

Abstract
Aims/Introduction
Peroxisome proliferator‐activated receptor (PPAR)‐γ2 is a transcription factor crucial for regulating adipogenesis and glucose/lipid metabolism, and synthetic PPARγ ligands, such as thiazolidinediones, are effective oral medication for type 2 diabetes. Sirtuin 7 (SIRT7), a nicotinamide adenine dinucleotide‐dependent deacetylase, also controls metabolism. However, it is not known whether SIRT7 regulates the function of PPARγ2 by its deacetylation.
Materials and Methods
Physical interaction between SIRT7 and PPARγ2, the effect of SIRT7 on PPARγ2 acetylation, and the deacetylation residue targeted by SIRT7 were investigated. The effects of PPARγ2 K382 acetylation on lipid accumulation, gene expression in C3H10T1/2 cell‐derived adipocytes, and ligand‐dependent transactivation activity were also evaluated.
Results
We demonstrated that SIRT7 binds to PPARγ2 and deacetylates PPARγ2 at K382. C3H10T1/2‐derived adipocytes expressing PPARγ2K382Q (a mimic of acetylated K) accumulated much less fat than adipocytes expressing wild‐type PPARγ2 or PPARγ2K382R (a mimic of nonacetylated K). Global gene expression analysis of adipocytes expressing PPARγ2K382Q revealed that K382Q caused the dysregulation of a set of genes involved in lipogenesis, including Srebp1c, Acaca, Fasn, and Scd1. The rosiglitazone‐dependent transcriptional activity of PPARγ2K382Q was reduced compared with that of PPARγ2K382R.
Conclusion
Our findings indicate that SIRT7‐dependent PPARγ2 deacetylation at K382 controls lipogenesis in adipocytes.
Keywords: SIRT7, PPARγ, acetylation
SIRT7 regulates lipogenesis in adipocytes through deacetylation of PPARgamma2.
INTRODUCTION
Peroxisome proliferator‐activated receptor (PPAR)‐γ is a transcription factor belonging to the nuclear receptor superfamily. Similar to other nuclear receptors, PPARγ has several functional domains, including an N‐terminal transactivation domain, a DNA‐binding domain (DBD), and a C‐terminal region that forms a ligand‐binding domain (LBD), which has a ligand‐dependent transactivation function. PPARγ binds to PPAR response elements (PPRE) with the retinoid X receptor to regulate the expression of various genes involved in adipogenesis, lipid metabolism, and insulin sensitivity 1 , 2 . PPARγ has two isoforms: PPARγ1 and PPARγ2. Whereas PPARγ1 is expressed in many tissues, PPARγ2 is selectively expressed at high levels in white adipose tissue (WAT). Endogenous ligands for PPARγ include unsaturated fatty acids and 15‐deoxy‐prostaglandin J2 3 . Ligand binding induces a conformational change in the receptor that allows for the differential recruitment of cofactors and subsequent modulation of PPARγ activity 1 . Synthetic PPARγ ligands (thiazolidinediones) are effective insulin sensitizers and they improve hyperinsulinemia and hyperglycemia in patients with type 2 diabetes 4 . The transcriptional activity of PPARγ is also regulated by post‐translational modifications, including phosphorylation, acetylation, and SUMOylation 1 , 4 .
Sirtuins (SIRT1‐7 in mammals) are evolutionarily conserved nicotinamide adenine dinucleotide‐dependent deacetylases/deacylases that regulate a large number of biological processes, including metabolism 5 , 6 . SIRT1 inhibits adipogenesis and enhances lipid mobilization from white adipocytes through the suppression of PPARγ activity by docking with nuclear receptor co‐repressor 7 . SIRT1 also promotes adipocyte browning through deacetylation of PPARγ2 at K268 and K293 8 . Although the physiological roles of SIRT7 are poorly defined, recent studies have revealed that it performs various roles in metabolism by deacetylating target proteins 9 , 10 , 11 , 12 . Fang et al. reported that SIRT7 promotes adipogenesis by inhibiting the autocatalytic activation of SIRT1 13 , indicating that SIRT7 indirectly regulates PPARγ activity. However, it is not known whether SIRT7 directly regulates PPARγ activity through its deacetylation.
In this report, we demonstrated that SIRT7 interacts with the LBD of PPARγ2 and deacetylates PPARγ2 at K382. Mouse mesenchymal C3H10T1/2 cell‐derived adipocytes expressing PPARγ2K382Q accumulated much less fat than adipocytes expressing wild‐type (WT) PPARγ2 or PPARγ2K382R. Global gene expression analysis of adipocytes expressing PPARγ2K382Q revealed that K382Q caused the dysregulation of a set of genes involved in lipogenesis, including Srebp1c, Acaca, Fasn, and Scd1. The rosiglitazone‐dependent transcriptional activity of PPARγ2K382Q was reduced compared with that of PPARγ2K382R. Our findings indicate that SIRT7‐dependent PPARγ2 deacetylation at K382 controls lipogenesis in adipocytes.
METHODS
Plasmids, antibodies, cell lines, and mice
Detailed information is provided in Appendix S1. The sequences of the primers used to amplify the PPARγ2 mutants and fragments are listed in Table S1.
Halo tag pull‐down assay
Halo or Halo‐SIRT7 proteins expressed in Escherichia coli K12 (KRX; Promega, Madison, WI) were purified with HaloLink resin 14 . The various expression plasmids were transfected into HEK293T cells using the jetPRIME transfection reagent (Polyplus, New York, NY). At 24 h after transfection, the cells were lysed in pull‐down buffer (10 mM Tris‐HCl [pH 7.4], 1mM NaF, 200 mM NaCl, 10 mM Na4P2O7, 1% NP‐40, 1 mM PMSF, protease inhibitor cocktail [Nacalai Tesque, Kyoto, Japan]) containing 10 mM nicotinamide (Sigma‐Aldrich) and 1 mM TSA (Wako Pure Chemical Industries, Ltd.) by sonication (Sonifier‐150; Branson, Cosmo Bio, Carlsbad, CA) at 4°C. Halo or Halo‐SIRT7 proteins (30 µg) fixed on HaloLink resin were incubated with HEK293T cell lysate (150 µg) overnight at 4°C, and the resins were washed 5 times with the pull‐down buffer. The bound proteins were detected by western blotting with the respective antibody as previously described 9 .
Co‐immunoprecipitation assay
A co‐immunoprecipitation assay was performed as previously described 14 . HEK293T cells transfected with the indicated expression plasmids by the jetPRIME reagent for 24 h were lysed in lysis buffer (20 mM Tris‐HCl [pH 7.4], 200 mM NaCl, 2.5 mM MgCl2, 0.5% NP‐40, 1 mM PMSF, protease inhibitor cocktail) containing 10 mM nicotinamide and 1 mM TSA by passing through a 29 G needle (Terumo, Tokyo, Japan) 6 times. After centrifugation at 14,000 g for 10 min at 4°C, cell lysate (1,000 µg) was subjected to immunoprecipitation overnight at 4°C with anti‐HA antibody beads (clone 4B2; Wako Pure Chemical Industries, Ltd.). To detect interactions between endogenous PPARγ and SIRT7, epididymal WAT (epiWAT) was homogenized with a Dounce homogenizer (Tight; ISIS Co., Ltd., Osaka, Japan) in lysis buffer containing 10 mM nicotinamide and 1 mM TSA on ice. After centrifugation at 14,000 g for 10 min at 4°C, cell lysate (1000 µg) was incubated with anti‐PPARγ antibody‐crosslinked resin, which was prepared using a Pierce Crosslink Immunoprecipitation Kit (Thermo Scientific, Rockford, IL), at 4°C overnight for immunoprecipitation. After washing 5 times with lysis buffer, precipitated proteins were eluted with the elution buffer (pH 2.8, containing primary amine) provided in the kit, and detected by western blotting with the respective antibody.
Detection of lysine acetylation
HEK293T cells transfected with the indicated plasmids by the jetPRIME reagent for 24 h were lysed in lysis buffer containing 10 mM nicotinamide and 1 µM TSA by sonication (Sonifier‐150; Branson) at 4°C. After centrifugation at 14,000 g for 10 min at 4°C, the cell lysates and HA‐tag antibody beads (Wako Pure Chemical Industries, Ltd.) were incubated overnight at 4°C. After washing 5 times with lysis buffer, precipitated proteins were eluted with 2× SDS sample buffer (100 mM Tris‐HCl [pH 6.8], 4% SDS, 20% glycerol, 0.2% bromophenol blue), and lysine acetylation was detected by western blotting with an anti‐acetyl lysine antibody (Cell Signaling Technology). To detect the endogenous acetylation of PPARγ, 350 µg lysate from epiWAT of WT and Sirt7 KO mice (described above) was incubated with anti‐PPARγ antibody‐crosslinked resin (described above) at 4°C overnight for immunoprecipitation. Proteins were eluted with the elution buffer provided in the Pierce Crosslink Immunoprecipitation Kit. Acetylation of lysine was detected by western blotting with an anti‐acetyl lysine antibody (Cell Signaling Technology).
Retroviral infection and adipocyte differentiation
For knockdown (KD) of Sirt7, pSIREN‐RetroQ‐Sirt7 12 and pSIREN‐RetroQ (negative control) vectors were transfected into Plat‐E cells by the jetPRIME reagent. At 48 h after transfection, the retrovirus‐containing medium was collected and filtered with a 0.2‐µM syringe filter. For PPARγ2 overexpression, pMXs‐Puro‐PPARγ2‐WT, pMXs‐Puro‐PPARγ2K382R, pMXs‐Puro‐PPARγ2K382Q, and pMXs (negative control) vectors were used. To generate a stable cell line, C3H10T1/2 cells were infected with these retroviruses for 8 h and selected by treatment with 3 µg/mL puromycin for 72 h. For adipocyte differentiation, at 2 days after reaching confluence, C3H10T1/2 cells were treated with 1 µM dexamethasone (Sigma‐Aldrich), 0.5 mM isobutyl‐methylxanthine (Sigma‐Aldrich), 1.5 µg/mL insulin (Wako Pure Chemical Industries, Ltd.), and 1 µM rosiglitazone (#R2408; Sigma‐Aldrich) in maintenance medium for 48 h. Then, the cells were cultured in the maintenance medium with 1.5 µg/mL insulin, which was replenished every 2 days thereafter.
RNA‐seq analysis
RNA was extracted using an RNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Sequencing libraries were prepared using a NEBNext Ultra II Directional RNA Library Prep Kit (New England Biolabs, Ipswich, MA) and samples were sequenced on an Illumina NextSeq 500 platform in 76‐bp single‐end reads. The reads were trimmed for universal Illumina adaptors with TrimGalore (ver 0.6.5) 15 and mapped to transcripts from GENCODE release M25 using salmon (ver 1.2.1) 16 with the default and “GC” parameters. Data were loaded into R using the tximport package (v1.16.0) 17 and aggregated to gene‐level abundance in TPM. Differentially expressed genes were determined using DESeq2 (v1.28.0) 18 . Gene ontology analysis was performed using DAVID software 19 , 20 .
Gene expression analysis
Total RNA was extracted from C3H10T1/2‐derived adipocytes and from epiWAT of WT and Sirt7 KO mice with the Sepasol RNA I Super reagent (Nacalai Tesque). Quantitative real time (qRT)‐PCR was performed as previously described 9 . The relative expression of each gene was normalized to that of Tbp. Primer sequences are listed in Table S2.
Chromatin immunoprecipitation (ChIP) assay
Differentiated C3H10T1/2 cells were fixed in 1% formaldehyde for 5 min at room temperature. Then, the ChIP assay was performed as described in Appendix S1 using an anti‐PPARγ antibody.
Luciferase assay
HEK293T cells were transfected with pBIND‐PPARγ2 LBDK382R or pBIND‐PPARγ2 LBDK382Q and pG5luc plasmids using the jetPRIME transfection reagent, followed by treatment with or without 2 µM rosiglitazone. At 18 h after transfection, the cells were lysed and assayed using the firefly and Renilla luciferase substrates in the Dual‐Luciferase Reporter Assay System (Promega).
Statistical analysis
All results are expressed as the mean ± the standard error of the mean. Statistical significance was determined using the two‐tailed Student’s t‐test. A P‐value < 0.05 was considered to indicate a significant difference.
RESULTS
SIRT7 interacts with PPARγ2
To investigate the direct regulation of PPARγ2 activity by SIRT7, we first examined whether SIRT7 and PPARγ2 physically interacted with each other. When we performed a Halo tag pull‐down assay using lysates from 3×HA‐PPARγ2‐overexpressing HEK293T cells, Halo‐SIRT7, but not Halo, interacted with PPARγ2 (Figure 1a). We also examined the interaction of SIRT7 with PPARγ2 in cultured cells. HEK293T cells were transfected with the 3×HA‐PPARγ2 expression plasmid alone or with FLAG‐SIRT7, and the resulting cell lysates were immunoprecipitated with anti‐FLAG antibody resins. As shown in Figure 1b, PPARγ2 co‐immunoprecipitated with SIRT7. The interaction between endogenous PPARγ and SIRT7 was also detected in epiWAT (Figure 1c).
Figure 1.
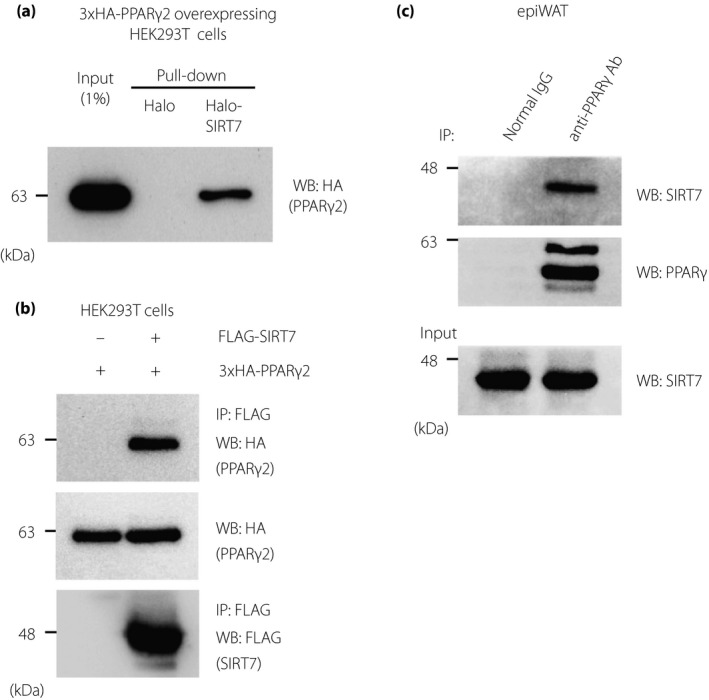
SIRT7 interacts with PPARγ2. (a) Halo‐SIRT7 pull‐down assay was performed using lysates from 3×HA‐PPARγ2‐overexpressing HEK293T cells to detect the binding between PPARγ2 and SIRT7. (b, c) Co‐immunoprecipitation assay between FLAG‐SIRT7 and 3×HA‐PPARγ2 in HEK293T cells (b) and between endogenous SIRT7 and PPARγ in epiWAT (c)
SIRT7 deacetylates PPARγ2
PPARγ is an acetylated protein 8 and SIRT7 is a deacetylase. Thus, we next assessed whether SIRT7 deacetylates PPARγ2. As shown in Figure 2a, PPARγ2 acetylation was detected in HEK293T cells, and SIRT7 overexpression decreased PPARγ2 acetylation, whereas SIRT7H188Y (a loss of function mutant) 9 did not reduce its acetylation. In addition, PPARγ acetylation was increased in epiWAT from Sirt7 KO mice (Figure 2b). These results indicate that SIRT7 exhibits deacetylation activity for PPARγ2.
Figure 2.
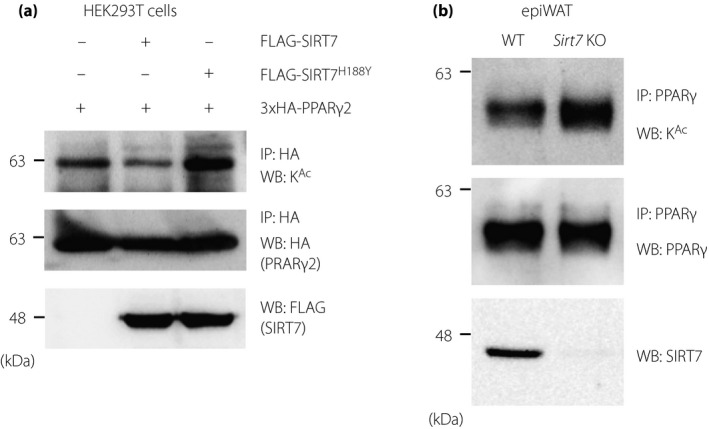
SIRT7 deacetylates PPARγ2. (a) Effect of SIRT7 overexpression on the acetylation of the PPARγ2 mutants. HEK293T cells were transfected with the 3×HA‐PPARγ2 and PCAF expression plasmids, as well as FLAG‐SIRT7 or FLAG‐SIRT7H188Y. The acetylation level of PPARγ2 was determined by immunoprecipitation and western blotting analysis. (b) Effect of SIRT7 deficiency on the endogenous acetylation of PPARγ. Protein lysates of epiWAT from WT and Sirt7 KO mice were subjected to immunoprecipitation, after which acetylated PPARγ was detected by western blotting analysis
SIRT7 deacetylates PPARγ2 at K382
To identify the SIRT7‐interacting region of PPARγ2, lysates from HEK293T cells expressing PPARγ2 deletion mutants (GAL4DBD‐PPARγ2‐M1 [1–200], GAL4DBD‐PPARγ2‐M2 [201–350], or GAL4DBD‐PPARγ2‐M3 [351–505]) were pull‐downed with Halo or Halo‐SIRT7 immobilized resin. As shown in Figure 3a, SIRT7 bound only to GAL4DBD‐PPARγ2‐M3. Further studies with additional deletion mutants (GAL4DBD‐PPARγ2‐M3A [351–439] and GAL4DBD‐PPARγ2‐M3B [440–505]) revealed that SIRT7 specifically bound to the M3A region, which lies in the LBD of PPARγ2 (Figure 3a) 19 . This M3A region contains 6 lysine residues (K364, K382, K386, K395, K401, and K432). To identify the residues targeted by SIRT7, we introduced a deacetylation‐mimicking K‐to‐R mutation into each of the 6 residues and examined the acetylation levels of these mutants in HEK293T cells. As shown in Figure 3b, the acetylation levels of the PPARγ2K364R, PPARγ2K395R, and PPARγ2K401R mutants were similar to those of PPARγ2 WT, whereas the PPARγ2K382R, PPARγ2K386R, and PPARγ2K432R mutants were less acetylated, indicating that K382, K386, and K432 are acetylated in the cells. We next examined whether SIRT7 deacetylates the K382R, K386R, and K432R mutants of PPARγ2. Although SIRT7 reduced the acetylation levels of PPARγ2K386R and PPARγ2K432R, it did not further deacetylate PPARγ2K382R (Figure 3c), indicating that K382 is targeted for deacetylation by SIRT7. This lysine residue is conserved in human, pig, mouse, chicken, frog, and zebrafish and is located in helix 6 of the LBD of PPARγ2 (Figure 3d) 21 .
Figure 3.
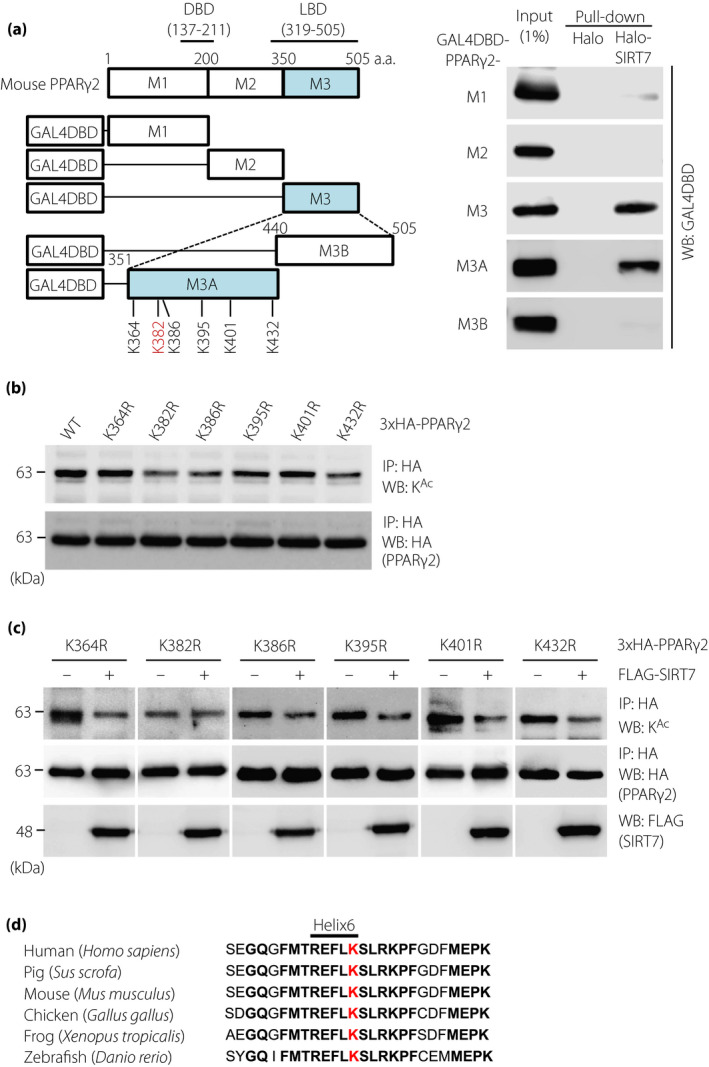
K382 is a target of the SIRT7‐mediated deacetylation of PPARγ2. (a) Mapping of SIRT7‐interacting sites in PPARγ2 by a pull‐down assay. Schematic diagrams of GAL4DBD‐fused mouse deletion mutants of PPARγ2, namely, M1 (1–200), M2 (201–350), M3 (351–505), M3A (351–439), and M3B (440–505), are illustrated on the left side. Halo‐SIRT7‐FLAG pull‐down assay with lysates from HEK293T cells expressing the indicated PPARγ2 deletion mutants fused with GAL4DBD (right). (b) Acetylation of KR mutants of PPARγ2. HEK293T cells were transfected with the indicated 3×HA‐PPARγ2 expression vectors. PPARγ2 acetylation was examined by immunoprecipitation and western blot analysis. (c) Effect of SIRT7 on the acetylation of the PPARγ2 KR mutants. HEK293T cells were transfected with PCAF and the indicated expression plasmids. PPARγ2 acetylation was examined by immunoprecipitation and western blot analysis. (d) Alignment of the PPARγ2 LBD from different species. The K382 of mouse PPARγ2 (red) is highly conserved in the indicated vertebrates
PPARγ2 acetylation at K382 regulates lipid accumulation in adipocytes
Previous studies have shown that SIRT1 attenuates adipogenesis, whereas SIRT7 promotes adipogenesis by inhibiting SIRT1 7 , 13 . We evaluated the role of SIRT7 in mouse mesenchymal C3H10T1/2 cells. Treatment of C3H10T1/2 cells with an adipocytic differentiation cocktail resulted in fat accumulation, as determined by Oil Red‐O staining of cellular lipids (Figure 4a). Sirt7 mRNA levels were significantly increased after day 5 of differentiation (Figure S1). Consistent with previous reports 13 , 22 , Sirt7 KD led to much less fat accumulation in C3H10T1/2‐derived adipocytes after differentiation (Figure 4a). The expression levels of PPARγ2 and its target genes, such as Ap2, Cd36, Adipoq, and Lpl, were significantly decreased in Sirt7 KD C3H10T1/2‐derived adipocytes, but the expression of Sirt1 mRNA was unchanged (Figure 4b). Then, we investigated the functional roles of PPARγ2 K382 acetylation using these cells. PPARγ2 WT, PPARγ2K382R, and PPARγ2K382Q (acetylation‐mimicking mutant) were retrovirally overexpressed in C3H10T1/2 cells and these cells were differentiated into adipocytes. Both PPARγ2 WT‐ and PPARγ2K382R‐expressing cells clearly differentiated into lipid‐filled adipocytes, whereas PPARγ2K382Q‐expressing cells accumulated less lipid, despite similar PPARγ mRNA expression (Figure 4c,d). Moreover, lipid accumulation was markedly increased by the PPARγ2K382R overexpression in Sirt7 KD C3H10T1/2‐derived adipocytes (Figure 4e). These results indicate that PPARγ2 K382 acetylation affects lipid accumulation in adipocytes. Interestingly, the expression of Adipoq and Lpl mRNA was lower in PPARγ2K382Q‐expressing adipocytes, but the expression levels of other PPARγ2 target genes (Cebpa and Ap2) were unchanged (Figure 4f), suggesting that the direct effect of SIRT7 on PPARγ2 (K382 deacetylation) is different from the indirect effect (PPARγ2 activation by suppressing SIRT1).
Figure 4.
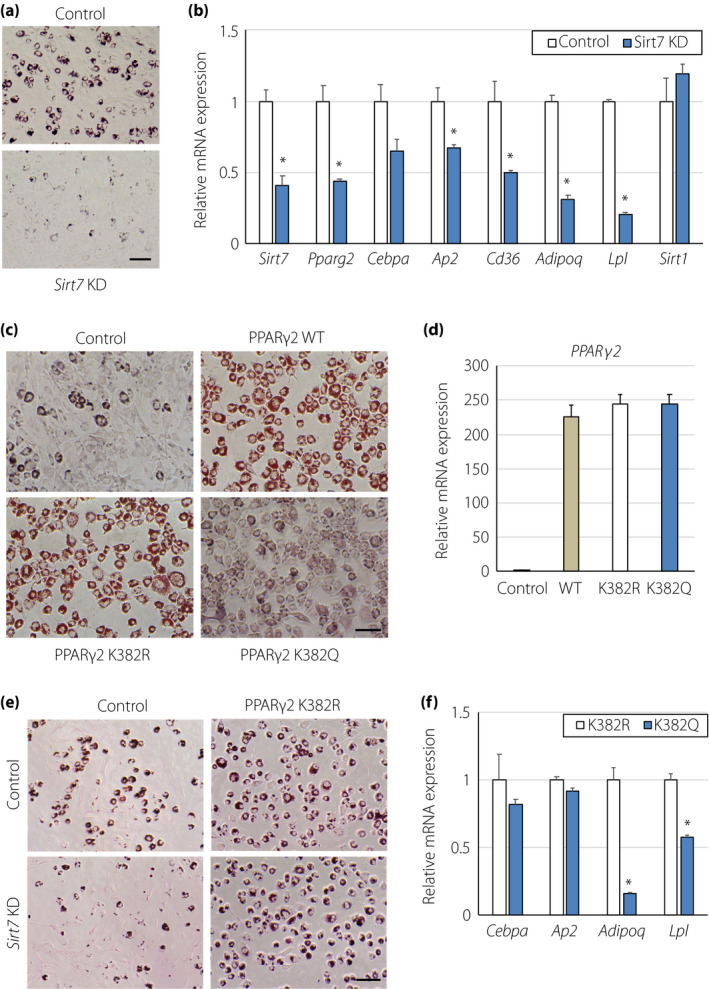
PPARγ2 acetylation at K382 regulates lipid accumulation. (a) Effect of Sirt7 KD on Oil Red‐O staining in adipocytes. C3H10T1/2 cells were infected with control and Sirt7 short hairpin RNA retrovirus. After selection by puromycin, C3H10T1/2 cells were differentiated into adipocytes for 5 days. Representative images of 3 independent experiments are shown. (b) Gene expression of Sirt1, Sirt7, Pparg2, and target genes for PPARγ2 in differentiated C3H10T1/2 adipocytes (n = 3). (c, d) Effect of PPARγ2 WT, PPARγ2K382R, and PPARγ2K382Q overexpression on Oil Red‐O staining in adipocytes. C3H10T1/2 cells were infected with control retrovirus or retroviruses expressing PPARγ2, PPARγ2K382R, and PPARγ2K382Q. After puromycin selection, the cells were differentiated into adipocytes for 5 days. Representative images (c) and expression of Pparg2 mRNA (d) from 3 independent experiments are shown. (e) Effect of PPARγ2K382R overexpression on Oil Red‐O staining in adipocytes. Control and Sirt7 KD‐C3H10T1/2 cells were infected with retroviruses expressing PPARγ2K382R, and the cells were differentiated into adipocytes for 5 days. (f) Expression of PPARγ2 target genes in differentiated C3H10T1/2‐derived adipocytes expressing PPARγ2K382R and PPARγ2K382Q (n = 3). Data are shown as the mean ± the standard error of the mean. *P < 0.05
To further investigate the roles of PPARγ2 K382 acetylation in lipid accumulation, we examined global gene expression in PPARγ2K382R‐ and PPARγ2K382Q‐expressing adipocytes by RNA‐seq analysis. This analysis revealed that the expression of 469 genes, including Adipoq (encoding adiponectin), Adipsin, and Fasn (encoding fatty acid synthase), was significantly downregulated (fold change > 2) in PPARγ2K382Q‐expressing cells compared with that in PPARγ2K382R‐expressing adipocytes (Figure 5a). Gene ontology analysis of the downregulated genes in PPARγ2K382Q‐expressing cells revealed their significant enrichment in the lipid metabolism process (Figure 5b). qRT‐PCR analysis confirmed that the expression levels of a number of genes involved in lipogenesis, such as Acaca (encoding acetyl CoA carboxylase α),Hacd2 (encoding 3‐hydroxyacyl CoA dehydratase 2), Fasn, Elovl7 (encoding elongation of very long chain fatty acids‐like 7), and Scd1 (encoding stearoyl CoA desaturase), were significantly lower in PPARγ2K382Q‐expressing adipocytes (Figure 5c). Sterol regulatory element‐binding protein‐1c (SREBP‐1c) plays a central role in lipogenesis 23 . The expression of Srebp1c mRNA was also lower in PPARγ2K382Q‐expressing cells (Figure 5c). PPARγ2K382Q overexpression led to the reduced expression of several PPARγ2 target genes, such as Adipoq, Pck1, Adipsin, and Lpl, but it had no effect on the expression of a number of PPARγ2 target genes involved in adipogenesis (such as Cebpa, Cebpb, Cebpd, Stat5a, and Stat5b) and lipid metabolism (such as Ap2, Acbp, Nr1h3, Cd36, and Acs1) (Figures 4e, 5c,d). Sirt1 mRNA expression was also unchanged in PPARγ2K382Q‐expressing cells (Figure 5d). Consistently, the expression of lipogenic genes, such as Fasn, Acaca, and Srebp1c, was significantly decreased in epiWAT of Sirt7 KO mice (Figure 5e).
Figure 5.
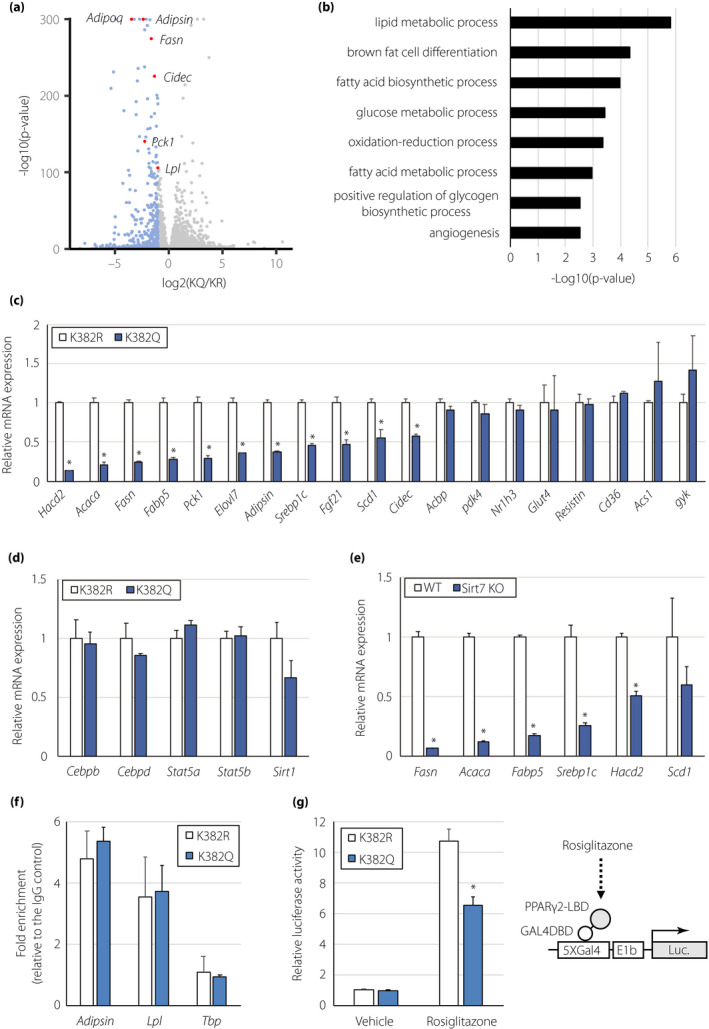
PPARγ2 acetylation at K382 regulates the expression of lipogenesis‐related genes. (a) Volcano plot derived from RNA‐seq analysis of PPARγ2K382R‐ and PPARγ2K382Q‐expressing adipocytes. Transcripts downregulated (fold change > 2, P < 0.05) in PPARγ2K382Q‐expressing adipocytes are in blue. (b) Gene ontology analysis of the downregulated genes in PPARγ2K382Q‐expressing cells. (c, d) Expression of genes involved in lipid metabolism (c) and adipocyte differentiation (d) in PPARγ2K382R‐ and PPARγ2K382Q‐expressing adipocytes (n = 3). (e) Expression of lipogenic genes in epiWAT of WT and Sirt7 KO mice (n = 4). (f) ChIP for the recruitment of PPARγ to the indicated genes in PPARγ2K382R‐ and PPARγ2K382Q‐expressing adipocytes (n = 3). Quantification of enrichment is represented as fold‐enrichment relative to IgG. (g) Effect of K382 acetylation on the ligand‐dependent activity of PPARγ2 in HEK293T cells. The cells were transfected with the GAL4DBD‐PPARγ2 LBDK382R or GAL4DBD‐PPARγ2 LBDK382Q expression plasmid, as well as the 5×GAL4‐luciferase reporter plasmid, followed by treatment with or without rosiglitazone. Luciferase activity was determined after 18 h (n = 4). Data are shown as the mean ± the standard error of the mean. *P < 0.05
To understand how K382 acetylation affects gene expression, we compared the binding of PPARγ2K382R and PPARγ2K382Q by a ChIP assay. The binding of PPARγ2K382R and PPARγ2K382Q to the promoter of the Adipsin and Lpl genes was similar (Figure 5f), suggesting that K382 acetylation does not affect the DNA‐binding ability of PPARγ2. We next investigated the influence of K382 acetylation on the ligand‐dependent activity of PPARγ2. HEK293T cells were transfected with an expression vector containing the PPARγ2 LBD fused with the GAL4 DBD and a luciferase reporter plasmid driven by GAL4 binding sites. The transcriptional activity of PPARγ2K382Q induced by rosiglitazone was significantly reduced by as much as 40% compared with that of PPARγ2K382R (Figure 5g), indicating that K382 acetylation alters ligand‐dependent PPARγ2 activity.
DISCUSSION
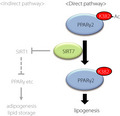
Lysine acetylation is a well‐known post‐translational modification that affects the function of a variety of proteins 24 . PPARγ is an acetylated protein, and SIRT1‐dependent deacetylation at K268 and K293 modulates PPARγ coactivator/corepressor exchange 8 . In the present study, we found that SIRT7 deacetylates PPARγ2 at K382 and enhances fat accumulation in adipocytes by regulating the expression of genes involved in lipogenesis (Figure 6). SIRT1 is activated upon fasting and promotes fat mobilization 7 . Thus, SIRT1 and SIRT7 exert clearly opposite roles in lipid accumulation. It is not presently known how the functions of SIRT1 and SIRT7 are integrated in vivo, but SIRT7 may be suppressed in a low‐energy state, as reported previously 25 .
Figure 6.
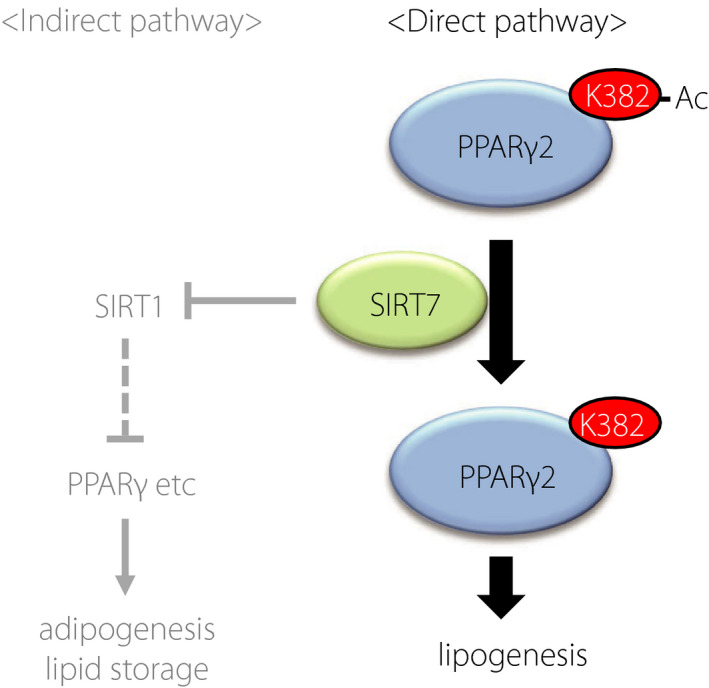
Schematic model of SIRT7‐mediated deacetylation of PPARγ2
The mechanism by which K382 acetylation controls lipid accumulation in adipocytes is unclear, but we found that the ligand‐dependent transcriptional activity of PPARγ2K382Q was reduced. K382 is located within the helix 6 region of the LBD, which forms the ligand‐binding pocket of PPARγ2 21 , 26 . Ligand binding or co‐regulator recruitment may be differentially regulated by K382 acetylation. More studies are needed to clarify the functional roles of PPARγ2 K382 acetylation.
SREBP‐1c plays an important role in lipogenesis 23 . We showed that the expression levels of Srebp1c and its target genes, including Acaca, Fasn, and Scd1, were significantly lower in PPARγ2K382Q‐expressing cells than in PPARγ2K382R‐expressing cells. Srebp1c transcription is regulated by liver X receptor α (LXRα), and Nr1h3 (encoding LXRα) is a target gene of PPARγ2 27 . However, the expression of Nr1h3 mRNA was unchanged in PPARγ2K382Q‐expressing cells. Thus, it is unlikely that PPARγ2K382Q regulates Srebp1c mRNA expression through the regulation of Nr1h3. Further studies are necessary to elucidate the mechanism.
Recent studies clarified that post‐translational modifications regulate the function of PPARγ2 1 , 3 . For example, phosphorylation of S273 in PPARγ2 alters the transcription of a distinct group of genes whose expression is altered in obesity, and non‐thiazolidinedione compounds that block PPARγ2 phosphorylation at S273 exhibit excellent anti‐diabetic effects 28 , 29 . Our findings suggest that low molecular weight compounds inhibiting the deacetylation of K382 in PPARγ2 may have a beneficial effect against metabolic syndrome and/or type 2 diabetes by decreasing the accumulation of fat in adipocytes.
In conclusion, we clarified that SIRT7 controls lipogenesis and lipid metabolism in adipocytes by directly regulating the acetylation of PPARγ2. Our findings may have significant implications for the development of novel drugs against obesity and type 2 diabetes.
DISCLOSURE
The authors declare no conflicts of interest associated with this manuscript.
Supporting information
Figure S1 | The expression of Sirt7 increases with the differentiation of adipocytes.
Table S1 | Primer sequences used in mutagenesis.
Table S2 | Primer sequences used in quantitative qRT‐PCR.
ACKNOWLEDGMENTS
This study was supported by a Grant‐in‐Aid for Scientific Research (B) (19H03711; K.Y., 20H04107; T.Y.), Challenging Research (Exploratory) (19K22639; K.Y.), and Scientific Research on Innovative Areas “LipoQuality” (18H04676; T.Y.) from MEXT, Japan; by a grant from the Japan Agency for Medical Research and Development under Grant Number (JP20gm5010002; K.Y.); and by grants from the Naito Foundation (K.Y.) and Takeda Science Foundation (K.Y., T.Y.).
J Diabetes Investig. 2021; 12: 1765–1774
REFERENCES
- 1. Ahmadian M, Suh JM, Hah N, et al. PPARγ signaling and metabolism: the good, the bad and the future. Nat Med 2013; 19: 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rangwala SM, Lazar MA. Peroxisome proliferator‐activated receptor γ in diabetes and metabolism. Trends Pharmacol Sci 2004; 25: 331–336. [DOI] [PubMed] [Google Scholar]
- 3. Sauer S. Ligands for the nuclear peroxisome proliferator‐activated receptor gamma. Trends Pharmacol Sci 2015; 36: 688–704. [DOI] [PubMed] [Google Scholar]
- 4. Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab 2014; 20: 573–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol cell biol 2012; 13: 225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yamagata K, Yoshizawa T. Transcriptional regulation of metabolism by SIRT1 and SIRT7. Int Rev Cell Mol Biol 2018; 335: 143–166. [DOI] [PubMed] [Google Scholar]
- 7. Picard F, Kurtev M, Chung N, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR‐γ. Nature 2004; 429: 771–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Qiang LI, Wang L, Kon N, et al. Brown remodeling of white adipose tissue by SirT1‐dependent deacetylation of Pparγ. Cell 2012; 150: 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yoshizawa T, Karim M, Sato Y, et al. SIRT7 controls hepatic lipid metabolism by regulating the ubiquitin‐proteasome pathway. Cell Metab 2014; 19: 712–721. [DOI] [PubMed] [Google Scholar]
- 10. Karim MF, Yoshizawa T, Sobuz SU, et al. Sirtuin 7‐dependent deacetylation of DDB1 regulates the expression of nuclear receptor TR4. Biochem Biophys Res Commun 2017; 490: 423–428. [DOI] [PubMed] [Google Scholar]
- 11. Ryu D, Jo YS, Sasso GL, et al. A SIRT7‐dependent acetylation switch of GABPβ1 controls mitochondrial function. Cell Metab 2014; 20: 856–869. [DOI] [PubMed] [Google Scholar]
- 12. Fukuda M, Yoshizawa T, Karim MF, et al. SIRT7 has a critical role in bone formation by regulating lysine acylation of SP7/Osterix. Nat Commun 2018; 9: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fang J, Ianni A, Smolka C, et al. Sirt7 promotes adipogenesis in the mouse by inhibiting autocatalytic activation of Sirt1. PNAS 2017; 114: E8352–E8361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sobuz SU, Sato Y, Yoshizawa T, et al. SIRT7 regulates the nuclear export of NF‐κB p65 by deacetylating Ran. Biochim Biophy Acta Mol Cell Res 2019; 1866: 1355–1367. [DOI] [PubMed] [Google Scholar]
- 15. http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/.
- 16. Patro R, Duggal G, Love MI, et al. Salmon provides fast and bias‐aware quantification of transcript expression. Nat Methods 2017; 14: 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Soneson C, Love MI, Robinson MD. Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000 2016; 4: 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 2014; 15: 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 2009; 37: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 2009; 4: 44. [DOI] [PubMed] [Google Scholar]
- 21. Chandra V, Huang P, Hamuro Y, et al. Structure of the intact PPAR‐γ–RXR‐α nuclear receptor complex on DNA. Nature 2008; 456: 350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cioffi M, Vallespinos‐Serrano M, Trabulo S, et al. MiR‐93 controls adiposity via inhibition of Sirt7 and Tbx3. Cell Rep 2015; 12: 1594–1605. [DOI] [PubMed] [Google Scholar]
- 23. Shao W, Espenshade PJ. Expanding roles for SREBP in metabolism. Cell Metab 2012; 16: 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choudhary C, Weinert BT, Nishida Y, et al. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol cell biol 2014; 15: 536–550. [DOI] [PubMed] [Google Scholar]
- 25. Chen S, Seiler J, Santiago‐Reichelt M, et al. Repression of RNA polymerase I upon stress is caused by inhibition of RNA‐dependent deacetylation of PAF53 by SIRT7. Mol Cell 2013; 52: 303–313. [DOI] [PubMed] [Google Scholar]
- 26. Shang J, Brust R, Mosure SA, et al. Cooperative cobinding of synthetic and natural ligands to the nuclear receptor PPARγ. Elife 2018; 7: e43320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seo JB, Moon HM, Kim WS, et al. Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator‐activated receptor γ expression. Mol Cell Biol 2004; 24: 3430–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Choi JH, Banks AS, Estall JL, et al. Anti‐diabetic drugs inhibit obesity‐linked phosphorylation of PPARγ by Cdk5. Nature 2010; 466: 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Choi JH, Banks AS, Kamenecka TM, et al. Antidiabetic actions of a non‐agonist PPARγ ligand blocking Cdk5‐mediated phosphorylation. Nature 2011; 477: 477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 | The expression of Sirt7 increases with the differentiation of adipocytes.
Table S1 | Primer sequences used in mutagenesis.
Table S2 | Primer sequences used in quantitative qRT‐PCR.