

Abstract
GDC‐0334 is a novel small molecule inhibitor of transient receptor potential cation channel member A1 (TRPA1), a promising therapeutic target for many nervous system and respiratory diseases. The pharmacokinetic (PK) profile and pharmacodynamic (PD) effects of GDC‐0334 were evaluated in this first‐in‐human (FIH) study. A starting single dose of 25 mg was selected based on integrated preclinical PK, PD, and toxicology data following oral administration of GDC‐0334 in guinea pigs, rats, dogs, and monkeys. Human PK and PK‐PD of GDC‐0334 were characterized after single and multiple oral dosing using a population modeling approach. The ability of GDC‐0334 to inhibit dermal blood flow (DBF) induced by topical administration of allyl isothiocyanate (AITC) was evaluated as a target‐engagement biomarker. Quantitative models were developed iteratively to refine the parameter estimates of the dose‐concentration‐effect relationships through stepwise estimation and extrapolation. Human PK analyses revealed that bioavailability, absorption rate constant, and lag time increase when GDC‐0334 was administered with food. The inhibitory effect of GDC‐0334 on the AITC‐induced DBF biomarker exhibited a clear sigmoid‐Emax relationship with GDC‐0334 plasma concentrations in humans. This study leveraged emerging preclinical and clinical data to enable iterative refinement of GDC‐0334 mathematical models throughout the FIH study for dose selection in subsequent cohorts throughout the study.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
GDC‐0334 is a novel, small molecule TRPA1 inhibitor and a pharmacokinetic‐pharmacodynamic (PK‐PD) modeling strategy could be implemented in a systematic and step‐wise manner to build and learn from emerging data for early clinical development.
WHAT QUESTION DID THIS STUDY ADDRESS?
Can noncompartmental and population‐based analyses be used to describe the PK and PD characteristics of GDC‐0334 in preclinical and clinical studies?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
GDC‐0334 exposure generally increased with dose in rats, dogs, and monkeys. The starting dose (25 mg) in the clinical study was determined based on the preclinical data. GDC‐0334 exhibited linear PK in humans and the bioavailability was increased with food. The inhibitory effect of GDC‐0334 on dermal blood flow induced by the TRPA1 agonist allyl isothiocyanate in humans indicates a clear PK‐PD relationship.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
The models developed based on TRPA1 agonist‐induced dermal blood flow inhibition data can be used to predict PK‐PD relationships in future preclinical and clinical studies evaluating new drug entities that target TRPA1.
INTRODUCTION
Model‐informed drug development (MIDD) involves modeling and simulation to leverage preclinical findings to predict the clinical outcomes for novel drugs. 1 Translational pharmacokinetic‐pharmacodynamic (PK‐PD) modeling can inform dose selection for first‐in‐human (FIH) studies, allow for better prediction of clinical efficacy earlier in the drug development process, and reduce attrition of drug candidates during clinical development. 2 However, direct translation based on preclinical data alone is not sufficient for accurate prediction of the clinical PK‐PD relationship. Incorporating available clinical data is necessary in order to refine dose prediction for the next stage of clinical development. 3
For this FIH study, we demonstrate the use of the “learn and confirm” paradigm of MIDD for evaluating the PK and PK‐PD relationship of GDC‐0334 (also known as RG6174), a novel small molecule inhibitor of transient receptor potential cation channel member A1 (TRPA1). The nonselective cation channel TRPA1 is a promising therapeutic target for treating pain, itch, and respiratory diseases. 4 TRPA1 is expressed by sensory neurons in the lungs, skin, and gastrointestinal tract, as well as airway smooth muscle cells in the lung. It is a polymodal irritant sensor of noxious exogenous chemicals, including cinnamaldehyde, allyl isothiocyanate (AITC), acrolein, and endogenous oxidative stress and proinflammatory mediators. 5 We have previously described a translational AITC skin challenge method that is suitable for use as a PD biomarker of TRPA1 activity and does not require target tissue sampling. 6 In this biomarker model, topical application of AITC in rats or humans transiently increases local dermal blood flow (DBF), as measured by noninvasive laser speckle contrast imaging. Metrics from the AITC‐induced DBF are highly reproducible and suitable for small cohort sizes making them ideal for deployment in phase I clinical trials searching for evidence of target engagement by TRPA1‐antagonists.
We used “learn and confirm” to project human PK and PK‐PD and inform dose selection of GDC‐0334 during the conduct of the FIH study. Initially, we developed a physiologically‐based PK (PBPK) model by incorporating the PK in relevant preclinical species (i.e., rats, dogs, and monkeys) and leveraged in vitro preclinical data and PD data in guinea pigs for predicting the PKs of GDC‐0334 and safe starting dose in humans. Once clinical data was available, our projections were updated by developing population PK (PopPK) and PK‐PD models to quantify and explain the variability in PK and PD in healthy volunteers. This allowed us to refine the dose selection for subsequent cohorts throughout the phase I study. The projections based on PopPK and PK‐PD models were updated iteratively by characterizing the human PK profile of GDC‐0334 in healthy volunteers as phase I clinical data became available. We developed PopPK and PK‐PD models to quantify and explain the variability in PK and PD responses among healthy individuals, allowing us to refine dose selection for subsequent cohorts throughout the study. In combination, these approaches provided us with a more holistic understanding of PK and PK‐PD properties for GDC‐0334 and helped inform decision making during early phase drug development.
METHODS
Preclinical study
PK and toxicokinetics in rats, dogs, and monkeys
Single‐dose PK of GDC‐0334 were determined in male Sprague‐Dawley rats (5 mg/kg by p.o.), beagle dogs and cynomolgus monkeys (1 mg/kg p.o. and 0.5 mg/kg by intravenous i.v.).
Toxicokinetics (TK) of GDC‐0334 in male and female animals of the aforementioned species were determined following repeat daily p.o. dosing over 4 weeks. GDC‐0334 was administered for up to 30 mg/kg (dogs), 200 mg/kg (monkeys), and 300 mg/kg (rats). Serial blood samples were collected for up to 24 h postdose.
Blood samples were centrifuged and plasma was harvested. PK and TK analyses were performed using noncompartmental analysis (NCA) methods. (See Data S1 for additional study details and bioanalytical methods.)
Clinical trial
GB40223 trial design and trial population
This was a double blind, randomized, placebo‐controlled phase I clinical trial of GDC‐0334 in healthy volunteers (ClinicalTrials.gov Identifier: NCT03381144) designed to evaluate safety, tolerability, PK, and exploratory PD effects of single ascending doses (SADs) and multiple ascending doses (MADs; Figure S1). In addition, the effect of food on the PK of GDC‐0334 was evaluated. The study was conducted at a single clinical research site (Quotient Clinical Ltd., Nottingham, UK). Informed consent was obtained from all subjects. The study was conducted in accordance with the ethical principles of the Declaration of Helsinki and Good Clinical Practice guidelines, and was approved by the appropriate institutional review boards. Additional trial design details are in the Data S1.
Dose selection
In accordance with European Medicines Agency (EMA) guidance, 7 a starting single dose of 25 mg in humans was selected to be below the minimal anticipated biological effect level (MABEL; i.e., anticipated to achieve PD‐associated target engagement in the AITC skin challenge). The maximum human dose was selected to maintain the anticipated steady‐state, 24‐h postdose plasma concentration above the maximum target engagement (i.e., human 95% inhibitory concentration [IC95] value based on an in vitro TRPA1 calcium flux assay), determined based on data from a separate guinea pig study. 8 The absorption, distribution, metabolism, and excretion properties of GDC‐0334 based on in vitro and preclinical in vivo studies were described previously. 8
PK collection and bioassay
Serial plasma samples were collected at multiple timepoints up to 35 days postdose for SAD cohorts and up to 35 days post the last dose for MAD cohorts (Table S1). Plasma GDC‐0334 concentrations were analyzed by Covance BioAnalytical Laboratories Ltd (Harrogate, UK) using a validated, supported‐liquid extraction/high performance liquid chromatography with tandem mass spectrometry. The lower limit of quantification is 1 ng/ml.
PD evaluation
The inhibitory effect of GDC‐0334 on AITC‐induced DBF on the forearm was assessed as an exploratory PD end point. Detailed methodologies for the skin challenge and for the summary calculations of the measurements have been described previously 6 and presented in the Data S1. For the initial cohort, the AITC skin challenge was timed to occur between 3 and 4 h postadministration of GDC‐0334 (25 mg) or placebo based on observations of the approximate time associated with the maximum plasma concentration (Tmax) in preclinical species. This was adjusted to the approximate Tmax following review of emerging PK data from later cohorts: between 6 and 8 h postdose for subsequent doses in SAD, and between 6 and 8 h postdose on the last dosing day for 100 mg once daily for 28 days in MAD.
Noncompartmental analysis
PK parameters were calculated by NCA methods for individual plasma concentrations of GDC‐0334 against actual sampling times using Phoenix WinNonlin software (version 8.0, Certara USA, Inc., USA). Dose proportionality following a single dose of GDC‐0334 in the fasted state was performed on the maximum plasma concentration (Cmax), plasma concentration‐time area under the curve during specific intervals (AUC) using a power model:
where µ and ß are the intercept and the slope of the model, respectively.
Population PK analysis
A structural model for describing the PopPK of GDC‐0334 was developed based on the characteristics of GDC‐0334 from graphical exploration. Model selection was based on objective function value, goodness‐of‐fit plots, successful termination of the estimation and covariance routines, and the physiological plausibility and precision of the parameter estimates.
The impacts of covariates on the PKs were evaluated using a forward addition and backward elimination approach. 9 The covariates included baseline body weight, sex, age, dose in mg, and food intake (fasted vs. fed‐state). Criteria for the covariate model development are shown in the Data S1.
Bayesian post hoc PK parameters of the final model were used to calculate the individual PK exposure for the subsequent PK‐PD modeling analysis. The PopPK analysis was conducted using the nonlinear mixed‐effects modeling software NONMEM version 7.3 (ICON Plc, Ireland) running under PsN (Perl speaks NONMEM) version 4.2.0. 10
Population PK‐PD analysis
General time‐DBF measurement profiles were first explored using skin challenge data collected from a previously published biomarker method‐development study in untreated, healthy subjects. 6 Preliminary population PD models were developed using both time‐course data and summarized values of baseline‐normalized DBF measurements and AUC at specific intervals after AITC application. The covariate effects of arm (right or left), AITC concentration (10% or 15%), occasion, and subject demographics were explored.
Data from the phase I clinical trial were used to develop a PK‐PD model for characterizing the relationship between GDC‐0334 exposure and DBF. Only subjects who had both PK and PD data from the phase I clinical trial were included in the model development (N = 50). Subject‐specific predictions of GDC‐0334 exposure metrics from the PopPK model were used to enable PK‐PD modeling. Individual demographics, arm, AITC concentration, and baseline DBF values were investigated as potential covariates to explain the inter‐ and intrasubject variabilities. A simulation (n = 1000) based on the final model was summarized using prediction‐corrected visual predictive check (pcVPC) plots. 11
The PopPK and PK‐PD models were developed iteratively throughout the clinical trial to refine the dose selection for the subsequent cohorts.
RESULTS
Preclinical studies
Preclinical PK and PD studies
The PK parameters of GDC‐0334 in rats, dogs, and monkeys are provided in Table 1 and Table S2. The bioavailability was moderate to high (68.7–85.0%) and Tmax ranged from 0.417 to 6.00 h following p.o. administration. A separate study in guinea pigs demonstrated that GDC‐0334 treatment prior to topical AITC challenge caused a dose‐dependent reduction in AITC‐induced DBF for the dose groups (5, 15, and 45 mg/kg p.o.) tested. 8 An increase of GDC‐0334 dose was associated with an increase in plasma exposure and a plateau of the PD effect at the higher dose groups. The minimum dose to achieve target engagement was 5 mg/kg, with an estimated target concentration above 171 ng/ml expected to show at least a 50% effect on AITC‐induced DBF based on the PD model in guinea pigs. At this dose in guinea pigs, numerically but not statistically significant suppression of AITC‐induced dermal blood perfusion was observed compared to vehicle control.
TABLE 1.
Plasma PK parameters (arithmetic mean ± SD) of GDC‐0334 following single oral administration in rats, dogs, and monkeys
|
Species n = 3/group |
Tmax (h) | Cmax (ng/ml) | AUC0–24 h (ng•hr/ml) |
Oral % (F) |
|---|---|---|---|---|
| Rat (5 mg/kg) | 6.00 ± 3.46 | 568 ± 85.9 | 9140 ± 1410 | 80.8 ± 12.7 |
| Dog (1 mg/kg) | 0.417 ± 0.144 | 291 ± 65.8 | 2030 ± 563 | 68.7 ± 19.0 |
| Monkey (1 mg/kg) | 0.833 ± 0.289 | 270 ± 47.5 | 849 ± 191 | 85.0 ± 20.0 |
The vehicle was DMSO (dimethyl sulfoxide)/0.5% methylcellulose with 0.2% polysorbate 80 (Tween 80) in water (15:85).
Abbreviations: AUC0–24 h, area under the concentration‐time curve from 0 to 24 h; Cmax, maximum concentration observed; F, bioavailability; PK, pharmacokinetic; SD, standard deviation; Tmax, time to maximum concentration.
Preclinical TK/toxicology studies
In the good laboratory practice (GLP) repeat‐dose toxicity studies, GDC‐0334 was well‐tolerated to the highest oral dose tested in each species when administered once daily for 28 days. The no observed adverse effect levels (NOAEL) were established to be 30 mg/kg (dogs), 200 mg/kg (monkeys), and 300 mg/kg (rats.)
The TK parameters of GDC‐0334 are shown in Table S3, Table S4, and Table S5 in the supplementary information. Exposure increased with dose in all species, but was less than dose proportional. There were no sex‐related differences in exposure. In dogs, the Day 26 to Day 1 accumulation ratio is ~ 2 to 3 for Cmax and ~ 3 to 6 for AUC0–24 h. No accumulation in exposure was observed in the rats or monkeys.
Clinical starting dose selection
The starting dose of 25 mg for the phase I clinical trial was selected based on the following considerations: (a) GDC‐0334 human PBPK model was developed in Simcyp (version 15) to predict the human PK based on human in vitro data (e.g., human hepatocyte stability, the plasma protein binding, and the blood to plasma ratio), physicochemical properties (e.g., acid dissociation constant and log of partition coefficient), human physiology, and the understanding of in vitro‐in vivo PK extrapolation gained from preclinical species; (b) Based on the PK‐PD observed in the guinea pig skin challenge model, it was projected that a PD effect on AITC‐induced DBF would be expected if the plasma concentration at 24 h postdose is greater than 61 ng/ml in humans, after adjusting for plasma protein binding and in vitro potency differences between humans and guinea pigs. Integrating the predicted PK‐PD relationship and assuming human bioavailability is 70%, a single dose of 75 mg was projected to be the minimum dose anticipated to achieve target engagement in humans. The proposed starting dose of 25 mg was expected to be ~ 3‐fold below the minimum dose to achieve target engagement; and (c) the MABEL approach was used because the guinea pig skin challenge model served as a preclinical PD model relevant to predict clinical PD activity and target engagement. The starting dose of 25 mg is ~ 4‐fold lower than the proposed starting dose using the traditional maximum recommended starting dose (MRSD) approach determined from the GLP toxicology studies. 12 In this approach, the NOAEL (mg/kg) from the most sensitive species was used to determine the MRSD (117 mg in dogs; Table 2).
TABLE 2.
NOAEL oral exposures and corresponding MRSD in rats, dogs, and monkeys
| Species | NOAEL (mg/kg/day) | Cmax (ng/ml) | AUC0–24 h (ng•h/ml) | MRSD mg/day (70 kg) |
|---|---|---|---|---|
| Rat | 300 | 8850 | 116,000 | 339 |
| Dog | 30 | 26,200 | 483,000 | 117 |
| Monkey | 60 a | 7920 | 65,300 | 135 |
The vehicle was DMSO (dimethyl sulfoxide)/0.5% methylcellulose with 0.2% polysorbate 80 (Tween 80) in water (15:85).
Abbreviations: AUC0–24 h, area under the concentration‐time curve from 0 to 24 hours; Cmax, maximum concentration observed; MRSD, maximum recommended starting dose; NOAEL, no observed adverse effect level.
The NOAEL was considered to be 200 mg/kg, although apparently higher exposures were achieved at 60 mg/kg.
Clinical trial
GDC‐0334 was safe and well‐tolerated in healthy subjects following administration as a single oral dose up to 600 mg in the fasted or high‐fat fed state and 1200 mg (as 400 mg 3 times daily) in the low‐fat fed state, and multiple oral doses for 28 days up to 125 mg in the low‐fat fed state. There were no serious treatment‐emergent adverse events (TEAEs), adverse events (AEs) of special interest, TEAEs leading to death, or TEAEs leading to study withdrawal. Overall, there were no clinically significant findings in the hematology or chemical parameters in the study.
Noncompartmental analysis
The Tmax of GDC‐0334 was variable with median Tmax at 3.5 to 12.0 h post‐single‐dose oral administration in the fasted state (Table S6). Plasma concentrations declined in a biphasic or triphasic manner and remained quantifiable for at least 168 h for all dose groups (Figure 1). A small second peak appeared at 24 h postdose after SAD in a number of subjects, likely due to colonic absorption. Exposure metrics of Cmax and AUC0‐24 h increased in a dose proportional manner from 75 mg to 600 mg (Table 3). Mean terminal half‐life (t1/2) elimination was ~ 7 days.
FIGURE 1.
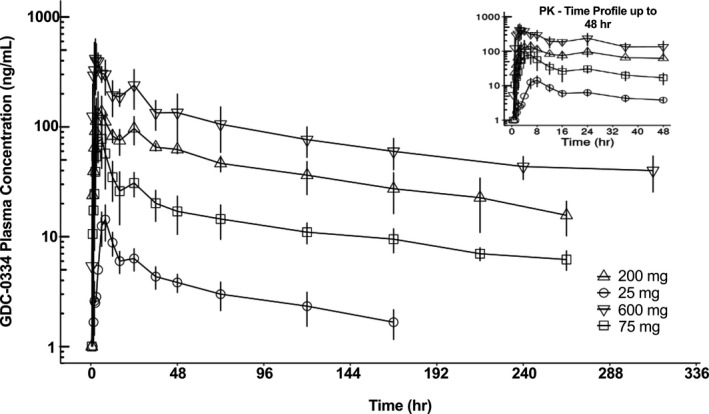
Plasma concentration‐time profiles of GDC‐0334 in humans after single ascending dose under fasting condition show increasing GDC‐0334 plasma concentrations associated with the ascending dose levels. Symbols = arithmetic mean +/‐ SD per nominal sampling time per treatment group; lines = PK‐time profile per treatment group. The inset figure only displays data up to 48 h postdose. PK, pharmacokinetic
TABLE 3.
GDC‐0334 dose proportionality assessment (geometric means) in humans
| Fasted state | 75 mg (n = 6) | 200 mg (n = 6) | 300 mg (n = 12) | 600 mg (n = 6) | Β (90% CI) |
|---|---|---|---|---|---|
| Cmax (ng/ml) | 81.5 | 155 | 321 | 499 | 0.92 (0.75–109) |
| AUC0–24 h (ng•h/mL) | 875 | 2100 | 4200 | 5640 | 0.96 (0.81–1.10) |
| AUC0–72 h (ng•h/ml) | 1750 | 5130 | 8420 | 11,900 | 0.97 (0.83–1.10) |
Abbreviations: AUC0–24 h, area under the plasma concentration‐time curve from time 0 to 24 h; AUC0–72 h, area under the plasma concentration‐time curve from time 0 to 72 h, used for dose‐escalation decisions; CI, confidence interval; Cmax, maximum (peak) plasma drug concentration.
For both high‐ and low‐fat diets, food intake increased GDC‐0334 exposure by 3‐fold in Cmax and 2‐fold in AUC0–24 h relative to the fasted state, but had no impact on t1/2 (Table S7).
Over 28 days of daily dosing, the steady‐state plasma concentration of GDC‐0334 was reached around day 15 (100 mg dose) in the fasted state and day 22 (125 mg dose) in the fed state based on assessment of trough plasma concentration . Accumulation ratios were ~ 3.6 (Cmax) and 5.1 (AUC0‐tau) in the fasted state, and 2.4 (Cmax) and 3.8 (AUC0‐tau) in the fed state (Table S8).
Population PK analysis
In total, 2116 PK samples were collected from 90 subjects enrolled in the study (Table S9). Subjects who received GDC‐0334 were included in the final PopPK analysis dataset (N = 67) were predominantly White (N = 62) and male (N = 43), between 19 and 56 years of age (mean age: 35.5 years), with body weights ranging between 51.2 and 101 kg. The concentration‐time profiles of plasma GDC‐0334 stratified by dose and fasted/fed state decline in a biphasic manner, and the PK can be described by a 2‐compartment structural model (Figure S2). A summary of the models explored is shown in Table S10. No evidence of nonlinear PKs was observed by visual inspection of the concentration‐time profiles. Dose‐normalized concentrations were reasonably superimposed. The proportion of GDC‐0334 plasma concentrations below the limit of quantification was low (5.7%). These PK records were treated as missing values and were not included in the analysis. The PK model was parameterized in terms of apparent clearance from the central compartment (CL/F), volume of distribution of the central compartment (Vc/F), intercompartmental clearance (Q/F), and volume of distribution of the peripheral compartment (Vp/F), with a first order absorption rate (Ka), absorption lag time (Tlag), and relative bioavailability of the food effect (Frel). Interindividual variability was estimated for CL/F, Vc/F, Q/F, Vp/F and Ka, with interaction between CL/F and Vc/F, and between Q/F and Vp/F. The residual error structure consisted of proportional and additive components.
Final model parameters are well‐estimated and are provided in Table 4. An estimated t1/2 of 149 h is consistent with the value determined using NCA. Moderate to high intersubject variability of GDC‐0334 (30.5% for CL/F and 113% for Vc/F) was estimated in healthy subjects. The model‐estimated correlation coefficients were 15.4% and 73.1%, between CL/F and Vc/F and between Q/F and Vp/F, respectively. PKs of GDC‐0334 was well‐described by the final PopPK model, with no model prediction bias in goodness‐of‐fit (Figure S3) or pcVPC (Figure S4) plots. The NONMEM model code for the final PopPK model is included in Data S1.
TABLE 4.
Parameter estimates of the final PopPK model for GDC‐0334 in humans
| Parameter | exp(θ) | 95% CI | %RSE | ω2 | %RSE | %Shrinkage |
|---|---|---|---|---|---|---|
| CL/F (L/h) | 16.0 | 14.4–17.8 | 5.49 | 0.0931 | 16.8 | 8.1 |
| Vc/F (L) | 141 | 102–196 | 16.7 | 1.28 | 20.5 | 3.4 |
| Q/F (L/h) | 62.1 | 55.5–69.4 | 5.7 | 0.104 | 24.5 | 12.8 |
| Vp/F (L) | 2650 | 2350–2980 | 6.04 | 0.120 | 17.8 | 9.7 |
| Ka_fasted (1/h) | 0.0932 | 0.0801–0.108 | 7.74 | 0.0405 | 72.1 | 47.2 |
| Ka_fed (1/h) | 0.201 | 0.170–0.238 | 8.53 | 0.0405 | 72.1 | 47.2 |
| Tlag_fasted (h) | 0.872 | 0.830–0.915 | 2.51 | – | – | – |
| Tlag_fed (h) | 0.959 | 0.935–0.983 | 1.26 | – | – | – |
| Frel_fed | 1.46 | 1.30–1.65 | 6.11 | – | – | – |
| CL/F:Vc/F | – | – | – | 0.0531 | 76.9 | – |
| Q/F:Vp/F | – | – | – | 0.0817 | 24.4 | – |
| Proportional error | 0.255 | 0.235–0.275 | 3.93 | – | – | – |
| Additive error (ng/ml) | 3.70 | 2.72–4.69 | 13.6 | – | – | – |
The 95% CI was obtained using 1000 sets of bootstrap samples.
Abbreviations: CI, confidence interval; CL/F, apparent clearance; Frel, relative bioavailability; Ka, absorption rate; Q/F, apparent inter‐compartment clearance; PopPK, population pharmacokinetic; RSE, relative standard error; Tlag, lag time; Vc/F, apparent central volume of distribution; Vp/F, apparent peripheral volume of distribution.
Food intake increased Ka, Tlag, and Frel of GDC‐0334, resulting in higher exposure (46% in plasma concentration AUC) in subjects who were administered GDC‐0334 with food. Subject demographics (sex, age, race, and weight) had no impact on the GDC‐0334 PK profile.
Population PK‐PD analysis
DBF time‐course profiles were complex and varied over time. None of the covariate effects explained the variability in the DBF‐time profiles from the preliminary PK‐PD model development using healthy subjects. Therefore, there was no predictive advantage of using time‐course data over summary metrics of human DBF data (N = 44), and the summary metrics of baseline‐normalized DBF values at specific time points and AUC at specific intervals after AITC application were used for GDC‐0334 PK‐PD model development.
Observed PD data from the phase I clinical trial show a clear dose relationship for GDC‐0334 after single (with or without high‐fat food intake) and multiple doses (Figure 2). The PK‐PD relationship between GDC‐0334 exposure and baseline‐normalized AUC0–10 from the skin challenge was modeled as a direct effect sigmoidal maximum effect (Emax) model. The structural model of the population PK‐PD model was described as:
where DBF is the baseline‐normalized DBF in terms of AUC0–10 (units), DE0 is the placebo effect on AITC‐induced DBF, DEmax is the maximum inhibition of GDC‐0334 on DBF, DEC50 is the GDC‐0334 exposure required to inhibit 50% of the AITC effect, γ is the Hill coefficient describing the steepness of the PK‐PD response, and PK is the time‐matched GDC‐0334 plasma concentration. Interindividual variability was placed on the baseline parameter. Semimechanistic direct‐ and indirect‐response models were not investigated due to the large discrepancy in time‐course scale between changes in GDC‐0334 plasma concentration (t1/2 of 149 h from PopPK model) and in DBF imaging measurement (20‐min duration). Additional details of the final PK‐PD model development are located in Data S1.
FIGURE 2.
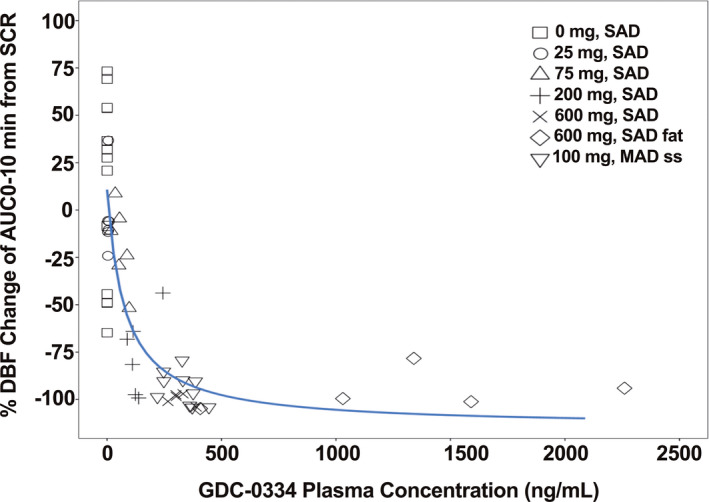
Observed pharmacokinetic‐pharmacodynamic (PK‐PD) relationship from the clinical study demonstrates the inhibition of allyl isothiocyanate (AITC)‐induced dermal blood is dose proportional to the plasma concentrations of GDC‐0334. DBF, dermal blood flow; AUC0–10 min, area under the curve from time 0 to 10 min; SCR, baseline data from screening period; SAD, single ascending dose; MAD, multiple ascending dose; ss, steady state; solid black line, nonlinear regression curve fit line. Data partially published previously: ©2020 Balestrini et al., J Exp Med. 2021; 218(4):e2021637 10
The subsequent univariate screening found that baseline DBF value was a statistically significant covariate for DE0 and DEmax, whereas the impact of AITC application location, AITC concentration, and demographics were not significant covariates. Table S11 in the Supplementary Information shows the summary statistics of covariates explored for the PK‐PD analysis. Parameter estimates for the PK‐PD model in Table S12 shows the parameters were estimated with reasonable precision (relative standard error [RSE] < 49%), with the exception of the typical value for DEC50 (RSE = 166%).
The inhibitory effect of GDC‐0334 on AITC‐induced PD exhibited a clear Emax relationship with GDC‐0334 plasma concentrations despite the small number of subjects and limited amount of PD response data collected. The final population PK‐PD model estimated a DEC50 of 1480 ng/ml, which is well within the GDC‐0334 exposures investigated in the phase I clinical trial. However, the RSE is large (166%), indicating a large uncertainty in the parameter estimation. The pcVPC plot (Figure 3) was used to assess model performance, and the final model showed an adequate fit to the phase I data, with the medians of the observed data well captured by the 95% prediction intervals of the PopPK model.
FIGURE 3.
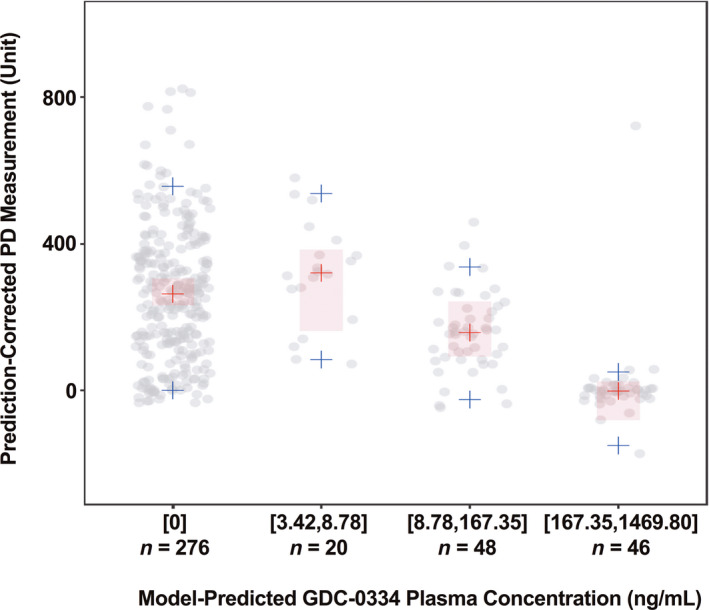
Prediction‐corrected visual predictive check of final pharmacokinetic‐pharmacodynamic (PK‐PD) model. Observed data (gray circles), observed median (red crosses), observed 5th and 95th percentiles (blue crosses); 95% prediction interval (PI; 1000 replicates) of median (red shaded areas); 95% PI for 5th and 95th percentiles not displayed due to large PI; n = sample size (by allyl isothiocyanate [AITC] application location, arm, occasion, and subject), bracket = inclusive, parenthesis = exclusive
DISCUSSION
This FIH study demonstrates the utility of integrating preclinical and clinical PK and PD data to inform the starting dose and dose selection in early phase clinical trials. We implemented this modeling strategy in a systematic and step‐wise manner in order to build and learn from emerging data and early analyses. The information generated by this series of analyses enabled more precise estimation of oral PK and PD profiles for GDC‐0334 and more efficient and effective decision making during early clinical development. The findings also support the use of AITC‐induced DBF from skin challenge as a reliable noninvasive metric for evaluating PK‐PD relationships for inhibitors of TRPA1.
We selected a starting dose of 25 mg for the FIH study based on the MABEL approach, which we anticipated would have no PD effect in the skin and would be well below the anticipated therapeutic dose. Results in humans confirmed that the 25 mg dose had minimal inhibitory effect on AITC‐induced DBF. This dose is approximately fourfold lower than the estimated starting dose using the traditional MRSD approach determined from the GLP toxicology studies. 12 In the latter approach, the NOAEL dose (mg/kg) from the most sensitive species (i.e., dog) was used to determine the MRSD.
We successfully predicted the PK‐PD relationship in humans based on preclinical animal studies and human PBPK to support GDC‐0334 dose selection in the FIH study. We developed models based on clinical PK and PD data obtained throughout the study to inform dose selection in subsequent cohorts. We found that GDC‐0334 had a clear dose‐response relationship with AITC‐induced DBF, demonstrating clinical target engagement, and mirroring findings in preclinical studies. 8
Cohort‐by‐cohort analysis that incorporated emerging safety, tolerability, and PK data guided our dose selection for later cohorts in the FIH study. PK samples were collected up to 168 h postdose in the initial cohort and extended to 840 h postdose in the later cohorts, after emerging data showed t1/2 in humans is longer than projected by the preclinical PBPK model, which was based on the overall t1/2 prediction from all preclinical species tested. The long t1/2 observed in humans is more in line with that observed from dogs than other preclinical species. We utilized both NCA and PopPK to analyze the PK data from the FIH study. NCA analysis showed that GDC‐0334 exhibited linear PK with a mean t1/2 greater than 9 days, and we observed accumulations in exposure following repeat dosing, consistent with our preclinical observation in dogs. 8 Tmax after oral administration in fasted condition was varied (3 to 7 h), with no trend based on dosage. Results from in vitro biopharmaceutical testing suggested that food has potential to increase GDC‐0334 exposure. Therefore, food effect was assessed at the anticipated therapeutic doses in the phase I study. The increase in GDC‐0334 exposures when dosed at 300 mg and 600 mg with either high‐ and/or low‐fat meal was more pronounced for the first 12 h postdosing, suggesting that food intake helps increase absorption in the upper gastrointestinal (GI) tract relative to the lower GI tract.
We used PopPK to determine the effect of food on GDC‐0334 PK, incorporating not only data from the dedicated food effect portion of the FIH study after a single dose, but the totality of PK data from the entire clinical trial. Higher shrinkage values and low RSE associated with Ka (fasted and fed) indicate the population values were precisely estimated, but data from some individual subjects might not be as informative, thus causing the individual estimates for Ka to shrink toward the population values. The PopPK results suggest GDC‐0334 disposition was dose‐proportional within the range of tested doses (i.e., 25 to 600 mg). We determined that all other investigated covariates are statistically insignificant, which we attribute to the FIH study’s limited heterogeneity and strict inclusion criteria.
We characterized the target engagement in humans using a population PK‐PD analysis, in order to accurately describe the typical PK‐PD relationship over a broad range of GDC‐0334 exposure. A similar strategy was implemented to provide target‐engagement information of calcitonin gene related peptide (CGRP) receptor antagonists in healthy volunteers and patients with migraine using a capsaicin‐induced DBF model. 13 We chose a priori a sigmoid Emax model because of obvious nonlinearity with dose after oral administration seen during exploratory graphical analysis and previous studies involving CGRP‐receptor antagonists that used capsaicin‐induced DBF as PD measurements. 14 , 15 As predicted by the preclinical PK‐PD and human PBPK models, the initial PD activity was observed at GDC‐0334 plasma concentrations greater than 30 ng/ml. The inhibitory effect was saturated at plasma concentrations greater than 250 ng/ml, underscoring the AITC‐skin challenge’s utility as a translational PD biomarker for TRPA1 activity. In humans, GDC‐0334 treatment approached complete inhibition of the AITC‐induced DBF, especially at concentrations greater than 250 ng/ml. This contrasts with observations in guinea pigs, in which maximal inhibition of DBF was saturated at ~ 70% of baseline, 8 which may be due to species differences in the TRPA1 specificity of the DBF response to AITC.
There are some limitations to the final PK‐PD model from the phase I trial. The model cannot assess hysteresis as we took only a single PD measurement after GDC‐0334 dosing. We did not collect DBF data at doses of GDC‐0334 between 200 and 600 mg. Furthermore, translatability of target engagement for GDC‐0334 in the skin may not imply target engagement at the site of action, and therefore the PD results do not imply efficacy for therapeutic indications, such as neuropathic pain, cough, or asthma. For comparison, results for CGRP receptor antagonists telcagepant and erenumab, which share a similar antineurogenic inflammation mechanism of action as TRPA1 inhibition, show that effective therapeutic doses required for efficacy in migraine were higher than those which saturated capsaicin‐induced DBF. 14 , 15
This study provides a blueprint for evaluating the PK and PK‐PD relationship of new therapies that target TRPA1. Although clinical development of GDC‐0334 was terminated earlier than anticipated due to toxicity finding in preclinical species, the population‐based models can be used in future clinical trials for predicting PopPK and PK‐PD relationships in future clinical trials, by simulating the therapeutic concentrations needed to achieve IC90 in the population of interest and by assuming DBF inhibition is predictive of efficacy with maximum DBF inhibition required for efficacy. This may expedite future therapies targeting TRPA1 into the clinic and allow them to move more efficiently through clinical development.
CONFLICT OF INTEREST
All authors are current or former employees and stockholders of Genentech/Roche.
AUTHOR CONTRIBUTIONS
P.C., H.T.D., B.M.L., J.M., L.C., S.S.G., V.J., X.Y., R.N.B., and L.P. wrote the manuscript. P.C., H.T.D., B.M.L., J.M., L.C., S.S.G., V.J., X.Y., J.S.L., P.B., M.S.M., W.S.P., A.Q., R.N.B., and L.P. designed the research. P.C., H.T.D., B.M.L., J.M., L.C., S.S.G., V.J., X.Y., J.S.L., P.B., M.S.M., R.N.B., and L.P. performed the research. P.C., H.T.D., B.M.L., J.M., L.C., S.S.G., V.J., X.Y., R.N.B., and L.P. analyzed the data.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Hanbin Li and Lu Liu of Certara for their contributions on the population PK analysis; Gregory Z. Ferl of Genentech for conducting the exploratory population PK‐PD model development; the following Genentech team members for support of the phase I study: Horace Rhee, Farah Gowgani, Joshana Amiel, Edward Cruz, Joshua Galanter, and Chandra Chopra; and Robby Weimer and Alex de Crespigny for input and advice on the skin challenge methodology and data analysis. We also thank colleagues and collaborators in the Safety Assessment and Drug Metabolism and Pharmacokinetics departments and at Covance and Ricerca Biosciences for contributions in generating data for this study. Editorial assistance was provided by Anshin Biosolutions, Corp.
Phyllis Chan, Han Ting Ding, and Bianca M. Liederer contributed equally to this research.
Rebecca N. Bauer and Lin Pan jointly led the strategy for the work.
Funding information
This work was supported by Genentech/Roche.
REFERENCES
- 1. EFPIA MID3 Workgroup , Marshall S, Burghaus R, et al. Good practices in model‐informed drug discovery and development: practice, application, and documentation. CPT Pharmacometrics Syst Pharmacol. 2016;5:93‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wong H, Bohnert T, Damian‐Iordache V, et al. Translational pharmacokinetic‐pharmacodynamic analysis in the pharmaceutical industry: an IQ Consortium PK‐PD Discussion Group perspective. Drug Dis Today. 2017;22:1447‐1459. [DOI] [PubMed] [Google Scholar]
- 3. Plock N, Vollert S, Mayer M, Hanauer G, Lahu G. Pharmacokinetic/pharmacodynamic modeling of the PDE4 inhibitor TAK‐648 in Type 2 diabetes: early translational approaches for human dose prediction. Clin Transl Sci. 2017;10:185‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Moran MM, McAlexander MA, Bíró T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Disc. 2011;10:601‐620. [DOI] [PubMed] [Google Scholar]
- 5. Verma VA, Shore DGM, Chen H et al. α‐Aryl pyrrolidine sulfonamides as TRPA1 antagonists. Bioorganic Med Chem Lett. 2016;26:495‐498. [DOI] [PubMed] [Google Scholar]
- 6. Joseph V, Yang X, Gao SS, et al. Development of AITC‐induced dermal blood flow as a translational in vivo biomarker of TRPA1 activity in human and rodent skin. Br J Clin Pharmacol. 2021;87(1):129‐139. [DOI] [PubMed] [Google Scholar]
- 7. European Medicines Agency, Committee for Medicinal Products for Human Use (CHMP) . Guideline on strategies to identify and mitigate risks for first‐in‐human and early clinical trials with investigational medicinal products, 2017. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐strategies‐identify‐mitigate‐risks‐first‐human‐early‐clinical‐trials‐investigational_en.pdf. Accessed September 15, 2020.
- 8. Balestrini A, Joseph V, Dourado M, et al. A TRPA1 inhibitor suppresses neurogenic inflammation and smooth muscle contraction in asthma. J Exp Med. 2021; 218(4):e2021637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mandema JW, Verotta D, Sheiner LB. Building population pharmacokinetic‐‐pharmacodynamic models. I. Models for covariate effects. J Pharmacokinet Biopharm. 1992;20:511‐528. [DOI] [PubMed] [Google Scholar]
- 10. Lindbom L, Pihlgren P, Jonsson N. PsN‐Toolkit—A collection of computer intensive statistical methods for non‐linear mixed effect modeling using NONMEM. Computer Met Prog Biomed. 2005;79:241‐257. [DOI] [PubMed] [Google Scholar]
- 11. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13:143‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Food and Drug Administration . FDA Guidance for Industry for Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers, 2005. https://www.fda.gov/media/72309/download. Accessed September 15, 2020.
- 13. Li C‐C, Vermeersch S, Denney WS, et al. Characterizing the PK/PD relationship for inhibition of capsaicin‐induced dermal vasodilatation by MK‐3207, an oral calcitonin gene related peptide receptor antagonist. Br J Clin Pharmacol. 2015;79:831‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vu T, Ma P, Chen JS, et al. Pharmacokinetic‐pharmacodynamic relationship of erenumab (AMG 334) and capsaicin‐induced dermal blood flow in healthy and migraine subjects. Pharmaceut Res. 2017;34:1784‐1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Buntinx L, Vermeersch S, De Hoon J. Development of anti‐migraine therapeutics using the capsaicin‐induced dermal blood flow model. Br J Clin Pharmacol. 2015;80:992‐1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material