

SUMMARY
Subtype C is the most prevalent clade of human immunodeficiency virus type 1 (HIV-1) worldwide. The reasons for this are poorly understood. Here, we demonstrate that a characteristic additional third nuclear factor κB (NF-κB) binding site in the long terminal repeat (LTR) promoter allows subtype C HIV-1 strains to evade restriction by nuclear PYHIN proteins, which sequester the transcription factor Sp1. Further, other LTR alterations are responsible for rare PYHIN resistance of subtype B viruses. Resistance-conferring mutations generally reduce the dependency of HIV-1 on Sp1 for virus production and render LTR transcription highly responsive to stimulation by NF-κB/p65. A third NF-κB binding site increases infectious virus yield in primary CD4+ T cells in an γ-interferon-inducible protein 16 (IFI16)-dependent manner. Comprehensive sequence analyses suggest that the frequency of circulating PYHIN-resistant HIV-1 strains is increasing. Our finding that an additional NF-κB binding site in the LTR confers resistance to nuclear PYHIN proteins helps to explain the dominance of clade C HIV-1 strains.
Graphical Absttract
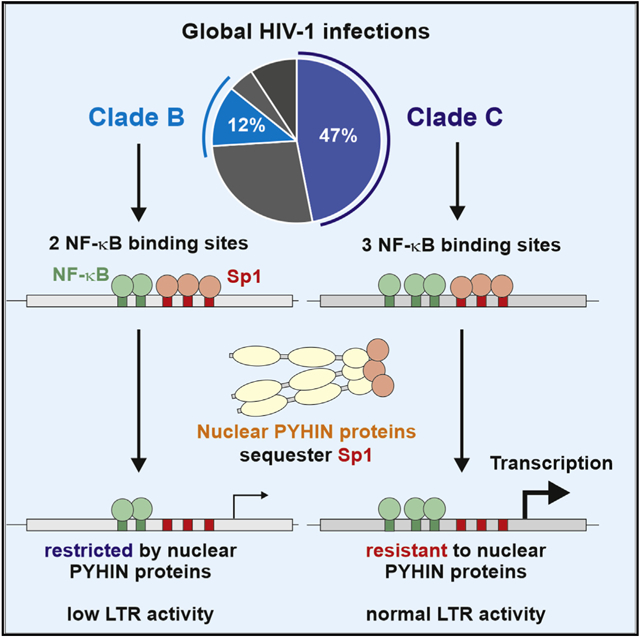
In brief
It is poorly understood why subtype C HIV-1 strains are most prevalent worldwide. Bosso et al. show that a characteristic additional NF-κB binding site in the long terminal repeat (LTR) promoter allows clade C HIV-1 strains to evade restriction by interferon-inducible nuclear PYHIN proteins.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) is the consequence of at least four independent zoonotic transmissions of simian immunodeficiency viruses (SIVs) infecting great apes giving rise to groups M, N, O, and P (Sauter and Kirchhoff, 2019; Sharp and Hahn, 2011). The prevalence of the four groups varies enormously, and HIV-1 group M (major) is responsible for the vast majority of all infections around the globe. Most likely, differences in the ability to evade or counteract antiviral innate defense mechanisms played a key role in the differential spread of the rare (N and P), endemic (O), and pandemic (M) HIV-1 strains (Sauter and Kirchhoff, 2019). The 10 different genetic subtypes (A–D, F–H, and J–L) and numerous circulating recombinant forms (CRFs) of pandemic HIV-1 group M also vary strongly in their distribution and prevalence. Subtype C viruses are most common and responsible for almost half of all global HIV-1 infections (Bbosa et al., 2019; Gartner et al., 2020; Hemelaar et al., 2019). Today, subtype C HIV-1 strains dominate in Southern and Eastern Africa, India, and parts of Brazil and China. It has been suggested that clade C strains may expand at a faster rate and thus outcompete other subtypes of HIV-1 (Locateli et al., 2007; Soares et al., 2003). However, whether specific features render clade C HIV-1 strains particularly fit for spread in human populations or introduction into high-risk groups and whether founder effects are responsible for their effective spread are under debate (Gartner et al., 2020).
One genetic feature that distinguishes HIV-1 subtype C strains from less-prevalent subtypes is the presence of an additional NF-κB target motif in the viral LTR (De Baar et al., 2000; Hunt and Tiemessen, 2000; Munkanta et al., 2005). This third NF-κB binding site is located upstream of the core enhancer/promoter region that usually consists of two identical canonical κB motifs and three conserved Sp1 interaction sites. It has been reported that the additional NF-κB site increases viral transcription and might thus contribute to the efficient spread and current dominance of subtype C HIV-1 strains (Bachu et al., 2012). However, the effects of an additional NF-κB site on LTR promoter activity and viral replication in immortalized cell lines were generally modest (Bachu et al., 2012; Lemieux et al., 2004; Montano et al., 1997; Naghavi et al., 1999). Thus, the relevance of the additional NF-κB site and the reasons for the effective spread of subtype C HIV-1 strains remain poorly understood.
We have previously shown that subtype C viruses are much less susceptible to inhibition by the γ-interferon (IFN)-inducible protein 16 (IFI16) compared with subtype A, B, and D viruses (Hotter et al., 2019). Functional analyses revealed that IFI16 interacts with Sp1 and inhibits viral gene expression by limiting the availability of this transcription factor. In contrast, SIVcpz, the direct precursor of HIV-1 M, was susceptible to IFI16 inhibition (Hotter et al., 2019), suggesting that the resistance of subtype C HIV-1 strains evolved during adaptation to humans. IFI16 belongs to the family of PYHIN proteins whose members are known to play key roles as IFN-inducible mediators and effectors of innate immune responses (Cridland et al., 2012; Schattgen and Fitzgerald, 2011). Recently, we have shown that two additional members of this family, MNDA and PYHIN1, share the antiviral activity and mechanism of IFI16 (Bosso et al., 2020a). In addition, we demonstrated that these nuclear PYHIN proteins inhibit HIV-1 clade A, B, and D replication in primary human macrophages and/or activated CD4+ T lymphocytes (Bosso et al., 2020a; Hotter et al., 2019). The only exception was a subtype B infectious molecular clone (IMC) termed THRO (Ochsenbauer et al., 2012), which shares the resistance phenotype of subtype C HIV-1 strains. Our previous studies suggested that resistance to inhibition by IFI16, MNDA, and PYHIN1 is associated with reduced viral dependency on Sp1 for efficient transcription (Bosso et al., 2020a; Hotter et al., 2019). However, the viral determinants of resistance remained unclear. Here, we demonstrate that the additional NF-κB site in the LTR of subtype C HIV-1 strains confers resistance to nuclear PYHIN proteins and strongly decreases dependency on Sp1 while increasing the responsiveness to stimulation by NF-κB. In addition, we show that the subtype B THRO HIV-1 strain acquired different resistance-conferring mutations in its LTR. Our results help to explain the efficient spread of subtype C HIV-1 strains and suggest that other subtypes of HIV-1 might also acquire increased transmission fitness by evading restriction imposed by nuclear PYHIN proteins.
RESULTS
Determinants of HIV-1 subtype B THRO resistance to nuclear PYHIN proteins
We have previously shown that IFI16, MNDA, and PYHIN1/IFIX inhibit transcription and infectious virus production by most subtype A, B, and D HIV-1 IMCs (Bosso et al., 2020a; Hotter et al., 2019). An exception was the subtype B HIV-1 THRO transmitted/founder (TF) IMC that was fully resistant to nuclear PYHIN proteins. HIV-1 transcription is determined by the 5′ long terminal repeat (LTR). As first step to map the determinants of HIV-1 resistance to inhibition by PYHIN proteins, we therefore replaced the LTRs at both the 5′ and 3′ ends of the resistant THRO strain by those of the sensitive subtype B CH058 IMC (Figure 1A) and determined infectious virus yields and p24 antigen production from HEK293T cells cotransfected with the proviral constructs and expression vectors for individual PYHIN proteins (Figures 1B and S1A). In agreement with the proposed mechanism that PYHIN proteins suppress proviral HIV-1 transcription by limiting the availability of the transcription factor Sp1 (Bosso et al., 2020a; Hotter et al., 2019), viral sensitivity to inhibition by IFI16, PYHIN1, and MNDA was determined by the LTR region (Figure 1B, lanes 1, 2, 8, and 9).
Figure 1. Determinants of HIV-1 clade B sensitivity to nuclear human PYHIN proteins.

(A) Alignment of HIV-1 CH058 and THRO LTRs. Dots indicate nucleotide identity, and dashes gaps to optimize the alignment. Some elements, such as the MFNLP, NF-κB, and Sp1 binding sites, are indicated. The blue arrow indicates the TATA box, i.e., the transcription start site. The areas used to generate chimeric LTRs are colored yellow (5′ half) and light blue (3′ half).
(B) Susceptibility of HIV-1 CH058 and THRO constructs differing in their LTR regions to inhibition by IFI16, MNDA, and IFIX. The upper panel provides the infectious virus yields (left) and levels of p24 antigen (right) in the supernatants of HEK293T cells cotransfected with the indicated HIV-1 constructs and vectors expressing the PYHIN proteins or an empty control vector. Each symbol represents the average values obtained in three independent experiments. The lower panel shows the mean values (±SEM) of infectious virus and p24 production measured in the presence of the indicated PYHIN proteins relative to the vector control (100%).
See also Figure S1.
The LTR of HIV-1 THRO differs from that of the PYHIN-sensitive HIV-1 CH058 strain by about 50 nt and a 34-nt duplication upstream of the viral enhancer element (Figure 1A). Such duplications at the 3′ end of the nef gene are known as “most frequent naturally occurring length polymorphism” (MFNLP). They are found in about 15%–40% of subtype B LTRs and contain potential binding sites for various transcription factors, including Ras-responsive binding factor 2 (RBF-2) and Ras-binding element III (RBE III) (Estable, 2007; Estable et al., 1996). Functional analyses of HIV-1 THRO and CH058 IMCs containing chimeric LTRs revealed that sensitivity to inhibition by nuclear human PYHIN is largely governed by the 3′ half of the LTR encompassing the tandem NF-κB and the three Sp1 binding sites (Figure 1B, lanes 4, 5, 11, and 12). The effect of the 34-nt MFNLP was dependent on the viral backbone. Its deletion completely inactivated the HIV-1 THRO IMC (Figure 1B, lane 7). In comparison, insertion of the 34 nt in the CH058 LTR still allowed significant p24 antigen production and reduced sensitivity to PYHIN inhibition but fully abrogated virion infectivity (Figure 1B, lane 14), suggesting maintenance of LTR promoter activity but disruption of RNA function. Prediction of the U3-R RNA structures indicates that both deletion of the MFNLP from the THRO LTR, as well as its insertion into the CH058 LTR, may cause major structural changes and results in a large number of unpaired nucleotides (Figure S1B). Accordingly, context-dependent effects on both LTR promoter function and viral RNA structures affected p24 production and infectious virus yield. HIV-1 THRO and CH058 IMCs containing the 5′ half plus MFNLP of the THRO LTR showed efficient p24 antigen production and modest sensitivity to PYHIN inhibition but strongly impaired infectious virus yields in both the absence and (even more severely) presence of PYHIN proteins (Figure 1B, lanes 3 and 10). Finally, deletion of the MFNLP from HIV-1 IMCs containing the 5′ half of the CH058 and 3′ half of the THRO LTR did not affect virus production in the absence of PYHIN protein expression but increased viral sensitivity to inhibition by IFI16, MNDA, and PYHIN1 (Figure 1B, lanes 6 and 13). Altogether, these results show that PYHIN resistance of HIV-1 THRO is largely determined by the 3′ half of the LTR. However, the 34-bp MFNLP duplication is required for full resistance.
To further define determinants of resistance, we performed comprehensive sequence-based phenotypic association analyses using 29 full-length proviral sequences of HIV-1 clade A, B, C, and D IMCs differing in their susceptibility to PYHIN inhibition (Bosso et al., 2020a; Hotter et al., 2019). These proviruses were ranked according to their residual infectious virus yield in the presence of IFI16 (Table S1). Subsequently, associations of IFI16 sensitivity with genotypes at specific nucleotide positions were determined using one-way analysis of variance (ANOVA). Analyses were performed without or with correcting for subtype-specific effects and resulted in the identification of 12 and 26 sites, respectively, that were significantly associated with IFI16 sensitivity (Figure S2A; Tables S2 and S3). Only two of these sites (alignment positions 414 and 10,000) were identified by both approaches (Tables S2 and S3). Interestingly, these sites represent identical positions in the 5′ and 3′ LTRs just upstream of the tandem NF-κB sites (position −1) (Figure 2A). This nucleotide is located in the 3′ half of the LTR that plays a key role in determining PYHIN sensitivity of HIV-1 clade B strains (Figure 1B). Although sensitive subtype A, B, and D HIV-1 clones harbor an A at the −1 position, resistant subtype C viruses contain a T (Figure 2A). The resistant THRO IMC was the only one of 17 subtype A, B, and D HIV-1 IMCs functionally analyzed in our previous studies (Bosso et al., 2020a; Hotter et al., 2019) containing a G instead of an A residue at the −1 position (Figures 2A and S2B). Further analyses showed that only 16 of 462 (3%) subtype B HIV-1 sequences with reported detectable viral load in the Los Alamos database had a G, while 436 (94%) had an A, similarly to the majority of clade A, B, and D HIV-1 strains (Figure 2B). Notably, one of the most prevalent CRFs of HIV-1 (CRF01_AE) also contains a T at the −1 position, although this was not the case for other CRFs, such as CFR02_AG (Figure 2B). HIV-1 CRF01_AE dominates in Southeast Asia and may be responsible for ~5% of global HIV-1 infections (Angelis et al., 2015).
Figure 2. Group- and subtype-specific differences in the −1 nucleotide position upstream of the tandem NF-κB element.

(A) The indicated regions of HIV-1 LTRs were sorted based on the sensitivity of the corresponding IMCs to inhibition by IFI16 from least sensitive at the top to most sensitive at the bottom (Hotter et al., 2019; McLaren et al., 2015) (Table S1). Dashes indicate gaps to optimize the alignment. NF-κB (gray) and Sp1 (water green) binding sites and the MFNLP in THRO (light purple) and the −1 nucleotide (yellow) are highlighted.
(B) Frequency plots indicating differences in the nucleotide at the −1 position relative to the conserved NF-κB enhancer element.
See also Figure S2.
To determine the impact on PYHIN sensitivity, we introduced substitutions of G to A and vice versa upstream of the tandem NF-κB elements into the subtype B THRO (G380A) and CH058 (A349G) LTRs, respectively. Unexpectedly, the single G380A change reduced infectious virus yield of HIV-1 THRO by about two orders of magnitude (Figure 3A) but had only modest effects on p24 antigen production (Figure 3B). In comparison, the A349G substitution in the HIV-1 CH058 LTRs moderately increased both infectious virus yield (Figure 3A) and p24 antigen production (Figure 3B). Molecular modeling analyses predicted that the G380A change causes major changes in the U3-R RNA secondary structure of the THRO LTR, while only minimal effects were predicted for the A349G mutation in CH058 (Figure S3A). This may explain why the G380A mutation strongly impaired infectious virus yield of HIV-1 THRO but had only modest effects on p24 antigen production. More importantly, the G380A change clearly increased and the A349G mutation significantly decreased HIV-1 sensitivity to inhibition by IFI16, MNDA, and IFIX. Consequently, the THRO G380A and CH058 A349G mutant viruses showed phenotypes intermediate between the two parental viruses (Figures 3A and 3B, right panels).
Figure 3. A “G” residue at the −1 position reduces HIV-1 subtype B susceptibility to restriction by human PYHIN proteins.

(A and B) HEK293T cells were cotransfected with the indicated proviral constructs (2.5 μg) and either a vector control or expression constructs for human nuclear PYHIN proteins (1 μg). Two days post-transfection, infectious virus yield was determined by TZM-bl cells infection (A), and p24 in the supernatant was assessed by ELISA (B). The left panels show absolute values and the right panels infectious virus and p24 production in the presence of the indicated PYHIN proteins relative to the vector control (100%). Shown are average values (±SD) obtained from four independent experiments.
(C–F) HEK293T cells were cotransfected with either an empty vector or expression plasmids for Sp1 (C and D) or NF-κB (E and F) and the indicated proviral HIV-1 constructs. Infectious virus yields (C and E) and p24 levels (D and F) were determined 2 days post-transfection. The left panel shows absolute and the right panel relative levels of production of described above. Curves were derived from three to four independent experiments and show average values (±SEM).
(G) Effect of Sp1 or NF-κB overexpression on infectious virus (top) and p24 antigen (bottom) yield of the wild-type and A349G HIV-1 CH058 IMCs. Values were derived from (C)–(F) at the highest concentration of Sp1 and NF-κB expression, respectively. **p < 0.01; ***p < 0.001.
See also Figure S3.
Previous results showed that nuclear PYHIN proteins inhibit HIV-1 by sequestering Sp1, and that sensitive viral strains are more dependent on this factor for efficient transcription than resistant HIV-1 strains (Bosso et al., 2020a; Hotter et al., 2019). In agreement with this, increasing levels of Sp1 expression (Figure S3B) enhanced infectious virus yield and p24 antigen production of the sensitive CH058 IMC up to 5-fold but had no significant effect on the resistant THRO IMC (Figures 3C and 3D). The A349G change substantially reduced the responsiveness of HIV-1 CH058 to Sp1 activation (Figures 3C and 3D). In contrast, it increased dose-responsive enhancement of infectious virus yields and p24 antigen production by NF-κB p65 overexpression (Figures 3E-3G). In comparison, wild-type (WT) THRO was generally highly active, while the G380A mutant was strongly attenuated. Notably, the WT HIV-1 CH058 IMC was most active in the presence of high levels of Sp1, while the A349G mutant was most effective in producing infectious virus in the presence of high levels of NF-κB (Figure 3G). Thus, the relative efficiencies of transcription of PYHIN-resistant and -sensitive HIV-1 strains depend on the levels of active Sp1 and NF-κB in the viral target cells. To further determine the role of the MFNLP and the A349G change on Sp1 dependency of HIV-1, we took advantage of the chronic myelogenous leukemia cell line HAP1 and a derivative thereof lacking Sp1 expression (Carette et al., 2009). We found that the A349G mutation increased infectious virus yield and p24 production of CH058, especially in the absence of Sp1 (Figures S3C and S3D, lanes 2 and 6). Lack of Sp1 reduced virus yield of WT HIV-1 CH058 by >80%, while the A349G mutant virus was inhibited by only ~50% (Figure S3E, lanes 2 and 6). The MFNLP was required for full activity of HIV-1 CH058 mutant LTR constructs in a host cell environment lacking Sp1 (Figure S3E, lanes 7 and 8). Altogether, these results showed that the nucleotide at position −1 upstream of the conserved tandem NF-κB and the MFNLP duplication determine the sensitivity of HIV-1 subtype B to inhibition by nuclear PYHIN proteins and dependency on Sp1 for efficient transcription.
An additional NF-κB site allows clade C HIV-1 to evade PYHIN-mediated restriction
Compared with HIV-1 subtypes A, B, and D, clade C viruses generally showed low susceptibility to inhibition by nuclear human PYHIN proteins (Bosso et al., 2020a; Hotter et al., 2019). In contrast with most other clades of HIV-1, subtype C strains usually contain a T nucleotide at the −1 position upstream of the tandem NF-κB sites (Figure 2B). In addition, HIV-1 subtype C differs from less prevalent subtypes of HIV-1 by an additional NF-κB site (Bachu et al., 2012; De Baar et al., 2000; Hunt and Tiemessen, 2000; Munkanta et al., 2005; Naghavi et al., 1999). To determine whether these characteristic features of subtype C viruses contribute to PYHIN resistance, we generated a set of mutant HIV-1 constructs differing specifically in the presence of the additional NF-κB site and in the nucleotide at the −1 position relative to the tandem NF-κB element (Figure 4A). Functional analyses showed that an additional NF-κB site in the LTR of the HIV-1 subtype B CH058 IMC does not significantly affect infectious virus yield in the absence of IFI16, MNDA, and IFIX (Figure 4B). However, it conferred almost full resistance to inhibition by the three nuclear PYHIN proteins (Figure 4C). Conversely, elimination of the third NF-κB site significantly increased the susceptibility of HIV-1 subtype C ZM247 and (more severely) CH185 to inhibition by IFI16, MNDA, and PYHIN1 (Figures 4A and 4B). In contrast with the −1 A-to-G mutation in clade B HIV-1 strains, the −1 T-to-A change in the subtype C ZM247 and CH185 IMCs had little effect on infectious virus yield and IFI16, MNDA, and PYHIN1 sensitivity (Figures 4B and 4C). Altogether, these results showed that the additional NF-κB site in the LTR of clade C HIV-1 is key to their resistance against human PYHIN proteins.
Figure 4. Effect of an additional NF-κB site on HIV-1 sensitivity to human PYHIN proteins.

(A) Mutations introduced into the LTRs of HIV-1 CH058, ZM247, and CH185 IMCs.
(B and C) HEK293T cells were transfected with either an empty vector or an expression plasmid for human nuclear PYHIN proteins (1 μg) and the indicated proviral constructs harboring the indicated mutations in both LTRs. Infectious virus yield was determined by TZM-bl cells infection assay. Shown are absolute infectious virus yields (B) and the levels relative to those obtained in the presence of the empty control constructs (C). Data represent the mean from three independent experiments, with each measured in triplicates ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
A third NF-κB site has opposing effects on LTR stimulation by Sp1 and NF-κB
Previous results showed that HIV-1 resistance to restriction by IFI16 correlated with reduced viral dependency on Sp1 for LTR-driven transcription (Hotter et al., 2019). To obtain further insights into the mechanisms underlying HIV-1 resistance to human PYHIN proteins and the consequences for the LTR promoter, we determined the responsiveness of WT and mutant HIV-1 B and C constructs to stimulation by increasing doses of Sp1 and NF-κB p65. We found that Sp1 dose-dependently increases HIV-1 CH058 subtype B infectious virus yield up to 5-fold but has only marginal effects on virus production by clade C HIV-1 ZM247 and CH185 IMCs (Figure 5A, upper). Conversely, WT subtype C HIV-1 constructs were more responsive to stimulation by increasing levels of NF-κB compared with the HIV-1 subtype B IMC (Figure 5A, lower). The A349G mutation or an additional NF-κB binding site significantly reduced the responsiveness of HIV-1 CH058 to Sp1 activation, while increasing the efficiency of virus production in the presence of increasing levels of NF-κB (Figure 5A). The combination of both conferred a subtype C-like phenotype to the subtype B CH058 strain. Unlike the A349G mutation in the CH058 LTR, insertion of A350 or mutation of T353A had little impact on the responsiveness of HIV-1 subtype C ZM247 and CH185 IMCs to Sp1 and NF-κB stimulation (Figure 5A). Mutation of the third NF-κB site, however, rendered both clade C viruses more responsive to Sp1 stimulation, while reducing the enhancing effect of NFκB/p65 overexpression (Figure 5A). On average, the subtype B HIV-1 CH058 IMC produced about 3-fold higher levels of infectious virus compared with the clade C ZM247 and CH185 strains in the presence of high levels of Sp1 (Figure 5B, upper). In contrast, the subtype C HIV-1 IMCs achieved about 2-fold higher infectious virus yields than CH058 in the presence of high levels of p65 NF-κB expression (Figure 5B, lower). Altogether, the parental subtype B HIV-1 CH058 IMC yielded the highest levels of infectious virus in the presence of high levels of Sp1, while the subtype C HIV-1 strains were most active in the presence of high levels of NF-κB Figure S4). These differences in the dependency and responsiveness on these two key transcription factors are largely determined by the additional third NF-κB binding site in subtype C LTRs.
Figure 5. Effect of an additional NF-κB binding site on HIV-1 responsiveness to Sp1 and p65 stimulation.

HEK293T cells were cotransfected with the indicated proviral HIV-1 constructs and increasing amounts of Sp1 (upper) or NF-κB p65 (lower) expression vectors and/or an empty vector. Infectious virus yields in the presence of increasing levels of Sp1 or p65 relative to the vector control (100%) (A) and absolute values (B) were determined by infection of TZM-bl cells. Shown are mean values (±SEM) derived from three experiments. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S4.
A third NF-κB site renders HIV-1 resistant to IFI16 restriction in CD4+ T cells
IFI16 is efficiently expressed in CD4+ T cells (Bosso et al., 2020a; Hotter et al., 2019) representing the main target cells for HIV-1 replication in vivo. Using a previously established protocol (Bosso et al., 2020b), we depleted IFI16 expression in CD4+ T cells by nucleofection of CRISPR-Cas9 complexes containing IFI16-specific guide RNAs (gRNAs) (Figure 6A). In agreement with previous data (Bosso et al., 2020a; Hotter et al., 2019), partial knockout (KO) of IFI16 in primary CD4+ T cells increased infectious yield of the HIV-1 subtype B CH058 IMC about 10-fold but had little effect on the HIV-1 subtype C CH185 IMC (Figure 6B). An additional NF-κB site rendered HIV-1 CH058 almost fully resistant to IFI16 inhibition. Measurement of p24 capsid antigen levels in the culture supernatants confirmed that endogenous IFI16 expression strongly inhibits WT HIV-1 CH058 production but has no significant effect on the otherwise isogenic construct containing an additional NF-κB site (Figure 6C). Deletion of the third NF-κB site strongly attenuated the subtype C HIV-1 CH185 IMC in primary CD4+ T cells (Figures 6A and 6B), although it did not impair infectious virus production from transfected HEK293T cells (Figure 4). This agrees with previous findings showing that NF-κB signaling is poorly active in HEK239T cells but plays a major role in primary CD4+ T cells (Oh and Ghosh, 2013). Partial KO of IFI16 increased infectious virus yield and p24 production of the NF-κB mutant HIV-1 clade C CH185 strain about 5-fold but had no significant effect on the parental virus (Figures 6B and 6C). Altogether, these data show that the additional NF-κB site in clade C LTRs confers resistance to inhibition by IFI16. In addition, our results demonstrate that evasion of this antiviral mechanism efficiently increases infectious HIV-1 production in activated primary human CD4+ T cells.
Figure 6. An additional NF-κB site renders HIV-1 CH058 resistant to IFI16 restriction in primary CD4+ T cells.

Activated CD4+ T cells from three blood donors were transfected with Cas9 and either a non-targeting (nt) or an IFI16-specific gRNA. 96 hours later, cells were transduced with the indicated vesicular stomatitis virus glycoprotein (VSV-G)-pseudotyped HIV-1 strains. Residual levels of IFI16 protein expression in infected cell cultures treated with IFI16 targeting gRNA (A), the infectious virus yields (B), and p24 antigen production (C) were determined by western blot, TZM-bl infection assay, and ELISA, respectively, at 3 days postinfection. Numbers above bars indicate n-fold change between cells treated with control or IFI16-specific gRNA. Data represent the mean from three different donors ± SEM. *p < 0.05, **p < 0.01.
Potential evolution of increased PYHIN resistance in HIV-1 clade B and C strains
If HIV-1 resistance to PYHIN proteins is advantageous for viral spread in vivo, the frequency of resistance-conferring properties should increase in viral populations. To assess this, we took advantage of HIV-1 clade B and C sequences sampled over the last 40 years available in the Los Alamos HIV database. We focused on the region corresponding to nt 9,391–9,444 in the reference HIV-1 HXB2 strain (Figure 7A; named US-NF-κB-II region hereafter; US, upstream) because it encompasses the MFNLP in clade B, as well as the additional NF-κB site in subtype C LTRs, respectively. In addition, the end of the nef gene and the conserved NF-κB-II site provide useful starting points and endpoints. Analysis of 2,347 sequences from subtype B HIV-1-infected individuals (one viral sequence from each person) revealed that the frequency of strains containing additional nucleotides in this region has significantly increased over the last 40 years (Figure 7B). Analyses of 2,367 clade C HIV-1 LTR sequences showed that the proportion of strains with longer sequences upstream the tandem NF-κB site also increased over the last decades, although less consistently than observed for clade B (Figures 7C and 7D). The proportion of clade C sequences above median length increased continuously from 12% to 44% (Figure 7D). For subtype B strains, increases in length were associated with a higher frequency of the LTRs containing the MFNLP and additional RBE III sites in the US-NF-κB-II region. In addition, the frequency of a “G” nucleotide at the −1 position increased slightly from 1.7% before 1991 to ~5% in clade B LTR sequences obtained during the last decade (data not shown). An increased length of the clade C HIV-1 US-NF-κB-II region was associated with an increased number of potential NF-κB binding sites (Figure S5). However, the number of NF-κB-like sites in HIV-1 clade C LTRs did not increase over time, while the proportion of strains containing an additional RBEIII site increased during the last decades. Altogether, the results of these sequence analyses indicate that the frequency of HIV-1 strains showing features associated with reduced sensitivity to PYHIN proteins increased over the last four decades.
Figure 7. Increasing length of the LTR region upstream of the tandem NF-κB sites.

(A) Sequence of the US-NF-κB-II region in the HIV-1 HXB2 reference strain.
(B and C) Length of the region corresponding to the US-NF-κB-II sequence shown in (A) in 2,347 sequences from subtype B and 2,367 sequences from clade C HIV-1-infected individuals (one viral sequence from each person) obtained in the years indicated.
(D) Changes in the length of US-NF-κB-II region (left) and the number of potential NF-κB (middle) and RBEIII sites (right) in the US-NF-κB-II region in subtype B (upper) and C (lower) LTRs. Asterisk (*) indicates that the actual number is one higher because the highly conserved NF-κB-I site is not included in the region analyzed (see A). For clade C, no sequences were available before 1985, and just five sequences were available for the 1986–1990 period.
See also Figure S5.
DISCUSSION
IFI16 and other nuclear human PYHIN proteins (i.e., PYHIN1/IFIX and MNDA) restrict subtype A, B, and D strains of HIV-1 in primary human T cells and/or macrophages by reducing the availability of Sp1 for proviral transcription (Bosso et al., 2020a; Hotter et al., 2019). Subtype C HIV-1 strains, however, evade this restriction because they are less dependent on Sp1 for proviral gene expression. Here, we demonstrate that the acquisition of an additional NF-κB site in the LTR promoter plays a key role in the resistance of subtype C HIV-1 strains to restriction by nuclear human PYHIN proteins. In addition, our results show that clade B HIV-1 strains may evade PYHIN restriction by different alterations in the LTR, i.e., a sequence duplication and an A-to-G change just upstream of the tandem NF-κB element. Analysis of HIV-1 sequences obtained over four decades indicates that the frequency of resistance-conferring changes in clade B and C LTRs is increasing.
Subtype C HIV-1 strains are currently responsible for almost half of all global HIV-1 infections, and thus are at least 4-fold more prevalent than any other subtype (Hemelaar et al., 2019). Whether or not specific viral features render subtype C viruses particularly fit for spread in the human population is under debate (Gartner et al., 2020). It has been reported that the additional NF-κB site increases viral gene expression, although the impact was usually modest, i.e., ~1.5- to 2-fold (Bachu et al., 2012; Jeeninga et al., 2000; Montano et al., 1997; Naghavi et al., 1999). We found that endogenous IFI16 expression reduces infectious HIV-1 CH058 yield in primary CD4+ T cells by more than an order of magnitude (Figure 6). This result most likely still underestimates the inhibitory effect of IFI16 on susceptible primary HIV-1 strains since the average frequency of IFI16 KO cells achieved by treatment with Cas9 and IFI16-specific gRNA was just ~70% (Bosso et al., 2020a, 2020b). Introduction of the additional NF-κB site that is characteristic for subtype C strains conferred almost full resistance to this inhibitory mechanism. IFI16, as well as MNDA and IFIX, which restrict HIV-1 by the same mechanism, are all known to be induced by IFNs (Schattgen and Fitzgerald, 2011). The levels of IFN are particularly high during the acute phase of HIV-1 infection, and it has been established that relative IFN resistance is a key determinant of HIV-1 transmission fitness (Iyer et al., 2017; Parrish et al., 2013). Notably, analyses of clade B HIV-1 sequences available in the Los Alamos HIV database obtained at different stages of infection showed that 3 of 65 LTRs (4.6%) from acutely infected individuals and 6 of 99 (6.1%) from AIDS patients, but only 1 of 146 (0.7%) LTRs from chronically infected individuals contain an additional NF-κB site, similarly to clade C viruses. This agrees with the possibility that an additional NF-κB site is particularly advantageous during the early and late stages of infection when the levels of immune activation are high. It is tempting to speculate that the ability to evade restriction by IFN-inducible PYHIN proteins may play a role in the efficient spread of HIV-1 subtype C strains. Notably, it has been reported that an SIVmac239 chimera containing the clade C LTR dominates during primary infection but is later outgrown by a chimera containing a subtype B LTR (Centlivre et al., 2005). Thus, subtype C LTRs might be specifically associated with a selective advantage during the earliest stage of HIV-1 infection when the virus is most vulnerable to elimination.
The additional NF-κB site is characteristic for subtype C HIV-1 strains, and our previous analyses of a set of 18 subtype A, B, and D strains identified only a single subtype B IMC (THRO) that was resistant to IFI16 inhibition (Hotter et al., 2019). Here, we show that the underlying determinants are distinct from that of clade C viruses and involve an A-to-G change just upstream (i.e., at the −1 position) of the tandem NF-κB sites and a sequence duplication, referred to as MFNLP because of its high frequency in clade B LTRs. Evasion of restriction by nuclear PYHIN proteins by at least two different mechanisms together with the global predominance of clade C viruses suggests that this feature may be advantageous for viral replication and spread in vivo. Further sequence and functional analyses are required to better assess the emergence of HIV-1 resistance to nuclear PYHIN proteins. We show that the MFNLP in clade B LTRs reduces susceptibility to PYHIN restriction and found that the frequency of viruses containing such duplications in the LTR is increasing (Figures 7B and S6). Similarly, we observed that the length of this part of clade C LTRs is also increasing, and that an increasing length is frequently associated with additional putative NF-κB sites. Notably, it has been reported that HIV-1 clade C strains containing an additional NF-κB, NF-κB-like, or RBEIII site in the viral promoter are replacing the prototype subtype C viruses containing three NF-κB sites in India (Bachu et al., 2012; Dave et al., 2020). Notably, RBEIII sites for RBF-2 binding are commonly found in the MFNLP region of clade B LTRs (Estable et al., 1998), and the frequency of LTR sequences containing additional RBEIII sites increased independently of their country of origin (data not shown). Altogether, these data support the possibility that resistance to nuclear PYHIN proteins is advantageous for HIV-1 and increasing over the last decades.
The present results agree with the previous finding that Sp1 is a limiting factor for efficient transcription of most subtypes of HIV-1 (Bosso et al., 2020a; Hotter et al., 2019). In addition, they further reveal subtype-specific differences in the effects of Sp1 and NF-κB for efficient viral gene expression. At high levels of Sp1, subtype B HIV-1 IMCs generated higher infectious virus yields than clade C viruses, while the opposite was observed at high levels of NF-κB (Figures 5 and S5). Our finding that the relative replication fitness of clade B and C viruses depends on the availability of Sp1 and NF-κB might explain some past controversial findings (Abraha et al., 2009; Ariën et al., 2005; Rodriguez et al., 2009). One interesting question is whether these distinct properties affect viral pathogenesis. Sp1 is almost ubiquitously expressed and constitutively active, while NF-κB is strongly activated upon T cell stimulation. It has been suggested that HIV-1 replication in highly stimulated but short-lived effector T cells might be less harmful for the host than virus infection of memory T cells (Descours et al., 2012; Klatt et al., 2014). It is tempting to speculate that clade C viruses may replicate particularly well in activated T cells showing high levels of NF-κB activation, while other subtypes of HIV-1 spread well in T cell subsets expressing high levels of Sp1. Although Sp1 is ubiquitously expressed, the available levels of this transcription factor may vary and become limited, especially upon sequestration by nuclear PYHIN proteins under inflammatory conditions. In addition, it has been reported that Sp1 is involved in tissue- and cell-type-specific regulation of cellular gene expression (McAllister et al., 2000; O’Connor et al., 2016). Thus, it will be interesting to further investigate how differential dependency on Sp1 affects the HIV-1 cell and tissue tropism. It has been reported that women infected with clade A, C, and D HIV-1 strains do not show differences in plasma viral loads, but those infected with subtype C strains showed slower declines in CD4+ T cell counts (Venner et al., 2016). However, comparison of disease progression characteristics between subtypes yielded contradictory data, and it is unclear whether subtype C viruses are less virulent than other subtypes of HIV-1 (Gartner et al., 2020).
In conclusion, we demonstrate that the additional NF-κB site reduces the dependency of subtype C HIV-1 strains on Sp1 for efficient restriction and consequently confers resistance to restriction by IFN-inducible nuclear human PYHIN proteins (IFI16, PYHIN1, and MNDA). We also show that subtype B viruses may achieve the same by mutating their LTR at different locations. This supports a role of this viral evasion mechanism in vivo and suggests that other subtypes of HIV-1 may occasionally evolve properties similar to those of clade C HIV-1 strains. Although further studies are required to obtain definitive proof, our finding that IFI16 efficiently restricts non-C transmitted-founder HIV-1 strains in primary CD4+ T cells and macrophages supports a key role of PYHIN resistance for viral transmission fitness. To which extent subtype-dependent differences in Sp1 and NF-κB dependency and responsiveness affect HIV-1 pathogenicity and the establishment of latent viral reservoirs in different T cell subsets and responsiveness to latency reversing agents warrants further study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Frank Kirchhoff (frank.kirchhoff@uni-ulm.de)
Materials availability
All unique reagents generated in this study are listed in the Key resources table and available from the Lead Contact.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-IFI16 (1G7) | Santa Cruz | Cat# sc-8023; RRID:AB_627775 |
| Rat monoclonal anti-GAPDH | Biolegend | Cat# 607902; RRID:AB_2734503 |
| Mouse monoclonal anti-GAPDH | Santa Cruz | Cat# sc-365062; RRID:AB_10847862 |
| Rabbit polyclonal anti-p65 | Santa Cruz | Cat# sc-372; RRID:AB_632037 |
| Mouse monoclonal anti-HA | Abcam | Cat# ab18181; RRID:AB_444303 |
| Rabbit polyclonal anti-Sp1 | Abcam | Cat# ab13370; RRID:AB_300283 |
| IRDye® 680RD Goat anti-Mouse IgG (H + L) | LI-COR | Cat# 926-68070; RRID:AB_10956588 |
| IRDye® 680RD Goat anti-Rabbit IgG (H + L) | LI-COR | Cat# 925-68071; RRID:AB_2721181 |
| IRDye® 800CW Goat anti-Rat IgG (H + L) | LI-COR | Cat# 926-32219; RRID:AB_1850025 |
| IRDye® 800CW Goat anti-Mouse IgG (H + L) | LI-COR | Cat# 926-32210; RRID:AB_621842 |
| Rabbit anti-p24 serum derived from immunized rabbits | Eurogentec | N/A |
| Peroxidase-AffiniPure goat anti-rabbit IgG, Fc fragment specific antibody |
Dianova | Cat# 111-035-008; RRID:AB_2337937 |
| Anti-HIV-1 p24 core antigen (MAK183) | ExBIO | Cat# 11-CM006-BULK |
| Bacterial strains | ||
| Escherichia coli XL-2 blue | Escherichia coli XL-2 blue | Escherichia coli XL-2 blue |
| XL2-Blue MRF’ TM Ultracompetent cells | Agilent Technologies | Cat# 200151 |
| Biological samples | ||
| Human: Peripheral blood mononuclear cells | DRK-Blutspendedienst BW-Hessen, Ulm, Germany |
N/A |
| Chemicals, peptides, and recombinant proteins | ||
| L-Glutamine | Pan Biotech | Cat# P04-80100 |
| Penicillin-Streptomycin | ThermoFisher | Cat# 15140122 |
| Recombinant human IL-2 | NIH AIDS Reagent | Cat# 136 |
| HiFi Cas9 nuclease V3 | IDT | Cat #1081061 |
| TransIT®-LT1 Transfection Reagent | Mirus | Cat# MIR 2305 |
| β-mercaptoethanol | Sigma Aldrich | Cat# M6250-100ML |
| HIV-1 p24 protein (ELISA standard) | Abcam | Cat# 43037 |
| KPL SureBlue TMB Microwell Peroxidase Substrate | Medac | Cat# 52-00-04 |
| Sulfuric acid concentrate for 1l standard solution 0.5 M H2SO4 | Sigma-Aldrich | Cat# 38294-1EA |
| 4X Protein Sample Loading Buffer | LI-COR | Cat# 928-40004 |
| Critical commercial assays | ||
| RosetteSep Human CD4+ T Cell Enrichment Cocktail | Stem Cell Technologies | Cat# 15062 |
| Amaxa™ 4D-Nucleofactor™ Human Activated Cell P3 Lonza Kit | Lonza | Cat# V4XP-3024 |
| Q5® High-Fidelity PCR Kit | New England Biolabs | Cat# E0555S |
| Phusion High-Fidelity PCR Kit | ThermoFisher | Cat# F553L |
| DNA Ligation Kit Ver. 2.1 | TaKaRa | Cat# 6022 |
| GalScreen | Applied Bioscience | Cat# T1027 |
| Experimental models: Cell lines | ||
| Human: HEK293T cells | ATCC | Cat# CRL-3216 RRID: CVCL_0063 |
| Human: TZM-bl cells | NIH AIDS Reagent Program | Cat# 8129 RRID: CVCL_B478 |
| Human: HAP1 cells | Horizon | Cat# HZGHC001141c002 RRID:CVCL_TQ04 |
| Human: Sp1 KO HAP1 cells | Horizon | Cat# HZGHC001141c002 RRID:CVCL_TQ04 |
| Oligonucleotides | ||
| IFI16 sgRNA: GACCAGCCCTATCAAGAAAG | IDT | N/A |
| Non-targeting control sgRNA: ACGGAGGCTAAGCGTCGCAA | IDT | N/A |
| Primers used for cloning (see Table S4) | Biomers.net | N/A |
| Recombinant DNA | ||
| Plasmid: pCG_IFI16 | (Hotter et al., 2019) | N/A |
| Plasmid: pCG_PYHIN1 | (Bosso et al., 2020a) | N/A |
| Plasmid: pCG_MNDA | (Bosso et al., 2020a) | N/A |
| Plasmid: p65 | This paper | N/A |
| Plasmid: pCG_Sp1 | (Hotter et al., 2019) | N/A |
| Plasmid: pCMV-VSV-G | Addgene | Cat# 8454 |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype B CH058.c (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pCR-XL-TOPO HIV-1 M subtype B pTHRO.c (transmitted founder virus) | B. H. Hahn (Ochsenbauer et al., 2012) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR of THRO | (Hotter et al., 2019) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M THRO with 3′ and 5′ LTR of CH058 | (Hotter et al., 2019) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR of THROlong-CH058 | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR of THROshort-CH058 | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR of CH058long-THRO | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR of CH058short-THRO | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M THRO with 5′ and 3′ LTR of THROlong-CH058 | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M THRO with 5′ and 3′ LTR of THROshort-CH058 | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M THRO with 5′ and 3′ LTR of CH058long-THRO | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M THRO with 5′ and 3′ LTR of CH058short-THRO | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR of CH058long-CH058 | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M THRO with 5′ and 3′ LTR of THROshort-THRO | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR A349G | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M THRO with 3′ and 5′ LTR G380A | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M subtype C ZM247Fv-1 (transmitted founder virus) | B. H. Hahn (Salazar-Gonzalez et al., 2009) | N/A |
| Plasmid: pCR-XL TOPO_HIV-1 M subtype C CH185 (transmitted founder) | B. H. Hahn (Parrish et al., 2013) | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M ZM247Fv-1 with 5′ and 3′ LTR +A350 | This paper | N/A |
| Plasmid: pCR-XL TOPO_HIV-1 M CH185 with 5′ and 3′ LTR T353A | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M ZM247Fv-1 with 5′ and 3′ LTR ΔNF-κB | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M ZM247Fv-1 with 5′ and 3′ LTR ΔNF-κB +A350 | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH185 with 5′ and 3′ LTR ΔNF-κB | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH185 with 5′ and 3′ LTR ΔNF-κB T353A | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR +NF-κB | This paper | N/A |
| Plasmid: pCR-XL-TOPO_HIV-1 M CH058 with 5′ and 3′ LTR +NF-κB A349G | This paper | N/A |
| Software and algorithms | ||
| Corel DRAW 2019 | Corel Corporation | https://www.coreldraw.com/en/ |
| GraphPad Prism Version 8 | GraphPad Software, Inc. | https://www.graphpad.com RRID: SCR_002798 |
| ImageJ | Open source | https://imagej.nih.gov/ij/ |
| LI-COR Image Studio Lite Version 5.0 | LI-COR | https://www.licor.com/ RRID: SCR_013715 |
| MEGA6 | (Tamura et al., 2013) | https://www.megasoftware.net/ |
Data and code availability
This study did not generate or analyze datasets or codes.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Primary Cultures
Experiments involving human blood, CD4+ T cells or macrophages, were reviewed and approved by the Institutional Review Board (i.e., the Ethics Committee of Ulm University). Individuals and/or their legal guardians provided written informed consent prior to donating blood. All human-derived samples were anonymized before use. CD4+ T cells were negatively isolated using the RosetteSep Human CD4+ T Cell Enrichment Cocktail (Stem Cell Technologies) according to the manufacturer’s instructions. Primary cells were cultured in RPMI-1640 medium containing 10% FCS, glutamine (2 mM), streptomycin (100 μg/ml), penicillin (100 U/ml) and interleukin 2 (IL-2) (10 ng/ml).
Cell lines
All cells were cultured at 37°C in a 5% CO2 atmosphere. HEK293T and TZM-bl cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal calf serum (FCS), L-glutamine (2 mM), streptomycin (100 μg/ml) and penicillin (100 U/ml). HEK293T cells (DuBridge et al., 1987) were provided and authenticated by the ATCC. TZM-bl cells were provided and authenticated by the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH from Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc (Platt et al., 1998). TZM-bl are derived from HeLa cells, which were isolated from a 30-year-old female. HAP1 cells were maintained in Iscove’s Modified Dulbecco’s Medium (IMDM) supplemented with 10% FCS, streptomycin (100 μg/mL) and penicillin (100 U/mL). HAP1 cells were provided by Horizon and represent a near-haploid human cell line that was derived from the male chronic myelogenous leukemia (CML) cell line KBM-7 (Carette et al., 2009).
METHOD DETAILS
Construction of the expression constructs
Expression vectors for IFI16 and Sp1 were generated by gene amplification via PCR using cDNA as template and subsequent cloning via unique XbaI and MluI restriction sites into a pCG expression vector. The constructs of PYHIN1 and MNDA were a kind gift of Professor Jakobsen and the p65 expression plasmid was kindly provided by Dr. B. Baumann. One internal XbaI restriction site was removed from MNDA via overlap-extension (OE) PCR prior to its subcloning into the pCG expression vector. Sequences of primers used for cloning are listed in Table S4.
Proviral HIV-1 constructs
Infectious molecular clones of HIV-1 CH058, THRO, ZM247 and CH0185 have been described previously (Ochsenbauer et al., 2012; Parrish et al., 2013). Overlap-extension (OE) PCR mutagenesis and standard cloning techniques were used to swap 5‘ and 3‘ LTR sequences of HIV-1 CH058 and THRO. OE-PCR was also employed to generate chimeric CH058 and THRO LTRs. Briefly, PCR fragments encompassing the fused hybrid 5‘ or 3‘ LTRs and single restriction sites at their termini were inserted into the CH058 proviral construct using NotI-BstZ17I and MluI-StuI and into THRO using SbfI-NotI and BamHI-BbvCI restriction sites, respectively. DNA fragments encompassing wild-type or mutant CH058, THRO, ZM247, CH185 LTRs were used as template for mutating the A349 in CH058, G380 in THRO, T353 in CH185 and inserting one additional “A” after T349 in ZM247. PCR-OE fragments containing the desired changes were used to replace the 5′ and 3′ LTRs in HIV-1 IMCs using NotI-BstZ17I and MluI-StuI (CH058), SbfI-NotI and BamHI-BbvCI (THRO), BamHI-AgeI and Pac-NotI (CH185) and MluI-AgeI and PacI-NotI (ZM247). The above-mentioned proviral HIV-1 constructs were used as basis for the generation of a final set of LTR mutants differing specifically in the number of NF-κB binding sites by using OE-PCR mutagenesis and standard cloning techniques. Sequences of primers used for cloning are listed in Table S4. All parental and mutant proviral HIV-1 constructs were sequenced to verify the presence of desired and absence of additional changes.
Transfections and production of virus stocks
HEK293T cells were transiently transfected using the calcium-phosphate precipitation method. One day before transfection, 5x105 HEK293T cells/well were seeded in 6-well plates in 2 mL medium to obtain a confluence of 50%–60% at the time of transfection. For transfection, DNA was mixed with 13 μL 2 M CaCl2 and filled up with water to 100 μl. Afterward, 100 μL 2 x HBS was added dropwise to this mixture, which was mixed by pipetting and added dropwise to the cells. To generate virus stocks, cells were transfected with proviral constructs (5 μg), in combination with an expression vector for the vesicular stomatitis virus glycoprotein (VSV-G) (1 μg) (Fouchier et al., 1997). Prior to spinoculation, virus stocks were concentrated by a 10-fold factor using Amicon Ultra 15 100 k Centrifugal Filters (Merck Millipore). To test the antiviral effect of different proteins and HIV-1 responsiveness to Sp1 and p65, pCG-based expression constructs were cotransfected with proviral constructs. Whenever different amounts of pCG expression vectors were used within an experiment, empty vector control plasmids were used to keep the total DNA amount constant. The transfected cells were incubated for 8-16 h before the medium was replaced by fresh supplemented DMEM. HAP1 cells were transfected using the TransIT-LT1 Transfection Reagent (Mirus). One day before transfection, 2.5*105 HAP1 cells/well were seeded in 12-well plates in 1 mL medium. For transfection, DNA and 4 μL of TransIT-LT1 Transfection Reagent were mixed with 50 μl Opti-MEM, each. These two solutions were then mixed, incubated at RT for 20 min and added dropwise to the cells. The transfected cells were incubated for 6 h before the medium was replaced by fresh supplemented IMDM. For both HEK293T and HAP1 cells, 48 h post transfection, cell culture supernatants were harvested, centrifuged at 1.000 x g for 3 min and transferred to fresh tubes to remove cell debris.
Transient expression of PYHIN protein or transcription factors
HEK293T cells in 6-well plates were transfected with pCG empty vector IRES GFP (control) or increasing concentrations (0.1 μg, 0.5μg, 1μg) of p65 expression vector or pCG IRES BFP vector expressing C-terminally HA-tagged IFI16, IFIX, MNDA or Sp1. The total amount of DNA was normalized to 5μg using pCG empty vector IRES BFP. Two days post-transfection, cells were harvested and processed for Western Blot analysis.
Viral infectivity
To determine infectious virus yield, 6,000 TZM-bl reporter cells/well were seeded in 96-well plates and infected with cell culture supernatants in triplicates on the following day. Three days post-infection, cells were lysed and β-galactosidase reporter gene expression was determined using the GalScreen Kit (Applied Bioscience) according to the manufacturer’s instructions with an Orion microplate luminometer (Berthold).
ELISA
HIV-1 p24 antigen levels in cell culture supernatants were determined using an in-house ELISA (Konvalinka et al., 1995). 96-well MaxiSorp microplates were coated with 500 ng/ml anti-HIV-1 p24 (100 μl/well) and incubated in a wet chamber at RT overnight. The next day, plates were washed 3 times with 350 μL PBS-T (PBS and 0.05% Tween 20) and incubated with 100 μL blocking solution (PBS and 10% (v/v) FCS) for 2 hours at 37°C. After washing, the plates were loaded with 100 μL serially diluted HIV-1 p24 protein as standard and dilutions of virus stocks lysed with 1% (v/v) Triton X-100. After overnight incubation in a wet chamber at RT, plates were washed to remove unbound capsid proteins. 100 μl/well polyclonal rabbit antiserum against p24 antigen (1:1,000 in PBS-T (PBS, 0.05% Tween 20 and 10% (v/v) FCS)) was added for 1 hour at 37°C and after another washing step, 100 μL of a goat anti-rabbit HRP-coupled antibody (1:2,000) was loaded on the plate and incubated for another hour at 37°C. Finally, the plate was washed and 100 μL SureBlue TMB 1-Component Microwell Peroxidase Substrate was added. After 20 minutes shaking at 450 rpm and RT, the reaction was stopped with 0.5 M H2SO4 (100 μl/well). The optic density, proportional to the p24 capsid antigen amount, was determined by comparing with a standard curve and measured at 450 nm and 650 nm with the Thermo Max microplate reader (Molecular devices, UK).
Western blot
To determine expression of cellular proteins, cells were washed in PBS and subsequently lysed in western blot lysis buffer (150 mM NaCl, 50 mM HEPES, 5 mM EDTA, 0.1% NP40, 500 μM Na3VO4, 500 μM NaF, pH 7.5). Lysates were mixed with 4x Protein Sample Loading Buffer (LI-COR, at a final dilution of 1x) supplemented with 10% β-mercaptoethanol (Sigma Aldrich), heated at 95°C for 5 min, separated on NuPAGE 4 ± 12% Bis-Tris Gels (Invitrogen) for 90 minutes at 130 V and blotted onto Immobilon-FL PVDF membranes (Merck Millipore or Thermo Fisher). Membranes were blocked in 5% milk and probed with mouse anti-IFI16 (Santa Cruz Biotechnology, sc-8023), mouse anti-GAPDH (Santa Cruz Biotechnology, sc-365062), rat anti-GAPDH (BioLegend, 607902), rabbit anti-NFκB p65 (Santa Cruz Biotechnology, sc-372), mouse anti-HA (Abcam, ab18181), rabbit anti-Sp1 (Abcam, ab13370), followed by fluorescent goat anti-rabbit, anti-rat and anti-mouse secondary antibodies (680RD or 800CW, LI-COR).
Nucleofection and transduction of CD4+ T lymphocytes
CD4+ T lymphocytes were isolated from healthy donors as described above. Cells were stimulated with IL-2 (10 ng/ml) (MACS Miltenyi Biotec) and with anti-CD3/CD28 beads (Life Technologies). Cells were cultured in RPMI-1640 medium containing 20% FCS. Three days later, 1*106 cells were nucleofected with the HiFi Cas9 Nuclease V3 (IDT)/gRNA complex (80 pmol/300 pml) (Lonza) using a non-targeting (IDT (ACGGAGGCTAAGCGTCGCAA)) or an IFI16-specific (IDT (GACCAGCCCTATCAAGAAAG)) sgRNA, using the Amaxa 4D-Nucleofector Human Activated T Cell P3 Lonza Kit (Lonza), pulse code EO115. At four-days post Cas9/sgRNA-transfection, 3*105 cells/sample were transduced with the indicated VSV-G pseudotyped HIV-1 strains by spinoculation and 1.5*106 uninfected cells were processed for western blot analysis. After three days, supernatants were harvested, diluted 1:10 in DMEM containing 10% FCS, L-glutamine (2 mM), streptomycin (100 μg/ml) and penicillin (100 U/ml), and used to quantify viral capsid protein p24 via ELISA and infectious virus yield via the TZM-bl reporter cells assay.
Association analyses of HIV-1 genome sequences and sensitivity to IFI16
A multiple sequence alignment of full-length HIV nucleotide sequences was constructed using MUSCLE in MEGA6. Of 10,308 aligned sites, 2,868 sites were regarded as informative sites. An informative site was defined as site with ≥ 2 genotypes that can be found in at least three HIV-1 clones each. For each informative site, associations of genotypes and residual median infectious virus yield in the presence of IFI16 (% of control without IFI16) were examined using one-way Anova. Family-wise error rate (FER) was calculated using Bonferroni correlation. 12 sites with FER ≤ 0.01 were regarded as statistically significant. To control for clade-dependent effects, the association analyses were repeated using a linear regression model with correlating subgroup effects. In this case, 26 sites (FER ≤ 0.01) were identified.
HIV-1 isolate sequence analysis
Sequences of HIV-1 isolates (one sequence per patient with 0% of non-ACGT) were obtained from the Los Alamos HIV database (https://www.hiv.lanl.gov/content/index). The RBEIII binding site was defined as 5′-ACTGCTGA-3′ and NF-κB-like sites were defined as those exactly or closely matching (with maximum 1 nt variation) the consensus interaction sequence 5′-GGGRNNYYCC-3′ where R is a purine, N is any nucleotide and Y is pyrimidine. Sequence logo plots were generated using WebLogo tool (https://weblogo.berkeley.edu/).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad PRISM 8 (GraphPad Software). P values were determined using a two-tailed Student’s t test. Quantification of the p24 concentration in the supernatants via ELISA was achieved with GraphPad PRISM 8, using the “Sigmoidal, 4PL, X is log(concentration)” analysis option. Unless otherwise stated, data are shown as the mean of at least three independent experiments ± SD. Significant differences are indicated as: *, p < 0.05; **, p < 0.01; ***, p < 0.001. Statistical parameters are specified in the figure legends.
Supplementary Material
Highlights.
A third NF-κB binding site renders HIV-1 clade C resistant to PYHIN restriction
Transcription of HIV-1 subtypes differs strongly in Sp1 and NF-κB dependency
The frequency of PYHIN-resistant HIV-1 clade B strains seems to be increasing
Resistance to PYHIN restriction may explain the dominance of subtype C HIV-1
ACKNOWLEDGMENTS
We thank Martha Mayer and Daniela Krnavek for technical assistance. TZM-bl cells were obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu, and Tranzyme Inc. This work was supported by grants from the National Institutes of Health (R01 AI 162646, UM1 AI 164570, and P30 AI 045008), the German Research Foundation (DFG CRC 1279 and SPP 1923), and the Baden-Württemberg Foundation (BWST-ISF2018-032). D.K.’s work was funded by the German Research Foundation (DFG KM 5/1-1). K.S. was supported by a grant from the German Federal Ministry of Research and Education (BMBF Junior Research Group IMMUNOMOD). D.S. was supported by the Heisenberg Program of the German Research Foundation and the Canon Foundation Europe. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109735.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abraha A, Nankya IL, Gibson R, Demers K, Tebit DM, Johnston E, Katzenstein D, Siddiqui A, Herrera C, Fischetti L, et al. (2009). CCR5- and CXCR4-tropic subtype C human immunodeficiency virus type 1 isolates have a lower level of pathogenic fitness than other dominant group M subtypes: implications for the epidemic. J. Virol 83, 5592–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelis K, Albert J, Mamais I, Magiorkinis G, Hatzakis A, Hamouda O, Struck D, Vercauteren J, Wensing AMJ, Alexiev I, et al. (2015). Global Dispersal Pattern of HIV Type 1 Subtype CRF01_AE: A Genetic Trace of Human Mobility Related to Heterosexual Sexual Activities Centralized in Southeast Asia. J. Infect. Dis 211, 1735–1744. [DOI] [PubMed] [Google Scholar]
- Ariën KK, Abraha A, Quiñones-Mateu ME, Kestens L, Vanham G, and Arts EJ (2005). The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. J. Virol 79, 8979–8990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachu M, Yalla S, Asokan M, Verma A, Neogi U, Sharma S, Murali RV, Mukthey AB, Bhatt R, Chatterjee S, et al. (2012). Multiple NF-κB sites in HIV-1 subtype C long terminal repeat confer superior magnitude of transcription and thereby the enhanced viral predominance. J. Biol. Chem 287, 44714–44735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bbosa N, Kaleebu P, and Ssemwanga D (2019). HIV subtype diversity worldwide. Curr. Opin. HIV AIDS 14, 153–160. [DOI] [PubMed] [Google Scholar]
- Bosso M, Prelli Bozzo C, Hotter D, Volcic M, Stürzel CM, Rammelt A, Ni Y, Urban S, Becker M, Schelhaas M, et al. (2020a). Nuclear PYHIN proteins target the host transcription factor Sp1 thereby restricting HIV-1 in human macrophages and CD4+ T cells. PLoS Pathog. 16, e1008752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosso M, Bozzo CP, Volcic M, and Kirchhoff F (2020b). IFI16 knockdown in primary HIV-1 target cells. STAR Protoc. 2, 100236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carette JE, Guimaraes CP, Varadarajan M, Park AS, Wuethrich I, Godarova A, Kotecki M, Cochran BH, Spooner E, Ploegh HL, and Brummelkamp TR (2009). Haploid genetic screens in human cells identify host factors used by pathogens. Science 326, 1231–1235. [DOI] [PubMed] [Google Scholar]
- Centlivre M, Sommer P, Michel M, Ho Tsong Fang R, Gofflo S, Valladeau J, Schmitt N, Thierry F, Hurtrel B, Wain-Hobson S, and Sala M (2005). HIV-1 clade promoters strongly influence spatial and temporal dynamics of viral replication in vivo. J. Clin. Invest 115, 348–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL, Ragan MA, Kassahn KS, and Stacey KJ (2012). The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol. Biol 12, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dave RS, Ali H, Sil S, Knight LA, Pandey K, Madduri LSV, Qiu F, Ranga U, Buch S, and Byrareddy SN (2020). NF-κB Duplications in the Promoter-Variant HIV-1C LTR Impact Inflammation Without Altering Viral Replication in the Context of Simian Human Immunodeficiency Viruses and Opioid-Exposure. Front. Immunol 11, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Baar MP, De Ronde A, Berkhout B, Cornelissen M, Van Der Horn KH, Van Der Schoot AM, De Wolf F, Lukashov VV, and Goudsmit J (2000). Subtype-specific sequence variation of the HIV type 1 long terminal repeat and primer-binding site. AIDS Res. Hum. Retroviruses 16, 499–504. [DOI] [PubMed] [Google Scholar]
- Descours B, Avettand-Fenoel V, Blanc C, Samri A, Mélard A, Supervie V, Theodorou I, Carcelain G, Rouzioux C, and Autran B; ALT ANRS CO15 Study Group (2012). Immune responses driven by protective human leukocyte antigen alleles from long-term nonprogressors are associated with low HIV reservoir in central memory CD4 T cells. Clin. Infect. Dis 54, 1495–1503. [DOI] [PubMed] [Google Scholar]
- DuBridge RB, Tang P, Hsia HC, Leong PM, Miller JH, and Calos MP (1987). Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol 7, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estable MC (2007). In search of a function for the most frequent naturally-occurring length polymorphism (MFNLP) of the HIV-1 LTR: retaining functional coupling, of Nef and RBF-2, at RBEIII? Int. J. Biol. Sci 3, 318–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estable MC, Bell B, Merzouki A, Montaner JS, O’Shaughnessy MV, and Sadowski IJ (1996). Human immunodeficiency virus type 1 long terminal repeat variants from 42 patients representing all stages of infection display a wide range of sequence polymorphism and transcription activity. J. Virol 70, 4053–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estable MC, Bell B, Hirst M, and Sadowski I (1998). Naturally occurring human immunodeficiency virus type 1 long terminal repeats have a frequently observed duplication that binds RBF-2 and represses transcription. J. Virol 72, 6465–6474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fouchier RA, Meyer BE, Simon JH, Fischer U, and Malim MH (1997). HIV-1 infection of non-dividing cells: evidence that the amino-terminal basic region of the viral matrix protein is important for Gag processing but not for post-entry nuclear import. EMBO J. 16, 4531–4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartner MJ, Roche M, Churchill MJ, Gorry PR, and Flynn JK (2020). Understanding the mechanisms driving the spread of subtype C HIV-1. EBioMedicine 53, 102682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemelaar J, Elangovan R, Yun J, Dickson-Tetteh L, Fleminger I, Kirtley S, Williams B, Gouws-Williams E, and Ghys PD; WHO–UNAIDS Network for HIV Isolation Characterisation (2019). Global and regional molecular epidemiology of HIV-1, 1990-2015: a systematic review, global survey, and trend analysis. Lancet Infect. Dis 19, 143–155. [DOI] [PubMed] [Google Scholar]
- Hotter D, Bosso M, Jønsson KL, Krapp C, Stürzel CM, Das A, Littwitz-Salomon E, Berkhout B, Russ A, Wittmann S, et al. (2019). IFI16 Targets the Transcription Factor Sp1 to Suppress HIV-1 Transcription and Latency Reactivation. Cell Host Microbe 25, 858–872.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt G, and Tiemessen CT (2000). Occurrence of additional NF-kappaB-binding motifs in the long terminal repeat region of South African HIV type 1 subtype C isolates. AIDS Res. Hum. Retroviruses 16, 305–306. [DOI] [PubMed] [Google Scholar]
- Iyer SS, Bibollet-Ruche F, Sherrill-Mix S, Learn GH, Plenderleith L, Smith AG, Barbian HJ, Russell RM, Gondim MVP, Bahari CY, et al. (2017). Resistance to type 1 interferons is a major determinant of HIV-1 transmission fitness. Proc. Natl. Acad. Sci. USA 114, E590–E599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeeninga RE, Hoogenkamp M, Armand-Ugon M, de Baar M, Verhoef K, and Berkhout B (2000). Functional differences between the long terminal repeat transcriptional promoters of human immunodeficiency virus type 1 subtypes A through G. J. Virol 74, 3740–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klatt NR, Bosinger SE, Peck M, Richert-Spuhler LE, Heigele A, Gile JP, Patel N, Taaffe J, Julg B, Camerini D, et al. (2014). Limited HIV infection of central memory and stem cell memory CD4+ T cells is associated with lack of progression in viremic individuals. PLoS Pathog. 10, e1004345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konvalinka J, Litterst MA, Welker R, Kottler H, Rippmann F, Heuser AM, and Kräusslich HG (1995). An active-site mutation in the human immunodeficiency virus type 1 proteinase (PR) causes reduced PR activity and loss of PR-mediated cytotoxicity without apparent effect on virus maturation and infectivity. J. Virol 69, 7180–7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemieux A-M, Paré M-È, Audet B, Legault E, Lefort S, Boucher N, Landry S, van Opijnen T, Berkhout B, Naghavi MH, et al. (2004). T-cell activation leads to poor activation of the HIV-1 clade E long terminal repeat and weak association of nuclear factor-kappaB and NFAT with its enhancer region. J. Biol. Chem 279, 52949–52960. [DOI] [PubMed] [Google Scholar]
- Locateli D, Stoco PH, de Queiroz ATL, Alcântara LCJ, Ferreira LGE, Zanetti CR, Rodrigues R, Grisard EC, and Pinto AR (2007). Molecular epidemiology of HIV-1 in Santa Catarina State confirms increases of subtype C in Southern Brazil. J. Med. Virol 79, 1455–1463. [DOI] [PubMed] [Google Scholar]
- McAllister JJ, Phillips D, Millhouse S, Conner J, Hogan T, Ross HL, and Wigdahl B (2000). Analysis of the HIV-1 LTR NF-kappaB-proximal Sp site III: evidence for cell type-specific gene regulation and viral replication. Virology 274, 262–277. [DOI] [PubMed] [Google Scholar]
- McLaren PJ, Gawanbacht A, Pyndiah N, Krapp C, Hotter D, Kluge SF, Götz N, Heilmann J, Mack K, Sauter D, et al. (2015). Identification of potential HIV restriction factors by combining evolutionary genomic signatures with functional analyses. Retrovirology 12, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montano MA, Novitsky VA, Blackard JT, Cho NL, Katzenstein DA, and Essex M (1997). Divergent transcriptional regulation among expanding human immunodeficiency virus type 1 subtypes. J. Virol 71, 8657–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munkanta M, Handema R, Kasai H, Gondwe C, Deng X, Yamashita A, Asagi T, Yamamoto N, Ito M, Kasolo F, and Terunuma H (2005). Predominance of three NF-kappaB binding sites in the long terminal repeat region of HIV Type 1 subtype C isolates from Zambia. AIDS Res. Hum. Retroviruses 21, 901–906. [DOI] [PubMed] [Google Scholar]
- Naghavi MH, Schwartz S, Sönnerborg A, and Vahlne A (1999). Long terminal repeat promoter/enhancer activity of different subtypes of HIV type 1. AIDS Res. Hum. Retroviruses 15, 1293–1303. [DOI] [PubMed] [Google Scholar]
- O’Connor L, Gilmour J, and Bonifer C (2016). The Role of the Ubiquitously Expressed Transcription Factor Sp1 in Tissue-specific Transcriptional Regulation and in Disease. Yale J. Biol. Med. 89, 513–525. [PMC free article] [PubMed] [Google Scholar]
- Ochsenbauer C, Edmonds TG, Ding H, Keele BF, Decker J, Salazar MG, Salazar-Gonzalez JF, Shattock R, Haynes BF, Shaw GM, et al. (2012). Generation of transmitted/founder HIV-1 infectious molecular clones and characterization of their replication capacity in CD4 T lymphocytes and monocyte-derived macrophages. J. Virol 86, 2715–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, and Ghosh S (2013). NF-κB: roles and regulation in different CD4(+) T-cell subsets. Immunol. Rev 252, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish NF, Gao F, Li H, Giorgi EE, Barbian HJ, Parrish EH, Zajic L, Iyer SS, Decker JM, Kumar A, et al. (2013). Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. USA 110, 6626–6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Wehrly K, Kuhmann SE, Chesebro B, and Kabat D (1998). Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol 72, 2855–2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MA, Ding M, Ratner D, Chen Y, Tripathy SP, Kulkarni SS, Chatterjee R, Tarwater PM, and Gupta P (2009). High replication fitness and transmission efficiency of HIV-1 subtype C from India: Implications for subtype C predominance. Virology 385, 416–424. [DOI] [PubMed] [Google Scholar]
- Salazar-Gonzalez JF, Salazar MG, Keele BF, Learn GH, Giorgi EE, Li H, Decker JM, Wang S, Baalwa J, Kraus MH, et al. (2009). Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med 206, 1273–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauter D, and Kirchhoff F (2019). Key Viral Adaptations Preceding the AIDS Pandemic. Cell Host Microbe 25, 27–38. [DOI] [PubMed] [Google Scholar]
- Schattgen SA, and Fitzgerald KA (2011). The PYHIN protein family as mediators of host defenses. Immunol. Rev 243, 109–118. [DOI] [PubMed] [Google Scholar]
- Sharp PM, and Hahn BH (2011). Origins of HIV and the AIDS pandemic. Cold Spring Harb. Perspect. Med 1, a006841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MA, De Oliveira T, Brindeiro RM, Diaz RS, Sabino EC, Brigido L, Pires IL, Morgado MG, Dantas MC, Barreira D, et al. ; Brazilian Network for Drug Resistance Surveillance (2003). A specific subtype C of human immunodeficiency virus type 1 circulates in Brazil. AIDS 17, 11–21. [DOI] [PubMed] [Google Scholar]
- Tamura K, Stecher G, Peterson D, Filipski A, and Kumar S (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol 30, 2725–2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venner CM, Nankya I, Kyeyune F, Demers K, Kwok C, Chen P-L, Rwambuya S, Munjoma M, Chipato T, Byamugisha J, et al. (2016). Infecting HIV-1 Subtype Predicts Disease Progression in Women of Sub-Saharan Africa. EBioMedicine 13, 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets or codes.