

Abstract
Cocaine use disorder (CUD) is a major public health issue associated with physical, social, and psychological problems. Excessive and repeated cocaine use induces oxidative stress leading to a systemic inflammatory response. Cannabidiol (CBD) has gained substantial interest for its anti-inflammatory properties, safety, and tolerability profile. However, CBD anti-inflammatory properties have yet to be confirmed in humans. This exploratory study is based on a single-site randomized controlled trial that enrolled participants with CUD between 18 and 65 years, randomized (1:1) to daily receive either CBD (800 mg) or placebo for 92 days. The trial was divided into a 10-day detoxification (phase I) followed by a 12-week outpatient follow-up (phase II). Blood samples were collected from 48 participants at baseline, day 8, week 4, and week 12 and were analyzed to determine monocytes and lymphocytes phenotypes, and concentrations of various inflammatory markers such as cytokines. We used generalized estimating equations to detect group differences. Participants treated with CBD had lower levels of interleukin-6 (p = 0.017), vascular endothelial growth factor (p = 0.032), intermediate monocytes CD14+CD16+ (p = 0.024), and natural killer CD56negCD16hi (p = 0.000) compared with participants receiving placebo. CD25+CD4+T cells were higher in the CBD group (p = 0.007). No significant group difference was observed for B lymphocytes. This study suggests that CBD may exert anti-inflammatory effects in individuals with CUD.
Subject terms: Predictive markers, Addiction, Drug development
Introduction
Cocaine has been used in the past year by more than 18 million people worldwide [1] and will trigger a cocaine use disorder (CUD) in 16% of users [2]. Excessive and repeated cocaine consumption is associated with psychological, social, and physiological problems and represents a major public health issue [3]. Chronic cocaine abuse and withdrawal lead to impaired immune system, with increased pro-inflammatory cytokines and lymphocytes [4–8]. These changes may be explained by the overdrive of the hypothalamic–pituitary–adrenal axis [9, 10] and the sympathetic nervous system [11, 12], generating oxidative stress over time and promoting a systemic inflammatory response [13–17]. Cocaine also alters tight junction proteins and cytoskeleton of the blood–brain barrier (BBB), which increases BBB permeability [18, 19]. This increased BBB permeability allows inflammatory cells to migrate across the BBB leading to neuroinflammation [20–22]. In addition, CUD is associated with serious comorbidities such as human immunodeficiency virus and hepatitis C virus infections, skin and soft tissue infections, cardiovascular disease, depression, and psychosis [3]. These comorbidities have an important inflammatory component [23, 24] although their causal link with CUD inflammatory process has not been established.
Cannabidiol (CBD), one of cannabis’s major cannabinoids, has gained substantial interest for its anti-inflammatory, anti-oxidative, and neuroprotective properties in preclinical studies [25–27] in addition to its favorable safety and tolerability profile [28–30]. Anti-inflammatory actions of CBD have been relatively well investigated in animals and in vitro. Cumulating preclinical evidence highlights that CBD decreases cytokines, monocytes, and lymphocytes by several hypothetical mechanisms such as cannabinoid receptor type 2 activation, peroxisome proliferator-activated receptor γ activation, and anandamide upregulation following fatty acid amide hydrolase inhibition [25, 27, 31]. Cytokines are a broad family of immune signaling molecules including interleukins (IL) and chemokines. Monocytes are precursors of antigen-presenting cells acting as messengers between the innate and adaptive immune system. Lymphocytes T and B are the principal components of the adaptative immune system while natural killer (NK) cells have an innate cytotoxic activity. All these immune molecules are key targets in the inflammatory process.
However, CBD anti-inflammatory properties have yet to be confirmed in humans. To our knowledge, only one clinical study in patients with type 2 diabetes assessed CBD effects on only three inflammatory markers: IL-6, C-reactive protein (CRP), and tumor necrosis factor-α (TNF-α) [32]. The authors found no significant difference between CBD and placebo groups on these markers with a 100 mg CBD dose twice daily for 13 weeks. No study has yet evaluated CBD anti-inflammatory action at higher dosage and in individuals with addiction, including CUD. Consequently, it remains unclear if and how CBD modulates inflammatory markers in individuals with CUD.
This study is part of a larger randomized controlled trial (RCT) assessing CBD effects on cocaine craving and relapse in individuals with CUD [33]. Our exploratory aim was to examine the association between CBD and inflammatory markers in individuals with CUD. In accordance with preclinical findings, we hypothesized that treatment with daily oral CBD (800 mg) would modulate inflammation by decreasing pro-inflammatory markers and increasing anti-inflammatory markers compared with placebo in individuals with CUD.
Patients and methods
Trial design
The single-site RCT was conducted at Centre Hospitalier de l’Université de Montréal (CHUM) in Montreal, Quebec, Canada, and is described in more details elsewhere [33]. The trial was approved by the CHUM research ethics committee and followed relevant ethical guidelines (Helsinki Declaration, International Standards of Good Clinical Practice, Tri-Council Policy Statement, Health Canada division 5 guidelines). All participants gave their written informed consent prior to enrollment. The trial was divided into two phases. Phase I consisted in an inpatient detoxification period lasting 10 days. Phase II involved an outpatient follow-up period lasting 12 weeks. Participants received up to $400 for their time.
Participants
Key inclusion criteria were adults aged between 18 and 65 years diagnosed with current CUD (Structured Clinical Interview [SCID] for DSM-V) who had consumed cocaine within 2 weeks prior to admission. Participants needed to speak English or French and be able to give a valid informed consent. Key exclusion criteria were severe and/or unstable medical or psychiatric conditions, immunodeficiency, other substance use disorder (except nicotine) that would require treatment during the study, hypersensitivity to cannabinoids, and use of medication that could interact with CBD. Participants needed to successfully complete phase I to become eligible for phase II.
Randomization and blinding
An independent biostatistician created the computer-generated randomization sequence to assign participants to CBD or placebo group in a 1:1 ratio. The stratification variables were sex [34] and baseline severity of cocaine dependence (< or ≥10) assessed by the Severity of Dependence Scale (SDS) [35]. Participants and research staff were blinded to treatment allocation. Blinding strategies are explained elsewhere [33].
Sample size
The sample size for the RCT was calculated based on the primary outcomes as reported elsewhere [33]. Full biobanking was done only for the first 48 participants enrolled before September 27, 2018 (CBD, n = 24; placebo, n = 24) due to funding restrictions.
Interventions
Participants were admitted at the CHUM for a 10-day detoxification (phase I). They received at 10 a.m. either CBD (400 mg/day) or placebo (Insys Therapeutics) on days 2 and 3. This dose was increased to 800 mg/day from day 4 until the end of the trial for all participants except one who experienced intolerable side effects. During phase II (weeks 1–12), participants were weekly given treatment bottles at each visit. The dosage selection was based on available safety and relevant clinical data for relapse and craving [36].
Data collection
Blood samples were collected at 9 a.m. at the CHUM from all participants on day 2 (baseline), day 8, week 4, and week 12 in ethylenediaminetetraacetic vacutainer tubes. Plasma was obtained by centrifugation (1500 g, 15 min) and stored at −80 °C. Peripheral blood mononuclear cells (PBMC) were isolated by density gradient centrifugation. Whole blood, diluted ½ with phosphate buffer saline (PBS), was layered over lymphocyte separation medium (Wisent) in a falcon tube and centrifuged 30 min at 300 g without brakes. PBMC were gently removed at the interface and washed twice with a large volume of PBS. The pelleted cells were counted, and the percentage of viability was estimated using trypan blue staining. PBMC were cryopreserved in liquid nitrogen until use.
Inflammatory markers selection
Due to funding restrictions, we did a discovery pre-analysis with the first 12 completed participants to select markers showing a differential expression between groups (p < 0.1). The inflammatory multiplex V-PLEX Human Biomarker 40-Plex Kit and six panels were used for the discovery phase (corresponding author can be contacted for more details). Analyses of markers showing a differential expression between groups were extended to all 48 participants and are listed in Tables S1 and S2. Inflammatory markers were analyzed using the following methods.
Flow cytometry
PBMCs were thawed and first stained with the Live/Dead Fixable cell Kit (Invitrogen). Then, nonspecific binding sites were blocked with human gamma globulin (Jackson Immunoresearch). Cells surface staining was performed using fluorescent dye-conjugated monoclonal antibodies (BD Biosciences, listed in Table S1) for 30 min at 4 °C in the dark. Cells were then washed with staining buffer (PBS-5% Bovine serum albumin-0.1% NaNO3) and resuspended in staining buffer. Flow cytometry data were acquired using a BDFortessa instrument (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Cytokines measurements
Peripheral soluble cytokines and chemokines were measured in cryopreserved plasma from participants using the V-plex Ultra-sensitive kit (Meso Scale Discovery). Table S2 lists all analytes. The kit was processed as per manufacturer’s instructions. Electroluminescent data were analyzed with a four-parameter logistic curve fit using MSD Discovery Workbench.
Outcomes
The concentrations of cytokines and the percentages of monocytes and lymphocytes were measured in each treatment group at baseline (day 2), day 8, week 4, and week 12. We also measured CBD blood concentration on day 8, week 4, and week 12. Anti-inflammatory drugs used during the trial were also recorded.
Statistical analyses
Between-group comparisons were performed using generalized estimating equations (GEE) with SPSS 26 software. Sex, baseline SDS score, and age were included as covariates. Considering our small sample size, we adjusted for levels of inflammatory markers at baseline (day 2) to account for possible imbalances between groups. Only p values < 0.05 were considered statistically significant. Quantitative data were normally distributed (D’Agostino–Pearson normality test). A posteriori sensitivity analyses were realized to assess the effect of anti-inflammatory drug use. First, we repeated the GEE for the five main significant inflammatory markers excluding all participants taking anti-inflammatory drugs at least once during the trial. Second, anti-inflammatory drug use was added as a binary covariate in our model. To verify if our fixed CBD dose may have affected CBD effect on inflammation, we performed a posteriori Pearson correlations between the five main significant inflammatory markers’ level variation from baseline and weight or CBD plasma concentration. Finally, while we did not find differences in craving between CBD and placebo groups as previously reported [33], hypothetical mechanisms suggest that neuroinflammation can lead to modulation of the drug-reward system and possibly of addictive behaviors such as craving [37]. We thus conducted a posteriori linear regressions to determine if the five main significant inflammatory markers’ level influenced drug-cue-induced craving by treatment group.
Results
Participant flow and numbers analyzed
Recruitment took place between July 20, 2016 and June 25, 2019. Biobanking stopped on September 27, 2018, due to funding restrictions. The first 48 (CBD, 24/40, 60.0%; placebo, 24/38, 63.2%) participants randomized before this date provided blood samples and were included in the analyses. Figure 1 presents participant’s flowchart.
Fig. 1. Consolidated Standards of Reporting Trials (CONSORT) flowchart of participants (adapted with permission from Mongeau-Pérusse et al. [33]).

*Other reasons for exclusion were male fertility issues (n = 2), immunosuppression (n = 2), and not currently moderate or severe CUD according to the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition criteria (n = 1). **Lost consent form and refusal to reconsent ended participation. The data safety and monitoring board requested that no data from this participant be used in the study. CBD cannabidiol, D day, n number of participants, W week.
Demographics and clinical characteristics
Table 1 presents participants’ demographics and baseline characteristics. Most participants were men (n = 39/48, 81.2%) with a mean (standard deviation, SD) age of 46.0 (11.0) years old. The mean body mass index was 25.7 (4.6) kg/m2. Most participants (n = 45/48, 93.8%) had a severe CUD based on the SCID. Last cocaine use was on average (SD) 2.9 (3.1) days before study initiation. Furthermore, 14/48 (29.2%) participants also had another substance use disorder. All demographics and baseline characteristics were balanced between groups.
Table 1.
Demographic and baseline characteristics of participants.
| Characteristics | Treatment group | Total (n = 48) | |
|---|---|---|---|
| CBD (n = 24) | Placebo (n = 24) | ||
| Age, mean (SD), y | 46.6 (10.1) | 45.4 (12.0) | 46.0 (11.0) |
| Female sex, n (%) | 4 (16.7) | 5 (20.8) | 9 (18.8) |
| Weight, mean (SD), kg | 74.9 (11.8) | 78.7 (20.4) | 76.8 (16.6) |
| Body mass index, mean (SD), kg/m2 | 25.0 (3.3) | 26.3 (5.6) | 25.7 (4.6) |
| Time between study initiation and last cocaine use, mean (SD), days | 2.1 (1.5) | 3.8 (3.9) | 2.9 (3.1) |
| Frequency of cocaine use 2 weeks before study initiation, mean (SD), days | 7.5 (4.5) | 6.0 (3.9) | 6.8 (4.2) |
| SDS total score, mean (SD) | 11.1 (2.3) | 11.9 (2.7) | 11.5 (2.5) |
| SDS group, n (%) | |||
| Low (SDS < 10) | 6 (25.0) | 5 (20.8) | 11 (22.9) |
| High (SDS ≥ 10) | 18 (75.0) | 19 (79.2) | 37 (77.1) |
| Severity of Cocaine Use Disorder based on the SCID, n (%) | |||
| Severe | 22 (91.7) | 23 (95.8) | 45 (93.8) |
| Moderate | 2 (8.3) | 1 (4.2) | 3 (6.3) |
| Preferred route of cocaine administration, n (%) | |||
| Nasal | 5 (20.8) | 10 (41.7) | 15 (31.3) |
| Smoking | 15 (62.5) | 13 (54.2) | 28 (58.3) |
| Non-intravenous injection | 1 (4.2) | 0 (0.0) | 1 (2.1) |
| Intravenous | 3 (12.5) | 1 (4.2) | 4 (8.3) |
| Highest level of schooling completed, n (%) | |||
| Less than high school | 8 (33.3) | 6 (25.0) | 14 (29.2) |
| High school | 7 (29.2) | 10 (41.7) | 17 (35.4) |
| More than high school | 9 (37.5) | 8 (33.3) | 17 (35.4) |
| Ethnicity, n (%) | |||
| White | 20 (83.3) | 21 (87.5) | 41 (85.4) |
| Other | 4 (16.7) | 3 (12.5) | 7 (14.6) |
| Employment status, n (%) | |||
| Full time | 7 (29.2) | 11 (45.8) | 18 (37.5) |
| Part time | 5 (20.8) | 6 (25.0) | 11 (22.9) |
| Disability or employment insurance | 2 (8.3) | 4 (16.7) | 6 (12.5) |
| Social welfare | 7 (29.2) | 2 (8.3) | 9 (18.8) |
| Unstable condition | 3 (12.5) | 1 (4.2) | 4 (8.3) |
| Marital status, n (%) | |||
| Married or common-law couple | 0 (0.0) | 2 (8.3) | 2 (4.2) |
| Single | 24 (100.0) | 22 (91.7) | 46 (95.8) |
| Housing status, n (%) | |||
| Stable housing | 21 (87.5) | 22 (91.7) | 43 (89.6) |
| Homeless | 3 (12.5) | 2 (8.3) | 5 (10.4) |
| Current substance use disorder, n (%) | 8 (33.3) | 6 (25.0) | 14 (29.2) |
| Cannabis | 3 (12.5) | 3 (12.5) | 6 (12.5) |
| Alcohol | 4 (16.7) | 3 (12.5) | 7 (14.6) |
| Stimulant | 1 (4.2) | 0 (0.0) | 1 (2.1) |
| Other | 2 (8.3) | 0 (0.0) | 2 (4.2) |
| Current and past medical conditions, n (%) | |||
| Cardiovascular | 4 (16.7) | 5 (20.8) | 9 (18.8) |
| Respiratory | 6 (25.0) | 6 (25.0) | 12 (25.0) |
| Hepatobiliary | 6 (25.0) | 5 (20.8) | 11 (22.9) |
| Gastroentorological | 2 (8.3) | 6 (25.0) | 8 (16.7) |
| Genital | 4 (16.7) | 2 (8.3) | 6 (12.5) |
| Hematological | 0 (0.0) | 1 (4.2) | 1 (2.1) |
| Musculoskeletal | 7 (29.2) | 4 (16.7) | 11 (22.9) |
| Neurological | 7 (29.2) | 4 (16.7) | 11 (22.9) |
| Endocrine | 0 (0.0) | 3 (12.5) | 3 (6.3) |
| Immunological | 0 (0.0) | 1 (4.2) | 1 (2.1) |
| Dermatological | 5 (2.8) | 2 (8.3) | 7 (14.6) |
| Allergy | 6 (25.0) | 1 (4.2) | 7 (14.6) |
| Otorhinolaryngology | 6 (25.0) | 4 (16.7) | 10 (20.8) |
| Neoplasia | 0 (0.0) | 1 (4.2) | 1 (2.1) |
| Other | 6 (25.0) | 3 (12.5) | 9 (18.8) |
Modified with permission from Mongeau-Pérusse et al. [33].
CBD cannabidiol, n number of participants, SCID structured clinical interview for the Diagnostic and Statistical Manual of Mental Disorders 5th edition (DSM-V), SD standard deviation, SDS Severity of Dependence Scale.
Cytokine profiles
Figure 2 presents the cytokine profiles in each treatment group. Both IL-6 (estimate (B) = −0.392, Wald = 5.674, p = 0.017, effect size (ES) = 0.347) and vascular endothelial growth factor-A (VEGF-A) (B = −14.417, Wald = 4.600, p = 0.032, ES = 0.313) levels were significantly lower in participants receiving CBD compared with those receiving placebo. No statistically significant group difference was observed for IL-16, macrophage inflammatory protein-1β nor for macrophage-derived chemokine (MDC). Extensive cytokines results are presented in Table S3.
Fig. 2. Cytokines profile of the cannabidiol group compared with the placebo group at baseline (day 2), day 8, week 4, and week 12.

Individual concentrations of interleukin-6 (IL-6), interleurkin-16 (IL-16), macrophage inflammatory protein-1β (MIP-1β), macrophage-derived chemokine (MDC), and vascular endothelial growth factor-A (VEGF-A) are presented in column scatter plots. The mean and standard deviation (SD) are expressed with horizontal and vertical bars, respectively. *Indicates p value < 0.05. D day, W week.
Monocyte profiles
Figure 3 illustrates the monocyte profiles in each treatment group. Intermediate monocytes (CD14+CD16+) were lower in the CBD group compared with the placebo group (B = −0.468, Wald = 5.082, p = 0.024, ES = 0.330). No statistically significant group difference was observed for total CD11c+ monocytes, classical (CD14+CD16−), and nonclassical (CD14loCD16+) monocytes, dendritic cells, and myeloid-derived suppressor cells. Extensive monocytes results are presented in Table S4.
Fig. 3. Monocytes profile of the cannabidiol group compared with the placebo group at baseline (day 2), day 8, week 4, and week 12.

Individual percentages of total CD11c+ monocytes, classical monocytes (CD14+CD16−), intermediate monocytes (CD14+CD16+), nonclassical monocytes (CD14loCD16+), dendritic cells (DC), myeloid dendritic cells (mDC), monocytic myeloid-derived suppressor cells (mMDSC), granulocytic myeloid-derived suppressor cells (gMDSC) are presented in column scatter plots. The mean and standard deviation (SD) are expressed with horizontal and vertical bars, respectively. *Indicates p value < 0.05. CD cluster of differentiation, D day, W week.
Lymphocyte profiles
Figure 4 shows lymphocytes profile in each treatment group. Subpopulations of CD4+T and CD8+T are presented in the Figs. S1 and S2, respectively. Total T cells, CD4+T, and CD8+T did not vary between groups. However, subpopulation of CD25+ CD4+T was significantly higher in the CBD group compared with the placebo group (B = 0.689, Wald = 7.220, p = 0.007, ES = 0.393). This increase was caused by higher levels of CD25+ CD4+Tem (B = 0.621, Wald = 6.026, p = 0.014, ES = 0.358), CD25+ CD4+Temra (B = 0.586, Wald = 6.740, p = 0.009, ES = 0.378) and CD25+ CD4+Tn (B = 0.511, Wald = 5.419, p = 0.020, ES = 0.340). Total B cells, early, and late B cells did not differ between groups. Extensive lymphocyte results are presented in Tables S5–S7.
Fig. 4. Lymphocytes profile of the cannabidiol group compared with the placebo group at baseline (day 2), day 8, week 4, and week 12.
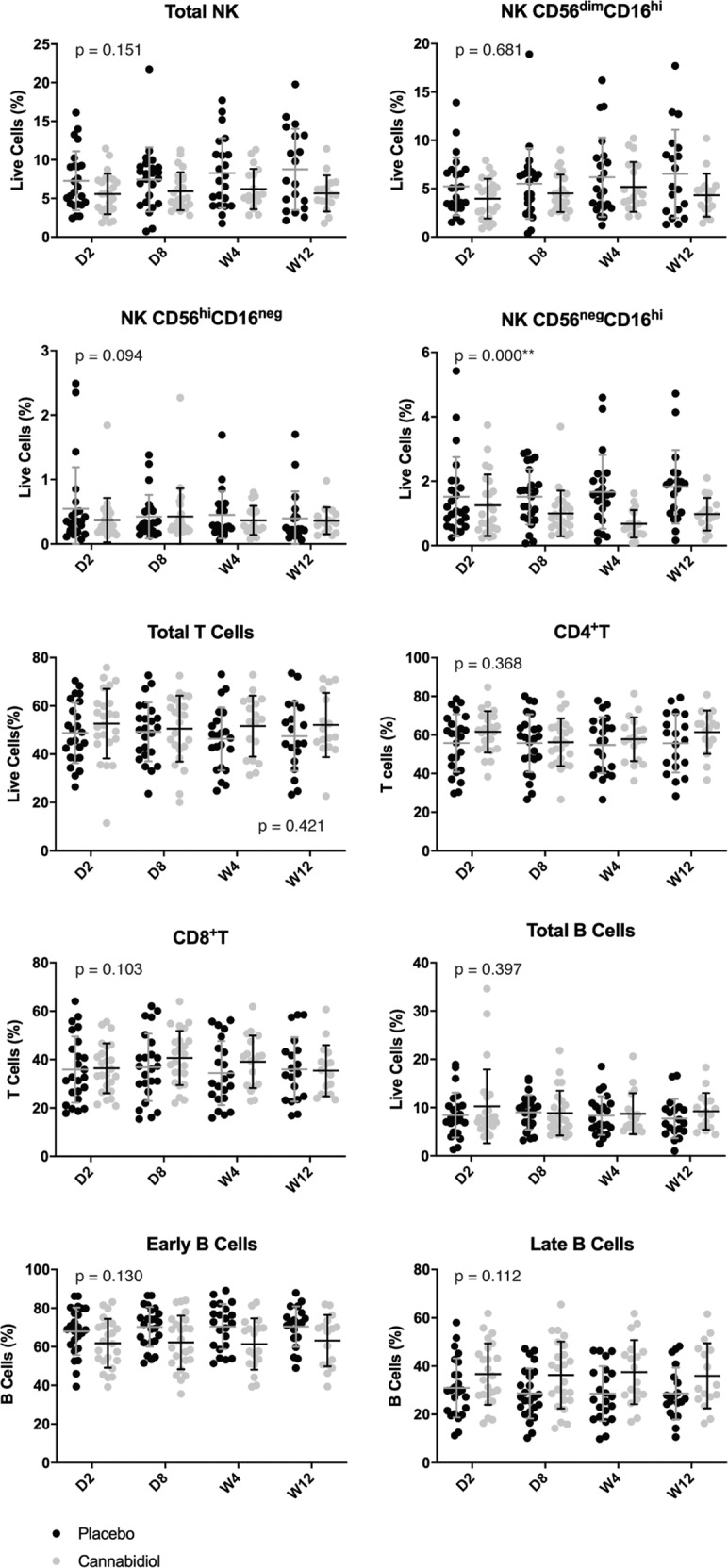
Percentages of total natural killer (NK) cells, NK CD56dimCD16hi, NK CD56hiCD16neg, NK CD56negCD16hi, total T cells, CD4+T, CD8+T, total B cells, early B cells, and late B cells are presented in column scatter plots. The mean and standard deviation (SD) are expressed with horizontal and vertical bars, respectively. *Indicates p value < 0.05; **Indicates p value < 0.01. CD cluster of differentiation, D day, W week.
Total NK cells did not differ between groups. However, a subpopulation of activated mature NK cells (CD56negCD16hi cells) with poor cytotoxic activity was significantly lower in the CBD group compared with the placebo group (B = −0.736, Wald = 19.552, p = 0.000, ES = 0.643). No statistically significant group difference was observed for the two other subpopulations of immature NK CD56hiCD16neg and mature NK CD56dimCD16hi cells.
CBD plasma concentrations
In the CBD group, mean (±SD) CBD blood concentration increased during the trial from 38.79 ± 15.73 ng/mL on day 8 to 54.37±43.28 ng/mL at week 4 and 88.11 ± 149.05 ng/mL at week 12. CBD blood concentrations are represented in box–whisker plots in Fig. S3. The placebo group had no detectable CBD blood concentration except for one participant having 0.06 ng/mL at week 12.
Anti-inflammatory drugs
Twelve (n = 12/48, 25%) participants (CBD, n = 6/24, 25%; placebo, n = 6/24, 25%) took an anti-inflammatory drug at least once during the trial. Categories of anti-inflammatory molecules used in each treatment group are reported in Table S8. The sensitivity analysis excluding those participants revealed similar results as shown in Table S9. Reduced IL-6 and NK CD56negCD16hi levels in the CBD group remained statistically significant. This was not the case for VEGF-A, intermediate monocytes CD14+CD16+ and CD25+CD4+T. However, their estimate values were similar. The sensitivity analysis in which anti-inflammatory drug use was added as a binary covariate revealed that this covariate was not significantly associated with outcomes, with p values ranging from 0.592 to 0.923. All p values associated with treatment group remained significant as shown in Table S10.
A posteriori weight correlations
Correlations between weight and inflammatory markers levels difference from baseline were assessed for five statistically significant inflammatory markers. The only statistically significant correlation was for VEGF-A levels (p = 0.027), for which weight had a weak effect (r = −0.397). Correlation coefficients are presented in Table S11.
A posteriori CBD plasma concentration correlations
Correlations between CBD plasma concentration and inflammatory markers levels difference from baseline were assessed for five statistically significant inflammatory markers. No correlation was statistically significant. Correlation coefficients are presented in Table S12.
A posteriori linear regression for craving
Table S13 presents the results of the five linear regressions conducted to assess the influence of the five main significant inflammatory markers’ level on drug-cue-induced craving scores by treatment group. No statistically significant association was observed.
Discussion
This RCT is the first to evaluate CBD immunomodulatory properties on cytokine levels and leukocyte phenotypes in a clinical population of people who use drugs, namely cocaine. Our findings suggest that CBD may reduce several pro-inflammatory cytokines/leukocytes and increase anti-inflammatory cells, which confirms our exploratory hypothesis. We observed that CBD stabilizes and/or reduces levels of VEGF-A, IL-6, weak cytotoxic NK cells, and intermediate monocytes compared with placebo. Moreover, CD25+CD4+T cells of various maturity stages, known for their regulatory properties, were higher in participants treated with CBD compared with those receiving placebo.
Our findings showed that IL-6 levels are lower following CBD administration compared with placebo, which is consistent with a recent review of preclinical studies [25]. This review showed that when animals with various inflammatory diseases were exposed to a dose range from 0.3 to 80 mg/kg/day during 1–46 days, all studies reported a decrease in IL-6 levels. More recent findings revealed an IL-6 production reduction in a dose-dependent manner after CBD exposure (10, 50, and 100 μg/mL) in vitro to whole human blood cells stimulated with lipopolysaccharide [38]. A near-complete inhibition of IL-6 levels was observed with CBD concentrations of 50 and 100 μg/mL [38]. Other recent studies also portrayed similar conclusion in favor of an IL-6 reduction following CBD administration [39, 40]. However, the only in vivo study evaluating the CBD effect on IL-6 in 13 diabetic participants in a 13-week trial found no difference in IL-6 concentration between CBD and placebo groups [32]. These negative results may be explained by the lower CBD dose (200 mg/day) compared with the one used in our study (800 mg/day). Considering that IL-6 is an acute phase reactant playing several roles such as immune cells’ communication differentiation and proliferation and protein synthesis’s induction [41], IL-6 is a key target to curb the inflammatory cascade from its initiation. Interestingly, Jadoon et al. results for TNF-α and CRP are in line with our results from the discovery phase where no difference was found between groups on TNF-α or CRP concentrations.
In our study, CBD reduced VEGF-A compared with placebo. Similar results were observed in the BBB of diabetic rats in vivo [42] and in Kaposi sarcoma-associated herpesvirus infected endothelial cells in vitro [43] exposed to CBD. VEGF promotes vascular permeability and angiogenesis [44]. VEGF increases the BBB permeability by disrupting tight junction proteins and occluding organization, promoting neuroinflammation [45–47], which may result in poor psychological outcomes such as depression, anxiety, psychosis [48, 49], and neurodegenerative disorders [50]. VEGF inhibition by anti-VEGF antibody has shown promising results to decrease BBB disruption in ischemic rats’ brain to reduce neuroinflammation [51, 52]. Considering that cocaine promotes neuroinflammation by similar mechanisms as VEGF activation, CBD capacity to decrease VEGF levels may promote BBB impermeability, thereby reducing neuroinflammation and associated psychiatric comorbidities.
Our study demonstrated a significant group difference for one of the three monocyte subtypes, intermediate monocytes, with CBD group having lower levels. Although preclinical studies did not assess monocyte phenotypes specifically, their results on total monocytes are in line with a decrease of monocytes induced by CBD. In vitro, CBD induces human monocyte apoptosis [53]. Furthermore, CBD decreases monocyte adhesion and monocyte transendothelial migration in human coronary artery endothelial cells exposed to high glucose concentrations [54]. Intermediate monocytes are involved in the production of various inflammatory molecules such as reactive oxygen species, pro-inflammatory lymphocytes [55–57] and have been related to atherosclerosis development [58, 59]. Reducing circulating intermediate monocytes number, which play key pro-inflammatory functions by their role in linking innate and adaptative immunity, may be a useful therapeutic target in CUD patients.
We observed no significant group difference on total B cells, as well as in their maturity stage. This is inconsistent with previous preclinical studies in an allergic model reporting a decrease in B cells in rats receiving CBD [60] and a decrease in antibodies immunoglobulins M and G in mice exposed to CBD prior to ovalbumin sensitization [61], which were not assessed in our study. Similarly, no group difference was observed in total T lymphocytes nor CD4+T and CD8+T cells. It is somehow contradictory with results in rodents showing that CBD induces T-cell apoptosis [62] and decreases number of total T cells as well as CD4+T and CD8+T subpopulations [60]. However, our results revealed significant higher levels of CD25+CD4+T cells of various maturity stages in the CBD group compared with the placebo group. This is consistent with a study demonstrating CBD induction of CD25+CD4+T in mice [63]. CD25+CD4+T are recently activated T cells including regulatory T cells (Tregs) in a significant proportion [64] as determined by intracellular expression of FoxP3, among others [65]. Tregs are known for their immunosuppressive activity that inhibits T-cell activation and proliferation both in vitro and in vivo [66, 67]. Tregs can also promote anti-inflammatory monocytes [68]. Furthermore, intermediate monocytes can also modulate the presence of Tregs in patients [69]. Therefore, this may suggest that CBD plays it immunosuppressive activity in humans through interactions between intermediate monocytes and Tregs. Future studies are needed to confirm the immunosuppressive properties of CBD-induced CD25+CD4+T.
We found that CBD diminished only the percentage of NK CD56negCD16hi. NK CD56negCD16hi is a subpopulation of NK that has a poor cytotoxic activity [70] compared with other NK populations. Although no other study investigated the effects of CBD on NK population CD56negCD16hi specifically, one study assessed the total number of NK in rats. After 14 days of treatment, the number of NK was not affected by a 5 mg/kg CBD dose but was increased by a 2.5 mg/kg CBD dose compared with placebo [60]. This suggests that NK cells react differently to CBD exposure depending on its dose. Our fixed 800 mg dose corresponds to a mean (SD) of 10.9 (1.8) mg/kg. This dose in humans seems to remove the weak NK cells without affecting the effective cytotoxic NK cells essential for immune surveillance [71].
This study has several limitations that should be considered when interpreting results. First, the discovery pre-analysis to select markers was only done for the first 12 participants. This may have underpowered our pre-analysis to detect group differences and some pertinent markers may have been excluded for the full analysis. Second, the small sample size for which we collected blood samples may have underpowered our full analysis. Third, our participants could use other drugs during the 12-week follow-up (phase II), which may have impacted inflammatory markers. As stated in the main article [33], a high relapse rate occurred in both groups for cocaine consumption and 14/48 (29.2%) participants also had another active substance use disorder. Concomitant use of other drugs may have masked CBD anti-inflammatory potential. However, cocaine use was similar in both groups during the trial and other substance use disorders were well balanced after randomization. Fourth, taking anti-inflammatory drugs was not prohibited during the trial, which may have decreased inflammatory markers levels. However, our sensitivity analyses revealed similar trends after excluding participants who took an anti-inflammatory drug at least once and after adding anti-inflammatory drug use as a binary covariate in our statistical model. Fifth, our high attrition rate (n = 9/48, 18.75%) may have affected our external validity although it is consistent with attrition rates reported in CUD population [72]. Sixth, we did not adjust for multiple comparison considering the exploratory nature of this analysis. Therefore, we should exercise caution when interpreting and generalizing our results. Seventh, mean CBD blood concentration raised over the trial, suggesting that bioavailable CBD increased with treatment duration. Despite satisfactory treatment adherence as previously reported [33], we observed relatively wide standard deviation, which suggests heterogenous individual CBD metabolism. Haney et al. also observed notable variability in CBD plasma concentrations after supervised administration of 800 mg oral CBD, with peak concentrations varying between 1.6 and 271.9 ng/mL (mean: 77.9 ng/mL) [73]. The CBD dose was fixed and a dose adjusted to participants’ weight may have been more appropriate to decrease variability in plasma concentrations between participants. However, our a posteriori analyses revealed that CBD plasma concentration did not correlate with levels of inflammatory markers. Last, we did not look at the effects of inflammatory levels on clinical correlates except for craving, which was not significantly associated with inflammation markers.
To our knowledge, this is the first study assessing CBD effects on a broad spectrum of inflammatory markers in a human population, namely individuals with a substance use disorder. Considering increased cannabis use and accessibility worldwide due to its legalization and growing therapeutic use, it is crucial to understand the biological effects of its consumption. Our results highlighted promising anti-inflammatory properties of 800 mg per day of CBD that should be replicated in future studies. Other dosage regimen should be tested to establish the optimal CBD dose for reducing inflammation. There is also a need to further explore the clinical benefits associated with CBD inflammatory reduction. Populations with substance use disorder have many comorbidities with an inflammatory component such as cardiovascular disease, psychosis, and depression [3]. CBD could potentially have long-term effects on inflammatory burden development and progression that should be explored in a harm reduction perspective. Considering its safety and tolerability profile [28–30], it could also be timely to assess CBD effects in other inflammatory conditions in humans.
Supplementary information
Acknowledgements
We would like to thank the CIHR (#125864), the Fonds de recherche du Québec en Santé, the Université de Montréal, the CRCHUM, the Institut du Cancer de Montréal, and the Institut universitaire sur les dépendances for their financial support. We thank the clinical immunomonitoring core facility of the CRCHUM for performing biobanking, immunophenotyping, and cytokine measurements. We are grateful to Léa Gagnon for reviewing and editing the manuscript, and Guillaume Gazil and his team (Unité de recherche clinique appliquée) for their support in data management and statistical analysis for the main trial. The study team also wants to extend special thanks to all participants for their engagement in this study.
Author contributions
FM, VM-P, ER, and PT wrote the original draft. FM and VM-P did the formal analysis. PT and SL performed biobanking, immunophenotyping, and cytokine measurements. FM and PT did the visualization. SB, JB, SD, ES, and DJ-A conceptualized the study. J-FC and DJ-A co-supervised the project. JB, ES, DJ-A obtained the funding. All authors reviewed the manuscript.
Funding
The Canadian Institutes of Health Research (CIHR) funded this study (#125864). Insys Therapeutics provided the investigational product. DJ-A holds a scholar award from the Fonds de recherche du Québec en Santé. FM received scholarships from the CIHR, the Institut Universitaire sur les Dépendances, the Université de Montréal, and the CRCHUM. JB holds the Canada Research Chair in Addiction Medicine and receives investigator-driven grants from Gilead Sciences and AbbVie for work outside this study.
Competing interests
VM-P, ER, PT, SL, SB, SD, ES, and J-FC declare no competing interests. The funders of the study and Insys Therapeutics had no role in study design, data collection, data analysis, data interpretation, writing of the report, or decision to publish.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Jean-François Cailhier, Didier Jutras-Aswad.
Supplementary information
The online version contains supplementary material available at 10.1038/s41386-021-01098-z.
References
- 1.United Nations Office on Drugs and Crime. World drug report 2019. Vienna, Austria: United Nations Publications; 2019. Sales No. E.19.XI.8.
- 2.Florez-Salamanca L, Secades-Villa R, Hasin DS, Cottler L, Wang S, Grant BF, et al. Probability and predictors of transition from abuse to dependence on alcohol, cannabis, and cocaine: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Am J Drug Alcohol Abus. 2013;39:168–79. doi: 10.3109/00952990.2013.772618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farrell M, Martin NK, Stockings E, Bórquez A, Cepeda JA, Degenhardt L, et al. Responding to global stimulant use: challenges and opportunities. Lancet. 2019;394:1652–67. doi: 10.1016/s0140-6736(19)32230-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zaparte A, Schuch JB, Viola TW, Baptista TAS, Beidacki AS, do Prado CH, et al. Cocaine use disorder is associated with changes in Th1/Th2/Th17 cytokines and lymphocytes subsets. Front Immunol. 2019;10:2435. doi: 10.3389/fimmu.2019.02435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Narvaez JC, Magalhaes PV, Fries GR, Colpo GD, Czepielewski LS, Vianna P, et al. Peripheral toxicity in crack cocaine use disorders. Neurosci Lett. 2013;544:80–4. doi: 10.1016/j.neulet.2013.03.045. [DOI] [PubMed] [Google Scholar]
- 6.Moreira FP, Medeiros JR, Lhullier AC, Souza LD, Jansen K, Portela LV, et al. Cocaine abuse and effects in the serum levels of cytokines IL-6 and IL-10. Drug Alcohol Depend. 2016;158:181–5. doi: 10.1016/j.drugalcdep.2015.11.024. [DOI] [PubMed] [Google Scholar]
- 7.Fox HC, D’Sa C, Kimmerling A, Siedlarz KM, Tuit KL, Stowe R, et al. Immune system inflammation in cocaine dependent individuals: implications for medications development. Hum Psychopharmacol. 2012;27:156–66. doi: 10.1002/hup.1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Levandowski ML, Viola TW, Wearick-Silva LE, Wieck A, Tractenberg SG, Brietzke E, et al. Early life stress and tumor necrosis factor superfamily in crack cocaine withdrawal. J Psychiatr Res. 2014;53:180–6. doi: 10.1016/j.jpsychires.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 9.Manetti L, Cavagnini F, Martino E, Ambrogio A. Effects of cocaine on the hypothalamic–pituitary–adrenal axis. J Endocrinol Invest. 2014;37:701–8. doi: 10.1007/s40618-014-0091-8. [DOI] [PubMed] [Google Scholar]
- 10.Sholar MB, Mendelson JH, Mello NK, Siegel AJ, Kaufman MJ, Levin JM, et al. Concurrent pharmacokinetic analysis of plasma cocaine and adrenocorticotropic hormone in men. J Clin Endocrinol Metab. 1998;83:966–8. doi: 10.1210/jcem.83.3.4654. [DOI] [PubMed] [Google Scholar]
- 11.Riezzo I, Fiore C, De Carlo D, Pascale N, Neri M, Turillazzi E, et al. Side effects of cocaine abuse: multiorgan toxicity and pathological consequences. Curr Med Chem. 2012;19:5624–46. doi: 10.2174/092986712803988893. [DOI] [PubMed] [Google Scholar]
- 12.Loftis JM, Huckans M. Substance use disorders: psychoneuroimmunological mechanisms and new targets for therapy. Pharm Ther. 2013;139:289–300. doi: 10.1016/j.pharmthera.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lopez-Pedrajas R, Ramirez-Lamelas DT, Muriach B, Sanchez-Villarejo MV, Almansa I, Vidal-Gil L, et al. Cocaine promotes oxidative stress and microglial-macrophage activation in rat cerebellum. Front Cell Neurosci. 2015;9:279. doi: 10.3389/fncel.2015.00279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cisneros IE, Erdenizmenli M, Cunningham KA, Paessler S, Dineley KT. Cocaine evokes a profile of oxidative stress and impacts innate antiviral response pathways in astrocytes. Neuropharmacology. 2018;135:431–43. doi: 10.1016/j.neuropharm.2018.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Picard M, Juster RP, McEwen BS. Mitochondrial allostatic load puts the ‘gluc’ back in glucocorticoids. Nat Rev Endocrinol. 2014;10:303–10. doi: 10.1038/nrendo.2014.22. [DOI] [PubMed] [Google Scholar]
- 16.Kovacic P. Role of oxidative metabolites of cocaine in toxicity and addiction: oxidative stress and electron transfer. Med Hypotheses. 2005;64:350–6. doi: 10.1016/j.mehy.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 17.Sajja RK, Rahman S, Cucullo L. Drugs of abuse and blood–brain barrier endothelial dysfunction: a focus on the role of oxidative stress. J Cereb Blood Flow Metab. 2016;36:539–54. doi: 10.1177/0271678X15616978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dhillon NK, Peng F, Bokhari S, Callen S, Shin SH, Zhu X, et al. Cocaine-mediated alteration in tight junction protein expression and modulation of CCL2/CCR2 axis across the blood–brain barrier: implications for HIV-dementia. J Neuroimmune Pharmacol. 2008;3:52–6. doi: 10.1007/s11481-007-9091-1. [DOI] [PubMed] [Google Scholar]
- 19.Pimentel E, Sivalingam K, Doke M, Samikkannu T. Effects of drugs of abuse on the blood–brain barrier: a brief overview. Front Neurosci. 2020;14:513. doi: 10.3389/fnins.2020.00513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kousik SM, Napier TC, Carvey PM. The effects of psychostimulant drugs on blood brain barrier function and neuroinflammation. Front Pharmacol. 2012;3:121. doi: 10.3389/fphar.2012.00121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark KH, Wiley CA, Bradberry CW. Psychostimulant abuse and neuroinflammation: emerging evidence of their interconnection. Neurotox Res. 2013;23:174–88. doi: 10.1007/s12640-012-9334-7. [DOI] [PubMed] [Google Scholar]
- 22.Moretti M, Belli G, Morini L, Monti MC, Osculati AMM, Visona SD. Drug abuse-related neuroinflammation in human postmortem brains: an immunohistochemical approach. J Neuropathol Exp Neurol. 2019;78:1059–65. doi: 10.1093/jnen/nlz084. [DOI] [PubMed] [Google Scholar]
- 23.Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. 2019;25:1822–32. doi: 10.1038/s41591-019-0675-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barron H, Hafizi S, Andreazza AC, Mizrahi R. Neuroinflammation and oxidative stress in psychosis and psychosis risk. Int J Mol Sci. 2017;18:651. doi: 10.3390/ijms18030651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nichols JM, Kaplan BLF. Immune responses regulated by cannabidiol. Cannabis Cannabinoid Res. 2020;5:12–31. doi: 10.1089/can.2018.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKenna M, McDougall JJ. Cannabinoid control of neurogenic inflammation. Br J Pharmacol. 2020;177:4386–99. doi: 10.1111/bph.15208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Atalay S, Jarocka-Karpowicz I, Skrzydlewska E. Antioxidative and anti-inflammatory properties of cannabidiol. Antioxidants. 2019;9:21. doi: 10.3390/antiox9010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lattanzi S, Brigo F, Trinka E, Zaccara G, Cagnetti C, Del Giovane C, et al. Efficacy and safety of cannabidiol in epilepsy: a systematic review and meta-analysis. Drugs. 2018;78:1791–804. doi: 10.1007/s40265-018-0992-5. [DOI] [PubMed] [Google Scholar]
- 29.Bonaccorso S, Ricciardi A, Zangani C, Chiappini S, Schifano F, Cannabidiol CBD. use in psychiatric disorders: a systematic review. Neurotoxicology. 2019;74:282–98. doi: 10.1016/j.neuro.2019.08.002. [DOI] [PubMed] [Google Scholar]
- 30.Larsen C, Shahinas J. Dosage, efficacy and safety of cannabidiol administration in adults: a systematic review of human trials. J Clin Med Res. 2020;12:129–41. doi: 10.14740/jocmr4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.de Almeida DL, Devi LA. Diversity of molecular targets and signaling pathways for CBD. Pharm Res Perspect. 2020;8:e00682. doi: 10.1002/prp2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jadoon KA, Ratcliffe SH, Barrett DA, Thomas EL, Stott C, Bell JD, et al. Efficacy and safety of cannabidiol and tetrahydrocannabivarin on glycemic and lipid parameters in patients with type 2 diabetes: a Randomized, Double-Blind, Placebo-Controlled, Parallel Group Pilot Study. Diabetes Care. 2016;39:1777–86. doi: 10.2337/dc16-0650. [DOI] [PubMed] [Google Scholar]
- 33.Mongeau-Perusse V, Brissette S, Bruneau J, Conrod P, Dubreucq S, Gazil G, et al. Cannabidiol as a treatment for craving and relapse in individuals with cocaine use disorder: a Randomized Placebo-Controlled Trial. Addiction. 2021 doi: 10.1111/add.15417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fox HC, Garcia M, Jr, Kemp K, Milivojevic V, Kreek MJ, Sinha R. Gender differences in cardiovascular and corticoadrenal response to stress and drug cues in cocaine dependent individuals. Psychopharmacology. 2006;185:348–57. doi: 10.1007/s00213-005-0303-1. [DOI] [PubMed] [Google Scholar]
- 35.Ferri CP, Dunn J, Gossop M, Laranjeira R. Factors associated with adverse reactions to cocaine among a sample of long-term, high-dose users in Sao Paulo, Brazil. Addict Behav. 2004;29:365–74. doi: 10.1016/j.addbeh.2003.08.029. [DOI] [PubMed] [Google Scholar]
- 36.Iffland K, Grotenhermen F. An update on safety and side effects of cannabidiol: a review of clinical data and relevant animal studies. Cannabis Cannabinoid Res. 2017;2:139–54. doi: 10.1089/can.2016.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui C, Shurtleff D, Harris RA. Neuroimmune mechanisms of alcohol and drug addiction. Int Rev Neurobiol. 2014;118:1–12. doi: 10.1016/B978-0-12-801284-0.00001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Szekely Y, Ingbir M, Bentur OS, Hochner O, Porat R. Natural cannabinoids suppress the cytokine storm in sepsis-like in vitro model. Eur Cytokine Netw. 2020;31:50–8. doi: 10.1684/ecn.2020.0445. [DOI] [PubMed] [Google Scholar]
- 39.Yeisley DJ, Arabiyat AS, Hahn MS. Cannabidiol-driven alterations to inflammatory protein landscape of lipopolysaccharide-activated macrophages in vitro may be mediated by autophagy and oxidative stress. Cannabis Cannabinoid Res. 2021 doi: 10.1089/can.2020.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lowin T, Tingting R, Zurmahr J, Classen T, Schneider M, Pongratz G. Cannabidiol (CBD): a killer for inflammatory rheumatoid arthritis synovial fibroblasts. Cell Death Dis. 2020;11:714. doi: 10.1038/s41419-020-02892-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tanaka T, Narazaki M, Kishimoto T. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6:a016295. doi: 10.1101/cshperspect.a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.El-Remessy AB, Al-Shabrawey M, Khalifa Y, Tsai NT, Caldwell RB, Liou GI. Neuroprotective and blood-retinal barrier-preserving effects of cannabidiol in experimental diabetes. Am J Pathol. 2006;168:235–44. doi: 10.2353/ajpath.2006.050500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maor Y, Yu J, Kuzontkoski PM, Dezube BJ, Zhang X, Groopman JE. Cannabidiol inhibits growth and induces programmed cell death in kaposi sarcoma-associated herpesvirus-infected endothelium. Genes Cancer. 2012;3:512–20. doi: 10.1177/1947601912466556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504. doi: 10.1038/nature03987. [DOI] [PubMed] [Google Scholar]
- 45.Argaw AT, Asp L, Zhang J, Navrazhina K, Pham T, Mariani JN, et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J Clin Invest. 2012;122:2454–68. doi: 10.1172/JCI60842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petty MA, Lo EH. Junctional complexes of the blood–brain barrier: permeability changes in neuroinflammation. Prog Neurobiol. 2002;68:311–23. doi: 10.1016/s0301-0082(02)00128-4. [DOI] [PubMed] [Google Scholar]
- 47.Suzuki Y, Nagai N, Umemura K. A review of the mechanisms of blood–brain barrier permeability by tissue-type plasminogen activator treatment for cerebral ischemia. Front Cell Neurosci. 2016;10:2. doi: 10.3389/fncel.2016.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lurie DI. An integrative approach to neuroinflammation in psychiatric disorders and neuropathic pain. J Exp Neurosci. 2018;12:1179069518793639. doi: 10.1177/1179069518793639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fourrier C, Singhal G, Baune BT. Neuroinflammation and cognition across psychiatric conditions. CNS Spectr. 2019;24:4–15. doi: 10.1017/S1092852918001499. [DOI] [PubMed] [Google Scholar]
- 50.Chen WW, Zhang X, Huang WJ. Role of neuroinflammation in neurodegenerative diseases (Review) Mol Med Rep. 2016;13:3391–6. doi: 10.3892/mmr.2016.4948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang HT, Zhang P, Gao Y, Li CL, Wang HJ, Chen LC, et al. Early VEGF inhibition attenuates blood–brain barrier disruption in ischemic rat brains by regulating the expression of MMPs. Mol Med Rep. 2017;15:57–64. doi: 10.3892/mmr.2016.5974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chi OZ, Hunter C, Liu X, Weiss HR. Effects of anti-VEGF antibody on blood–brain barrier disruption in focal cerebral ischemia. Exp Neurol. 2007;204:283–7. doi: 10.1016/j.expneurol.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 53.Wu HY, Huang CH, Lin YH, Wang CC, Jan TR. Cannabidiol induced apoptosis in human monocytes through mitochondrial permeability transition pore-mediated ROS production. Free Radic Biol Med. 2018;124:311–8. doi: 10.1016/j.freeradbiomed.2018.06.023. [DOI] [PubMed] [Google Scholar]
- 54.Rajesh M, Mukhopadhyay P, Batkai S, Hasko G, Liaudet L, Drel VR, et al. Cannabidiol attenuates high glucose-induced endothelial cell inflammatory response and barrier disruption. Am J Physiol Heart Circ Physiol. 2007;293:H610–9. doi: 10.1152/ajpheart.00236.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dhanda AD, Williams EL, Yates E, Lait PJP, Schewitz-Bowers LP, Hegazy D, et al. Intermediate monocytes in acute alcoholic hepatitis are functionally activated and induce IL-17 expression in CD4(+) T cells. J Immunol. 2019;203:3190–8. doi: 10.4049/jimmunol.1800742. [DOI] [PubMed] [Google Scholar]
- 56.Gaur P, Myles A, Misra R, Aggarwal A. Intermediate monocytes are increased in enthesitis-related arthritis, a category of juvenile idiopathic arthritis. Clin Exp Immunol. 2017;187:234–41. doi: 10.1111/cei.12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.O’Brien EC, Abdulahad WH, Rutgers A, Huitema MG, O’Reilly VP, Coughlan AM, et al. Intermediate monocytes in ANCA vasculitis: increased surface expression of ANCA autoantigens and IL-1beta secretion in response to anti-MPO antibodies. Sci Rep. 2015;5:11888. doi: 10.1038/srep11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Franca CN, Izar MCO, Hortencio MNS, do Amaral JB, Ferreira CES, Tuleta ID, et al. Monocyte subtypes and the CCR2 chemokine receptor in cardiovascular disease. Clin Sci. 2017;131:1215–24. doi: 10.1042/CS20170009. [DOI] [PubMed] [Google Scholar]
- 59.Wolf AA, Yanez A, Barman PK, Goodridge HS. The ontogeny of monocyte subsets. Front Immunol. 2019;10:1642. doi: 10.3389/fimmu.2019.01642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ignatowska-Jankowska B, Jankowski M, Glac W, Swiergel AH. Cannabidiol-induced lymphopenia does not involve NKT and NK cells. J Physiol Pharmacol. 2009;60(Suppl 3):99–103. [PubMed] [Google Scholar]
- 61.Jan TR, Su ST, Wu HY, Liao MH. Suppressive effects of cannabidiol on antigen-specific antibody production and functional activity of splenocytes in ovalbumin-sensitized BALB/c mice. Int Immunopharmacol. 2007;7:773–80. doi: 10.1016/j.intimp.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 62.Wu HY, Chu RM, Wang CC, Lee CY, Lin SH, Jan TR. Cannabidiol-induced apoptosis in primary lymphocytes is associated with oxidative stress-dependent activation of caspase-8. Toxicol Appl Pharmacol. 2008;226:260–70. doi: 10.1016/j.taap.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 63.Dhital S, Stokes JV, Park N, Seo KS, Kaplan BL. Cannabidiol (CBD) induces functional Tregs in response to low-level T cell activation. Cell Immunol. 2017;312:25–34. doi: 10.1016/j.cellimm.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bahador A, Hadjati J, Hassannejad N, Ghazanfari H, Maracy M, Jafari S, et al. Frequencies of CD4+ T regulatory cells and their CD25(high) and FoxP3(high) subsets augment in peripheral blood of patients with acute and chronic Brucellosis. Osong Public Health Res Perspect. 2014;5:161–8. doi: 10.1016/j.phrp.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Santegoets SJ, Dijkgraaf EM, Battaglia A, Beckhove P, Britten CM, Gallimore A, et al. Monitoring regulatory T cells in clinical samples: consensus on an essential marker set and gating strategy for regulatory T cell analysis by flow cytometry. Cancer Immunol Immunother. 2015;64:1271–86. doi: 10.1007/s00262-015-1729-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shevach EM. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol. 2002;2:389–400. doi: 10.1038/nri821. [DOI] [PubMed] [Google Scholar]
- 67.Corthay A. How do regulatory T cells work? Scand J Immunol. 2009;70:326–36. doi: 10.1111/j.1365-3083.2009.02308.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Romano M, Fanelli G, Tan N, Nova-Lamperti E, McGregor R, Lechler RI, et al. Expanded regulatory T cells induce alternatively activated monocytes with a reduced capacity to expand T helper-17 cells. Front Immunol. 2018;9:1625. doi: 10.3389/fimmu.2018.01625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Guo N, Liu L, Yang X, Song T, Li G, Li L, et al. Immunological changes in monocyte subsets and their association with Foxp3(+) regulatory T cells in HIV-1-infected individuals with syphilis: a brief research report. Front Immunol. 2019;10:714. doi: 10.3389/fimmu.2019.00714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muller-Durovic B, Grahlert J, Devine OP, Akbar AN, Hess C. CD56-negative NK cells with impaired effector function expand in CMV and EBV co-infected healthy donors with age. Aging. 2019;11:724–40. doi: 10.18632/aging.101774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol. 2019;10:3038. doi: 10.3389/fimmu.2019.03038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chan B, Kondo K, Freeman M, Ayers C, Montgomery J, Kansagara D. Pharmacotherapy for cocaine use disorder—a systematic review and meta-analysis. J Gen Intern Med. 2019;34:2858–73. doi: 10.1007/s11606-019-05074-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haney M, Malcolm RJ, Babalonis S, Nuzzo PA, Cooper ZD, Bedi G, et al. Oral cannabidiol does not alter the subjective, reinforcing or cardiovascular effects of smoked cannabis. Neuropsychopharmacology. 2016;41:1974–82. doi: 10.1038/npp.2015.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.