

Abstract
Islatravir (MK‐8591) is a nucleoside analogue in development for the treatment and prevention of HIV‐1. Two phase 1 trials were conducted during initial evaluation of islatravir: rising single doses (Study 1) and rising multiple doses (Study 2) of oral islatravir in male and female participants without HIV (aged 18–60 years). Safety, tolerability, and pharmacokinetics of islatravir (plasma) and islatravir‐triphosphate (peripheral blood mononuclear cells) were assessed. In Study 1, 24 participants, assigned to 1 of 3 panels, received alternating single doses of islatravir in a fasted state from 5 mg to 400 mg, or placebo, over 3 dosing periods; a 30 mg dose was additionally assessed following a high‐fat meal. In Study 2, 8 participants per dose received 3 once‐weekly doses of 10, 30, or 100 mg islatravir or placebo in a fasted state. For each panel in both trials, 6 participants received active drug and 2 received placebo. Islatravir was generally well‐tolerated, with no serious adverse events or discontinuations due to adverse events. Islatravir was rapidly absorbed (median time to maximum plasma concentration 0.5 hours); plasma half‐life was 49–61 h; intracellular islatravir‐triphosphate half‐life was 118–171 h. Plasma exposure increased in an approximately dose‐proportional manner; there was no meaningful food effect. There was a modest degree of intracellular islatravir‐triphosphate accumulation after multiple weekly dosing. After single oral doses of islatravir greater than or equal to 5 mg, intracellular islatravir‐triphosphate levels were comparable to levels associated with efficacy in preclinical studies. These results warrant continued clinical investigation of islatravir.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
Current HIV treatment and prevention strategies have limitations, and novel agents that offer improved safety and tolerability, a high barrier to HIV resistance, and more convenient dosing regimens are required.
WHAT QUESTION DID THIS STUDY ADDRESS?
Two phase 1 studies in participants without HIV assessed safety and pharmacokinetics of rising single and multiple doses of oral islatravir, a nucleoside analogue, to support continued development for the treatment and prevention of HIV‐1 infection.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Islatravir was generally well‐tolerated at single doses up to 400 mg. Oral doses of islatravir greater than or equal to 10 mg resulted in intracellular peripheral blood mononuclear cell levels of the active form, islatravir‐triphosphate, comparable to those associated with antiviral efficacy in preclinical studies.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
These studies provide important safety and pharmacokinetic information about islatravir in adults without HIV, which will be used to support further clinical investigation of islatravir for the treatment and prevention of HIV‐1 infection.
INTRODUCTION
Advances in antiretroviral therapy (ART) have contributed significantly to the reduction in morbidity, mortality, and transmission of HIV‐1. 1 However, the global burden of HIV‐1 infection remains substantial, with an estimated 38 million people living with HIV‐1 in 2019 and ~1.7 million new infections and 690,000 HIV‐related deaths in the same year. 2 US and European guidelines recommend ART regimens that generally consist of 3 drugs from at least 2 drug classes for the treatment of HIV‐1 infection and 2‐drug regimens as pre‐exposure prophylaxis (PrEP) for prevention of HIV‐1. However, the advent of new agents with improved effectiveness against HIV‐1 is resulting in the emergence of more compact regimens for both the treatment and prevention of HIV‐1, including 2‐drug regimens for treatment and single agents for prophylaxis. 1 , 3 , 4 The recommended HIV‐1 treatment and prevention strategies rely on good adherence to daily administration; thus, agents with more favorable and extended dosing schedules would be of benefit. Novel agents with improved safety and tolerability profiles and a high barrier to the selection of resistance are needed, as existing agents can be associated with long‐term toxicities, as well as a potential for selection of drug‐resistant HIV‐1 variants. 1 , 3 , 5
Islatravir (MK‐8591) is a nucleoside analogue in development for the treatment and prevention of HIV‐1 infection. 6 Due to its novel structure, islatravir inhibits reverse transcriptase (RT) by multiple mechanisms, contributing to its high affinity for RT and high barrier to resistance. 7 , 8 , 9 , 10 , 11 In vitro, islatravir inhibits multiple strains of HIV‐1, including common nucleoside reverse transcriptase inhibitor (NRTI)‐resistant variants, and has a high barrier to the development of resistance. 7 , 12 , 13 , 14 In addition, islatravir has demonstrated robust antiviral efficacy in preclinical animal models of HIV‐1 infection, including HIV infection in humanized mice 15 and simian immunodeficiency virus (SIV) infection in rhesus macaques. 16 , 17
When administered enterally in a rhesus macaque model, islatravir was rapidly absorbed, and peak plasma concentrations occurred ~90 min postdose. 15 Islatravir is rapidly cleared from plasma, and adenosine deaminase appears to be an important contributor to islatravir metabolism. 18 Islatravir is taken up into cells, where it is converted to islatravir‐triphosphate (TP) by endogenous intracellular kinases; 12 preclinically, islatravir‐TP persists for long periods intracellularly (>72 h). 15 Given the intracellular persistence of islatravir‐TP, once‐weekly oral islatravir was tested in SIV‐infected rhesus macaques. An intracellular islatravir‐TP trough concentration (C168 h) of 0.53 pmol/106 cells was shown to result in rapid and robust declines in SIV RNA for at least 7 days. 17 In a model of HIV prophylaxis, weekly oral islatravir protected rhesus macaques from infection via an intrarectal SIV challenge. 19
The high antiviral potency of islatravir and favorable profiles for toxicity and pharmacokinetics in preclinical studies supported further evaluation of islatravir in humans. Here, we report the results of 2 phase I clinical trials in participants without HIV designed to assess the initial safety and pharmacokinetic profile of oral islatravir after rising single and multiple doses in humans, which were used to inform selection of doses in clinical trials enrolling participants living with HIV‐1.
METHODS
Study design
Study 1 (protocol MK‐8591–001) was a double‐blind, randomized, placebo‐controlled, 3‐period, 3‐panel, alternating panel, rising single‐dose study in participants without HIV (Figure 1a). The study included a washout interval of at least 27 days between periods for any given participant, and there was a minimum of 10 days preceding dose escalation between panels to fully assess safety. Islatravir was dosed as an oral suspension. The suspension vehicle alone served as the placebo.
FIGURE 1.
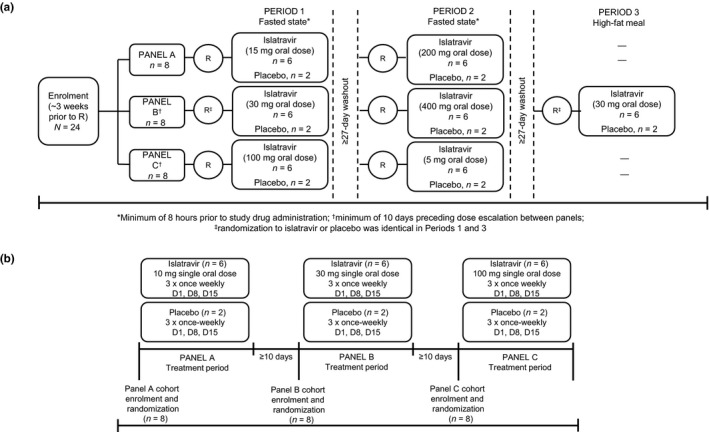
Study designs. (a) Study 1 design. The participants receiving placebo or active treatment varied by period, except for the 30 mg dose in which the same participants received placebo in periods 1 and 3 to allow for food effect assessment. High‐fat breakfast included 55.6 g total fat, 55 g total carbohydrate and 31.1 g total protein; total calories: 500.4 in fat, 220 in carbohydrates; and 124.4 in protein. R, randomization. (b) Study 2 design. D, day
Study 2 (protocol MK‐8591–002) was a double‐blind, randomized, placebo‐controlled, serial panel, rising multiple‐dose study in participants without HIV (Figure 1b). Islatravir or placebo were administered as a capsule once weekly for 3 weeks. Following administration of the final multiple dose of islatravir or placebo at each dose level, there was a minimum of 10 days preceding administration of the next higher dose in the subsequent panel.
In both studies, safety and pharmacokinetic data were reviewed on an ongoing basis and informed escalation to the next highest dose level. Individuals participated in only one panel. In all panels and throughout treatment periods, the participants and the investigator were blinded to the allocation of active treatment (islatravir) or placebo. Islatravir and matching placebo were prepared by an unblinded pharmacist and then dispensed in a blinded fashion by qualified trial site personnel. Participants were randomly assigned according to a computer‐generated allocation schedule. The number of participants in each panel (n = 8, allocated n = 6 to islatravir, and n = 2 to placebo) was chosen for these early phase I studies to balance scientific requirements for statistical significance with the trials’ objectives regarding the ethical use of human participants in a clinical study. For safety and tolerability analyses, data were pooled from participants in the placebo groups of each panel for both studies.
Both trials were conducted at the SGS Clinical Pharmacology Unit, Antwerp, Belgium, in conformance with applicable local requirements regarding ethical committee review, informed consent, and other statutes or regulations regarding the protection of the rights and welfare of humans participating in biomedical research. The Independent Ethics Committee for both studies was de Commissie voor Medische Ethiek – Ziekenhuisnetwerk Antwerpen (The Committee for Medical Ethics – Hospital Network Antwerp).
Participants
Eligible participants were men or women of nonchildbearing potential without HIV, 18 to 60 years of age, and with a body mass index less than or equal to 32 kg/m2 at the screening visit. Key exclusion criteria included creatinine clearance less than or equal to 90 ml/min (Study 1) or less than or equal to 80 ml/min (Study 2), a history of current significant abnormalities or diseases, history of tobacco use, and an inability to refrain from the use of any medication, including prescription and nonprescription, and herbal remedies.
Assessments
Safety and tolerability
Safety and tolerability were assessed by clinical evaluation, including vital signs, 12‐lead electrocardiograms (ECGs), and laboratory safety tests (hematology and blood chemistry) obtained at prespecified time points. Participants were also monitored for adverse events from the time the consent form was signed through 21 days following cessation of treatment.
Pharmacokinetics
Blood samples for the determination of plasma islatravir concentrations were collected predose and at specific time points up to ~96 h post dose in study 1 and up to ~168 h postdose (following dosing on days 7, 14, and 21) in study 2. Peripheral blood mononuclear cells (PBMCs) were collected for the assessment of pharmacokinetics for islatravir‐TP at specific time points up to ~336 h (~14 days) after the last dose of islatravir in both studies.
Islatravir was extracted from plasma by protein precipitation and analyzed by reversed phase chromatographic separation coupled with tandem mass spectrometric detection. The liquid chromatography‐tandem mass spectrometry system consisted of a Waters ACQUITY Ultra Performance Liquid Chromatography system (Waters Corporation, Milford, MA, USA) coupled with an AB Sciex API 5500 triple quadrupole mass spectrometer (AB Sciex, Framingham, MA, USA) with an electrospray ionization source. The lower limit of quantitation was 1.00 ng/ml (0.00341 µmol/L) with a linear calibration range from 1.00 ng/ml to 1000.00 ng/ml (0.00341 µmol/L to 3.41 µmol/L), using 50 μl of plasma. Assay intra‐run accuracy was between 97.5% and 112.0%, and precision was between 1.0% and 12.9% (n = 5).
Islatravir and islatravir‐TP from PBMC lysates were extracted using protein precipitation and analyzed by reversed phase chromatography for islatravir and ion exchange chromatography for TPs, coupled with tandem mass spectrometry. 20 Methanol was included in the lysis solvent to efficiently lyse all PBMCs and to stabilize anabolites from degradation. 20 The lower limit of quantitation was 0.25 ng/ml (0.000852 µmol/L) for islatravir in PBMCs, with a linear curve range from 0.25 ng/ml to 250.00 ng/ml (0.000852 µmol/L to 0.852 µmol/L). Assay accuracy was between 95.4% and 96.8%, and precision was between 1.5% and 3.5% (n = 3). The lower limit of quantitation was 0.50 ng/ml (0.000938 µmol/L) for islatravir‐TP in PBMCs, with a linear curve range from 0.50 ng/ml to 200.00 ng/ml (0.000938 µmol/L to 0.375 µmol/L). Assay accuracy for islatravir‐TP was between 94.0% and 99.3%, and precision was between 2.7% and 7.7% (n = 3).
Statistical analyses
Pharmacokinetics
Pharmacokinetic parameter values were calculated using Phoenix WinNonlin (Certara, Princeton, NJ, USA). The maximum concentration (Cmax), time to maximum concentration (Tmax), and trough concentration at day 7 (C168 h) were obtained from the observed concentration–time data. The area under the concentration–time curve from 0 to 168 h (AUC0–168) and from 0 to infinity (AUC0–∞) were estimated using the linear trapezoidal method for ascending concentrations and the log trapezoidal method for descending concentrations. The slope of the terminal phase λz was calculated by regression of the terminal log‐linear portion of the concentration–time profile; this was used to extrapolate beyond the last measured concentration to estimate AUC0–168 h (Study 1) and AUC0–∞. The apparent terminal half‐life (t½) was calculated as the quotient of natural log of two and λz. The following pharmacokinetic parameters were calculated for islatravir plasma concentrations and intracellular islatravir‐TP concentrations: AUC0–∞, AUC0–168 h, Cmax, Tmax, and apparent t½. In addition, C168 h for islatravir‐TP was calculated. In Study 1, doses for which there was carryover in islatravir‐TP from the previous dosing period (defined as a predose concentration (C0) >5% of Cmax of the current dose), the concentration of the current dose was modified using the equation:
where Ct was the concentration at time t due to the current dose, Cobserved was the measured concentration at time t, and k was the slope of the terminal phase from the preceding dose. This effectively removes the contribution of the previous dose.
Plasma islatravir and intracellular islatravir‐TP AUC0–∞, AUC0–168 h, Cmax, and C168 h (islatravir‐TP only), were natural log‐transformed and evaluated with a linear mixed effects model containing a fixed effect for treatment and random effect for subject. In study 2, the linear mixed effects model also contained a fixed effect for day (day 1, day 8, and day 15 for C168 h and day 1 and day 15 for AUC0–168 h and Cmax) and treatment by day interaction. Least squares means and 95% confidence intervals (CIs) were obtained from the model and back‐transformed to obtain geometric means and 95% CIs for each treatment; and for C168 h, 90% CIs were also obtained from the model.
The same model was used to estimate the effect of food on plasma islatravir and intracellular islatravir‐TP pharmacokinetics; the geometric mean ratio (fed/fasted) and 90% CI at the 30 mg dose level were obtained for AUC0–∞, Cmax, AUC0–168 h, and C168 h (islatravir‐TP only).
The posterior probability that the true islatravir‐TP C168 h exceeded the concentration target of 0.53 pmol/106 cells associated with efficacy in SIV‐infected rhesus macaques was calculated at each dose level, using the least squares estimates and total variance from the above models, assuming a noninformative prior and normality on the log scale.
RESULTS
Participants
A total of 24 participants were enrolled in and completed each study between January 29, 2014, and May 27, 2014 (Study 1) and May 15, 2014, and January 12, 2015 (Study 2). Participant demographics for both studies are summarized in Table S1.
Safety and tolerability
Single and multiple oral dose administration of islatravir were generally well‐tolerated. There were no serious adverse events and no participants discontinued due to an adverse experience in either study. In Study 1, 16 participants (66.7%) reported a total of 30 nonserious adverse events; none were considered related to islatravir. All adverse events were mild to moderate in intensity. The most commonly reported adverse events (>2 participants) were headache (n = 9) and nasopharyngitis (n = 4). In Study 2, 9 participants (37.5%) reported a total of 12 nonserious adverse events; all were mild to moderate in intensity. The most commonly reported adverse event (>2 participants) was headache (n = 3).
No clinically meaningful relationships were observed for changes in clinical laboratory values, vital signs, or ECG safety parameter values following treatment with islatravir in either study.
Pharmacokinetics
Rising single‐dose clinical trial (Study 1)
Pharmacokinetic parameter values of plasma islatravir following administration of single oral doses of 5 to 400 mg islatravir in the fasted state and 30 mg in the fed state are summarized in Table 1. In the fasted state, islatravir was rapidly absorbed, with a median Tmax of 0.5 h (Figure 2a) and plasma exposure appeared to increase in an approximately dose‐proportional manner between 5 and 400 mg. Plasma concentrations decreased in a biphasic manner, with a rapid initial phase (within the first 6–12 h) and a slow terminal phase with an apparent t½ of 49–61 h.
TABLE 1.
Summary of pharmacokinetic parameter values of plasma islatravir and intracellular islatravir‐TP a in Study 1 following administration of single oral doses of 5–400 mg islatravir in the fasted state (n = 6 per dose) and 30 mg after a standard high‐fat meal (n = 6) to participants without HIV
| Pharmacokinetic parameter | Least squares geometric mean (95% CI) | |||||||
|---|---|---|---|---|---|---|---|---|
| 5 mg fasted | 15 mg fasted | 30 mg fasted | 30 mg fed | 100 mg fasted | 200 mg fasted | 400 mg fasted | ||
| Plasma islatravir | Cmax b (nmol/L) | 147 (120–181) | 390 (317–480) | 1070 (867–1310) | 542 (441–666) | 3200 (2600–3930) | 6180 (5030–7600) | 9910 (8070–12,200) |
| Tmax c (h) | 0.50 (0.50–0.50) | 0.50 (0.25–1.00) | 0.50 (0.25–0.50) | 1.75 (0.50–4.02) | 0.50 (0.25–0.50) | 0.50 (0.50–0.50) | 0.50 (0.50–1.00) | |
| Apparent t1/2 d (h) | – e | 51.4 (16.6) | 48.6 (13.0) | 63.6 (20.8) | 58.9 (7.6) | 61.4 (8.2) | 48.5 (7.2) | |
| AUC0–168 h b (h*µM/L) | 0.37 (0.32–0.41) | 1.41 (1.25–1.59) | 3.74 (3.31–4.21) | 3.91 (3.47–4.41) | 10.9 (9.69–12.3) | 24.1 (21.4–27.2) | 47.2 (41.9–53.1) | |
| AUC0–∞ b (h*µM/L) | – e | 1.58 (1.41–1.78) | 3.91 (3.48–4.39) | 4.30 (3.82–4.83) | 11.70 (10.3–13.2) | 25.60 (22.8–28.8) | 48.30 (43.1–54.2) | |
| Intracellular islatravir‐TP | Cmax b (pmol/106cells) | 1.06 (0.83–1.37) | 3.65 (2.85–4.69) | 8.21 (6.39–10.50) | 7.85 (6.11–10.1) | 13.8 (10.7–17.7) | 38.4 (29.9–49.3) | 63.6 (49.6–81.6) |
| C168 h b (pmol/106 cells) | 0.29 (0.19–0.45) | 1.24 (0.80–1.92) | 2.76 (1.78–4.29) | 2.71 (1.67–4.40) f | 4.79 (3.09–7.43) | 7.73 (4.98–12.0) | 18.3 (11.8–28.4) | |
| Tmax c (h) | 9.00 (6.00–24.25) | 6.00 (6.00–48.07) | 24.00 (6.00–48.00) | 17.97 (6.00–24.00) | 24.00 (12.00–48.00) | 6.00 (6.00–12.00) | 12.08 (6.08–24.13) | |
| Apparent terminal t1/2 d (h) | 126.00 (50.3) | 161.45 (29.7) | 118.17 (23.8) | 144.65 (62.6) g | 171.28 (80.5) | 159.64 (27.4) | 160.32 (16.6) | |
| AUC0–168 h b (h*pmol/106cells) | 91.6 (73.5–114) | 341 (274–425) | 871 (700–1080) | 660 (520–838) f | 1260 (1010–1560) | 3100 (2490–3860) | 5300 (4260–6580) | |
| AUC0–∞ (h*pmol/106cells) b | 162 (120–217) | 670 (499–900) | 1420 (1060–1900) | 1370 (975–1920) g | 2480 (1850–3330) | 5180 (3850–6960) | 9320 (6950–12500) | |
Abbreviations: AUC0–∞, area under the concentration–time curve and from 0 to infinity; AUC0–168 h, area under the concentration–time curve from 0 to 168 h; CI, confidence interval; Cmax, maximum concentration; GCV, geometric coefficient of variation; PBMC, peripheral blood mononuclear cell; t½, half‐life; Tmax, time to maximum concentration; TP, triphosphate.
The reported concentrations of islatravir‐TP were potentially underestimated due to pre‐analytical sample handling issues.
Back‐transformed least squares mean and CI from linear mixed effects model performed on natural log‐transformed values.
Median (min, max).
Geometric mean (%GCV).
Not determined due to insufficient data at the terminal phase.
N = 5 for AUC0–168 h and C168 h.
N = 4 for AUC0–∞ and apparent t1/2.
FIGURE 2.
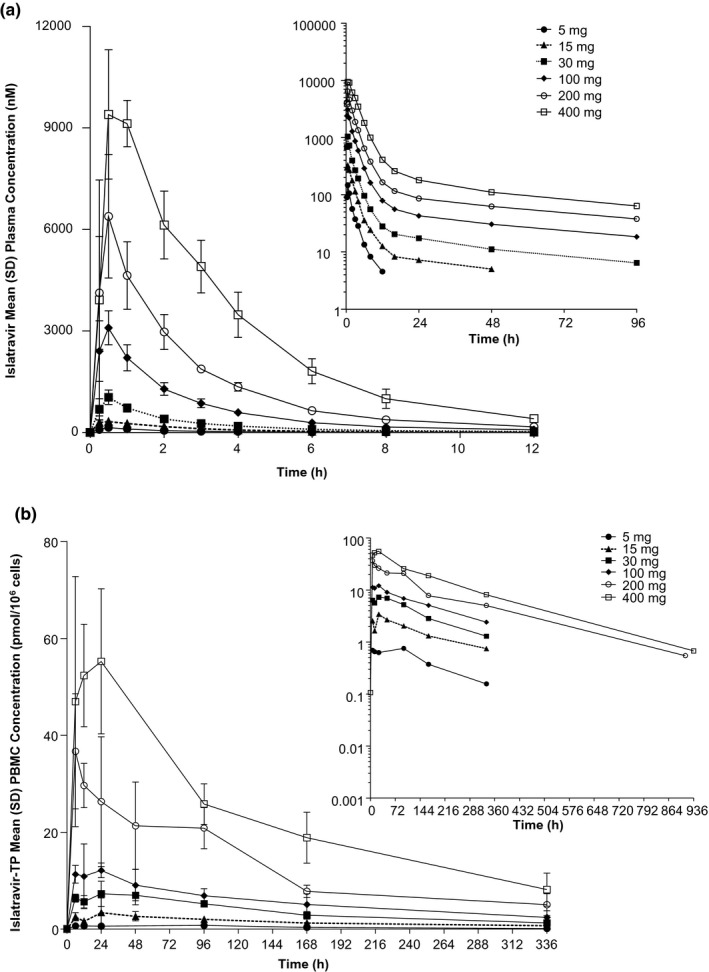
Concentration‐time profiles. (a) Arithmetic mean (±SD) plasma concentration versus time profiles of plasma islatravir following administration of single oral doses of 5–400 mg islatravir in the fasted state to participants without HIV (n = 6/panel); inset: semi‐log plot. (b) Arithmetic mean (±SD) PBMC concentration versus time profiles of islatravir‐TP following administration of single oral doses of 5–400 mg islatravir in the fasted state to participants without HIV (n = 6/panel); inset: semi‐log plot. PBMC, peripheral blood mononuclear cell; SD, standard deviation; TP, triphosphate
Pharmacokinetic parameter values of islatravir‐TP in the fasted and fed state are summarized in Table 1. Doses of 15 mg and above achieved a greater than 99% posterior probability that the true mean intracellular islatravir‐TP C168 h is greater than 0.53 pmol/106 cells (from 1.24 pmol/106 cells at 15 mg to 18.3 pmol/106 cells at 400 mg). In the fasted state, islatravir‐TP reached intracellular Cmax at a median Tmax of 6–24 h (range 6–48 h) and the concentrations declined with an apparent t½ of 118–171 h (Figure 2b). The reported concentrations of islatravir‐TP were potentially underestimated due to pre‐analytical sample handling issues.
Following administration of 30 mg islatravir with a high‐fat meal, islatravir had a slightly delayed absorption with a median plasma Tmax of 1.75 h. There was a 49% decrease in plasma islatravir Cmax and a 10% increase in AUC0–∞. Despite the decrease in plasma islatravir Cmax, islatravir‐TP AUC, Cmax, and C168 were largely unchanged.
Rising multiple‐dose clinical trial (Study 2)
Pharmacokinetic parameter values of plasma islatravir following administration of once‐weekly oral doses of islatravir for 3 weeks are summarized in Table 2. There was little to no accumulation of islatravir in plasma. All participants who received 10 mg islatravir had plasma concentrations below the lower limit of quantitation of 1.00 ng/ml (0.00341 µmol/L) at 48 h postdose in week 1 and at 96 h postdose in week 3.
TABLE 2.
Summary of pharmacokinetic parameter values of plasma islatravir and intracellular islatravir‐TP following administration of once‐weekly oral doses of islatravir for 3 weeks to participants without HIV
| Pharmacokinetic parameters | 10 mg (n = 6) | 30 mg (n = 6) | 100 mg (n = 6) | |
|---|---|---|---|---|
| Week 1 | ||||
| Plasma islatravir | AUC0–168 h a (h*μmol/L) | 0.713 (0.606, 0.840) | 3.26 (2.77, 3.84) | 9.62 (8.17, 11.3) |
| Cmax a (nmol/L) | 193 (133, 280) | 647 (445, 940) | 1470 (1010, 2140) | |
| Tmax b (h) | 1.00 (0.50, 2.00) | 1.00 (0.50, 1.00) | 2.00 (1.00, 6.00) | |
| Islatravir‐TP | AUC0–168 h a (h*pmol/106 cells) | 341 (256, 454) | 1160 (872, 1550) | 3150 (2370, 4200) |
| Cmax a (pmol/106 cells) | 3.79 (2.68, 5.34) | 13.2 (9.34, 18.6) | 31.3 (22.2, 44.2) | |
| C168 a (pmol/106 cells) | 0.988 (0.761, 1.28) | 3.67 (2.83, 4.76) | 13.5 (10.4, 17.6) | |
| Tmax b (h) | 48.00 (24.00, 96.03) | 9.00 (6.00, 24.13) | 18.00 (6.00, 48.00) | |
| Week 3 e | ||||
| Plasma islatravir | AUC0–168 h a (h*μmol/L) | 0.867 (0.736, 1.02) | 3.98 (3.38, 4.68) | 12.0 (10.2, 14.1) |
| Cmax a (nmol/L) | 241 (166, 350) | 637 (439, 926) | 1850 (1270, 2680) | |
| Tmax b (h) | 1.00 (0.50, 1.00) | 1.00 (0.50, 1.12) | 1.00 (1.00, 6.00) | |
| Apparent t1/2 c (h) | 16.48 (157.32) | 74.21 (14.20) | 87.14 (9.61) | |
| Islatravir‐TP | AUC0–168 h a (h*pmol/106 cells) | 476 (357, 634) | 1570 (1180, 2100) | 4740 (3560, 6320) |
| Cmax a (pmol/106 cells) | 6.38 (4.52, 9.00) | 19.6 (13.9, 27.7) | 46.7 (33.1, 65.9) | |
| C168 a (pmol/106 cells) | 1.30 (0.740, 2.29) | 5.36 (4.13, 6.96) | 14.3 (11.0, 18.6) | |
| Tmax b (h) | 18.00 (6.00, 48.00) | 15.00 (6.00, 24.00) | 24.00 (12.00, 48.00) | |
| Apparent t1/2 c (h) | 91.71 (17.76) | 136.72 (25.38) | 153.14 (20.37) | |
| Accumulation ratio: week 3 / week 1 | ||||
| Plasma islatravir | AUC0–168 h d (h*μmol/L) | 1.22 (1.08, 1.36) | 1.22 (1.09, 1.37) | 1.25 (1.11, 1.40) |
| Cmax d (nmol/L) | 1.25 (0.82, 1.92) | 0.99 (0.64, 1.51) | 1.26 (0.82, 1.93) | |
| Islatravir‐TP | AUC0–168 h d (h*pmol/106 cells) | 1.40 (1.04, 1.87) | 1.36 (1.01, 1.82) | 1.50 (1.12, 2.02) |
| Cmax d (pmol/106 cells) | 1.68 (1.17, 2.43) | 1.49 (1.03, 2.14) | 1.49 (1.03, 2.15) | |
| C168 d (pmol/106 cells) | 1.32 (0.82, 2.12) | 1.46 (1.17, 1.83) | 1.06 (0.85, 1.33) | |
AUC0–168 h, area under the concentration–time curve from 0 to 168 h; Cmax, maximum concentration; GCV, geometric coefficient of variation; CV, coefficient of variation; t½, half‐life; Tmax, time to maximum concentration; TP, triphosphate.
Back‐transformed least squares mean and 95% confidence interval from linear mixed effects model performed on natural log‐transformed values.
Median (min, max).
Geometric mean (%GCV).
Back‐transformed least squares mean difference and 90% confidence interval from mixed effects model performed on natural log‐transformed values.
N = 1 for C168 h; N = 3 for apparent t1/2.
Pharmacokinetic parameter values of intracellular islatravir‐TP are summarized in Table 2. Based on posterior probability calculation, the islatravir‐TP pharmacokinetic target of 0.53 pmol/106 cells was exceeded after the first weekly dose at all dose levels. Following multiple doses of 30 and 100 mg islatravir, there appeared to be a modest degree of accumulation of islatravir‐TP. Accumulation ratios for the 10 mg dose could not be calculated due to loss of the week 3 PBMC samples.
Pharmacokinetic parameters for both islatravir‐TP in PBMCs and plasma islatravir were comparable following administration of islatravir as an oral suspension or a capsule (Tables 1 and 2).
DISCUSSION
Islatravir is a nucleoside analogue with potential to be an effective agent for the treatment and prevention of HIV‐1 infection. Preclinical studies demonstrated its high potency and potential for extended duration efficacy. 21 The safety and pharmacokinetic profiles of islatravir were assessed following single‐ and multiple‐dose administration over a dose range of 5 to 400 mg in participants without HIV. The dose range was selected to cover exposures projected to be efficacious based on preclinical in vitro and in vivo data, while maintaining adequate safety margins from preclinical toxicology assessments. Islatravir was generally well‐tolerated, with no serious adverse events experienced at single doses up to 400 mg or following multiple dosing up to 100 mg once weekly for 3 weeks. There was no apparent association between dose and frequency or severity of adverse effects.
Pharmacokinetic analysis of single oral doses of islatravir of 5 to 400 mg demonstrated that islatravir and islatravir‐TP exhibit approximately linear pharmacokinetic behavior in plasma and PBMCs, respectively. As suggested by preclinical data, islatravir has a long intracellular half‐life, supportive of extended dosing schedules. Preclinical studies in rhesus macaques showed that the antiviral efficacy of islatravir is related to the trough concentration of islatravir‐TP in PBMCs, rather than to trough concentrations in plasma, which is the pharmacokinetic parameter of interest for a number of HIV‐1 antiretrovirals. 15 Pharmacokinetic/pharmacodynamic modeling of SIV‐infected rhesus macaques treated with once‐weekly islatravir showed an intracellular C168 h of greater than or equal to 0.53 pmol/106 cells, 17 , 22 which was used as the intracellular pharmacokinetic threshold in these trials. This was successfully achieved at doses of 10 mg and higher. The results support the further exploration of islatravir for extended duration dosing.
PBMC stabilization procedures were updated for Study 2 based on learnings from Study 1, which resulted in Study 2 islatravir‐TP concentrations being more accurate. Extremely precise sampling handling and addition of the appropriate amount of stabilizing agent is required to prevent islatravir‐TP from rapidly converting to the diphosphate (DP), monophosphate, and islatravir forms upon cell lysis. 20 Comparison of the ratios of DP to TP in Study 1 to those in Study 2 (data not shown) indicated that improvements to sample handling procedures implemented between studies resulted in better stabilization of islatravir‐TP. Therefore, the islatravir‐TP pharmacokinetic results from Study 2 are considered more accurate.
Consistent with the half‐life of islatravir in plasma, minimal accumulation was observed with weekly administration. There was evidence of modest accumulation of intracellular islatravir‐TP on the order of 1.5‐fold, which is consistent with the extended half‐life. Despite leading to a reduction in plasma islatravir Cmax, food did not affect intracellular islatravir‐TP levels. Accordingly, in the absence of a clinically meaningful effect of a high‐fat meal on the pharmacokinetics of islatravir, the impact of a high‐fat meal was not investigated further in Study 2.
Study 1 had limitations in the pharmacokinetic assessment of islatravir t½ due to plasma sampling to 96 h, which did not sufficiently span an adequate timespan (>2.5‐times the actual half‐life). Based on data from Study 1, plasma pharmacokinetic sampling was extended to 168 h in Study 2 to ensure that measurements were made across an adequate timeframe to sufficiently assess islatravir t½.
The studies described demonstrate that the safety and pharmacokinetic profile of islatravir are sufficient to continue clinical investigation. The potency of islatravir‐TP, coupled with its long intracellular half‐life, suggest potential for a variety of dosing regimens and formulations, including longer‐acting formulations for both the treatment and prevention of HIV‐1 infection. 21 Based on the favorable safety and pharmacokinetic profile described in the single‐dose and multiple rising‐dose studies, additional clinical investigation has progressed, including a phase 1b monotherapy trial in treatment‐naïve adults living with HIV‐1, 23 a phase 2 HIV‐1 prevention trial with monthly dosing (ClinicalTrials.gov ID: NCT04003103), a phase 2 HIV‐1 treatment trial with weekly dosing in combination with the long‐acting oral NNRTI MK‐8507 (ClinicalTrials.gov ID: NCT04564547), and phase 2 24 (ClinicalTrials.gov ID: NCT03272347, NCT04295772), and phase 3 HIV‐1 treatment trials with daily dosing in combination with doravirine (ClinicalTrials.gov ID: NCT04223778, NCT04223791, NCT04233879, and NCT04233216). In the phase 1b trial in treatment‐naïve adults living with HIV‐1, single oral doses of islatravir as low as 0.5 mg resulted in significant suppression of HIV‐1 RNA by greater than 1.0 log at day 7. Islatravir was generally well‐tolerated. Of note, the pharmacokinetic profile in adults living with HIV‐1 23 was similar to the pharmacokinetic profile observed in study participants without HIV‐1 in the 2 studies described here. Results from a phase 2 dose‐ranging trial in treatment‐naïve adults living with HIV‐1 showed that once daily dosing of islatravir in combination with doravirine maintained HIV‐1 viral suppression for up to 96 weeks. 25 The islatravir + doravirine combination was generally well‐tolerated and the rate of drug‐related adverse events was lower than the control treatment arm of doravirine/lamivudine/tenofovir disoproxil fumarate. 24 Additional dosing regimens and programs are being considered to meet the diverse needs of populations of people living with HIV‐1.
CONFLICT OF INTEREST
R.P.M., W.A., E.F., D.J.R., Y.L., R.M., D.P., I.D.L., J.A.G., S.A.S., and M.I. are current or former employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA and may own stock and/or options in Merck & Co., Inc., Kenilworth, NJ, USA. M.P. declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
All authors wrote the manuscript. E.F., J.A.G., S.A.S., and M.I. designed the research. I.D.L. and M.P. performed the research. R.P.M., E.F., S.A.S., and M.I. analyzed the data.
Supporting information
Table S1
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Valerie Schulz, Candice Smith, and Sabrina Fox‐Bosetti for excellent operational support, and acknowledge Dr. J. Leempoels as an investigator for the single‐dose study. The authors thank the participants and clinical research staff who participated in this study. Medical writing and editorial assistance, under the direction of the authors, were provided by Yee‐Man Ching, PhD, Apothecom (UK), in accordance with Good Publication Practice (GPP3) guidelines. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
CLINICAL TRIAL REGISTRATION: EudraCT Numbers: 2013–004683–60 (Study 1); 2014–000635–16 (Study 2).
Funding information
Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA (MSD).
DATA AVAILABILITY STATEMENT
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA’s data sharing policy, including restrictions, is available at http://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.
REFERENCES
- 1. DHHS . Panel on Antiretroviral Guidelines for Adults and Adolescents Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents with HIV. Department of Health and Human Services, 2019. https://clinicalinfo.hiv.gov/sites/default/files/inline‐files/AdultandAdolescentGL.pdf. Accessed October 2020.
- 2. UNAIDS . Joint United Nations Programme on HIV/AIDS (UNAIDS) FactSheet ‐ Global AIDS Update 2020, 2020. https://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf. Accessed October 2020.
- 3. EACS . Guidelines for the management of people living with HIV (PLWH) in Europe. Version 10.1. 2020. https://www.eacsociety.org/files/guidelines‐10.1.finalsept2020.pdf. Accessed October 2020.
- 4. CDC . Centers for Disease Control and Prevention (CDC) US Public Health Service: Preexposure prophylaxis for the prevention of HIV infection in the United States ‐ 2017 Update. A Clinical Practice Guideline. 2018. https://www.cdc.gov/hiv/pdf/risk/prep/cdc‐hiv‐prep‐guidelines‐2017.pdf. Accessed October 2020.
- 5. Aves T, Tambe J, Siemieniuk RA, Mbuagbaw L. Antiretroviral resistance testing in HIV‐positive people. Cochrane Database Syst Rev. 2018;11:CD006495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Markowitz M, Grobler JA. Islatravir for the treatment and prevention of infection with the human immunodeficiency virus type 1. Curr Opin HIV AIDS. 2020;15:27‐32. [DOI] [PubMed] [Google Scholar]
- 7. Kawamoto A, Kodama E, Sarafianos SG, et al. 2'‐deoxy‐4'‐C‐ethynyl‐2‐halo‐adenosines active against drug‐resistant human immunodeficiency virus type 1 variants. Int J Biochem Cell Biol. 2008;40:2410‐2420. [DOI] [PubMed] [Google Scholar]
- 8. Markowitz M, Sarafianos SG. 4'‐Ethynyl‐2‐fluoro‐2'‐deoxyadenosine, MK‐8591: a novel HIV‐1 reverse transcriptase translocation inhibitor. Curr Opin HIV AIDS. 2018;13:294‐299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Michailidis E, Marchand B, Kodama EN, et al. Mechanism of inhibition of HIV‐1 reverse transcriptase by 4'‐Ethynyl‐2‐fluoro‐2'‐deoxyadenosine triphosphate, a translocation‐defective reverse transcriptase inhibitor. J Biol Chem. 2009;284:35681‐35691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Michailidis E, Huber AD, Ryan EM, et al. 4'‐Ethynyl‐2‐fluoro‐2'‐deoxyadenosine (EFdA) inhibits HIV‐1 reverse transcriptase with multiple mechanisms. J Biol Chem. 2014;289:24533‐24548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Salie ZL, Kirby KA, Michailidis E, et al. Structural basis of HIV inhibition by translocation‐defective RT inhibitor 4'‐ethynyl‐2‐fluoro‐2'‐deoxyadenosine (EFdA). Proc Natl Acad Sci USA. 2016;113:9274‐9279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nakata H, Amano M, Koh Y, et al. Activity against human immunodeficiency virus type 1, intracellular metabolism, and effects on human DNA polymerases of 4'‐ethynyl‐2‐fluoro‐2'‐deoxyadenosine. Antimicrob Agents Chemother. 2007;51:2701‐2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maeda K, Desai DV, Aoki M, Nakata H, Kodama EN, Mitsuya H. Delayed emergence of HIV‐1 variants resistant to 4'‐ethynyl‐2‐fluoro‐2'‐deoxyadenosine: comparative sequential passage study with lamivudine, tenofovir, emtricitabine and BMS‐986001. Antivir Ther. 2014;19:179‐189. [DOI] [PubMed] [Google Scholar]
- 14. Grobler J, Huang Q, Hazuda D, Lai M. Efficacy of MK‐8591 against diverse HIV‐1 subtypes and NRTI‐resistant clinical isolates. J Int AIDS Soc. HIV Glasgow, October 28–31, 2018, Glasgow, UK; p. e25187.
- 15. Stoddart CA, Galkina SA, Joshi P, et al. Oral administration of the nucleoside EFdA (4'‐ethynyl‐2‐fluoro‐2'‐deoxyadenosine) provides rapid suppression of HIV viremia in humanized mice and favorable pharmacokinetic properties in mice and the rhesus macaque. Antimicrob Agents Chemother. 2015;59:4190‐4198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murphey‐Corb M, Rajakumar P, Michael H, et al. Response of simian immunodeficiency virus to the novel nucleoside reverse transcriptase inhibitor 4'‐ethynyl‐2‐fluoro‐2'‐deoxyadenosine in vitro and in vivo. Antimicrob Agents Chemother. 2012;56:4707‐4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grobler J, Friedman E, Barrett SE, et al. Long‐Acting Oral and Parenteral Dosing of MK‐8591 for HIV Treatment or Prophylaxis. Conference on Retroviruses and Opportunistic Infections (CROI), 22–25 February 2016, Boston, MA, USA.
- 18. Kirby KA, Michailidis E, Fetterly TL, et al. Effects of substitutions at the 4’ and 2 positions on the bioactivity of 4'‐ethynyl‐2‐fluoro‐2'‐deoxyadenosine. Antimicrob Agents Chemother. 2013;57:6254‐6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Markowitz M, Gettie A, St. Bernard L, et al. Once‐weekly oral dosing of MK‐8591 protects male rhesus macaques from intrarectal challenge With SHIV109CP3. J Infect Dis. 2020;221:1398‐1406. [DOI] [PubMed] [Google Scholar]
- 20. Sun L, Chavez‐Eng C, Fillgrove KL, et al. Toward highly sensitive and reproducible LC‐MS/MS analysis of MK‐8591 phosphorylated anabolites in human peripheral blood mononuclear cells. Bioanalysis. 2019;11(4):233‐250. [DOI] [PubMed] [Google Scholar]
- 21. Grobler JA, Fillgrove K, Hazuda D. MK‐8591 Potency and PK Provide High Inhibitory Quotients at Low Doses QD and QW. Conference on Retroviruses and Opportunistic Infections (CROI), March 4–7, 2019, Seattle, WA, USA.
- 22. Grobler J, Friedman E, Barrett SE, et al. Long‐acting oral and parenteral dosing of MK‐8591 for HIV treatment or prophylaxis. Conference on Retroviruses and Opportunistic Infections (CROI), February 22–25, 2016: Boston, MA, USA., 2016.
- 23. Schürmann D, Jackson Rudd D, Zhang S, et al. Safety, pharmacokinetics, and antiretroviral activity of islatravir (ISL, MK‐8591), a novel nucleoside reverse transcriptase translocation inhibitor, following single‐dose administration to treatment‐naive adults infected with HIV‐1: an open‐label, phase 1b, consecutive‐panel trial. Lancet HIV. 2020;7:e164‐e172. [DOI] [PubMed] [Google Scholar]
- 24. Molina JM, Yazdanpanah Y, Afani Saud A, et al. MK‐8591 at doses of 0.25 to 2.25 mg QD, in combination with doravirine establishes and maintains viral suppression through 48 weeks in treatment‐naïve adults with HIV‐1 infection. 10th International AIDS Society Conference on HIV Science (IAS). July 21–24, 2019, Mexico City, Mexico.
- 25. Molina JM, Yazdanpanah Y, Afani Saud A. Islatravir in combination with doravirine maintains HIV‐1 viral suppression through 96 weeks. HIV Glasgow, October 5–8, 2020, Glasgow, Scotland, UK. Abstract 4913173.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Supplementary Material
Supplementary Material
Supplementary Material
Data Availability Statement
Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA’s data sharing policy, including restrictions, is available at http://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.