

Abstract
Background
Globally, about 6% of children are born with a serious birth defect of genetic or partially genetic origin. Carrier screening or testing is one way to identify couples at increased risk of having a child with an autosomal recessive condition. The most common autosomal recessive conditions are thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease, with higher carrier rates in high‐risk populations of specific ancestral backgrounds. Identifying and counselling couples at genetic risk of the conditions before pregnancy enables them to make fully informed reproductive decisions, with some of these choices not being available if testing is only offered in an antenatal setting. This is an update of a previously published review.
Objectives
To assess the effectiveness of systematic preconception genetic risk assessment to enable autonomous reproductive choice and to improve reproductive outcomes in women and their partners who are both identified as carriers of thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease in healthcare settings when compared to usual care.
Search methods
We searched the Cochrane Cystic Fibrosis and Genetic Disorders Group's Trials Registers. Date of latest search of the registers: 04 August 2021.
In addition, we searched for all relevant trials from 1970 (or the date at which the database was first available if after 1970) to date using electronic databases (MEDLINE, Embase, CINAHL, PsycINFO), clinical trial databases (National Institutes of Health, Clinical Trials Search portal of the World Health Organization, metaRegister of controlled clinical trials), and hand searching of key journals and conference abstract books from 1998 to date (European Journal of Human Genetics, Genetics in Medicine, Journal of Community Genetics). We also searched the reference lists of relevant articles, reviews and guidelines and also contacted subject experts in the field to request any unpublished or other published trials. Date of latest search of all these sources: 25 June 2021.
Selection criteria
Any randomised controlled trials (RCTs) or quasi‐RCTs (published or unpublished) comparing reproductive outcomes of systematic preconception genetic risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease when compared to usual care.
Data collection and analysis
We identified 37 papers, describing 22 unique trials which were potentially eligible for inclusion in the review. However, after assessment, we found no RCTs of preconception genetic risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease.
Main results
No RCTs of preconception genetic risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease are included. A trial identified earlier has published its results and has subsequently been listed as excluded in this review.
Authors' conclusions
As there are no RCTs of preconception genetic risk assessment for thalassaemia, sickle cell disease, cystic fibrosis, or Tay‐Sachs disease included in either the earlier or current versions of this review, we recommend considering potential non‐RCTs studies (for example prospective cohorts or before‐and‐after studies) for future reviews. While RCTs are desirable to inform evidence‐based practice and robust recommendations, the ethical, legal and social implications associated with using this trial design to evaluate the implementation of preconception genetic risk assessment involving carrier testing and reproductive autonomy must also be considered. In addition, rather than focusing on single gene‐by‐gene carrier testing for specific autosomal‐recessive conditions as the intervention being evaluated, preconception expanded genetic screening should also be included in future searches as this has received much attention in recent years as a more pragmatic strategy.
The research evidence for current international policy recommendations is limited to non‐randomised studies.
Plain language summary
Identifying carrier status for thalassaemia, sickle cell disease, cystic fibrosis, or Tay‐Sachs disease in non‐pregnant women and their partners
Review question
We looked for evidence to show whether identifying people who are carriers for thalassaemia, sickle cell disease, cystic fibrosis, or Tay‐Sachs disease, before pregnancy leads to improving reproductive choice and pregnancy outcomes.
Background
Across the world, about 6% of children are born with a birth defect of genetic or partially genetic origin. Many of these conditions can be passed down from parent to child. There are tests to identify the genetic risk of the most common genetic conditions (thalassaemia, sickle cell disease, cystic fibrosis, or Tay‐Sachs disease) before pregnancy. In these conditions, called autosomal recessive conditions, the parents of affected children are 'carriers' of the condition, which means they do not usually have symptoms. All 'carrier' couples will have a 25% chance of having an affected child. Risk assessment for these genetic conditions before getting pregnant would benefit potential parents who may be carriers. This information would give the at‐risk couple the opportunity to make fully informed decisions about family planning. However, genetic risk assessment before pregnancy may potentially have a negative psychological impact. This is an updated version of the original review.
Search date
We last looked for evidence on 04 August 2021.
Study characteristics
We did not find any trials that we could include in this review. In an earlier version of this review, we had already found the protocol for a trial that has now published its results, but we have excluded the trial in this version of the review because it did not look at the right topic after all.
Key results
Although no trials were identified in which people taking part would have equal chances of being in either group, there are several studies which are not so strictly designed which support current policy recommendations for genetic risk assessment prior to pregnancy in routine clinical practice. We recommend considering potential observational studies in future reviews as well as looking at ‘expanded carrier screening’ before pregnancy and not just screening for one condition. Any future trials need to consider legal, ethical and cultural barriers to implementing genetic risk assessment before pregnancy.
Background
A glossary of terms is available as an appendix (Appendix 1).
Description of the condition
Genetic medicine is expanding into almost every aspect of health care; reproductive risk assessment during the preconception period is a prime example. Identifying genetic risks before pregnancy or conception might produce significant benefits, such as providing information about the risk of having children with genetic conditions and thus giving couples or prospective parents the opportunity to make more informed reproductive decisions. It has been estimated that a couple has a baseline risk of 2% to 3% of having a child with a congenital or genetic disorder (Teeuw 2010). The probability of having an affected child increases when there is a family history of genetic disorders (Shapira 2006; Teeuw 2010). Globally, about 6% of children are born with a serious birth defect of genetic or partially genetic origin (March of Dimes 2006) with over 1800 genes linked to recessively inherited disorders (Antonarakis 2019).
Preconception risk assessment for autosomal recessive or X‐linked recessive genetic disorders would benefit couples who may be carriers. The most common examples of these autosomal recessive disorders are thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease. In these disorders, such carriers are usually asymptomatic; however, their child will be affected if he or she inherits the affected genes from both parents. All carrier couples have a 25% chance of having an affected child. These conditions have a high morbidity risk, are potentially life‐threatening and have a significant psychological impact not only on the affected child but also on their families or care givers. These diseases are also more prevalent in individuals of particular ethnic backgrounds (WHO 2000). X‐linked recessive genetic disorders include haemophilia A and B, and Duchenne muscular dystrophy. In these disorders, if the mother is a carrier, she has a 25% chance of having a son with the condition in each pregnancy (Haque 2016).
In some countries, the need for medical care for children with these diseases, as well as psychological interventions to offer behavioural and emotional support, imposes a potentially high economic and public health burden (Cornel 2021). In view of the magnitude of these conditions and their implications for children and their families, there have been considerable efforts to identify the genetic reproductive risk for the four specified conditions and offer counselling and support to potential parents before the birth of an affected child. Women and couples at increased genetic risk, as well as healthcare professionals, have recognised the importance of preconception assessment to increase reproductive autonomy (Boulton 1996; Henneman 2001; Holtkamp 2017; Janssens 2014; Locock 2008; Massie 2014; Mennie 1998; Poppelaars 2004; Watson 1999; Wille 2004). To date, the practical experience of reproductive genetic risk assessment for autosomal recessive disorders focuses mainly on the antenatal and newborn periods (Qureshi 2004). Identifying genetic reproductive risk during the antenatal period leaves the couple a short period of time to make difficult or limited choices, such as terminating the pregnancy or continuing with the pregnancy and caring for the affected child (Dormandy 2008). Identifying couples who have confirmed genetic carrier status before conception provides an opportunity for individuals or couples to make fully informed reproductive choices including not having children, PGT using donor gametes, arranging early prenatal diagnosis and antenatal care and also considering adoption of a child (Jones 2002; Wille 2004).
Thalassaemia
According to the March of Dimes Birth Defects Foundation, an estimated 307,900 children are born annually with major haemoglobin disorders, the most common being thalassaemia and sickle cell disease (March of Dimes 2006). Thalassaemia is characterised by the defects or absence of synthesis in one of the two globin chains (α or β) which form the normal adult human haemoglobin molecule; this leads to haemolytic anaemia (Peters 2012). Thalassaemia can be diagnosed by measuring fractions of haemoglobin A and haemoglobin F with high‐performance liquid chromatography (HPLC) or electrophoresis. In addition, DNA analysis is required to detect an α or β globin chain mutation (Peters 2012). It is estimated that between two and five per cent of the world's population are carriers and this is more prevalent in the Mediterranean, the Middle East, South and East Asia, and the Pacific, with carrier rates ranging from two per cent to 19 per cent (Modell 2001; March of Dimes 2006). Because of founder effect, carrier rates have also increased in countries that previously had low prevalences (Cousens 2010). Morbidity is related to severe anaemia and an affected child will require lifelong blood transfusions. Multiple blood transfusions may eventually result in iron overload and potentially cause heart failure, infection, hypogonadism, infertility, diabetes mellitus, and hypothyroidism. Affected individuals may die prematurely unless given optimal medical management. In individuals with thalassaemia and their families or caregivers, psychosocial problems have also been reported, for example, stigmatisation, isolation, family adjustment, coping with school and education, and social interaction (Gharaibeh 2009; Ratip 1996; Telfer 2005).
Sickle cell disease
Sickle cell disease is caused by a mutation in the haemoglobin gene (βS) which individuals inherit from both parents (Weatherall 1997). The WHO estimates that sickle cell disease affects 275,000 conceptions each year globally (Modell 2008; Yusuf 2011). Diagnosis is confirmed using HPLC or electrophoresis with the detection of haemoglobin S and C fraction. It affects mainly individuals of African origin but is also found in Indian and some Mediterranean populations. The reported prevalence of carrier frequency ranges from one to 40 per cent, depending on the population group. The condition causes the red blood cells to have a sickle shape which results in premature haemolysis and can lead to life‐threatening acute and chronic vaso‐occlusion, including renal and cardiovascular complications. Individuals with this condition are also susceptible to serious septicaemia. Like thalassaemia, individuals and their families are also confronted with psychosocial challenges which include the disruption of school and work, social isolation and loneliness, stigmatisation, bullying, and rejection by peers (Barbarin 1999). Recently, there has been a new development in the treatment of sickle cell disease, somatic genome editing which is currently being studied (Bonham 2019).
Cystic fibrosis
Cystic fibrosis is caused by a mutation in the gene cystic fibrosis transmembrane conductance regulator (CFTR); more than 2000 CFTR mutations have now been identified (Bareil 2020). Diagnosis of cystic fibrosis is indicated by phenotypic features (chronic sino‐pulmonary disease, gastrointestinal and nutritional abnormalities, obstructive azoospermia and salt‐loss syndromes), a family history of cystic fibrosis or a positive newborn screening test, together with laboratory evidence of a CFTR abnormality. Abnormalities in the CFTR genes can be identified by elevated sweat chloride concentrations (sweat test), identification of two CFTR mutations, or in vivo demonstration of characteristic abnormalities in ion transport across the nasal epithelium. Carriers are confirmed by the identification of a CFTR mutation from the blood or saliva (CDC 2004).
Cystic fibrosis is most common among people of European descent with a carrier frequency of 1 in 25 (Murray 1999). This condition is commonly associated with recurrent pulmonary infections, which potentially lead to bronchiectasis and atelectasis, and also pancreatic exocrine insufficiency. There is currently no cure for the disease, with treatment mainly aimed at improving a person's quality of life. However, in the past years, significant progress has been made, in particular, regarding CFTR‐directed therapeutics (Patel 2020) The need for emotional and social adjustment is a significant psychosocial consequence for people with cystic fibrosis (Bregnballe 2007; Glasscoe 2008).
Tay‐Sachs disease
Tay‐Sachs disease is caused by a genetic mutation in the α chains of the hexosaminidase A (Hex A) isozyme in the gangliosides in nerve cells of the brain (Bach 2001). The disease is diagnosed by measuring the activity of hexosaminidase A and further identification of a genetic mutation in Hex A (ACOG Committee Opinion 2017). It is most prevalent in the Ashkenazi Jewish populations, with a carrier frequency of around 1 in 30 (ACOG Committee Opinion 2017). The condition leads to a progressive deterioration of mental and physical abilities. Death usually occurs before five years of age. At present, there is no cure or available treatment.
Description of the intervention
Women and their partners can be assessed during the preconception period to identify if they are carriers of one of these four autosomal recessive conditions, which represent the most common autosomal recessive conditions globally. Cystic fibrosis is most common in Northern European populations; sickle cell disease and thalassaemia are most common non‐Northern European populations, and Tay‐Sachs disease is most common in individuals of Ashkenazi Jewish ancestry. Approaches to improve health outcomes and reproductive choice in couples who carry these genetic conditions should be generalisable to other, but rarer, autosomal recessive conditions. In populations with high carrier rates or significant burden of affected individuals, or both, carrier screening may be offered during preconception to all women in some healthcare settings (ACOG Committee Opinion 2017). More commonly, women and their partners may be assessed on the need for carrier testing. This assessment would be based firstly on a review of the family history for any of the autosomal recessive conditions or their carrier status; and, secondly, on the ethnic origin of the woman and her partner (Dyson 2006). This assessment of ancestry will identify if the individual originates from an ethnic group with a greater probability of being a healthy carrier of any of the four autosomal recessive disorders; for example, those with Ashkenazi Jewish ancestry are more likely to carry Tay‐Sachs disease, whilst those of African descent may carry sickle cell trait. The benefits of recording family history as one of the components of preconception health checks have been reported in previous observational community‐based studies for a broad range of genetic conditions (Czeizel 2012; Shaw 2020).
Overall, previous interventions have involved genetic carrier testing or screening (with or without educational support and genetic counselling) or both. There is often confusion between the terms genetic carrier testing and screening (Nuffield Council on Bioethics 1993). Genetic carrier testing refers to the testing of individuals to determine the presence or absence of the carrier status (Human Genetics Commission 2011). This testing could, for example, be in the context of a family history of the autosomal recessive condition or relevant ethnicity. On the other hand, genetic carrier screening involves offering or testing the whole population group irrespective of individual risk (Castellani 2010; Human Genetics Commission 2011). Both genetic carrier testing and screening involve the analysis of blood, tissue or bodily fluid samples.
With regards to the actual genetic carrier tests, currently either HPLC or electrophoresis is used to detect haemoglobin variants and to confirm carrier status for thalassaemia and sickle cell disease (NHS SCT Screening Programme 2013). Carrier status for cystic fibrosis is confirmed by analysing the mutations in the gene CFTR, using DNA commonly obtained from white blood cells, mouthwashes and buccal swabs (Murray 1999). Confirmation of Tay‐Sachs disease carrier status comprises of molecular analysis to detect genetic mutations in the α chains of the hexosaminidase A (Hex A) isozyme. To improve the detection rate, this should be combined with biochemical tests (ACOG Committee Opinion 2017).
For each condition, as well as confirmed carrier status identified by genetic carrier tests, there are other laboratory investigations that could indicate a probable carrier state. A microcytic or hypochromic blood picture, or both, without anaemia suggests a probable thalassaemia carrier, whilst a probable sickle cell carrier is indicated by a positive sickle solubility test. Elevated sodium chloride levels in sweat can indicate a probable cystic fibrosis carrier state.
At present, there is no formal or standard recommendation that fully addresses preconception genetic risk assessment (NHS SCT Screening Programme 2013). There is variability in how preconception genetic risk assessment is offered across countries. For example, screening for haemoglobinopathies is offered in pre‐marital clinics (Cousens 2010; Samavat 2004), whereas, screening for any reproductive genetic disorder may be offered opportunistically in a range of settings such as family planning clinics (Delatycki 2019; Watson 1999). Similarly, in current clinical practice, preconception risk assessment is not offered systematically, but most commonly offered opportunistically, for example when the issue is brought up by the couple or through family history (Heyes 2004).
Preconception expanded carrier screening
In recent years, next‐generation sequencing has enabled the screening of hundreds of genetic conditions in one panel simultaneously, including the four conditions described above, as compared to single gene‐by‐gene carrier screening offered traditionally (ACOG Committee Opinion 2017 (reaffirmed 2020); Grody 2013). Expanded carrier screening allows testing of all individuals regardless of ancestry, thereby potentially reducing the risk of stigmatisation of specific groups and a more pragmatic approach to preconception carrier screening. The Superior Health Council of Belgium has proposed an expanded carrier screening for autosomal and X‐linked recessive conditions to be offered preconceptionally (Superior Health Council Belgium 2017). Although there is no firm or standard recommendation for preconception expanded carrier screening, professional bodies including the European Society of Human Genetics have published recommendations on how to responsibly implement expanded carrier screening (ACOG Committee Opinion 2017 (reaffirmed 2020)). Currently, the Australian Reproductive Genetic Carrier Screening Project (“Mackenzie’s Mission”) is developing a carrier screening model which involves gene selection for carrier screening panel, evaluation of uptake, reproductive decisions, ethical and psychosocial aspects as well as cost‐effectiveness (Delatycki 2019; Kirk 2021).
How the intervention might work
In the specified autosomal recessive disorders (thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease), preconception genetic risk assessment ensures at‐risk couples, in which both the women and her partner are carriers of the specified conditions, are aware that they have a one‐in‐four chance of an affected child prior to pregnancy, enabling them to achieve fully informed reproductive autonomy (Cannon 2019; Christie 2009; Czeizel 2012; Lena‐Russo 2002; Massie 2009). This offers the at‐risk couples the opportunity to consider the full range of reproductive options (Borry 2011); for instance, couples may choose to have in vitro fertilisation (IVF) with pre‐implantation genetic testing (PGT), use donor gametes, adopt a child or remain childless (Human Genetics Commission 2011; Jones 2002; Wille 2004). These options are not available to couples who are only made aware of their reproductive genetic risk during the pregnancy. Of equal importance, if couples who have already been informed of their risk, decide to carry on with pregnancy they may consider and be offered prenatal diagnosis earlier in pregnancy. This enables the option of termination in early gestation or can enhance preparation for foetal and maternal well‐being throughout pregnancy, preparation following the birth of an infant, and postnatal support. (Wille 2004). If the carrier testing is implemented in the antenatal period, all of these decisions are delayed (Qureshi 2004). With regards to family history assessment, participants have acknowledged that this intervention enables pregnancy planning (Rose 1999) and early identification of couples at reproductive genetic risk (Czeizel 2012).
While the aim of preconception genetic risk assessment is to enable informed reproductive choices, in communities where families are affected by a high burden of disease, prevention of the birth prevalence of disease seemed appropriate (de Wert 2012). At a societal level, preconception carrier state identification has reduced the rate of affected births (Angastiniotis 1998; Cannon 2019; Samavat 2004). Although it is estimated that preconception screening programmes worldwide have caused a small decrease in affected births for haemoglobin disorders from 2.7 per 1000 conceptions to 2.55 per 1000 conceptions over a five‐year period from 1998 to 2003, more data and across all common autosomal recessive conditions need to be explored (Modell 2008). Similarly, early observational studies involving genetic carrier screening programmes for Tay‐Sachs disease and thalassaemia in Canada and France carried out in high school students were associated with an increased rate of early prenatal diagnosis and termination of affected pregnancies (Lena‐Russo 2002; Mitchell 1996; Zeesman 1984). In Cyprus and Iran, the prevalence of thalassaemia has decreased tremendously with the introduction of mandatory pre‐marital genetic carrier screening programmes (Alswaidi 2009; Angastiniotis 1998; Samavat 2004). In Hungary, preconception screening has resulted in improved identification of carrier couples and access to genetic counselling services (Czeizel 2012). The introduction of preconception expanded genetic screening allows for the detection of more carriers and carrier couples and potentially maximises opportunities for reproductive autonomy with a much wider array of reproductive risks; however, it has its own ethical, legal and social challenges (van der Hout 2016).
Why it is important to do this review
While a number of observational studies have reported the potential benefits of preconception risk assessment for genetic conditions in general (Czeizel 2012), and specifically for cystic fibrosis (Christie 2009; Massie 2009), haemoglobinopathies (Cao 1996) and Tay‐Sachs disease (Mitchell 1996), as with other programmes for genetic testing or screening, this has potential adverse effects. Genetic assessment for reproductive risk has been linked to psychological distress such as anxiety; however, the raised anxiety was a transient phenomenon (Archibald 2011; Bekker 1994). Further, it has been reported that carrier status may be associated with a poor perception of health (Henneman 2001) and may have an impact on the carrier's relationships with their partner (Fanos 1995). Although the occurrence is low, social impacts such as stigmatisation and discrimination have been reported with mandatory carrier screening (Bonham 2010; Kenen 1978; Whitten 1973). Despite these reported adverse effects, there are numerous psychological benefits including the opportunity for informed decision‐making and reproductive autonomy in prospective parents (Anido 2005; Archibald 2011; Cannon 2019; Lewis 2011).
With regards to the economic implications, as for other programmes for genetic testing and screening, there is an opportunity cost for redistributing resources from medical care to preconception risk assessment (WHO 1968). Addressing cost‐effectiveness in preconception carrier screening can be ethically sensitive (Cornel 2021). Several economic appraisals of haemoglobinopathies screening in the antenatal and neonatal settings have nevertheless indicated that these strategies are cost‐effective (Davies 2000; Zeuner 1999). A review of existing screening programmes in Australia has shown that targeted preconception screening in certain ethnic groups demonstrates both clinical and cost‐effectiveness (Lew 2014).
At a policy level, preconception genetic risk assessment has been recommended in clinical practice in the Netherlands, the USA and the UK (ACOG Committee Opinion 2009; Health Council of Netherlands 2007; Human Genetics Commission 2011). However, a comprehensive review of the current evidence still needs to be undertaken to directly inform healthcare practice.
This review will explore if robust trial evidence exists on the effect of preconception genetic risk assessment for genetic disorders, particularly before its widespread routine implementation in current healthcare settings. This is an update of a previously published review (Hussein 2015; Hussein 2018).
Objectives
The purpose of this review is to assess the effectiveness of systematic preconception genetic risk assessment to enable autonomous reproductive choice and to improve reproductive outcomes in women and their partners who are identified as carriers of thalassaemia, sickle cell disease, cystic fibrosis or Tay‐Sachs disease in healthcare settings when compared to usual care.
Methods
Criteria for considering studies for this review
Types of studies
We planned for this review to include all relevant randomised controlled trials (RCTs) and quasi‐RCTs.
Types of participants
Women and their partners of reproductive age (aged 16 to 50 years old) who are carriers for thalassaemia, sickle cell disease, cystic fibrosis or Tay‐Sachs disease, accessing any healthcare services which include hospitals and community‐based healthcare settings. Community‐based healthcare settings include family or general practices, community health centres, community health services, community or outpatient clinics and ambulatory care services. Settings outside of healthcare do not directly inform healthcare practice, and thus will be excluded as being outside the scope of this review. If trials contain both eligible and ineligible participants, they will be included if data on eligible participants can be extracted.
Types of interventions
We planned to assess the effects of systematic preconception genetic risk assessment for thalassaemia, sickle cell disease, cystic fibrosis or Tay‐Sachs disease, in any healthcare setting. We define systematic preconception genetic risk assessment as a package of risk assessment including one or more of these components:
family history assessment;
assessment of ethnicity background;
genetic carrier testing;
genetic carrier screening.
Risk assessment can be offered at anytime prior to conception.
We planned to compare systematic preconception genetic risk assessment with standard care. We define standard care as where people receive usual or alternative care in any healthcare setting, that does not involve a specific systematic offer or approach to preconception genetic risk assessment.
Types of outcome measures
The listed outcomes do not form part of the eligibility criteria for the included trials.
Primary outcomes
-
Reproductive outcomes in women and their partners who are carriers of thalassaemia, sickle cell disease, cystic fibrosis or Tay‐Sachs disease identified during or after pregnancy
number of infants born with genetic conditions
number of infants born with congenital anomalies
number of infants born with low birth weight
number of infants born prematurely
-
Decisions about future conception and pregnancy in women and their partners who are carriers for thalassaemia, sickle cell disease, cystic fibrosis or Tay‐Sachs disease
number of women or couples who would make use of prenatal diagnosis
number of women or couples who would make use of prenatal diagnosis and consider termination of pregnancy if the child is affected
number of women or couples who would consider PGT and IVF
number of women or couples who would conceive using donated gametes
number of women or couples who would consider adoption
number of women or couples who would refrain from having any children
Number of women or couples who make an informed choice measured by tools such as the Multidimensional Measure of Informed Choice (MMIC)
Secondary outcomes
-
During pregnancy following intervention
gestational date of prenatal diagnosis in at‐risk women
gestational date of prenatal counselling in at‐risk women or couples
-
Self‐reported measures (short‐term change from baseline)
any objective measures of health‐related quality of life resulting from preconception genetic risk assessment, using validated tools such as Short Form Health Survey 36 (SF36) and Health Questionnaire EQ‐5D
any objective measures of quantifying psychological or social outcomes or both resulting from preconception genetic risk assessment using validated tools such as Spielberger State‐Trait Anxiety Inventory (STAI), Perceived Stress Questionnaire (PSQ)
knowledge (any measures of the women's or couples' or both, knowledge of reproductive genetic risk associated with carrier status for thalassaemia, sickle cell disease, cystic fibrosis or Tay‐Sachs disease using validated self‐reported questionnaire)
satisfaction (any measures of the women's or couples' or both, satisfaction with the intervention using validated self‐reported questionnaire)
Cost of the intervention (including follow‐up visits and tests)
Search methods for identification of studies
We searched for all relevant published and unpublished trials without restrictions on language, year or publication status. If we identify potentially eligible non‐English language trials in future searches, we will source a person who can read the language in order to assess these trials for possible inclusion and data extraction.
Electronic searches
We sought trials from the relevant Cystic Fibrosis and Genetic Disorders Group's Trials Registers using the terms: ((carrier* OR trait OR risk assessment OR Tay‐Sachs):kw) AND ((cystic fibrosis OR haemoglobinopathies OR Tay‐Sachs):kw). For full details of all searching activities for the registers, please see the relevant section of the Cystic Fibrosis and Genetic Disorders Group's website.
Date of latest search: 04 August 2021.
In addition, we searched all relevant trials from the following databases:
Ovid MEDLINE (1970 until 25 June 2021);
Ovid Embase ((1974 until 25 June 2021);
CINAHL (1970 until 25 June 2021);
Ovid PsycINFO (1970 until 25 June 2021).
Date of latest search: 25 June 2021.
The search strategies are available in the appendices (Appendix 2). Start dates for database searches are set to when carrier screening or testing was first available. Based on WHO reports, earliest carrier status assessment was introduced for Tay‐Sachs disease and haemoglobinopathies from the early 1970s (Angastiniotis 1995; Kaback 2000). We searched for relevant trials in the databases from 1970 or from when the date of the database was first available if after 1970.
We searched the following clinical trial databases for ongoing and unpublished trials:
National Institutes of Health database;
Clinical Trials Search Portal of the World Health Organization;
Current Controlled Trials in the metaRegister of controlled clinical trials
Date of latest search: 25 June 2021.
Searching other resources
We planned to examine the reference lists of eligible published trials to identify further relevant trials. We handsearched the key journals European Journal of Human Genetics,Genetics in Medicine and the Journal of Community Genetics from 1998 to June 2021. We complemented the search by contacting subject experts or centres in the field to request any unpublished or other published trials that we may not have identified.
Data collection and analysis
Selection of studies
We saved the results of the searches in the Endnote reference managing software (EndNote X9). Two review authors (one content expert and one methodologist) independently screened the citations and article abstracts of every retrieved record. We would have resolved any disagreements on eligibility by discussion and if doubt remained, we would have acquired the relevant full article(s) for further inspection. Two review authors independently screened all full‐text articles of the eligible trials. We aimed to resolve any disagreement by discussion. If required, we would have consulted a third review author. If necessary, we planned to contact the authors of the articles for further information and clarification of trials. We have reported reasons for excluding trials and provided a PRISMA flowchart (Figure 1).
1.
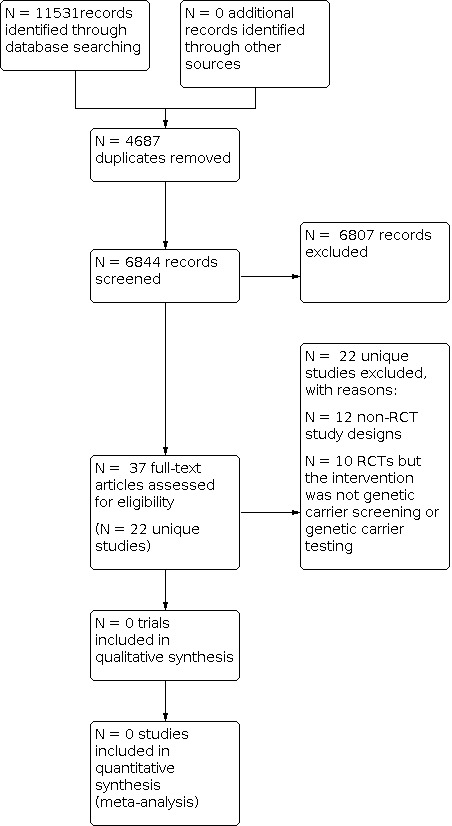
Study flow diagram
We did not identify any trials for inclusion in this version of the review. However, if we identify any trials for future updates of the review, we plan to undertake the following.
Data extraction and management
Two review authors will independently extract data from each included trial using an agreed data extraction form. We will collect data on trial population characteristics (including sample size, participants' ethnic or cultural characteristics, geographic locations), intervention characteristics (including process and duration of intervention) and primary and secondary outcome measures of interest. We plan to report short‐term outcomes post intervention up to six months. We plan to report long‐term outcomes post intervention from six months up to 12 months, and then annually thereafter.
We will settle any disagreements about the data extracted through discussion by the two review authors, and if necessary by arbitration with a third author. We will enter all the data into the Review Manager software (RevMan 2014).
Assessment of risk of bias in included studies
We will construct a risk of bias table for each trial as outlined in chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Two review authors will independently assess and record the following six domains in the risk of bias table:
random sequence generation;
allocation sequence concealment;
blinding of participants, trial personnel, outcome assessors;
incomplete outcome data;
selective outcome reporting;
other sources of bias.
We will judge the methods used in the trials for each domain as having either a low, high or unclear risk of bias. Two review authors will aim to resolve any disagreements in the judgement of the domains through discussion. If no agreement can be reached, then they will consult a third author and aim to resolve the disagreement by consensus.
We will record the information in the 'Risk of bias' tables in Review Manager (RevMan 2014). We aim to resolve any disagreement by consensus or arbitration by a third author. We will use the results of the risk of bias assessment to provide an evaluation of the overall risk of bias of the included trials based on the approach outlined in the chapter 8 (Table 8.7a) of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Measures of treatment effect
We will extract all the main results of the included trials as mentioned below. We will contact relevant authors of the original reports for data or any missing relevant information or when clarification is needed. We will settle any disagreements about the data extracted through discussion and if necessary by arbitration by a third author. We will enter all the data into the Review Manager software (RevMan 2014).
Continuous data
For scale‐derived data, we will include continuous data from rating scales only if the measuring instrument has been validated. We will include endpoint data and only use change data if the former are not available. For continuous outcomes we will record mean, standard deviation (SD) and number of participants for each group and report effect size using the mean difference (MD) for the same units of measurement or the standardised mean difference (SMD) when different scales are used to evaluate the same outcome, with 95% confidence intervals (CI). The MD measures the absolute difference between the means in two groups, whereas the SMD is the MD relative to variability observed in that trial.
Dichotomous data
We will report dichotomous data using the risk ratio (RR) and the corresponding 95% CIs.
Unit of analysis issues
We anticipate cluster‐randomised designs to be used in the included trials; for example, groups of patients of a single doctor or practice. If available, we will extract the direct estimate of the effect (RR with CI) that accounts for a cluster design. We will contact the primary authors of the included trials to obtain the intra‐cluster correlation coefficient (ICC) which will describe the relative variability within and between clusters, to adjust for clustering effect (Donner 1980). We will meta‐analyse the appropriate analyses of cluster randomised trials using the generic inverse variance method. Alternatively, we will estimate an ICC to describe the relative variability within and between clusters (Donner 1980). An ICC usually derives from the trial or from other sources (ICC from a similar trial in an existing database) (Ukoumunne 1999). If the ICC is derived from other sources, we will report this and conduct a sensitivity analysis. If the trials were analysed as if the randomisation was performed on the individuals rather than the clusters, we will re‐calculate the correct analysis if we are able to extract the following information: the number of clusters randomised to each intervention group; the mean size of each cluster; and the outcome data ignoring the cluster design for the total number of individuals.
If we identify more than one intervention group of interest in a trial, we will analyse the effect of the additional intervention group using pair‐wise comparisons. If the additional intervention group is irrelevant, we will not reproduce the data.
Dealing with missing data
Whenever possible, we will contact the original investigators and the authors of the included trials to request any missing data. If this is unsuccessful we will deal with missing data as mentioned below.
Overall loss of credibility
We will choose that, if for any particular outcome there is a high risk bias for missing data according to the risk of bias assessment, we will not use these data in the analyses and will present the results in the form of a narrative synthesis.
Continuous data
If SDs are not reported or available, we will first look for statistics that allow the calculation of the SD (for example, the CI and the standard error (SE) of group means, as well as P values and T values for the differences in means). If this is not possible, we will consider imputing SDs of other included trials. We will examine the consequences of imputations in a sensitivity analysis.
Assessment of heterogeneity
Clinical heterogeneity
We will consider clinical heterogeneity which can result from differences between trials in characteristics of the populations, interventions and outcomes. We will fully discuss the influence of clinical heterogeneity on the observed effects.
Methodological heterogeneity
We will assess for methodological heterogeneity, which can result from differences in characteristics of the trial designs. We will fully discuss the influence of methodological heterogeneity on the observed effects.
Statistical heterogeneity
We plan to examine graphs or summary tables of the trials to investigate the possibility of statistical heterogeneity. We plan to consider the I2 statistic which estimates the proportion of variability in effect estimates that is due to heterogeneity (Higgins 2002). We will determine the level of heterogeneity by the following reference ranges: low 0% to 40%; moderate 41% to 75%; and high 76% to 100%. We also plan to use the Chi2 statistic and if the P value is less than 0.10 it will be considered an indication of heterogeneity. If there is a high level of heterogeneity between trials, it may not be appropriate to conduct a meta‐analysis, thus we will present results in a qualitative analysis. These trials will be entered into RevMan and presented on a forest plot with their individual effect sizes, but with no combined effect to give an overall picture of evidence (RevMan 2014).
Assessment of reporting biases
If we include and combine more than 10 trials, we will create a funnel plot to investigate the possibility of small trial effects (a tendency for the intervention effects estimated in smaller trials to differ from those estimated in larger trials) (Sterne 2011).
Data synthesis
We will summarise all trials using narrative synthesis methods. This will involve the use of narrative text and tables to summarise data, consider outcomes in the light of differences in trial designs and address potential sources of bias for each of the trials being reviewed. We will group trials according to types of genetic conditions, and then organise them in terms of intervention and outcomes. We will summarise the results of the trials, including the range and size of any reported associations and important trial characteristics. We will also include a detailed commentary on the major methodological problems or biases affecting the trials, together with a description of how these may have affected the individual trial results.
We will use a random‐effects model to conduct the meta‐analysis due to anticipated differences between trial location and population. If there is substantial variation in results, particularly if there is inconsistency in the direction of effect, we will not perform a meta‐analysis.
Subgroup analysis and investigation of heterogeneity
The authors will perform subgroup analyses where sufficient data are available. In the subgroup analyses, the authors will analyse the data in pre‐specified subgroups of trials that share characteristics of interest, to see whether the intervention effect remains consistent or whether it varies for particular characteristics of trials. For this review, the authors aim to compare the effects of interventions on outcome measures in the following groups by:
healthcare setting (primary, secondary, tertiary care or other);
intensity of the intervention (number or duration of intervention sessions);
nature of carrier status testing (confirmed genetic carrier status compared to probable carrier status);
type of condition.
Sensitivity analysis
If there is a spread of bias across the trials, we will provide two estimates of the intervention effect; firstly for all included trials, and secondly only including trials with an overall assessment of a low risk of bias.
Summary of findings and assessment of the certainty of the evidence
For future updates of this review, we will present the main results of the included trials in a summary of findings table for each comparison we report. We will group the trials according to types of genetic conditions, interventions (versus usual care) and outcomes. We will include the following outcomes in each summary of findings table.
Number of women or couples who refrain from having biological children with their current partner ‐ up to 24 months post intervention
Number of women or couples undergoing IVF with PGT or using donor gametes ‐ up to 24 months post intervention
Number of women or couples undergo prenatal diagnosis and consider termination of pregnancy if the child is affected ‐ up to 24 months post intervention
Number of women or couples who make an informed choice measured by tools such as the MMIC ‐ at 0 month (at intervention)
Psychological outcomes resulting from preconception genetic risk assessment quantified by validated tools such as STAI, PSQ and health perception questionnaires ‐ at three months post intervention
Psychological outcomes resulting from preconception genetic risk assessment quantified by validated tools such as STAI, PSQ and health perception questionnaires ‐ at six months post intervention
Self‐reported satisfaction score at 0 month (at intervention)
Overall grading of the evidence related to each of the outcome will use the GRADE (Grades of Recommendation, Assessment, Development and Evaluation) approach. We will grade the certainty of the evidence as high, moderate, low or very low, based on the five GRADE domains: risk of bias, inconsistency, indirectness, imprecision, and publication bias (Schünemann 2021). We will produce the summary of the findings tables using the GRADEpro software.
Results
Description of studies
Results of the search
Database searching identified 11,531 records. After we screened 6844 unique records, we retrieved 37 full‐text articles describing 22 unique trials for further analysis. We found no RCTs that were eligible for inclusion in the review. A flow diagram illustrates the search flow process (Figure 1).
Included studies
No RCTs were found to be eligible for inclusion in the review.
Excluded studies
We excluded a total of 22 trials. We excluded 12 studies due to their non‐RCT study designs (Alhamdan 2007; Archibald 2017; Bekker 1993; Childs 1976; Clayton 1996; Hegwer 2006; Henneman 2001; Honnor 2000; Payne 1997; Sallevalt 2021; Tambor 1994; Watson 1991), while 10 RCTs were excluded because the intervention was not preconception genetic carrier testing or genetic carrier screening for any of the four specified genetic conditions (Castellani 2011; Cheuvront 1998; Fan 2018; Fisher 1981; Moudi 2016; Punj 2018; Quigley 2018; Rémus 2020; Temme 2015 Wilkie 2013). One of these 10 RCTs was previously listed as ongoing (protocol by Kauffman from 2017 identified in earlier search); on assessing the published paper containing the full results, we excluded the trial because the intervention was genome sequencing (Punj 2018). The tables summarise the study details and reasons for exclusions (Characteristics of excluded studies).
Risk of bias in included studies
No trials were included in this review.
Allocation
No trials were included in this review.
Blinding
No trials were included in this review.
Incomplete outcome data
No trials were included in this review.
Selective reporting
No trials were included in this review.
Other potential sources of bias
No trials were included in this review.
Effects of interventions
No trials were eligible for inclusion in this review.
Discussion
To date, in many countries, reproductive genetic risk assessment for autosomal recessive disorders has focused on the antenatal period and carrier status that has emerged as an incidental finding in neonatal screening. In the antenatal period, carrier status is identified either through formal screening programmes, or opportunistically during antenatal follow up in women at increased risk based on ancestry. During the antenatal period, if both parents are found to be carriers of the genes (at‐risk couples), prenatal diagnostic tests, such amniocentesis, may only be available either late in the first trimester or in the second trimester of pregnancy, which leaves the couple only a short period of time to make limited and difficult choices about termination or continuation of the pregnancy. This limits reproductive choices, with the potential of increased psychological distress in at‐risk couples (Modell 1980a; Modell 1980b). The incidental finding of the carrier state during neonatal screening for the specific disorders has highlighted concerns from at‐risk couples about the failure to offer this information prior to pregnancy (Locock 2008).
Identifying those couples before pregnancy, who have a confirmed genetic carrier status for thalassaemia, sickle cell disease, cystic fibrosis, or Tay‐Sachs disease may provide the opportunity for individuals or couples to make fully informed reproductive choices such as avoiding pregnancy, pre‐implantation diagnosis, in vitro fertilisation, arranging early prenatal diagnosis, or consideration of adoption.
Summary of main results
There is currently no evidence from RCTs for the impact of genetic risk assessment for these conditions in non‐pregnant women on pregnancy outcomes, informed reproductive choices or psychological adverse effects.
We identified a number of observational studies that were excluded from the review (Alhamdan 2007; Archibald 2017; Bekker 1993; Childs 1976; Clayton 1996; Hegwer 2006; Henneman 2001; Honnor 2000; Payne 1997; Sallevalt 2021; Tambor 1994; Watson 1991). The majority of these observational studies used before and after intervention designs (Bekker 1993; Clayton 1996; Hegwer 2006; Henneman 2001; Honnor 2000; Payne 1997; Tambor 1994; Watson 1991), while four studies utilised cross‐sectional designs (Alhamdan 2007; Archibald 2017; Childs 1976; Sallevalt 2021). We also identified 10 RCTs; however, nine of these did not evaluate preconception genetic risk assessment for the four specified genetic conditions (Castellani 2011; Cheuvront 1998; Fan 2018; Fisher 1981; Moudi 2016; Quigley 2018; Rémus 2020; Temme 2015; Wilkie 2013) and one study involved genome sequencing as the intervention (Punj 2018). Only a few studies have assessed reproductive intentions (Cheuvront 1998; Henneman 2001; Watson 1991), whilst no studies have assessed actual reproductive outcomes. Most of the above studies have assessed psychological, attitudes, or knowledge outcomes, but there was some heterogeneity in these outcomes between and within studies. Further, none of the outcome measures for knowledge had used validated instruments. Although study participants recognised the importance of identifying genetic carrier states before pregnancy, different attitudes towards genetic testing were elicited and reproductive intentions varied following positive test results. In the Netherlands, study participants would consider prenatal diagnosis and abortion if an affected foetus is identified (Henneman 2001). In contrast, in a study of cystic fibrosis screening conducted in the state of Tennessee in the USA, reproductive intentions were limited by cultural and socio‐political factors, such as insurability, being labelled as 'at risk', a lack of understanding, and religious beliefs about abortion (Clayton 1996). In addition, barriers to implementation include legal discrimination (Lapham 1996) and religious restrictions on abortion (Fowzan 2001). Previously there were also concerns about fears of stigma (Kenen 1978).
Overall completeness and applicability of evidence
We did not identify any evidence for inclusion in our review.
Quality of the evidence
We did not identify any evidence for inclusion in our review.
Potential biases in the review process
The review authors have attempted to limit the bias in the review process through multiple authors and non‐author contributors who independently searched for trials, screened titles and abstracts, selected full‐text articles and extracted data. Any disagreements were resolved by group discussion and consensus, and therefore it was unlikely that trials have been incorrectly excluded. Although all clinical trials should be registered, there is always the potential of publication bias. However, attempts have been made to minimise publication bias through searching the grey literature and contacting key experts in the field.
In addition, while not of concern for the current review version, for future updates, rather than focusing on single gene‐by‐gene carrier testing for specific autosomal‐recessive conditions as the intervention under study, preconception expanded genetic screening should also be included in searches as this has replaced the single gene intervention in recent years.
Agreements and disagreements with other studies or reviews
This is the only systematic review looking at preconception genetic risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease and there were no randomised controlled trials eligible for inclusion, and therefore no comparisons could be made to other reviews or studies.
Authors' conclusions
Implications for practice.
We have not identified any relevant trials up to the 2021 update and this systematic review shows that there is a complete lack of randomised controlled trials (RCTs) in the field of preconception genetic risk assessment for autosomal recessive conditions.
As such, healthcare providers need to assess whether the information provided in published policy recommendations (see below) and non‐randomised studies (for example prospective cohorts or before‐and‐after studies) is relevant to inform their preconception screening practice. While RCTs are theoretically desirable to inform evidence‐based practice and robust recommendation, such trials must also consider the ethical, legal and social implications associated with implementation of preconception genetic risk assessment involving carrier testing and reproductive autonomy. Perhaps it is time to rethink whether using RCTs to explore the evidence for reproductive genetic risk assessment is the way to move forward when recommending policy? Furthermore, to consider whether it is ethically acceptable to involve women and men preconceptionally for genetic carrier testing in prospective randomised trials? Healthcare providers have to balance the benefits of increasing reproductive choice against the potential psychological adverse effects from preconception genetic risk assessment, whilst taking into account the legal and socia‐cultural context of their healthcare setting and patient population.
Despite the lack of RCT evidence and the research evidence for current policy recommendations being limited to non‐randomised studies, a number of international organisations have recommended offering preconception genetic risk assessment routinely at the population level (ACOG Committee Opinion 2009; ACOG Committee Opinion 2017; ACOG Committee Opinion 2017 (reaffirmed 2020); Health Council of Netherlands 2007; Human Genetics Commission 2011; Johnson 2006; March of Dimes 2006). In the USA, the recommendations to improve preconception healthcare were developed through collaborative efforts of the Centres for Disease Control and Prevention (CDC), March of Dimes and the American College of Obstetrics and Gynaecology (ACOG) (ACOG Committee Opinion 2017; Johnson 2006). For instance, the ACOG Committee has recommended that for couples planning pregnancy to identify if either member of the couple are of Eastern European (Ashkenazi) Jewish ancestry or have a family history of relevant recessive genetic diseases (such as Tay‐Sachs disease and Cystic Fibrosis), and furthermore that such couples should be offered carrier screening before conception or early in pregnancy (ACOG Committee Opinion 2009; ACOG Committee Opinion 2017).
Similarly, the Health Council of Netherlands has recognised the seriousness of these conditions and high prevalence in local population groups, advocating preconception genetic risk assessment for cystic fibrosis, sickle cell and thalassaemia (Health Council of Netherlands 2007). However, despite local initiatives, large‐scale studies have not been implemented in the Netherlands (Delatycki 2019).
The WHO's Regional Office of Eastern Mediterranean recommends preconception genetic risk assessment for sickle cell and thalassaemia ideally before marriage, taking account of the socio‐cultural issues in the region, in particular religious reservations towards the termination of pregnancy (Alwan 1997). Since the 1970s, the Cyprus Thalassaemia Control Programme has been at the forefront of premarital genetic screening and this has contributed to a fall in the prevalence of thalassaemia in the country (Angastiniotis 1981). This universal premarital approach to thalassaemia carrier screening has also been adopted by Sardinia, Italy (Cao 1996) and Greece (Loukopoulos 1996).
In line with international policy recommendations, the UK Human Genetics Commission has recognised that since antenatal screening is currently already offered for genetic conditions such as sickle cell disease and thalassaemia, there are no ethical, legal or social issues with regards to the implementation of a preconception screening programme which would provide the advantage of improving reproductive choices (Human Genetics Commission 2011).
In South‐East Asia, the Family Planning Association of Hong Kong has recognised the benefits of preconception screening of genetic risk due to the high prevalence of thalassaemia carriers, accounting for up to eight per cent of the local population (Lau 1997).
Implications for research.
It has been suggested that the optimum evidence to evaluate the reproductive and psychological outcomes as a result of preconception screening compared to standard practice is a systematic review of RCTs, or a high‐quality RCT with a large enough sample size to ensure the control of potential confounding factors (UK National Screening Committee 2003). Such trials address methodological issues that are particularly associated with screening interventions such as ascertainment bias due to non‐randomisation, with individuals joining screening programmes tending to have healthier lifestyles and better adherence to interventions (Smith 2003).
Previous observational studies and RCTs on preconception genetic risk assessment for thalassaemia, sickle cell disease, cystic fibrosis, or Tay‐Sachs disease have been limited by the duration of follow‐up and restricted to the assessment of psychological or knowledge outcomes. Indeed, none of the excluded studies identified in the searches for this review has evaluated reproductive outcomes. This is possibly related to the limited duration of follow‐up in these studies. Although preconception genetic carrier tests and screening have been shown to be highly accurate and efficient in determining carrier status (Bach 2001; CDC 2004; Peters 2012; Weatherall 1997), the effectiveness of such interventions is to enable reproductive choice for carrier couples, which may, as a consequence, lead to reduced morbidity and mortality of the diseases. Therefore, reproductive outcomes are essential to addressing this question. Adequately‐powered RCTs assessing reproductive outcomes (number of affected children born with genetic conditions) and reproductive decision outcomes on future conception (termination, in vitro fertilisation, use of donor gametes, adoption, or refraining from having children) are ideal to better inform recommendations for clinical practice. Any self‐reported secondary outcome measures need to use validated instruments. These trials will require longer durations of follow‐up than previous studies, starting from pre‐pregnancy and lasting into the post‐natal period.
What's new
| Date | Event | Description |
|---|---|---|
| 16 August 2021 | New search has been performed | A search of the Group's Trials Registers identified seven new references potentially eligible for inclusion in the review; three of these were additional references to an already excluded study (Wilkie 2013) and the remaining four were not even suitable to be listed as excluded studies and were immediately discarded. Additional planned searches of databases and key journals identified nine new references potentially eligible for inclusion in the review. One of these was the full paper to a study previously listed as ongoing, but which has now been excluded (Punj 2018). All of the remaining eight references to six studies were excluded as they did not meet the review's eligibility criteria (Archibald 2017; Fan 2018; Moudi 2016; Quigley 2018; Rémus 2020; Sallevalt 2021). We have added plans for generating a summary of findings table for each comparison, which we may be able to present in future updates of the review, to the Methods section in line with current Cochrane guidance. We have added a third primary outcome to assess the number of women or couples who make an informed choice measured by tools such as the Multidimensional Measure of Informed Choice (MMIC). |
| 16 August 2021 | New citation required but conclusions have not changed | No new data have been added to the review. However, we have added suggestion for future searches in our conclusions. A new author, Professor Lidewij Henneman, has joined the team; Professor Jos Kleijnen and Dr Stephen Weng have left the review team. |
History
Protocol first published: Issue 12, 2013 Review first published: Issue 8, 2015
| Date | Event | Description |
|---|---|---|
| 25 January 2018 | New citation required but conclusions have not changed | No new data have been added to the review so our conclusions remain the same. |
| 25 January 2018 | New search has been performed | A search of the Cystic Fibrosis and Genetic Disorders Review Group's trials registers identified two references to a single trial which has been excluded (Temme 2015). A search from MEDLINE identified one reference which was potentially eligible for inclusion in the review and has been listed as ongoing until completed (Kauffman 2017a). |
Acknowledgements
The authors would like to acknowledge and thank Stephen Weng and Jos Kleijnen for contributing to the development and early phase of the review, and Luke Robles and Richard Birnie for their help in screening and identifying full‐text articles for this review.
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Appendices
Appendix 1. Glossary
| Term | Explanation |
| Antenatal | A period during pregnancy and before birth of the child. |
| Ancestry | A person's ethnic origin or descent. |
| Atelectasis | A collapsed portion of the lung which does not contain air. This can be caused by excessive accumulations of mucous secretions, inhaled foreign bodies or bronchial cancers. |
| Autosomal recessive genetic disorders | A genetic trait or disorder which appears only when an individual inherits a pair of chromosomes, each containing the gene for the trait. One chromosome of the pair comes from the father and the other from the mother. Autosomal recessive disorders can occur only if both parents are carriers of the trait. |
| Bronchiectasis | Persistent and progressive dilation of bronchi (branches from the trachea which lead to the lungs) often as a consequence of inflammatory disease (lung infections). |
| Carrier (in genetics) | An individual who possesses one copy of a mutated allele that causes disease only when two copies are present (an autosomal recessive genetic disorders). A carrier is not affected by the disease, but two carriers can produce a child with the disease. |
| Chronic vaso‐occlusion | Blockage of arteries marked by long duration, by frequent recurrence over a long time, and often by slowly progressing deterioration; having a slow progressive course of indefinite duration. |
| Cystic fibrosis transmembrane conductance regulator (CFTR) | A protein, involved in the movement of salt across cell membranes, which is lacking or does not function normally in people with cystic fibrosis. |
| Diabetes mellitus | A pancreatic disorder that causes abnormal insulin production. This affects the body's ability to utilise sugar and other food substances and is usually treated by diet modification (restricted sugar intake) and use of insulin. |
| DNA (Deoxyribonucleic acid) | The chemical coding for a gene. DNA determines the 'genetic message' within each cell, organ, and organism. |
| Electrophoresis | A method of separating particles relative to a fluid under the influence of a spatially uniform electric field. |
| Ethnicity | Common characteristics of people of a distinct national, racial or cultural group. |
| Gangliosides | A group of glysolipid cells that are found in the brain. |
| Gene | The functional and physical unit of heredity passed from parent to offspring. Genes are pieces of DNA, and most genes contain the information for making a specific protein. |
| Globin chains | Blood proteins found in red blood cells that are combined to make haemoglobin. They are α or β globin chains. |
| Haemoglobin A | Normal adult haemoglobin. |
| Haemoglobin F | A kind of haemoglobin usually present during fetal (intrauterine) life, which has a different chemical structure from normal adult haemoglobin. After birth, the fetal haemoglobin in the red blood cells is gradually replaced by the adult type of haemoglobin, this process is usually complete during the first six months of life. |
| Haemolysis | Breaking of the red cell membrane causing release of haemoglobin. |
| Haemolytic anaemia | A condition where there are fewer red blood cells than average circulating in the blood stream due to breaking of the red cell membrane causing release of haemoglobin. |
| Hexosaminidase A isozyme | A protein found in the nerve cells of the brain which does not function normally in people with Tay‐Sachs disease. |
| High performance liquid chromatography (HPLC) | A method that is used to separate a mixture of compounds to identify and quantify the individual components of the mixture. |
| Hypothyroidism | Results from a deficiency of thyroid hormone, and is characterized by a decrease in basal metabolic rate and by tiredness, lethargy and sensitivity to cold. |
| In vitro fertilization | A technique by which eggs are collected from a woman and fertilised with a man's sperm outside the body. Usually one or two resulting embryos are then transferred to the womb. If one or more of them implants successfully in the womb it results in a pregnancy. |
| In vivo | Inside the living body. |
| Mutation | A change or alteration of the DNA sequence within a gene. |
| Nasal epithelium | The tissue that covers and lines the surface of the nose. |
| Obstructive azoospermia | A condition where there is no measurable sperm detected in the semen due to ejaculatory dysfunction or ductal blockage. This condition can occur in people with cystic fibrosis. |
| Pancreatic exocrine insufficiency | A condition characterized by deficiency of the pancreatic enzymes, resulting in the inability to digest food properly, or maldigestion. |
| Salt‐loss syndromes | A condition found in people with cystic fibrosis where there is loss of salt resulting in depletion of salt in the body. |
| Septicaemia | A condition characterized by the widespread destruction of tissues due to absorption of disease containing bacteria or their toxins from the bloodstream. |
For further statistical terms, please refer to the The Cochrane Collaboration Glossary (http://cochrane.org/glossary).
For technical or clinical terms, please refer to The Human Genetics Commission Glossary (http://webarchive.nationalarchives.gov.uk/20100419143351/hgc.gov.uk/client/content.asp?contentid=729).
Appendix 2. Search strategies
| Database or resource | Date searched | Search strategy |
| Ovid MEDLINE(R) Daily Update Ovid MEDLINE(R) In-Process & Other Non-Indexed Citations and Ovid MEDLINE(R) |
1970 to 25 June 2021 | 1. exp Thalassemia/ 2. thalass?emia.ti,ab,ot,hw. 3. ((erythroblastic or erythro‐blastic or hypochromic or cooley$ or mediterranean) adj2 an?emia$).ti,ab,ot,hw. 4. (h?emoglobin adj2 disease$).ti,ab,ot,hw. 5. exp Hemoglobinopathies/ 6. hereditary persistence of f?etal h?emoglobin.ti,ab,ot,hw. 7. (h?emoglobin adj2 (H or F or D or E) adj2 disease$).ti,ab,ot,hw. 8. 1 or 2 or 3 or 4 or 5 or 6 or 7 9. exp Anemia, Sickle Cell/ 10. Sickle Cell Disease.ti,ab,ot,hw. 11. (sickle cell adj2 (an?emia$ or disease$ or disorder$)).ti,ab,ot,hw. 12. (h?emoglobin adj2 (S or C or SC)).ti,ab,ot,hw. 13. ((drepanocytosis or drepanocytic) adj2 an?emia).ti,ab,ot,hw. 14. 9 or 10 or 11 or 12 or 13 15. Cystic Fibrosis/ 16. cystic fibrosis.ti,ab,ot,hw. 17. CF.ti,ab. 18. mucoviscidosis.ti,ab,ot,hw. 19. (fibrocystic adj3 disease$).ti,ab,ot,hw. 20. (pancreas$ adj2 (fibrosis or cystic disease$)).ti,ab,ot,hw. 21. 15 or 16 or 17 or 18 or 19 or 20 22. Tay‐Sachs Disease/ 23. Tay Sachs.ti,ab,ot,hw. 24. ((familial or infantile) adj2 amaurotic idiocy).ti,ab,ot,hw. 25. TSD.ti,ab. 26. (GM2 adj2 gangliosidosis).ti,ab,ot,hw. 27. 22 or 23 or 24 or 25 or 26 28. Heterozygote/ 29. trait$.ti,ab,ot,hw. 30. carrier$.ti,ab,ot,hw. 31. 28 or 29 or 30 32. 8 or 14 or 21 or 27 or 31 33. (Preconcept$ or Pre‐concept$ or Prepregnan$ or Pre‐pregnan$).ti,ab,ot,hw. 34. Maternal Health Services/ 35. ((pregnan$ or conception or family) adj3 plan$).ti,ab,ot,hw. 36. (Pre‐marital or Premarital or Pre‐marriage or Premarriage).ti,ab,ot,hw. 37. ((Preconcept$ or Pre‐concept$ or Prepregnan$ or Pre‐pregnan$) adj2 (care or counsel$ or advice$ or advise or inform$)).ti,ab,ot,hw. 38. ((Pre‐marital or Premarital or Pre‐marriage or Premarriage) adj2 (care or counsel$ or advice$ or advise or inform$)).ti,ab,ot,hw. 39. 33 or 34 or 35 or 36 or 37 or 38 40. (carrier$ adj3 (screen$ or test$ or counsel$ or assess$ or detect$ or diagnos$ or inform$ or analys$)).ti,ab,ot,hw. 41. (genetic$ adj3 (screen$ or test$ or counsel$ or assess$ or detect$ or diagnos$ or inform$ or analys$)).ti,ab,ot,hw. 42. (heterozygot$ adj3 (screen$ or test$ or counsel$ or assess$ or detect$ or diagnos$ or inform$ or analys$)).ti,ab,ot,hw. 43. Genetic Services/ 44. family history.ti,ab,ot,hw. 45. 40 or 41 or 42 or 43 or 44 46. (h?emoglobin adj2 electrophoresis).ti,ab,ot,hw. 47. Cystic Fibrosis Transmembrane Conductance Regulator/ or sweat test.ti,ab,ot,hw. 48. ((CFTR gene mutation$ or CFTR mutation$ or Hexoaminidase‐A or Hexoaminidase A or HEX‐A or H?emoglobin F or H?emoglobin A2 or H?emoglobin S) adj3 (test$ or analys$ or screen$ or profil$)).ti,ab,ot,hw. 49. 46 or 47 or 48 50. 32 or 45 or 49 51. 39 and 50 52. exp animals/ not humans.sh. 53. 51 not 52 54. limit 53 to yr="1970‐Current" |
| PsycINFO | 1970 to 25 June 2021 | 1. thalassemia.ti,ab,ot,hw. 2. thalassaemia.ti,ab,ot,hw. 3. ((erythroblastic or erythro‐blastic or hypochromic or cooley* or mediterranean) adj2 anaemia*).ti,ab,ot,hw. 4. ((erythroblastic or erythro‐blastic or hypochromic or cooley* or mediterranean) adj2 anemia*).ti,ab,ot,hw. 5. ((haemoglobin or hemoglobin) adj2 disease*).ti,ab,ot,hw. 6. ((haemoglobin or hemoglobin) adj2 (H or F or D or E) adj2 disease*).ti,ab,ot,hw. 7. 1 or 2 or 3 or 4 or 5 or 6 8. Sickle Cell Disease/ 9. (sickle cell adj2 (anaemia* or disease* or disorder*)).ti,ab,ot,hw. 10. ((haemoglobin or hemoglobin) adj2 (S or C or SC)).ti,ab,ot,hw. 11. 8 or 9 or 10 12. Cystic Fibrosis/ 13. cystic fibrosis.ti,ab,ot,hw. 14. CF.ti,ab. 15. mucoviscidosis.ti,ab,ot,hw. 16. (fibrocystic adj3 disease*).ti,ab,ot,hw. 17. 12 or 13 or 14 or 15 or 16 18. Tay Sachs Disease/ 19. Tay Sachs.ti,ab,ot,hw. 20. ((familial or infantile) adj2 amaurotic idiocy).ti,ab,ot,hw. 21. TSD.ti,ab. 22. (GM2 adj2 gangliosidosis).ti,ab,ot,hw. 23. 18 or 19 or 20 or 21 or 22 24. heterozygote.ti,ab,ot,hw. 25. trait*.ti,ab,ot,hw. 26. carrier*.ti,ab,ot,hw. 27. 24 or 25 or 26 28. 7 or 11 or 17 or 23 or 27 29. (Preconcept* or Pre‐concept* or Prepregnan* or Pre‐pregnan*).ti,ab,ot,hw. 30. (Pre‐marital or Premarital or Pre‐marriage or Premarriage).ti,ab,ot,hw. 31. maternal health service*.ti,ab,ot,hw. 32. maternal care.ti,ab,ot,hw. 33. ((pregnan* or conception or family) adj3 plan*).ti,ab,ot,hw. 34. ((Preconcept* or Pre‐concept* or Prepregnan* or Pre‐pregnan*) adj2 (care or counsel* or advice* or advise or inform*)).ti,ab,ot,hw. 35. ((Pre‐marital or Premarital or Pre‐marriage or Premarriage) adj2 (care or counsel* or advice* or advise or inform*)).ti,ab,ot,hw. 36. 29 or 30 or 31 or 32 or 33 or 34 or 35 37. (genetic* adj3 (screen* or test* or counsel* or assess* or detect* or diagnos* or inform* or analys*)).ti,ab,ot,hw. 38. (carrier* adj3 (screen* or test* or counsel* or assess* or detect* or diagnos* or analys*)).ti,ab,ot,hw. 39. (heterozygot* adj3 (screen* or test* or counsel* or assess* or detect* or diagnos* or analys*)).ti,ab,ot,hw. 40. genetic service*.ti,ab,ot,hw. 41. family history.ti,ab,ot,hw. 42. 37 or 38 or 39 or 40 or 41 43. ((haemoglobin or hemoglobin) adj2 electrophoresis).mp. [mp=title, abstract, heading word, table of contents, key concepts, original title, tests & measures] 44. Cystic Fibrosis Transmembrane Conductance Regulator/ or sweat test.mp. 45. 43 or 44 46. 28 or 42 or 45 47. 36 and 46 48. exp Animals/ 49. human.mp. 50. 48 and 49 51. 48 not 50 52. 47 not 51 53. limit 52 to yr="1970‐Current" |
| Embase | 1974 to 25 June 2021 | 1. exp thalassemia/cn, di, ep, et, pc [Congenital Disorder, Diagnosis, Epidemiology, Etiology, Prevention] 2. exp delta thalassemia/ or exp beta thalassemia/ or exp thalassemia major/ or exp alpha thalassemia/ or exp thalassemia intermedia/ or exp sickle cell beta thalassemia/ or exp thalassemia minor/ 3. thalass?emia.ti,ab,ot,hw. 4. ((erythroblastic or erythro‐blastic or hypochromic) adj2 an?mia$).ti,ab,ot,hw. 5. (h?emoglobin adj2 disease$).ti,ab,ot,hw. 6. hemoglobinopathy/cn, di, ep, et, pc [Congenital Disorder, Diagnosis, Epidemiology, Etiology, Prevention] 7. hereditary persistence of f?etal h?emoglobin.ti,ab,ot. 8. (h?emoglobin adj2 (h or d or e) adj2 disease$).ti,ab,ot,hw. 9. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 10. exp sickle cell anemia/cn, di, ep, et, pc [Congenital Disorder, Diagnosis, Epidemiology, Etiology, Prevention] 11. sickle cell disease.ti,ab,ot,hw. 12. (h?emoglobin adj2 (s or c)).ti,ab,ot,hw. 13. 10 or 11 or 12 14. exp cystic fibrosis/cn, di, ep, et, pc [Congenital Disorder, Diagnosis, Epidemiology, Etiology, Prevention] 15. cystic fibrosis.ti,ab,ot,hw. 16. CF.ti,ab. 17. 14 or 15 or 16 18. exp Tay Sachs disease/cn, di, ep, et, pc [Congenital Disorder, Diagnosis, Epidemiology, Etiology, Prevention] 19. Tay Sachs.ti,ab,ot,hw. 20. ((familial or infantile) adj2 amaurotic idiocy).ti,ab,ot,hw. 21. TSD.ti,ab. 22. 18 or 19 or 20 or 21 23. exp heterozygote/ or exp heterozygote detection/ 24. trait$.ti,ab,ot,hw. 25. carrier$.ti,ab,ot,hw. 26. 23 or 24 or 25 27. 9 or 13 or 17 or 22 or 26 28. (Preconcept$ or Pre‐concept$ or Prepregnan$ or Pre‐pregnan$).ti,ab,ot,hw. 29. (Pre‐marital or Premarital or Pre‐marriage or Premarriage).ti,ab,ot,hw. 30. ((pregnan$ or conception or family) adj3 plan$).ti,ab,ot,hw. 31. ((Preconcept$ or Pre‐concept$ or Prepregnan$ or Pre‐pregnan$) adj2 (care or counsel$ or advice$ or advise or inform$)).ti,ab,ot,hw. 32. ((Pre‐marital or Premarital or Pre‐marriage or Premarriage) adj2 (care or counsel$ or advice$ or advise or inform$)).ti,ab,ot,hw. 33. 28 or 29 or 30 or 31 or 32 34. (carrier$ adj3 (screen$ or test$ or counsel$ or assess$ or detect$ or diagnos$ or inform$ or analys$)).ti,ab,ot,hw. 35. (genetic$ adj3 (screen$ or test$ or counsel$ or assess$ or detect$ or diagnos$ or inform$ or analys$)).ti,ab,ot,hw. 36. (heterozygot$ adj3 (screen$ or test$ or counsel$ or assess$ or detect$ or diagnos$ or inform$ or analys$)).ti,ab,ot,hw. 37. Genetic Service$.ti,ab,ot,hw. 38. family history.ti,ab. 39. 34 or 35 or 36 or 37 or 38 40. (h?emoglobin adj2 electrophoresis).ti,ab,ot,hw. 41. Cystic Fibrosis Transmembrane Conductance Regulator.ti,ab,ot,hw. 42. sweat test.ti,ab. 43. ((CFTR gene mutation$ or CFTR mutation$ or Hexoaminidase‐A or Hexoaminidase A or HEX‐A or H?emoglobin F or H?emoglobin A2 or H?emoglobin S) adj3 (test$ or analys$ or screen$ or profil$)).ti,ab,ot,hw. 44. 40 or 41 or 42 or 43 45. 27 or 39 or 44 46. 33 and 45 47. animal/ 48. human/ 49. 47 and 48 50. 47 not 49 51. 46 not 50 52. limit 51 to yr="1970‐Current" |
| CINAHL | 1970 to 25 June 2021 | SI. (MH "Thalassemia") OR (MH "beta‐Thalassemia") OR (MH "alpha‐Thalassemia") OR (MH "delta‐Thalassemia") S2. (MH "Hemoglobinopathies") S3. (MM "Anemia, Hypochromic") S4. (MH "Anemia, Sickle Cell") OR (MH "Sickle Cell Trait") S5. (MH "Cystic Fibrosis") OR "mucoviscidosis" S6. (MH "Tay‐Sachs Disease") S7. (MH "Prepregnancy Care") S8. (MH "Genetic Screening") S9. (MH "Family Assessment") OR (MH "Family History") S10. "hemoglobin electrophoresis" S11. "cystic fibrosis transmembrane conductance regulator" S12. "sweat test" S13. S1 OR S2 OR S3 OR S4 OR S5 OR S6 OR S8 OR S9 OR S10 OR S11 OR S12 S14. S7 AND S13 S15. Limiters ‐ Published Date from: 19700101‐current |
| National Institutes of Health database (clinicaltrials.gov/) | 2005 to 25 June 2021 | preconception OR prepregnancy OR premarital |
| Clinical Trials Search Portal of the World Health Organization (apps.who.int/trialsearch/) | 2004 to 25 June 2021 | preconcep* OR prepregnan* OR premarital |
| Current Controlled Trials in the metaRegister of controlled clinical trials (www.controlled-trials.com/) | 2004 to 25 June 2021 | "preconception OR prepregnancy OR premarital" |
Characteristics of studies
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alhamdan 2007 | Participants: couples planning to marry and applying for marriage licence Intervention: premarital screening programme for sickle cell and beta‐thalassemia Comparator: none Outcome: number confirmed sickle cell and beta thalassemia carriers, decision for marriage Design: observational, cross‐sectional study Excluded due to non‐RCT design |
| Archibald 2017 | Participants: woman prior to pregnancy or early in pregnancy (recommended ≤ 12 weeks gestation) Intervention: simultaneous genetic carrier testing for CF, FXS and SMA Comparator: none Outcome: number of carriers for CF, FXS and SMA Design: observational, cross‐sectional Excluded due to non‐RCT study design |
| Bekker 1993 | Participants: adults 18 ‐ 45 years Intervention: genetic carrier testing for cystic fibrosis Comparator: none Outcome: anxiety Design: observational, before and after intervention study Excluded due to non‐RCT study design |
| Castellani 2011 | Participants: infertile couples undergoing cystic fibrosis screening as part of assisted reproduction process Intervention: genetic counselling via computer program Comparator: standard care genetic counselling session Outcome: knowledge Design: RCT Excluded because intervention was method of delivering genetic counselling and not preconception genetic carrier testing or screening compared to standard care |
| Cheuvront 1998 | Participants: relatives of people with CF Intervention: home‐based pretest education from pamphlet with genetic test Comparator: clinic based pretest education via genetic counselling with genetic test Outcome: anxiety, knowledge, satisfaction, reproductive intent Design: RCT Excluded because intervention was method of delivering genetic counselling and not preconception genetic carrier testing or screening compared to standard care |
| Childs 1976 | Participants: carriers of Tay‐Sachs disease identified prospectively or retrospectively during population screening Intervention: genetic carrier population screening Comparator: none Outcomes: knowledge, attitudes, anxiety, concerns, satisfaction Design: observational, cross‐sectional Excluded due to non‐RCT study design |
| Clayton 1996 | Participants: non‐pregnant adults visiting clinical and non‐clinical sites Intervention: genetic carrier testing for CF Comparator: none Outcome: attitudes, beliefs Design: observational, before and after intervention design Excluded due to non‐RCT study design |
| Fan 2018 | Participants: adults > 18 years old with no known carriers status Intervention: educational online module for carrier screening for Tay Sach disease Comparator: in person genetic counselling Outcome: post‐interventional genetics knowledge, perception of genetic risk score and anxiety score Design: RCT Excluded due intervention is not preconception genetic carrier testing or screening and participants are not known carrier status |
| Fisher 1981 | Participants: adults carriers of beta‐thalassaemia 18 ‐ 65 years in a HMO Intervention: genetic counselling through video Comparator: conventional counselling by a trained physician Outcome: knowledge, sexual activity, mood change, behaviour, anxiety Design: RCT Excluded because the intervention is not preconception genetic carrier testing or screening |
| Hegwer 2006 | Participants: adults of Ashkenazi Jewish background in prenatal and preconception settings Intervention: genetic carrier screening and education programme for Tay‐Sachs disease Comparator: none Outcome: knowledge, concern, attitudes, perceptions of genetic risk Design: observational, before and after intervention Excluded due to non‐RCT study design |
| Henneman 2001 | Participants: adults aged 20 ‐ 35 years invited through Municipal Health Service or General Practitioner Intervention: genetic carrier screening for cystic fibrosis Comparator: none Outcome: knowledge, attitudes, understanding, satisfaction, psychological well‐being, uptake, worry, reproductive intentions, sharing of information Design: observational, before and after intervention Excluded due to non‐RCT study design |
| Honnor 2000 | Participants: adults 18 ‐ 50 years in a primary care setting Intervention: genetic carrier testing and counselling for cystic fibrosis Comparator: none Outcome: anxiety, knowledge Design: observational, before and after intervention Excluded due to non‐RCT study design |
| Moudi 2016 | Participants: couples went for pre‐marital counselling center Intervention: motivational interviewing before carrier screening for thalassaemia Comparator: usual care Outcome: screening rate Design: RCT Excluded due intervention is not preconception genetic carrier testing or screening and partcipants are not known carrier status |
| Payne 1997 | Participants: adults 16 ‐ 45 years in a primary care practice in South Wales Intervention: genetic carrier testing for CF Comparator: none Outcome: knowledge, anxiety Design: observational, before and after intervention Excluded due to non‐RCT study design |
| Punj 2018 | Participants: women planning a pregnancy Intervention: genome sequencing of the expanded genetic screening program Comparator: screening for CF Outcome: number of variants reported Design: RCT Excluded due intervention is not preconception genetic carrier testing or screening and participants are not known carrier status |
| Quigley 2018 | Participants: parents of children who were identified as increased risk of CF following newborn screening programme Intervention: information pack on CF Comparator: no information pack on CF Outcome: knowledge and stress score Design: RCT Excluded due intervention is not preconception genetic carrier testing or screening |
| Rémus 2020 | Participants: parents of children who were identified SCD carrier following newborn screening programme Intervention: methods for invitation to come for screening: letter and a follow‐up phone call; letter and 3 follow‐up text messages within 5 days. Comparator: invitation by letter only Outcome: screening rate Design: RCT Excluded due intervention is not preconception genetic carrier testing or screening |
| Sallevalt 2021 | Participants: consanguineous couples Intervention: exome sequencing of preconception carrier testing Comparator: none Outcome: variants from exome‐sequencing and preconception carrier testing gene panel analysis Design: cross‐sectional study Excluded due to non‐RCT study design and intervention is not preconception genetic carrier testing or screening |
| Tambor 1994 | Participants: adults 18 ‐ 44 years in a HMO Intervention: invitation offering CF carrier screening and information giving either by personal education on‐site or by mailed brochure Comparator: none Outcome: attitudes, tolerance, utilization Design: observational, before and after intervention Excluded due to non‐RCT study design |
| Temme 2015 | Participants: parents of infants with positive newborn screening results for CF and one identified CFTR mutation Intervention: genetic counselling plus a 4‐minute video on CF Comparator: genetic counselling only Outcome: knowledge: understanding of carrier status, autosomal recessive inheritance, the newborn screening process, and symptoms of CF Design: RCT Excluded due to intervention being the method of delivering genetic counselling and education and not preconception genetic carrier testing or screening |
| Watson 1991 | Participants: adults 16 ‐ 44 years from primary care practices and family planning clinics Intervention: genetic carrier testing for CF Comparator: none Outcome: anxiety, response to positive results, knowledge, reproductive intentions, behaviour Design: observational, before and after intervention Excluded due to non‐RCT study design |
| Wilkie 2013 | Participants: adults 18 ‐ 35 years with sickle cell disease or sickle cell trait from clinics and community settings Intervention: web‐based multimedia educational intervention Comparator: usual care information e‐book Outcome: knowledge, reproductive intent and behaviour Design: RCT Excluded because the intervention was the delivery of education and not preconception genetic carrier testing or screening |
CF: cystic fibrosis FXS: fragile X syndrome SCD: sickle cell disease HMO: health maintenance organisation RCT: randomised controlled trial SMZ: spinal muscular atrophy
Differences between protocol and review
Handsearching of key journals was originally specified from 1984 to date in the protocol. In the review, handsearching was conducted from 1998 to date for the European Journal of Human Genetics and Genetics in Medicine and from 2010 to date for the Journal of Community Genetics. This change was made because these were the earliest dates that the online tables of contents were accessible for these key journals.
We have added plans for summary of findings tables to the Methods.
We have added a third primary outcome to assess the number of women or couples who make an informed choice measured by tools such as the Multidimensional Measure of Informed Choice (MMIC).
Contributions of authors
Writing the protocol: all authors Developing the search strategy: NH, NQ, JK and other (not review authors) Searching for trials: NH, LH and NQ Selection of trials: NH, LH, NQ, and other (not review authors) Data entry: NH Analysis: NH, LH and NQ Interpret analysis: all authors Draft final review: all authors Update the review: NH, LH and NQ
Sources of support
Internal sources
-
University of Nottingham, UK
The university provided computer and internet access.
External sources
-
National Institute for Health Research, UK
This systematic review was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Cystic Fibrosis and Genetic Disorders Group.
Declarations of interest
Dr Norita Hussein has no known interest to declare.
Dr Nadeem Qureshi is an investigator on a UK National Institute of Health research project evaluating preconception screening in primary care and plan to pursue further research in this area. Dr Qureshi is also collaborating on a project to evaluate the evidence base relevant to NICE guidelines behind primary care. In February 2019, he received an honorarium and travel expenses for a lecture from Amgen Inc.
Professor Hennemann declares she has worked on several pilot studies for carrier screening, which are not eligible for inclusion in the review. She received a research grant from the Netherlands Organisation for Health Research and Development (ZonMw) to study: "Preconception carrier screening in the Netherlands: Advantages and consequences, societal support and ethical framework". She is an unpaid member of the Dutch Working Group for Preconception Carrier Screening and a member of the Advisory Board for the Association of Clinical Genetics Netherlands (VKGN). She is affiliated to the University Hospital Amsterdam UMC that offers expanded carrier in a non‐commercial setting.
Dr Joe Kai was an investigator on the UK National Institute of Health research project evaluating preconception screening in primary care up to December 2019, but currently has no known interest to declare.
New search for studies and content updated (no change to conclusions)
References
References to studies excluded from this review
Alhamdan 2007 {published data only}
- Alhamdan NA, Almazrou YY, Alswaidi FM, Choudhry AJ. Premarital screening for thalassemia and sickle cell disease in Saudi Arabia. Genetics in Medicine 2007;9(6):372-7. [DOI] [PubMed] [Google Scholar]
Archibald 2017 {published data only}
- Archibald AD, Smith MJ, Burgess T, Scarff KL, Elliott J, Hunt CE, et al. Reproductive genetic carrier screening for cystic fibrosis, fragile X syndrome, and spinal muscular atrophy in Australia: outcomes of 12,000 tests. Genetic Medicine 2018;20(5):513-23. [DOI: 10.1038/gim.2017.134] [DOI] [PubMed] [Google Scholar]
Bekker 1993 {published data only}
- Bekker H, Modell M, Denniss G, Silver A, Mathew C, Bobrow M, Marteau T. Uptake of cystic fibrosis testing in primary care: supply push or demand pull? British Medical Journal 1993;306(6892):1584-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Castellani 2011 {published data only}
- Castellani C, Perobelli S, Bianchi V, Seia M, Melotti P, Zanolla L, et al. An interactive computer program can effectively educate potential users of cystic fibrosis carrier tests. American Journal of Medical Genetics 2011;155A(4):778-85. [DOI] [PubMed] [Google Scholar]
Cheuvront 1998 {published data only}
- Callanan NP, Cheuvront B, Sorenson JR. CF Carrier testing in a high risk population: Anxiety,risk perceptions, and reproductive plans of carrier by "non-carrier'' couples. Genetics in Medicine 1999;1(7):323-7. [DOI] [PubMed] [Google Scholar]
- Cheuvront B, Sorenson JR, Callanan NP, Stearns SC, DeVellis BM. Psychosocial and educational outcomes associated with home- and clinic-based pretest education and cystic fibrosis carrier testing among a population of at-risk relatives. American Journal of Medical Genetics 1998;75(5):461-8. [PubMed] [Google Scholar]
Childs 1976 {published data only}
- Childs B, Gordis L, Kaback MM, Kazazian HH. Tay-Sachs screening: motives for participating and knowledge of genetics and probability. American Journal of Human Genetics 1976;28(6):537-49. [PMC free article] [PubMed] [Google Scholar]
- Childs B, Gordis L, Kaback MM, Kazazian HH. Tay-Sachs screening: social and psychological impact. American Journal of Human Genetics 1976;28(6):550-8. [PMC free article] [PubMed] [Google Scholar]
Clayton 1996 {published data only}
- Clayton EW, Hannig VL, Pfotenhauer JP, Parker RA, Campbell PW, Phillips JA. Lack of interest by nonpregnant couples in population-based cystic fibrosis carrier screening. American Journal of Human Genetics 1996;58(3):617-27. [PMC free article] [PubMed] [Google Scholar]
Fan 2018 {published data only}
- Fan CW, Castonguay L, Rummell S, Lévesque S, Mitchell JJ, Sillon G. Online module for carrier screening in Ashkenazi Jewish individuals compared with in-person genetics education: a randomized controlled trial. Journal of Genetic Counseling 2018;27:426-38. [DOI: 10.1007/s10897-017-0133-4] [DOI] [PubMed] [Google Scholar]
- NCT01999257. Efficacy study of an online educational module before carrier genetic screening in persons of Ashkenazi Jewish descent. clinicaltrials.gov/ct2/show/NCT01999257 (first posted 03 December 2013).
Fisher 1981 {published data only}
- Fisher L, Rowley PT, Lipkin JR. Genetic counselling for beta-thalassemia trait following health screening in a health maintenance organization:comparison of programmed and conventional counselling. American Journal of Human Genetics 1981;33(6):987-94. [PMC free article] [PubMed] [Google Scholar]
- Rowley PT, Fisher L, Lipkin JR. Screening and genetic counselling for thalassemia trait in a population unselected for interest: effects on knowledge and mood. American Journal of Human Genetics 1979;31(6):718-30. [PMC free article] [PubMed] [Google Scholar]
- Rowley PT, Lipkin M Jr, Fisher L. Screening and genetic counselling for beta-thalassemia trait in a population unselected for interest: comparison of three counselling methods. American Journal of Human Genetics 1984;36(3):677-89. [PMC free article] [PubMed] [Google Scholar]
Hegwer 2006 {published data only}
- Hegwer G, Fairley C, Charrow J, Ormond KE. Knowledge and attitudes toward a free education and Ashkenazi Jewish carrier testing program. Journal of Genetic Counseling 2006;15(1):61-70. [DOI] [PubMed] [Google Scholar]
Henneman 2001 {published data only}
- Henneman L, Bramsen I, van der Ploeg, ten Kate, LP. Preconception cystic fibrosis carrier couple screening: impact, understanding, and satisfaction. Genetic Testing 2002;6(3):195-202. [DOI] [PubMed] [Google Scholar]
- Henneman L, Bramsen I, Ploeg HM, Ader HJ, Horst HE, Gille JJP, et al. Participation in preconceptional carrier couple screening: characteristics, attitudes, and knowledge of both partners. Journal of Medical Genetics 2001;38(10):695-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneman L, Bramsen I, Kempen L, Acker MB, Pals G, Horst HE, et al. Offering preconceptional cystic fibrosis carrier couple screening in the absence of established preconceptional care services. Community Genetics 2003;6(1):5-13. [DOI] [PubMed] [Google Scholar]
- Lakeman PL, Plass AMC, Henneman L, Bezemer PD, Cornel MC, ten Kate LP. Three-month follow-up of Western and non-Western participants in a study on preconceptional ancestry based carrier couple screening for cystic fibrosis and hemoglobinopathies in the Netherlands. Genetics in Medicine 2008;10(11):820-30. [DOI] [PubMed] [Google Scholar]
Honnor 2000 {published data only}
- Honnor M, Zubrick SR, Walpole I, Bower C, Goldblatt J. Population screening for cystic fibrosis in Western Australia: community response. American Journal of Medical Genetics 2000;93(3):198-204. [DOI] [PubMed] [Google Scholar]
Moudi 2016 {published data only}
- IRCT2015061022651N1. The study of effect of motivational interviewing on adherence to iron supplementation and prenatal diagnostic test in couples suspected to have ß- thalassemia, referring to pemarital counseling center, Zahedan. www.who.int/trialsearch/Trial2.aspx?TrialID=IRCT2015061022651N1 (first registered 01 August 2015). [CFGD REGISTER: TH194]
- Moudi Z, Chermahini ED, Miri Moghaddam E, Navidian A. Motivational interviewing and compliance with carriers screening for beta-thalassemia trait in Zahedan premarital counseling center, Iran. Shiraz e Medical Journal 2016;17(10):e41381. [CFGD REGISTER: TH164] [DOI: 10.17795/semj41381] [DOI] [Google Scholar]
Payne 1997 {published data only}
- Payne Y, Williams M, Cheadle J, Stott NC, Rowlands M, Shickle D, et al. Carrier screening for cystic fibrosis in primary care: evaluation of a project in South Wales. The South Wales Cystic Fibrosis Carrier Screening Research Team. Clinical Genetics 1997;51(3):153-63. [DOI] [PubMed] [Google Scholar]
Punj 2018 {published data only}
- Kauffman TL, Wilfond BS, Jarvik GP, Leo MC, Lynch FL, Reiss JA et al. Design of a randomized controlled trial for genomic carrier screening in healthy patients seeking preconception genetic testing. Contemporary Clinical Trials 2017;53:100-5. [DOI: 10.1016/j.cct.2016.12.007] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punj S, Akkari Y, Huang J, Yang F, Creason A, Pak C, et al. Preconception carrier screening by genome sequencing: results from the clinical laboratory. American Journal of Human Genetics 2018;102(6):1078-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
Quigley 2018 {published data only}
- Quigley SJ, Linnane B, Connellan S, Ward A, Ryan P. Psychosocial distress and knowledge deficiencies in parents of children in ireland WHO carry an altered cystic fibrosis gene. Journal of Genetic Counseling 2018;27(3):589-96. [CFGD REGISTER: MH75] [DOI: 10.1007/s10897-017-0150-3] [DOI] [PubMed] [Google Scholar]
Rémus 2020 {published data only}
- Rémus C, Stanislas A, Bouazza N, Gauthereau V, Polak M, Blanche S, et al. An evaluation of three ways of communicating carrier status results to the parents of children in a neonatal sickle cell screening programme. Frontiers in Pediatrics 2020;19(8):300. [CFGDREGISTER: SC420] [DOI: 10.3389/fped.2020.00300] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sallevalt 2021 {published data only}
- Sallevelt SC, Stegmann AP, Koning B, Velter C, Steyls A, Esch M, et al. Diagnostic exome-based preconception carrier testing in consanguineous couples: results from the first 100 couples in clinical practice. Genetic Medicine 2021;23:1125–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tambor 1994 {published data only}
- Tambor ES, Bernhardt BA, Chase GA, Faden RR, Geller G, Hofman KJ, et al. Offering cystic fibrosis carrier screening to an HMO population: factors associated with utilization. American Journal of Human Genetics 1994;55:626-37. [PMC free article] [PubMed] [Google Scholar]
Temme 2015 {published data only}
- Temme R, Gruber A, Johnson M, Read L, Liu M, Lu Y, et al. Quality improvement in genetic counseling following false positive newborn screen results for cystic fibrosis: assessment of parental knowledge with the use of an educational video. Pediatric Pulmonology 2013;48 Suppl 36:374. [ABSTRACT NO.: 461] [CFGD REGISTER: PC14a] [Google Scholar]
- Temme R, Gruber A, Johnson M, Read L, Lu Y, McNamara J. Assessment of parental understanding of positive newborn screening results and carrier status for cystic fibrosis with the use of a short educational video. Journal of Genetic Counseling 2015;24(3):473-81. [CFGD REGISTER: PC14b] [DOI: 10.1007/s10897-014-9767-7] [DOI] [PubMed] [Google Scholar]
Watson 1991 {published data only}
- Watson EK, Mayall ES, Chapple J, Dalziel M, Harrington K, Williams C, et al. Screening for carriers of cystic fibrosis through primary health care services. British Medical Journal 1991;303(6801):504-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson EK, Mayall ES, Lamb J, Chapple J, Williamson R. Psychological and social consequences of community carrier screening programme for cystic fibrosis. Lancet 1992;340(8813):217-20. [DOI] [PubMed] [Google Scholar]
Wilkie 2013 {published data only}
- Gallo AM, Wilkie DJ, Wang E, Labotka RJ, Molokie RE, Stahl C, et al. Evaluation of the SCKnowIQ tool and reproductive CHOICES intervention among young adults with sickle cell disease or sickle cell trait. Clinical Nurse Research 2014;23(4):421-41. [CFGD REGISTER: SC237d] [DOI: 10.1177/1054773813479377] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo AM, Wilkie DJ, Yao Y, Molokie RE, Stahl C, Hershberger PE, et al. Reproductive health CHOICES for young adults with sickle cell disease or trait: randomized controlled trial uutcomes over two years. Journal of Genetic Counseling 2016;25(2):325-36. [CFGD REGISTER: SC237c] [DOI: 10.1007/s10897-015-9874-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger PE, Gallo AM, Molokie R, Thompson AA, Suarez ML, Yao Y, Wilkie DJ, . Perception of young adults with sickle cell disease or sickle cell trait about participation in the CHOICES randomized controlled trial. Journal of Advanced Nursing 2016;72(6):1430-40. [CFGD REGISTER: SC237b] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie DJ, Gallo AM, Yao Y, Molokie RE, Stahl C, Hershberger PE et al. Reproductive health choices for young adults with sickle cell disease or trait: randomized controlled trial immediate posttest effects. Nursing Research 2013;62(5):352-61. [CFGD REGISTER: SC237a] [DOI: 10.1097/NNR.0b013e3182a0316b] [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
ACOG Committee Opinion 2009
ACOG Committee Opinion 2017
- American College of Obstetricians and Gynecologists. Carrier screening for genetic conditions. ACOG Committee Opinion No. 691 2017;129(3):41-55. [Google Scholar]
ACOG Committee Opinion 2017 (reaffirmed 2020)
- American College of Obstetricians and Gynecologists. Carrier screening in the age of genomic medicine. ACOG Committee Opinion No. 690 2017;129(3):595-6. [DOI] [PubMed] [Google Scholar]
Alswaidi 2009
- Alswaidi FM, O'Brien SJ. Premarital screening programmes for haemoglobinopathies, HIV and hepatitis viruses: review and factors affecting their success. Journal of Medical Screening 2009;16(1):22-8. [DOI] [PubMed] [Google Scholar]
Alwan 1997
- Alwan A, Modell B. EMRO Technical Publication 24. Alexandria: Eastern Mediterranean Regional Office: World Health Organization, 1997. [Google Scholar]
Angastiniotis 1981
- Angastiniotis MA, Hadjiminas MG. Prevention of thalassaemia in Cyprus. Lancet 1981;1(8216):369-71. [DOI] [PubMed] [Google Scholar]
Angastiniotis 1995
- Angastiniotis M, Modell B, Englezos P, Boulyjenkov V. Prevention and control of haemoglobinopathies. Bulletin of the World Health Organization 1995;73(3):375-86. [PMC free article] [PubMed] [Google Scholar]
Angastiniotis 1998
- Angastiniotis M, Modell B. Global epidemiology of haemoglobin disorders. Annals of the New York Academy of Sciences 1998;850:251-69. [DOI] [PubMed] [Google Scholar]
Anido 2005
- Anido A, Carlson LM, Taft L, Sherman SL. Women's attitudes toward testing for fragile X carrier status: a qualitative analysis. Journal of Genetic Counselling 2005;14(4):295-306. [DOI] [PubMed] [Google Scholar]
Antonarakis 2019
- Antonarakis SE. Carrier screening for recessive disorders. Nature Revires Genetics 2019;20(9):549-61. [DOI: 10.1038/s41576-019-0134-2] [DOI] [PubMed] [Google Scholar]
Archibald 2011
- Archibald AD, Wilford BS. Population carrier screening: psychological impact. Encyclopedia of Life Sciences 2011. [DOI: 10.1002/9780470015902.a0005641.pub2] [DOI]
Bach 2001
- Bach G, Tomczak J, Risch N, Ekstein J. Tay-Sachs screening in the Jewish Ashkenazi population: DNA testing is the preferred procedure. American Journal of Medical Genetics 2001;99(1):70-5. [DOI] [PubMed] [Google Scholar]
Barbarin 1999
- Barbarin OA, Whitten CF, Bond S, Conner-Warren R. The social and cultural of coping with sickle cell disease: the role of financial hardship in adjustment to sickle cell disease. Journal of Black Psychology 1999;23(3):294-315. [Google Scholar]
Bareil 2020
- Bareil C, Bergougnoux A. CFTR gene variants, epidemiology and molecular pathology,. Archives de Pédiatrie 2020;27(1):8-12. [DOI: 10.1016/S0929-693X(20)30044-0.] [DOI] [PubMed] [Google Scholar]
Bekker 1994
- Bekker H, Denniss G, Modell M, Bobrow M, Marteau T. The impact of population based screening for carriers of cystic fibrosis. Journal of Medical Genetics 1994;31(5):364-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bonham 2010
- Bonham VL, Dover GJ, Brody LC. Screening student athletes for sickle cell trait: a social and clinical experiment. New England Journal of Medicine 2010;363(11):997-9. [DOI] [PubMed] [Google Scholar]
Bonham 2019
- Bonham VL, Smilan LE. Somatic Genome Editing in Sickle Cell Disease: Rewriting a More Just Future. North Carolina Law Review 2019;97:1093. [Google Scholar]
Borry 2011
- Borry P, Henneman L, Lakeman P, ten Kate LP, Cornel MC, Howard HC. Preconceptional genetic carrier testing and the commercial offer directly-to-consumers. Human Reproduction 2011;26(5):972-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boulton 1996
- Boulton M, Cummings C, Williamson R. The views of general practitioners on community carrier screening for cystic fibrosis. British Journal of General Practice 1996;46(406):299-301. [PMC free article] [PubMed] [Google Scholar]
Bregnballe 2007
- Bregnballe V, Thastum M, Schiøtz PO. Psychosocial problems in children with cystic fibrosis. Acta Paediatrica 2007;96(1):58-61. [DOI] [PubMed] [Google Scholar]
Cannon 2019
- Cannon J, Van Steijvoort E, Borry P, Chokoshvili D. How does carrier status for recessive disorders influence reproductive decisions? A systematic review of the literature. Expert Review of Molecular Diagnostic 2019;19(12):1117-29. [DOI: 10.1080/14737159.2020.1690456] [DOI] [PubMed] [Google Scholar]
Cao 1996
- Cao A, Rosatelli MC, Gallanello R. Control of beta-thalassaemia by carrier screening, genetic counselling, and prenatal diagnosis: the Sardinian experience. Ciba Foundation Symposium 1996;197:137-51. [DOI] [PubMed] [Google Scholar]
Castellani 2010
- Castellani C, Macek M Jr, Cassiman JJ, Duff A, Massie J, ten Kate LP, et al. Benchmarks for cystic fibrosis carrier screening: a European consensus document. Journal of Cystic Fibrosis 2010;9(3):165-78. [DOI] [PubMed] [Google Scholar]
CDC 2004
- Grosse SD, Boyle CA, Botkin JR, Comeau AM, Kharrazi M, Rosenfeld M, et al. Newborn screening for cystic fibrosis. Evaluation of benefits and risks and recommendations for state newborn screening programs. MMWR Recommendations Report 2004;53(RR-13):1-36. [PubMed]
Christie 2009
- Christie LM, Ingret AJ, Turner GM. Outcomes of a cystic fibrosis carrier testing clinic for couples. Medical Journal of Australia 2009;191(9):499-501. [DOI] [PubMed] [Google Scholar]
Cornel 2021
- Cornel MC, Rigter T, Jansen ME, Henneman L. Neonatal and carrier screening for rare diseases: how innovation challenges screening criteria worldwide. Journal of Community Genetics 2021;12(2):257-65. [DOI: 10.1007/s12687-020-00488-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cousens 2010
- Cousens NE, Gaff CL, Metcalfe SA, Delatycki MB. Carrier screening for beta-thalassaemia: a review of international practice.. European Journal of Human Genetics 2010;18(10):1077-83. [DOI: 10.1038/ejhg.2010.90] [DOI] [PMC free article] [PubMed] [Google Scholar]
Czeizel 2012
- Czeizel AE. Experience of the Hungarian Preconception Service between 1984 and 2010. European Journal of Obstetrics & Gynecology and Reproductive Biology 2012;161(1):18-25. [DOI] [PubMed] [Google Scholar]
Davies 2000
- Davies SC, Cronin E, Gill M, Greengross P, Hickman M, Normand C. Screening for sickle cell disease and thalassaemia: a systematic review with supplementary research. Health Technology Assessment 2000;4(3):1-99. [PubMed] [Google Scholar]
Delatycki 2019
- Delatycki, MB, Alkuraya, F, Archibald, A, et al. International perspectives on the implementation of reproductive carrier screening.. Prenatal Diagnosis 2020;20:301-10. [DOI: 10.1002/pd.5611] [DOI] [PubMed] [Google Scholar]
de Wert 2012
- De Wert GM, Dondorp WJ, Knoppers BM. Preconception care and genetic risk: ethical issues. Journal of Community Genetics 2012;3(3):221-28. [DOI: 10.1007/s12687-011-0074-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Donner 1980
- Donner A, Koval JJ. The estimation of intraclass correlation in the analysis of family data. Biometrics 1980;36(1):19-25. [PubMed] [Google Scholar]
Dormandy 2008
- Dormandy E, Gulliford MC, Reid EP, Brown K, Marteau TM. Delay between pregnancy confirmation and sickle cell and thalassaemia screening: a population-based cohort study. British Journal of General Practice 2008;58(548):154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Dyson 2006
- Dyson SM, Culley LA, Gill C, Hubbard S, Kennefick A, Morris P, et al. Ethnicity questions and antenatal screening for sickle cell/thalassaemia [EQUANS] in England: A randomized controlled trial of two questionnaires. Ethnicity and Health 2006;11(2):169-89. [DOI] [PubMed] [Google Scholar]
EndNote X9 [Computer program]
- EndNote X9. Clarivate, 2009.
Fanos 1995
- Fanos JH, Johnson JP. Barriers to carrier testing for adult cystic fibrosis sibs: the importance of not knowing. American Journal of Medical Genetics 1995;59(1):85-91. [DOI] [PubMed] [Google Scholar]
Fowzan 2001
- Fowzan S, Alkuraya, Ramzi AK. Attitude of Saudi families affected with hemoglobinopathies towards prenatal screening and abortion and the influence of religious ruling (Fatwa). Prenatal Diagnosis 2001;21(6):448-51. [DOI] [PubMed] [Google Scholar]
Gharaibeh 2009
- Gharaibeh H, Amarneh BH, Zamzam SZ. The psychological burden of patients with beta thalassemia major in Syria. Pediatrics International 2009;51(5):630-6. [DOI] [PubMed] [Google Scholar]
Glasscoe 2008
- Glasscoe CA, Quittner AL. Psychological interventions for people with cystic fibrosis and their families. Cochrane Database of Systematic Reviews 2008, Issue 3. Art. No: CD003148. [DOI: 10.1002/14651858.CD003148.pub2] [DOI] [PubMed] [Google Scholar]
Grody 2013
- Grody WW, Thompson BH, Gregg AR, Bean LH, Monaghan KG, Schneider A, et al. ACMG position statement on prenatal/preconception expanded carrier screening. Genetic Medicine 2013;15(6):482-83. [DOI: 10.1038/gim.2013.47] [DOI] [PubMed] [Google Scholar]
Haque 2016
- Haque IS, Lazarin GA, Kang HP, Evans EA, Goldberg JD, Wapner RJ. Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA 2016;316(7):734-42. [DOI] [PubMed] [Google Scholar]
Health Council of Netherlands 2007
- Health Council of Netherlands. Preconception care: A good beginning. The Hague: Health Council of Netherlands 2007. [ISBN: 978-90-5549-678-5]
Heyes 2004
- Heyes T, Long S, Mathers N. Preconception care. Practice and beliefs of primary care workers. Family Practice 2004;21(1):22-7. [DOI] [PubMed] [Google Scholar]
Higgins 2002
- Higgins JP, Thompson SG. Quantifying heterogeneity in a meta-analysis. Statistics in Medicine 2002;21(11):1539-58. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Altman DG, Sterne JA on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.handbook-5-1.cochrane.org/.
Holtkamp 2017
- Holtkamp KC, Vos EM, Rigter T, Lakeman P, Henneman L, Cornel MC . Stakeholder perspectives on the implementation of genetic carrier screening in a changing landscape.. BMC Health Services Research 2017;17(1):146. [DOI: 10.1186/s12913-017-2083-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Human Genetics Commission 2011
- Human Genetics Commission. Increasing options, informing choice: A report on preconception genetic testing and screening. Human Genetics Commission 2011.
Janssens 2014
- Janssens S, De Paepe A, Borry P. Attitudes of health care professionals toward carrier screening for cystic fibrosis. A review of the literature. Journal of Community Genetics 2014;5(1):13-29. [DOI: 10.1007/s12687-012-0131-z] [DOI] [PMC free article] [PubMed] [Google Scholar]
Johnson 2006
- Johnson K, Posner SF, Biermann J, Cordero JF, Atrash HK, Parker CS, et al. Recommendations to improve preconception health and health care--United States. A report of the CDC/ATSDR Preconception Care Work Group and the Select Panel on Preconception Care. MMWR Recommendation Report 2006;55(RR-6):1-23. [PubMed]
Jones 2002
- Jones SL, Fallon LA. Reproductive options for individuals at risk for transmission of a genetic disorder. Journal of Obstetric, Gynecologic, & Neonatal Nursing 2002;31(2):193-9. [DOI] [PubMed] [Google Scholar]
Kaback 2000
- Kaback MM. Population-based genetic screening for reproductive counselling: the Tay-Sachs disease model. European Journal of Pediatrics 2000;159(3):192-5. [DOI] [PubMed] [Google Scholar]
Kenen 1978
- Kenen RH, Schmidt RM. Stigmatization of carrier status: social implications of heterozygote genetic screening programs. American Journal of Public Health 1978;68(11):1116-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kirk 2021
- Kirk EP, Ong R, Boggs K, Hardy T, Righetti S, Kamien B, et al. Gene selection for the Australian Reproductive Genetic Carrier Screening Project (“Mackenzie’s Mission”). European Journal of Human Genetics 2021;29:79-87. [DOI: 10.1038/s41431-020-0685-x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lapham 1996
- Lapham EV, Kozma C, Weiss JO. Genetic discrimination: perspectives of consumers. Science 1996;274(5287):621-4. [DOI] [PubMed] [Google Scholar]
Lau 1997
- Lau YL, Chan LC, Chan YY, Ha SY, Yeung CY, Waye JS, et al. Prevalence and genotypes of alpha- and beta-thalassemia carriers in Hong Kong -- implications for population screening. New England Journal of Medicine 1997;336(18):1298-301. [DOI] [PubMed] [Google Scholar]
Lena‐Russo 2002
- Lena-Russo D, Badens C, Aubinaud M, Merono F, Paolasso C, Martini N, et al. Outcome of a school screening programme for carriers of haemoglobin disease. Journal of Medical Screening 2002;9(2):67-9. [DOI] [PubMed] [Google Scholar]
Lew 2014
- Lew RM, Burnett L, Proos AL, Barlow-Stewart K, Delatychi MB, Bankier A, et al. Ashkenazi Jewish population screening for Tay–Sachs disease: the international and Australian experience. Journal of Paediatrics and Child Health 2014;51(3):271-9. [DOI] [PubMed] [Google Scholar]
Lewis 2011
- Lewis C, Skirton H, Jones R. Reproductive empowerment: The main motivator and outcome of carrier testing. Journal of Health Psychology 2011;17(4):567-78. [DOI] [PubMed] [Google Scholar]
Locock 2008
- Locock L, Kai J. Parents' experiences of universal screening for haemoglobin disorders: implications for practice in a new genetics era. British Journal of General Practice 2008;58(548):161-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Loukopoulos 1996
- Loukopoulos D. Current status of thalassemia and the sickle cell syndromes in Greece. Seminars in Hematology 1996;33(1):76-86. [PubMed] [Google Scholar]
March of Dimes 2006
- Christianson A, Howson CP, Modell B. Global Report on Birth Defects. The hidden toll of dying and disabled children. Available from www.marchofdimes.org/materials/global-report-on-birth-defects-the-hidden-toll-of-dying-and-disabled-children-executive-summary.pdf 2006 (accessed 28 July 2021).
Massie 2009
- Massie J, Petrou V, Forbes R, Curnow L, Ioannou L, Bankier A, et al. Population-based carrier screening for cystic fibrosis in Victoria: The first three years experience. Australian and New Zealand Journal of Obstetrics and Gynaecology 2009;49(5):484-9. [DOI] [PubMed] [Google Scholar]
Massie 2014
- Massie J, Ioannou L, Delatycki M. Prenatal and preconception population carrier screening for cystic fibrosis in Australia: where are we up to? Australia and New Zealand Journal of Obstetric and Gynaecology 2014;54(6):503-09. [DOI: 10.1111/ajo.12255] [DOI] [PubMed] [Google Scholar]
Mennie 1998
- Mennie M, Campbell H, Liston WA, Brock DJ. Attitudes of general practitioners to screening for cystic fibrosis. Journal of Medical Screening 1998;5(1):11-5. [DOI] [PubMed] [Google Scholar]
Mitchell 1996
- Mitchell JJ, Capua A, Clow C. Twenty-year outcome analysis of genetic screening programs for Tay-Sachs and β-Thalassemia disease carriers in high schools. American Journal of Medical Genetics 1996;59:793-8. [PMC free article] [PubMed] [Google Scholar]
Modell 1980a
- Modell B, Mouzouras M, Camba L, Ward RHT, Fairweather DVI. Population screening for carriers of recessively inherited disorders. Lancet 1980;316(8198):806. [DOI] [PubMed] [Google Scholar]
Modell 1980b
- Modell B, Ward RHT, Fairweather DVI. Effect of introducing antenatal diagnosis on reproductive behaviour of families at risk for thalassaemia major. BMJ 1980;280(6228):1347-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Modell 2008
- Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bulletin of the World Health Organization 2008;86(6):480-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Murray 1999
NHS SCT Screening Programme 2013
- NHS Sickle Cell and Thalassaemia Screening Programme. United Kingdom National Screening Programmes Information Strategy. http://sct.screening.nhs.uk (accessed 8 January 2013).
Nuffield Council on Bioethics 1993
- Nuffield Council on Bioethics. Genetic screening: ethical issues. Nuffield Council on Bioethics 1993. [ISBN: 0 9522701 0 2]
Patel 2020
- Patel SD, Bono TR, Rowe SM, Solomon GM. CFTR targeted therapies: recent advances in cystic fibrosis and possibilities in other diseases of the airways. European Respiratory Review 2020;29(156):190068. [DOI: 10.1183/16000617.0068-2019] [DOI] [PMC free article] [PubMed] [Google Scholar]
Peters 2012
- Peters M, Heijboer H, Smiers F, Giordano PC. Diagnosis and management of thalassaemia. BMJ 2012;344:e228. [DOI] [PubMed] [Google Scholar]
Poppelaars 2004
- Poppelaars FAM, Ader HJ, Cornel MC, Henneman L, Hermens RP, Wal G, et al. Attitudes of potential providers towards preconceptional cystic fibrosis carrier screening. Journal of Genetic Counseling 2004;13(1):31-44. [DOI] [PubMed] [Google Scholar]
Qureshi 2004
- Qureshi N, Modell B, Modell M. Raising the profile of genetics in primary care. Nature Reviews Genetics 2004;5(10):783-90. [DOI] [PubMed] [Google Scholar]
Ratip 1996
- Ratip S, Modell B. Psychological and sociological aspects of the thalassemias. Seminars in Hematology 1996;33(1):53-65. [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rose 1999
- Rose P, Humm E, Hey K, Jones L, Huson SM. Family history taking and genetic counselling in primary care. Family Practice 1999;16(1):78‐83. [DOI] [PubMed] [Google Scholar]
Samavat 2004
- Samavat A, Modell B. Iranian national thalassaemia screening programme. BMJ 2004;329(7475):1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2021
- Schünemann HJ, Higgins JP, Vist GE, Glasziou P, Akl EA, Skoetz N, et al. Chapter 14: Completing ‘Summary of findings’ tables and grading the certainty of the evidence. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA (editors). Cochrane Handbook for Systematic Reviews of Interventionsversion 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook.
Shapira 2006
- Shapira SK, Dolan S. Genetic risks to the mother and the infant: assessment, counselling, and management. Maternal & Child Health Journal 2006;10(5):143-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shaw 2020
- Cornel MC, Goodman S, Henneman L. Genetic health care before conception. In: Shawe J, Steegers E, Verbiest S, editors(s). Preconception Health and Care: A Life Course Approach. 1st edition. Springer International Publishing, 2020:35-52. [DOI: 10.1007/978-3-030-31753-9_4] [DOI] [Google Scholar]
Smith 2003
- Smith RA, Mettlin CJ, Eyre H. Methodologic issues in the evaluation of early detection programs. In: Kufe DW, Pollock RE, Weichselbaum RR, editors(s). Holland-Frei Cancer Medicine. 6th edition. Hamilton (ON): BC Decker, 2003. [Google Scholar]
Sterne 2011
- Sterne JA, Egger M, Moher D on behalf of the Cochrane Bias Methods Group (editors). Chapter 10: Addressing reporting bias. In; Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.handbook-5-1.cochrane.org/.
Superior Health Council Belgium 2017
- Superior Health Council Belgium. Advisory report of the superior health council: expanded carrier screening in a reproductive context. Towards a Responsible Implementation in the Healthcare System. Available from www.health.belgium.be/fr/avis‐9240 2017;9240:1-26. [Google Scholar]
Teeuw 2010
- Teeuw ME, Henneman L, Bochdanovits Z, Heutink P, Kuik DJ, Corneland MC, et al. Do consanguineous parents of a child affected by an autosomal recessive disease have more DNA identical-by-descent than similarly-related parents with healthy offspring? Design of a case-control study. BMC Medical Genetics 2010;11:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Telfer 2005
- Telfer P, Constantinidou G, Andreou P, Christou S, Modell B, Angastiniotis M. Quality of life in thalassemia. Annals of the New York Academy of Sciences 2005;1054(1):273-82. [DOI] [PubMed] [Google Scholar]
UK National Screening Committee 2003
- UK National Screening Committee. Criteria for screening programmes. www.screening.nhs.uk/criteria (accessed 22 July 2014).
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JA, Burney PG. Methods for evaluating area-wide and organisation-based interventions in health and health care: a systematic review. Health Technology Assessment 1999;3(5):iii-92. [PubMed] [Google Scholar]
van der Hout 2016
- Hout S, Holtkamp KC, Henneman L, Wert G, Dondorp WJ. Advantages of expanded universal carrier screening: what is at stake? European Journal of Human Genetics 2016;1:17-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Watson 1999
- Watson EK, Shickle D, Qureshi N, Emery J, Austoker J. The 'new genetics' and primary care: GPs' view on their role and their educational needs. Family Practice 1999;16(4):420-5. [DOI] [PubMed] [Google Scholar]
Weatherall 1997
- Weatherall D. ABC of clinical haematology: the hereditary anaemias. BMJ 1997;314(7079):492-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Whitten 1973
- Whitten CF. Sickle-cell programming: an imperilled promise. New England Journal of Medicine 1973;288(6):318-9. [DOI] [PubMed] [Google Scholar]
WHO 1968
- Wilson JMG, Jungner G. Principles and practice of screening for disease. Geneva: World Health Organization, 1968. [Google Scholar]
WHO 2000
- World Health Organization. Genes and Human Disease. www.who.int/genomics/public/geneticdiseases/en/index2.html (accessed 8 May 2013);(Publication no. 2007/19E). [ISBN: 978-90-5549-678-5]
Wille 2004
- Wille MC, Weitz B, Kerper P, Frazier S. Advances in preconception genetic counselling. Journal of Perinatal & Neonatal Nursing 2004;18(1):28-40. [DOI] [PubMed] [Google Scholar]
Yusuf 2011
- Yusuf HR, Lloyd-Puryear MA, Grant AM, Parker CS, Creary MS, Atrash HK. Sickle cell disease: the need for a public health agenda. American Journal of Preventive Medicine 2011;41(6):376-83. [DOI] [PubMed] [Google Scholar]
Zeesman 1984
- Zeesman S, Clow CL, Cartier L, Scriver CR. A private view of heterozygosity: eight-year follow-up study on carriers of the Tay-Sachs gene detected by high school screening in Montreal. American Journal of Medical Genetics 1984;18(4):769-78. [DOI] [PubMed] [Google Scholar]
Zeuner 1999
- Zeuner D, Ades AE, Karnon J. Antenatal and neonatal haemoglobinopathy screening in the UK: review and economic analysis. Health Technology Assessment 1999;3(11):1-186. [PubMed] [Google Scholar]
References to other published versions of this review
Hussein 2015
- Hussein N, Weng SF, Kai J, Kleijnen J, Qureshi N. Preconception risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay-Sachs disease. Cochrane Database of Systematic Reviews 2015, Issue 8. Art. No: CD010849. [DOI: 10.1002/14651858.CD010849.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hussein 2018
- Hussein N, Weng SF, Kai J, Kleijnen J, Qureshi N. Preconception risk assessment for thalassaemia, sickle cell disease, cystic fibrosis and Tay‐Sachs disease. Cochrane Database of Systematic Reviews 2018, Issue 3. Art. No: CD010849. [DOI: 10.1002/14651858.CD010849.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]