

Abstract
Historically, administration of dacarbazine to sarcoma patients was limited by frequent treat-ment-related nausea/vomiting and neutropenia. These toxicities are now largely preventable with contemporary antiemetics and growth factor support. In this single-arm, phase II study, dacarbazine 850 mg/m2 was given on day 1 of each 3-week cycle until disease progression or intolerance with prophylactic serotonin-3 receptor, neurokinin-1 antagonists, corticosteroids, and pegfilgrastim. Coprimary endpoints included clinical benefit rate (CBR), and any grade of nausea/vomiting and/or grade 3–4 neutropenia. With a sample size of 80 patients, >24 patients with clinical benefit would indicate that the CBR exceeds the historical (<20%) [Power 0.80; alpha 0.05]. In addition, we hypothesized that the rates of nausea/vomiting would be 27% and grade 3–4 neutropenia would be 1% (historical: 90% and 36%, respectively) [power 0.95; alpha 0.05]. The CBR was 30% (24 patients: PR-2 and stable-22). The rate of nausea/vomiting was 37.5% (31 patients) and grades 3–4 neutropenia was 10% (8 patients). Median time-to-progression was 8.1 weeks (95% CI 8–9.7) and median overall survival was 35.8 weeks (95% CI 26.2–55.4). PET scans demonstrated no association with response. Modern prophylactic anti-emetics and pegfilgrastim given with dacarbazine reduced the rates of treatment related nausea/vomiting and serious neutropenia.
Keywords: Soft tissue sarcoma, bone sarcoma, dacarbazine, nausea, vomiting, PET
Introduction
Soft tissue sarcomas (STS) and bone sarcomas are a rare group of heterogeneous tumors of mesenchymal cell origin. Currently, over 150 subtypes of sarcoma have been identified. Sarcomas have an expected incidence of ~18,000 in 2020 in the US 1 and account for approximately 1% of all cancer diagnoses. For disease that is localized, surgery with or without radiation therapy is the standard approach. For patients with metastatic disease, systemic chemotherapy is the mainstay of care with a median overall survival (OS) of 20 months.2–6 In the 1970’s, dacarbazine was one of the first chemotherapeutic agents found to have anti-tumor activity in the treatment of metastatic sarcoma. 7 In an era without potent prophylactic anti-emetics and leukocyte growth factors, the effectiveness of dacarbazine was limited by treatment-related nausea/vomiting, which occurred in 90% of patients, and grade 3–4 neutropenia, which occurred in 36% of patients. These toxicities frequently resulted in early discontinuation of the drug even before first tumor response assessment or disease progression. Improvement in response rates were seen when dacarbazine was combined with ifosfamide and doxorubicin 8 ; however, randomized trials were not clearly or consistently able to document an improvement in OS with multi-agent chemotherapy.
In the late 1990s, the use of dacarbazine fell out of favor among many investigators with the prevailing belief that it was less effective than either ifosfamide or doxorubicin, and caused significant toxicity, namely nausea, vomiting, and myelosuppresion, which at the time had few effective prevention strategies. In current practice, when dacarbazine is utilized in the first line setting, it is often given in combination with anthracyclines in patients with a sensitive histologic subtype, or in instances where the use of ifosfamide is contraindicated. In later-line settings, dacarbazine is utilized as a single agent. In 2007, a retrospective analysis of single-agent dacarbazine given in the second/third-line setting to 40 patients with refractory disease demonstrated a clinical benefit rate (CBR) of 20% and a median progression-free survival (PFS) of 2 months. 9
In recent years, dacarbazine has been used as a reference arm for new investigational treatments in clinical trials of patients with anthracycline-refractory disease, such as in the phase III evaluations of trabectedin 10 or eribulin. 11 Across these two trials of nearly 1000 patients, dacarbazine resulted in objective response rates of 5%–7%, median PFS of 1.5–2.6 months, and median OS of 11.5–12.9 months. The proportion of patients who were given dacarbazine that experienced nausea/vomiting was 47%–49%, and grade 3–4 neutropenia was 16%–22%. The antiemetics administered before dacarbazine included only corticosteroids, and prophylactic leukocyte growth factors were not routinely used.
In this trial, we hypothesized that administration of potent prophylactic anti-emetics and growth factors with dacarbazine would markedly reduce the proportion of patients who experience treatment-related nausea, vomiting, and neutropenia and would result in a CBR better than historical data.
Materials and methods
This was a single-arm, single-institution, prospective phase II trial (NCT00802880) of patients 18 years of age or older with a histologically proven diagnosis of metastatic or locally recurrent STS or bone sarcoma that had progressed after one or more prior chemotherapy regimens (excluding adjuvant chemotherapy). Patients were required to have measurable disease by CT, FDG-avid disease (SUVmax ⩾ 3) on FDG-PET/CT, Eastern Cooperative Oncology Group (ECOG) performance status of 0–2 and adequate marrow and organ function (ANC ⩾1000/µL, hemoglobin ⩾8 g/dL, platelets ⩾100,000/dL, serum creatinine ⩽2.0 mg/dL, total bilirubin ⩽2.0, and AST or ALT <3× ULN). Patients were ineligible if they had chemotherapy or radiation within the last 21 days or if any investigational agent had been given within the last 30 days. The study was conducted at Washington University in St. Louis and was approved by the Institutional Review Board and Radioactive Drug Research Committee, as well as the Protocol Review and Monitoring Committee of the Siteman Cancer Center. All patients provided written informed consent.
Baseline assessments, including medical history, physical examination, vital signs, clinical laboratory tests including complete blood count and metabolic profile, and baseline imaging including CT and FDG-PET/CT (skull vertex to thighs), were performed within 21 days of Cycle 1.
Dacarbazine was administered intravenously (IV) at a dose of 850 mg/m2 over 1 h on day 1 of each 3-week cycle. The prophylactic anti-emetic regimen given IV prior to dacarbazine included three drugs: palonosetron (0.25 mg) or ondansetron (32 mg), aprepitant (150 mg), and dexamethasone (20 mg). Pegfilgrastim (6 mg subcutaneously) was administered on day 2 of each cycle. Treatment continued until disease progression, unacceptable toxicity, or withdrawal of consent. On treatment, assessments included physical examination and laboratory studies on day 1 of each cycle, and tumor response assessments every three cycles.
Tumor response was assessed on CT of the chest, abdomen, and pelvis by RECIST 1.0 12 and metabolic response was assessed on FDG-PET/CT by a modification of the EORTC criteria. 13 These imaging assessments were obtained at baseline, and planned on days 12–21 after the start of cycle 3 and every three cycles thereafter unless clinical progression occurred earlier (Schema in Figure 1). Disease progression was defined based on RECIST. FDG-PET/CT was utilized for metabolic response assessment using the SUVmax within metastatic tumor sites. Up to a maximum of three lesions having the greatest FDG uptake were identified as target lesions on baseline PET. If more than one target lesion was identified, the average change in SUVmax was used to determine the metabolic response.
Figure 1.

Schema of the phase II dacarbazine trial. Baseline assessments included medical history, physical examination, vital signs, clinical laboratory tests including complete blood count and metabolic profile, and baseline imaging including CT and FDG-PET/CT (skull vertex to thighs), were performed within 21 days of cycle 1.
Safety was assessed by monitoring for treatment-emergent adverse events (TEAEs) and graded using NCI CTCAE version 3.0. Dose reductions by 15% of the original dacarbazine dose (850 mg/m2) were required for grade 3 and 4 neutropenia, thrombocytopenia, nausea/emesis, diarrhea, hepatic and renal toxicity, hypocalcemia, or hypersensitivity. A maximum of three dose reductions were permissible, after which the drug was discontinued.
Co-primary endpoints included CBR (the proportion of patients with complete or partial response [PR] or stable disease [SD]), and the frequency of any grade of nausea/vomiting or grade 3–4 neutropenia. When our trial was developed, the historical CBR with dacarbazine monotherapy was reported to be ⩽20%. 9 Data from the phase III trials of eribulin or trabectedin versus dacarbazine were reported several years later.10,11 With a sample size of 80 patients, ⩾24 patients with clinical benefit resulted in a CBR that exceeded the historical rate (20%) [power 0.80; alpha 0.05]. Also, a sample size of 80 patients was able to detect a reduction in the rates of any grade nausea/vomiting from the historical of 90% to 27% and in the rates of grade 3–4 neutropenia from the historical of 36% to 1% (power 0.95; alpha 0.05).
Kaplan-Meier models were used to estimate median time-to-progression (TTP: time from study enrollment to tumor progression) and median OS (time from the start of treatment to death from any cause) with 95% confidence intervals. The log-rank test was used to compare TTP and OS among categories of each response endpoint.
Results
Patients
From March 2009 to December 2014, 80 patients were enrolled. Baseline characteristics are listed in Table 1. The majority of patients had ECOG performance scores of 0–1. All but one patient had metastatic disease, and more than half of patients had received three or greater lines of prior systemic therapy. The most common histologic subtypes were leiomyosarcoma and liposarcoma. The number of STS tumors was 75, the number of bone tumors was 5 (Table 2).
Table 1.
Patient characteristics.
| Characteristic | No. | % |
|---|---|---|
| Sex | ||
| Female | 40 | 50 |
| Male | 40 | 50 |
| Age, years | ||
| Median | 53 | |
| Range | 20–83 | |
| ECOG performance status | ||
| 0 | 45 | 56 |
| 1 | 32 | 40 |
| 2 | 3 | 3 |
| Site of primary | ||
| Extremity | 37 | 46 |
| Retroperitoneum/uterus | 27 | 33 |
| Head and neck | 6 | 7 |
| Other | 10 | 12 |
| Extent of disease | ||
| Locally advanced | 1 | 1 |
| Metastatic | 79 | 98 |
| Pulmonary metastases only | 11 | 13 |
| Liver metastases | 18 | 22 |
| No. of previous therapies | ||
| 1 | 1 | 1 |
| 2 | 28 | 35 |
| 3 | 22 | 27 |
| >3 | 29 | 36 |
Table 2.
Histologic subtypes.
| Histologic subtype | #Included |
|---|---|
| Leiomyosarcoma | 19 |
| Liposarcoma | 10 |
| Malignant fibrous histiocytoma | 7 |
| Peripheral nerve sheath | 6 |
| Osteosarcoma | 5 |
| Hemangiopericytoma/solitary fibrous tumor | 5 |
| Synovial sarcoma | 5 |
| Pleomorphic sarcoma | 5 |
| Chondrosarcoma | 4 |
| Ewing’s | 3 |
| Paraganglioma | 2 |
| Fibrosarcoma | 2 |
| Sarcoma – spindle cell | 2 |
| High grade undifferentiated | 2 |
| Endometrial stromal cell | 1 |
| Adenocarcinoma | 1 |
| Desmoid/small round cell tumor | 1 |
Treatment administered
Patients received a median of three cycles of dacarbazine (range 1–36). Seven of the 80 patients received 10 or more cycles. The most common reason for therapy discontinuation was disease progression (78%). Other reasons included drug toxicity, patient noncompliance, and patient death.
Primary end points
The CBR was 30% (24 of 80 patients). Two patients had a PR (3%), and 22 patients had SD (28%). The overall number of patients with PD was 56 patients (55%): 30 had PD by RECIST and 26 were considered to have PD because they did not undergo a tumor response assessment, as CT and PET/CT cans were not obtained per protocol on patients who progressed or died before week 9 and thus were deemed to be off trial. The two patients with objective tumor response had leiomyosarcoma.
The rate of any grade nausea/vomiting was 37.5% (31 patients) and grade 3–4 neutropenia was 10% (8 patients).
Secondary efficacy endpoints
Metabolic tumor response was assessed with FDG-PET/CT performed at baseline and after every three cycles of treatment. The numbers of patients evaluable for metabolic response at baseline and after cycles 3, 6, 9, and 12 were 51, 50, 20, 7, and 5, respectively; 29 patients were not evaluable because a second FDG/PET scan was not performed, most often because of early progression. Of the 50 evaluable patients, partial metabolic response (PMR) occurred in 4, stable metabolic disease (SMD) in 13 and progressive metabolic disease (PMD) in 34. Tumor metabolic response rates were significantly correlated to the anatomic response rate in 49 patients as shown in Table 3.
Table 3.
Correlation of metabolic to anatomic responses.
| Partial metabolic response + stable metabolic disease | Progressive metabolic disease | Total | Cohen’s Kappa | Fisher exact test p value | |
|---|---|---|---|---|---|
| PR + SD | 13 | 9 | 22 | ||
| PD | 3 | 24 | 27 | ||
| Total | 16 | 33 | 49 | 0.49 (0.25–0.73) | 0.0005876 |
Seventy-nine patients were included in the OS analysis as one patient withdrew consent: the median OS was 8.09 months (95% CI 5.72–12.7; Figure 2(a)). Median TTP was 2.7 months (95% CI 1.84–2.3; Figure 2(b)). OS and TTP were reported by best anatomic response (Figure 2(c) and (d), respectively). OS and TTP were also reported by best metabolic response (Figure 2(e) and (f), respectively).
Figure 2.

Response data. Demonstrates overall survival (a), time to progression (b), times to progression by best anatomic (c) or best metabolic (d) response, as well as overall survival by best anatomic response (e), and best metabolic response (f).
PD: progressive disease; PMD: partial metabolic response; PMR: partial metabolic response; PR: partial response; SD: stable disease; SMD: stable metabolic response.
Mean SUVs were compared at baseline and at cycle 3 in patients with PR + SD versus those with PD. At baseline there was no significant difference in the mean SUV for those with PD versus PR + SD, but at cycle 3 there was a significant difference in the SUV between these two groups (p = 0.01; Figure 3).
Figure 3.
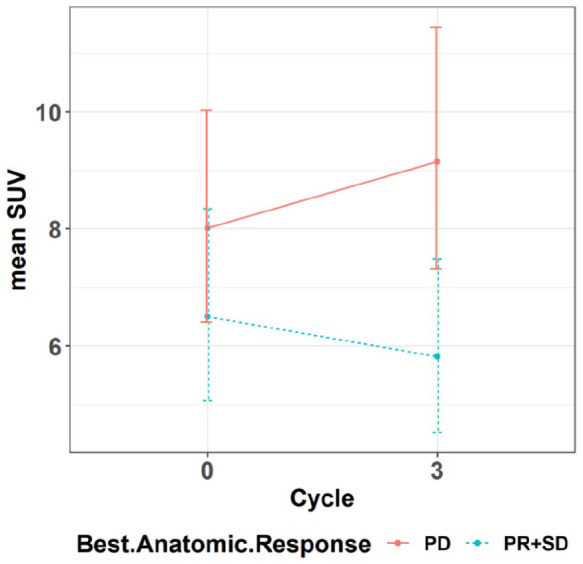
Demonstrates mean PET SUV from baseline to cycle 3 by response.
In the 51 patients who received a PET at cycle 3, the responses of PD versus PR + SD were correlated to OS (Figure 4(a)) and TTP (Figure 4(b)).
Figure 4.

(a) Mean OS and (b) TTP based on metabolic responses at cycle 3.
Adverse events
The most frequent AEs (Table 4) of any grade included anemia (72%), lymphopenia (67%), and thromobocytopenia (35%). The most frequent grade 3–4 AEs included lymphopenia (26%) and fatigue (12%).
Table 4.
Adverse events reported in ⩾10% of participants.
| Grade 1 | Grade 2 | Grade 3 | Grade 4 | Grade 5 | |
|---|---|---|---|---|---|
| Constitutional | |||||
| Edema | 14 | 7 | 1 | 0 | 0 |
| Fatigue | 13 | 10 | 10 | 0 | 0 |
| Anorexia | 6 | 5 | 1 | 0 | 0 |
| Gastrointestinal | |||||
| Constipation | 13 | 5 | 1 | 0 | 0 |
| Diarrhea | 11 | 2 | 0 | 1 | 0 |
| Nausea | 11 | 2 | 5 | 0 | 0 |
| Vomiting | 7 | 2 | 3 | 0 | 0 |
| Hematologic | |||||
| Hemoglobin | 29 | 18 | 9 | 2 | 0 |
| Leukocytes (WBC) | 12 | 7 | 4 | 1 | 0 |
| Lymphopenia | 11 | 22 | 18 | 3 | 0 |
| Neutrophils (ANC) | 4 | 3 | 4 | 4 | 0 |
| Platelets | 18 | 6 | 4 | 6 | 0 |
| Hepatic function | |||||
| Alkaline phosphatase | 13 | 4 | 2 | 0 | 0 |
| SGOT (AST) | 12 | 0 | 2 | 2 | 0 |
| SGPT (ALT) | 11 | 2 | 1 | 1 | 0 |
| Metabolic/laboratory | |||||
| Low albumin | 15 | 10 | 2 | 0 | 0 |
| Hypocalcemia | 19 | 9 | 0 | 1 | 0 |
| Hyperglycemia | 8 | 9 | 5 | 0 | 0 |
| Hypokalemia | 8 | 0 | 3 | 1 | 0 |
| Hyponatremia | 16 | 2 | 3 | 0 | 0 |
| Pain | |||||
| Abdominal | 4 | 3 | 2 | 1 | 0 |
| Bone | 14 | 4 | 5 | 0 | 0 |
| Disease pain | 4 | 6 | 7 | 0 | 0 |
| Pulmonary | |||||
| Cough | 7 | 5 | 0 | 0 | 0 |
| Dyspnea (SOB) | 6 | 6 | 6 | 0 | 1 |
| Renal/genito-urinary | |||||
| Creatinine | 7 | 4 | 0 | 0 | 0 |
Discussion
In this trial, we administered a prophylactic modern three-drug anti-emetic regimen and pegfilgrastim with each cycle of dacarbazine. Using this approach, the rates of treatment-related nausea/vomiting (37.5%) and grade 3–4 neutropenia (10%) were lower than historical and contemporary reports of dacarbazine monotherapy. Historical reports conducted in an era of poorly effective anti-emetics and lack of clinically available growth factors showed that the rate of dacarbazine-related nausea/vomiting was 90% and grade 3–4 neutropenia was 36%. 9 Contemporary reports used only corticosteroids as the anti-emetic regimen and no prophylactic growth factors and showed rates of nausea/vomiting and grade 3–4 neutropenia were 47%–49% and 16%–22%, respectively.10,11 Based on the results of our trial, modern multi-agent anti-emetics and leukocyte growth factors should be prophylactically administered with dacarabazine to reduce the risk of these AEs.
When our trial was developed, historical reports showed that the CBR with dacarbazine monotherapy was 20%. 9 We showed that the CBR assessed by RECIST with dacarbazine was 30%. Clinical benefit (PR or SD) occurred in 24 of 80 patients, meeting the pre-specified threshold of superiority of the CBR compared to historical (20%) reports. 9 The patients who had clinical benefit that lasted over 6 months had varying histologic subtypes, including fibrosarcoma, malignant fibrous histiocytoma, and leiomyosarcoma. All five patients with osteosarcoma experienced disease progression as the best response to dacarbazine, suggesting a lack of efficacy of dacarbazine in this subtype.
After our trial was initiated, the phase III trials of trabectedin or eribulin versus dacarbazine were reported.10,11 Interestingly, the CBR of dacarbazine in these reports were 42%–53%, higher than historical reports and higher than we observed in this trial. Differences in tumor characteristics and prior therapy likely contributed to these observations. For example, all patients in the trabectedin and eribulin trials had leiomyosarcoma and liposarcoma, subtypes know to be chemosensitive; whereas, in our trial, only 29 of 80 patients (36%) had these subtypes.
In the dacarbazine arms of the phase III trabectedin or eribulin versus dacarbazine trials, the ORRs were 5%–7%, the median PFS was 1.5–2.6 months, and the median OS was 11.5–12.9 months.10,11 In our trial, the ORR with dacarbazine was 3%, the median TTP was 8.14 weeks and median OS was 35.8 weeks. Given the differences in histologic subtypes and prior therapy, it is surprising that the ORR and TTP or PFS were similar across these three trials.
This is the first report that describes the metabolic tumor response to dacarbazine in patients with STS. The role of FDG-PET/CT in predicting response in sarcoma is currently not clear.14–16 Table 3 illustrates the significant correlation between anatomic and metabolic responses in the evaluable patients. Our data showed no significant difference in anatomic responses based on mean SUV at baseline, but did show a significant difference at cycle 3; where those with PR + SD had a lower mean SUV at this time point as compared to those with PD. The difference in overall survival based on those with PMD versus those with PMR + SMD at cycle 3 was not significant; however, TTP was trending toward significance in those with PMR + SMD at cycle 3 versus those with PMD. Taken together, these data suggest that FDG-PET/CT may have some utility as a predictive tool for response, but larger scale studies are needed.
While our trial included a large sample size for a phase 2 trial, there was no parallel control arm. Additionally, histologic subtypes were heterogeneous, as was number of lines of prior therapy, which makes generalizability of these data difficult. Procedures and grading and assessment systems for AEs and tumor response in our trial varied from the historical report due to the large gap in time between the two trials. 9 However, these issues were similar to those used in contemporary reports.10,11 Doses of dacarbazine used across the trials varied, although the range of doses were all within what is clinically relevant to practice patterns.
Conclusions
We conclude from our trial that a modern prophylactic three-drug anti-emetic regimen and pegfilgrastim given with dacarbazine reduced the rates of treatment-related nausea/vomiting and serious neutropenia. The CBR was modestly improved. Prophylactic, potent anti-emetics, and pegfilgrastim should be routinely administered with dacarbazine.
Footnotes
Author contributions: Conceptualization: Brian A Van Tine, Barry A Siegel, and Douglas R Adkins; Methodology: Brian A Van Tine, Kathryn Trinkaus Farrokh Dehdashti, and Barry A Siegel; Formal analysis: Kathryn Trinkaus and Jingqin Luo; Investigation: Brian A Van Tine, Marilyn J Siegel, Farrokh Dehdashti, Barry A Siegel, and Douglas R Adkins; Data Curation: Sarah Abaricia, Shellie Berry, Tyler Ruff, Cheryl Callahan, Jacqui Toensikoetter, and Jessica Ley; Writing—Original Draft Preparation: Mia C Weiss; Writing—Review and Editing: Brian A Van Tine, Mia C Weiss, Barry A Siegel, Douglas R Adkins, Angela C Hirbe, and Peter J Oppelt. All authors have read and agreed to the published version of the manuscript.
Declaration of conflicting interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Brian A Van Tine declares grants from Merck; grants and personal fees from Pfizer; grants from TRACON Pharmaceuticals; grants, personal fees, and other from GlaxoSmithKline; personal fees from Polaris Inc.; personal fees from Lilly; personal fees from Caris Life Sciences; personal fees from Novartis; personal fees from CytRX; personal fees from Plexxikon; personal fees from Epizyme; personal fees from Daiichi Sankyo; personal fees from Adaptimmune; personal fees from Immune Design; personal fees from Bayer; personal fees from Cytokinetics; personal fees from Deciphera; and has a patent issued for the use of ME1 as a biomarker and ACXT3102. Mia C Weiss: None. Angela C Hirbe: Consultant for AstraZeneca and Springworks. Peter J Oppelt: Speaking fees from Merck, BMS, EISAI. Kathryn Trinkaus: None. Tyler Ruff: None. Cheryl Callahan: None. Jessica Ley: None. Marilyn J Siegel: declares personal fees from Siemens Healthineers outside the submitted work. See also spouse disclosures for Barry A Siegel. Farrokh Dehdashti: None. Barry A Siegel: declares personal fees from Avid Radiopharmaceuticals, Inc., Capella Imaging, LLC, Curium Pharma, General Electric Healthcare and Imaginab, Inc.; grants and personal fees from Progenics Pharmaceuticals; grants from the ECOG-ACRIN Medical Research Foundation; grants and personal fees from American College of Radiology; and personal fees from the American Medical Foundation for Peer Review and Education outside the submitted work. See also spouse disclosure for Marilyn J Siegel. Douglas R Adkins: declares consulting or scientific advisory board support from Pfizer, Eli Lilly, Merck, Celgene, Cue Biopharma, and institutional research support from Pfizer, Eli Lilly, Merck, Celgene/BMS, Novartis, AstraZeneca, Atara Bio, Blueprint Medicine, Celldex, Enzychem, Kura, Exelixis, Innate, Sensei, and Matrix Biomed.
Ethical approval: The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of Washington University in St. Louis (protocol 08-1922, 11/16/2012).
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Division of Medical Oncology support was provided by John D DiPersio, MD, PhD. This study used the Siteman Cancer Center Imaging and Response Assessment Core supported in part by NCI Grant number P30 CA91842.
Informed consent: Informed consent was obtained from all subjects involved in the study.
Trial registration: ClinicalTrials.gov Identifier: NCT00802880.
ORCID iDs: Brian A Van Tine  https://orcid.org/0000-0003-4572-6668
https://orcid.org/0000-0003-4572-6668
Angela C Hirbe
https://orcid.org/0000-0003-1719-0771
References
- 1. Cancer Facts & Figures [Webpage]. Key statistics for soft tissue sarcomas, https://www.cancer.org/cancer/soft-tissue-sarcoma/about/key-statistics.html#references (2020, accessed 16 August 2020).
- 2. Nielsen OS, Judson I, van Hoesel Q, et al. Effect of high-dose ifosfamide in advanced soft tissue sarcomas. A multicentre phase II study of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer 2000; 36(1): 61–67. [DOI] [PubMed] [Google Scholar]
- 3. Palumbo R, Palmeri S, Antimi M, et al. Phase II study of continuous-infusion high-dose ifosfamide in advanced and/or metastatic pretreated soft tissue sarcomas. Ann Oncol 1997; 8(11): 1159–1162. [DOI] [PubMed] [Google Scholar]
- 4. Maki RG, Wathen JK, Patel SR, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007; 25(19): 2755–2763. [DOI] [PubMed] [Google Scholar]
- 5. Harris SJ, Maruzzo M, Thway K, et al. Metastatic soft tissue sarcoma, an analysis of systemic therapy and impact on survival. J Clin Oncol 2015; 33(15_suppl): 10545. [Google Scholar]
- 6. Leahy M, Garcia Del Muro X, Reichardt P, et al. Chemotherapy treatment patterns and clinical outcomes in patients with metastatic soft tissue sarcoma. The SArcoma treatment and Burden of Illness in North America and Europe (SABINE) study. Ann Oncol 2012; 23(10): 2763–2770. [DOI] [PubMed] [Google Scholar]
- 7. Gottlieb JA, Benjamin RS, Baker LH, et al. Role of DTIC (NSC-45388) in the chemotherapy of sarcomas. Cancer Treat Rep 1976; 60(2): 199–203. [PubMed] [Google Scholar]
- 8. Gottlieb JA, Baker LH, Quagliana JM, et al. Chemotherapy of sarcomas with a combination of adriamycin and dimethyl triazeno imidazole carboxamide. Cancer 1972; 30(6): 1632–1638. [DOI] [PubMed] [Google Scholar]
- 9. Zucali PA, Bertuzzi A, Parra HJ, et al. The “old drug” dacarbazine as a second/third line chemotherapy in advanced soft tissue sarcomas. Investig New Drugs 2008; 26(2): 175–181. [DOI] [PubMed] [Google Scholar]
- 10. Demetri GD, von Mehren M, Jones RL, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol 2016; 34(8): 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schöffski P, Maki RG, Italiano A, et al. Randomized, open-label, multicenter, phase III study of eribulin versus dacarbazine in patients (pts) with leiomyosarcoma (LMS) and adipocytic sarcoma (ADI). J Clin Oncol 2015; 33(18_suppl): LBA10502. [Google Scholar]
- 12. Therasse P, Arbuck SG, Eisenhauer EA, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000; 92(3): 205–216. [DOI] [PubMed] [Google Scholar]
- 13. Young H, Baum R, Cremerius U, et al. Measurement of clinical and subclinical tumour response using [18F]-fluorodeoxyglucose and positron emission tomography: review and 1999 EORTC recommendations. Eur J Cancer 1999; 35(13): 1773–1782. [DOI] [PubMed] [Google Scholar]
- 14. Lim HJ, Johnny Ong CA, Tan JW, et al. Utility of positron emission tomography/computed tomography (PET/CT) imaging in the evaluation of sarcomas: a systematic review. Crit Rev Oncol Hematol 2019; 143: 1–13. [DOI] [PubMed] [Google Scholar]
- 15. Angelini A, Castellucci P, Ceci F. Future perspective of the application of positron emission tomography-computed tomography-MR imaging in musculoskeletal disorders. PET Clin 2019; 14(1): 183–191. [DOI] [PubMed] [Google Scholar]
- 16. Muheremu A, Ma J, Amudong A, et al. Positron emission tomography/computed tomography for osseous and soft tissue sarcomas: a systematic review of the literature and meta-analysis. Mol Clin Oncol 2017; 7(3): 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]