

Abstract
Nonclinical testing has served as a foundation for evaluating potential risks and effectiveness of investigational new drugs in humans. However, the current two‐dimensional (2D) in vitro cell culture systems cannot accurately depict and simulate the rich environment and complex processes observed in vivo, whereas animal studies present significant drawbacks with inherited species‐specific differences and low throughput for increased demands. To improve the nonclinical prediction of drug safety and efficacy, researchers continue to develop novel models to evaluate and promote the use of improved cell‐ and organ‐based assays for more accurate representation of human susceptibility to drug response. Among others, the three‐dimensional (3D) cell culture models present physiologically relevant cellular microenvironment and offer great promise for assessing drug disposition and pharmacokinetics (PKs) that influence drug safety and efficacy from an early stage of drug development. Currently, there are numerous different types of 3D culture systems, from simple spheroids to more complicated organoids and organs‐on‐chips, and from single‐cell type static 3D models to cell co‐culture 3D models equipped with microfluidic flow control as well as hybrid 3D systems that combine 2D culture with biomedical microelectromechanical systems. This article reviews the current application and challenges of 3D culture systems in drug PKs, safety, and efficacy assessment, and provides a focused discussion and regulatory perspectives on the liver‐, intestine‐, kidney‐, and neuron‐based 3D cellular models.
INTRODUCTION
Traditional in vitro assays, that use recombinant enzymes, liver microsomes, and two‐dimensional (2D) cell cultures, such as primary hepatocytes and Caco‐2 cells, have been routinely used in the pharmaceutical industry to evaluate absorption, distribution, metabolism, and excretion (ADME) properties and drug risk assessments of novel molecular candidates in drug discovery and development. 1 However, these assays have difficulty mimicking the physiological environment of organs and therefore limited utilities in the extrapolation of data to complex in vivo environments. Over the past decades, researchers have developed better models to evaluate and promote the use of improved cell‐ and organ‐based assays for more accurate representation of human physiology and better prediction of drug response. 2 , 3 , 4 Currently, there are numerous developments of three‐dimensional (3D) cell culture systems, from simple spheroids to more complicated organs‐on‐chips and from single‐cell type static 3D models to cell co‐culture 3D models equipped with microfluidic flow control (Figure 1). Each provides different advantages and limitations (Table 1). Compared to 2D cell culture models, the emerging 3D models appear to have a better representation of the natural environment, experienced by cells under physiological and/or pathophysiological conditions, and offer a greater potential in assessing drug disposition and pharmacokinetics (PKs) that influence drug safety and efficacy at an early stage of drug development. 5 , 6 , 7 These systems offer complex microphysiological environments of human organs than 2D systems and allow for drug safety evaluation of low clearance drugs and multiple dosing studies, collection of critical PK and biomarker data for mechanistic modeling, and in‐depth evaluation of disease models from patient‐derived cells. However, the phenotypical performance and potential implications of these systems, due to their early stage in development, have not been fully illustrated. Moreover, how different types of cells communicate with each other under the 3D conformation and whether drugs can efficiently reach cells in the inner part of the 3D models are poorly understood. In August 2020, representatives from academic institutions, pharmaceutical industry, and federal government agencies convened an educational workshop for a scientific exchange about considerations and challenges in the use of novel 3D cell and tissue models in the evaluation of drug PKs, efficacy, and safety, as well as pathways to communicate with the US Food and Drug Administration (FDA) on the application of these models in early drug development. This article highlights the top‐line discussions from the workshop.
FIGURE 1.
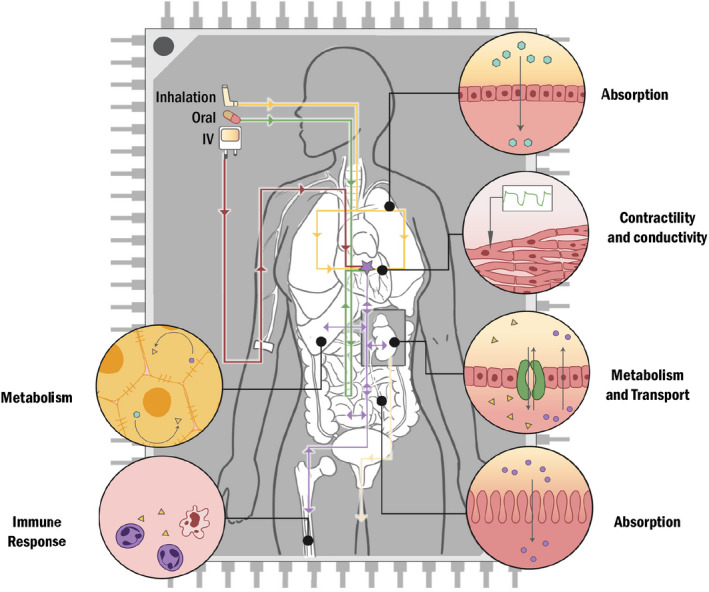
A general scheme of areas of 3D cell culture systems in development. The 3D cell culture systems are being developed to mimic physiological organs in the body to evaluate drug absorption, distribution, metabolism and excretion, cardiac function in the heart, and immune response in the lymphatic system. The optimal goal is to integrate these organ‐specific 3D models into a multiple interconnected microphysiological system, which allows the evaluation of different routes of administration of drugs on drug pharmacokinetics and to improved drug safety and efficacy assessment, and disease modeling
TABLE 1.
Summary of general characteristics of 3D cell culture systems
| Cell type | Purpose/advantages | Limitations a | |
|---|---|---|---|
| Liver models | |||
| Liver‐on‐chip | Cryopreserved primary liver cells |
|
|
| Spheroid co‐culture |
Cryopreserved primary cells |
|
|
| HepaRG spheroids | Differentiated HepaRG cells |
|
|
| Hepatocyte spheroids |
Cryopreserved or fresh hepatocytes |
|
|
| Micropatterned co‐culture | Cryopreserved or fresh hepatocytes |
|
|
| Intestinal models | |||
| Duodenum intestine‐chip |
Human intestinal organoids |
|
|
| Cryopreserved human intestinal mucosa |
Cryopreserved primary cells |
|
|
| Intestinal organoids | Freshly primary cells |
|
|
| Patient‐derived intestinal organoids | Patient‐derived primary cells |
|
|
| Kidney models | |||
| Kidney microphysiological system | Primary cells or iPSC‐derived kidney cells |
|
|
| Kidney organoids | iPSC‐derived renal cells or patient‐derived primary cells |
|
|
| Other models | |||
| Neuronal multi‐organs model | Primary cells or patient‐derived primary cells |
|
|
| Patient‐derived 3D tumor organoids | Patient‐derived tumor cells |
|
|
Abbreviations: iPSC, induced pluripotent stem cell; PBPK, physiologically‐based pharmacokinetic; PD, pharmacodynamic; PK, pharmacokinetic.
In this table, we intended to summarize the major limitations of each 3D system. Shortages, such as limited data on functional interplay between drug transporters and metabolism and unestablished in vitro to in vivo extrapolation, which are common to all 3D systems have not been listed under individual models.
3D IN VITRO LIVER MODELS
Liver‐on‐chip model for toxicity and PK evaluation
Modeling drug metabolism, transport, and other drug interactions in the liver is central for drug development because this first pass organ is responsible for the formation of most drug metabolites, contributes to drug clearance and bioavailability, and is sensitive to drug‐induced hepatotoxicity, which is among the main causes for drug attrition. 8 , 9 , 10 , 11 Cellular microsystems that enhance the physiology of hepatocytes or cocultured nonparenchymal cells (NPC) may predict liver effects early in drug development and further increase development efficiency by replacing, reducing, or refining animal studies or clinical trials. 2 , 5 , 12 A diversity of liver models have been developed in the last decade with primary or induced pluripotent stem cell (iPSC)‐derived human liver cells (Figure 2). 13 , 14 Liver on‐a‐chip or microphysiological systems (MPS) showed high potential for drug PKs and toxicity studies 15 , 16 , 17 through pilot studies that suggested their use in the early stages of drug discovery, and led industry stakeholders to produce guidelines for risk assessment and ADME contexts. 3 , 4 , 18 In addition to these performance guidelines, reliability and robustness of systems must be ensured for use on the late stages of drug development, regulatory context, or supporting clinical trials. Applied research is being developed in the FDA Division of Applied Regulatory Science in the Center for Drug Evaluation and Research (CDER), 19 where a commercially available liver MPS developed at the Massachusetts Institute of Technology 20 is being characterized for studying reliability and robustness of its use in drug toxicity and PK applications.
FIGURE 2.
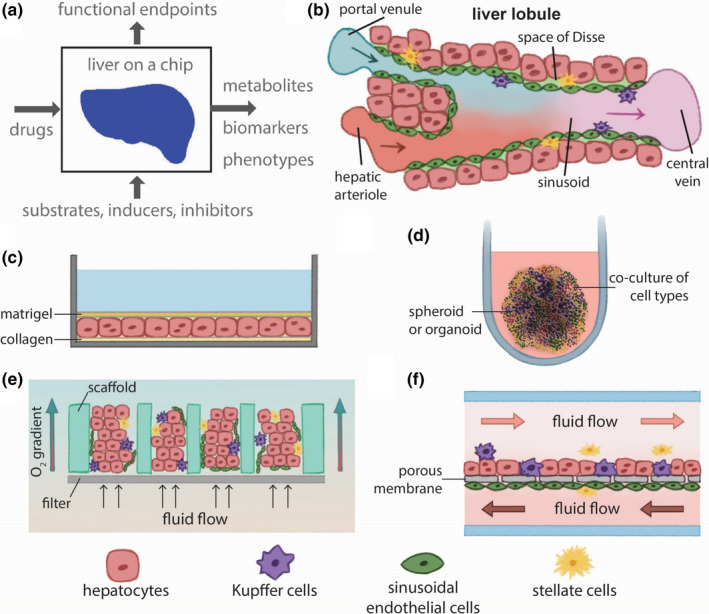
Structural characteristics and applications of liver microphysiological systems (MPS). (a) Metabolism, drug–drug interactions, biomarkers, transport, structure, and toxicity can be assayed from MPS samples. (b) The microenvironment of the liver lobule is multicellular, three‐dimensional, under flow, and defines the sinusoid. (c) Culturing hepatocytes in 2D between a bottom collagen layer and a top layer of Matrigel as sandwich cultures. (d) Different types of hepatic cells can be cultured as spheroids or organoids in 3D to recreate more physiological intracellular interactions. (e, f) Liver MPS has been designed in various configurations to maintain co‐cultures of hepatic cells under fluid flow. Scaffolds support the formation of microtissues, and oxygen gradients can be controlled by regulating the rate of flowing medium in (e). A porous membrane in a microfluidic chamber induces a barrier function by separating layers of endothelial cells from hepatocytes (f)
The characterized liver system cultures hepatic cells in a 3D platform exposed to flowing culture medium, 20 , 21 which enhances and prolongs metabolic activity. The system also uses large cell quantities (>500,000) and high volumes of medium (milliliter range), enabling the generation of replicate test samples for bioanalytical assays in PK studies. 4 This effort follows the Division’s mission of closing the gap between scientific innovation and drug review. 19 System robustness and reliability is being evaluated from the outcome of experiments assaying: (i) metabolism and albumin production relative to other hepatocyte culture platforms; and (ii) the reproducibility of results from experiments conducted in different laboratories and using distinct batches of cells. Lack of reproducibility of results is among the main hurdles in reliably translating MPS 22 and their robustness can be tested when being compared with in vitro gold standards. 23 , 24 Cell types selection, drug class, and specific drug concentrations, and sample/data collection schedules of functional end points or biomarkers should be planned depending on the intended context of use. For example, coculturing Kupffer cells with hepatocytes can recreate inflammatory factors that induce liver toxicity, 20 as known to be caused by trovafloxacin, an antibiotic. 25 , 26 In addition to coculturing Kupffer cells, inclusion of other hepatic cell types, like sinusoidal endothelial cells, stellate cells, and cholangiocytes can be of relevance when aiming to test drug effects that target mechanisms where these cell types are involved. 27 , 28 , 29 Access to primary sources of hepatic cell types can present difficulties, 30 but the iPSC field is evolving toward providing differentiation protocols that can eventually originate pure or complex mixed populations of these types of hepatic cells. 14 In regard to pharmacology context, given the enhanced stability and duration of its function, 31 the system can address opportunities to eventually improve on the scope of current in vitro approaches by enabling the detection of drug metabolites formed within days of drug exposure and the study of compounds with low clearance. Future studies can address this opportunity because the functional end points measured from microsomes or primary hepatocytes do not last long in vitro and can limit the evaluation of drugs with low clearance. 32 , 33
The ability to derive drug ADME data from liver MPS that has higher functional stability, robustness, and physiological relevance than traditional 2D cultures or other in vitro methods has been noted to potentially having the ability to improve the development of physiologically based pharmacokinetic models early in drug development and in advance of clinical studies. 4 , 34 Pilot studies with interconnected organ systems have demonstrated the ability to predict key PK parameters, such as compound terminal half‐life, peak serum concentration, and others from the quantification of parent drug and metabolites in samples (media, lysate, adsorbed to materials) collected from MPS at different times of drug incubation. 17 , 35 For this purpose, liver systems have been demonstrated in early pilot studies to estimate intrinsic hepatic clearance and elimination rate constant, which are key properties for modeling drug PKs. 15 , 20 , 36 Despite this demonstrated use of liver MPS, the added benefit of these systems relative to traditional 2D platforms is still unclear for this application. To further elucidate the value of liver MPS in predicting clinical drug PKs, initial efforts should be continued while including additional types of drugs, comparing data with clinical results, and with the results of traditional 2D culture platforms. Overall, MPS can prolong and mature hepatocyte function and enhance the physiological relevance of their extracellular microenvironment, which are the advantages of these systems relative to 2D platforms in contexts of use to be established.
Physiological transport function in several organ models like liver, kidney, gut, or blood‐brain barrier is a fundamental requirement for predicting drug pharmacology. 9 , 34 Caetano‐Pinto and Stahl 34 have reviewed initial characterization efforts in the field around the activity of drug transporters in several MPS models, including the liver, which depends on the types of used cells, but also on exposure to fluid flow and culture in 3D settings. In addition to increased expression levels of relevant transporters, functional activity also relies on the cellular localization of transporters related to hepatocyte polarity in primary hepatocytes gold standard. 37 Despite the initial characterization of transport function, additional comparative studies between culture platforms should be done to comprehensively define how liver MPS models can improve the physiological relevance of drug transport of cultured cells relative to traditional 2D culture methods. Furthermore, studies using the liver system demonstrated that quality control criteria for cells and performance standards are sorely needed and critical for ensuring reproducibility.
Prediction of drug‐induced liver injury risk using hepatic spheroid co‐culture models
Despite many recent advances to improve hazard identification and risk mitigation, drug‐induced liver injury (DILI) continues to be a significant source of preclinical and clinical drug attrition. This is due to the many different etiologies of DILI, general insensitivity of preclinical animal models to detect many human hepatotoxicants, and poor understanding of the drivers for patient susceptibility. There is a continued need to identify new approaches that can better identify and characterize the risk for DILI during drug discovery. The 3D hepatic spheroids or human liver microtissues (hLiMTs), comprising self‐aggregated primary human hepatocytes cocultured with NPC, have emerged that have the potential to improve the ability to assess mechanisms of hepatotoxicity. They have demonstrated enhanced liver phenotype, stability in culture not attainable with conventional hepatic models, and increased sensitivity to drug‐induced cytotoxicity of a relatively small panel of hepatotoxicants. 38 , 39 , 40 However, comprehensive assessments of the predictivity of hLiMTs to identify known hepatotoxic drugs has been generally lacking. In a collaboration between AstraZeneca and Genentech, the predictive value of hLiMTs and 2D hepatocytes to identify known hepatotoxicants was assessed using a panel of 110 drugs with and without clinical DILI. Using cytotoxicity as an end point, the group evaluated the responses in 2D hepatocytes after 2‐day single‐dose treatment and in 3D hLiMTs following 14‐day repeat‐dose treatment and compared the predictive value in isolation or when correcting the cytotoxicity half‐maximal inhibitory concentration (IC50) values to the clinically relevant plasma exposures to determine an in vitro margin of safety (MOS). In this study, hLiMTs demonstrated increased sensitivity in identifying known hepatotoxicants in comparison to the 2D assay, whereas high specificity (>85–90%) was consistent across both assays regardless of comparing cytotoxicity IC50 values alone or exposure‐corrected MOS values. 41 In particular, hLiMTs had approximately double the sensitivity to identify known hepatotoxicants when applying a 25 times MOS threshold than the 2D primary plated hepatocytes assay (48% vs. 27%, respectively), while maintaining nearly identical specificity (93% vs. 95%, respectively). 41 This finding was significant as high assay specificity is an important criterion when supporting drug discovery, where there is a need to minimize deprioritizing otherwise good compounds unnecessarily (e.g., “throwing the baby out with the bathwater”).
Although there is evidence supporting the utility of hLiMTs to predict DILIs, significant effort is still needed to qualify these tools to support drug discovery and development. In this retrospective study, hLiMTs failed to identify roughly 40–60% of known hepatotoxicants depending on the thresholds applied. 41 It is likely that particular mechanisms that were responsible for the clinical hepatotoxicity observed for those compounds were not recapitulated by hLiMTs, the primary human hepatocytes or NPC used, or by the end point assessed (e.g., cytotoxicity). For example, varying the source of NPC affected the degree of nanoparticle‐induced toxicity in hLiMTs. 42 Moreover, more comprehensive computational approaches incorporating population‐relevant responses from hLiMTs may provide further insight into both the predictive power of hLiMTs for assessing DILIs broadly and for elucidating mechanisms of DILIs observed for a particular compound or drug. This approach was taken by Tsamandouras et al. in 2017 in the context of assessing population variability in metabolism to predict model plasma PKs. In this report, the authors were successful in recapitulating the observed clinical concentration‐time profiles and the associated population variability of lidocaine by quantitative systems pharmacology modeling with data from the variable responses in metabolism observed in perfused 3D hepatic spheroids generated from multiple different human hepatocyte donor lots. 36 Moving forward, continued work assessing additional end points, patient/population variability in responses, and computational methods for integrating these data into predictive DILI models are needed. Last, it is important to closely examine the chemical properties profiled in these retrospective studies and address any gaps or imbalances related to the contemporary chemical space. Multiparametric approaches to assesses DILI risk are becoming more common across the industry. 43
HepaRG spheroids and 2D models in metabolism and toxicity prediction
Recent advances with free‐floating 3D hepatocyte spheroid screening models have revealed remarkably enhanced cellular function and differentiation that have begun to translate into an explosion of applied research. 44 Hallmarks of functionality in comparison with conventional 2D configurations reveal drug metabolism enzymatic activities on par with suspensions of primary human hepatocytes directly isolated from human liver. Typical gel‐based spheroid models or 2D dynamic flow models increase metabolism, but generally do not achieve these levels of metabolic competence, suggesting free‐floating spheroids display a unique combination of 3D architecture and nutrient/waste exchange. In addition, unlike 2D HepaRG cultures that require extremely high concentrations of DMSO (e.g., 2%) and only induce a subset of drug metabolizing enzymes that preclude their proficiency for liver enzyme induction, 45 3D HepaRG spheroids are markedly inducible by activators of major nuclear receptor pathways over and above their physiologically relevant baseline metabolism. 46 In combination, these functional characteristics rival zone‐2 hepatocyte functionality not achieved with conventional 2D culture models. Furthermore, compounds known to require metabolic activation of hepatotoxicity (e.g., troglitazone, acetaminophen, valproic acid, and cyclophosphamide) cause markedly more severe cytotoxicity in 3D spheroid over 2D configurations following repeated exposures. 38 Given their highly efficient use of hepatocytes, compatibility with higher throughput screening, remarkable balance of baseline and inducible metabolism, and utility for modeling metabolically activated liver toxicity, 3D HepaRG spheroids represent an excellent system for routine screening within a consistent genetic background.
In addition to xenobiotic metabolism, Ramaiahagari et al., 47 further evaluated 2D versus 3D HepaRG cultures using various techniques including high‐throughput transcriptomics. Direct lysis and highly multiplexed gene expression assays enable efficient survey of biological response “space,” and reveal characteristic responses with DILI compounds that are consistent with clinical observations and significantly differ from their conventional 2D culture counterparts.
There are three major limitations with free‐floating HepaRG spheroid screening models. (1) Exchange of cell culture medium requires liquid handling to minimize spheroid loss and reduce variability, (2) small biomass (i.e., 1000–2000 cells) optimized to eliminate the formation of necrotic centers over time that creates challenges for certain assays (e.g., metabolic clearance, RNA isolations, and lower sensitivity protein secretion assays), and (3) the ability to clearly image cell morphology using conventional laboratory systems. For each of these challenges, solutions are available, and their implementation will depend on the relative requirements for various applications.
Micropatterned hepatocyte coculture for drug metabolism and toxicity study
Liver architecture has been maintained using microfluidic chips, formation of spheroids, and the bioprinting of 3D tissues, however, there is a tradeoff among complexity, assayable end points, and throughput. A micropatterned coculture (MPCC) system, which allows for the control of the hepatocyte homotypic interactions and heterotypic interactions with stroma cells, was developed to maintain cultures that would provide improved in vivo predictivity. 48 This model commercialized as HEPATOPAC, consisted of multiple islands of hepatocytes surrounded by stromal cells patterned in a standard multi‐well plate with island size and spacing controlled. This allowed for maintenance of hepatic function, including phase I and II drug‐metabolizing enzymes, over 4 weeks extending the utility of isolated hepatocytes.
The maintenance of high‐level hepatic function over long durations in the HEPATOPAC system has allowed for the determination of hepatic clearance and metabolic fate of compounds whose rate of turnover may not be accurately predicted in the short duration of standard suspension metabolism assays, typically hours. This system was able to elucidate accurate metabolic stability, the metabolite profile, and route of excretion of faldaprevir, a low turnover compound with high levels of transporter interaction. The compound’s fate included uptake transport, metabolism by P450, and glucuronidation enzymes, as well as excretion into the biliary system all which could be recapitulated in the HEPATOPAC model. 49 The advantages of biological complexity offered in microtissues and spheroids, however, comes with a limitation in ease of use or throughput. Utilizing a standard multi‐well plate format with imageable hepatic islands allows for the MPCC to be utilized in automated and high‐content imaging (HCI) systems, incorporating multiple end points with quantitation down to the single cell level. This high assay density and throughput of HCI methods allow for the elucidation of mechanism as well as potency of toxic compounds. 50 The maintenance of in vivo like regulation of transcriptional pathways over long periods of culture was key to generating a repeat dose toxicity test in HEPATOPAC for determining the DILI potential. Recently, an assay was developed by scientists at Merck incorporating a 9‐day drug exposure and analysis of transcriptional profiles of the NRF1/NRF2 pathways to predict DILI potential. 51 In rat HEPATOPAC, an 81% sensitivity and 90% specificity correlating to in vivo toxicity was achieved showing the utility of the model and a transcriptomic signature‐based approach. Incorporation of Kupffer cells for the investigation of immune interactions and their contribution to toxicity or drug‐drug interactions (DDIs) have shown promise to elucidate behavior of large molecules, such as therapeutic proteins. 52 Meanwhile, limitations of the MPCC system, including relatively lower cell number than traditional multi‐well cultures as well as the coculture, contains nonhepatic cells call for further optimizations. Clearly, choosing appropriately complex models that recapitulate hepatic architecture while maintaining usability are key to accurate predictions of in vivo behavior for drug development in an efficient and cost‐effective manner.
3D IN VITRO INTESTINE MODELS
Intestine‐chip for drug metabolism and toxicity evaluation
Through a combination of design and engineering, Organ‐Chips recreate the epithelial‐endothelial interface, provide mechano‐actuation to emulate breathing or peristalsis, and utilize microfluidics to develop shear stress while maintaining nutrient‐waste homeostasis. Together, these features offer an unprecedented insight into biological mechanisms and have been shown to more accurately recreate in vivo relevant phenotypes, hence becoming attractive tools to the pharmaceutical industry and beyond. 53
The intestine is a common site for drug toxicity with gastrointestinal adverse effects frequently reported in clinical trials. 54 Although not usually life‐threatening, these toxicities impact significantly on quality of life and can affect drug efficacy as well as reduce patient compliance. Improved in vitro models are required to increase the probability of drug success in the clinical trial process through earlier identification of toxic liability in addition to the development of mitigation strategies.
Drug absorption is a major function of the intestine, especially in the regions of the duodenum and jejunum, making it susceptible to drug toxicity. Kasendra et al. 55 have developed and characterized a duodenum Organ‐Chip model using Emulate’s Human Emulation System. Briefly, dissociated human duodenum intestinal organoids were seeded on the top channel of a chip coated with extracellular matrix. In the lower parallel channel of the chip, primary human small intestinal microvascular endothelial cells (siHIMECs) were seeded. The cells were perfused at 30 µL/h and exposed to a 10% cyclic mechanical strain at a frequency of 0.2 Hz (Figure 3). Such configurations enable the multilineage differentiation of epithelial cells into absorptive enterocytes, goblet cells, enteroendocrine cells, and Paneth cells at ratios comparable to those found in the native human intestine. Transcriptomic analysis revealed a common subset of 305 genes between the Organ‐Chips and human tissue, but not with organoids alone, with overlapping genes associated with drug metabolism, digestion, nutrient transport, and detoxification. Gene expression of the major efflux and uptake transporters as well as the metabolizing enzyme CYP3A4 and nuclear hormone receptors 1,25‐(OH)2‐vitamin D3 receptor and pregnane X receptor, were also more comparable to adult human duodenum. These data propose that the duodenum Organ‐Chip could be used for prediction of drug‐induced toxicity, drug absorption, and metabolism. 55
FIGURE 3.
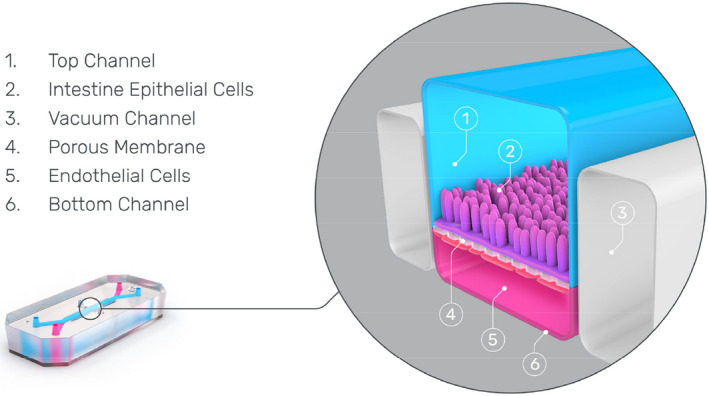
A cross‐sectional view through an Intestinal Organ‐Chip that contains two parallel microfluidic channels. The upper luminal channel is where fragmented intestinal organoids are seeded and the lower vascular channel is where intestine‐specific endothelial cells are seeded. The grey channels on either side of the chip enable the vacuum to be applied, emulating the peristaltic motion of the intestine
Currently, unpublished data using indomethacin, a common toxicant of the gastrointestinal tract, were shared during a recent workshop (https://www.emulatebio.com/intestine‐chip‐toxicity‐application‐note#download). In brief, following 3 days treatment with indomethacin (0.5 mM), epithelial cells appeared stressed, whereas no morphological changes were observed in the endothelial cells. Accumulation of intestinal fatty‐acid binding protein, a marker of absorptive enterocyte viability, was measured in chips exposed to indomethacin from day 1 but declined on day 3. This decline was reflected by a large increase in lactate dehydrogenase, indicative of cell death. Barrier function, essential for normal functionality of the gastrointestinal tract, demonstrated a lower integrity following three days of indomethacin treatment, indicative of cellular injury. Taken together, these data propose that the intestinal Organ‐Chip model is suitable for assessing potential drug‐induced toxicity.
Novel in vitro enteric models for DDIs and safety evaluation
A novel in vitro enteric experimental system, cryopreserved human intestinal mucosal epithelium (CHIM), has been developed for the evaluation of enteric drug metabolism, DDI, drug toxicity, and pharmacology. CHIM are isolated from the small intestines of human donors via collagenase digestion of the intestinal lumens 56 followed by gentle homogenization to yield 3D, multiple cellular fragments retaining cell junctions, and multiple cell types as that in vivo. Upon isolation, CHIM are cryopreserved and stored in liquid nitrogen. After recovery from cryopreservation, CHIM are found to retain high viability and robust drug‐metabolizing enzyme activity. Drug‐metabolizing enzyme activities quantified in CHIM include P450 isoforms CYP1A1, CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1, CYP2J2, and CYP3A4, with CYP2A6 as the only isoform with negligible activity. Non‐P450 activities expressed in CHIM include UGT, SULT, FMO, MAO, NAT1, NAT2, and AO. Inter‐regional and interindividual evaluation of enteric drug metabolism have been evaluated with CHIM, with the proximal jejunum in general exhibiting higher drug metabolizing enzyme activities than those in duodenum and ileum, and with significant interindividual variations, especially for the inducible P450 isoforms. 57 , 58 CHIM is found to be a useful experimental model for the evaluation of first‐pass intestinal metabolism of orally administered drugs for the estimation of the fraction escaping gut metabolism, especially for moderately and rapidly metabolized drugs. 59 CHIM has been applied toward the evaluation of DDI potential of commercial formulations of herbal supplements, identifying green tea extract, St. John’s wort, valerian root, horehound, and grapefruit juice as having potent CYP3A inhibition/induction potentials, and suggesting potential for herb‐drug interaction with orally administered drugs that are CYP3A substrates. 60 CHIM as an experimental tool for the evaluation of intestinal drug toxicity is illustrated by dose‐dependent cytotoxicity of the nonsteroidal anti‐inflammatory drugs acetaminophen and naproxen, with naproxen exhibiting a higher enterotoxicity than acetaminophen as in humans in vivo. 56 The robust drug‐metabolizing enzyme activity is a major advantage of CHIM over crypt cell/stem cell‐derived and cell line‐based in vitro enteric models. 61 The overall results suggest that CHIM represents a convenient and effective 3D in vitro experimental model, which, together with results obtained with other metabolically competent enteric models, including human intestinal slices, 62 , 63 cryopreserved human enterocytes 64 and permeabilized, cofactor‐supplemented (MetMax) cryopreserved human enterocytes 65 can be used for the assessment of human enteric drug properties, including drug metabolism, DDIs, and drug toxicity. Future developments with CHIM include the establishment of experimental approaches for the evaluation of transporter‐mediated efflux and uptake, which are mainly evaluated with cell lines, such as Caco2 cells that are deficient in drug metabolizing enzyme activities. 66
Patient‐derived 3D intestine model for drug evaluation
Patient‐derived intestinal organoid technologies provide a novel platform to investigate drug treatment of enteric diseases including cystic fibrosis, immunological disease, and cancers. Specifically, these cultures have been implemented to investigate mechanistic alterations in cells contributing to these disease states and as screening tools for potential therapies. The ability to expand patient‐derived intestinal organoids in vitro long‐term allows for advanced laboratory‐based disease modeling and treatment screening.
Cystic fibrosis (CF) has been modeled using intestinal organoids isolated from rectal biopsies of patients with CF. CF is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene, which mediates fluid and electrolyte homeostasis. Activation of CFTR results in the rapid swelling of organoids generated from a healthy rectum, colon, ileum, or duodenum tissue, whereas rectal organoids derived from patients with CF show diminished response to treatment with the CFTR activator forskolin. 67 These healthy and diseased organoids have been utilized to predict which drugs will be most effective in patients with various mutations in the CFTR gene and as an alternative tool to measure functional drug levels in blood versus standard PK monitoring. In an encouraging application of organoid technology paired with gene therapy, CRISPR/Cas9 gene editing has been used to correct a CFTR gene mutation in small intestinal and colorectal organoids isolated from two patients with CF. Using the above‐described swelling assay, Schwank et al. 68 demonstrated that the corrected allele was fully functional.
High‐throughput screening (HTS) assays using cancer‐derived cell lines have been used to derive correlations between drug sensitivity and the genetic and mutational background of the source tumor. However, immortalized cell lines often do not faithfully recapitulate human tumor histology and gene expression, and the generation of these cell lines is costly and inefficient. Organoid cultures generated from patient‐derived gastrointestinal tissue may provide a platform to improve patient‐specific treatment strategies. The study by van der Wetering et al. successfully isolated organoids from cancerous colorectal tissue alongside healthy tissue in 2015. These isolated organoids were utilized in an HTS assay to assess their sensitivity to a range of antineoplastic drugs. 69 Later, Sato and colleagues established a bank of organoids from 55 colorectal tumors representing a range of histological subtypes. Importantly, the organoids isolated from colorectal tumors maintained the histological and genetic tumor characteristics native to the source tissue. 70
Patient‐derived intestinal organoids are also being optimized as HTS tools. In 2016, Velasco and colleagues developed an automated 384‐well platform, which subjected organoids isolated from tissue samples from patients with colorectal cancer to treatment with various drugs and the system was validated for robustness and reproducibility. 71 This technology provides a platform, which provides increased physiological relevance than existing 2D cultures without sacrificing screening throughput. These cultures provide a useful tool to investigate the underlying correlation between tumor genetics and drug response as well as performing ex vivo drug testing on cultured tumor tissue from individual patients, another key step toward precision treatment of gastrointestinal cancers.
Regulation of gut epithelia renewal and barrier function in intestinal organoids
The mammalian intestinal epithelium constantly self‐renews throughout adult life and acts as a physical barrier between the luminal microbiota and the body. Nonetheless, disrupted integrity of the intestinal epithelium occurs commonly in critically ill patients, leading to the translocation of luminal bacteria to the bloodstream and, in some instances, resulting in multiple organ dysfunction syndrome and death. 72 , 73 Primary cultured intestinal organoids are an excellent ex vivo model for studying gut epithelium homeostasis and have provided an enormous stimulus to the fields of gut mucosal biology and pharmacology. The following studies, using intestinal organoids as a model, advance our knowledge about the roles of long noncoding RNAs (lncRNAs) and RNA‐binding proteins (RBPs) in the regulation of intestinal epithelial renewal and barrier function.
LncRNAs are defined as transcripts spanning greater than 200 nucleotides in length, capable of regulating a variety of cellular processes. 73 Uc.173 is a member of the novel class of lncRNAs transcribed from genomic ultraconserved regions. Results obtained from studies conducted in cultured intestinal epithelial cells (IECs), in ex vivo‐prepared organoids, and in animals revealed that uc.173 is essential for normal epithelial renewal in the small intestine. 74 An intestinal organoid could be initiated from a single proliferating cell, but by 3 or 4 days after culture, the structures of organoids consist of multiple cells and buds in both control and uc.173‐transfected organoids. Strikingly, however, ectopic expression of uc.173 activates DNA synthesis in multiple cells markedly, and augments the surface area of intestinal organoids. The numbers of cells per organoid also increase after uc.173 overexpression. In contrast, uc.173 silencing inhibits the growth of intestinal organoids and disrupts the epithelial barrier function. 75 These results indicate that uc.173 is a critical regulator of the gut epithelium homeostasis and its induction enhances epithelial renewal and barrier function.
Many RBPs associate with specific subsets of mRNAs and control gene expression in response to pathophysiological stresses. HuR is among the most prominent sequence‐specific translation and turnover regulatory RBPs. Recently, HuR was found to regulate gut epithelium defense and barrier function by altering Paneth cell (lysozyme‐positive cell) function. 76 Loss of HuR in mice leads to lysozyme granule abnormalities in Paneth cells and it also results in gut barrier dysfunction and autophagy inactivation. Paneth cells in the organoids isolated from control mice are highly enriched, but they decrease dramatically and are almost undetectable in the organoids generated from HuR−/− mice. HuR deletion also inhibits growth of the intestinal organoids. HuR−/− mice do not have the apical distribution of Toll‐like receptor 2 in the intestinal mucosal tissues and organoids as observed in control mice. Together with results from cultured IECs, these findings strongly suggest that both specific LncRNAs and RBPs contribute to gut barrier dysfunction in intestinal pathologies.
3D IN VITRO KIDNEY MODELS
Kidney organoids and MPS for disease modeling and HTS
Kidneys maintain the precise balance of solutes in the blood and eliminate toxins through the urine. Kidney tissue is complex, and its fundamental subunit—the nephron—cannot undergo epimorphic regeneration. Nephron loss leads to kidney disease, affecting up to 10% of the world’s population. Therapy is largely limited to dialysis or transplantation, which have substantial limitations and side effects.
Several years ago, the first techniques were innovated to differentiate human pluripotent stem cells into the kidney lineage, culminating in kidney organoids, complex structures that resemble primitive nephron arrays. The hallmark of kidney organoids is formation of distal tubules, proximal tubules, and podocytes (filtering cells) in contiguous segments along a distal‐to‐proximal axis. 77 In total, organoids contain ~16 cell types, by single cell RNA sequencing. 78
Kidney organoids can be readily applied to understand pathophysiology and discover candidate therapeutics. A major advantage is that these organoids are of human origin, whereas mouse models may not replicate human disease or its treatment. Moreover, organoids are higher‐throughput and less expensive compared with animals. An impressive example is polycystic kidney disease, in which tiny tubules swell to form balloon‐like sacs of fluid (cysts). Kidney organoids with knockout mutations in the PKD1 or PKD2 disease genes form large cysts, which are not observed in isogenic controls and resemble human cysts phenotypically. 79 , 80 Organoids can also model glomerulosclerosis and even coronavirus infection. 79 , 81
To facilitate drug discovery, organoid differentiation has been adapted for 96‐ and 384‐well plate formats amenable to HTS using liquid handling robots (Figure 4a). 78 A dose‐dependent pilot screen of eight microenvironment‐related factors identified an inhibitor of myosin II as a positive modulator of kidney cystogenesis. 78 As myosin was not previously implicated in this disease, this finding suggests new mechanisms and therapeutic approaches.
FIGURE 4.
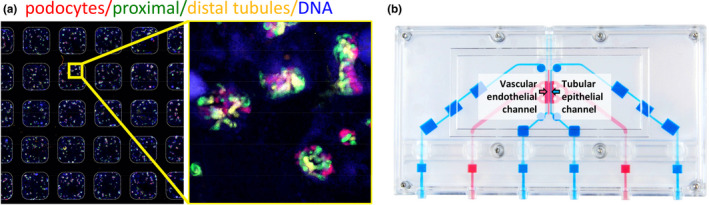
Kidney microphysiological systems. (a) Kidney organoids in a 384‐well plate generated with liquid handling robots, with immunofluorescence co‐localization of nephron segment markers. (b) Kidney on a chip device. Blue channel is seeded with primary proximal tubular epithelial cells and includes luminal perfusion. Pink channel is seeded with human umbilical vein endothelial cells and perfused with the drug to reflect basolateral transport
Approximately 10% of drugs fail because of kidney toxicities, and this occurs disproportionately in phase III clinical trials. Existing platforms based on primary or immortalized cells in vitro frequently fail to accurately predict nephrotoxicity in vivo. Kidney organoids are sensitive to nephrotoxic chemicals, and can express the biomarker, kidney injury molecule‐1. 79 A current challenge is to optimize organoid nephrotoxicity assays for HTS to test whether organoids are predictive of toxicity in the clinic.
It remains critical to establish the relevance of organoids for drug safety assessments for regulatory bodies, such as the FDA. Likewise, studies of drug metabolism and PKs in organoids would be valuable for understanding their predictive value. There is still ample room to improve this technology. Batch‐to‐batch and well‐to‐well variability can creep in during the weeks required to differentiate these structures, raising challenges for reproducibility. 78 Organoids remain less mature than bona fide tissues, contain non‐kidney contaminants, and lack integrated vasculature, rendering them unsuitable for transplantation. 82 Although organoids have an impressive degree of organization and cell type diversity, there is no filtration through the podocytes or perfusion through the tubules of these structures. These remain significant limitations for modeling physiological functional metrics of the kidneys, such as urine output or glomerular filtration rate.
Kidney MPS present a complementary technology to organoid cultures, which addresses some of these limitations. Typically, these MPS consist of bioengineered cylindrical molds that can be seeded with kidney tubular cells, enabling microfluidic perfusion of the resultant tubules via input and output ports. This system is lower throughput, compared to organoids, but incorporates flow through the tubule. As with organoids, treatments or compounds can be added to kidney MPS to study transport processes and features of disease. For example, human serum albumin in a guanidinylated state, found in patients with chronic kidney disease, shows reduced capacity to promote indoxyl sulfate clearance (secretion) via OAT1‐overexpressing cells in a kidney MPS, compared to unmodified albumin. 83 Kidney MPS can also be coupled to other MPS in a fluidic series to produce system‐wide insights into disease processes. For instance, coupling of a liver‐on‐a‐chip MPS to a kidney‐on‐a‐chip MPS enables reconstitution and elucidation of aristolochic acid bioactivation, transport, and nephrotoxicity in vitro. 84
Kidney MPS are typically seeded with primary cells harvested from fresh kidney tissue, which retain differentiated characteristics for a few passages in culture. Such cells are of limited availability, and their features can vary substantially from one patient to the next. As primary kidney cells are not immortal, they are less suitable for genome editing, compared to pluripotent stem cells. Currently, these challenges surrounding cell sourcing are a substantial impediment to the application of MPS for kidney disease modeling. The absence of podocytes in kidney MPS also limits the field’s ability to study the filtration mechanisms of the kidney glomerulus.
For kidney disease modeling, the relative advantages and disadvantages of MPS versus organoids are largely complementary. Either of these systems can be productive, depending on the question at hand. There is also potential for organoids and MPS to be combined into “organoid on a chip” systems with the benefits of both technologies. Despite limitations, human organoids and MPS have emerged as go‐to models for the kidneys, with vast potential for new disease models and complex architectures that inch ever closer to the real thing.
Human kidney MPS to generate data for physiologically‐based pharmacokinetic modeling
Prediction of renal clearance of drugs that are subject to active transport in the kidneys is challenging, and, at present, no reliable system for in vitro to in vivo quantitative predictions of renal clearance have been established. Although transporter expression in proximal tubule cells has been quantified and transporter quantification can be done by liquid chromatography tandem mass spectrometry, 85 , 86 expression can vary between in vitro systems with culture time and conditions and the localization of the transporters within the cells can be challenging to determine. 87 , 88 The lack of quantitative knowledge of uptake and efflux transporter expression in the proximal tubular cells in vivo and lack of quantitative knowledge of contribution of individual uptake and efflux transporters makes scaling of transport clearance from in vitro to in vivo challenging. Similarly, whereas Caco‐2 and MDCK cells can be used to elucidate the passive permeability of drugs, these cell systems may not reflect the permeability of human proximal tubule or the extent of microvilli in the proximal tubule cells. Many 2D culture systems that model proximal tubular cells, including primary cells, lack transporter expression and localization and therefore are not useful for predicting renal secretion clearance. Recently, MPS of proximal tubule (proximal tubule on a chip) have been developed to enable characterization of renal secretory clearance in the kidney, 89 , 90 although these systems still have challenges with potential flow limitations in drug transport and lack of fully physiological arrangement of the kidneys, including filtration, tubular reabsorption, and active transport. For some applications, the low cell numbers used can also be a challenge. However, MPS that include tubular flow, facilitate appropriate transporter localization and activity. As such, an MPS with a known content of primary proximal tubular cells can be used to determine the passive permeability clearance and active transport (secretion) clearance of drugs in a dynamic system. The transporters contributing to the renal clearance of the drug of interest can generally be identified via the use of transporter inhibitors and by measuring the impact of inhibitors on substrate secretion, although these experiments can be challenging due to the lack of specific probes and inhibitors of renal transporters. In essence, the kidney tubule MPS can be used to define the tubular secretion and permeability clearances at individual tubule scale. These clearances can then be scaled to the whole kidney via the knowledge of the number of proximal tubular cells in vitro and in vivo. However, such scaling alone is not sufficient to predict overall renal clearance as it does not incorporate glomerular filtration and passive reabsorption that occur in the kidneys. Notably, the secretion and passive permeability data defined in the chip system can be incorporated together with plasma protein binding information into a physiologically based mechanistic model of the human kidneys. 91 Such combination of in vitro data generated using the MPS and physiologically‐based pharmacokinetic (PBPK) modeling provides a cutting edge powerful tool to predict renal clearance of drugs and metabolites and DDIs that may involve renal transporters. In addition, once the in vitro data are generated, PBPK modeling can be further used to predict how renal clearance of the drug of interest declines in renal impairment or with progression of chronic kidney disease. 92 Taken together the combination of the proximal tubule on a chip system with PBPK modeling (Figure 4b) provides great promise to predict renal clearance, that offers hope to bring renal clearance predictions to par with predictions of metabolic, hepatic clearance.
OTHER 3D MODELS FOR DRUG EVALUATION
Neuronal multi‐organ‐on‐chip model for disease and risk assessment
One of the primary limitations in drug discovery and toxicology research is the lack of reliable model systems between the single cell level and up to in vivo animal or human systems. This is especially true for neurodegenerative diseases, such as amyotrophic lateral sclerosis (ALS) and Alzheimer’s disease (AD) as well as many rare diseases. In addition, with the banning of animals for toxicology testing in many industries, body‐on‐a‐chip systems could be used to replace animals with human mimics for product development and safety testing as well as augment their use in preclinical drug discovery. One specific research focus is on the establishment of functional in vitro systems to address this deficit by creating organs and subsystems to model motor control, muscle function, myelination, and cognitive function, as well as cardiac and liver subsystems. The idea is to integrate microsystems fabrication technology and surface modifications with protein and cellular components, for initiating and maintaining self‐assembly and growth into biologically, mechanically, and electronically interactive functional multicomponent systems. 93 A specific embodiment of this technology is the creation of a functional human neuromuscular junction (NMJ) system to understand ALS (Figure 5). The primary mutations found in patients with ALS included SOD1, FUS, TDP43, and C9ORF72. Guo et al. 94 recently published a functional evaluation for SOD1 and FUS where deficits were reversed by a therapeutic treatment. The ALS‐MNs reproduced pathological phenotypes, including increased axonal varicosities, reduced axonal branching and elongation, and increased excitability. These MNs form functional NMJs with wild type muscle, but with significant deficits in NMJ quantity, fidelity, and fatigue index. Furthermore, treatment with the Deanna protocol was found to correct the NMJ deficits in all the ALS mutant lines tested. Quantitative analysis also revealed the variations inherent in each mutant line. This functional NMJ system now provides a platform for the study of both fALS and sALS and has the capability of being adapted into subtype‐specific or patient‐specific models for ALS etiological investigation and patient stratification for drug testing.
FIGURE 5.
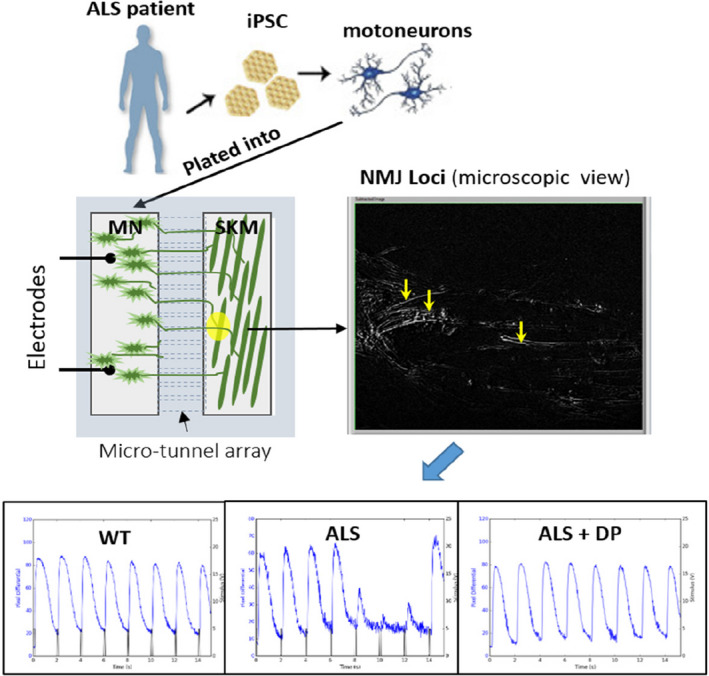
A functional neuromuscular junction (NMJ) model for amyotrophic lateral sclerosis (ALS). Motoneurons (MNs) differentiated from patients with ALS induced pluripotent stem cells (iPSCs) were introduced into a chambered NMJ system and, cocultured with primary myoblasts derived from healthy subjects to encourage NMJ formation. Functional NMJs were detected by recording myofiber contractions (by phase contrast differentials) while electrically stimulating the MNs. NMJs containing ALS MNs demonstrated increased failure of muscle contractions upon repetitive MN stimulations compared to WT controls. This defect was rescued by the application of Deanna Protocol (DP). SKM, skeletal muscle
To study AD, a functional in vitro assay based on long‐term potentiation (LTP‐correlated to learning and memory) was used to demonstrate that exposure to amyloid beta (Aβ42) and tau oligomers led to prominent changes in human induced pluripotent stem cells‐derived cortical neurons, notably, without cell death. Impaired information processing was demonstrated by treatment of neuron‐microelectrode array systems with the oligomers by reducing the effects of LTP induction. These data confirm the neurotoxicity of molecules linked to AD pathology in a functional model and indicate the utility of this human‐based system to model aspects of AD in vitro and study LTP deficits without loss of viability; a phenotype that more closely models the preclinical or early stage of AD. 95 A model has also shown PK/pharmacodynamic (PD) models of an in vitro human heart/liver system can be used to predict in vivo outcomes. 96 In addition, a multi‐organ innate immune system‐on‐a‐chip indicated that this platform can be extended to evaluate circulating monocytes that reproduce responses for both restorative (M2) and inflammatory (M1) phenotypes. 97
Patient‐derived 3D organoids for tumor growth and drug response
The complexity of human tumors underscores the need for relevant ex vivo models for drug discovery and drug efficacy studies. Despite therapeutic advancements made from cancer drug screening using 2D monolayer cell cultures, there remains questions as to whether traditional cancer models and measures of cellular toxicity accurately capture heterogeneous patient responses to drugs. Developing models that can more closely mimic the cellular and microenvironmental heterogeneity of tumors will assist in translating preclinical findings to clinical efficacy. The development and application of 3D models in the cancer field has significantly increased over the last few years to help fill this void. Initial evaluations have already begun to show promise in significantly improving the drug discovery pipeline.
The patient‐derived tumor organoid (PDTO) model has gained significant traction as a valuable biomimetic model in cancer research. 98 PDTOs are 3D multicellular, self‐assembling structures that have been shown to recapitulate the molecular and phenotypic heterogeneity of the patient tissue they were derived from. Moreover, PDTOs are amenable to expansion and biobanking making this an attractive and scalable model for drug screening. Several prominent studies have shown PDTOs are able to predict patient drug response. 99 , 100 These studies are paving the way for the use of PDTOs as a precision medicine tool.
Despite the excitement and promise surrounding the PDTO model, it is not without limitations. Traditional PDTO culture methods often select for epithelial cells, with minimal remnants of stromal cells observed after several passages in culture. It is appreciated that cell‐cell crosstalk between epithelial cancer cells and cells in the microenvironment can significantly impact tumor growth and drug response. Therefore, certain drug classes may not be relevant to test in the traditional organoid model based on the incomplete microenvironment. However, new methods are being developed, such as the air‐liquid interface system, to support stromal cell diversity, including immune, fibroblast, and endothelial cells, in organoid cultures.
Another important consideration for drug screening is intra‐ and interpatient organoid variability, including size differences, cellular heterogeneities, and temporal response dynamics. Although a majority of PDTO drug testing is performed using biochemical ATP assays as a readout for viability, sophisticated imaging‐based approaches are emerging to detangle the aforementioned complexities that influence drug response. Confocal live cell imaging coupled with data analysis techniques can capture a vast array of parameters that can be used to determine cell behavior and drug efficacy in PDTOs. For example, we have shown the ability to distinguish between cytotoxic and cytostatic drug effects using high content PDTO imaging. 101
The aspiration for 3D cancer models, as well as for other disease models, is to predict clinical response; serving as a better drug screening model to improve the translatability of compounds during preclinical drug testing. Although this promise is beginning to become a reality, important work remains to improve standardization, reproducibility, and broad adoption by the cancer community.
REGULATORY CONSIDERATION OF 3D CULTURE MODELS
The FDA has been involved in the development of in vitro MPS since 2010 when the FDA and National Institutes of Health (NIH) awarded grant support to the Wyss Institute for Biologically Inspired Engineering to develop a heart‐lung micromachine. The goal of the Common Fund Program is to accelerate the development and use of new tools, standards, and approaches to efficiently develop medical products and to more effectively evaluate medical product safety, efficacy, and quality. A major focus of the program became the development of cutting edge MPS.
In 2011, DARPA approached the FDA Office of the Chief Scientist for the collaboration of development of a human body on a chip for medical countermeasures. DARPA involved the FDA from the beginning of this MPS program to help ensure that regulatory challenges of reviewing drug safety and efficacy were considered during development of the MPS platform. DARPA had also coordinated efforts with the NIH, which was conducting separate but parallel research. This program ended in 2017.
In 2017, the FDA launched its Predictive Toxicology Roadmap, which highlighted the FDA’s goal of reducing the use of animal testing. The report discussed strategies for using 21st‐century science to foster the development and evaluation of emerging toxicological methods as well as new technologies and to incorporate them into the FDA regulatory review. The roadmap emphasized that the end users of new technology, namely the FDA regulators, should be involved up front as these technologies evolve for them to be effective. The FDA subsequently recognized that it needed to create a platform available to stakeholders and the public that would inform them of the FDA’s progress in promoting the development and implementation of alternative test methods. To that end, the FDA established an Alternative Methods Working Group, which is developing a targeted strategy for moving toward the use of alternative methods for regulatory testing.
Since the successful completion of the FDA DARPA NCATS MPS program, the FDA has continued its engagement in chip technology in a myriad of ways. Chips from several academic and commercial vendors have been deployed in the FDA research laboratories and the FDA MPS researchers meet monthly to discuss the advances in this rapidly evolving technology space. The goal is to eventually help regulators formulate expectations for regulator performance criteria. This hands‐on experience with developing technology, such as described above related to a liver‐on‐chip model for toxicity and PK evaluation, is a critical component of the FDA’s strategy to understand how eventually MPS data may move into regulatory decisions.
The FDA has also drafted a definition for MPS and for organs on a chip; and is requesting comments through its email address at alternatives@fda.hhs.gov.
The Microphysiological System is an in vitro platform composed of cells, explants derived from tissues/organs, and/or organoid cell formations of human or animal origin in a micro‐environment that provides and supports biochemical/electrical/mechanical responses to model a set of specific properties that define organ or tissue function.
Organ‐on‐a‐chip is a miniaturized physiological environment engineered to yield and/or analyze functional tissue units capable of modeling specified/targeted organ‐level responses.
The FDA is working to advance MPS and other alternative technologies that may help better predict the safety and efficacy of the products it oversees. The FDA reminds its stakeholders that change takes time, but it will happen if we all work together toward progress to developing MPS for regulatory use (Figure 6).
FIGURE 6.
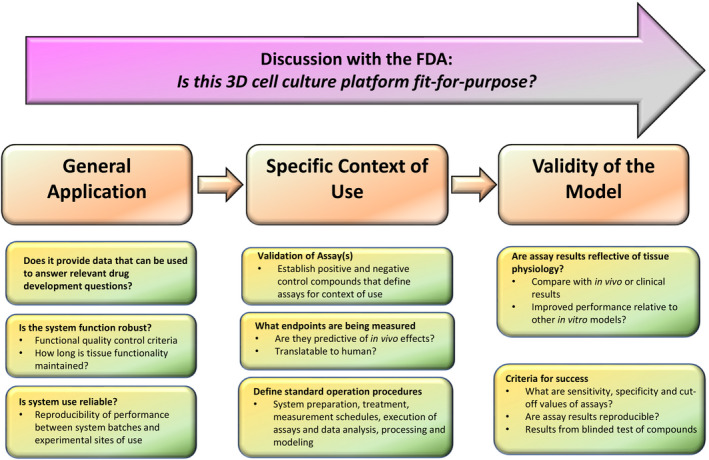
Regulatory considerations for the use of 3D cell culture models. Schematic illustration of specific questions about 3D model selections, including general application, specific context of use, and the validity of the model, to help facilitate discussion with the US Food and Drug Administration (FDA)
Nonclinical safety evaluation
Several topics need to be considered when thinking about how 3D culture models may be incorporated into regulatory uses related to nonclinical safety evaluation of human pharmaceuticals. The regulatory framework for human pharmaceuticals is inherently flexible and allows for the submission of a wide range of study types. For example, the section of the Code of Federal Regulations describing the regulations governing Investigational New Drug Applications states that the pharmacology and toxicology information in an application should include “Adequate information about pharmacological and toxicological studies of the drug involving laboratory animals or in vitro, on the basis of which the sponsor has concluded that it is reasonably safe to conduct the proposed clinical investigations.” Regulations for New Drug Applications have similar wording. Indeed, pivotal information related to drug safety evaluation is currently collected from in vitro systems, such as some genotoxicity studies, safety pharmacology studies, and special toxicity studies of effects, such as ocular and skin irritation.
The Agency publishes guidance documents that interpret regulations and provide recommendations to industry on the types of nonclinical information needed to support development of human pharmaceuticals. Some of these guidances are developed through the International Council for Harmonization of Technical Requirements for Pharmaceuticals for Human Use (ICH) and others are developed solely by the FDA. These guidances are also flexible and allow for the use of alternative approaches. The FDA encourages sponsors to consult with review divisions when considering a nonanimal testing method believed to be suitable, adequate, and feasible. The FDA will consider whether the alternative method is adequate to meet the nonclinical regulatory need.
Some guidances explicitly include alternative approaches. For example, the ICH S3A guidance on toxicokinetics has a question‐and‐answer document that describes the use of microsampling techniques that can reduce the number of animals used in toxicity studies and minimize pain and distress in animals that are used. ICH S5(R3) on Detection of Reproductive and Developmental Toxicity for Human Pharmaceuticals has extensive language describing potential scenarios in which alternative approaches to assessing malformations and embryofetal lethality might be used and on the basic principles that would assist in the development and qualification of such alternative approaches. ICH S10 on Photosafety Evaluation of Pharmaceuticals includes discussion of in chemico photoreactivity assays and in vitro phototoxicity assays. Even guidances that do not describe particular alternative approaches usually include language allowing the potential use of such assays when adequate and appropriate.
The FDA’s experience with complex in vitro systems and 3D culture models in regulatory submissions is relatively limited to date. Of those submitted, many addresses potential pharmacologic activity and for the most part have not played a pivotal role in safety decisions. For example, pharmacology studies using in vitro studies have been submitted with organoids based on bronchial epithelium, intestinal cells (including from patients with disease), and retinal cells.
To move 3D culture models toward regulatory use, several questions need to be addressed. The Agency is interested in knowing whether a new approach can provide data that can be used to answer fundamental drug development questions, such as “what are potential human target organs,” “what dose can be safely administered to humans,” and “what biomarkers should be monitored in humans?” These types of questions should help define the context of use of the approach, which must be clearly described. The assessment of a new approach methodology must include considering whether the assay is stable and reproducible, whether end points are predictive of in vivo effects and translatable to humans, whether appropriate test compounds have been assessed, whether the applicability domain of the approach has been adequately determined, and what the performance of the approach is (i.e., accuracy, sensitivity, specificity, etc.)
The FDA notes that the use of many 3D culture models early in drug discovery may provide unique opportunities outside the regulatory jurisdiction of the Agency. Although no formal validation is required before a New Approach Methodology (NAM) is submitted in a drug application and the Agency will review such information, the FDA encourages sponsors to discuss the potential use of NAMs with the Agency when moving into the regulatory space.
Evaluation of ADME and PK‐PD relationship
As described earlier, applied research is being developed in CDER, 19 using a commercially available liver MPS for studying reliability and robustness of its use in PK applications. The potential utility of MPS in the evaluation of ADME and PK‐PD relationship of new drug products has been reviewed. 102 The authors indicated that MPS systems can enhance the understanding of physiology, pathology, and pharmacology (Figure 7) and described specific applications of the MPS systems in the areas of biomarker development; demonstrating proof‐of‐concept, elucidating the mechanism of drug toxicity, and characterizing the complex physiologic changes that occur in disease states that can provide the necessary information to advance the role of quantitative clinical pharmacology models in drug development. 102
FIGURE 7.
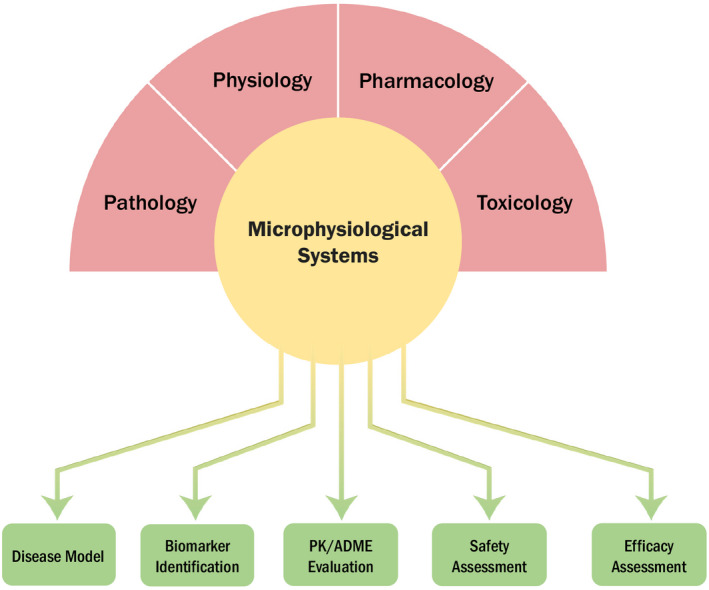
The scientific background and application of microphysiological Systems (MPS). MPS are viewed as innovative scientific tools that allow for collection of specific and critical physiological, pharmacological, pathological, and toxicological parameters to better understand and develop systems pharmacology models. Specific applications of MPS include areas, such as physiological understanding and characterization of rare disease, biomarker development of target therapies, pharmacokinetic (PK) evaluation of novel drugs, and elucidating mechanism of action for drug efficacy and safety
As model‐informed drug development (MIDD) was formally recognized in Prescription Drug User Fee Act (PDUFA) VI, it is increasingly acknowledged as a critical component of drug development and regulatory review. 103 PBPK is one critical MIDD approach that has been applied in the evaluation of how various intrinsic (age, sex, genetics, race, and comorbidity) and extrinsic factors (concomitant medications) can affect the PK, PD, and thereby drug response. One frequent application of PBPK is in the prediction of DDIs. 104 As pointed out in this paper that reviewed PBPK applications submitted to the US FDA between 2018 and 2019, one of the deficiencies in the PBPK models not being accepted by the Agency was “the elimination or disposition pathways of the probe substrate model were not well characterized.” Appropriate applications of MPS systems could potentially fill the data void and contribute to the understanding of the ADME of new drug products. As stated in the section above related to 3D kidney models, the combination of critically needed in vitro data generated using MPS and the careful use of PBPK modeling provides an essential tool to predict clearance of drugs and metabolites and DDIs.
When MPS is used for clinical pharmacology evaluations related to regulatory decision making, it is important to clearly state the question of interest, define the context of use, and discuss the validity of the assay (e.g., sensitivity, specificity, and reproducibility). It is critical to make sure that the MPS used is fit‐for‐purpose (Figure 6).
CONCLUSION
In order to enhance drug development, regulatory review, and to inform the safe and optimal use of medications, continued development and use of novel technologies are needed. MPS can play a pivotal role in advancing development of innovative new medicines by incorporating state‐of‐the‐art preclinical models with regulatory science and clinical pharmacology principles, and to promote therapeutic optimization and individualization through best practices. It is evident now that novel 3D cell culture models hold great potential in assessing drug safety and efficacy, as well as modeling various disease conditions. Significant progress has been achieved in the development of various MPS. However, each of these models presents inherent advantages and limitations that call for further model improvement and careful data interpretation. Continual public‐private partnerships are critical to advance research in this field and to promote the application of these new technologies in drug development and regulatory review.
DISCLAIMER
As an Associate Editor of Clinical and Translational Science, Nina Isoherranen was not involved in the review or decision process for this paper. The opinions expressed in this review are those of the authors and should not be interpreted as the position of the US Food and Drug Administration.
CONFLICT OF INTEREST
L.E. is an employee of Emulate Inc. and holds stock in the company. B.S.F. is an inventor on pending patent applications for kidney organoid technology, and receives royalties. W.H. is an employee of Bristol‐Myers Squibb Company. S.H. is an employee of BioIVT. J.H. is Chief Scientist of Hesperos, Inc. A.P.L. is President and CEO of In Vitro ADMET Laboratories Inc. W.R.P. is an employee of Genentech, Inc. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
H.W and S.H conceptualized the review article. All authors involved in writing the manuscript.
ACKNOWLEDGEMENTS
The authors are grateful to Ms. Giang Ho and Kimberly Bergman from the US FDA for their assistance in graphic design of Figures 1 and 6. We apologize to the scientists who have made contributions to the field, but were not cited due to space limitations.
Funding information
This work was supported in part by The University of Maryland's Center of Excellence in Regulatory Science and Innovation (M‐CERSI) funded by the U.S. Food and Drug Administration (2U01FD0059462). Effort of Freedman laboratory related to this work is supported by NIH R01DK117914, U01DK127553, UG3TR002158, UC2DK126006, and UG3TR003288, Department of Defense W81XWH2110007, Allen Institute for Cell Science Collaborative Research Award, and Lara Nowak Macklin Fund.
Contributor Information
Hongbing Wang, Email: hongbing.wang@rx.umaryland.edu.
Shiew‐Mei Huang, Email: ShiewMei.Huang@fda.hhs.gov.
REFERENCES
- 1. Huang SM, Strong JM, Zhang L, et al. New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol. 2008;48:662‐670. [DOI] [PubMed] [Google Scholar]
- 2. Ribeiro AJS, Yang X, Patel V, Madabushi R, Strauss DG. Liver microphysiological systems for predicting and evaluating drug effects. Clin Pharmacol Ther. 2019;106:139‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baudy AR, Otieno MA, Hewitt P, et al. Liver microphysiological systems development guidelines for safety risk assessment in the pharmaceutical industry. Lab Chip. 2020;20:215‐225. [DOI] [PubMed] [Google Scholar]
- 4. Fowler S, Chen WLK, Duignan DB, et al. Microphysiological systems for ADME‐related applications: current status and recommendations for system development and characterization. Lab Chip. 2020;20:446‐467. [DOI] [PubMed] [Google Scholar]
- 5. Dame K, Ribeiro AJ. Microengineered systems with iPSC‐derived cardiac and hepatic cells to evaluate drug adverse effects. Exp Biol Med (Maywood). 2021;246:317‐331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breslin S, O'Driscoll L. Three‐dimensional cell culture: the missing link in drug discovery. Drug Discov Today. 2013;18:240‐249. [DOI] [PubMed] [Google Scholar]
- 7. Mittal R, Woo FW, Castro CS, et al. Organ‐on‐chip models: implications in drug discovery and clinical applications. J Cell Physiol. 2019;234:8352‐8380. [DOI] [PubMed] [Google Scholar]
- 8. Almazroo OA, Miah MK, Venkataramanan R. Drug metabolism in the liver. Clin Liver Dis. 2017;21:1‐20. [DOI] [PubMed] [Google Scholar]
- 9. Li R, Barton HA, Varma MV. Prediction of pharmacokinetics and drug‐drug interactions when hepatic transporters are involved. Clin Pharmacokinet. 2014;53:659‐678. [DOI] [PubMed] [Google Scholar]
- 10. Li Y, Meng Q, Yang M, et al. Current trends in drug metabolism and pharmacokinetics. Acta Pharm Sin B. 2019;9:1113‐1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weaver RJ, Blomme EA, Chadwick AE, et al. Managing the challenge of drug‐induced liver injury: a roadmap for the development and deployment of preclinical predictive models. Nat Rev Drug Discov. 2020;19:131‐148. [DOI] [PubMed] [Google Scholar]
- 12. Hughes DJ, Kostrzewski T, Sceats EL. Opportunities and challenges in the wider adoption of liver and interconnected microphysiological systems. Exp Biol Med (Maywood). 2017;242:1593‐1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Beckwitt CH, Clark AM, Wheeler S, et al. Liver ‘organ on a chip'. Exp Cell Res. 2018;363:15‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cotovio JP, Fernandes TG. Production of human pluripotent stem cell‐derived hepatic cell lineages and liver organoids: current status and potential applications. Bioengineering (Basel). 2020;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsamandouras N, Chen WLK, Edington CD, et al. Integrated gut and liver microphysiological systems for quantitative in vitro pharmacokinetic studies. AAPS J. 2017;19:1499‐1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jang KJ, Otieno MA, Ronxhi J, et al. Reproducing human and cross‐species drug toxicities using a Liver‐Chip. Sci Transl Med. 2019;11(517):eaax5516. [DOI] [PubMed] [Google Scholar]
- 17. Herland A, Maoz BM, Das D, et al. Quantitative prediction of human pharmacokinetic responses to drugs via fluidically coupled vascularized organ chips. Nat Biomed Eng. 2020;4:421‐436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Fabre K, Berridge B, Proctor WR, et al. Introduction to a manuscript series on the characterization and use of microphysiological systems (MPS) in pharmaceutical safety and ADME applications. Lab Chip. 2020;20:1049‐1057. [DOI] [PubMed] [Google Scholar]
- 19. Rouse R, Kruhlak N, Weaver J, et al. Translating new science into the drug review process: the US FDA's Division of Applied Regulatory Science. Ther Innov Regul Sci. 2018;52:244‐255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sarkar U, Ravindra KC, Large E, et al. Integrated assessment of diclofenac biotransformation, pharmacokinetics, and omics‐based toxicity in a three‐dimensional human liver‐immunocompetent coculture system. Drug Metab Dispos. 2017;45:855‐866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Domansky K, Inman W, Serdy J, et al. Perfused multiwell plate for 3D liver tissue engineering. Lab Chip. 2010;10:51‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dehne EM, Hasenberg T, Marx U. The ascendance of microphysiological systems to solve the drug testing dilemma. Future Sci OA. 2017;3:Fso185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bell CC, Dankers ACA, Lauschke VM, et al. Comparison of hepatic 2D sandwich cultures and 3D spheroids for long‐term toxicity applications: a multicenter study. Toxicol Sci. 2018;162:655‐666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ortega‐Prieto AM, Skelton JK, Wai SN, et al. 3D microfluidic liver cultures as a physiological preclinical tool for hepatitis B virus infection. Nat Commun. 2018;9:682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaplowitz N. Drug‐induced liver injury. Clin Infect Dis. 2004;38(Suppl 2):S44‐S48. [DOI] [PubMed] [Google Scholar]
- 26. Luyendyk JP, Roth RA, Ganey PE. 9.13 ‐ Inflammation and hepatotoxicity. In: McQueen CA, ed. Comprehensive Toxicology. 2nd ed. Elsevier, Oxford; 2010:295‐317. [Google Scholar]
- 27. Chang W, Song L, Chang X, et al. Early activated hepatic stellate cell‐derived paracrine molecules modulate acute liver injury and regeneration. Lab Invest. 2017;97:318‐328. [DOI] [PubMed] [Google Scholar]
- 28. McCuskey RS. The hepatic microvascular system in health and its response to toxicants. Anat Rec (Hoboken). 2008;291:661‐671. [DOI] [PubMed] [Google Scholar]
- 29. Visentin M, Lenggenhager D, Gai Z, Kullak‐Ublick GA. Drug‐induced bile duct injury. Biochim Biophys Acta Mol Basis Dis. 2018;1864:1498‐1506. [DOI] [PubMed] [Google Scholar]
- 30. Zeilinger K, Freyer N, Damm G, Seehofer D, Knöspel F. Cell sources for in vitro human liver cell culture models. Exp Biol Med (Maywood). 2016;241:1684‐1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vivares A, Salle‐Lefort S, Arabeyre‐Fabre C, et al. Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device. Xenobiotica. 2015;45:29‐44. [DOI] [PubMed] [Google Scholar]
- 32. Di L, Obach RS. Addressing the challenges of low clearance in drug research. AAPS J. 2015;17:352‐357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hutzler JM, Ring BJ, Anderson SR. Low‐turnover drug molecules: a current challenge for drug metabolism scientists. Drug Metab Dispos. 2015;43:1917‐1928. [DOI] [PubMed] [Google Scholar]
- 34. Caetano‐Pinto P, Stahl SH. Perspective on the application of microphysiological systems to drug transporter studies. Drug Metab Dispos. 2018;46:1647‐1657. [DOI] [PubMed] [Google Scholar]
- 35. Schurdak M, Vernetti L, Bergenthal L, et al. Applications of the microphysiology systems database for experimental ADME‐Tox and disease models. Lab Chip. 2020;20:1472‐1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tsamandouras N, Kostrzewski T, Stokes CL, et al. Quantitative assessment of population variability in hepatic drug metabolism using a perfused three‐dimensional human liver microphysiological system. J Pharmacol Exp Therap. 2017;360:95‐105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bachour‐El Azzi P, Sharanek A, Burban A, et al. Comparative localization and functional activity of the main hepatobiliary transporters in HepaRG cells and primary human hepatocytes. Toxicol Sci. 2015;145:157‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bell CC, Hendriks DF, Moro SM, et al. Characterization of primary human hepatocyte spheroids as a model system for drug‐induced liver injury, liver function and disease. Sci Rep. 2016;6:25187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Messner S, Agarkova I, Moritz W, Kelm JM. Multi‐cell type human liver microtissues for hepatotoxicity testing. Arch Toxicol. 2013;87:209‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Messner S, Fredriksson L, Lauschke VM, et al. Transcriptomic, proteomic, and functional long‐term characterization of multicellular three‐dimensional human liver microtissues. Appl In Vitro Toxicol. 2018;4:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Proctor WR, Foster AJ, Vogt J, et al. Utility of spherical human liver microtissues for prediction of clinical drug‐induced liver injury. Arch Toxicol. 2017;91:2849‐2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kermanizadeh A, Brown DM, Moritz W, Stone V. The importance of inter‐individual Kupffer cell variability in the governance of hepatic toxicity in a 3D primary human liver microtissue model. Sci Rep. 2019;9:7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Weaver RJ, Blomme EA, Chadwick AE, et al. Managing the challenge of drug‐induced liver injury: a roadmap for the development and deployment of preclinical predictive models. Nat Rev Drug Discov. 2020;19(2):131‐148. [DOI] [PubMed] [Google Scholar]
- 44. Ramaiahgari SC, Waidyanatha S, Dixon D, et al. Three‐dimensional (3D) HepaRG spheroid model with physiologically relevant xenobiotic metabolism competence and hepatocyte functionality for liver toxicity screening. Toxicol Sci. 2017;160:189‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gripon P, Rumin S, Urban S, et al. Infection of a human hepatoma cell line by hepatitis B virus. Proc Natl Acad Sci USA. 2002;99:15655‐15660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramaiahgari SC, Waidyanatha S, Dixon D, et al. From the cover: three‐dimensional (3D) HepaRG spheroid model with physiologically relevant xenobiotic metabolism competence and hepatocyte functionality for liver toxicity screening. Toxicol Sci. 2017;159:124‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ramaiahgari SC, Auerbach SS, Saddler TO, et al. The power of resolution: contextualized understanding of biological responses to liver injury chemicals using high‐throughput transcriptomics and benchmark concentration modeling. Toxicol Sci. 2019;169:553‐566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Khetani SR, Bhatia SN. Microscale culture of human liver cells for drug development. Nat Biotechnol. 2008;26:120‐126. [DOI] [PubMed] [Google Scholar]
- 49. Ramsden D, Tweedie DJ, Chan TS, Taub ME, Li Y. Bridging in vitro and in vivo metabolism and transport of faldaprevir in human using a novel cocultured human hepatocyte system. HepatoPac Drug Metab Dispos. 2014;42:394‐406. [DOI] [PubMed] [Google Scholar]
- 50. Trask OJ Jr, Moore A, LeCluyse EL. A micropatterned hepatocyte coculture model for assessment of liver toxicity using high‐content imaging analysis. Assay Drug Dev Technol. 2014;12:16‐27. [DOI] [PubMed] [Google Scholar]
- 51. Kang W, Podtelezhnikov AA, Tanis KQ, et al. Development and application of a transcriptomic signature of bioactivation in an advanced in vitro liver model to reduce drug‐induced liver injury risk early in the pharmaceutical pipeline. Toxicol Sci. 2020;177:121‐139. [DOI] [PubMed] [Google Scholar]
- 52. Nguyen TV, Ukairo O, Khetani SR, et al. Establishment of a hepatocyte‐Kupffer cell coculture model for assessment of proinflammatory cytokine effects on metabolizing enzymes and drug transporters. Drug Metab Dispos. 2015;43:774‐785. [DOI] [PubMed] [Google Scholar]
- 53. Low LA, Mummery C, Berridge BR, Austin CP, Tagle DA. Organs‐on‐chips: into the next decade. Nat Rev Drug Discov. 2021;20(5):345‐361. [DOI] [PubMed] [Google Scholar]
- 54. Monticello TM, Jones TW, Dambach DM, et al. Current nonclinical testing paradigm enables safe entry to first‐in‐human clinical trials: the IQ consortium nonclinical to clinical translational database. Toxicol Appl Pharmacol. 2017;334:100‐109. [DOI] [PubMed] [Google Scholar]
- 55. Kasendra M, Luc R, Yin J, et al. Duodenum intestine‐chip for preclinical drug assessment in a human relevant model. eLife. 2020;9:e50135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li AP, Alam N, Amaral K, et al. Cryopreserved human intestinal mucosal epithelium: a novel in vitro experimental system for the evaluation of enteric drug metabolism, cytochrome P450 induction, and enterotoxicity. Drug Metab Dispos. 2018;46:1562‐1571. [DOI] [PubMed] [Google Scholar]
- 57. Li AP, Ho M‐CD, Alam N, et al. Inter‐individual and inter‐regional variations in enteric drug metabolizing enzyme activities: Results with cryopreserved human intestinal mucosal epithelia (CHIM) from the small intestines of 14 donors. Pharmacol Res Perspect. 2020;8:e00645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zhang H, Wolford C, Basit A, et al. Regional proteomic quantification of clinically relevant non‐cytochrome P450 enzymes along the human small intestine. Drug Metab Dispos. 2020;48:528‐536. [DOI] [PubMed] [Google Scholar]
- 59. Davies M, Peramuhendige P, King L, et al. Evaluation of in vitro models for assessment of human intestinal metabolism in drug discovery. Drug Metab Dispos. 2020;48(11):1169‐1182. [DOI] [PubMed] [Google Scholar]
- 60. Loretz C, Ho MD, Alam N, Mitchell W, Li AP. Application of cryopreserved human intestinal mucosa and cryopreserved human enterocytes in the evaluation of herb‐drug interactions: evaluation of CYP3A inhibitory potential of grapefruit juice and commercial formulations of twenty‐nine herbal supplements. Drug Metab Dispos. 2020;48:1084‐1091. [DOI] [PubMed] [Google Scholar]
- 61. Li AP. In vitro human cell‐based experimental models for the evaluation of enteric metabolism and drug interaction potential of drugs and natural products. Drug Metab Dispos. 2020;48:980‐992. [DOI] [PubMed] [Google Scholar]
- 62. Li M, Vokral I, Evers B, et al. Human and rat precision‐cut intestinal slices as ex vivo models to study bile acid uptake by the apical sodium‐dependent bile acid transporter. Eur J Pharm Sci. 2018;121:65‐73. [DOI] [PubMed] [Google Scholar]
- 63. Pham BT, van Haaften WT, Oosterhuis D, et al. Precision‐cut rat, mouse, and human intestinal slices as novel models for the early‐onset of intestinal fibrosis. Physiol Rep. 2015;3:e12323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ho MD, Ring N, Amaral K, Doshi U, Li AP. Human enterocytes as an in vitro model for the evaluation of intestinal drug metabolism: characterization of drug‐metabolizing enzyme activities of cryopreserved human enterocytes from twenty‐four donors. Drug Metab Dispos. 2017;45:686‐691. [DOI] [PubMed] [Google Scholar]
- 65. Li AP, Amaral K, Ho MD. A novel in vitro experimental system for the evaluation of enteric drug metabolism: cofactor‐supplemented permeabilized cryopreserved human enterocytes (MetMax cryopreserved human enterocytes). Drug Metab Lett. 2018;12:132‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hayeshi R, Hilgendorf C, Artursson P, et al. Comparison of drug transporter gene expression and functionality in Caco‐2 cells from 10 different laboratories. Eur J Pharm Sci. 2008;35:383‐396. [DOI] [PubMed] [Google Scholar]
- 67. Dekkers JF, Wiegerinck CL, de Jonge HR, et al. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med. 2013;19:939‐945. [DOI] [PubMed] [Google Scholar]
- 68. Schwank G, Koo B‐K, Sasselli V, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13:653‐658. [DOI] [PubMed] [Google Scholar]
- 69. van de Wetering M, Francies HE, Francis JM, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933‐945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fujii M, Shimokawa M, Date S, et al. A colorectal tumor organoid library demonstrates progressive loss of niche factor requirements during tumorigenesis. Cell Stem Cell. 2016;18:827‐838. [DOI] [PubMed] [Google Scholar]
- 71. Boehnke K, Iversen PW, Schumacher D, et al. Assay establishment and validation of a high‐throughput screening platform for three‐dimensional patient‐derived colon cancer organoid cultures. J Biomol Screen. 2016;21:931‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Rao JN, Xiao L, Wang JY. Polyamines in gut epithelial renewal and barrier function. Physiology. 2020;35:328‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Xiao L, Gorospe M, Wang JY. Long noncoding RNAs in intestinal epithelium homeostasis. Am J Physiol Cell Physiol. 2019;317:C93‐C100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Xiao L, Wu J, Wang J‐Y, et al. Long noncoding RNA uc.173 promotes renewal of the intestinal mucosa by inducing degradation of MicroRNA 195. Gastroenterology. 2018;154:599‐611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang JY, Cui YH, Xiao L, et al. Regulation of intestinal epithelial barrier function by long noncoding RNA uc.173 through interaction with MicroRNA 29b. Mol Cell Biol. 2018;38(13):e00010‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Xiao L, Li XX, Chung HK, et al. RNA‐binding protein HuR regulates paneth cell function by altering membrane localization of TLR2 via post‐transcriptional control of CNPY3. Gastroenterology. 2019;157:731‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Little MH, Combes AN. Kidney organoids: accurate models or fortunate accidents. Genes Dev. 2019;33:1319‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Czerniecki SM, Cruz NM, Harder JL, et al. High‐throughput screening enhances kidney organoid differentiation from human pluripotent stem cells and enables automated multidimensional phenotyping. Cell Stem Cell. 2018;22:929‐940 e924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Freedman BS, Brooks CR, Lam AQ, et al. Modelling kidney disease with CRISPR‐mutant kidney organoids derived from human pluripotent epiblast spheroids. Nat Commun. 2015;6:8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cruz NM, Song X, Czerniecki SM, et al. Organoid cystogenesis reveals a critical role of microenvironment in human polycystic kidney disease. Nat Mater. 2017;16:1112‐1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Monteil V, Kwon H, Prado P, et al. Inhibition of SARS‐CoV‐2 infections in engineered human tissues using clinical‐grade soluble human ACE2. Cell. 2020;181:905‐913 e907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Nam SA, Seo E, Kim JW, et al. Graft immaturity and safety concerns in transplanted human kidney organoids. Exp Mol Med. 2019;51:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. van der Made TK, Fedecostante M, Scotcher D, et al. Quantitative translation of microfluidic transporter in vitro data to in vivo reveals impaired albumin‐facilitated indoxyl sulfate secretion in chronic kidney disease. Mol Pharm. 2019;16:4551‐4562. [DOI] [PubMed] [Google Scholar]
- 84. Chang SY, Weber EJ, Sidorenko VS, et al. Human liver‐kidney model elucidates the mechanisms of aristolochic acid nephrotoxicity. JCI Insight. 2017;2(22):e95978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Prasad B, Achour B, Artursson P, et al. Toward a consensus on applying quantitative liquid chromatography‐tandem mass spectrometry proteomics in translational pharmacology research: a white paper. Clin Pharmacol Ther. 2019;106:525‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Prasad B, Johnson K, Billington S, et al. Abundance of drug transporters in the human kidney cortex as quantified by quantitative targeted proteomics. Drug Metab Dispos. 2016;44:1920‐1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sweet DH, Eraly SA, Vaughn DA, Bush KT, Nigam SK. Organic anion and cation transporter expression and function during embryonic kidney development and in organ culture models. Kidney Int. 2006;69:837‐845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Motohashi H, Sakurai Y, Saito H, et al. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol. 2002;13:866‐874. [DOI] [PubMed] [Google Scholar]
- 89. Weber EJ, Chapron A, Chapron BD, et al. Development of a microphysiological model of human kidney proximal tubule function. Kidney Int. 2016;90:627‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Chapron A, Chapron BD, Hailey DW, et al. An improved vascularized, dual‐channel microphysiological system facilitates modeling of proximal tubular solute secretion. ACS Pharmacol Transl Sci. 2020;3:496‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Huang W, Isoherranen N. Development of a dynamic physiologically based mechanistic kidney model to predict renal clearance. CPT Pharmacometrics Syst Pharmacol. 2018;7:593‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Huang W, Isoherranen N. Novel mechanistic PBPK model to predict renal clearance in varying stages of CKD by incorporating tubular adaptation and dynamic passive reabsorption. CPT Pharmacomet Syst Pharmacol. 2020;9(10):571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Shuler ML, Hickman JJ. Toward in vitro models of brain structure and function. Proc Natl Acad Sci USA. 2014;111:13682‐13683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Guo X, Smith V, Jackson M, et al. A human‐based functional NMJ system for personalized ALS modeling and drug testing. Adv Therap. 2020;3(11):2000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Caneus J, Akanda N, Rumsey JW, et al. A human induced pluripotent stem cell‐derived cortical neuron human‐on‐a chip system to study Aβ42 and tau‐induced pathophysiological effects on long‐term potentiation. Alzheimers Dement (NY). 2020;6:e12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. McAleer CW, Pointon A, Long J, et al. On the potential of in vitro organ‐chip models to define temporal pharmacokinetic‐pharmacodynamic relationships. Nat Sci Rep. 2019;9:9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sasserath T, Rumsey JW, McAleer CW, et al. Differential monocyte actuation in a three‐organ functional innate immune system‐on‐a‐chip. Adv Sci (Weinh). 2020;7:2000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Tuveson D, Clevers H. Cancer modeling meets human organoid technology. Science. 2019;364:952‐955. [DOI] [PubMed] [Google Scholar]
- 99. Vlachogiannis G, Hedayat S, Vatsiou A, et al. Patient‐derived organoids model treatment response of metastatic gastrointestinal cancers. Science. 2018;359:920‐926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ooft SN, Weeber F, Dijkstra KK, et al. Patient‐derived organoids can predict response to chemotherapy in metastatic colorectal cancer patients. Sci Transl Med. 2019;11(513):eaay2574. [DOI] [PubMed] [Google Scholar]
- 101. Kim S, Choung S, Sun RX, et al. Comparison of cell and organoid‐level analysis of patient‐derived 3D organoids to evaluate tumor cell growth dynamics and drug response. SLAS Discov. 2020;25:744‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Isoherranen N, Madabushi R, Huang SM. Emerging role of organ‐on‐a‐chip technologies in quantitative clinical pharmacology evaluation. Clin Transl Sci. 2019;12:113‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Wang Y, Zhu H, Madabushi R, et al. Model‐informed drug development: current US Regulatory Practice and Future Considerations. Clin Pharmacol Ther. 2019;105:899‐911. [DOI] [PubMed] [Google Scholar]
- 104. Zhang X, Yang Y, Grimstein M, Fan J, Grillo JA, Huang SM, Zhu H, Wang Y. Application of PBPK modeling and simulation for regulatory decision‐making and its impact on the US prescribing information: an update on the 2018–2019 submissions to the US FDA's Office of Clinical Pharmacology. J Clin Pharmacol. 2020;60:S160‐S178. [DOI] [PubMed] [Google Scholar]