

Abstract
Safe and effective new oral therapies for autoimmune, allergic, and inflammatory conditions remain a significant therapeutic need. Here, we investigate the human pharmacokinetics, pharmacodynamics (PDs), and safety of the selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor, remibrutinib. Study objectives were explored in randomized single and multiple ascending dose (SAD and MAD, respectively) cohorts with daily doses up to 600 mg, and a crossover food effect (FE) cohort, in adult healthy subjects without (SAD [n =80]/FE [n =12]) or with asymptomatic atopic diathesis (MAD [n =64]). A single oral dose of remibrutinib (0.5‒600 mg) was rapidly absorbed (time to maximum concentration = 0.5 h‒1.25 h) with an apparent blood clearance of 280‒560 L/h and apparent volume of distribution of 400‒15,000 L. With multiple doses (q.d. and b.i.d.), no pronounced accumulation of remibrutinib was detected (mean residence time was <3 h). Food intake showed no clinically relevant effect on remibrutinib exposure suggesting no need for dose adaptation. With remibrutinib doses greater than or equal to 30 mg, blood BTK occupancy was greater than 95% for at least 24 h (SAD). With MAD, remibrutinib reached near complete blood BTK occupancy at day 12 predose with greater than or equal to 10 mg q.d. Near complete basophil or skin prick test (SPT) inhibition at day 12 predose was achieved at greater than or equal to 50 mg q.d. for CD63 and at greater than or equal to 100 mg q.d. for SPT. Remibrutinib was well‐tolerated at all doses without any dose‐limiting toxicity. Remibrutinib showed encouraging blood and skin PDs with a favorable safety profile, supporting further development for diseases driven by mast cells, basophils, and B‐cells, such as chronic spontaneous urticaria, allergic asthma, or Sjögren’s syndrome.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Bruton’s tyrosine kinase (BTK) inhibition is being actively explored as a promising new pharmacological approach for the treatment of various autoimmune, allergic, and inflammatory conditions.
WHAT QUESTION DID THIS STUDY ADDRESS?
This study investigated the human pharmacokinetics (PKs), pharmacodynamics (PDs), and safety of the selective, covalent BTK inhibitor, remibrutinib.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Remibrutinib, a potent oral covalent BTK inhibitor with a favorable safety profile, shows convincing blood and tissue PDs in skin and is a promising drug development candidate for allergy‐ or antibody‐driven diseases.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
This study demonstrates the importance of PK and PD evaluation in dose selection for treatments such as remibrutinib, which is currently being investigated in phase II trials for diseases, such as chronic spontaneous urticaria (NCT03926611) and Sjögren’s syndrome (NCT04035668).
INTRODUCTION
Despite recent progress, significant medical need remains in the treatment of several autoimmune, allergic, and inflammatory conditions. Because of its central role in B‐cell receptor (BCR) signaling and pro‐inflammatory Fc‐receptor function, Bruton’s tyrosine kinase (BTK) inhibition has recently emerged as an attractive approach for selective immune modulation. 1 , 2 BTK is a protein tyrosine kinase of the TEC kinase family and expressed in selected cells of the adaptive and innate immune system, including B cells, macrophages, mast cells, basophils, and thrombocytes. 1 , 3 , 4 The role of BTK in regulating BCR signaling strength is the rationale to explore BTK inhibitors in B‐cell‐driven diseases, such as rheumatoid arthritis, Sjögren’s syndrome, and systemic lupus erythematosus (SLE), as well as B‐cell lymphomas, like chronic lymphocytic leukemia, leading to the approval of ibrutinib, acalabrutinib, and zanubrutinib by several regulatory agencies, including the US Food and Drug Administration. 5 , 6 BTK is also an indispensable downstream effector for signaling through the high‐affinity immunoglobulin‐E (IgE) receptor, FcεRI. 3 , 7 Inhibition of IgE‐mediated allergic skin reaction has been clinically demonstrated with ibrutinib, validating BTK as a potential target for drug development in asthma, atopic dermatitis, and chronic spontaneous urticaria (CSU). 8 , 9
Remibrutinib (LOU064) is an oral selective, covalent BTK inhibitor with a potential best‐in‐class profile. 10 , 11 Here, we explore the safety, pharmacokinetics (PKs), and pharmacodynamics (PDs) of remibrutinib for the first time in healthy human subjects, including subjects with atopic diathesis (clinicaltrials.gov: NCT03918980).
METHODS
Study design
The aim of this first‐in‐human, single center (Parexel EPCU Berlin), randomized, double‐blinded study (conducted between August 18, 2016, and June 27, 2018) was to assess the safety and tolerability, PK, and blood and tissue PD of single and multiple (q.d./b.i.d.) doses of remibrutinib oral administration. The study was conducted in three separate parts: a set of single ascending dose (SAD) cohorts and a food effect (FE) cohort in healthy subjects, and a set of multiple ascending dose (MAD) cohorts in healthy subjects with asymptomatic atopic diathesis (Figure 1). All cohorts were fasted with the exception of the FE cohort. Randomization of subjects was done by Novartis Drug Supply Management using a validated automated system for random assignment of treatment arms. Assignment of randomization numbers to subjects was done by investigator staff at Parexel EPCU Berlin. Subjects, investigator staff, persons performing assessments, and data analysts remained blinded to study treatments. The study protocol and all amendments were reviewed by an independent ethics committee, and the study was conducted according to International Conference on Harmonization (ICH) E6 Guideline for Good Clinical Practice that has their origin in the Declaration of Helsinki.
FIGURE 1.
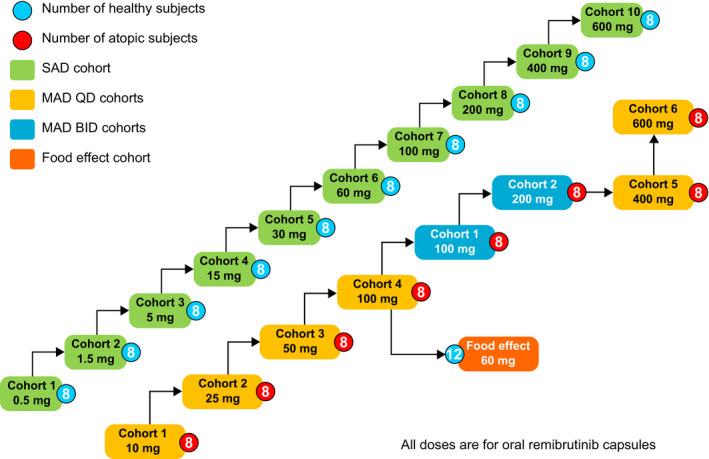
Study design of the first‐in‐human assessment of safety, PK and PD effects of remibrutinib. MAD, multiple ascending dose; PD, pharmacodynamic; PK, pharmacokinetic; SAD, single ascending dose
The SAD part was a randomized (3:1) double‐blind, placebo‐controlled, dose‐escalation study in 10 cohorts of 8 subjects each (remibrutinib at doses of 0.5, 1.5, 5, 15, 30, 60, 100, 200, 400, and 600 mg). With a sample size of 6 subjects receiving remibrutinib per cohort, there was at least 80% probability for an adverse event (AE) with an underlying occurrence rate of 24% or higher to be observed at least once for the given dose. Eligible subjects received a single dose of remibrutinib or placebo under fasting conditions. MAD cohorts were randomized (3:1), double‐blind, placebo‐controlled, dose‐escalations with 8 subjects in each cohort. Six cohorts had a q.d. schedule (remibrutinib doses of 10, 25, 50, 100, 400, and 600 mg; 12 doses over 12 days), and two cohorts had b.i.d. dosing (remibrutinib 100 and 200 mg, 23 doses over 12 days). Each subject was followed for safety and PK/PD assessments for 21 days postdose. A complete and blinded safety assessment of all participants was conducted by the investigator and the sponsor before escalating to a higher dose. The lowest MAD dose cohort was initiated after the 200 mg SAD cohort was confirmed safe.
The FE cohort was a single dose, open‐label, randomized, two‐way, cross‐over study arm to investigate the PK of remibrutinib 60 mg under fasted and fed conditions. Each subject participated in 2 treatment periods, fed and fasted, separated by a washout period of 18 (±1) days. In total, six amendments were introduced to the protocol, of which three amendments were considered important. The b.i.d. dosing regimen was added to the study design considering the safety summary from the SAD cohorts (May 30, 2017). The highest investigated dose was revised from 300 mg to 400 mg (August 11, 2017) and then to 600 mg (January 26, 2018), respectively, based on emerging safety and PK data.
Study population
Healthy male and female subjects, aged 18–65 years, were recruited for the study determined by medical history, physical examination, vital signs, electrocardiogram (ECG), and laboratory tests at screening, with a body mass index of 18–30 kg/m2. Subjects with a history of bleeding or a positive Hemoccult test were excluded. For the MAD cohorts, healthy subjects with asymptomatic atopic diathesis were recruited if they showed a positive skin prick test (SPT) to a known allergen at screening.
Subjects were excluded from the study if they had used other investigational drugs within 5 (terminal) half‐lives or 30 days (whichever was longer) before enrollment. Any history of hypersensitivity to any of the study drugs or to drugs of similar chemical classes, long QT syndrome, clinically significant ECG abnormalities, clinically significant arrhythmias, and use of concomitant medications were criteria for exclusion. Women of childbearing potential, including pregnant or lactating women, were also excluded.
Study end points
The primary end point of the study was assessment of the safety and tolerability of single and multiple ascending oral doses of remibrutinib via physical examination and medical history, vital signs, ECG, laboratory assessments, and reported AEs and serious AEs (SAEs). Secondary end points included assessing blood PKs of single and multiple doses of remibrutinib. Exploratory end points included assessment of PDs of single and multiple doses of remibrutinib.
Assessments
Pharmacokinetics
For SAD cohorts, blood collection for PK analysis was scheduled on the day of dosing at predose and at predefined timepoints, as specified in the protocol, up to day 8. Similarly, for MAD cohorts, blood samples were collected at predose and predefined timepoints up to 24 h on day 1, and on subsequent days at trough and 2 h postdose. On day 12 (full profile) of the MAD cohorts, the blood collection schedule was similar to that on the first day of dosing, and at 36, 48, 72, 96, and 168 h after the last dose. MAD cohorts (b.i.d.) samples were collected in the same way up to 12 h. Respective urine samples were collected to probe for direct renal excretion of remibrutinib. Remibrutinib blood concentrations were determined by a validated liquid chromatography‐tandem mass spectrometry analysis. A summary of the PK methodology is given in Appendix S1.
Pharmacodynamics
BTK occupancy
The primary PD response to remibrutinib was assessed by measuring BTK occupancy by remibrutinib in blood, as described previously with some modifications. 11 , 12 A summary of the BTK occupancy assay is described in Appendix S1. Free (unoccupied) and total BTK were measured by immunoassays on the Meso Scale Discovery platform.
Basophil activation
For distal mechanistic PD response, basophil activation was assessed using a modified commercially available kit (BASOTEST; Glycotope Biotechnology, Germany). Briefly, heparinized whole blood collected at predetermined timepoints was stimulated ex vivo with an anti‐IgE for 25 minutes at 37°C/5%/CO2 (incubator). Activation was evaluated by monitoring the percentage of CD63+ and CD203c+ basophils using flow cytometry. Results are expressed as percentage inhibition relative to baseline; data from subjects with CD63+ basophil stimulation values less than 10% at baseline were excluded.
Skin prick test
In subjects with atopic diathesis in the MAD cohorts, repeated SPTs to known allergens determined from a battery of 7 undiluted allergens (grass pollen mix, birch tree pollen, alder tree pollen, hazelnut tree pollen, house dust mite, cat dander, and horse dander) were performed prior to any dosing at baseline (day −1), at day 1 (predose, and 2 and 8 h postdose), day 2 (predose), and around the last dose at day 12 (predose and 8, 24, and 96 h postdose) for assessing PD effects in the skin. The difference in wheal diameter determined by the average of the longest diameter of a wheal and the maximal diameter perpendicular to the first one between the two predose (average of day −1 and day 1 predose) and all postdose (average of all 7 measurements) tests was assessed.
Safety
Safety assessments consisted of collecting all AEs and SAEs, and their severity and relationship to the study drug. Safety assessments included the regular monitoring of hematology, blood chemistry and coagulation, urine analysis, vital signs, physical examination, 12‐lead ECGs, 24‐h 12‐lead Holter ECG, meal record, and bodyweight performed at the study center. Each subject was also followed up with a bruising log for potential bleeding events.
Statistical analysis
Data were analyzed grouped by cohort and dose group. Subjects on placebo were pooled separately from the SAD and MAD cohorts. The safety analysis set consisted of all subjects who received any study drug. The dose‐exposure relationship was assessed using a regression of log‐transformed peak plasma concentration (Cmax) and area under the curve (AUC) parameters versus log‐transformed dose and an estimate of the slope together with a 90% confidence interval (CI). For FE, point estimate ratio of geometric means (fed/fasted) were provided together with a 90% CI back‐transformed from log‐scale. The PK analysis set consisted of all subjects with available PK data and no protocol deviations with relevant impact on PK data. The PD analysis set consisted of all subjects with available PD data and no protocol deviations with relevant impact on PD data. No formal statistical hypotheses for safety or tolerability were tested for this study, and descriptive summary statistics were provided for blood and urine PK parameters.
RESULTS
Subject demographics
Of 156 healthy subjects who participated in the 3 study parts, 80 were randomized to the 10 cohorts of the SAD part (remibrutinib n = 60, placebo n = 20), 64 to the 6 cohorts of the MAD q.d. part and 2 cohorts of the MAD b.i.d. part (remibrutinib n = 48, placebo n = 16), and 12 participated in the FE cohort. All analysis sets included all participating subjects, except for subjects on placebo who were not included in the PK analysis set. Baseline characteristics of subjects by investigational cohorts are given in Table 1. No discontinuations were reported from any study arm.
TABLE 1.
Baseline characteristics of subjects by cohorts
| SAD cohorts | MAD cohorts | Food effect cohort | |||
|---|---|---|---|---|---|
|
Placebo N = 20 n (%) |
Remibrutinib N = 60 n (%) |
Placebo N = 16 n (%) |
Remibrutinib N = 48 n (%) |
Remibrutinib N = 12 n (%) |
|
| Age in years, median (range) | 46 (22–63) | 51 (18–64) | 46.5 (23–59) | 37 (23–63) | 39 (22–65) |
| Males, n (%) | 15 (75.0) | 44 (73.3) | 12 (75.0) | 47 (97.9) | 9 (75.0) |
| Race, n (%) | |||||
| White | 20 (100) | 58 (96.7) | 16 (100) | 47 (97.9) | 10 (83.3) |
| Asian | – | 2 (3.3) | – | 1 (2.1) | 1 (8.3) |
| Black | – | – | – | – | 1 (8.3) |
| Ethnicity, n (%) | |||||
| Non‐Hispanic or Latino | 19 (95.0) | 59 (98.3) | 16 (100) | 48 (100) | 11 (91.7) |
| Hispanic or Latino | 1 (5.0) | 1 (1.7) | – | – | 1 (8.3) |
| Weight in kg, mean (SD) | 74.0 (11.43) | 77.9 (10.23) | 76.5 (13.08) | 79.9 (10.34) | 82.9 (14.77) |
| Height in cm, mean (SD) | 174.4 (9.91) | 174.8 (8.30) | 177.9 (9.00) | 179.4 (7.79) | 178.0 (7.45) |
| BMI, kg/m2, mean (SD) | 24.2 (2.33) | 25.5 (2.81) | 24.0 (2.36) | 24.8 (2.56) | 26.0 (2.93) |
Abbreviations: BMI, body mass index; MAD, multiple ascending dose; SAD, single ascending doses.
Pharmacokinetics
Absorption of remibrutinib was fast (median time to maximum concentration [Tmax]: 0.5–1.25 h for all tested doses Table S1). At low doses (<30 mg), remibrutinib blood levels rapidly fell below the lower limit of quantification (1 ng/mL) compromising the determination of terminal half‐life. Most of the compound was cleared under the initial elimination phase with an apparent half‐life of 1–2 h. The terminal elimination phase only became apparent at doses greater than or equal to 100 mg (Figure 2a). For the remibrutinib dose range tested, an apparent blood clearance of 280‒560 L/h and apparent volume of distribution of 400‒15,000 L was observed. Dose proportionality over the full dose range was not demonstrated for Cmax, AUCinf, or AUClast (Figure 3a). However, in the pharmacologically relevant range (10–100 mg), a dose‐dependent increase in exposure was observed. Details of PK of the SAD cohorts are given in Table S1.
FIGURE 2.
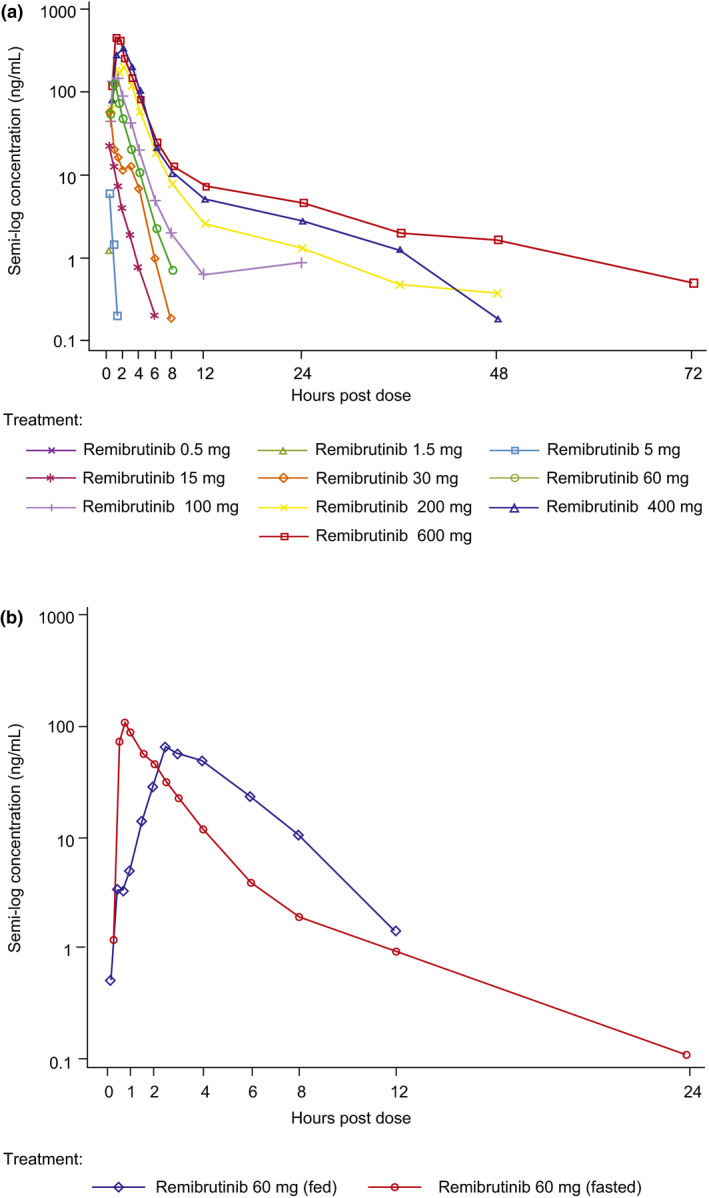
Pharmacokinetics and pharmacodynamics of remibrutinib after single oral administration. (a) Blood concentration‐time profiles of single dose remibrutinib 0.5–600 mg over 72 h. (b) Blood concentration‐time profiles of remibrutinib 60 mg given fasted versus fed. BTK, Bruton’s tyrosine kinase; SAD, single ascending dose
FIGURE 3.
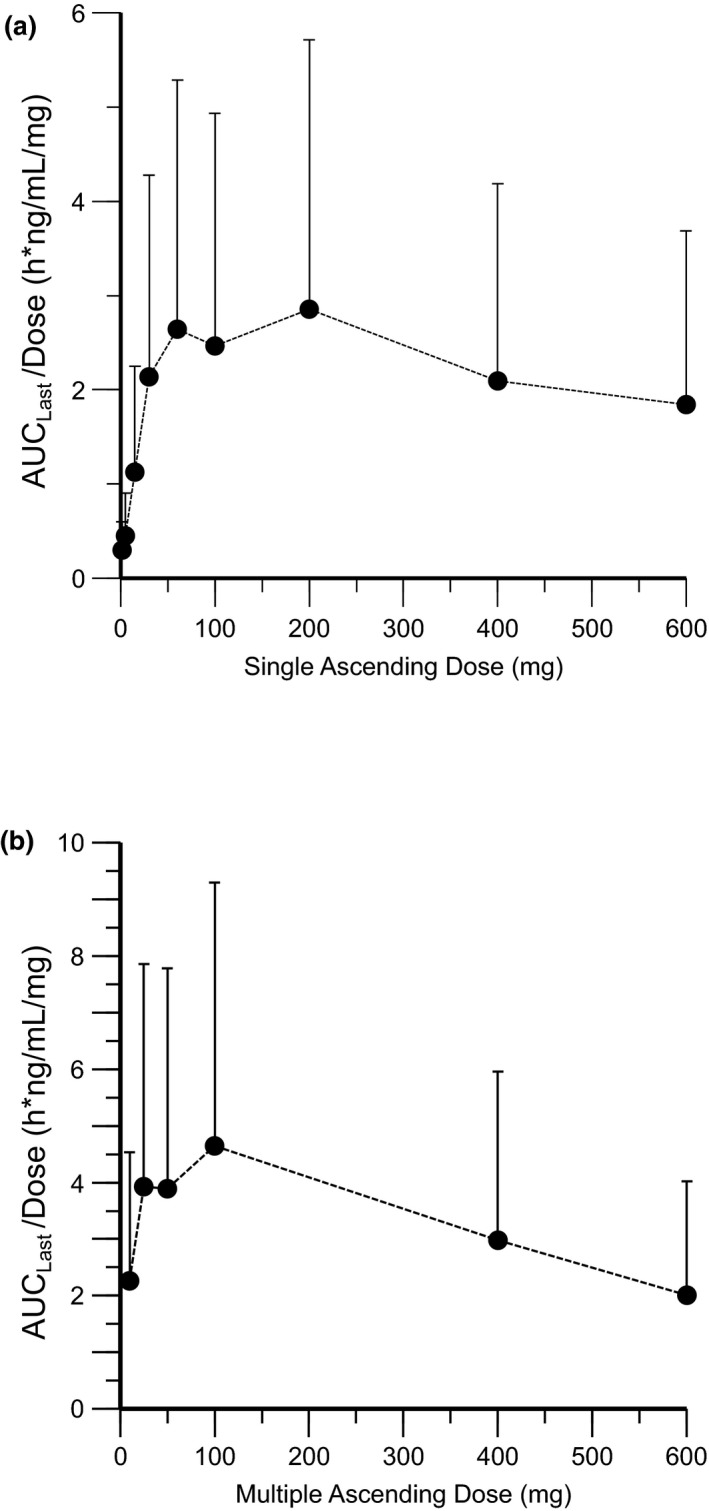
Dose‐exposure relationship of remibrutinib for dose normalized AUClast (Geo‐mean ±SD). (a) Single ascending dose cohorts, and (b) multiple ascending dose cohorts (q.d. only). AUC, area under the curve
In the MAD cohorts, remibrutinib absorption and elimination was fast (median Tmax: 0.5–1.5 h for q.d./b.i.d. doses). Blood concentrations declined rapidly, resulting in a mean residence time (MRT) of ~3 h. There was no marked accumulation with b.i.d. dosing and steady‐state was reached as early as the third day of dosing. In line with SAD results, dose proportional increases of Cmax and AUC were not observed over the full dose range (Figure 3b). A small increase in Cmax and AUC after q.d. and b.i.d. dosing (Table 2) was observed at steady‐state. Direct renal excretion of remibrutinib was low and represented a negligible contribution (~1%) to the total apparent blood clearance across doses in all MAD cohorts.
TABLE 2.
Summary statistics of MAD cohorts (100–600 mg) PK parameter values at days 1 and 12
| Remibrutinib MAD cohorts | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 100 mg q.d. | 400 mg q.d. | 600 mg q.d. | 100 mg b.i.d. | 200 mg b.i.d. | ||||||
| Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | Day 1 | Day 12 | |
| Cmax, ng/mL | 187 (85.0) | 233 (84.1) | 518 (89.1) | 551 (263) | 550 (87.6) | 563 (229) | 180 (83.4) | 306 (202) | 236 (134) | 347 (112) |
| Tmax, h | 0.742 (0.50‒1.95) | 0.867 (0.73‒1.50) | 0.750 (0.50‒1.50) | 0.758 (0.70‒1.50) | 0.750 (0.50–3.00) | 0.883 (0.50‒3.00) | 1.50 (0.47–2.97) | 0.775 (0.50–2.00) | 0.875 (0.50–1.52) | 0.992 (0.52–2.50) |
| AUClast, h*ng/mL | 311 (89.1) | 488 (172) | 973 (379) | 1300 (602) | 1080 (377) | 1240 (341) | 334 (146) | 518 (334) | 464 (211) | 963 (439) |
| AUC0–24 h, h*ng/mL | 315 (91.5) | 485 (179) | 977 (378) | 1280 (577) | 1080 (377) | 1230 (356) | – | – | – | – |
| AUC0–12 h, h*ng/mL | – | – | – | – | – | – | 337 (147) | 496 (306) | 464 (211) | 890 (416) |
| T1/2, h | – | 1.41 (1.41‒11.9) | – | 8.51 (1.22‒22.3) | – | 8.29 (4.69‒17.3) | – | 2.84 (2.15–18.9) | – | 12.4 (2.26–26.3) |
| T1/2,acc, h | – | 1.01 (1.00–1.73) | – | 1.18 (1.00–1.90) | – | 1.16 (1.03–1.62) | – | – | – | – |
| CLss/F, L/h | – | 198 (88.7) | – | 366 (150) | – | 521 (131) | – | 255 (153) | – | 267 (126) |
| MRT, h | 2.79 (1.28) | – | 3.02 (1.11) | – | 3.14 (0.58) | – | – | – | – | – |
Data are presented as mean (SD). For Tmax, T1/2, and T1/2,acc, data are presented as median (minimum‐maximum).
Abbreviations: AUC, area under the curve; CLss/F, apparent total body clearance of drug from blood at steady state; Cmax, maximum concentration; inf, infinity; MAD, multiple ascending dose; MRT, mean residence time; PK, pharmacokinetic; T1/2, terminal half‐life; T1/2,acc, the effective half‐life determined based on drug accumulation ratio at steady‐state; Tmax, time to reach maximum drug concentration in blood.
After food intake of a standardized high‐fat breakfast, the PK parameters of remibrutinib were only slightly affected compared with subjects who were assessed under fasted conditions. Although Cmax showed a trend to slightly decrease under fed conditions (Cmax‐fed/Cmax‐fasted: 0.801; 90% CI, 0.638–1.01), a slight increase in AUClast (AUClast‐fed/AUClast‐fasted: 1.30; 90% CI, 1.14–1.47) was observed (Table S2). The mean ±SD MRT for remibrutinib was higher on fed (4.17 ± 1.20 hours) compared with fasted conditions (2.29 ± 0.72 h). A notable change was observed for the median Tmax of remibrutinib, which was delayed by ~2 h (fasted: 0.767 h; fed: 2.78 h; Figure 2b and Table S3).
Pharmacodynamics
In the SAD cohorts, no BTK occupancy was observed in blood at the lowest dose of 0.5 mg. At doses greater than or equal to 5 mg, BTK occupancy increased within 30 min in a dose‐dependent fashion and maintained some level of BTK‐occupancy for several days. At doses of greater than or equal to 30 mg, complete BTK occupancy (>95%) was observed for at least 24 h and declined after 48 h (Figure 4a).
FIGURE 4.
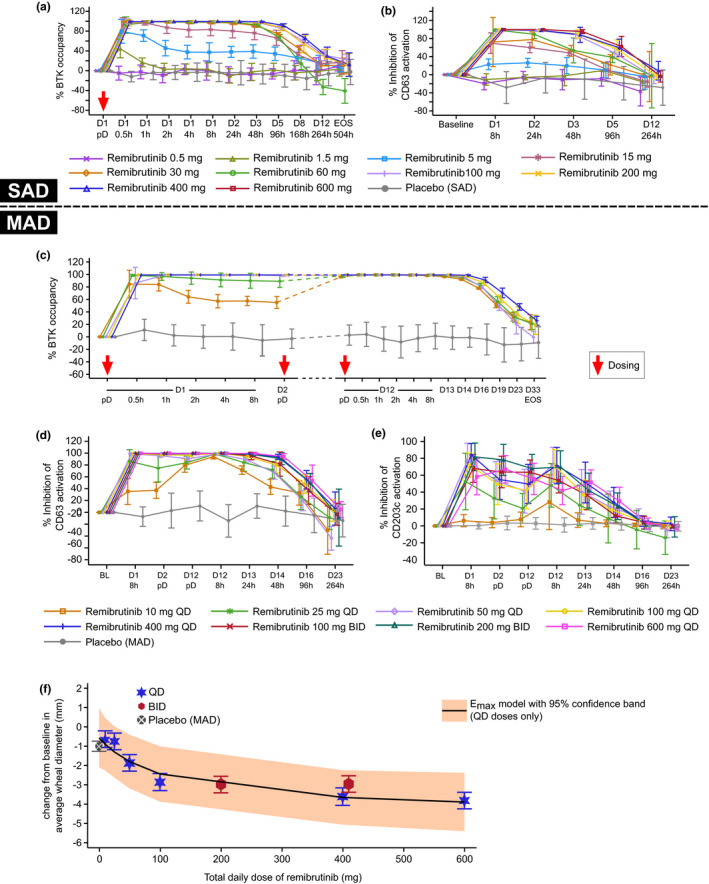
Remibrutinib pharmacodynamics in SAD and MAD cohorts. (a) BTK occupancy over complete study duration in the SAD cohorts, (b) basophil inhibition assessed by CD63 expression over 12 days postdose in the SAD cohorts, (c) PD of remibrutinib after multiple oral doses (10–400 mg q.d.) over 12 days, (d) inhibition of basophil activation by CD63 expression in the MAD cohorts, (e) inhibition of basophil activation by CD203c expression in the MAD cohorts, and (f) change from baseline in average wheal size after SPT in subjects with atopic diathesis (MAD‐part). BL, baseline; BTK, Bruton’s tyrosine kinase; D, day; Emax maximum effect model, nonlinear model; MAD, multiple ascending dose; pD, predose; PD, pharmacodynamic; SAD, single ascending dose; SPT, skin prick test
Inhibition of ex vivo anti‐IgE activated basophils was observed with an activation marker‐specific kinetics and magnitude. Dose‐dependent inhibition of the CD63 expression was observed starting from a single dose of 5 mg. Close to 90% basophil inhibition was achieved with remibrutinib 60 mg after 24 h, and doses greater than or equal to 100 mg demonstrated almost complete inhibition (Figure 4b). With remibrutinib doses of 100 mg and higher, basophil inhibition was sustained for 24 h and longer, declining to near baseline values after 12 days (not shown). The other basophil activation marker, CD203c, showed inhibition up to 79.4% at 8 h in the 200 mg cohort.
In the MAD cohorts, complete BTK occupancy (>95%) was achieved within 30 min of administration of the first dose of greater than or equal to 25 mg. At the time of the last dose at day 12, all doses (including the 10 mg q.d. dose) achieved complete BTK occupancy at the predose timepoint; this was sustained for at least 24 h after the last dose and exhibited gradual decline over several days thereafter. Doses greater than or equal to 50 mg maintained complete BTK occupancy for the measured dosing period and declined 48 h after the last dose (Figure 4c).
Full inhibition of blood basophil activation measured by CD63 expression was achieved over the entire treatment period (day 1–12), and several days beyond, with a daily remibrutinib dose of greater than or equal to 50 mg (Figure 4d). With multiple doses, trough level inhibition of CD63 on the last day of dosing was 80% with the lowest tested dose of 10 mg q.d.; this was stronger with any higher dose. Dose‐dependent inhibition was observed with CD63 marker reaching greater than 97% trough inhibition with greater than or equal to 100 mg q.d. at the last day of dosing. Inhibition of CD203c+ basophils was highest in the 400 mg q.d. and 200 mg b.i.d. cohorts, peaking at 84% and 81.8% at 8 h, respectively, on day 1. Maximum trough inhibition of CD203c at day 12 was achieved with 100 and 200 mg b.i.d. remibrutinib at 63% and 67%, respectively (Figure 4e).
SPT analysis with undiluted allergen was used in all MAD cohorts; dose‐dependent decrease in wheal size was observed up to 100 mg dose. Beyond 100 mg, a dose‐dependent trend was unclear, although the highest decrease was observed with the highest tested dose (600 mg q.d.). Across all tests, participants showed an average ~3 mm decrease in wheal size at doses of greater than or equal to 100 mg q.d. (Figure 4f). There was no indication for a stronger decrease in wheal size with b.i.d. versus q.d. dosing.
Safety and tolerability
No clinically significant changes in hematology, biochemistry, or urinalysis parameters were reported, nor were any clinically significant changes in vital signs and ECG parameters (Table 3). In the SAD cohorts, 20 subjects (25%) experienced at least 1 AE; 17 received remibrutinib (28.3%) and 3 received placebo (15.0%). The most common AEs reported were headache (8.8%) and nasopharyngitis (6.3%). Eighteen subjects reported AEs with mild severity (22.5%) and two (2.5%) with moderate severity. No SAE was reported in any SAD cohort. No AEs were reported in the 100 and 200 mg cohorts. The incidence of AEs reported in all other cohorts was balanced and was not dose‐dependent. Two mild (nasopharyngitis and skin irritation) and 1 moderate (headache) AEs were reported in 3 of 20 subjects on placebo. AEs in 7 (all on remibrutinib) of the 80 subjects in the SAD cohorts were suspected to be related to the study drug by the investigator. In the MAD cohorts, 32 of 64 subjects experienced at least 1 AE; 24 were on remibrutinib and 8 on placebo (incidence of AEs 50.0% each). Similar to the SAD cohorts, the 2 most common AEs experienced by the subjects were headache (9.4%) and nasopharyngitis (6.3%). Altogether, 28 (43.8%) subjects reported AEs of mild severity, 8 (12.5%) reported AEs of moderate severity (headache, nausea, toothache, back pain, and catheter site phlebitis), and 1 (1.6%) reported an SAE (syncope; in 25 mg dose group), which was not considered related to the study medication by the investigator (Table 3). A total of 8 of 64 (12.5%) subjects in the MAD cohorts were reported with AEs suspected to be related to the study drug by the investigator (remibrutinib, 5/48 [10.4%]; placebo 3/16 [18.8%]). Of the 12 subjects in the FE cohort, 7 (58.3%) experienced at least 1 AE. A higher incidence of AEs was reported in subjects under fed versus fasted condition (50% vs. 16.7%), with headache the most common AE (2/12 [16.7%]); all AEs were mild or moderate. In the context of the low severity and the safety data seen in the MAD cohort, with up to 10‐times higher doses given over several days, this difference was not considered clinically meaningful.
TABLE 3.
Incidence of AEs by preferred term and their severity and causality for all doses within different cohorts (IR ≥5% in any cohort)
| SAD cohorts a | MAD cohorts b | Food effect cohort c | |||
|---|---|---|---|---|---|
|
Placebo N = 20 n (%) |
Remibrutinib N = 60 n (%) |
Placebo N = 16 n (%) |
Remibrutinib N = 48 n (%) |
Remibrutinib N = 12 n (%) |
|
| Subjects with at least 1 AE | 3 (15.0) | 17 (28.3) | 8 (50.0) | 24 (50.0) | 7 (58.3) |
| Headache | 1 (5.0) | 6 (10.0) | 1 (6.3) | 5 (10.4) | 2 (16.7) |
| Nasopharyngitis | 1 (5.0) | 4 (6.7) | 1 (6.3) | 3 (6.3) | 1 (8.3) |
| Toothache | – | – | 2 (12.5) | 1 (2.1) | – |
| AE severity | |||||
| Mild | 2 (10.0) | 16 (26.7) | 6 (37.5) | 22 (45.8) | 7 (58.3) |
| Moderate | 1 (5.0) | 1 (1.7) | 3 (18.8) | 5 (10.4) | 1 (8.3) |
| Severe | – | – | – | 1 (2.1) | – |
| Study drug‐related AEs | – | 7 (11.7) | 3 (18.8) | 5 (10.4) | 3 (25.0) |
| SAEs | – | – | – | – | – |
| AEs leading to discontinuation | – | – | – | – | – |
| Death | – | – | – | – | – |
Other AEs with n (%) IR ≥5% included.
Abbreviations: AE, adverse event; IR, incident rate; MAD, multiple ascending dose; SAD, single ascending doses; SAE, serious adverse event.
Skin irritation in placebo group at 1 (5.0).
Dysphonia, oral herpes, sleep disorder, agitation, dizziness, hot flush, rash, and rhinitis in placebo group at 1 (6.3) each.
Alopecia areata, hematoma, myalgia, nasopharyngitis, orthostatic hypotension, pain in extremity, palpitations, and phlebitis at 1 (8.3) each.
All subjects were carefully investigated for any potential bleeding events using a bruising log. Two subjects in the 600 mg MAD group had an event of self‐limiting epistaxis not requiring any specific medical intervention; both had a history of repeated epistaxis. Furthermore, two hematomas (1 after an adequate trauma and 1 after a vessel puncture) were reported, both occurring in the 100 mg MAD cohort; both resolved spontaneously.
Overall, remibrutinib was well‐tolerated at all evaluated doses. The most frequently reported AEs suspected to be related to study medication were headache and diarrhea. No clinically significant changes were observed in the hematology, biochemistry, or urinalysis parameters. No SAEs or deaths were reported, and no AEs or dose‐limiting toxicity led to study treatment discontinuation in any cohort (Table 3).
DISCUSSION
Apart from the established use in hematological malignancies, inhibition of BTK is being actively explored as a promising new pharmacological approach for the treatment of various autoimmune, allergic, and inflammatory conditions, including CSU, rheumatoid arthritis, Sjögren’s syndrome, SLE, and multiple sclerosis. 13 , 14 , 15 , 16 Remibrutinib belongs to a new generation of designed covalent enzyme inhibitors and it shows potentially best‐in‐class selectivity and high potency; therefore remibrutinib may have fewer off target effects versus earlier BTK inhibitors, such as ibrutinib. 10 , 11 Recently, the hypothesis that BTK inhibition may be relevant in allergic or atopic skin pathology was verified by testing allergic subjects before and after initiation of treatment with the BTK inhibitor ibrutinib. 8 , 9
The benefit–risk ratio of covalent enzyme inhibitors strongly depends on their PK/PD properties requiring balance of (i) high selectivity to the target and (ii) fast target inactivation kinetics. Remibrutinib demonstrates a favorable profile as it has a fast absorption (Tmax <1.5 h) and an observed apparent blood clearance leading to a short MRT (<3 hours), limiting duration of systemic exposure and ensures steady state achievement with less than 5 doses and minimal accumulation. Due to covalent binding to its target, remibrutinib may provide a potential advantage over reversible BTK inhibitors, because it is expected to maintain full BTK‐inhibition over the dosing interval.
Remibrutinib showed no dose‐proportional exposure over the full dose range, and it is assumed that multiple factors affect the dose exposure response, in addition to contributing to the time dependence of PK parameters. A major contribution is probably due to the covalent binding of remibrutinib to its target, which leads to the overall high apparent clearance, especially at low doses and at the start of multiple dosing. As saturation of systemic blood and tissue bound BTK is reached with increasing doses of remibrutinib, the impact of covalent binding may become negligible. In addition, the low solubility of remibrutinib may contribute to an under‐proportional dose‐exposure relationship with increasing doses, as the extent of absorption (fa) becomes the limiting factor. Further, time‐dependent auto‐inhibition of CYP3A4 may counterbalance solubility and contribute to time dependence of PK parameters, such as clearance. The same applies to auto‐induction, which is expected to reduce exposure at the highest doses as observed in animal studies. The interplay of these parameters leads to a complex PK profile and dose‐exposure relationship, with maximum impact of individual factors observed at different doses.
Despite the short residence time, the PD effect of remibrutinib in blood (BTK occupancy and basophil inhibition) is sustained much longer due to the covalent binding to the target. Short systemic exposure of remibrutinib was sufficient for blood PD effects lasting more than 24 h and may limit potential off‐target effects. This represents a desired drug profile and should allow q.d./b.i.d. dosing despite a drug residence time of less than 3 h. The duration of PD effects for a covalent BTK inhibitor is, however, determined by the replenishment of inactivated BTK protein by newly synthesized protein, which has been shown to range from 20 to 40% after 24 h. 13 , 17 , 18 The BTK occupancy in blood was near complete for more than 24 h with a remibrutinib dose of greater than or equal to 30 mg q.d., demonstrating a potent PD response. The almost complete BTK occupancy observed for doses as low as 10 mg between days 1 and 12 in the MAD cohorts suggests sustained BTK saturation with continuous q.d. dosing. However, tissue PD effects may require higher doses, as evidenced by the higher doses required for a full or maximal effect on basophils via CD63 and CD203c inhibition and the partial inhibition of the SPT. Evobrutinib, a different covalent BTK inhibitor, also shows complete blood BTK saturation. However, the effect was achieved at higher doses. 19
BTK‐occupancy in blood and basophil inhibition assessed via CD63 showed a sustained effect for higher doses over 24 h after q.d. dosing. For fenebrutinib, more than 90% CD63 inhibition was not sustained beyond 8 h with doses greater than or equal to 200 mg, probably due to reversible BTK inhibition resulting in declining PD with decreasing drug concentration. 16 Consequently, unlike remibrutinib, persistent systemic exposure of reversible inhibitors would be required for sustained inhibition of BTK. Basophil inhibition by remibrutinib was also assessed via CD203c inhibition, demonstrating stronger effects during b.i.d. dosing, possibly because CD203c is a basophil lineage marker with kinetics different from the CD63 degranulation marker. 20
As discussed above, PD response is driven by BTK turnover. Therefore, maximal PD effects may likely be achieved with repeated dosing during the day rather than by increasing a q.d. dose. Because the kinetics of BTK‐remibrutinib complex in tissues are unknown, it would be rational to test a b.i.d. regimen for a PD effect required for an entire day. Remibrutinib administered in a fed state led to a small decrease in Cmax and an increase in Tmax and AUC. However, the relatively small increase of AUC observed in the fed state with a reduction of Cmax is within the intersubject concentration range seen in the fasted state and may be clinically inconsequential.
The reduction of wheal size in response to SPT in subjects with atopic diathesis indicates a good distribution of remibrutinib in the skin and suggests potential implications for patients with basophil‐ and mast‐cell‐driven dermatological disorders, such as CSU. Consistent with the irreversible inhibition, sustained occupancy of BTK by remibrutinib was associated with strong and prolonged inhibition of basophil activation by CD63 marker. The PD response onset was dose‐dependent, with 30 mg being the lowest dose showing both complete BTK occupancy and almost complete basophil inhibition. In the MAD cohorts, both PD responses showed good association, peaking in parallel and remaining persistent at all doses. For SPT, there was a dose‐dependent response, which may almost saturate beyond the 100 mg dose, which is higher than that achieved by full BTK‐occupancy and CD63 suppression. Therefore, the doses tested in any dose‐ranging trial should cover the response range across all observed PD markers, including a 100 mg dose arm. The PD effects observed in skin may not be generalized to all tissue compartments because the duration and the magnitude of effect would be determined by the tissue distribution of remibrutinib, as well as the turnover of BTK in that particular tissue. Measuring BTK occupancy in tissue rather than whole blood would have been more informative and may be undertaken in subsequent studies.
Oral administration of up to 600 mg remibrutinib was well‐tolerated and was not associated with any clinical or laboratory safety signals of concern in healthy human subjects with or without atopic diathesis. Due to the established role of BTK in platelet biology, 6 specific assessments to detect an increased bleeding risk were implemented in this study, including a bruising log and coagulation testing. However, no increased risk of bleeding was observed with any dose of remibrutinib. Almost all AEs were mild or moderate in intensity. No dose‐limiting toxicity was observed, even with the highest dose tested. A further increase from the tested supratherapeutic doses was not required.
Overall, remibrutinib demonstrated a favorable safety profile and strong BTK inhibition in blood with evident effects in the skin. However, because the duration of the study drug dosing was limited to 12 days, no long‐term safety in humans can be inferred from the present data. Nevertheless, these data provide a rational basis for remibrutinib to be further explored for the treatment of patients with CSU, asthma, and B‐cell dependent diseases, such as Sjögren’s syndrome.
CONFLICT OF INTEREST
M.Ka., P.E., M.C., A.J., M.Ki., A.K., A.M., and B.C. are shareholders and employees of Novartis, the company which is developing remibrutinib as pharmacological treatment in relevant indications. C.S. is an employee of GCE Solutions, the Clinical Research Organization sponsored by Novartis for the analysis of the study data. A.S. and R.F. are employees of Parexel, the Clinical Research Organization sponsored by Novartis for the execution of this study.
AUTHOR CONTRIBUTIONS
M.Ka., P.E., and B.C. wrote the manuscript. M.Ka., M.C., C.S., A.J., and B.C. designed the research. M.Ki., A.K., A.M., A.S., and R.F. performed the research. C.S., A.K., and A.M. analyzed the data.
Supporting information
Supplement S1
Supplement S2
ACKNOWLEDGMENTS
Medical writing and editorial support were provided to the authors by Mohammad Fahad Haroon and Avishek Anant of Novartis Healthcare Pvt. Ltd. India, which was funded by Novartis Pharma AG in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Funding information
This study was funded by Novartis Pharma AG, Switzerland.
REFERENCES
- 1. Liang C, Tian D, Ren X, et al. The development of Bruton's tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini‐review. Eur J Med Chem. 2018;151:315‐326. [DOI] [PubMed] [Google Scholar]
- 2. Norman P. Investigational Bruton's tyrosine kinase inhibitors for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs. 2016;25:891‐899. [DOI] [PubMed] [Google Scholar]
- 3. Smiljkovic D, Blatt K, Stefanzl G, et al. BTK inhibition is a potent approach to block IgE‐mediated histamine release in human basophils. Allergy. 2017;72:1666‐1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rawlings DJ, Witte ON. The Btk subfamily of cytoplasmic tyrosine kinases: structure, regulation and function. Semin Immunol. 1995;7:237‐246. [DOI] [PubMed] [Google Scholar]
- 5. Aalipour A, Advani RH. Bruton tyrosine kinase inhibitors: a promising novel targeted treatment for B cell lymphomas. Br J Haematol. 2013;163:436‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feng Y, Duan W, Cu X, Liang C, Xin M. Bruton's tyrosine kinase (BTK) inhibitors in treating cancer: a patent review (2010–2018). Expert Opin Ther Pat. 2019;29:217‐241. [DOI] [PubMed] [Google Scholar]
- 7. Hata D, Kawakami Y, Inagaki N, et al. Involvement of Bruton's Tyrosine kinase in FcεRI‐dependent mast cell degranulation and cytokine production. J Exp Med. 1998;187:1235‐1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dispenza MC, Pongracic JA, Singh AM, Bochner BS. Short‐term ibrutinib therapy suppresses skin test responses and eliminates IgE‐mediated basophil activation in adults with peanut or tree nut allergy. J Allergy Clin Immunol. 2018;141:1914‐1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Regan JA, Cao Y, Dispenza MC, et al. Ibrutinib, a Bruton's tyrosine kinase inhibitor used for treatment of lymphoproliferative disorders, eliminates both aeroallergen skin test and basophil activation test reactivity. J Allergy Clin Immunol. 2017;140:875‐9 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gabizon R, London N. A fast and clean BTK inhibitor. J Med Chem. 2020;63:5100‐5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Angst D, Gessier F, Janser P, et al. Discovery of LOU064 (Remibrutinib), a potent and highly selective covalent inhibitor of Bruton’s Tyrosine kinase. J Med Chem. 2020;63(10):5102–5118. [DOI] [PubMed] [Google Scholar]
- 12. Pulz R, Angst D, Dawson J, et al. Design of potent and selective covalent inhibitors of Bruton’s tyrosine kinase targeting an inactive conformation. ACS Med Chem Lett. 2019;10:1467‐1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Haselmayer P, Camps M, Liu‐Bujalski L, et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J Immunol. 2019;202:2888‐2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Crawford JJ, Johnson AR, Misner DL, et al. Discovery of GDC‐0853: a potent, selective, and noncovalent Bruton's tyrosine kinase inhibitor in early clinical development. J Med Chem. 2018;61:2227‐2245. [DOI] [PubMed] [Google Scholar]
- 15. Montalban X, Arnold DL, Weber MS, et al. Placebo‐controlled trial of an oral BTK inhibitor in multiple sclerosis. N Engl J Med. 2019;380:2406‐2417. [DOI] [PubMed] [Google Scholar]
- 16. Cohen S, Tuckwell K, Katsumoto TR, et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: a randomized, double‐blind. Phase II trial. Arthritis Rheumatol. 2020;72:1435‐1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Watterson SH, Liu Q, Beaudoin Bertrand M, et al. Discovery of branebrutinib (BMS‐986195): a strategy for identifying a highly potent and selective covalent inhibitor providing rapid in vivo inactivation of Bruton’s tyrosine kinase (BTK). J Med Chem. 2019;62:3228‐3250. [DOI] [PubMed] [Google Scholar]
- 18. Evans EK, Tester R, Aslanian S, et al. Inhibition of Btk with CC‐292 provides early pharmacodynamic assessment of activity in mice and humans. J Pharmacol Exp Ther. 2013;346:219‐228. [DOI] [PubMed] [Google Scholar]
- 19. Becker A, Martin EC, Mitchell DY, et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration‐QT analysis of the novel BTK inhibitor evobrutinib in healthy volunteers. Clin Transl Sci. 2020;13(2):325‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. MacGlashan D Jr. Expression of CD203c and CD63 in human basophils: relationship to differential regulation of piecemeal and anaphylactic degranulation processes. Clin Exp Allergy. 2010;40:1365‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement S1
Supplement S2