

Graphical abstract

Keywords: neurodegeneration, protein aggregation, crystal structure, HDX-MS, SAXS
Abbreviations: AXH domain, Ataxin-1 and HMG-box protein 1 domain; CIC, Capicua; DSS, disuccinimidyl suberate; DSG, disuccinimidyl glutarate; HDX-MS, Hydrogen deuterium exchange mass spectrometry; ITC, Isothermal Titration Calorimetry; NLS, nuclear localization signal; polyQ, polyglutamine; pS, phosphorylated Serine; SAXS, small-angle X-ray scattering; SCA1, spinocerebellar ataxia type 1; SUMO, small ubiquitin-related modifier
Highlights
-
•
14-3-3 was postulated to prevent cytoplasmic aggregation of Ataxin-1.
-
•
Experimental support for an anti-aggregation effect of 14-3-3 on Ataxin-1 is provided.
-
•
Structural studies suggest 14-3-3 reduces Ataxin-1 dimerisation and further self-association.
-
•
Modulation of the 14-3-3/Ataxin-1 interaction could provide a treatment for SCA1.
Abstract
Expansion of the polyglutamine tract in the N terminus of Ataxin-1 is the main cause of the neurodegenerative disease, spinocerebellar ataxia type 1 (SCA1). However, the C-terminal part of the protein – including its AXH domain and a phosphorylation on residue serine 776 – also plays a crucial role in disease development. This phosphorylation event is known to be crucial for the interaction of Ataxin-1 with the 14-3-3 adaptor proteins and has been shown to indirectly contribute to Ataxin-1 stability. Here we show that 14-3-3 also has a direct anti-aggregation or “chaperone” effect on Ataxin-1. Furthermore, we provide structural and biophysical information revealing how phosphorylated S776 in the intrinsically disordered C terminus of Ataxin-1 mediates the cytoplasmic interaction with 14-3-3 proteins. Based on these findings, we propose that 14-3-3 exerts the observed chaperone effect by interfering with Ataxin-1 dimerization through its AXH domain, reducing further self-association. The chaperone effect is particularly important in the context of SCA1, as it was previously shown that a soluble form of mutant Ataxin-1 is the major driver of pathology.
Introduction
Spinocerebellar ataxia type 1 (SCA1) is an inherited neurodegenerative disease, for which a cure so far remains elusive. A few early symptoms are gait disturbance and balance problems, brisk deep tendon reflexes, slurred speech, difficulty to swallow and rapid uncontrolled eye movements. In later stages, symptoms include problems with timing and judging distances, muscle atrophy, cognitive impairment and bulbar dysfunction.1 Ultimately, the main cause of death for SCA1 patients is respiratory failure. The disease typically manifests itself from the third or fourth decade, with an average survival period of fifteen years after onset. However, in cases of juvenile onset before the age of thirteen, disease symptoms and progression are more severe, and the young patients die before they reach sixteen years of age.2 The disease symptoms are explained by a degeneration of cerebellar Purkinje cells, inferior olive neurons and neurons within brainstem cranial nerve nuclei.3
On a molecular level, SCA1 is caused by an anomalous expansion of the polyglutamine (polyQ) tract in the N terminus of the Ataxin-1 protein. Native Ataxin-1 proteins have between six and forty-two glutamine repeats, with repeats exceeding twenty-one glutamines interrupted by one-to-three histidines. In contrast, the toxic Ataxin-1 protein has pure polyQ tracts of between thirty-nine and eighty-two units.4 The normal cellular function of Ataxin-1 is not fully understood, but it is known to play a role in regulating transcription and RNA metabolism, through nuclear interactions with other proteins such as the transcriptional repressor Capicua (CIC), and splicing factors RBM17 and U2AF65.5 PolyQ expansion has now been shown to contribute to SCA1 pathology by gain-of-function mechanisms on the Ataxin-1/RBM17 and Ataxin-1/CIC interactions.6, 7
Interestingly, an expanded polyQ tract in the N terminus of Ataxin-1 is not sufficient for disease onset. Indeed, the following regions located in its C-terminal half were also shown to be crucial: the AXH domain (residues 567–689), the nuclear localisation signal (residues 771–774) and finally the serine residue at position 776 (S776), specifically upon its phosphorylation (further referred to as pS776) in the cytoplasm by protein kinase A (PKA).8, 9, 10, 11, 12 A scheme with the domain architecture of Ataxin-1 is shown in Figure 5(A). After translocation to the nucleus, pS776 is important for the interaction of Ataxin-1 with splicing factor RBM17.6 However, pS776 also plays a crucial role in the Ataxin-1 life cycle in the cytoplasm, where it is required for binding of Ataxin-1 to the 14-3-3 adaptor proteins.13
Figure 5.

Analysis of SAXS data for AXH-C, 14-3-3 and their complex. (A) schematic structure of the Ataxin-1 protein. The full-length Ataxin-1 protein has 816 amino acids, with a central AXH domain. In this study, a shorter version of the protein including the AXH domain and C terminus was used. It is referred to as AXH-C. The 14-3-3 binding site created upon phosphorylation of S776 (pS776) is marked. (B) Distance distribution functions for 14-3-3, AXH-C and their complex are shown in green, blue and purple respectively. (C) Dimensionless Kratky plots, with same colour coding as in panel B. The grey lines mark the characteristic maximum for globular proteins of 1.107 at sRg = √3. (D) Ensemble model of AXH-C generated by EOM. The AXH dimer is coloured in shades of blue. Various conformations of the disordered C-termini on each monomer are shown as wire model. (E) Crysol fit of the AXH-C ensemble model (black line) to the experimental AXH-C scattering data (blue dots).
14-3-3 proteins are a highly conserved protein family among eukaryotes. Humans have seven isoforms, denoted by the Greek letters β, ε, γ, η, σ, τ, and ς. These are expressed ubiquitously.14 Besides Ataxin-1, 14-3-3 proteins are known to interact with several hundred other proteins.15, 16 It is therefore no surprise that besides its involvement in SCA1, 14-3-3 proteins are also involved in many diseases including cancer, metabolic diseases, epilepsy, immune disorders, infectious diseases, and other neurodegenerative diseases such as Alzheimer’s and Parkinson’s.17
For Ataxin-1 specifically, binding to 14-3-3 proteins regulates its subcellular localization. Since pS776 lies adjacent to the nuclear localization signal (NLS), binding to 14-3-3 proteins impedes the transport of Ataxin-1 to the nucleus.18 While the actual trigger for releasing 14-3-3 and nuclear translocation of Ataxin-1 yet remains unknown, Lai et al. suggested it might be a second, yet unknown post-translational modification on Ataxin-1.18
In addition, the binding of 14-3-3 proteins has been shown to stabilize Ataxin-1 and promote its accumulation in transfected cells and transgenic flies.13 Also, SCA1 flies showed increased neurodegeneration upon overexpression of 14-3-3 epsilon.13 Furthermore, in one mouse model, haplo-insufficiency of 14-3-3ε rescued cerebellar pathology and motor phenotypes.19 In another mouse model expressing a phospho-unreceptive and thus 14-3-3 protein binding-defective S776A Ataxin-1 mutant, cerebellar extracts showed significantly reduced Ataxin-1 levels compared to mice expressing wild-type Ataxin-1.20 Lai et al. showed that 14-3-3 proteins block S776 dephosphorylation in the cytoplasm. Therefore, they suggested that binding to 14-3-3 proteins indirectly protects Ataxin-1 from cytoplasmic proteolytic degradation, by blocking its dephosphorylation at S776.18
Besides the effect of 14-3-3 on Ataxin-1’s cellular localisation and stability, previous reports on Ataxin-1 and 14-3-3 strongly suggest the existence of a cytoplasmic anti-aggregation or “chaperone effect” of 14-3-3 on Ataxin-1.9, 13 However, experimental evidence to its existence has not yet been provided. Here, we show first that 14-3-3 and Ataxin-1 form soluble, cytoplasmic complexes in mammalian cells. In addition, we show that 14-3-3 reduces aggregation of Ataxin-1 into inclusion bodies in an E. coli model system. Then, supported by a combination of several biophysical methods providing structural information about the interaction of Ataxin-1 with 14-3-3, we provide a possible explanation for this effect on a molecular level.
Results
14-3-3 acts as chaperone for Ataxin-1 in the cytoplasm
Previously, Chen et al. showed that over-expression of 14-3-3 leads to increased levels of Ataxin-1, which eventually leads to Ataxin-1 aggregation in the nucleus.13 In contrast, it was shown that when transport of Ataxin-1 to the nucleus is prevented, Ataxin-1 is non-toxic and does not aggregate in the cytoplasm, even when overexpressed.9 Combining these observations, we reasoned that 14-3-3 exerts a chaperone activity on Ataxin-1, keeping it soluble in the cytoplasm. If this were the case, we expect the two proteins to interact in the cytoplasm of mammalian cells. To test this, we generated three inducible cell lines expressing FLAG-Atxn1[2Q], FLAG-Atxn[30Q] and FLAG-Atxn1[82Q] Using these cell lines, we showed that 14-3-3 and Ataxin-1 form soluble complexes in the cytoplasm (Figure 1). Hence, this interaction may play an important role in mediating Ataxin-1 solubility in the cytoplasm. Figure 1 also shows that 14-3-3 levels are much higher in the cytoplasm compared to the nucleus. Thus, as more polyglutamine Ataxin-1 accumulates in the nucleus over time, this eventually saturates the chaperone activity of 14-3-3 and leads to the formation of nuclear aggregates. In contrast, in the cytoplasm 14-3-3 acts as a chaperone for Ataxin-1, keeping it soluble. Interestingly, expression levels of 14-3-3 proteins are particularly high in the brain, totalling about 1% of the soluble protein fraction.14
Figure 1.

Ataxin-1 interacts with 14-3-3 in the cytoplasm. α-FLAG co-immunoprecipitation was performed on nuclear and cytoplasmic fractions of doxycycline inducible cell lines stably expressing FLAG-Ataxin-1 [2Q], FLAG-Ataxin-1 [30Q] and FLAG-Ataxin-1 [82Q] LAMIN A and GAPDH were used as nuclear and cytoplasmic markers, respectively.
We provide further support for the cytoplasmic chaperone effect of 14-3-3 on Ataxin-1, using an E. coli model system. E. coli cells naturally lack 14-3-3 proteins, allowing to study its effect on Ataxin-1 solubility more easily. In mammalian cells, a complete knock-out of all seven isoforms would have to be created, as loss of an isoform could be alleviated by others.21 However, a complete knock-out of all isoforms is expected to cause viability issues, as observed in S. cerevisiae.21 Figure 2 shows the results of expressing Ataxin-1 and 14-3-3 in E. coli: upon inducing expression of full-length polyQ expanded (84Q) Ataxin-1, there is virtually no protein in the soluble fraction (SF, lane 5), as most of it ends up in the insoluble fraction (IF, lane 6). Even using the N-terminal SUMO tag – which is known to enhance solubility of fusion proteins22 – in our expression construct could not prevent this. The same is observed for Ataxin-1 co-expressed with PKA to phosphorylate S776 (SF lane 9 and IF lane 10). However, co-expression of Ataxin-1 with both PKA and 14-3-3ζ rescues a significant amount of Ataxin-1 to the soluble fraction of the cell lysate (SF lane 13), revealing the chaperone effect of 14-3-3. As expected, co-expression of Ataxin-1 with just 14-3-3ζ did not rescue Ataxin-1 to the soluble fraction (SI Figure 1), since the interaction requires S776 to be phosphorylated.13 While we have not evaluated the effect of the other isoforms on Ataxin-1 solubility here, Ataxin-1 has been shown to interact with the ε, η, γ and β 14-3-3 isoforms as well.13
Figure 2.
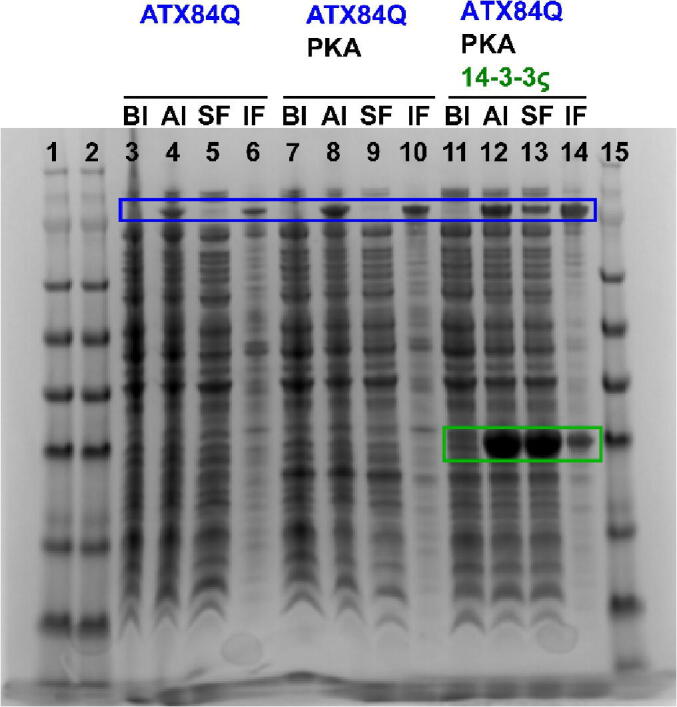
SDS-PAGE Solubility analysis of full-length 84Q Ataxin-1 in the E. coli cytoplasm upon co-expression of protein kinase A and 14-3-3. BI, before IPTG induction. AI, after induction. SF, soluble fraction. IF, insoluble fraction. The blue box highlights Ataxin-1 bands, and the green box highlights 14-3-3ζ bands. PKA bands are not visible, due to the weak promotor driving its expression, as opposed to the strong T7 promoter driving expression of Ataxin-1 and 14-3-3.
Crystal structure of an Ataxin-1 pS776 peptide bound to 14-3-3 sigma
To understand the mechanism behind the chaperone effect of 14-3-3 on Ataxin-1, we wanted to obtain structural insight into this protein–protein interaction. Until now, all structural characterization of Ataxin-1 has been limited to its AXH domain,23, 24, 25, 26, 27 most likely because it is the only known folded part of the protein. Indeed, the N and C termini were predicted to be disordered.28 To increase chances of crystallisation, we omitted the N terminus and focused our effort on a construct further referred to as AXH-C (Figure 5(A)). Unlike full-length Ataxin-1, this construct does not aggregate into inclusion bodies upon expression in E. coli, and still allows the role of the AXH domain and phosphorylated S776 in the C terminus – two crucial contributors to the SCA1 disease development – to be investigated, when interacting with 14-3-3. ITC experiments showed that the affinity of the AXH-C protein construct for the seven human 14-3-3 isoforms varied between 66 and 660 nM (SI Figure 2). Initially, we attempted to crystallize in vitro phosphorylated AXH-C construct in complex with 14-3-3ζ, with the rationale being that the latter might induce sufficient disorder-to-order transition in the Ataxin-1C terminus to promote crystallisation. However, no hits were obtained after extensive crystallisation trials, suggesting too many disordered regions remain present.
Therefore, as a substitute for the AXH-C construct, we instead used a synthetic peptide (AcNH-RKRRWpSAPESR-NH2) containing the anchor site for Ataxin-1 binding to 14-3-3 proteins, i.e. phosphorylated S776.13 Here, the sigma isoform was chosen for structure determination since we have extensive experience obtaining crystals of this isoform with phosphorylated peptides. Moreover, all 14-3-3 isoforms show a high degree of amino acid conservation in the phosphopeptide binding pocket. The data collection and refinement statistics are summarised in Table 1. Strong electron density was seen for nine out of the 11 amino acids (R773 through to R781) of the Ataxin-1 pS776 peptide (Figure 3(A)). As observed for other 14-3-3 mediated protein interactions, the phosphorylated S776 on Ataxin-1 is anchored directly to the 14-3-3σ protein through a hydrogen bond with Y130 and ionic interactions with R56 and R129. Intra-molecularly, R773 and R774 on Ataxin-1 also contribute to the coordination of pS776 through water-mediated and direct ionic interactions respectively. Besides helping to coordinate pS776, R774 contributes to 14-3-3σ binding by forming a salt bridge with E182. At the top of 14-3-3σ’s central binding groove, W775 on Ataxin-1 makes a hydrophobic contact with L222 as well as a hydrogen bond to D225. As is the case for about 50% of the interaction partners of 14-3-3 proteins, the pS776 on Ataxin-1 is followed by a +2 proline. This proline can contort the binding peptide away from the central binding groove of the 14-3-3 protein. However, this appears not to be the case for Ataxin-1 as instead three more residues – E779, S780 and R781 – stretch further into the binding groove. The side chain carboxyl group of Ataxin-1 residue E779 acts as a hydrogen bond acceptor for residue S45 on the surface of the 14-3-3σ protein. Finally, the side chain guanidinium group of R781 can be seen making ionic interactions with both E14 and E39 on 14-3-3σ (Figure 3(B)). A 2D plot of the interactions, created using MOE,29 is shown in SI Figure 3.
Table 1.
Crystallographic statistics.
| Data collection | |
| Wavelength (Å) | 1.54 |
| Resolution (Å) | 45.64–1.80 (1.84–1.80) |
| Space group | C 2 2 21 |
| Cell parameters | a = 82.66 Å |
| b = 111.95 Å | |
| c = 62.74 Å | |
| CC1/2 (%)a,b | 0.999 (0.969) |
| Rsym (%)a,c | 4.9 (21.6) |
| Rmeas (%)a,d | 5.3 (23.8) |
| Average I/σ(I)a | 34.0 (7.72) |
| Completeness (%)a | 97.2 (88.0) |
| No. of unique reflectionsa | 26,505 (2351) |
| Redundancya | 6.4 (5.4) |
| Wilson B-factor (Å2) | 11.57 |
| Refinement | |
| Number of non-hydrogen protein/solvent atoms | 2128/435 |
| Rwork (%) | 14.95 |
| Rfree (%) | 19.32 |
| No. of reflections in the 'free' set | 1306 |
| R.m.s. deviations from ideal values | |
| bond lengths (Å)/bond angles (°) | 0.01/0.926 |
| Average protein/solvent B-factor (Å2) | 12.2/27.7 |
| Ramachandran plot: favoured/outlier residuese (%) | 98.3/0 |
| Molprobity overall/clash score | 1.31/5.07 |
Number in parentheses is for the highest resolution shell used in the refinement
CC1/2 = Pearson's intra-dataset correlation coefficient, as described by Karplus and Diederichs.82
Rsym = ∑h∑l|Ihl − <Ih>|/∑h∑l<Ih>, where Ihl is the intensity of the lth observation of reflection h and < Ih > is the average intensity of reflection h.
Rmeas = ∑h|√(nh/(nh − 1))∑l|Ihl-<Ih>||/∑h∑l<Ih>, where nh is the number of observations of reflection h.
As calculated using Molprobity.83
Figure 3.

Binding mechanism of the Ataxin-1 pS776 to the 14-3-3 sigma protein. A. Crystal structure of the Ataxin-1 phosphopeptide including pS776 (cyan), bound to the 14-3-3σ isoform (green). The 14-3-3σ protein exists as a dimeric protein, but only the monomer was observed in the crystal asymmetric unit. The grey mesh shows the electron density for the Ataxin-1 peptide at 1.2σ. B. Interactions between the Ataxin-1 peptide and the 14-3-3σ protein. Dashed lines indicate hydrogen bonds and ionic interactions. Labels for the 14-3-3σ protein and Ataxin-1 residues are shown in black and blue respectively.
HDX-MS analysis of the 14-3-3/AXH-C complex shows the AXH domain becomes more solvent exposed upon interacting with 14-3-3
Since the inherent disorder of the protein prohibited crystallisation of AXH-C in complex with 14-3-3ζ, we studied the interaction further using hydrogen–deuterium exchange MS (HDX-MS). We were particularly interested in probing the Ataxin-1 AXH domain, since if it were to participate in the interaction with 14-3-3, it would provide the best potential target for the development of interaction inhibitors. Indeed, from the 6 only structure reports of 14-3-3 in complex with larger protein constructs,30, 31, 32, 33, 34, 35 it is clear that secondary interfaces outside of 14-3-3’s central binding groove may exist, depending on the binding partner. It has also been shown these can be targeted with small molecule ligands.36
HD exchange kinetics were measured for 14-3-3ζ, AXH-C and the 14-3-3ζ/AXH-C protein complex. For the 14-3-3ζ sample, deuterium uptake was lower at longer reaction time points – likely due to protein stability or aggregation issues – so statistical analysis was not performed. However, the exchange kinetics for AXH-C versus the AXH-C in complex with 14-3-3ζ could still be compared. 61 peptides were identified with high confidence, covering 80% of sequence of AXH-C. At significance level p < 0.001, the only region of AXH-C showing decreased exchange in the presence of 14-3-3ζ was a peptide consisting of L709-E728, which is located in the disordered C terminus. This result suggests that this region of AXH-C either interacts with the 14-3-3ζ protein or undergoes a conformational change to a state less prone to HD exchange. Interestingly, several regions of increased HD exchange were observed for AXH-C while in complex with 14-3-3ζ, predominantly in the ordered AXH domain and adjacent to it (Figure 4(A)). These regions on the AXH domain – i.e. S563-F575, A584-L594, L609-A628, A633-V643 and F670-C683 – could be considered as more solvent accessible or dynamic upon formation of the 14-3-3ζ/Ataxin-1 protein complex.
Figure 4.

Mapping of HD exchange in the presence of 14-3-3ζ, on the sequence of the Ataxin-1 AXH-C construct and the structure of its AXH domain. (A) Amino acid sequence of the AXH-C construct (Ataxin-1 residues 563–816). Red = increased HD exchange in the 14-3-3ζ/AXH-C complex vs AXH-C. Blue = decreased HD exchange in complex vs AHX-C. Black = no change in HD exchange. Grey = no data available. (B) Crystal structure of the AXH domain (Ataxin-1 residues 567–689, PDB ID 4APT), in the absence of 14-3-3ζ. An AXH dimer is shown in cartoon representation with one monomer shown in grey and the other in black. Two other dimers related by crystallographic symmetry are also shown in surface representations. (C) This panel zooms in on panel B, with amino acids with increased HDX exchange (p < 0.001) in the presence of 14-3-3ζ, shown in red on one the AXH molecules (i.e. the one in black cartoon representation). These regions are clearly present in the AXH dimer interface, as well as the interfaces with the two dimers related by crystallographic symmetry, suggesting these regions become more solvent exposed upon the interaction of AXH-C with 14-3-3ζ. The yellow circle highlights residue R638 on the AXH domain, which has been suggested to play a role in Ataxin-1 self-association and fibril formation.46
SAXS and AUC analysis of the AXH-C construct and its interaction with 14-3-3
To further complement our crystallographic and HDX-MS data, size-exclusion SAXS (SEC-SAXS) data were collected for AXH-C, 14-3-3ζ, and their complex. These were plotted as a distribution function P(r) (Figure 5(B)), and a dimensionless Kratky Plot (Figure 5(C)) to assess and compare the level of disorder in the three samples. The derived structural parameters are summarised in Table 2 and Guinier plots are shown in SI Figure 4.
Table 2.
Structural parameters derived from SAXS data.
Our data now provide the first biophysical evidence that the C terminus of Ataxin-1 is indeed disordered: The P(r) function (Figure 5(B)) for AXH-C shows an extended tail which is characteristic for partially unfolded proteins. In contrast, the P(r) for 14-3-3ζ shows a symmetric bell-shaped curve, as is expected for globular proteins. Similarly, in a dimensionless Kratky plot (Figure 5(C)), 14-3-3 behaves as expected for globular protein (showing a characteristic maximum value of 1.107 for sRg = √3) while the AXH-C plot is typical for that of a partially unfolded protein.37 Comparing the AXH-C construct with the 14-3-3ζ/AXH-C complex, the dimensionless Kratky Plot reveals that 14-3-3 only partially reduces the disorder in the C terminus of Ataxin-1, explaining the difficulty in crystallising this complex.
The SAXS data was also used to evaluate atomic models of AXH-C and its complex with 14-3-3. To generate an atomic model for the AXH-C construct, we used the Ensemble Optimisation Method (EOM).38 Using the previously published crystal structure of the AXH domain;26 we let EOM generate a pool of ten thousand random conformations of the C-terminal residues (563–816) while treating the AXH domain as a dimeric rigid body. An ensemble of six conformers was then selected from the pool which fits best with the experimental data. Figure 5(D) shows the selected ensemble, revealing the structural variability of the C-termini in the modelled conformers. As shown in Figure 5(E), the ensemble has a very good fit to the experimental scattering data, with a χ2 of 1.08.
Starting from a previously published structure of the AXH domain26 as well as our structure of 14-3-3σ with the pS776 peptide, we modelled the missing residues in the disordered C terminus of AXH-C in complex with 14-3-3ζ, using CORAL (COmplexes with RAndom Loops)39 A total of 236 flexible amino acids in the 964 amino acid 14-3-3ζ/AXH-C complex had to be modelled. As the HDX-MS data indicated the AXH domain becomes more solvent exposed when AXH-C binds to 14-3-3, we decided to model two types of conformations of the complex. In the first conformation, two AXH-C molecules were bound to a 14-3-3 dimer through pS776 at their C-termini, but their AXH domains were allowed to move independently as monomers. In contrast, in the second conformation, the AXH domains were forced to remain dimeric. While the model in which the AXH-C dimer was forced to remain intact has a χ2 fit of 8.38 (Figure 6(A), SI Figure 5(C)) with the experimental data, the model with two monomeric AXH-C molecules bound to a 14-3-3 dimer shows a better fit with χ2 = 4.96. (Figure 6(A), SI Figure 5(A)). Repeating this CORAL modelling ten times, led to similar outcomes. (SI Table 1)
Figure 6.

Modelling of the 14-3-3ζ/AXH-C complex. (A) On the left, a 1-state model with two AXH-C molecules (cartoon representation, shades of blue) in a monomeric state, bound to a 14-3-3 dimer (green, surface representation). On the right, a 1-state model with two AXH-C molecules in a dimeric state, bound to a 14-3-3 dimer. The models were generated using CORAL.39 The χ2 values are scores reported by FoXS40, 84 for the quality of fit of the models to the experimental SAXS data for the complex. (B) Analytical ultracentrifugation results for the 14-3-3/AXH-C complex. The purple curve shows the c(s) distribution analysis of the 14-3-3ζ/AXH-C complex at 0.5 mg/mL. Sedimentation (s) on x axis represent s apparent only. The blue and cyan lines respectively represent where the 14-3-3ζ/AXH-C complexes, containing either monomeric or dimeric AXH-C molecules bound to the 14-3-3 dimer, would theoretically sediment. (C) Table containing experimentally determined S values (Sedfit method) and Frictional ratios (2DSA analysis) which is the measure of the shape and size of the macromolecule. The 2DSA analyses reveals multiple species sedimenting between 5.0 S and 5.67 S, inferring the 14-3-3ζ/AXH-C complex adopts multiple conformations, predominantly one with a dissociated AXH-C dimer. (D) Mixed 3-state model for the 14-3-3ζ/AXH-C complex, containing monomeric and dimeric AXH-C. (E) 3-state model for the 14-3-3ζ/AXH-C complex containing only monomeric AXH-C conformations. (F) 3-state model for the 14-3-3ζ/AXH-C complex containing only monomeric AXH-C conformations. χ2 values in panels (D, E and F) report on the quality of fit of the respective models to the experimental SAXS data for the complex, as determined by MultiFoXS.40 For all models shown in this figure, the fits to the 14-3-3ζ/AXH-C SAXS data is shown in SI Figure 5.
We also performed a sedimentation velocity (SV) experiment by analytical ultracentrifugation (AUC). From the SV-AUC data, the sedimentation coefficient of the 14-3-3ζ/AXH-C complex was determined to be 5.29 S (Sedfit column in Figure 6(C)). The S20,w calculations from the 14-3-3ζ/AXH-C models with the AXH-C molecules in monomeric or dimeric states, revealed that the experimentally determined sedimentation coefficient value corresponds best to the models containing monomeric AXH-C molecules (Figure 6(A, B)). Nonetheless, the models with the dimeric AXH-C molecules bound to 14-3-3, still crossed the continuous distribution of sedimentation coefficients on the tail-end of the curve (Figure 6(A and B)). This is shown for one model with monomeric AXH-C molecules (theoretical S20,w = 5.24 S) and one model with dimeric AXH-C molecules (theoretical S20,w = 5.61 S) bound to 14-3-3. Again, similar results were obtained for the other generated models, as reported in SI Table 1. This suggests the 14-3-3ζ/AXH-C complex is dynamic and samples an ensemble of conformations, possibly still including conformations with an intact AXH-C dimer. To investigate this further, we used the 2DSA method where various conformations of the complex were separated into discrete species, all sedimenting at marginally different rates with varying frictional ratios. This analysis demonstrated that 88.3% of the complex corresponds to the monomeric AXH-C state, and only 5.6% corresponds to the more compact dimeric AXH-C state (Figure 6(C)). Therefore, the AUC experiments suggest that, although both conformations potentially exist in solution, the 14-3-3ζ/AXH-C complex spends most of the time sampling the monomeric AXH-C state. This finding agrees with the better fits of those models to the experimental SAXS data.
Since the C-termini of AXH-C are disordered, and the AUC data suggests that a AXH-C molecules could be present as a mix of monomeric (in majority) and dimeric (in minority) states, the single state models shown in Figure 6(A) are probably too simplistic. This is also reflected by their high χ2 scores. Therefore, we have generated a multi-state model using MultiFoXS.40 Provided with 100 models of 14-3-3/AXH-C complex containing monomeric AXH-C, as well as 100 models containing dimeric AXH-C, MultiFoXS selected a three-state model (Figure 6(D), SI Figure 5(E)) with a much better fit to the SAXS data, as suggested by its χ2 score of 2.04. This model contained two states of the 14-3-3/AXH-C complex with monomeric AXH-C and one state with the complex containing dimeric AXH-C. The latter had a weight of 24.6%, while the two 14-3-3/AXH-C states containing monomeric AXH-C had weights of 42.4% and 33% respectively. A three-state model containing only monomeric AXH-C states had a similar fit, with χ2 = 2.24 (Figure 6(E), SI Figure 5(B)). In contrast, the fit of a three-state model containing only dimeric AXH-C was considerably worse with a χ2 = 5.28 (Figure 6(E), SI Figure 5(D)). Using the BILBOMD webserver, we generated four-state models of the 14-3-3/AXH-C complex with AXH-C in monomeric or dimeric conformations. They have virtually identical fits to the experimental data, with χ2 = 1.21 and χ2 = 1.12 for monomeric and dimeric AXH-C respectively. These 4-state models are shown in SI Figure 6. We point out that this shows that SAXS is probably not capable of discriminating between both conformations, however it shows that both these conformations can indeed exist in solution.
Cross-linking MS of study of the 14-3-3ζ/AXH-C complex
Finally, we studied the 14-3-3ζ/AXH-C complex by cross-linking MS. An SDS-PAGE gel showing the cross-linking of 14-3-3 and AXH-C using DSS and DSG is shown in SI Figure 7. Bands corresponding to cross-linked 14-3-3/AXH-C complex were excised from the gel and analysed by mass spectrometry. Six cross-linked peptides could be identified. On the 14-3-3ζ side, they all included lysine 212 (K212) which is located at the start of Helix 9 in the central phosphopeptide binding groove of 14-3-3. On AXH-C, lysine residues 750, 766, 782, 785, 796 and 816 were identified as the cross-linked amino acids (SI Figure 2). These are all located in the flexible C terminus of AXH-C. Figure 7(A and B) show the identified cross-links on a model of the 14-3-3/AXH-C complex. For simplicity, a single state model is shown with the AXH-C molecules in a monomeric conformation. The measured distances in the model between cross-linked lysine residues are sensible to allow for DSS and DSG mediated crosslinking. The fact that no cross-linking was obtained between 14-3-3ζ and any of the AXH domain lysine residues is consistent with the fact that in our HDX-MS experiments we did not observe HDX protected regions in the AXH domain. Finally, no crosslinks between AXH domains were observed. This further supports the hypothesis in which the AXH-C molecules are predominantly monomeric when bound to the 14-3-3 dimer.
Figure 7.

Cross-linking MS results for the 14-3-3ζ/AXH-C complex. (A) For simplicity, the complex is shown as a single state model with AXH-C molecules (shades of blue) in monomeric conformation, bound to a 14-3-3 dimer (shades of green). Lysine 112 on 14-3-3 partakes in all observed cross-links (black lines) with AXH-C. Panel. (B) Zooming on panel A, the dashed lines show crosslinks of 14-3-3 residue K212 with lysine residues 750, 766, 782, 785, 796 and 816 on AXH-C. The black numbers are the distances (Nε-Nε) measured in Å.
Discussion
Soluble vs aggregated nuclear Ataxin-1 in SCA1
Aggregation of polyQ expanded Ataxin-1 into nuclear inclusions is a pathological hallmark of SCA1. For a long time, these deposits have been considered as the cause of neurodegeneration. However, more recent findings revealed that they mainly accumulate in brain regions which do not degenerate, in contrast to the cerebellar Purkinje cells which are the first to lose viability. Therefore, Cummings et al. suggested that these deposits might have a protective role instead, possibly by sequestering the mutant protein.41 In fact, Lasagna-Reeves et al. showed that in SCA1 mice, a soluble form of polyQ expanded Ataxin-1 can be detected in brain regions showing neurodegeneration. Moreover, this soluble form appeared to be stabilised by nuclear association with CIC. Given that CIC levels are coincidently high in the cerebellum, this finding provided a first potential explanation for why this brain region is more susceptible to neurodegeneration.42 This hypothesis was further supported by recent findings showing that the nuclear interaction of polyQ expanded Ataxin-1 with CIC in cerebellar Purkinje cells is a major driver of pathology, through a gain-of-function mechanism..7
Support for a cytoplasmic chaperone effect of 14-3-3 on Ataxin-1
As early as 1998, Klement et al. studied transgenic mice expressing polyQ Ataxin-1K772T, a mutant incapable of translocating to the nucleus. This mutant was non-pathogenic, demonstrating that – besides polyQ expansion – nuclear translocation is critical for pathogenesis. Interestingly, no significant cytoplasmic aggregates could be observed either. Therefore, Klement et al. suggested the cytoplasm might have chaperones capable of handling large amounts of polyQ expanded Ataxin-1 to prevent aggregation.9 Later, Chen et al. showed that 14-3-3 proteins positively influence Ataxin-1 levels and aggravate neurodegeneration in a SCA1 transgenic fly model. They also showed that transgenic mice overexpressing polyQ expanded Ataxin-1 show a mostly unaffected cellular distribution of 14-3-3, without its sequestration into nuclear inclusions. Therefore, they stated that “it is likely that 14-3-3 and Ataxin-1 preferentially form soluble complexes in vivo”.13
Here we provided in vitro evidence corroborating the theory of Klement et al.9 and Chen et al.13 First, by co-IP on cytoplasmic and nuclear cell fractions, we show that Ataxin-1 and 14-3-3 indeed mostly interact in the cytoplasm. Then, we co-expressed full length, polyQ expanded Ataxin-1 with PKA and 14-3-3 in E. coli, which naturally lack 14-3-3 proteins and Ataxin-1. 14-3-3 co-expression rescues a significant amount of phosphorylated Ataxin-1 from forming insoluble inclusion body aggregates (Figure 2). Further support for a cytoplasmic chaperone effect of 14-3-3 comes from its cellular distribution profile: Lai et al reported that in the cerebellum, cytoplasmic levels of 14-3-3 isoforms β, ε and ζ are significantly higher than their nuclear levels.18
Besides Ataxin-1, 14-3-3 proteins have been shown to act as a chaperone for several other proteins. For more information on the subject, we would like to refer to an extensive review by Sluchanko and Gusev.43 A particularly interesting parallel with Ataxin-1, is a finding by Ichimura et al. showing that 14-3-3 proteins sequester a soluble pool of TRIM32 (an E3 ubiquitin-protein ligase), by disrupting the propensity of TRIM32 to engage in higher-order self-association and form cytoplasmic bodies.44
Structural analysis of the 14-3-3/AXH-C complex suggests that the presence of 14-3-3ζ reduces Ataxin-1 aggregation by preventing self-association through its AXH domain
Besides polyQ expansion in the N terminus, the AXH domain has been shown to contribute to the Ataxin-1 aggregation process.45 Crystal structures of the AXH domain have shown that it forms dimers, and two crystallographic dimer-dimer interfaces were also observed (Figure 4(B)). In solution, the AXH domain was also shown to form an equilibrium between monomer, dimer, tetramer and higher order molecular weight species. De Chiara et al. speculated that this monomer/dimer/tetramer equilibrium of the AXH domain could contribute to the Ataxin-1 aggregation process.26
Interestingly, in our HDX-MS data, the regions of increased HD exchange on AXH-C can be mapped to residues in the AXH dimer interface (residues S563-F575), as well as residues belonging to the two dimer-dimer interfaces observed in the crystal structure (Figure 4(B and C)). This includes R638, an Ataxin-1 residue which was recently implicated by molecular dynamics data in Ataxin-1 self-association and fibril formation through stabilisation of the AXH domain tetramers.46 The reduction of inter-molecular interactions would result in an increase in HD exchange being observed in those regions. Therefore, we considered that 14-3-3 proteins may capture monomeric Ataxin-1, shifting the monomer–dimer equilibrium of its AXH domain towards the monomeric state. Consequentially, this would reduce further oligomerization – and ultimately aggregation – through the Ataxin-1 AXH dimer-dimer interactions.
Because of the high degree of disorder in the AXH-C terminus, we could not confirm this by X-ray crystallography using our AXH-C construct. Still, our crystal structure of the pS776 peptide with 14-3-3σ showed how phosphorylation of Ataxin-1 allows it to anchor onto 14-3-3 proteins. Besides the disorder, the size of the 14-3-3ζ /AXH-C complex would make it poorly amenable to alternatives such as cryo-EM or NMR studies. Therefore, we modelled the complex and verified the models against experimental data obtained from SAXS, AUC and crosslinking MS. Using CORAL39 (COmplexes with RAndom Loops) we created models with either 2 monomeric AXH-C molecules bound to a 14-3-3 dimer, or models with an intact AXH-C dimer bound to 14-3-3. In general, models with two monomeric AXH-C molecules bound to 14-3-3 had better fits to the experimental SAXS data (Figure 6(A) and SI Table 1). Our AUC data showed that both kinds of complexes could exist, but with a majority of AXH-C binding as two monomers to 14-3-3 (88.3%), rather than an intact AXH-C dimer (5.6%) (Figure 6(C)). Multistate modelling of the SAXS data using MultiFoXS showed a similar trend, with 75.4% of the complex having AXH-C in a monomeric state (Figure 6(D)). Further support for the model with monomeric AXH-C came from the crosslinking MS data (SI Table 2), where we could not observe crosslinks between residues in the AXH domain, but only between 14-3-3K212 and lysine residues in the disordered C terminus of Ataxin-1 (Figure 7).
In summary, our structural analysis leads us to hypothesize that 14-3-3 proteins exert their observed anti-aggregation effect on Ataxin-1 by reducing Ataxin-1 dimerization through its AXH domain, and thus reducing the amount of Ataxin-1 entering further self-association and aggregation pathways. In comparison, while cytoplasmic 14-3-3 proteins appear to affect the AXH domain’s monomer/dimer equilibrium by “spatial separation”, CIC in the cerebellar nuclei stabilizes soluble Ataxin-1 through a different mechanism. Indeed, crystallographic information showed how CIC directly interacts with the AXH domain. This blocks Ataxin-1 homo-dimerization through the AXH domain, by forming a hetero-tetrameric Ataxin-1/CIC complex.27
Modulating the 14-3-3/Ataxin-1 interaction for treatment of SCA1
Figure 8 shows an overview of the cytoplasmic role of 14-3-3 in Ataxin-1 biology, and the nuclear interactions of Ataxin-1 which have been shown to contribute to neurodegeneration. Since a soluble form of polyQ expanded Ataxin-1 needs to reach the nucleus for pathology to develop, modulation of the 14-3-3/Ataxin-1 interaction could provide a new strategy in the treatment of SCA1. There are two ways of modulating the 14-3-3/Ataxin-1 interaction, namely inhibition or stabilisation. In theory, both strategies could have the desired effect of reducing the levels of soluble Ataxin-1 reaching the nucleus: Inhibition of the interaction is expected to be beneficial since the interaction with 14-3-3 has been shown to stabilize and protect Ataxin-1 from degradation.13, 18 Also, in both fly and mouse models the interaction with 14-3-3 has been shown to contribute to pathology.19, 20 Nonetheless, the opposite modulation strategy of stabilising the interaction could be beneficial as well, since the interaction with 14-3-3 masks the nuclear localisation signal. Hence, by stabilizing the interaction one could envision forming a “dead-end trap” for soluble Ataxin-1, preventing its translocation to the nucleus. Interestingly, an Ataxin-1K772T mutant – which is incapable of nuclear translocation – was indeed shown to be non-pathogenic.9 Therefore, we suggest that chemical biology tools should be developed, which allow to further investigate which approach – inhibition or stabilisation – is most preferable for the 14-3-3/Ataxin-1 interaction specifically. In general, stabilizing protein interactions using “molecular glues” is a far less commonly used approach than inhibiting them. However, as stabilisers would simultaneously bind both proteins in the complex, they hold great potential in terms of selectivity. For 14-3-3 proteins, successful stabilization of several of its PPIs has already been described.47, 48
Figure 8.

Overview of cytoplasmic and nuclear protein interactions contributing to Ataxin-1 solubility and pathology. SCA1 pathology is associated with the formation insoluble deposits in the nucleus. However, they also occur in brain regions which do not degenerate. The current opinion in the field is that these nuclear inclusions have a protective role, while a soluble form of Ataxin-1 is the major driver of pathology.7 In the cytoplasm, phosphorylated Ataxin-1 (blue, with an ordered AXH domain flanked by disordered N- and C-termini) interacts with 14-3-3 (green). This interaction protects Ataxin-1 against proteasomal degradation13 and aggregation. Upon release from 14-3-3, Ataxin-1 can translocate to the nucleus and – when polyQ expanded – exert its neurodegenerative effect by altering nuclear protein interactions. Capicua (purple) has been shown to stabilise Ataxin-1 by forming soluble oligomeric complexes. The higher abundance of CIC in the cerebellum potentially explains why this region is more vulnerable to neurodegeneration than other regions in the brain.42 Another nuclear interaction that has been shown to contribute to SCA1 pathology is that between polyQ expanded, phosphorylated Ataxin-1 and RBM17 (orange).6
In conclusion, this work reports on the interaction between the C terminus of Ataxin-1 and 14-3-3 proteins. Firstly, we have provided evidence that the C terminus of Ataxin-1 is indeed disordered and solved a crystal structure showing how pS776 anchors Ataxin-1 onto 14-3-3 proteins. Secondly, we have modelled the 14-3-3ζ/AXH-C structure combining data from HDX-MS, SAXS, previously solved crystal structures of the Ataxin-1 AXH domain, and our crystal structure of the 14-3-3σ protein and the Ataxin-1 pS776 peptide. Based on these results, we hypothesize that the binding of Ataxin-1 (wild-type or polyQ expanded) onto the antiparallel-dimeric 14-3-3 proteins causes a shift of the Ataxin-1 monomer/dimer equilibrium towards the monomeric state. In this way, 14-3-3 acts as a chaperone in the cytoplasm, preventing further Ataxin-1 self-association and aggregation before it can translocate to the nucleus. This is an important proposition since recent findings suggest that a soluble form of Ataxin-1 in the nucleus makes a major contribution to disease onset. Therefore, we believe that modulation of the 14-3-3/Ataxin-1 protein interaction, aiming to reduce amounts of soluble Ataxin-1 reaching the nucleus, is worth investigating as a novel therapeutic strategy. It must be noted this will be a challenging endeavour since our data show that Ataxin-1 interacts with 14-3-3 proteins through its disordered C terminus, with no detectable contribution from its ordered AXH domain. However, as our HDX-MS data shows that the folded AXH domain remains accessible when Ataxin-1 interacts with 14-3-3, other approaches – such as reducing Ataxin-1 levels through proteolysis targeting chimeras (PROTACs)49, 50 – could be considered. Finally, an alternative, indirect approach to reduce the chaperone effect of 14-3-3 would be to block phosphorylation of S776 on Ataxin-1.
Materials and methods
Protein production and purification
A synthetic construct encoding His-tagged SUMO-AXH-C (Ataxin-1 residues 563–816) from the pCDFDuet-1 plasmid was ordered from Genscript. The plasmid was transformed by heat shock at 42 °C to NiCo21(DE3) cells. A single colony was used to inoculate 30 mL 2TY medium supplemented with Streptomycin at 100 μg/mL and grown overnight at 37 °C. 20 mL of this culture was used to inoculate 500 mL of 2TY medium and incubated at 37 °C until an A600nm of 4 was obtained. At that point, protein expression was induced by adding IPTG to a final concentration of 200 μM. Also, the temperature was lowered to 18 °C, and the growth medium was supplemented with 25 mL of a feed solution. A 20× stock of this feed solution has the following composition: 40% v/v glycerol, 1 M MOPS, 2 mM MgCl2 and 2 mM MgSO4, at pH7.4. After further overnight incubation, the cells were harvested by centrifugation for 30 min at 4000 rpm and dissolved in lysis buffer IMAC12.5 containing 20 mM Tris, 300 mM NaCl, 12.5 mM Imidazole and 2 mM 2-Mercaptoethanol at pH 7.5. Per gram of cell pellet, 100 mL lysis buffer was used. Before lysis, the cell suspension was supplemented with DNaseI and MgCl2, at concentrations of 1 μg/mL and 5 mM respectively. Then, the cells were lysed using a cell disruptor (Constant Systems). The cell lysate was centrifuged for 30 minutes at 20000 rpm, and the supernatant was loaded on a 5 mL HisTrap HP column (GE Life Sciences). After washing the column with 10 column volumes of lysis buffer, the protein was eluted with the lysis buffer containing 250 mM imidazole (IMAC250). The His-SUMO tag was then removed from AXH-C by adding recombinant SUMO protease51 to the fusion protein in a 1/500 w/w ratio, during overnight dialysis to IMAC12.5. Untagged AXH-C was then collected in the flow-through upon passing the mixture a second time over the 5 mL nickel column. To remove all imidazole, AXH-C was dialysed to 20 mM Tris pH7.5, 300 mM NaCl and 2 mM DTT. AXH-C was phosphorylated overnight by adding 20 mM MgCl2, a 3-fold molar excess of ATP and recombinant protein kinase A (Promega). Finally, the protein was purified by size-exclusion chromatography on a Superdex200 26/600 pg column (GE Life Sciences), equilibrated in 20 mM Tris pH7.5, 150 mM NaCl and 2 mM DTT.
14-3-3σΔC protein – with C terminal truncation after T231 to enhance crystallization – was expressed mainly as described by previously,52 except NiCO21(DE3) cells were used. In short, the cells were grown in TB medium at 37 °C, and protein expression was induced at A600nm = 0.6 by adding IPTG to a final concentration of 400 μM. At that point, the temperature was lowered to 18 °C. The cells were collected by centrifugation and were dissolved in IMAC12.5, supplemented with 1 μg/mL DnaseI and 5 mM MgCl2. After cell lysis using an Emulsiflex-C3 homogeniser (Avestin), the cell lysate was cleared by centrifugation and the His-tagged protein was captured using 10 mL his60 Ni Superflow resin (Takara). After elution with IMAC250, the his-tag was removed during overnight dialyis at 4 °C to IMAC12.5, by adding 1 mg TEV protease for every 100 mg of 14-3-3σΔC. Uncleaved protein was removed by flowing the dialysed protein for a second time through the 10 mL Nickel resin. As a last purification step, the protein was loaded on a Superdex75 26/600 pg gel filtration column (GE Life Sciences) equilibrated with 25 mM HEPES pH 7.5, 100 mM NaCl, 10 mM MgCl2, 250 μM TCEP. 14-3-3σΔC was concentrated to 60 mg/mL and aliquots were flash-frozen for storage at −80 °C.
14-3-3ζΔC – with C-terminal truncation at T234 – was expressed and purified as described above for 14-3-3σΔC.
SAXS experiments
In-line SEC-SAXS data for 14-3-3ζΔC (11.6 mg/mL), phosphorylated AXH-C (12.8 mg/mL) and their complex (11 mg/mL) were collected at Beamline B21 of the Diamond Light Source using an Agilent 1200 HPLC system and 2.4 mL column, equilibrated with 20 mM HEPES, 150 mM NaCl, 2 mM DTT. Injection volumes of 45 μl were used. Frames were collected for 3 s per frame, at 25 °C. X-ray scattering was recorded using a Pilatus 2 M Shodex-kw403 detector, with a fixed camera length of 4.014 m at 12.4 keV. The data were analysed using the ATSAS software package53: the scattering data were averaged using PRIMUS.54 Rg and forward scattering I(0) were calculated using AUTORG.55 Distance distribution functions were generated using GNOM.56 An ensemble model containing the disordered C terminus of the AXH-C construct was generated with EOM;38 using the crystal structure of the AXH dimer from PDB ID 4APT. CORAL39 was used to model the complex of AXH-C and 14-3-3ζ, based on our structure of 14-3-3σΔC with the Ataxin-1 pS776 peptide, the 14-3-3ζ dimer from PDB ID 4IHL57 and the structure of the AXH domain from PDB ID 4APT.26 10 models were created with AXH-C in a monomeric state in the 14-3-3 complex, and 10 with AXH-C in the dimeric state. As CORAL produces glycine linkers for the missing amino acids, Modeller58 was used to change the linker residues to the Ataxin-1 amino acid sequence, creating 10 models starting from each CORAL model. Using these models as input for MultiFoXS,40 3-state models were selected. BILBOMD59 was also used to create 4-state models of the 14-3-3/AXH-C complex, with AXH-C in monomeric or dimeric conformations. 200 conformations per Rg were sampled, with Rg ranging from 40 to 70 Å.
Peptide synthesis
The Ataxin-1 pS776 peptide (AcNH-RKRRWpSAPESR-NH2) was synthesized by manual solid-phase peptide synthesis (SPPS).60 The synthesis was performed on 50 µmol scale using a polystyrene-based Rink Amide resin (100–200 mesh, 0.59 mmolg−1 loading) employing a Fmoc-strategy61 and with HCTU as coupling reagent.62 In short, the N-terminal Fmoc-protecting group was removed using 20% piperidine in NMP and the next amino acid introduced using a combination of Fmoc-amino acid/HCTU/DIPEA (1:1:2) in NMP. The resin was washed with NMP between deprotection and coupling steps. Before peptide cleavage, the resin was rinsed in alternate fashion with CH2Cl2 and diethyl ether and dried under vacuum. Peptide cleavage and side-chain deprotection were performed in one pot by treating with a mixture of TFA/EDT/water/TIS (92.5:2.5:2.5:2.5) and incubating for 3 h. The crude peptide was isolated by precipitation into ice-cold ether, then redissolved H2O/acetonitrile and lyophilized. The crude peptide was purified using a Shimadzu HPLC on a C18 preparative column from Phenomenex (21.20 × 150 mm) and a flow rate of 15 mL/min. The products were eluted by using a solvent gradient consisting of solvents A and B, where solvent A = 0.1% FA/H2O; solvent B = 0.1% FA/CH3CN). UV signal at 210 nm was used for detection. The final pure peptide was characterized for molecular integrity and purity by reverse-phase liquid chromatography-mass spectrometry (LC-MS) analysis.
Crystallisation and structure determination
The pS776 peptide was dissolved at 10 mM in buffer containing 20 mM Hepes pH 7.4, 2 mM MgCl2 and 2 mM DTT. 14-3-3σΔC, at 377 μM (10 mg/mL), was mixed in the same buffer with the Ataxin pS776 peptide at a 1:2 molar stoichiometry and incubated overnight at 4 °C. Crystals were then obtained by hanging drop vapor diffusion in EasyXtal plates (Qiagen) by screening an in-house grid of crystallisation solutions containing 0.095 M Hepes (pH 7.1, 7.3, 7.5, 7.7), 0.19 M CaCl2, 5% glycerol, 24–29% PEG 400. Crystals were directly flash-frozen in liquid nitrogen. Diffraction data were collected using an in-house MicroMax-003 sealed tube X-ray generator and Pilatus 200 K detector (Rigaku). Data were indexed an integrated using XDS,63 and then scaled using AIMLESS.64, 65 The structure was then solved by molecular replacement using Phaser.66, 67 Phenix.refine67, 68 and Coot69 were used in alternating cycles of automatic and manual structure refinement. Figures were prepared using PyMOL.70
HDX-MS experiments
HDX-MS analysis was performed on 16 μM solutions of AXH-C alone and a purified 14-3-3ζ/AXH-C complex. Liquid handling steps were performed by a PAL robotic system (LEAP technologies). 4 µL of protein solution was mixed with 56 µL of either equilibration buffer (10 mM potassium phosphate in H2O, pH 7.0) for an unlabelled control, or labelling buffer (10 mM potassium phosphate in D2O, pH 7.0). The labelling reaction was incubated at 20 °C for 30 s, 2 min, 15 min, 1 h or 4 h, followed by 1:1 mixing with a cold quench solution (0 °C, 100 mM potassium phosphate in H2O, pH 2.4). 50 µL was then injected into a Waters HDX module connected to an M-Class Acquity UPLC, and washed over a Waters Enzymate pepsin column for 3 mins at 100 µL/min flow (0.02% HCOOH in H2O, pressure approximately 6500 psi) at 25 °C. Resulting peptides were trapped on a C18 VanGuard column. Peptides were washed off the trap column and through an analytical Waters BEH C18 column (100 × 1.0 mm, 1.7 µM particle), using the following gradient: 0 min, 5% B; 6 min, 35% B; 7 min, 40% B; 8 min, 95% B followed by two wash gradient steps (Eluent A: 0.02% HCOOH in H2O, Eluent B: 0.02% HCOOH in MeCN). MS data were collected on a Waters Synapt G2Si, with m/z range 50–2000 Th and scan rate 0.3 s. MSe data acquisition was used to identify peptides in the unlabelled sample, with collision energy ramp in the trap cell of 18–40 V for the high energy function, and lockmass correction using GluFib.
The resulting data (collected in triplicate) were processed using Waters PLGS v3.0.2 and DynamX v3.0 software packages. After processing in DynamX, all assignments were interrogated manually, and only peptides with a suitable signal to noise ratio and unambiguous peak-picking were taken forward. 2-tailed students’ t-tests were applied to identify peptides with statistically significant (p < 0.001) differences in deuterium uptake between samples.
Analytical ultracentrifugation experiments
400 μl sample was loaded into an Epon Charcoal-Filled centrepiece with twelve mm optical path length and sapphire windows. The corresponding buffer was loaded into the reference channel of each double sectored cell (the instrument functions like a dual beam spectrometer). These loaded cells were then placed into an AN-60Ti rotor, loaded into the Beckman-coulter proteome Lab XL-I analytical ultracentrifuge and equilibrated to 20°C. The rotor was then brought to 3,000 rpm and the samples were scanned at 280 nm to confirm proper cell loading and appropriate adjustment of the laser, via the laser delay settings. The rotor was then brought to the final run speed of 45,000 rpm. Scans were recorded every 6.2 minutes for 12 hours. Radial scans ranged from 5.8 cm to 7.2 cm.
The data were analysed using the c(s) method implemented in SEDFIT version 14.6e,71, 72 using 35,000 data points for each sample. The f/f0 values were varied to find the best overall fit of the data for each sample. A maximum entropy regularization probability of 0.95 was used and time invariant noise was removed. The coefficients were converted to standardised s20,w units using the calculated partial specific volume of 0.73, and the experimentally determined density and viscosity of the buffer measured as density 1.0060 g/mL and viscosity 1.02 cP at 20.2 °C. The analysis was performed using the standard solvent model.
The data was also analysed by the UltraScan two-dimensional spectrum analysis (2DSA) method implemented in the UltraScan II software (version 3.9, revision 2339).73 For analysis, the s-value and frictional ratio ranges were set to 1–15 S and 1–4, respectively. The grid settings were 10 × 10, and 28 grid repetitions were performed. Time-invariant noise correction, radial invariant noise and iterative optimization approach with the 5 iterations were enabled. The meniscus position was fitted. The obtained 2DSA distributions were then refined with parsimonious regularization using Genetic Algorithm analysis. Sedimentation Coefficients for the best fit scattering models were calculated directly from the atomic coordinates using HYDROPRO shell modelling program.74 The default value of 0.31 nm for the atomic element radius for all atoms was used to represent the hydration shell.
Crosslinking MS
Equimolar mixtures of phosphorylated AXH-C with 14-3-3ζ were cross-linked using disuccinimidyl suberate (DSS) and disuccinimidyl glutarate (DSG) to identify intermolecular cross-links. Cross-linking reactions were performed in buffer containing 10 mM HEPES (pH 7.5), 150 mM NaCl, 0.1 mM TCEP. The reactions were initiated by adding a freshly prepared solution of DSS or DSG (10 mg/mL) in a twenty-fold molar excess to protein solutions, followed by a 2 h incubation at room temperature. Following cross-linking, proteins were separated by reducing gel-electrophoresis on a NuPAGE 4–12% Bis-Tris gel using MES running buffer. Bands containing cross-linked protein were excised and analyzed for intermolecular cross-links using high resolution MS as previously described.75 All identified cross-links were manually checked in the raw data to remove false positives.
Testing solubility of Ataxin-1 in the E. coli cytoplasm
Plasmid pCDFDuet-1-SUMO-84Q-Ataxin-1 was transformed by heat shock to NiCo21(DE3), by itself or in combinations with pACYC-SUMO-PKA and pETDuet-1-14-3-3ζFL. Earlier studies have used PKA co-expression to phosphorylate 14-3-3 binding partners in E. coli.31, 76, 77 The plasmids carry Streptomycin, Chloramphenicol and Ampicillin resistance respectively and have compatible origin of replications. Typical working concentrations for these antibiotics are 100 μg/mL, 25 μg /mL and 100 μg/mL respectively. For transformations with two or three plasmids, these concentrations were lowered accordingly to half or one third. For transformation with three plasmids, cells were grown for 2 hours at 37 °C before plating the cells. Cultures were grown as described above for expression of the AXH-C construct. At A600nm of 4.0, a 500 μL “before induction” sample was taken and protein expression was induced by adding IPTG to a final concentration of 200 μM while lowering the temperature to 18 °C. The culture was incubated further overnight. The “before induction” samples were centrifuged for five minutes at 5000 rpm, after which the pellet was stored at −30 °C. The next morning, optical density was measured again, and an “after induction” sample was taken, with a volume ensuring a roughly equal number of cells as in the “before induction” sample. The cells were harvested by centrifugation and the growth medium was removed. The bacterial pellets, obtained before and after induction of protein expression, were lysed by adding 200 μL BugBuster Protein Extraction Reagent (Merck). Samples with volumes of 30 μL were kept aside for analysis by polyacrylamide gel electrophoresis. These samples corresponded to the whole cell lysates, before and after induction (WCL-BI and WCL-AI). The remainder of the WCL-AI was then centrifuged at 13,000 rpm for twenty minutes. Again, a 30 μL sample was taken from the supernatant (soluble fraction). After removing the remaining supernatant, the pellet (insoluble fraction) was re-dissolved in 170 μL of 1X NuPAGE LDS sample buffer (Invitrogen Life Sciences) with 25 mM DTT. To the other samples, 10 μL of 4× sample buffer containing 100 mM DTT was added before incubating all samples at 95 °C for 2 minutes. Finally, 8 μL of each sample was loaded on a NuPAGE™ 4–12% Bis-Tris Protein Gel (Invitrogen Life Sciences). After running the gel using MES running buffer, the gel was stained for 15 minutes using InstantBlue™ (Expedeon Protein Solutions) to analyse the whole cell lysates and their soluble and insoluble fractions for the presence of (SUMO-fused) 84Q polyglutamine Ataxin-1 and 14-3-3.
Mammalian cell culture experiments
DAOY cells stably expressing ATXN1 [2Q], ATXN1 [30Q], and ATXN1[82Q] fused with an N-terminal Flag tag were generated using the pINDUCER system.78, 79 For lentiviral production, 4 × 106 HEK239T cells were seeded in a 10-cm dish containing DMEM supplemented with 10% FBS (Atlanta Biological). The next day, 8 µg of each pINDUCER_Flag_ATXN1 vector, 6 µg of psPAX2 vector (Addgene 12260), and 2 µg of pMD2.G vector (Addgene 12259) were transfected using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. 24 hours following transfection, an additional 5 mL of fresh medium was added to the dish and all media were collected the following day. For lentiviral transduction, 2 × 105 DAOY cells were seeded in 6-well plates. 24 hours after seeding, 50 µL of the lentiviral-containing medium was added to the cells. The next day, the medium was changed to selection medium containing DMEM supplemented with 10% FBS (Atlanta Biological), 1% antibiotic/antimycotic (Invitrogen), and 350 ng/mL Geneticin (Invitrogen). The cells were passaged for a minimum of three generations in geneticin-containing medium to ensure complete selection.
For nuclear and cytoplasmic fractionation, 2 × 106 cells were plated in a 15-cm dish. 24 hours later ATXN1 expression was induced using 1,000 μg/ml doxycycline for the Flag-ATXN1 [2Q] cell line and 350 μg/ml doxycycline for the Flag-ATXN1 [30Q] and Flag-ATXN1 [82Q] cell lines. 48 hours after induction, the cells were washed with PBS and harvested. Nuclear cytoplasmic fractionation was performed using the NE-PER Nuclear and Cytoplasmic Extraction kit with the following amendments. Pierce universal nuclease (Thermo Fisher) was added to the NER buffer (1:2,000). Xpert Protease Inhibitor Cocktail Solution (1:100) (GenDEPOT) and Xpert Phosphatase Inhibitor Cocktail Solution (1:100) were added to the CER-I and NER buffer. The nuclear pellet was washed with 100 μl ice cold CER-I before lysis was performed in NER buffer. During the first incubation on ice during lysis in NER buffer, the nuclear pellet was passed through an 18-gauge needle (BD) 7 times to improve protein extraction. The Pierce BCA Protein Assay Kit (Thermo Fisher) was used to determine protein concentrations of the nuclear and cytoplasmic fractions. 200 μg total protein from each sample was transferred to 50 μl Anti-FLAG® M2 Magnetic beads (Sigma Aldrich) pre-washed with PBS. Co-immunoprecipitation (co-IP) was performed while rotating for 1 hour at 4 °C. 3 washes were performed with 150 μl ice cold PBS supplemented with XPert Protease and Phosphatase Inhibitor Cocktails (1:100) (GenDEPOT). Elution was performed by incubating the beads in 40 μl NuPAGE™ LDS buffer (Thermo Fisher) supplemented with NuPAGE™ Sample Reducing Agent at 65 °C for 5 minutes. The eluted samples were immediately removed from the beads and stored at 4 °C. The next day, input and co-IP elutions were run on a Bolt™ 4–12%, 1.0 mm Mini Protein Gel (Thermo Fisher) and were transferred for 2.5 hours at 0.32 Amps in 4 °C using tris-glycince transfer buffer with 15% methanol. The membranes were blocked for 1 hour at room temperature with 5% skim milk, then incubated at room temperature for 4 hours with the following primary antibodies diluted in 5% skim milk: mouse monoclonal α-FLAG® M2-Peroxidase (HRP) (1:2,500, Sigma Aldrich A8592), Rabbit polyclonal α-14-3-3 (pan) (1:1,000, Cell Signaling Technology #8312), Rabbit polyclonal α-Lamin A (1:1,000, Abcam 26300) and mouse monoclonal α-GAPDH (1:50,000). Following incubation with primary antibody, the blots were washed 3 times in TBST and incubated at room temperature for 2 hours with the following secondary antibodies diluted in 5% skim milk: Peroxidase Affipure Donkey Anti-Mouse IgG (H + L) (1:10,000, Jackson ImmunoResearch Laboratories, Inc 715–035-150) and Immun-Star Goat Anti-Rabbit (GAR)-HRP Conjugate (1:10,000, BIORAD #1705046). Each blot was washed 3 times in TBST then incubated in Amersham ECL Prime Reagent (VWR/GE) for 30 seconds before imaging. Chemiluminescence was captured with the GE Amersham Imager 680 blot and gel imager system and exposure time was automatically determined using the ‘high dynamic range’ setting. Blot images were prepared using Image Studio Lite.
Accession numbers
The crystal structure of the pS776 peptide bound to 14-3-3σΔC were submitted to the Protein Data Bank80 with PDB ID: 6QIU.
SAXS data for 14-3-3ζΔC, phosphorylated AXH-C, and their complex were submitted to the SASBDB81 with accession codes SASDL56, SASDLT3 and SASDL46 respectively.
Acknowledgments
Acknowledgements
We wish to thank Dr. Claire Pizzey for collection of SAXS data at the Diamond Light Source and Dr. Petr Pompach at The Czech Academy of Sciences for providing cross-linking/MS data. Initial Training Network TASPPI (H2020 Marie Curie Actions of the European Commission, Grant Agreement 675179); and the Netherlands Organization for Scientific Research (NWO) (Gravity Program 024.001.035 and Vici grant 016.150.366).
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article.
Author contributions
S.L., C.O, L.B., T.C. and J.M.D initiated the project. S.L. purified proteins, prepared protein crystals, solved and processed crystal structures, processed SAXS data and modelled protein structures. R.J.B. performed and analysed HDX-MS experiments. E.R. performed and analysed AUC experiments. L.G.M. synthesized and purified the ATX pS776 peptide. L.S. performed the ITC measurements. C.A. performed the co-IP experiments. L.N. made the stable DAOY cell lines expressing 2Q, 30Q and 82Q Ataxin-1. S.L. wrote the manuscript with essential input and feedback from all other authors.
Edited by J. Buchner
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2021.167174.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- 1.Opal P., Ashizawa T. In: GeneReviews((R)) Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Stephens K., editors. University of Washington; Seattle (WA): 1993. Spinocerebellar Ataxia Type 1. [PubMed] [Google Scholar]
- 2.Wagner J.L., O'Connor D.M., Donsante A., Boulis N.M. Gene, Stem Cell, and Alternative Therapies for SCA 1. Front. Mol. Neurosci. 2016;9:67. doi: 10.3389/fnmol.2016.00067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cummings C.J., Orr H.T., Zoghbi H.Y. Progress in pathogenesis studies of spinocerebellar ataxia type 1. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1999;354:1079–1081. doi: 10.1098/rstb.1999.0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zoghbi H.Y., Orr H.T. Pathogenic mechanisms of a polyglutamine-mediated neurodegenerative disease, spinocerebellar ataxia type 1. J. Biol. Chem. 2009;284:7425–7429. doi: 10.1074/jbc.R800041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Chiara C., Pastore A. Kaleidoscopic protein-protein interactions in the life and death of ataxin-1: new strategies against protein aggregation. Trends Neurosci. 2014;37:211–218. doi: 10.1016/j.tins.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lim J., Crespo-Barreto J., Jafar-Nejad P., Bowman A.B., Richman R., Hill D.E. Opposing effects of polyglutamine expansion on native protein complexes contribute to SCA1. Nature. 2008;452:713–718. doi: 10.1038/nature06731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rousseaux M.W.C., Tschumperlin T., Lu H.C., Lackey E.P., Bondar V.V., Wan Y.W. ATXN1-CIC Complex Is the Primary Driver of Cerebellar Pathology in Spinocerebellar Ataxia Type 1 through a Gain-of-Function Mechanism. Neuron. 2018;97 doi: 10.1016/j.neuron.2018.02.013. 1235–43.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Emamian E.S., Kaytor M.D., Duvick L.A., Zu T., Tousey S.K., Zoghbi H.Y. Serine 776 of ataxin-1 is critical for polyglutamine-induced disease in SCA1 transgenic mice. Neuron. 2003;38:375–387. doi: 10.1016/s0896-6273(03)00258-7. [DOI] [PubMed] [Google Scholar]
- 9.Klement I.A., Skinner P.J., Kaytor M.D., Yi H., Hersch S.M., Clark H.B. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 10.Tsuda H., Jafar-Nejad H., Patel A.J., Sun Y., Chen H.K., Rose M.F. The AXH domain of Ataxin-1 mediates neurodegeneration through its interaction with Gfi-1/Senseless proteins. Cell. 2005;122:633–644. doi: 10.1016/j.cell.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 11.Park J., Al-Ramahi I., Tan Q., Mollema N., Diaz-Garcia J.R., Gallego-Flores T. RAS-MAPK-MSK1 pathway modulates ataxin 1 protein levels and toxicity in SCA1. Nature. 2013;498:325–331. doi: 10.1038/nature12204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perez Ortiz J.M., Mollema N., Toker N., Adamski C.J., O'Callaghan B., Duvick L. Reduction of protein kinase A-mediated phosphorylation of ATXN1-S776 in Purkinje cells delays onset of Ataxia in a SCA1 mouse model. Neurobiol. Dis. 2018 doi: 10.1016/j.nbd.2018.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen H.K., Fernandez-Funez P., Acevedo S.F., Lam Y.C., Kaytor M.D., Fernandez M.H. Interaction of Akt-phosphorylated ataxin-1 with 14-3-3 mediates neurodegeneration in spinocerebellar ataxia type 1. Cell. 2003;113:457–468. doi: 10.1016/s0092-8674(03)00349-0. [DOI] [PubMed] [Google Scholar]
- 14.Berg D., Holzmann C.F., Riess O. 14-3-3 proteins in the nervous system. Nature Rev. Neurosci. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- 15.van Hemert M.J., Steensma H.Y., van Heusden G.P. 14-3-3 proteins: key regulators of cell division, signalling and apoptosis. BioEssays: News Rev. Mol. Cell. Dev. Biol. 2001;23:936–946. doi: 10.1002/bies.1134. [DOI] [PubMed] [Google Scholar]
- 16.Kleppe R., Martinez A., Døskeland S.O., Haavik J. The 14-3-3 proteins in regulation of cellular metabolism. Semin. Cell Dev. Biol. 2011;22:713–719. doi: 10.1016/j.semcdb.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 17.Kaplan A., Ottmann C., Fournier A.E. 14-3-3 adaptor protein-protein interactions as therapeutic targets for CNS diseases. Pharmacol. Res. 2017;125:114–121. doi: 10.1016/j.phrs.2017.09.007. [DOI] [PubMed] [Google Scholar]
- 18.Lai S., O'Callaghan B., Zoghbi H.Y., Orr H.T. 14-3-3 Binding to ataxin-1(ATXN1) regulates its dephosphorylation at Ser-776 and transport to the nucleus. J. Biol. Chem. 2011;286:34606–34616. doi: 10.1074/jbc.M111.238527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jafar-Nejad P., Ward C.S., Richman R., Orr H.T., Zoghbi H.Y. Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14-3-3epsilon haploinsufficiency in mice underscores complex pathogenicity in neurodegeneration. PNAS. 2011;108:2142–2147. doi: 10.1073/pnas.1018748108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jorgensen N.D., Andresen J.M., Lagalwar S., Armstrong B., Stevens S., Byam C.E. Phosphorylation of ATXN1 at Ser776 in the cerebellum. J. Neurochem. 2009;110:675–686. doi: 10.1111/j.1471-4159.2009.06164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Heusden G.P., Griffiths D.J., Ford J.C., Chin A.W.T.F., Schrader P.A., Carr A.M. The 14-3-3 proteins encoded by the BMH1 and BMH2 genes are essential in the yeast Saccharomyces cerevisiae and can be replaced by a plant homologue. Eur. J. Biochem. 1995;229:45–53. [PubMed] [Google Scholar]
- 22.Marblestone J.G., Edavettal S.C., Lim Y., Lim P., Zuo X., Butt T.R. Comparison of SUMO fusion technology with traditional gene fusion systems: enhanced expression and solubility with SUMO. Protein Sci. 2006;15:182–189. doi: 10.1110/ps.051812706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y.W., Allen M.D., Veprintsev D.B., Lowe J., Bycroft M. The structure of the AXH domain of spinocerebellar ataxin-1. J. Biol. Chem. 2004;279:3758–3765. doi: 10.1074/jbc.M309817200. [DOI] [PubMed] [Google Scholar]
- 24.de Chiara C., Kelly G., Menon R.P., McCormick J., Pastore A. Chemical shift assignment of the ataxin-1 AXH domain in complex with a CIC ligand peptide. Biomol. NMR Assign. 2014;8:325–327. doi: 10.1007/s12104-013-9509-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Chiara C., Menon R.P., Kelly G., Pastore A. Protein-protein interactions as a strategy towards protein-specific drug design: the example of ataxin-1. PLoS ONE. 2013;8 doi: 10.1371/journal.pone.0076456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.de Chiara C., Rees M., Menon R.P., Pauwels K., Lawrence C., Konarev P.V. Self-assembly and conformational heterogeneity of the AXH domain of ataxin-1: an unusual example of a chameleon fold. Biophys. J. 2013;104:1304–1313. doi: 10.1016/j.bpj.2013.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim E., Lu H.C., Zoghbi H.Y., Song J.J. Structural basis of protein complex formation and reconfiguration by polyglutamine disease protein Ataxin-1 and Capicua. Genes Dev. 2013;27:590–595. doi: 10.1101/gad.212068.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Darling A.L., Uversky V.N. Intrinsic Disorder in Proteins with Pathogenic Repeat Expansions. Molecules. 2017;22 doi: 10.3390/molecules22122027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.ULC CCG. Molecular Operating Environment (MOE). (2019).
- 30.Obsil T., Ghirlando R., Klein D.C., Ganguly S., Dyda F. Crystal structure of the 14-3-3zeta:serotonin N-acetyltransferase complex. a role for scaffolding in enzyme regulation. Cell. 2001;105:257–267. doi: 10.1016/s0092-8674(01)00316-6. [DOI] [PubMed] [Google Scholar]
- 31.Sluchanko N.N., Beelen S., Kulikova A.A., Weeks S.D., Antson A.A., Gusev N.B. Structural Basis for the Interaction of a Human Small Heat Shock Protein with the 14-3-3 Universal Signaling Regulator. Structure (London, England) 1993;2017(25):305–316. doi: 10.1016/j.str.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ottmann C., Marco S., Jaspert N., Marcon C., Schauer N., Weyand M. Structure of a 14-3-3 coordinated hexamer of the plant plasma membrane H+ -ATPase by combining X-ray crystallography and electron cryomicroscopy. Mol. Cell. 2007;25:427–440. doi: 10.1016/j.molcel.2006.12.017. [DOI] [PubMed] [Google Scholar]
- 33.Kondo Y., Ognjenović J., Banerjee S., Karandur D., Merk A., Kulhanek K. Cryo-EM structure of a dimeric B-Raf:14-3-3 complex reveals asymmetry in the active sites of B-Raf kinases. Science (New York, NY) 2019;366:109–115. doi: 10.1126/science.aay0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taoka K., Ohki I., Tsuji H., Furuita K., Hayashi K., Yanase T. 14-3-3 proteins act as intracellular receptors for rice Hd3a florigen. Nature. 2011;476:332–335. doi: 10.1038/nature10272. [DOI] [PubMed] [Google Scholar]
- 35.Alblova M., Smidova A., Docekal V., Vesely J., Herman P., Obsilova V. Molecular basis of the 14-3-3 protein-dependent activation of yeast neutral trehalase Nth1. PNAS. 2017;114 doi: 10.1073/pnas.1714491114. E9811-e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sijbesma E., Skora L., Leysen S., Brunsveld L., Koch U., Nussbaumer P. Identification of Two Secondary Ligand Binding Sites in 14-3-3 Proteins Using Fragment Screening. Biochemistry. 2017;56:3972–3982. doi: 10.1021/acs.biochem.7b00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Receveur-Brechot V., Durand D. How random are intrinsically disordered proteins? A small angle scattering perspective. Curr. Protein Pept. Sci. 2012;13:55–75. doi: 10.2174/138920312799277901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tria G., Mertens H.D.T., Kachala M., Svergun D.I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ. 2015;2:207–217. doi: 10.1107/S205225251500202X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petoukhov M.V., Franke D., Shkumatov A.V., Tria G., Kikhney A.G., Gajd New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012;45:342–350. doi: 10.1107/S0021889812007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016;44:W424–W429. doi: 10.1093/nar/gkw389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cummings C.J., Reinstein E., Sun Y., Antalffy B., Jiang Y., Ciechanover A. Mutation of the E6-AP ubiquitin ligase reduces nuclear inclusion frequency while accelerating polyglutamine-induced pathology in SCA1 mice. Neuron. 1999;24:879–892. doi: 10.1016/s0896-6273(00)81035-1. [DOI] [PubMed] [Google Scholar]
- 42.Lasagna-Reeves C.A., Rousseaux M.W., Guerrero-Munoz M.J., Park J., Jafar-Nejad P., Richman R. A native interactor scaffolds and stabilizes toxic ATAXIN-1 oligomers in SCA1. eLife. 2015;4 doi: 10.7554/eLife.07558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sluchanko N.N., Gusev N.B. Moonlighting chaperone-like activity of the universal regulatory 14-3-3 proteins. FEBS J. 2017;284:1279–1295. doi: 10.1111/febs.13986. [DOI] [PubMed] [Google Scholar]
- 44.Ichimura T., Taoka M., Shoji I., Kato H., Sato T., Hatakeyama S. 14-3-3 proteins sequester a pool of soluble TRIM32 ubiquitin ligase to repress autoubiquitylation and cytoplasmic body formation. J. Cell Sci. 2013;126:2014–2026. doi: 10.1242/jcs.122069. [DOI] [PubMed] [Google Scholar]
- 45.de Chiara C., Menon R.P., Dal Piaz F., Calder L., Pastore A. Polyglutamine is not all: the functional role of the AXH domain in the ataxin-1 protein. J. Mol. Biol. 2005;354:883–893. doi: 10.1016/j.jmb.2005.09.083. [DOI] [PubMed] [Google Scholar]
- 46.Grasso G., Morbiducci U., Massai D., Tuszynski J., Danani A., Deriu M. Destabilizing the AXH Tetramer by Mutations: Mechanisms and Potential Antiaggregation Strategies. Biophys. J. 2018;114:323–330. doi: 10.1016/j.bpj.2017.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ballone A., Centorrino F., Ottmann C. 14-3-3: A Case Study in PPI Modulation. Molecules. 2018;23 doi: 10.3390/molecules23061386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stevers L.M., Sijbesma E., Botta M., MacKintosh C., Obsil T., Landrieu I. Modulators of 14-3-3 Protein-Protein Interactions. J. Med. Chem. 2018;61:3755–3778. doi: 10.1021/acs.jmedchem.7b00574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lai A.C., Crews C.M. Induced protein degradation: an emerging drug discovery paradigm. Nature Rev. Drug Discov. 2017;16:101–114. doi: 10.1038/nrd.2016.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ottis P., Crews C.M. Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS Chem. Biol. 2017;12:892–898. doi: 10.1021/acschembio.6b01068. [DOI] [PubMed] [Google Scholar]
- 51.Weeks S.D., Drinker M., Loll P.J. Ligation independent cloning vectors for expression of SUMO fusions. Protein Expr. Purif. 2007;53:40–50. doi: 10.1016/j.pep.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schumacher B., Skwarczynska M., Rose R., Ottmann C. Structure of a 14-3-3sigma-YAP phosphopeptide complex at 1.15 A resolution. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2010;66:978–984. doi: 10.1107/S1744309110025479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017;50:1212–1225. doi: 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Konarev P.V., Volkov V.V., Sokolova A.V., Koch M.H.J., Svergun D.I. PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003;36:1277–1282. [Google Scholar]
- 55.Petoukhov M.V., Konarev P.V., Kikhney A.G., Svergun D.I. ATSAS 2.1 - towards automated and web-supported small-angle scattering data analysis. J. Appl. Crystallogr. 2007;40:s223–s228. [Google Scholar]
- 56.Svergun D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992;25:495–503. [Google Scholar]
- 57.Molzan M., Kasper S., Röglin L., Skwarczynska M., Sassa T., Inoue T. Stabilization of physical RAF/14-3-3 interaction by cotylenin A as treatment strategy for RAS mutant cancers. ACS Chem. Biol. 2013;8:1869–1875. doi: 10.1021/cb4003464. [DOI] [PubMed] [Google Scholar]
- 58.Webb B., Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protocols Bioinf. 2016;54:5.6.1–5.6.37. doi: 10.1002/cpbi.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pelikan M., Hura G.L., Hammel M. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 2009;28:174–189. doi: 10.4149/gpb_2009_02_174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Merrifield R.B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963;85:2149–2154. [Google Scholar]
- 61.Chan W., White P. Oxford University Press; 1999. Fmoc Solid Phase Peptide Synthesis. [Google Scholar]
- 62.Hood C.A., Fuentes G., Patel H., Page K., Menakuru M., Park J.H. Fast conventional Fmoc solid-phase peptide synthesis with HCTU. J. Pept. Sci. 2008;14:97–101. doi: 10.1002/psc.921. [DOI] [PubMed] [Google Scholar]
- 63.Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Evans P.R., Murshudov G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013;69:1204–1214. doi: 10.1107/S0907444913000061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Adams P.M.D., W M.D., Storoni L.C. Phaser crystallographic software. J. Appl. Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.M., Mustyakimov M. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. Sect. D. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Emsley P., Lohkamp B., Scott W.G., Cowtan K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.L.L.C. Schrodinger, The PyMOL Molecular Graphics System, Version 1.8. (2015).
- 71.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown P.H., Schuck P. Macromolecular size-and-shape distributions by sedimentation velocity analytical ultracentrifugation. Biophys. J. 2006;90:4651–4661. doi: 10.1529/biophysj.106.081372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brookes E., Demeler B. Genetic Algorithm Optimization for obtaining accurate Molecular Weight Distributions from Sedimentation Velocity Experiments. Analytical Ultracentrifugation VIII. Prog. Colloid Polym. Sci. 2006:78–82. [Google Scholar]
- 74.Garcia de la Torre J. Hydration from hydrodynamics. General considerations and applications of bead modelling to globular proteins. Biophys. Chem. 2001;93:159–170. doi: 10.1016/s0301-4622(01)00218-6. [DOI] [PubMed] [Google Scholar]
- 75.Macakova E., Kopecka M., Kukacka Z., Veisova D., Novak P., Man P. Structural basis of the 14-3-3 protein-dependent activation of yeast neutral trehalase Nth1. Biochim. Biophys. Acta, Mol. Cell. Biol. Lipids. 2013;10:29. doi: 10.1016/j.bbagen.2013.05.025. [DOI] [PubMed] [Google Scholar]
- 76.Tugaeva K.V., Tsvetkov P.O., Sluchanko N.N. Bacterial co-expression of human Tau protein with protein kinase A and 14-3-3 for studies of 14-3-3/phospho-Tau interaction. PLoS ONE. 2017;12 doi: 10.1371/journal.pone.0178933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Stevers L.M., Lam C.V., Leysen S.F., Meijer F.A., van Scheppingen D.S., de Vries R.M. Characterization and small-molecule stabilization of the multisite tandem binding between 14-3-3 and the R domain of CFTR. PNAS. 2016;113:E1152–E1161. doi: 10.1073/pnas.1516631113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meerbrey K.L., Hu G., Kessler J.D., Roarty K., Li M.Z., Fang J.E. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. PNAS. 2011;108:3665–3670. doi: 10.1073/pnas.1019736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nitschke L., Tewari A., Coffin S.L., Xhako E., Pang K., Gennarino V.A. miR760 regulates ATXN1 levels via interaction with its 5' untranslated region. Genes Dev. 2020;34:1147–1160. doi: 10.1101/gad.339317.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Berman H.M., Battistuz T., Bhat T.N., Bluhm W.F., Bourne P.E., Burkhardt K. The Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2002;58:899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 81.Kikhney A.G., Borges C.R., Molodenskiy D.S., Jeffries C.M., Svergun D.I. SASBDB: Towards an automatically curated and validated repository for biological scattering data. Protein Sci. 2020;29:66–75. doi: 10.1002/pro.3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karplus P.A., Diederichs K. Linking crystallographic model and data quality. Science (New York, NY) 2012;336:1030–1033. doi: 10.1126/science.1218231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chen V.B., Arendall W.B., 3rd, Headd J.J., Keedy D.A., Immormino R.M., Kapral G.J. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013;105:962–974. doi: 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.