen for anti-CYP2A5 IgG, Dr. Jerome Lasker for anti-CYP2E1 IgG.
This work was supported by the National Institutes of Alcoholism and Alcohol Abuse [grant numbers R01AA024723 to YL] and a pilot award to YL from the West Virginia IDeA Network of Biomedical Research Excellence (WV-INBRE) program which is funded by National Institute of General Medical Sciences (NIGMS) Award Number P20GM103434.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest: None
REFERENCES
- 1.Lieber CS. (2005). Metabolism of alcohol. Clin Liver Dis. 9, 1–35. [DOI] [PubMed] [Google Scholar]
- 2.Lu Y, Cederbaum AI. (2008). CYP2E1 and oxidative liver injury by alcohol. Free Radio Biol Med. 44, 723–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu Y, Zhuge J, Wang X, Bai J, Cederbaum AI. Cytochrome P450 2E1 contributes to ethanol-induced fatty liver in mice. Hepatology. 2008. May;47(5): 1483–94 [DOI] [PubMed] [Google Scholar]
- 4.Lu Y, Wu D, Wang X, Ward SC, Cederbaum AI. Chronic alcohol-induced liver injury and oxidant stress are decreased in cytochrome P4502E1 knockout mice and restored in humanized cytochrome P4502E1 knock-in mice. Free Radio Biol Med. 2010. November 15;49(9): 1406–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lu Y, Zhang XH, Cederbaum AI. (2012). Ethanol induction of CYP2A5: role of CYP2E1-ROS-Nrf2 pathway. Toxicol Sci. 128, 427–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu Y, Zhuge J, Wu D, Cederbaum AI. (2011). Ethanol induction of CYP2A5: permissive role for CYP2E1. Drug Metab Dispos. 39, 330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Messina ES, Tyndale RF, Sellers EM. (1997). A major role for CYP2A6 in nicotine C-oxidation by human liver microsomes. J Pharmacol Exp Ther. 282, 1608–1614. [PubMed] [Google Scholar]
- 8.Nakajima M, Yamamoto T, Nunoya K, Yokoi T, Nagashima K, Inoue K, Funae Y, Shimada N, Kamataki T, Kuroiwa Y. (1996). Role of human cytochrome P4502A6 in C-oxidation of nicotine. Drug Metab Dispos. 24, 1212–1217. [PubMed] [Google Scholar]
- 9.Raunio H, Pokela N, Puhakainen K, Rahnasto M, Mauriala T, Auriola S, Juvonen RO. (2008). Nicotine metabolism and urinary elimination in mouse: in vitro and in vivo. Xenobiotica. 38, 34–47. [DOI] [PubMed] [Google Scholar]
- 10.Zhou X, Zhuo X, Xie F, Kluetzman K, Shu YZ, Humphreys WG, Ding X. (2010). Role of CYP2A5 in the clearance of nicotine and cotinine: insights from studies on a Cyp2a5-null mouse model. J Pharmacol Exp Ther. 332, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu Y, Ward SC, Cederbaum AI. Nicotine enhances ethanol-induced fat accumulation and collagen deposition but not inflammation in mouse liver. Alcohol. 2013. August;47(5):353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X, Owoseni E, Salamat J, Cederbaum AI, Lu Y. Nicotine enhances alcoholic fatty liver in mice: Role of CYP2A5. Arch Biochem Biophys. 2018. November 1;657:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Wang K, Cederbaum AI, Lu Y. Suppressed hepatocyte proliferation via a ROS-HNE-P21 pathway is associated with nicotine- and cotinine-enhanced alcoholic fatty liver in mice. Biochem Biophys Res Commun. 2019. April 23;512(1): 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reddy JK, Hashimoto T. Peroxisomal beta-oxidation and peroxisome proliferator-activated receptor alpha: an adaptive metabolic system. Annu Rev Nutr 21: 193–230, 2001. [DOI] [PubMed] [Google Scholar]
- 15.Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979. July;59(3):527–605. [DOI] [PubMed] [Google Scholar]
- 16.Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995; 15(6):3012–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li G, Brocker CN, Xie C, Yan T, Noguchi A, Krausz KW, Xiang R, Gonzalez FJ. Hepatic peroxisome proliferator-activated receptor alpha mediates the major metabolic effects of Wy-14643. J Gastroenterol Hepatol. 2018. May;33(5):1138–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol 2015; 62:720–733. [DOI] [PubMed] [Google Scholar]
- 19.Fischer M, You M, Matsumoto M, Crabb DW. Peroxisome proliferator-activated receptor alpha (PPARalpha) agonist treatment reverses PPARalpha dysfunction and abnormalities in hepatic lipid metabolism in ethanol-fed mice. J Biol Chem. 2003. July 25;278(30):27997–8004. [DOI] [PubMed] [Google Scholar]
- 20.Bradford BU, Enomoto N, Ikejima K, Rose ML, Bojes HK, Forman DT, Thurman RG. Peroxisomes are involved in the swift increase in alcohol metabolism. J Pharmacol Exp Ther. 1999. January;288(1):254–9. [PubMed] [Google Scholar]
- 21.Bradford BU. Role of peroxisomes in the swift increase in alcohol metabolism. J Gastroenterol Hepatol. 2007. June;22 Suppl 1:S28–30. [DOI] [PubMed] [Google Scholar]
- 22.Oshino N, Oshino R, Chance B. The characteristics of the “peroxidatic” reaction of catalase in ethanol oxidation. Biochem J. 1973. March;131(3):555–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bradford BU, Seed CB, Handler JA, Forman DT, Thurman RG. Evidence that catalase is a major pathway of ethanol oxidation in vivo: dose-response studies in deer mice using methanol as a selective substrate. Arch Biochem Biophys. 1993;303(1):172–6. [DOI] [PubMed] [Google Scholar]
- 24.Handler JA, Thurman RG. Catalase-dependent ethanol oxidation in perfused rat liver. Requirement for fatty-acid-stimulated H2O2 production by peroxisomes. Eur J Biochem. 1988. September 15;176(2):477–84. [DOI] [PubMed] [Google Scholar]
- 25.Handler JA, Thurman RG. Hepatic ethanol metabolism is mediated predominantly by catalase-H2O2 in the fasted state. FEBS Lett. 1988. September 26;238(1): 139–41. [DOI] [PubMed] [Google Scholar]
- 26.Handler JA, Thurman RG. Fatty acid-dependent ethanol metabolism. Biochem Biophys Res Commun. 1985. November 27;133(1):44–51. [DOI] [PubMed] [Google Scholar]
- 27.Girnun GD, Domann FE, Moore SA, Robbins ME. Identification of a functional peroxisome proliferator-activated receptor response element in the rat catalase promoter. Mol Endocrinol. 2002;16:2793–2801. [DOI] [PubMed] [Google Scholar]
- 28.Nemali MR, Usuda N, Reddy MK, Oyasu K, Hashimoto T, Osumi T, Rao MS, Reddy JK. Comparison of constitutive and inducible levels of expression of peroxisomal beta-oxidation and catalase genes in liver and extrahepatic tissues of rat. .Cancer Res. 1988. September 15;48(18):5316–24. [PubMed] [Google Scholar]
- 29.Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 2006. December; 1763(12): 1755–66. [DOI] [PubMed] [Google Scholar]
- 30.Martin GG; Danneberg H; Kumar LS; Atshaves BP; Erol E; Bader M; Schroeder F; Binas B. 2003. Decreased liver fatty acid binding capacity and altered liver lipid distribution in mice lacking the liver fatty acid-binding protein gene. J Biol Chem 278(24):21429–38 [DOI] [PubMed] [Google Scholar]
- 31.Eagon PK. Alcoholic liver injury: influence of gender and hormones. World J Gastroenterol. 2010. March 21; 16(11): 1377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertola A, Mathews S, Ki SH, Wang H, Gao B. Mouse model of chronic and binge ethanol feeding (the NIAAA model). Nat Protoc. 2013. March;8(3):627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu Y, Wang X, Cederbaum Al. Lipopolysaccharide-induced liver injury in rats treated with the CYP2E1 inducer pyrazole. Am J Physiol Gastrointest Liver Physiol. 2005. August;289(2):G308–19. [DOI] [PubMed] [Google Scholar]
- 34.Gorrod JW, Hibberd AR. The metabolism of nicotine-delta 1’(5’)-iminium ion, in vivo and in vitro. Eur J Drug Metab Pharmacokinet. 1982. Oct-Dec;7(4):293–8. [DOI] [PubMed] [Google Scholar]
- 35.Brandänge S, Lindblom L. The enzyme “aldehyde oxidase” is an iminium oxidase. Reaction with nicotine delta 1’(5’) iminium ion. Biochem Biophys Res Commun. 1979. December 14;91 (3): 991–6. [DOI] [PubMed] [Google Scholar]
- 36.Nakajima T, Kamijo Y, Tanaka N, Sugiyama E, Tanaka E, Kiyosawa K, Fukushima Y, Peters JM, Gonzalez FJ, Aoyama T. Peroxisome proliferator-activated receptor alpha protects against alcohol-induced liver damage. Hepatology. 2004. October; 40(4):972–80. [DOI] [PubMed] [Google Scholar]
- 37.Hong F, Liu X, Ward SS, Xiong H, Cederbaum AI, Lu Y. Absence of cytochrome P450 2A5 enhances alcohol-induced liver injury in mice. Dig Liver Dis. 2015. June;47(6):470–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen X, Ward SC, Cederbaum AI, Xiong H, Lu Y. Alcoholic fatty liver is enhanced in CYP2A5 knockout mice: The role of the PPARα-FGF21 axis. Toxicology. 2017. March 15;379:12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang G, Gong Y, Anderson J, Sun D, Minuk G, Roberts MS, Burczynski FJ. Antioxidative function of L-FABP in L-FABP stably transfected Chang liver cells. Hepatology. 2005. October;42(4):871–9. [DOI] [PubMed] [Google Scholar]
- 40.Gómez-Sámano MÁ, Grajales-Gómez M, Zuarth-Vázquez JM, Navarro-Flores MF, Martínez-Saavedra M, Juárez-León ÓA, Morales-García MG, Enríquez-Estrada VM, Gómez-Pérez FJ, Cuevas-Ramos D. Fibroblast growth factor 21 and its novel association with oxidative stress. Redox Biol. 2017. April;11:335–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu Y, Cederbaum AI. Cytochrome P450s and Alcoholic Liver Disease. Curr Pharm Des. 2018;24(14): 1502–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ischiropoulos H Biological tyrosine nitration: a pathophysiological function of nitric oxide and reactive oxygen species. Arch Biochem Biophys 356: 1–11, 1998. [DOI] [PubMed] [Google Scholar]
- 43.Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha-dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003; 38: 123–132. [DOI] [PubMed] [Google Scholar]
- 44.Jiang Y, Zhang T, Kusumanchi P, Han S, Yang Z, Liangpunsakul S. Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease. Biomedicines. 2020. March 4;8(3):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keilin D, Hartree EF. Properties of catalase. Catalysis of coupled oxidation of alcohols. Biochem J. 1945;39(4):293–301 [PMC free article] [PubMed] [Google Scholar]
- 46.Keilin David and Hartree EF. Coupled oxidation of alcohol. Proceedings of the Royal Society B, 1936, 119(813): 141–159. [Google Scholar]
- 47.Handler JA, Thurman RG. Redox interactions between catalase and alcohol dehydrogenase pathways of ethanol metabolism in the perfused rat liver. J Biol Chem. 1990. January 25;265(3):1510–5. [PubMed] [Google Scholar]
- 48.Nicholls P The reaction between aminotriazole and catalase. Biochim Biophys Acta. 1962. May 21 ;59:414–20. [DOI] [PubMed] [Google Scholar]
- 49.De Duve C, Baudhuin P. Peroxisomes (microbodies and related particles). Physiol Rev. 1966. April;46(2):323–57. [DOI] [PubMed] [Google Scholar]
- 50.Reddy JK. Peroxisome proliferators and peroxisome proliferator-activated receptor alpha: biotic and xenobiotic sensing. Am J Pathol. 2004. June;164(6):2305–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shaw S, Jayatilleke E. The role of aldehyde oxidase in ethanol-induced hepatic lipid peroxidation in the rat. Biochem J. 1990. June 15;268(3):579–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Obach RS and van Vunakis H. Radioimmunoassay of nicotine-delta 1’(5’)-iminium ion, an intermediate formed during the metabolism of nicotine to cotinine. Drug Metabolism and Disposition July 1990, 18 (4) 508–513. [PubMed] [Google Scholar]
- 53.Kato S, Kawase T, Alderman J, Inatomi N, Lieber CS. Role of xanthine oxidase in ethanol-induced lipid peroxidation in rats. Gastroenterology. 1990. January;98(1):203–10. [DOI] [PubMed] [Google Scholar]