

Abstract
Background
A biallelic intronic AAGGG repeat expansion in the Replication Factor C subunit 1 (RFC1) gene has been recently associated with Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome, a disorder often presenting as a slowly evolving sensory neuropathy at the onset. “Chronic Idiopathic Axonal Polyneuropathy” (CIAP) is a common indolent axonal neuropathy of adulthood which remains without an identifiable cause despite thorough investigations.
Methods
We screened 234 probands diagnosed with CIAP for a pathogenic biallelic RFC1 AAGGG repeat expansion. Patients were selected from 594 consecutive patients with neuropathy referred to our tertiary-care center for a sural nerve biopsy over 10 years.
Results
The RFC1 AAGGG repeat expansion was common in patients with pure sensory neuropathy (21/40, 53%) and less frequent in cases with predominantly sensory (10/56, 18%, P < 0.001) or sensorimotor (3/138, 2%, P < 0.001) neuropathy. The mutation was associated with sensory ataxia (τb = 0.254, P < 0.001), autonomic disturbances (35% vs 8%, Prevalence Odds Ratio—POR 6.73 CI 95% 2.79–16.2, P < 0.001), retained deep tendon reflexes (score 18.0/24 vs 11.5/24, R = 0.275, P < 0.001). On pathology, we observed absent/scant regenerative changes (τb = − 0.362, P < 0.001), concomitant involvement of large (100% and 99%, n.s.), small myelinated (97% vs 81%, POR 7.74 CI 95% 1.03–58.4, P = 0.02) and unmyelinated nerve fibers (85% vs 41%, POR 8.52 CI 95% 3.17–22.9, P < 0.001). Cerebellar or vestibular involvement was similarly rare in the two groups.
Conclusions
This study highlights the frequent occurrence of the RFC1 AAGGG repeat expansion in patients diagnosed with CIAP and characterizes the clinical and pathological features of the related neuro(no)pathy.
Supplementary Information
The online version contains supplementary material available at 10.1007/s00415-021-10552-3.
Keywords: Chronic Idiopathic Axonal Neuropathy, Replication factor C subunit 1, Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome
Introduction
Chronic polyneuropathy is a common neurological disorder, occurring in at least 4% of the middle-aged and elderly population and increasing in prevalence with age. Current estimates report a missing etiology in as many as 11–45% of patients with chronic polyneuropathy, especially in axonal forms [1, 2]. Chronic Idiopathic Axonal Polyneuropathy (CIAP) is formally identified in the presence of (a) clinical signs of distal sensory or sensorimotor neuropathy with little progression over a 6-month time window, (b) large fiber axonal neuropathy as observed on nerve conduction studies (NCS) and (c) absence of an identifiable underlying etiology. Although a lack of a clear inheritance pattern is currently assumed for a CIAP diagnosis [3], recent reports indicated that ‘idiopathic’ late-onset axonal neuropathies do have a mendelian inheritance in some cases [4].
Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome (CANVAS) is a complex and presumably rare ataxic disorder occurring in middle-aged or older individuals, recently associated to a biallelic intronic pentanucleotide repeat expansion of a mutated AluSx3 element (AAGGGexp) of the Replication Factor C subunit 1 (RFC1) gene (Online Resource 1) [5]. First identified by neurotologists as a combined degenerative disorder of cerebellum and vestibulum [6], the disease was later renamed to encompass the frequent peripheral sensory disorder [7], an ubiquitous feature of the full presentation as shown in the largest retrospective CANVAS case series available to date [8].
Here we report a retrospective cross-sectional study examining the prevalence of the RFC1 expansion in biopsy-investigated CIAP patients according to their clinical presentations. As a secondary analysis, we determined the differences of clinical features, instrumental investigations and sural-nerve biopsy findings between RFC1 cases and non-mutated patients.
Materials and methods
Recruitment criteria
Two readers (M.T. and D.C.) reviewed the pathology register at Neuropathology laboratory, Azienda Ospedaliera Universitaria Integrata, Verona, for patients who underwent sural nerve biopsy between 01.01.2007 and 31.12.2016 and were older than 18 years at the time of biopsy. In accordance with clinical practice, thorough investigations for sensory and sensorimotor neuropathies are required before referral (nerve conduction studies and blood panel for rheumatologic, immune, infectious, metabolic and hematologic causes of neuropathy) [9]. Two neurologists (T.C and S.F.) evaluated every patient at the time of sural nerve biopsy.
To only include patients receiving a final diagnosis of CIAP, historical records were scrutinized to exclude patients with, in hierarchical order, (a) a definite alternative diagnosis; (b) acute onset; presence on pathology studies of (c) inflammatory or (d) demyelinating changes; (e) focal alterations on clinical examination and/or pathology; (f) absence of relevant alterations on pathology. We further excluded CIAP patients without an available sample for genetic analysis. Biopsy of multiple individuals from the same family are a very rare occurrence in our practice and were not observed in this particular sample.
Data collection
Each reader independently abstracted the following data from the clinical records: sex, age at onset, apparent sporadic or familial occurrence, disease duration, age at biopsy and patterns of clinical involvement. “Sensorimotor” cases were defined by a Medical Research Council (MRC) score ≤ 3/5 in at least one muscle group bilaterally, not otherwise explained; “predominantly sensory” cases had evident motor involvement not fulfilling the previous definition; “pure sensory” cases had no evident motor involvement. Sensory impairment was graded according to Sensory Modality Sum Score (SMSS) [10, 11] (pinprick sensation was amended as it is not systematically assessed at our Clinic). Ataxia was evaluated according to an in-house impairment scale (0 = normal, 1 = mild ataxic signs, 2 = overt ataxic gait, 3 = uses unilateral walking aid, 4 = uses bilateral walking aids/wheelchair). Ankle jerks, patellar, brachioradialis and biceps deep tendon reflexes (DTR) were graded as follows: 0 = normal, 1 = just elicitable, 2 = dull, 3 = normal, 4 = brisk, 5 = frank hyperreflexia or clonus. The presence of paresthesias/tingling in any site, Romberg sign, cerebellar signs, nystagmus, vestibulopathy, dysphagia, cough, restless legs syndrome, pes cavus, hammer-toes, neuropathic pain, autonomic disturbances were also investigated, either reported as suggestive symptoms or detected on dedicated testing.
Last NCS available prior to biopsy was assessed for the pattern of involvement (axonal/demyelinating/mixed) and for sural sensory nerve action potential (SNAP) amplitude of the biopsied nerve. If available, CSF results were collected.
All the biopsy specimens had been previously analyzed during routine neuropathological practice by the same trained neuropathologist (T.C.). Historical reports were abstracted for the involvement of large and small myelinated fibers (MF) (apparent reduction in fiber density on a qualitative analysis of toluidine blue semithin sections observed by light microscopy) and unmyelinated fibers (apparent reduction in fiber density, presence of collagen pockets and denervated Remak cells on a qualitative analysis of ultrathin sections assessed by transmission electron microscopy). Likert-scales were used to quantify degenerating fibers, regenerative changes (0–3 points) and microangiopathy (0–2 points). All aforementioned data extraction was done previous to genetic analysis, and discordance between the two readers was resolved by consensus.
Genetic analysis
Repeat expansion analysis in candidate probands was performed by a single operator (M.F.), blinded to clinical data, as described in a previous study [5]. Genomic DNA from peripheral blood leukocytes was extracted by the standard salting out procedure. At first, all specimens were tested by flanking PCR to investigate the presence of a 348 base-pairs product corresponding to at least one not-expanded allele in the RFC1 gene (Online Resource 2). Patients that failed to amplify were further analyzed by three distinct repeat-primed PCR (RP-PCR) for repeat expansions associated with the prototypical pathogenic (AAGGG) motif or with the (AAAAG) and (AAAGG) motifs (Online Resource 3). RP-PCR were performed using a GoTaqR Long PCR Master Mix (Promega); fragment length analysis was done on a CEQ 8800 DNA analyzer with Fragments software (Beckman Coulter); a “sawtooth” pattern is observed only in the RP-PCR reaction specific for the (AAGGG) motif identifying patients with the biallelic AAGGGexp associated with CANVAS, herein abbreviated ‘RFC1-positive’ (Online Resource 4). In RFC1-positive patients, a Next-Generation Sequencing (NGS) panel was used to rule out concomitant mutations in genes associated with hereditary neuropathies (Online Resource 5).
Exploratory morphometric analysis
Sural nerve biopsies of RFC1-positive cases were subsequently reviewed. Semithin (1.0 µm thick), cross-sural nerve sections stained with toluidine blue were assessed on an optical set (ZEISS Axioscope 5 light microscope). Images were acquired using a coupled ZEISS Axiocam 208 color digital camera on a final optical magnification of 50 × for nerve fascicles and 400 × for nerve fibers evaluation (additional digital magnification set by the operator), and analyzed using ZEN 3.2 software by a single operator (M.T.).
Total cross fascicular area was defined as the area enclosed by the inner perineurial sheath. Axonal area of each fiber was defined as the area enclosed by the inner perimeter of the myelin sheath, excluding and counting separately regenerative clusters, degenerating fibers and non-measurable fibers. We analyzed at least three nerve fascicles from each sample to cover at least 30% of total endoneurial area and 100 measurable fibers, up to the whole nerve. To harmonize with previous literature, axon size is presented as the diameter required to describe a round axon of the same area.
The total number of MF was estimated by multiplying fiber density and total cross fascicular area. We opted to analyze the total number of MF instead of fiber density as the former variable is less influenced by healthy aging [12, 13].
On a Gaussian mixture assumption, an Expectation–Maximization iterative algorithm can be applied to obtain the characteristic parameters of the theoretical bimodal distribution of MF axon diameters in adult sural nerves. However, extremely low nerve fiber counts in our population prevented this direct approach. Instead, four control specimens from patients with acute mononeuritis multiplex deemed normal on pathology (a 48 and a 65-year-old male; a 50 and a 72-year-old female) were sampled with a multiple windows approach to obtain data on a pooled population of ~ 2000 fiber. The algorithm was applied to this population to compute the five descriptive parameters (two means μ1–2, two standard deviations s1–2 and the mixture proportion λ). Two axon size intervals (μ1 ± s1 and μ2 ± s2) were then identified and adopted to estimate the two subpopulations of small and large MF in pathologic cases, as a tradeoff between data consistency and minimal subpopulation overlap.
Data analysis
We computed the following variables: proportion of total, small and large MF in RFC1-positive compared to reference values. Numerical variables were assessed with histograms, Q-Q plots and Kolmogorov–Smirnov’s test for normality. If appropriated, quadratic or logarithmic transformations were adopted.
First, patients were categorized according to clinical presentation. Group differences in demographical variables were checked using χ2 or Kruskal–Wallis one-way ANOVA non-parametric test followed by post hoc Mann–Whitney’s U as appropriated. Association between clinical phenotype and mutation status was assessed by Fisher’s exact test, followed by a post-hoc pairwise z-test for independent proportions; Bonferroni method was adopted to control for type I error inflation.
To explore clinical and instrumental elements suggestive for RFC1 expansion mutation, patients were grouped according to the presence of biallelic AAGGGexp and tested for differences (a) in frequencies on sex, clinical, instrumental and pathology variables with z-test for independent proportions and (b) in median age at onset, disease duration, SMSS, DTR score, semiquantitative pathology scales with Mann–Whitney’s U test, as normality could not be assumed. Effect size were described as (a) prevalence odds ratio (POR) for binomial variables; (b) Cramer’s V for categorial variables; (c) Kendall’s Tau b for ordinal variables; (d) Spearman’s Rho for continuous variables, as normality could not be assumed.
Relationships between morphometry and clinico-demographical variables were analyzed on scatterplots; linear relationships were then tested with Pearson’s correlation coefficient for normally distributed variables or alternatively Spearman’s rank correlation test. Difference between upper and lower limb involvement was tested with Wilcoxon signed-rank test. Significance was set at α < 0.05, each analysis being two-tailed. Blinding during data analysis was deemed unlikely due to extreme proportions.
Results
Sample definition and identification of RFC1 expansion in CIAP
Consent to research was available in 564 out of 594 screened patients, and inclusion criteria were not met in further 316 patients (Fig. 1). Absence of a stocked DNA sample led to exclusion of 14 CIAP patients. Ultimately, 234 CIAP patients were included in the analysis. Birthplace was Northern Italy in 189, Central Italy in 17, Southern Italy in 22, other european states in 3 and extra-european in 3 (one each from Northern Africa, South America and India). Forty patients presented with pure sensory, 56 with predominantly sensory and 138 with mixed sensorimotor symptoms. The three groups were similar regarding disease duration at biopsy and sex distribution, although a non-significant lower age at onset in the pure sensory group compared to sensorimotor cases was observed (Table 1).
Fig. 1.

Study flow diagram
Table 1.
Demographic and RFC1 intron repeat expansion mutation status according to clinical phenotype
| Pure sensory (n = 40) | Predominantly sensory (n = 56) |
Sensorimotor (n = 138) | P value | Effect size | |
|---|---|---|---|---|---|
| Age at onset, median (IQR), years | 58.3 (50.5–65.0) | 62.5 (52.0–69.0) | 64.0 (55.0–70.0) | 0.05a | |
| Disease duration, median (IQR), months | 36.0 (24.0–66.0) | 24.0 (18.0–60.0) | 48.0 (24.0–72.0) | 0.23 | |
|
Females, n (%, CI 95%) |
11 (28%, 15–44%) | 23 (41%, 28–55%) | 36 (26%, 19–34%) | 0.11 | φc = 0.137 |
|
AAAAGexp, n (%, CI 95%) |
1 (3%, 0–13%) | 3 (5%, 1–15%) | 5 (4%, 1–8%) | 0.80 | φc = 0.049 |
|
AAAGGexp, n (%, CI 95%) |
0 (0–9%) | 0 (0–6%) | 1 (1%, 0–4%) | > 0.99 | φc = 0.055 |
|
AAGGGexp, n (%, CI 95%) |
21 (53%, 36–68%) | 10 (18%, 9–30%) | 3 (2%, 0–6%) | < 0.001b | φc = 0.523 |
|
Compound or non-canonical expansion, n (%, CI 95%) |
1 (3%, 0–13%) | 4 (7%, 2–17%) | 4 (3%, 1–7%) | 0.32 | φc = 0.096 |
Tests reaching statistical significance are presented in bold
IQR interquartile range
aOn Dunn-Bonferroni post-hoc test no pairwise comparison between groups reached the significance value
bOn post-hoc z-test for independent proportions with Bonferroni correction each pairwise comparison between groups reached the significance value
Clinical presentation was associated with RFC1 expansion mutation status (φc = 0.381, P < 0.001): the RFC1-positive/biallelic AAGGGexp occurred in 21/40 (53%, CI 95% 36–68%) pure sensory CIAP, 10/56 (18%, CI 95% 9–30%) predominantly sensory and 3/138 (2%, CI 95% 0–6%) sensorimotor neuropathy cases (Fig. 2). The same was not observed in cases with biallelic AAAAGexp, AAAGGexp and compound or non-canonical expansions. All AAGGGexp and all but three non-mutated patients were Caucasian.
Fig. 2.

Prevalence of RFC1 expansion mutations according to clinical presentation. Bar charts showing carriers of biallelic a AAAAG, b AAAGG, c AAGGG and d compound or non-canonical pentanucleotide RFC1 expansion mutations in patients with Chronic Idiopathic Axonal Polyneuropathy grouped by clinical presentation. Error bar: 95% CI for within-group proportions
Clinical characteristics of RFC1-positive patients
Compared to non-mutated (Table 2), RFC1-positive patients showed a similar age at onset (median 61.0 vs 63.0 years, R = − 0.091, P = 0.17), disease duration at biopsy (median 48.0 vs 36.0 months, R = 0.052, P = 0.43) and sex distribution (female 38% vs 29%, POR 1.55 CI 95% 0.73–3.31, P = 0.25). Only a minority of cases appeared to be familiar (13% vs 18%, POR 0.72 CI 95% 0.23–2.23, P = 0.56). Consanguinity up to the third degree was not reported in any patient. RFC1-positive patients showed more often a positive Romberg sign (85% vs 60%, POR 3.95 CI 95% 1.47–10.6, P = 0.004) associated with sensory ataxia (τb = 0.254, P < 0.001) and relatively spared DTR (median total score 18.0/24 vs 11.5/24, R = 0.275, P = 0.001) compared to non-mutated, even though clinical examination did not disclose a relevant difference in tested sensory modalities (median SMSS 28.5/40 vs 30.0/40, R = − 0.054, P = 0.80). In RFC1-positives, a lower limb greater than upper limb involvement was evident in both sensory (median SMSS upper limbs 19.0 vs lower limbs 9.5, R = 0.873, P < 0.001) and DTR examination (median DTR score upper limbs 12.0 vs lower limbs 6.0, R = 0.738, P < 0.001).
Table 2.
Selected demographic, clinical and instrumental data according to pathogenetic RFC1 repeat expansion mutation status
|
RFC1-positive (n = 34) |
Non-mutated (n = 200) | P value | Effect size | |
|---|---|---|---|---|
| Age at onset, median (IQR), years | 61.0 (52.5–65.0) | 63.0 (53.5–70.0) | 0.17 | R = − 0.091 |
| Disease duration, median (IQR), months | 48.0 (24.0–72.0) | 36.0 (24.0–72.0) | 0.43 | R = 0.052 |
| Female, n (%) | 13 (38%) | 57 (29%) | 0.25 | POR 1.55 (0.73–3.31) |
| Familiar cases, n/n (%) | 4/30 (13%) | 27/153 (18%) | 0.56 | POR 0.72 (0.23–2.23) |
| Same generation | 3/30 (10%) | 9/153 (6%) | a | |
| Multiple generations | 1/30 (3%) | 18/153 (12%) | a | |
| Clinical features, n (%) | ||||
| Paresthesias | 27 (79%) | 118 (59%) | 0.02 | POR 2.68 (1.11–6.45) |
| Romberg sign | 29 (85%) | 119 (60%) | 0.004 | POR 3.95 (1.47–10.6) |
| Cerebellar signs | 3 (9%) | 8 (4%) | a | |
| Nystagmus | 2 (6%) | 0 | a | |
| Vestibulopathy | 0 | 1 (1%) | a | |
| Autonomic disturbances | 12 (35%) | 15 (8%) | < 0.001 | POR 6.73 (2.79–16.2) |
| Dysphagia | 2 (6%) | 8 (4%) | a | |
| Pyramidal signs | 5 (15%) | 20 (10%) | 0.41 | POR 1.55 (0.54–4.46) |
| Cough | 3 (9%) | 0 | a | |
| Restless legs syndrome | 2 (6%) | 1 (1%) | a | |
| Pes cavus | 12 (35%) | 94 (47%) | 0.20 | POR 0.62 (0.29–1.31) |
| Hammer toes | 5 (15%) | 52 (26%) | 0.16 | POR 0.49 (0.18–1.33) |
| Pain | 20 (59%) | 106 (53%) | 0.53 | POR 1.27 (0.61–2.65) |
| DTR sum, median (IQR), points | 18.0 (11.8–24.0) | 11.5 (4.0–16.0) | 0.001 | R = 0.275 |
| Ataxia score, n (%) | ||||
| 0 | 5 (15%) | 81 (41%) | < 0.001 | τb = 0.254 |
| 1 | 4 (12%) | 60 (30%) | ||
| 2 | 22 (65%) | 47 (24%) | ||
| 3 | 2 (6%) | 9 (5%) | ||
| 4 | 1 (3%) | 3 (2%) | ||
| SMSS, median/max (IQR), points | 28.5/40 (22.0–32.0) | 30.0/40 (26.0–32.0) | 0.80 | R = − 0.054 |
| Superficial sensation | 10.0/16 (10.0–12.0) | 12.0/16 (10.0–14.0) | 0.09 | R = − 0.123 |
| Vibration perception | 11.5/16 (7.0–14.0) | 10.0/16 (8.0–12.0) | 0.54 | R = 0.037 |
| Joint position | 8.0/8 (5.0–8.0) | 8.0/8 (6.0–8.0) | 0.29 | R = − 0.072 |
| Nerve conduction studies, n/n (%) | ||||
| Axonal | 34/34 (100%)b | 135/194 (70%)b | < 0.001 | φc = 0.247 |
| Demyelinating | 0 | 8/194 (4%) | ||
| Mixed | 0b | 51/194 (26%)b | ||
| Sural SNAP, median (IQR), μV | 0 (0–0) | 0 (0–3.20) | 0.003 | R = − 0.199 |
| Cerebrospinal fluid, n/n (%) | ||||
| Hyperproteinorrachia | 1/21 (5%) | 54/113 (48%) | < 0.001 | POR 0.05 (0.01–0.42) |
| White blood cells > 5 | 1/21 (5%) | 0/113 | a |
Tests reaching statistical significance are presented in bold
SMSS Sensory Modality Sum Score, DTR Deep Tendon Reflexes, SNAP Sensory Nerve Action Potential, IQR interquartile range; for categorial variables, the denominator is mentioned only in case of missing data at a group level, elsewhere assume the group total as denominator
aNot tested due to extreme counts;
bOn Bonferroni post-hoc test P < 0.05 on pairwise comparison between groups
Among frequently reported features, paresthesias (79% vs 59%, POR 2.68 CI 95% 1.11–6.45, P = 0.02) and autonomic disturbances of sort (35% vs 8%, POR 6.73 CI 95% 2.79–16.2, P < 0.001) seemed to be characteristic in RFC1-positive patients, whereas neuropathic pain was observed in similar numbers in both groups (59% vs 53%, POR 1.27 CI 95% 0.61–2.65, P = 0.53). Other distinctive CANVAS features, such as cerebellar signs, vestibulopathy, chronic cough, dysphagia and restless legs syndrome were present only in a small number of cases. On the other hand, foot deformities were frequently observed in both groups (see Table 2). RFC1-positive consistently presented axonal neuropathy on nerve conduction studies (φc = 0.247, P < 0.001) and severely reduced sural nerve SNAP (R = − 0.199, P = 0.003). Cerebrospinal fluid analysis was usually within limits; contrariwise mild hyperproteinorrachia was often observed in non-mutated (5% vs 48%, POR 0.05 CI 95% 0.01–0.42, P < 0.001).
Pathology findings in RFC1-positive patients
Sural nerve morphometry revealed a severe loss of MF (median total surviving fibers absolute count 678.9 IQR 412.9–918.8; proportion of surviving fiber relative to controls 12.8% IQR 7.8–17.3%) with a tendency for relevant although milder involvement of small MF compared to large MF (median relative surviving small MF 10.3% IQR 6.6–18.6%; large MF 5.4% IQR 3.8–8.6%) (Fig. 3). Accordingly, pathology records described an involvement of large, small MF and unmyelinated nerve fibers, respectively, in 100%, 97% and 85% of RFC1-positive patients, and 99%, 81% and 41% in other neuropathies (Table 3). Parcel involvement with substantial sparing of small MF and unmyelinated fibers was thus more frequent in non-mutated patients (group difference in the involvement of unmyelinated fibers POR 8.52 CI 95% 3.17–22.9, P < 0.001; small MF POR 7.74 CI 95% 1.03–58.4, P = 0.02; large MF: not tested due to extreme values). Nerve morphometry did not differ between patients presenting with pain, paresthesias or motor symptoms.
Fig. 3.
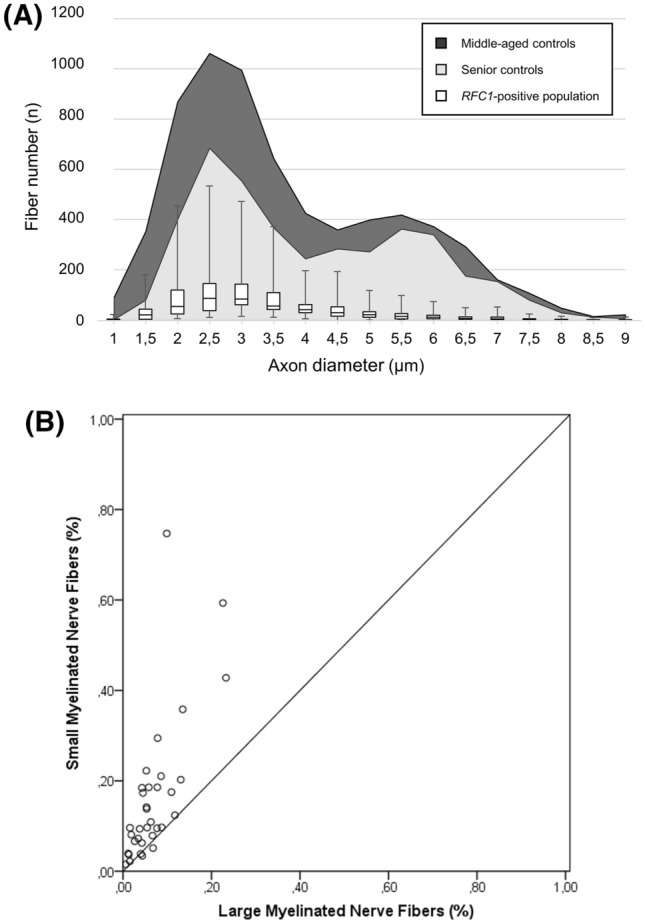
Nerve fiber distribution according to axon diameter. a The bimodal distribution observed in middle-aged (dark shade) and elderly controls (grey shade) is lost in RFC1-positive neuropathy cases (box: first and third quartile; middle bar: median; upper/lower whisker: either 1.5 interquartile range or maximum/minimum observed values); b scatterplot of estimated surviving large and small myelinated fibers: extreme loss of large fibers but variable involvement of small myelinated fibers is observed
Table 3.
Neuropathological features according to pathogenetic RFC1 repeat expansion mutation status
|
RFC1-positive (n = 34) |
Non-mutated (n = 200) | P value | Effect size | |
|---|---|---|---|---|
| Involvement of selected nerve fibers, n (%) | ||||
| Large myelinated nerve fibers | 34 (100%) | 198 (99%) | a | |
| Small myelinated nerve fibers | 33 (97%) | 162 (81%) | 0.02 | POR 7.74 (1.03–58.4) |
| Unmyelinated nerve fibers | 29 (85%) | 81 (41%) | < 0.001 | POR 8.52 (3.17–22.9) |
| Active degeneration, n (%) | ||||
| Absent | 16 (47%) | 83 (42%) | 0.47 | τb = − 0.046 |
| Rare | 17 (50%) | 106 (53%) | ||
| Occasional | 1 (3%) | 9 (5%) | ||
| Diffuse | 0 | 2 (1%) | ||
| Regeneration clusters, n (%) | ||||
| Absent | 17 (50%) | 20 (10%) | < 0.001 | τb = − 0.362 |
| Rare | 16 (47%) | 92 (46%) | ||
| Occasional | 1 (3%) | 64 (32%) | ||
| Diffuse | 0 | 24 (12%) | ||
| Microangiopathy, n (%) | ||||
| Absent | 17 (50%) | 117 (59%) | 0.34 | τb = − 0.060 |
| Occasional | 11 (32%) | 54 (27%) | ||
| Diffuse | 6 (18%) | 28 (14%) | ||
Tests reaching statistical significance are presented in bold
aNot tested due to extreme counts
Surviving total, small and large MF correlated with age at biopsy (ρ = − 0.557 P = 0.001, ρ = − 0.534 P = 0.001, ρ = − 0.634 P < 0.001 for total, small and large MF), amended SMSS (ρ = 0.366 P = 0.03, ρ = 0.338 P = 0.05, ρ = 0.301 P = 0.08) and ataxia severity (ρ = − 0.430 P = 0.01, ρ = − 0.465 P = 0.006, ρ = − 0.357 P = 0.04), but not DTR score. Both active degeneration (median 0.9% IQR 0.2–1.4%) and regeneration clusters (1.0% IQR 0–2.1%) were an uncommon finding, the latter being less represented than in non-mutated neuropathies (τb = − 0.362, P < 0.001). In a single instance, a 68-years-old male with a 36-month history of progressive frank sensory ataxia and moderate sensory impairment (SMSS 32) and past, non-active history of alcohol abuse, we observed frank regenerative changes associated with moderate axonal loss (estimated total MF 2941, surviving large MF 9.9%, small MF 74.7%) (Fig. 4a).
Fig. 4.
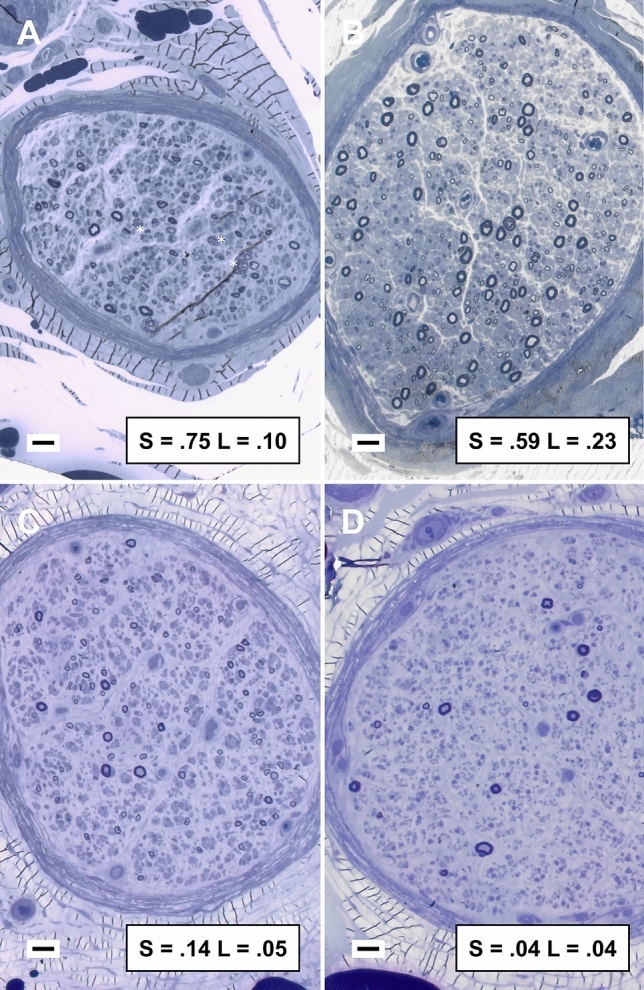
Representative cases from sural nerve pathology studies. Sural nerve biopsy revealed a diffuse depletion of large myelinated fibers that was similar in severity between patients, instead small myelinated fibers appeared relatively preserved in less affected nerves (a–b). Regeneration clusters were observed in a single instance and associated with mild disease (asterisks, a). Active degeneration was uncommon. Toluidine blue-stained semithin cross sections of sural nerve, bar = 20 µm; inbox: fraction of surviving small (S) and large (L) myelinated fibers compared to control average
Discussion
The distinctive biallelic RFC1 intronic mutation and expansion of the pentanucleotide AAGGG associated with CANVAS is a striking finding in this study investigating a cohort of selected CIAP patients. The mutation was identified in 15% of the selected patients’ population; it occurred in the majority (53%) of patients with pure sensory forms and was not uncommon (18%) even in cases with minor motor involvement. The clinical picture of a slowly progressive sensory ataxia associated with a length-dependent neuropathy with hypoesthesia, neuropathic pain and autonomic disturbances observed in our RFC1-positive population shares remarkable similarity to the peripheral involvement described in previous CANVAS series [8, 14], although frequently reported also in other neuropathies. Paradoxical retention of DTR in face of relevant sensory impairment, on the other hand, is a peculiar occurrence observed in our series as well as in other CANVAS cases [8]. Muscle spindle afferents are responsible for DTR and play a major role in proprioception, in concert with cutaneous and Golgi tendon organ afferents [15, 16]. A dissociated involvement of sensory afferents with greater impairment of cutaneous compared to muscle spindle innervation has been previously described [17, 18], and possibly underlies the paradoxical DTR retention in spite of frank rombergism. A similar dissociate impairment of sensory modalities has been proved also for vestibular responses [19].
Cerebellar and vestibular symptoms, the remaining features of the variegate CANVAS clinical phenotype, were otherwise absent at pre-biopsy visit, even though a considerable lapse of time had passed since onset.
In the largest retrospective cohort of RFC1-mutated CANVAS patients described so far, features of sensory neuro(no)pathy were accordantly observed in most patients and often reported since onset, while the involvement of other systems manifested later in the history of the disease or emerged only from extensive instrumental investigations [8]. An archetypic pattern of progression could then explain the lower yield of genetic investigations for RFC1 mutation expansion observed in previous reports on patients with adult-onset ataxia of unknown etiology [20–26] or Multiple System Atrophy [20, 23] compared to ours, even though the differences in study design prevent valid comparisons.
On pathology, we observed a marked and diffuse loss of large MF in all patients and, to a milder degree, of small MF. On the contrary, unmyelinated nerve fibers involvement was less frequently reported on our qualitative ultrastructural studies. Similar findings were reported on qualitative [14] and morphometry analysis [8] in smaller case series. Greater involvement of large MF suits both the clinical presentation as an ataxic neuropathy and the decrease in SNAP amplitude observed even in paucisymptomatic patients. Notably, the normal sural nerve rarely harbors muscle spindle afferents as it commonly lacks muscular branches, so that its biopsy does not provide information on group Ia sensory fibers. Demonstration of survival of these fibers relies on clinical and neurophysiological studies in life, while necropsy examinations will be required for pathological confirmation.
Absence of relevant signs of axonal regeneration points to a neuronopathy as the more probable mechanism of degeneration, in agreement with two histopathological case series reporting neuronal loss in dorsal root ganglia [27] and in vestibular, geniculate and trigeminal ganglia [28], although at odd with the length-dependent characteristic observed.
Based on these marked similarities, we confidently conclude that we identified either an early manifestation of full CANVAS or, less likely, a forme-fruste variant. Follow-up is needed to address the development of the full-blown syndrome along the disease course.
Our findings suggest that, even in the absence of the classic CANVAS triad, several elements should alert the clinician in middle-aged patients with a history of a slowly progressive sensory neuro(no)pathy, in particular: (a) sensory axonal neuropathy on NCS; (b) paradoxical retention of DTR in face of relevant sensory impairment; (c) dramatic involvement of both myelinated and unmyelinated nerve fibers on sural nerve biopsy. On the other hand, lack of a definite family history, the presence of minor motor deficits or foot deformities should not avert from investigating a patient for the RFC1 expansions.
Strengths and limitations
In our setting, heterogeneity in clinical assessment and pathology evaluation of a large cohort of patients has been minimized by means of operator consistency and shared practices. Neurophysiological evaluation, on the other hand, had been assessed by multiple neurophysiologists in different clinics and thus we cannot rule out inconsistencies on NCS conclusions. Although extensive, in this real-world scenario routine neurological examination was still aimed at the referral question and could have led to underrepresentation of ancillary clinical features. Advanced bedside clinical maneuvers to test the vestibular system were not customary and, similarly, assessment of autonomic disturbances was often limited to an evaluation by interview, leading to the identification of patients with remarkable symptoms only. Moreover, inquiry for chronic cough became relevant just recently after being described as a defining symptom in CANVAS. The authors acknowledge that implementation of further testing could possibly lead the wary clinician to identify preclinical involvement in part of these patients. Regarding our study, blinding during data extraction still secured an unbiased distribution of these characteristics between RFC1-positive and non-mutated participants.
The addition of pathological criteria to routine examinations in selecting CIAP patients may represent a double-edged sword: on the one hand, it may have ruled out other confounding acquired or genetic causes of neuropathy; on the other, it may have introduced a bias in the selection of the study population. A sural nerve biopsy is more likely offered to patients with a suspected treatable cause, absence of an overt family history, younger onset, more severe or rapidly progressive course; however, the relevance of this potential bias is probably limited because those features do not characterize the RFC1-positive patients depicted by previous works [8].
Clinical and/or neurophysiological involvement of the sural nerve is required for the bioptic procedure and could have biased the phenotypical characterization due to an enrichment in patients with distal involvement. However, we note that previous works on full CANVAS also showed a similar presence of length-dependent neuro(no)pathy [14].
Conclusions
The biallelic RFC1 AAGGG repeat expansion is a frequent cause of adult-onset, slowly progressive, pure or prevalently sensory axonal neuro(no)pathies of unknown cause. Future natural history studies will clarify whether the chronic axonal neuro(no)pathy necessarily heralds the development of a full-blown CANVAS or if it may remain for a long time as the dominant phenotype associated to the RFC1 AAGGG repeat expansion.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Online Resource 1. Schematic representation of RFC1 intron 2 (DOCX 36 KB)
Supplementary Online Resource 2. Flanking standard PCR (DOCX 55 KB)
Supplementary Online Resource 3. Primer sequences used in RFC1 gene analysis (DOCX 19 KB)
Supplementary Online Resource 4. Repeat primed PCR (DOCX 180 KB)
Supplementary Online Resource 5. Genes included in the Next Generation Sequencing panel (DOCX 20 KB)
Acknowledgements
The authors wish to thank Dr. Andrea Garbo (German Aerospace Center, Brunswick, Germany) for its valuable assistance in mathematical modeling.
Author contributions
All authors contributed to the study conception and design. Clinical evaluation was performed by SF and TC. Pathology evaluation was performed by TC and morphometry by MT. Genetic analysis was performed by MF and ST. Data collection and analysis were performed by MT and DC. The first draft of the manuscript was written by MT, DC, SM and GMF and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Funding
Open access funding provided by Università degli Studi di Verona within the CRUI-CARE Agreement. This work was partially sponsored by Akcea (GMF). The funder did not take any part in the definition of the study model, analysis, writing or in the decision to publish.
Data availability
Anonymized data will be shared on request from qualified investigators.
Declarations
Conflicts of interest
M. Tagliapietra reports a training grant from Pfizer and received support for attending scientific meetings from Alnylam. S. Ferrari received support for attending scientific meetings from Sanofi-Genzyme, Merck and Euroimmun. G.M. Fabrizi served on Advisory Boards for Alnylam, Akcea and Pharnext-Vitaccess, received speaker fees from Akcea and support for attending scientific meetings by Pfizer and Akcea. D. Cardellini, M. Ferrarini, S. Testi, S. Monaco and T. Cavallaro report no disclosures.
Ethical approvals
This retrospective study involving human participants was in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by the local Bioethics Committee (Comitato Etico per la Sperimentazione Clinica, Azienda Ospedaliera Universitaria Integrata di Verona) (BIOB-NEU-DNA-2014, protocol 13582).
Informed consent
Written informed consent for future research was obtained from all participants at the time of biopsy.
References
- 1.Hanewinckel R, Drenthen J, van Oijen M, Hofman A, van Doorn PA, Ikram MA. Prevalence of polyneuropathy in the general middle-aged and elderly population. Neurology. 2016;87:1892–1898. doi: 10.1212/WNL.0000000000003293. [DOI] [PubMed] [Google Scholar]
- 2.Zis P, Sarrigiannis PG, Rao DG, Hewamadduma C, Hadjivassiliou M. Chronic Idiopathic Axonal Polyneuropathy: a systematic review. J Neurol. 2016;263:1903–1910. doi: 10.1007/s00415-016-8082-7. [DOI] [PubMed] [Google Scholar]
- 3.Visser NA, Vrancken AFJE, van der Schouw YT, van den Berg LH, Notermans NC. Chronic Idiopathic Axonal Polyneuropathy is associated with the metabolic syndrome. Diabetes Care. 2013;36:817–822. doi: 10.2337/dc12-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Senderek J, Lassuthova P, Kabzińska D, Abreu L, Baets J, Beetz C, Braathen GJ, Brenner D, Dalton J, Dankwa L, Deconinck T, De Jonghe P, Dräger B, Eggermann K, Ellis M, Fischer C, Stojkovic T, Herrmann DN, Horvath R, Høyer H, Iglseder S, Kennerson M, Kinslechner K, Kohler JN, Kurth I, Laing NG, Lamont PJ, Löscher W, Ludolph A, Marques W, Nicholson G, Ong R, Petri S, Ravenscroft G, Rebelo A, Ricci G, Rudnik-Schöneborn S, Schirmacher A, Schlotter-Weigel B, Schoels L, Schüle R, Synofzik M, Francou B, Strom TM, Wagner J, Walk D, Wanschitz J, Weinmann D, Weishaupt J, Wiessner M, Windhager R, Young P, Züchner S, Toegel S, Seeman P, Kochański A, Auer-Grumbach M. The genetic landscape of axonal neuropathies in the middle-aged and elderly: focus on MME. Neurology. 2020;95:e3163–e3179. doi: 10.1212/WNL.0000000000011132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cortese A, Simone R, Sullivan R, Vandrovcova J, Tariq H, Yau WY, Humphrey J, Jaunmuktane Z, Sivakumar P, Polke J, Ilyas M, Tribollet E, Tomaselli PJ, Devigili G, Callegari I, Versino M, Salpietro V, Efthymiou S, Kaski D, Wood NW, Andrade NS, Buglo E, Rebelo A, Rossor AM, Bronstein A, Fratta P, Marques WJ, Züchner S, Reilly MM, Houlden H. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019;51:649–658. doi: 10.1038/s41588-019-0372-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Migliaccio AA, Halmagyi GM, McGarvie LA, Cremer PD. Cerebellar ataxia with bilateral vestibulopathy: description of a syndrome and its characteristic clinical sign. Brain. 2004;127:280–293. doi: 10.1093/brain/awh030. [DOI] [PubMed] [Google Scholar]
- 7.Szmulewicz DJ, Waterston JA, MacDougall HG, Mossman S, Chancellor AM, McLean CA, Merchant S, Patrikios P, Halmagyi GM, Storey E. Cerebellar Ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci. 2011;1233:139–147. doi: 10.1111/j.1749-6632.2011.06158.x. [DOI] [PubMed] [Google Scholar]
- 8.Cortese A, Tozza S, Yau WY, Rossi S, Beecroft SJ, Jaunmuktane Z, Dyer Z, Ravenscroft G, Lamont PJ, Mossman S, Chancellor A, Maisonobe T, Pereon Y, Cauquil C, Colnaghi S, Mallucci G, Curro R, Tomaselli PJ, Thomas-Black G, Sullivan R, Efthymiou S, Rossor AM, Laurá M, Pipis M, Horga A, Polke J, Kaski D, Horvath R, Chinnery PF, Marques W, Tassorelli C, Devigili G, Leonardis L, Wood NW, Bronstein A, Giunti P, Züchner S, Stojkovic T, Laing N, Roxburgh RH, Houlden H, Reilly MM. Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome due to RFC1 repeat expansion. Brain. 2020;143:480–490. doi: 10.1093/brain/awz418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.England JD, Gronseth GS, Franklin G, Carter GT, Kinsella LJ, Cohen JA, Asbury AK, Szigeti K, Lupski JR, Latov N, Lewis RA, Low PA, Fisher MA, Herrmann DN, Howard JF, Lauria G, Miller RG, Polydefkis M, Sumner AJ. Practice parameter: evaluation of distal symmetric polyneuropathy: role of laboratory and genetic testing (an evidence-based review). Report of the American Academy of Neurology, American Association of Neuromuscular and Electrodiagnostic Medicine, and American Academy of Physical Medicine and Rehabilitation. Neurology. 2009;72:185–192. doi: 10.1212/01.wnl.0000336370.51010.a1. [DOI] [PubMed] [Google Scholar]
- 10.Teunissen L. Differences between hereditary motor and sensory neuropathy type 2 and chronic Idiopathic Axonal Neuropathy. A clinical and electrophysiological study. Brain. 1997;120:955–962. doi: 10.1093/brain/120.6.955. [DOI] [PubMed] [Google Scholar]
- 11.Erdmann PG, Teunissen LL, van Genderen FR, Notermans NC, Lindeman E, Helders PJM, van Meeteren NLU. Functioning of patients with Chronic Idiopathic Axonal Polyneuropathy (CIAP) J Neurol. 2007;254:1204–1211. doi: 10.1007/s00415-006-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schellens RLLA, van Veen BK, Gabreëls-Festen AAWM, Notermans SLH, Hof MA, van’t, Stegeman DF, A statistical approach to fiber diameter distribution in human sural nerve. Muscle Nerve. 1993;16:1342–1350. doi: 10.1002/mus.880161212. [DOI] [PubMed] [Google Scholar]
- 13.Jacobs JM, Love S. Qualitative and quantitative morphology of human sural nerve at different ages. Brain. 1985;108:897–924. doi: 10.1093/brain/108.4.897. [DOI] [PubMed] [Google Scholar]
- 14.Szmulewicz DJ, Waterston JA, Halmagyi GM, Mossman S, Chancellor AM, McLean CA, Storey E. Sensory neuropathy as part of the Cerebellar Ataxia Neuropathy Vestibular Areflexia Syndrome. Neurology. 2011;76:1903–1910. doi: 10.1212/WNL.0b013e31821d746e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proske U, Gandevia SC. The proprioceptive senses: their roles in signaling body shape, body position and movement, and muscle force. Physiol Rev. 2012;92:1651–1697. doi: 10.1152/physrev.00048.2011. [DOI] [PubMed] [Google Scholar]
- 16.Bosco G, Poppele RE. Proprioception from a spinocerebellar perspective. Physiol Rev. 2001;81:539–568. doi: 10.1152/physrev.2001.81.2.539. [DOI] [PubMed] [Google Scholar]
- 17.Burke D, Halmagyi GM. Normal tendon reflexes despite absent sensory nerve action potentials in CANVAS: a neurophysiological study. J Neurol Sci. 2018;387:75–79. doi: 10.1016/j.jns.2018.01.023. [DOI] [PubMed] [Google Scholar]
- 18.Infante J, García A, Serrano-Cárdenas KM, González-Aguado R, Gazulla J, de Lucas EM, Berciano J. Cerebellar Ataxia, Neuropathy, Vestibular Areflexia Syndrome (CANVAS) with chronic cough and preserved muscle stretch reflexes: evidence for selective sparing of afferent Ia fibres. J Neurol. 2018;265:1454–1462. doi: 10.1007/s00415-018-8872-1. [DOI] [PubMed] [Google Scholar]
- 19.Yacovino DA, Zanotti E, Hain TC. Is Cerebellar Ataxia, Neuropathy, and Vestibular Areflexia Syndrome (CANVAS) a vestibular ganglionopathy? J Int Adv Otol. 2019;15:304–308. doi: 10.5152/iao.2019.7068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan L, Chen Z, Wan N, Liu M, Xue J, Chen H, Zhang Y, Peng Y, Tang Z, Gong Y, Yuan H, Wang S, Deng Q, Hou X, Wang C, Peng H, Shi Y, Peng L, Lei L, Duan R, Xia K, Qiu R, Shen L, Tang B, Ashizawa T, Jiang H. Biallelic intronic AAGGG expansion of RFC1 is related to multiple system atrophy. Ann Neurol. 2020;88:1132–1143. doi: 10.1002/ana.25902. [DOI] [PubMed] [Google Scholar]
- 21.Tsuchiya M, Nan H, Koh K, Ichinose Y, Gao L, Shimozono K, Hata T, Kim Y-J, Ohtsuka T, Cortese A, Takiyama Y. RFC1 repeat expansion in Japanese patients with late-onset cerebellar ataxia. J Hum Genet. 2020;65:1143–1147. doi: 10.1038/s10038-020-0807-x. [DOI] [PubMed] [Google Scholar]
- 22.Gisatulin M, Dobricic V, Zühlke C, Hellenbroich Y, Tadic V, Münchau A, Isenhardt K, Bürk K, Bahlo M, Lockhart PJ, Lohmann K, Helmchen C, Brüggemann N. Clinical spectrum of the pentanucleotide repeat expansion in the RFC1 gene in ataxia syndromes. Neurology. 2020;95:e2912–e2923. doi: 10.1212/WNL.0000000000010744. [DOI] [PubMed] [Google Scholar]
- 23.Fan Y, Zhang S, Yang J, Mao C, Yang Z, Hu Z, Wang Y, Liu Y, Liu H, Yuan Y, Shi C, Xu Y. No biallelic intronic AAGGG repeat expansion in RFC1 was found in patients with late-onset ataxia and MSA. Parkinsonism Relat Disord. 2020;73:1–2. doi: 10.1016/j.parkreldis.2020.02.017. [DOI] [PubMed] [Google Scholar]
- 24.Daele SHV, Vermeer S, Eesbeeck AV, Lannoo L, Race V, Pvan D, Claeys K, Vandenberghe W. Diagnostic yield of testing for RFC1 repeat expansions in patients with unexplained adult-onset cerebellar ataxia. J Neurol Neurosurg Psychiatry. 2020;91:1233–1234. doi: 10.1136/jnnp-2020-323998. [DOI] [PubMed] [Google Scholar]
- 25.Aboud Syriani D, Wong D, Andani S, De Gusmao CM, Mao Y, Sanyoura M, Glotzer G, Lockhart PJ, Hassin-Baer S, Khurana V, Gomez CM, Perlman S, Das S, Fogel BL. Prevalence of RFC1-mediated spinocerebellar ataxia in a North American ataxia cohort. Neurol Genet. 2020;6:e440. doi: 10.1212/NXG.0000000000000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montaut S, Diedhiou N, Fahrer P, Marelli C, Lhermitte B, Robelin L, Vincent MC, Corti L, Taieb G, Gebus O, Rudolf G, Tarabeux J, Dondaine N, Canuet M, Almeras M, Benkirane M, Larrieu L, Chanson J-B, Nadaj-Pakleza A, Echaniz-Laguna A, Cauquil C, Lannes B, Chelly J, Anheim M, Puccio H, Tranchant C. Biallelic RFC1-expansion in a French multicentric sporadic ataxia cohort. J Neurol. 2021 doi: 10.1007/s00415-021-10499-5. [DOI] [PubMed] [Google Scholar]
- 27.Szmulewicz DJ, McLean CA, Rodriguez ML, Chancellor AM, Mossman S, Lamont D, Roberts L, Storey E, Halmagyi GM. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology. 2014;82:1410–1415. doi: 10.1212/WNL.0000000000000352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szmulewicz DJ, Merchant SN, Halmagyi GM. Cerebellar Ataxia with Neuropathy and bilateral Vestibular Areflexia Syndrome: a histopathologic case report. Otol Neurotol. 2011;32:e63–65. doi: 10.1097/MAO.0b013e318210b719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Online Resource 1. Schematic representation of RFC1 intron 2 (DOCX 36 KB)
Supplementary Online Resource 2. Flanking standard PCR (DOCX 55 KB)
Supplementary Online Resource 3. Primer sequences used in RFC1 gene analysis (DOCX 19 KB)
Supplementary Online Resource 4. Repeat primed PCR (DOCX 180 KB)
Supplementary Online Resource 5. Genes included in the Next Generation Sequencing panel (DOCX 20 KB)
Data Availability Statement
Anonymized data will be shared on request from qualified investigators.