

Abstract
Allosteric modulation offers an alternate approach to target the cannabinoid type-1 receptor (CB1) for therapeutic benefits. Examination of the two widely studied prototypic CB1 negative allosteric modulators (NAMs) Org27569 and PSNCBAM-1 revealed structural resemblance and similar structure-activity relationships (SARs). In silico docking and dynamics simulation studies using the crystal structure of CB1 co-bound with CP55,940 and Org27569 suggested that Org27569 and PSNCBAM-1 occupied the same binding pocket and several common interactions were present in both series with the CB1 receptor. A new scaffold was therefore designed by merging the key structural features from the two series and the hybrids retained these binding features in the in silico docking studies. In addition, one such hybrid displayed similar functions to Org27569 in dynamic simulations by preserving a key R2143.50-D3386.30 salt bridge and maintaining an antagonist-like Helix3-Helix6 interhelical distance. Based on these results, a series of hybrids were synthesized and assessed in calcium mobilization, [35S]GTPγS binding and cAMP assays. Several compounds displayed comparable potencies to Org27569 and PSNCBAM-1 in these assays. This work offers new insight of the SAR requirement at the allosteric site of the CB1 receptor and provides a new scaffold that can be optimized for the development of future CB1 allosteric modulators.
Keywords: CB1 receptor, Allosteric modulator, Hybrid, Structure-activity relationship, Molecular modeling
1. Introduction
The endocannabinoid system comprises the cannabinoid receptors 1 and 2 (CB1 and CB2), their endogenous ligands (endocannabinoids), anandamide (AEA) and 2-arachidonoylglycerol (2-AG), and enzymes responsible for the synthesis, transport, and inactivation of these endocannabinoids.1–4 CB1 is one of the most abundantly expressed receptors in the human brain, while CB2 is detected mainly in immune cells.5–7 CB1 has been shown to play important roles in many physiological functions including pain, learning and memory, analgesia, appetite/feeding behaviors, anxiety/depression, and rewarding processes of many commonly abused drugs.7–9
The CB1 antagonists/inverse agonists rimonabant (SR141716A) and AM251 reduced self-administration of several drugs of abuse in rodents including cocaine, heroin, nicotine, and alcohol,10–12 demonstrating that CB1 is a viable target to develop therapies for substance use disorders. However, rimonabant, the first-in-class CB1 antagonist/inverse agonist, showed serious psychiatric side effects while it was used as an anti-obesity drug, including depression, anxiety, and a risk of suicidal ideation.13 Rimonabant was subsequently withdrawn from the market and Sanofi-Aventis, Pfizer and others ended their programs on CB1 antagonists/inverse agonists. As CB1 is known to possess constitutive activity that is crucial to maintain homeostasis,14 inverse agonists which disrupt this constitutive activity could lead to clinical ramifications.15 Therefore, many research groups have devised strategies to overcome this drawback by developing CB1 neutral antagonists, peripherally restricted antagonists and allosteric modulators.14–19
As part of a longstanding program aimed at identifying agents to treat substance use disorders, we have investigated CB1 allosteric modulation as an alternate approach to treat addictions to drugs such as cocaine.17 Allosteric modulators bind to allosteric binding sites which are topologically distinct from and often less highly conserved than the orthosteric binding site(s), where orthosteric ligands such as AEA, 2-AG, and CP55,940 bind. Allosteric modulators offer several advantages compared to orthosteric ligands, including lower risk of overdosing due to the “ceiling” effect and preservation of the temporal characteristics of receptor signaling due to the saturable nature of allosteric interactions which is determined by the concentration of the orthosteric ligand.17, 20, 21 CB1 allosteric modulators Org27569 (1) and PSNCBAM-1 (2) have been shown to increase agonist and decrease antagonist binding and decrease agonist signaling in a number of functional assays, thus commonly referred to as negative allosteric modulators (NAMs).17, 22, 23 We have reported that the Org27569 and PSNCBAM-1 and its analogues (e.g. 3, RTICBM-74, Figure 1) attenuated cue- and/or drug-induced reinstatement of extinguished cocaine-and/or methamphetamine-seeking behaviors, demonstrating their efficacy in blocking abuse-related effects of these drugs.24, 25
Figure 1.
Representative CB1 NAMs.
Close examination of the structures of these two series revealed that Org27569 and PSNCBAM-1 share a high degree of structural similarity, both with a chloro-substituted aromatic ring on the left, a urea moiety in PSNCBAM-1 that mimics the combination of amide and indole nitrogen in Org27569, and an aromatic ring (phenyl vs. pyridyl) on the right side (Figure 1). In addition, studies from our laboratory and others suggest similar structural-activity relationships (SARs) between these two scaffolds.25–33 For example, the piperidinyl ring on Org27569 can be replaced with a dialkylamino group (such as 4, Figure 1), or other smaller substituents.26–29 Similarly, we found that the pyrrolidinyl ring on the pyridyl ring of PSNCBAM-1 could be replaced with smaller groups such as a dimethylamino or removed completely without affecting activity (e.g. 3 and 5, Figure 1).25 The pyridyl ring itself could be replaced with a pyrimidine (6, Fig. 1)31 or five-membered heterocycles.33 Finally, the central phenyl ring of PSNCBAM-1 could be replaced with other heterocyclic rings such as a thiophene (e.g. 7, Fig. 1),32 implying that this site may act mostly as a spacer and is amenable to modifications.
In the present study, we first performed in silico studies of Org27569 and PSNCBAM-1 using the recently solved crystal structure of CB1 co-bound with Org27569 and the orthosteric agonist CP55,940 (RSCB: 6KQI).34 Our results revealed that PSNCBAM-1 bound in the same binding pocket as that of Org27569 in the crystal structure and had similar interactions with the CB1 receptor. On the basis of these observations, we designed a new scaffold by merging the key moieties from each molecule, namely the 4-chlorophenyl urea of PSNCBAM-1 on the left and the phenethyl group of Org27569 on the right and obtained a series of hybrid compounds such as 8 (Figure 2). Such changes will disrupt the planarity in the structure of PSNCBAM-1 and increase the three-dimensionality, a parameter that considered important in drug discovery.35, 36 Herein we report the design, synthesis and pharmacological characterization of these hybrid compounds in calcium mobilization, GTPγS and cAMP assays.
Figure 2.

Org27569, PSNCBAM-1 and design of the hybrid compound 8.
2. Results and discussion
2.1. Molecular Modeling.
Org27569 and PSNCBAM-1 were overlapped, employing a flexible shape/heteroatom scoring approach by Schrödinger’s flexible ligand superposition method, revealing excellent alignment of the indole with the chlorophenyl group, as well as the urea moiety with the carboxamide and the indole nitrogen from the two molecules (Figure 3). In addition, the pyridine ring of PSNCBAM-1 and the phenyl ring of Org27569 on the right side are positioned at approximately the same distance from the urea/carboxamide moieties. These commonalities suggest that these groups are possibly key components for interaction of these two NAMs with the CB1 receptor.
Figure 3.

Overlapping Org27569 (pink) and PSNCBAM-1 (cyan) illustrates scaffold commonalities.
We then performed docking studies of Org27569 and PSNCBAM-1 using our CB1 receptor model established based on the crystal structure of the CB1 co-bound with CP55,940 and Org27569 (RCSB: 6KQI) recently reported by Shao and colleagues.34 The crystal structure incorporates a CB1-PGSCM fusion protein, wherein ICL3 is fused to Pyrococcus abysii glycogen synthase, and a total of 5 thermostabilizing mutations in the CB1 portion of the protein which are not at sites of interactions with either CP55,940 or Org27569. In this crystal structure, Org27569 occupied the extrahelical site in the inner leaflet of the membrane, making most of the interactions with the transmembrane domain IV (TM4) and stretching a lipid-facing hydrophobic surface to contact the transmembrane domain II (TM2).34 This binding pocket of Org27569 overlapped the cholesterol binding region37 and in the same region predicted by the mutagenesis mass spectrometry study of a covalent Org27569 derivative by Stornaiuolo and colleagues.38
Following initial protein preparation steps of the CB1 receptor including energy minimization, refinement and protonation assignment, we explored the interactions of Org27569 with the CP55,940 bound CB1 receptor using GLIDE-XP docking, followed by Schrodinger’s Induced Fit redocking, allowing flexibility and rotamer explorations in a 5 Å window around the binding pocket. CP55,940 is present in the orthosteric binding site in all subsequent docking/induced fit and dynamics studies performed. This approach allowed probing allosteric ligand-receptor interactions as a function of minor orientational changes in residues in the binding pocket prior to molecular dynamics studies. This is important because the specific interactions may vary in the top scoring induced fit poses and in the time evolution of the poses in molecular dynamics trajectories, although the recurring themes of interactions key for ligand-receptor binding are salient. LIGPLOT representations are shown to illustrate examples of these key allosteric interactions (Figure 4). The receptor residues are numbered according to the Ballesteros-Weinstein system as AXY.Z in which A is one letter symbol of the amino acid residue, X is the absolute number in the whole receptor protein sequence, Y is the number of the transmembrane domain and Z indicates the position of the residue in relation to the most highly conserved class A residue in each transmembrane helix.39
Figure 4.

Representative top scored configurations of A) Org27569 and B) PSNCBAM-1 to the CB1 receptor using Schrödinger Emodel Induced Fit/GLIDE XP
When docked to the CB1 receptor model using this approach, the best scoring poses of Org27569 were highly similar to the location of Org27569 in the crystal structure, forming π-stacking with H1542.41 via indole (green lines) and the phenyl ring at the opposite end interacting with W2414.50, the most conserved residue in helix IV, at a perpendicular centroid distance of ca. 3.5 Å (Figure 4A). We observed that the flexibility at residues W2414.50 and H1542.41 in the induced fit allowed conformational changes leading to improved π-stacking interactions. Additionally, interactions with R2304.39 either via hydrogen bonding with the Cl (distances of 2.6–2.9 Å measured between Arg guanidinium hydrogens and the chloro) or π-cation interactions with the chlorophenyl group were also observed. We then employed a second computational approach using Autodock-VINA/MMGBSA, which utilizes a more exhaustive whole ligand configuration sampling as compared to the “anchor-and-grow” approach in GLIDE-XP and found the best MMGBSA rescored VINA-poses lay in the same region of the helical facets, providing independent support for the Induced Fit results. In all these approaches, π-stacking interactions were clearly of importance and were the recurring theme.
To confirm that this was the lowest energy regime for Org27569 allosteric binding, blind docking of Org27569 into a large (40 × 20 × 40 Å) intracellular region contacting all 7 helices of the CB1 receptor was performed using the Autodock VINA scoring function, which identified three low energy regions (Supplementary Figure S1). We then re-scored the initial Autodock VINA poses from these three regions using AMBER 18/MMGBSA multi-trajectory molecular dynamics (MD).40–42 Rescoring using such an approach provides flexible receptor response and stabilization of the initial pose, checks short-time pose dynamical/temporal interaction stability, and often has better prediction of native poses from PDB-BIND test set assessments and improved affinity correlations,41 as has been previously reported.43 We found that the lowest multi-trajectory average MMGBSA poses of all three regions had their locations in the cleft with an Org27569 ligand long-axis orientation akin the crystallographic structure (Supplementary Figure S2). Further, we performed a molecular dynamics study in a DOPC/KCl/water environment and found that Org27569 maintained occupancy in the pocket adjacent to W2414.50/H1542.41/G1572.44/S1582.45/R2304.39, with persistent π-stacking interactions with W2414.50/H1542.41 and transient hydrogen bonding with H1542.41/G1572.44/S1582.45/R2304.39. While the interactions of Org27569 with these residues time-evolved in a manner that interaction partners changed even over a short 300 ns simulation (Supplementary Figure S3), they were sufficient to maintain occupancy long enough to bias the ensemble of conformations of the receptor, maintaining conformations akin to antagonist bound states as evident structurally and dynamically.34
Similarly, we next examined the interactions of PSCNBAM-1 with CB1 by performing GLIDE XP and Induced Fit docking using the same docking grid positioned on the crystallographic binding site of Org27569 and a similar blind docking VINA-AMBER-MMGBSA/MMPBSA multi-trajectory probe as was used for Org27569. These two independent approaches confirmed that in top ranked poses PSNSBAM-1 bound in the same pocket as Org27569 with the chlorophenyl ring helping to orient PSNCBAM-1 to maximize π-stacking interactions with H1542.41 and the pyridyl group making interactions with W2414.50 (Figure 4B).
2.2. Design and docking studies of hybrids.
Molecular docking and dynamics simulation studies on both Org27569 and PSNCBAM-1 clearly pointed to similar interactions of these two compounds with CB1, including interactions of the indole or chlorophenyl with H1542.41 and the phenyl (Org27569) or pyridyl (PSCNBAM-1) interacting with W2414.50. We hypothesized that if the central phenyl group of PSNCBAM-1 was replaced with a more flexible linker, such as an ethyl group as in Org27569, these key elements (aromatic rings and urea functionality) would be retained in similar spatial arrangement to maintain these molecular interactions. Indeed, when examined in induced fit studies, compound 8 which had a phenyl group linked to the urea functionality via a flexible ethyl group (like Org27569) adopted a configuration and orientation similar to PSCNBAM-1 and Org27569, with the analogous π-stacking interactions with H1542.41 and/or W2414.50 and hydrogen bonding with H1542.41 or W241 via the urea functionality (Figure 5A-B). Interestingly, we also observed poses with the reversed orientation, in which the π-stacking was present, although interaction partners had changed (Figure 5C). Such poses appeared to be favored because of the chlorophenyl interaction with the W2414.50 indole ring, where quantum chemical benchmark model calculations indicated ca. 75% increase of the π-stacking energy with aromatic ring chlorination (B3LYP-D3//6–31G** phenyl/chlorophenyl:−2.91kcal vs. phenyl-phenyl: −1.67 kcal). In all poses, the persistent π-stacking at either end of the ligand with the additional hydrogen bonding interactions served as a key recognition element for this new scaffold. These results suggest that replacement of the central phenyl group with a flexible alkyl group of appropriate length will maintain most key interactions with CB1, thus retaining its activity.
Figure 5.

Interactions of hybrid 8 with the CB1 receptor showing receptor-ligand interaction maps (left panels) and 3D-illustrations (right panels). A, B) Representative high ranked poses with π-stacking and hydrogen bonding with H1542.41 and/or W2414.50, and C) representative top ranked pose with reversed orientation with π-stacking with H1542.41 and W2414.50 and hydrogen bonding of the urea functionality with G1572.44 and S1582.45.
2.3. Molecular Dynamics Simulations.
In an attempt to further examine the similarity between Org27569 and our designed hybrids, we performed dynamics simulation to study the impact of the allosteric modulated agonist bound state with Org27569 and 8 (RSCB: 6KQI).34 Both Org27569 and PSNCBAM-1 have been shown to act as NAMs in most functional assays.17, 44, 45 As expected, the crystal structure of CB1-PGSCM fusion protein showed the activation microswitch condition of CB1 co-bound with CP55,940 and Org27569 differed from the crystal structures co-bound with agonists such as tetrahydrocannabinol analog AM11542 and hexahydrocannabinol analog AM841,46 but was more reminiscent of the antagonist/inverse agonist bound structures with 5TGZ (AM6538),47 and 5U09 (Taranabant).48 The key features included an intact R2143.50-D3386.30 salt bridge linking helices III and VI and the dual aromatic toggle residues at the base of the orthosteric binding site W3566.48 and F2003.36 which were in an “inactivation” state similar to the antagonist bound structures. Salt bridges have been demonstrated to play important roles for G protein coupling, ligand specificity, and protein trafficking.49–51 Ahn and colleagues, through theoretical models prior to solutions of the CB1 crystal structure, have identified R1482.37-D3386.30 and D1762.63-K1923.28 as key salt bridges that are critical for the inactive and active states of the CB1 receptor, respectively.52 It is possible that Org27569 and 8 stabilized the distinct R2143.50-D3386.30 salt bridge located at the allosteric binding site of the native CB1, thus acting as NAMs.
In agreement with the structural observation by Shao et al,34 the Org27569 kept the CB1 receptor in the antagonist state in the presence of the orthosteric agonist CP55,940. The dynamics simulation reveals that after ca. 50 ns equilibration without coordinate constraints, Org27569 and 8 reduced the fluctuations induced between the intracellular helices such that the R2143.50-D3386.30 salt bridge, between helices III and VI, stayed intact for most of the simulation, as illustrated by the stable distances between the R2143.50-guanidiium hydrogens and the D3386.30 carboxyl oxygen (Fig. 6A-B). However, removal of the allosteric modulator resulted in transient activation and salt bridge breaking in the presence of CP55,940 during a 100–800 nanosecond simulations, as is required to reach an activated state for G protein binding and recognition (Fig. 6C), which leads to G protein facilitated greater spread in the intrahelical domain. These results revealed that 8 was effective in preserving an antagonist like salt-bridge state distance similar to Org27569, preventing CP55,940 signaling by locking in place the D2143.50-R3386.30 salt bridge (Fig. 6D), consistent with the results from the docked/induced fit modeling.52
Figure 6.

Activation R2143.50-D3386.30 salt bridge (Arg-NH ----O2C-Asp) time series for CP55,940 agonist bound CB1 in the presence of: A) Org27569, B) Compound 8, C) no NAM (i.e. only CP55,940 is present). D1-D8 each represents the distance between a hydrogen on the guanidine group of R2143.50 side chain and an oxygen of the carboxylic group of D3386.30 side chain and D) Relative positions of Org27569 to CP55,940 and the R214 (blue)-D338 (red) salt-bridge at the bottom.
2.4. Chemistry.
To test the in silico predictions, we synthesized and investigated the structure-activity relationships (SARs) surrounding this novel CB1 NAM scaffold. Compounds 8 – 31 were obtained by coupling the corresponding aliphatic primary amines to 4-chlorophenyl isocyanate as shown in Scheme 1. Amines 32, 35, 36, 38, 41 and 42 were prepared as depicted in Scheme 1, whereas the other amines were purchased from commercial vendors or prepared according to previous procedures.29 Amines 32 and 36 were prepared from the reduction of corresponding benzonitrile or carboxamide by borane dimethyl sulfide. Alkylation of the enolate anion of methyl isobutyrate with benzyl bromide afforded the intermediate 33, which was hydrolyzed to give acid 34. Acid 34 was converted to the corresponding amine 35 by Curtius rearrangement in the presence of diphenylphosphoryl azide.53 Conversion of the trans mixture of 2-phenyl-1-cyclopropanecarboxylic acid to carboxamide 37 by coupling with ammonia followed by reduction using borane dimethyl sulfide yielded amine 38. Henry reaction between corresponding thiophene carboxaldehydes and nitromethane gave intermediates 39 and 40 which were subsequently reduced by LiAlH4 to amines 41 and 42.
Scheme 1. Synthesis of compounds 8 – 31.

Reagents and conditions: a) (i) BH3.Me2S, THF, 0 °C to rt, 16 h, (ii) 2 N HCl/Et2O, rt, 5 min, 58% to quant. yield b) (i) LDA, THF, −78 °C, 1 h (ii) BnBr, THF, −78 °C, 1.5 h quant. over 2 steps c) LiOH, MeOH, H2O, rt, 3 h, quant. d) (i) DPPA, Et3N, MeCN, 50 °C (ii) aq. 1 N HCl, reflux, 16 h, 88% for 2 steps e) (i) COCl2, DMF, DCM, rt, 3 h (ii) aq. NH4OH, MeCN, 0 °C to rt, 16 h, quant. f) MeNO2, NH4OAc, reflux, 2 h, 86–95% g) LiAlH4, THF, 0 °C to rt, 3 h, 80–84% h) corresponding amine, CHCl3, 60 °C, 16 h, 12– 91%.
2.5. Biological Evaluations
We first screened the activity of our compounds at CB1 and CB2 in FLIPR-based calcium mobilization functional assays as described previously.25, 29, 32, 33 Briefly, CHO cells that overexpress the promiscuous Gα16 proteins (RD-HGA16 cells, Molecular Devices) were engineered to stably express the human CB1 or CB2. Compounds were evaluated for their ability to inhibit the mobilization of intracellular calcium levels stimulated by the orthosteric agonist CP55,940. Select compounds were subsequently evaluated in the [35S]GTPγS binding assay to determine the inhibitory activity against G protein activation following the agonist occupation and in the BRET CAMYEL cAMP assay to assess the ability of compounds to reverse the inhibitory effect of CP55,940 on forskolin-mediated cAMP production.26, 54
2.5.1. CB1 allosteric modulatory activities in calcium mobilization assays.
We first examined all compounds for their potency to reduce CP55,940-induced CB1-mediated calcium mobilization in CHO-RD-HGA16 cells overexpressing human CB1 and the promiscuous Gα16 protein, which enables CB1 to signal through mobilization of internal calcium as previously described.25, 29, 33, 55 As predicted in the in silico model, 8 showed good allosteric modulatory activity with an pIC50 value of 6.7, comparable with Org27569 and PSNCBAM-1. Inclusions of a piperidinyl ring in 9 or a pyrrolidinyl moiety in 10 resulted in a slightly reduced potency, implying that bulky substituents at the 4-position of the phenyl ring are not preferred, as is seen in our SAR studies on the PSNCBAM-1 and diarylurea series.25 Interestingly, while the diethylamino analogue 11 which is the ring-opened analogue of 9 lost considerable potency, its smaller dimethylamino homologue 12 had improved potency by approximately 3-fold (pIC50 = 6.6). This is consistent with previous findings by us and others that dimethylamino is well tolerated in both the PSNCBAM-1 and Org2769 series.27, 30
Next, we investigated the structural requirement at the ethyl linker between the phenyl ring and the urea moiety by varying the length from one to three carbons. Our modeling studies showed that an ethyl linker maintained all key interactions with CB1, particularly the π-stacking on either end of the molecule. As seen in Table 1, the 2-carbon ethyl linker in 8 appeared to be the optimal length, as shortening the linker to methyl in 13 or lengthening to 1,3-propyl in 14 resulted in loss of potency. We then probed the effects of substituents on the ethyl linker. Additions of two methyl groups in 15 and 16 on the Cα or Cβ resulted in complete abolishment of CB1 antagonism. Presence of the difluoro group on the Cβ of 17 weakened the potency albeit to a lesser extent, indicating that additional bulk at these positions was not favorable. Similarly, rigidifying the ethyl linker with the presence of a cyclopropyl group in 18 and 19 also reduced the potency. Lastly, 20 with a 1,2-cyclopropylmethyl linker, displayed weaker activity than the ethyl linker in 8 but was more potent than the 1,3-propyllinker in 14. This may be the result of the strain of the three-membered ring which shortened the length between the two carbons. This is consistent with our previous SAR studies of Org27569, where constraint of the ethyl group by replacement with proline or piperidone groups resulted in a loss of potency.29
Table 1.
Allosteric Antagonist Potencies of Org27569, PSNCBAM-1, and 8-20 in the hCB1 Calcium Mobilization.
| Compound | Structure | pIC50a |
|---|---|---|
| Org27569 |
|
6.1 ± 0.07 |
| PSNCBAM-1 |
|
7.5 ± 0.1 |
| 8 |
|
6.7 ± 0.09 |
| 9 |

|
6.1 ± 0.05 |
| 10 |

|
6.1 ± 0.05 |
| 11 |
|
5.5 ± 0.03 |
| 12 |
|
6.6 ± 0.14 |
| 13 |

|
< 5 |
| 14 |

|
5.4 ± 0.06 |
| 15 |

|
< 5 |
| 16 |
|
< 5 |
| 17 |

|
5.8 ± 0.08 |
| 18 |
|
5.3 ± 0.09 |
| 19 |
|
< 5 |
|
20 (trans mixture) |
|
5.6 ± 0.09 |
Compounds tested against EC80 (100 nM) of CP55,940. Values are the mean ± SEM of at least three independent experiments in duplicate.
The pyridine ring of PSNCBAM-1 has previously been replaced by other heterocycles, such as pyrimidine and pyrrole.31, 33 We therefore sought to investigate if the phenyl ring in 8 could be replaced with other heterocyclic rings or aliphatic rings. Table 2 shows that replacement of the phenyl group with a 2-pyridyl ring in 21 significantly reduced potency, whereas substitution with 3-pyridinyl in 22 or 4-pyridinyl in 23 ring resulted in inactive compounds. The 3-thiophenyl analogue in 24 displayed low potency, whereas the 2-thiophenyl analogue 25 (pIC50 = 6.1) possessed similar potency to Org27569. The 5‐methylfuran‐2‐yl analogue 26 (pIC50 = 6.6) showed the highest potency among thisgroup, comparable to the phenyl analogue 8. Replacement of the phenyl ring by nonaromatic rings such as piperazine, piperidine, morpholine, pyrrolidine or the linear acetylamino group in 27 – 31 completely abolished or significantly dampened the CB1 inhibitory potency. These results suggest that an aromatic group is required at this position to interact with CB1, most likely by π-π interactions.
Table 2.
Allosteric Antagonist Potencies of Compounds 21-31 in the hCB1 Calcium Mobilization Assay.

| |||||
|---|---|---|---|---|---|
| Compound | Structure | pIC50a | Compound | Structure | pIC50a |
| 21 |

|
5.4 ± 0.09 | 27 |
|
< 5 |
| 22 |

|
< 5 | 28 |

|
< 5 |
| 23 |
|
< 5 | 29 |

|
< 5 |
| 24 |

|
5.8 ± 0.03 | 30 |

|
< 5 |
| 25 |

|
6.1 ± 0.01 | 31 | NHCOMe | 5.8 ± 0.09 |
| 26 |

|
6.6 ± 0.06 | |||
Compounds tested against EC80 (100 nM) of CP55,940. Values are the mean ± SEM of at least three independent experiments in duplicate.
In general, these SAR results are consistent with the computational findings. Our modeling studies showed that the π-stacking interactions with W2414.50 and H1542.41 and/or hydrogen bonding between the urea functionality and G1572.44 and S1582.45 are key interactions. Figure 5 illustrates the optimal positioning of the aromatic regions in our scaffold in compound 8 to capitalize on interactions with W2414.50/H1542.41 in some of the top score poses. Alterations of the number of −(CH2)n− linkages in our SAR modify the distance been the Cl-phenyl and the urea group, which forces the compound to compromise between finding optimal urea hydrogen bonding and forming the best π stacking overlaps. When both were not possible, as with the methyl linker in 13 or the propyl linker in 14, the potency was greatly diminished. In addition, inclusion of substituents on the ethyl linker such as difluoro in 17 and dimethyl in 15 and 16 or replacement with a cyclopropyl in 18 and 19 modify the conformation sufficiently to reduce these interactions, resulting in reduced interactions between urea and G1572.44/S1582.45 and/or Cl-phenyl with W2414.50. These substitutions therefore reduce the allosteric modulatory potency compared to 8. While introduction of the pyridyl groups in 22 and 23 retained the π-stacking interactions, the desolvation costs of the pyridyl groups may have contributed to their reduced allosteric potency,41, 56 with considerably higher computed solvation energies for 22 (−17.3kcal/mol) and 23 (−17.4kcal/mol) compared to 8 (−13.6kcal/mol). Replacement of the phenyl with non-aromatic groups in 27-31 abolished the π-stacking interactions with H1542.41 and/or W2414.50 and thus the total loss of potencies.
All compounds were screened for agonist activity at the CB1 in the calcium mobilization assay and no significant agonist effects (<30% of CP55,940 Emax) were observed for any of the compounds. These compounds were also screened for agonist and antagonist activity at CB2 to determine receptor subtype selectivity in our CB2 calcium mobilization assay.25, 30 None of the compounds had significant CB2 agonist activity (<10% of CP55,940 Emax) or CB2 antagonist activity (<50% inhibition of CP55,940 EC80 concentration at 10 μM or pIC50 values < 5).
2.5.2. Evaluation of select compounds in [35S]GTPγS and cAMP assays.
Several compounds that showed good CB1 modulatory activity in the calcium mobilization assay were further characterized in [35S]GTPγS binding and/or cAMP assays using HEK293 cells overexpressing human CB1. 25, 33 Overall, compounds exhibited lower potency in both the [35S]GTPγS and cAMP assays as compared with the calcium mobilization assay. This could be due to overexpression of the promiscuous Gα16 in the cells used in the calcium assay which may shift more receptors into a coupled state with high affinity for agonists, thereby increasing the sensitivity of the calcium mobilization assay.57 PSNCBAM-1 exhibited the greatest inhibitory potency in both the [35S]GTPγS and calcium mobilization assays. In the [35S]GTPγS assay, the phenyl analogue 8 possessed a pIC50 of 5.2 whereas the two thiophenyl analogues (24 and 25) displayed similar pIC50 values, all lower than Org27569 or PSNCBAM-1. Compound 9 demonstrated comparable potencies in both calcium mobilization and [35S]GTPγS assays. Other analogues had weaker potency in the [35S]GTPγS assay (pIC50 < 5). In the cAMP assay, compounds 8, 12, and 26 demonstrated potency in the low micromolar range. The differences in the magnitude of rank order potencies between these assays could be due to differences in their affinities for the different conformations being promoted by each cell type’s cellular milieu, in particular the respective G protein content (i.e. a preponderance of Gα16 in the calcium assay).
Interestingly, 9 appeared to produce incomplete antagonism of CP55,940’s effect in [35S]GTPγS binding and calcium mobilization with the best-fit parameters with the curve bottoms being greater than zero ([35S]GTPγS: 64.6% ± 6.4, 95% CI: 36.9 – 92.3; calcium: 26.4% ± 4.0, 95% CI: 13.7 – 39.1). To determine whether compound 9 would produce incomplete insurmountable antagonism, we examined CP55,940’s concentration-response in [35S]GTPγS binding in the presence of multiple concentrations of compound 9 and fit the operational model of allosterism (equation 1, Experimental Section). While compound 9 (logKb = −4.4 ± 0.5; Kb = 38 µM) did not reach saturation at the highest concentration (100 µM), best-fit allosteric parameters suggested that it was a PAM-antagonist (logalpha = 0.7 ± 0.1; alpha = 5.1), exhibiting positive cooperativity with the probe CB1 agonist CP55,940 and incomplete antagonism (logbeta = −1.1 ± 0.1; beta = 0.09). Predicted curves for CP55,940 in the presence compound 9 (316 µM and 1 mM) based on simulations using the best-fit allosteric parameters are visible in Figure 7 as dashed lines and depict the saturation of effect. Such an allosteric effect would be promising as it would allow for partial, and possibly biased, antagonism, restricting activation of particular G protein subtypes while permitting the cycling of others. However, it is possible that limited solubility at the high concentrations used in the studies is a contributing factor to the observed affinity and beta parameters and cannot be ruled out as cannabinoid ligands are known with solubility challenges.17
Figure 7.

Compound 9 exhibited insurmountable and incomplete antagonism of CP55,940-stimulated [35S]GTPγS binding in hCB1 HEK293 membranes. Dashed lines depict simulated curves based upon best-fit allosteric parameters of the allosteric operational model (see methods). Each point represents the mean±SEM of at least N=3.
3. Conclusions
Negative allosteric modulation offers a promising approach targeting the CB1 receptor for obesity, drug addictions and other therapeutic applications while possibly limiting the side effects of orthosteric antagonists. Consistent with the similarities in the structures of Org27569 and PSNCBAM-1 as well as their SARs, computational modeling studies suggested that these NAMs preferentially bind to the same binding pocket of Org27569 in the recently solved crystal structure of CB1 co-bound with Org27569 and CP55,940, occupying the space between TM2 and TM4 and overlapping the cholesterol binding region. Key interactions between these NAMs with CB1 include π-stacking interactions with W2414.50 and H1542.41 and/or hydrogen bonding between the urea functionality and H1542.41. Based on these findings, we rationally designed hybrids of Org27569 and PSNCBAM-1 as CB1 NAMs such as 8 by merging the key structural components from each series. Compound 8 bound to the same binding pocket as Org27569 and PSCNBAM-1 with the same π-stacking interactions with W2414.50 and/or H1542.41. Similar to Org27569, 8 preserved an antagonist-like state of CB1 with the key R2143.50-D3386.30 salt bridge intact during dynamics simulations, whereas salt bridge repeatedly breaks in the agonist-alone models during microsecond simulations, suggesting that the functional effect of allosteric ligands may be predicted with short time dynamical simulations. A series of CB1 NAMs based on this novel hybrid scaffold were then synthesized and evaluated for their biological activity in several in vitro assays. Compound 8 showed similar potency to Org27569 and PSNCBAM-1 in calcium mobilization and slightly reduced potencies in GTPγS and cAMP, whereas 9 had pIC50 similar to Org27569 in the GTPγS assay. Consistent with computational findings highlighting the importance of linker length, shortening or elongation of the ethyl linker or substitution on this linker comprised one or both of these interactions, thus leading to reduced potency. In addition, substitution of the phenyl ring on the right-hand side by other non-aromatic rings resulted in abolishment of CB1 allosteric modulatory activity, confirming importance of π-stacking of this aromatic ring with CB1. Together, these findings provide a useful computational tool in predicting the potential functional effect (activation vs inactivation) of an allosteric ligand to be designed based on whether it retains the critical R2143.50-D3386.30 salt bridge. Our work expands the current SAR understanding of CB1 allosteric modulators and provides a new scaffold that can be further optimized for CB1 modulation.
4. Experimental
4.1. Chemistry.
All solvents and chemicals were reagent grade. Unless otherwise mentioned, all reagents and solvents were purchased from commercial vendors and used as received. Flash column chromatography was carried out on a Teledyne ISCO CombiFlash Rf system using prepacked columns. Solvents used include hexane, ethyl acetate (EtOAc), dichloromethane, methanol, and chloroform/methanol/ammonium hydroxide (80:18:2) (CMA-80). Purity and characterization of compounds were established by a combination of HPLC, TLC, mass spectrometry, and NMR analyses. Melting point was recorded by the Mel-Temp II instrument (Laboratory Devices Inc. , U.S.). 1H and 13C NMR spectra were recorded on a Bruker Avance DPX-300 (300 MHz) spectrometer and were determined in chloroform-d, DMSO-d6, or methanol-d4 with tetramethylsilane (TMS) (0.00 ppm) or solvent peaks as the internal reference. Chemical shifts are reported in ppm relative to the reference signal, and coupling constant (J) values are reported in hertz (Hz). Thin layer chromatography (TLC) was performed on EMD precoated silica gel 60 F254 plates, and spots were visualized with UV light or iodine staining. High resolution mass spectra were obtained using Agilent 1290 Infinity UHPLC-6230 TOF system (ESI). The calculated and found monoisotopic masses which are the sum of the accurate masses of the most abundant naturally occurring stable isotope of each atom in the molecule were reported. All test compounds were greater than 95% pure as determined by HPLC on an Agilent 1100 system using an Agilent Zorbax SB-Phenyl, 2.1 mm × 150 mm, 5 μm column using a 15-minute gradient elution of 5–95% solvent B at 1 mL/min followed by 10 minutes at 95% solvent B (solvent A, water with 0.1% TFA; solvent B, acetonitrile with 0.1% TFA and 5% water; absorbance monitored at 220 and 280 nm).
General Procedure A: To a solution of amine (1 eq) in anhydrous chloroform (0.04 M) was added 4-chlorophenyl isocyanate (1 eq) at room temperature. The reaction mixture was then heated at 60 °C for 16 h. The precipitated product was filtered and thoroughly washed with dichloromethane.
3‐(4‐Chlorophenyl)‐1‐(2‐phenylethyl)urea (8) was prepared from phenethylamine (0.04 ml, 0.32 mmol) following the general procedure A as white solid (0.06 g, 63%). 1H NMR (300 MHz, CDCl3) δ 7.29 – 7.35 (m, 2H), 7.15 – 7.25 (m, 7H), 6.12 (br. s., 1H), 4.58 (br. s., 1H), 3.53 (q, J = 6.6 Hz, 2H), 2.85 (t, J = 6.7 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 155.1 (1C), 138.9 (1C), 137.0 (1C), 129.2 (2C), 128.9 (1C), 128.8 (2C), 128.7 (2C), 126.6 (1C), 122.0 (2C), 41.5 (1C), 36.0 (1C). HRMS (ESI) m/z [M+H]+ calcd: 275.0946; found: 275.0936.
1-(4-Chlorophenyl)-3-{2-[4-(piperidin-1-yl)phenyl]ethyl}-urea (9) was prepared from 2-[4-(piperidin-1-yl)phenyl]ethanamine hydrochloride which was prepared as described previously29 (0.10 g, 0.42 mmol) following the general procedure A as white solid (0.12 g, 79%). 1H NMR (300 MHz, CDCl3) δ 7.19 (m, 4 H), 7.05 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 8.7 Hz, 2H), 6.34 (s, 1 H), 4.72 (t, J = 5.6 Hz, 1H), 3.45 (q, J = 6.4 Hz, 2H), 3.11 (m, 4 H), 2.73 (t, J = 6.7 Hz, 2H), 1.71 (quin, J = 5.6 Hz, 4H), 1.57 (m, 2 H). 13C NMR (75 MHz, CDCl3) δ 155.3 (1C), 151.0 (1C), 137.2 (1C), 129.4 (2C), 129.2 (1C), 129.1 (2C), 128.6 (1C), 121.8 (2C), 116.8 (2C), 50.8 (1C), 41.6 (1C), 35.1 (1C), 25.8 (1C), 24.2 (1C). HRMS (ESI) m/z [M+H]+ calcd: 358.1681; found: 358.1674.
1-(4-Chlorophenyl)-3-{2-[4-(pyrrolidin-1-yl)phenyl]ethyl}-urea (10) was prepared from 2-[4-(pyrrolidin-1-yl)phenyl]ethanamine hydrochloride (0.07 g, 0.44 mmol) which was prepared as described previously29 following the general procedure A as white solid (0.12 g, 79%). 1H NMR (300 MHz, CDCl3) δ 7.13 – 7.24 (m, 4H), 7.04 (d, J = 8.5 Hz, 2H), 6.51 (d, J = 8.5 Hz, 2H), 6.11 (s, 1H), 4.61 (br. s., 1H), 3.46 (q, J = 6.5 Hz, 2H), 3.23 – 3.29 (m, 4H), 2.73 (t, J = 6.6 Hz, 2H), 2.00 (td, J = 3.30, 6.59 Hz, 4H). 13C NMR (75 MHz, CDCl3) δ 155.2 (1C), 146.9 (1C), 137.2 (1C), 129.5 (2C), 129.1 (2C), 128.6 (1C), 125.1 (1C), 121.9 (2C), 111.9 (2C), 47.7 (1C), 41.9 (1C), 35.0 (1C), 25.4 (1C). HRMS (ESI) m/z [M+H]+ calcd: 344.1524; found: 344.1523.
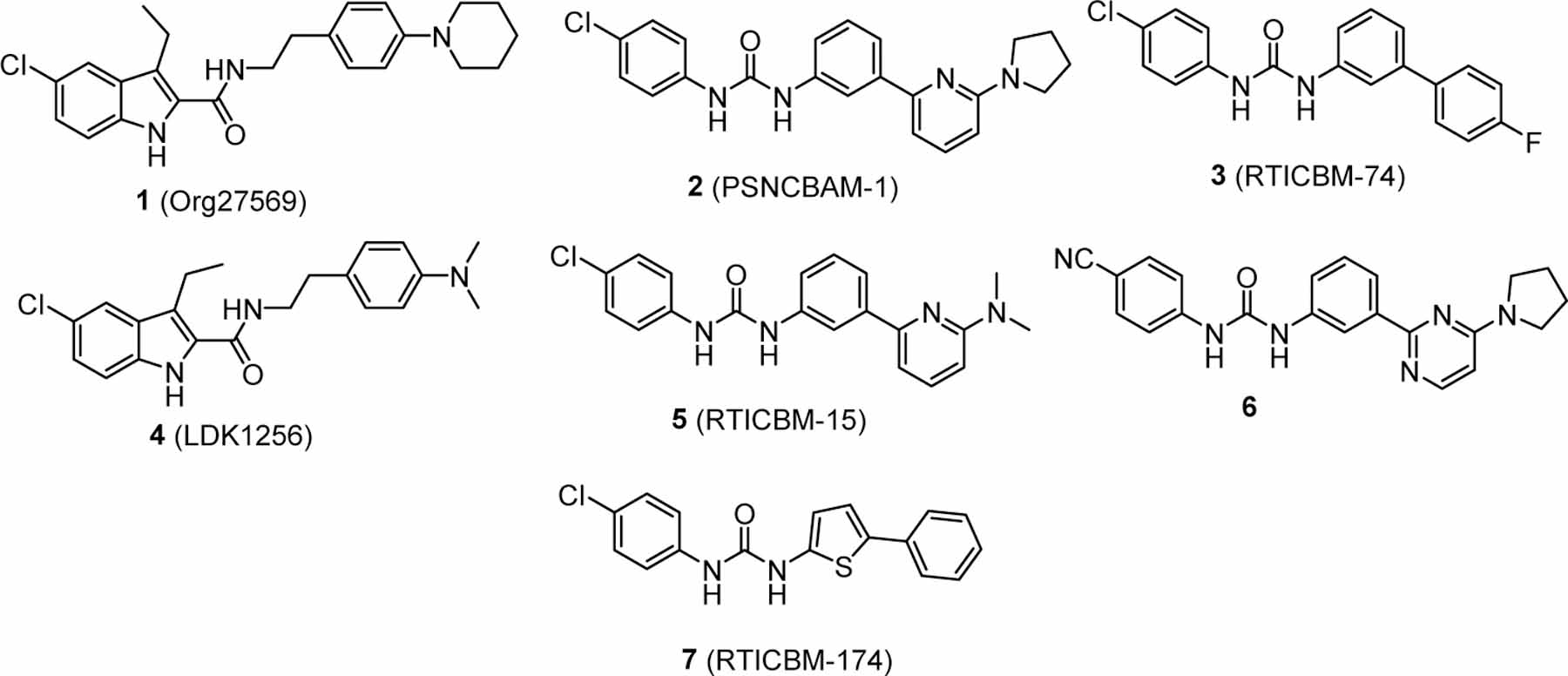
1-(4-Chlorophenyl)-3-{2-[4-(diethylamino)phenyl]ethyl}-urea (11) was prepared from 4-(2-aminoethyl)-N,N-diethylaniline hydrochloride (0.04 g, 0.28 mmol) which was prepared as described previously29 following the general procedure A as white solid (0.80 g, 82%). 1H NMR (300 MHz, CDCl3) δ 7.09 – 7.19 (m, 4H), 7.01 (br. s., 1H), 6.98 (d, J = 8.5 Hz, 2H), 6.58 (d, J = 8.5 Hz, 2H), 5.28 (d, J = 5.3 Hz, 1H), 3.38 (q, J = 6.7 Hz, 2H), 3.29 (q, J = 7.0 Hz, 4H), 2.65 (t, J = 6.9 Hz, 2H), 1.12 (t, J = 7.0 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 156.0 (1C), 146.6 (1C), 137.5 (1C), 129.6 (2C), 129.0 (2C), 128.2 (1C), 125.3 (1C), 121.6 (2C), 112.2 (2C), 44.3 (1C), 41.8 (1C), 35.1 (1C), 12.6 (1C). HRMS (ESI) m/z [M+H]+ calcd: 346.1681; found: 346.1675.
3‐(4‐Chlorophenyl)‐1‐{2‐[4‐(dimethylamino)phenyl]ethyl}-urea (12) was prepared from 4-dimethylaminophenethylamine (0.03 g, 0.18 mmol) following the general procedure A as white solid (0.01 g, 17%). 1H NMR (300 MHz, DMSO-d6) δ 8.63 – 8.71 (m, 1H), 7.41 (d, J = 9.0 Hz, 2H), 7.24 (d, J = 8.9 Hz, 2H), 7.04 (d, J = 8.7 Hz, 2H), 6.68 (d, J = 8.7 Hz, 2H), 6.08 – 6.16 (m, 1H), 3.26 (d, J = 6.2 Hz, 2H), 2.85 (s, 6H), 2.61 (t, J = 7.2 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 154.9 (1C), 149.1 (1C), 139.5 (1C), 129.0 (2C), 128.4 (2C), 126.9 (1C), 124.3 (1C), 119.0 (2C), 112.7 (2C), 40.9 (1C), 40.3 (1C), 34.8 (1C). HRMS (ESI) m/z [M+H]+ calcd: 318.1367; found: 318.1370.
3‐(4‐Chlorophenyl)‐1‐(3‐phenylmethyl)urea (13) was prepared from benzylamine (0.05 ml, 0.32 mmol) following the general procedure A as white solid (0.06 g, 71%). 1H NMR (300 MHz, DMSO-d6) δ 8.71 (s, 1H), 7.44 (d, J = 8.9 Hz, 2H), 7.20 – 7.37 (m, 7H), 6.66 (t, J = 5.8 Hz, 1H), 4.30 (d, J = 6.0 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 155.0 (1C), 140.2 (1C), 139.4 (1C), 128.4 (2C), 128.3 (2C), 127.1 (2C), 126.7 (1C), 124.5 (1C), 119.2 (2C), 42.7 (1C). HRMS (ESI) m/z [M+H]+ calcd: 261.0789; found: 261.0797.
3‐(4‐Chlorophenyl)‐1‐(3‐phenylpropyl)urea (14) was prepared from 3-phenylpropylamine (0.05 ml, 0.32 mmol) following the general procedure A as white solid (0.08 g, 82%). 1H NMR (300 MHz, CDCl3) δ 7.17 – 7.31 (m, 7H), 7.09 (d, J = 6.8 Hz, 2H), 5.42 (t, J = 5.4 Hz, 1H), 3.18 (q, J = 6.8 Hz, 2H), 2.57 (t, J = 7.6 Hz, 2H), 1.70 – 1.78 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 156.1 (1C), 141.3 (1C), 137.4 (1C), 129.1 (2C), 128.5 (2C), 128.4 (1C), 128.3 (2C), 126.0 (1C), 121.5 (2C), 39.9 (1C), 33.1 (1 C), 31.6 (1C). HRMS (ESI) m/z [M+H]+ calcd: 289.1102; found: 289.1100.
1-(4-Chlorophenyl)-3-(2-methyl-2-phenylpropyl)urea (15) was prepared from 3-phenylpropylamine (32) (0.10 g, 0.56 mmol) following the general procedure A as white solid (0.14 g, 81%). 1H NMR (300 MHz, CDCl3) δ 7.34 (d, J = 4.1 Hz, 4H), 7.20 – 7.25 (m, 1H), 7.16 – 7.20 (m, 2H), 7.05 – 7.11 (m, 2H), 6.14 (br. s., 1H), 4.37 (br. s., 1H), 3.45 (d, J = 6.0 Hz, 2H), 1.35 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 155.4 (1C), 146.6 (1C), 137.2 (1C), 129.0 (2C), 128.6 (2C), 128.5 (1C), 126.3 (1C), 126.0 (2C), 121.6 (2C), 51.7 (1C), 38.9 (1C), 30.9 (1C), 26.6 (1C). HRMS (ESI) m/z [M+H]+ calcd: 303.1259; found: 303.1259.
1-(4-Chlorophenyl)-3-(2-methyl-1-phenylpropan-2-yl)urea (16) was prepared from 2-methyl-1-phenylpropan-2-amine (35) (0.06 g, 0.38 mmol) following the general procedure A as white solid (0.07 g, 57%). 1H NMR (300 MHz, CDCl3) δ 7.20 – 7.27 (m, 5H), 7.12 – 7.19 (m, 4H), 6.21 (s, 1H), 4.41 (s, 1H), 3.03 (s, 2H), 1.33 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 154.4 (1C), 138.1 (1C), 137.4 (1C), 130.6 (2C), 129.2 (2C), 128.7 (1C), 128.1 (2C), 126.4 (1C), 122.0 (2C), 53.7 (1C), 45.5 (1C), 27.9 (2C). HRMS (ESI) m/z [M+H]+ calcd: 303.1259; found: 303.1255.
1-(4-Chlorophenyl)-3-(2,2-difluoro-2-phenylethyl)urea (17) was prepared from 2,2-difluoro-2-phenylethanamine (36) (0.06 g, 0.29 mmol) following the general procedure A as white solid (0.05 g, 52%). 1H NMR (300 MHz, DMSO-d6) δ 8.72 (s, 1H), 7.49 – 7.62 (m, 5H), 7.36 – 7.43 (m, 2H), 7.24 – 7.30 (m, 2H), 6.57 (t, J = 6.2 Hz, 1H), 3.88 (dt, J = 6.2, 14.98 Hz, 2H). 13C NMR (75 MHz, CD3OD) δ 157.5 (1C), 139.6 (1C), 136.6 (t, J = 25 Hz, 1C), 131.4 (1C), 129.7 (d, J = 5 Hz, 2C), 128.4 (1C) 126.4 (t, J = 6 Hz, 2C), 125.3 (1C), 122.1 (1C), 121.5 (2C), 118.9 (1C), 46.8 (t, J = 30.7 Hz, 1C). HRMS (ESI) m/z [M+H]+ calcd: 311.0757; found: 311.0768.
1-(4-Chlorophenyl)-3-[(1-phenylcyclopropyl)methyl]urea (18) was prepared from 1-phenylcyclopropanemethylamine (0.03 g, 0.20 mmol) following the general procedure A as white solid (0.04 g, 68%). 1H NMR (300 MHz, CDCl3) δ 7.28 – 7.39 (m, 4H), 7.10 – 7.24 (m, 5H), 6.22 (br. s., 1H), 4.70 (br. s., 1H), 3.43 (d, J = 5.5 Hz, 2H), 0.89 (s, 4H). 13C NMR (75 MHz, CDCl3) δ 155.2 (1C), 142.8 (1C), 137.1 (1C), 129.1 (2C), 128.6 (2C), 128.5 (2C), 126.7 (1C), 121.8 (2C), 49.2 (1C), 29.7 (1C), 25.8 (1C), 12.3 (1C). HRMS (ESI) m/z [M+H]+ calcd: 301.1102; found: 301.1096.
1-(1-Benzylcyclopropyl)-3-(4-chlorophenyl)urea (19) was prepared from (1-benzylcyclopropyl)amine (0.04 g, 0.20 mmol) following the general procedure A as white solid (0.03 g, 48%). 1H NMR (300 MHz, CDCl3) δ 7.29 – 7.34 (m, 2H), 7.27 – 7.28 (m, 1H), 7.20 – 7.24 (m, 2H), 7.17 – 7.20 (m, 2H), 7.10 – 7.15 (m, 2H), 6.34 (s, 1H), 4.86 (s, 1H), 2.86 (s, 2H), 0.91 – 0.98 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 153.5 (1C), 136.9 (1C), 135.5 (1C), 127.9 (2C), 127.3 (2C), 127.2 (2C), 126.8 (1C), 125.5 (1C), 119.6 (2C), 41.5 (1C), 33.3 (1C), 29.3 (1C), 13.0 (1C). HRMS (ESI) m/z [M+H]+ calcd: 301.1102; found: 301.1100.
trans-1-(4-Chlorophenyl)-3-[(2-phenylcyclopropyl)methyl-]urea (20) was prepared from trans-1-(2-phenylcyclopropyl)methanamine (38) (0.08 g, 0.45 mmol) following the general procedure A as white solid (0.07 g, 51%). 1H NMR (300 MHz, DMSO-d6) δ 8.62 (s, 1H), 7.38 – 7.46 (m, 2H), 7.20 – 7.29 (m, 4H), 7.11 – 7.15 (m, 1H), 7.04 – 7.10 (m, 2H), 6.32 (t, J = 5.6 Hz, 1H), 3.16 – 3.28 (m, 1H), 2.99 – 3.11 (m, 1H), 1.79 – 1.88 (m, 1H), 1.21 – 1.35 (m, 1H), 0.83 – 0.96 (m, 2H). 13C NMR (75 MHz, DMSO-d6) δ 155.0 (1C), 142.8 (1C), 139.5 (1C), 128.4 (2C), 128.1 (2C), 125.5 (2C), 125.2 (1C), 124.4 (1C), 119.1 (2C), 42.9 (1C), 23.4 (1C), 21.3 (1C), 14.3 (1C). HRMS (ESI) m/z [M+H]+ calcd: 301.1102; found: 301.1099.
3‐(4‐Chlorophenyl)‐1‐[2‐(pyridin‐2‐yl)ethyl]urea (21) was prepared from 2-(2-aminoethyl)pyridine (0.04 g, 0.32 mmol) following the general procedure A as white solid (0.08 g, 84%). 1H NMR (300 MHz, CDCl3) δ 8.38 (d, J = 4.1 Hz, 1H), 7.87 (br. s., 1H), 7.55 – 7.62 (m, 1H), 7.10 – 7.24 (m, 6H), 6.15 – 6.24 (m, 1H), 3.62 (q, J = 5.8 Hz, 2H), 2.94 – 3.01 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 159.6 (1C), 156.0 (1C), 148.8 (1C), 137.8 (1C), 136.9 (1C), 129.0 (2C), 128.0 (1C), 123.6 (1C), 121.7 (1C), 121.3 (2C), 39.6 (1C), 37.6 (1C). HRMS (ESI) m/z [M+H]+ calcd: 276.0898; found: 276.0895.
3‐(4‐Chlorophenyl)‐1‐[2‐(pyridin‐3‐yl)ethyl]urea (22) was prepared from 3-(2-aminoethyl)pyridine (0.04 g, 0.32 mmol) following the general procedure A as white solid (0.08 g, 91%). 1H NMR (300 MHz, CDCl3) δ 8.30 – 8.38 (m, 2H), 7.53 – 7.63 (m, 2H), 7.18 – 7.26 (m, 5H), 5.49 (t, J = 5.5 Hz, 1H), 3.52 (q, J = 5.7 Hz, 2H), 2.78 – 2.87 (m, 2H). 13C NMR (75 MHz, CDCl3) δ 155.6 (1C), 149.8 (1C), 147.6 (1C), 137.5 (1C), 136.8 (1C), 135.0 (1C), 129.1 (2C), 128.2 (1C), 123.9 (1C), 121.0 (2C), 40.7 (1C), 33.3 (1C). HRMS (ESI) m/z [M+H]+ calcd: 276.0898; found: 276.0894.
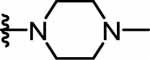
3‐(4‐Chlorophenyl)‐1‐[2‐(pyridin‐4‐yl)ethyl]urea (23) was prepared from 4-(2-aminoethyl)pyridine (0.04 ml, 0.32 mmol) following the general procedure A as white solid (0.03 g, 34%). 1H NMR (300 MHz, DMSO-d6) δ 8.62 (s, 1H), 8.48 (d, J = 5.8 Hz, 2H), 7.40 (d, J = 8.8 Hz, 2H), 7.22 – 7.30 (m, 4H), 6.19 (t, J = 5.7 Hz, 1H), 3.35 – 3.43 (m, 2H), 2.77 (t, J = 6.9 Hz, 2H). 13C NMR (75 MHz, DMSO-d6) δ 154.9 (1C), 149.5 (2C), 148.4 (1C), 139.4 (1C), 128.4 (2C), 124.4 (2C), 124.2 (1C), 119.1 (2C), 34.9 (1C). HRMS (ESI) m/z [M+H]+ calcd: 276.0898; found: 276.0899.
1-(4-Chlorophenyl)-3-[2-(thiophen-3-yl)ethyl]urea (24) was prepared from 41 (0.08 g, 0.54 mmol) following the general procedure A as white solid (0.13 g, 86%). 1H NMR (300 MHz, CDCl3) δ 7.30 (dd, J = 2.8, 4.9 Hz, 1H), 7.18 – 7.23 (m, 3H), 7.01 (d, J = 2.8 Hz, 1H), 6.96 (dd, J = 1.1, 4.9 Hz, 1H), 6.25 (s, 1H), 4.68 (br. s., 1H), 3.51 (q, J = 6.7 Hz, 2H), 2.87 (t, J = 6.7 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 155.1 (1C), 139.2 (1C), 137.1 (1C), 129.2 (2C), 128.0 (1C), 126.1 (1C), 122.0 (2C), 121.5 (1C), 121.0 (1C), 40.8 (1C), 30.6 (1C). HRMS (ESI) m/z [M+H]+ calcd: 281.0510; found: 281.0506.
1-(4-Chlorophenyl)-3-[2-(thiophen-2-yl)ethyl]urea (25) was prepared from 42 (0.03 g, 0.21 mmol) following the general procedure A as white solid (0.03 g, 50%). 1H NMR (300 MHz, CDCl3) δ 7.19 – 7.24 (m, 3H), 7.15 – 7.18 (m, 2H), 6.95 (dd, J = 3.4, 5.1 Hz, 1H), 6.84 (d, J = 2.8 Hz, 1H), 6.31 (s, 1H), 4.82 (br. s., 1H), 3.53 (q, J = 6.3 Hz, 2H), 3.06 (t, J = 6.5 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 155.1 (1C), 141.4 (1C), 137.0 (1C), 129.2 (2C), 127.1 (1C), 125.5 (1C), 124.0 (1C), 122.1 (2C), 121.5 (1C), 41.7 (1C), 30.4 (1C). HRMS (ESI) m/z [M+H]+ calcd: 281.0510; found: 281.0509.
1‐(4‐Chlorophenyl)‐3‐[2‐(5‐methylfuran‐2‐yl)ethyl]urea (26) was prepared from 2-(5-methyl-2-furyl)ethanamine (0.04 ml, 0.32 mmol) following the general procedure A as white solid (0.04 g, 44%). 1H NMR (300 MHz, CDCl3) δ 7.18 – 7.25 (m, 4H), 6.34 (br. s., 1H), 5.84 – 5.96 (m, 2H), 4.90 (t, J = 5.8 Hz, 1H), 3.51 (q, J = 6.2 Hz, 2H), 2.80 (t, J = 6.4 Hz, 2H), 2.23 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 155.2 (1C), 151.2 (1C), 137.1 (1C), 129.2 (2C), 128.9 (1C), 122.1 (2C), 107.2 (2C), 106.1 (1C), 39.1 (1C), 28.6 (1C), 13.5 (1C). HRMS (ESI) m/z [M+H]+ calcd: 279.0895; found: 279.0891.
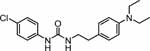
3‐(4‐Chlorophenyl)‐1‐[2‐(4‐methylpiperazin‐1‐yl)ethyl]-urea (27) was prepared from (4-methylpiperazin-1-yl)ethanamine (0.05 ml, 0.32 mmol) following the general procedure A as white solid (0.04 g, 43%). 1H NMR (300 MHz, CD3OD) δ 7.31 – 7.38 (m, 2H), 7.18 – 7.24 (m, 2H), 4.83 – 4.88 (m, 2H), 2.34 – 2.77 (m, 10H), 2.28 (s, 3H). 13C NMR (75 MHz, CD3OD) δ 158.0 (1C), 140.0 (1C), 129.7 (2C), 128.1 (1C), 121.3 (2C), 58.6 (1C), 55.8 (2C), 53.7 (2C), 46.0 (1C), 37.8 (1C). HRMS (ESI) m/z [M-H]‒ calcd: 297.1477; found: 297.1486.
3‐(4‐Chlorophenyl)‐1‐[2‐(piperidin‐1‐yl)ethyl]urea (28) was prepared from 1-(2-aminoethyl)piperidine (0.05 ml, 0.32 mmol) following the general procedure A as white solid (0.06 g, 66%). 1H NMR (300 MHz, CDCl3) δ 8.53 (br. s., 1H), 7.24 – 7.33 (m, 2H), 7.12 – 7.22 (m, 2H), 6.13 (br. s., 1H), 3.97 – 4.12 (m, 1H), 3.26 – 3.39 (m, 2H), 2.35 – 2.60 (m, 6H), 1.53 – 1.66 (m, 4H), 1.46 (d, J = 4.7 Hz, 2H). 13C NMR (75 MHz, CDCl3) δ 156.8 (1C), 138.1 (1C), 128.8 (2C), 127.5 (1C), 121.0 (2C), 58.8 (1C), 54.5 (2C), 37.3 (1C), 25.3 (2C), 23.8 (1C). HRMS (ESI) m/z [M+H]+ calcd: 282.1368; found: 282.1366.
3‐(4‐Chlorophenyl)‐1‐[2‐(morpholin‐4‐yl)ethyl]urea (29) was prepared from 4-(2-aminoethyl)morpholine (0.04 ml, 0.32 mmol) following the general procedure A as white solid (0.08 g, 82%). 1H NMR (300 MHz, CDCl3) δ 7.56 (br. s., 1H), 7.19 – 7.32 (m, 4H), 5.52 – 5.64 (m, 1H), 3.62 – 3.77 (m, 4H), 3.34 (q, J = 5.5 Hz, 2H), 2.36 – 2.56 (m, 6H). 13C NMR (75 MHz, CDCl3) δ 156.0 (1C), 137.6 (1C), 129.0 (2C), 128.3 (1C), 121.4 (2C), 66.8 (2C), 58.0 (1C), 53.5 (2C), 36.9 (1C). HRMS (ESI) m/z [M+H]+ calcd: 284.1160; found: 284.1161.
1‐(4‐Chlorophenyl)‐3‐[2‐(pyrrolidin‐1‐yl)ethyl]urea (30) was prepared from 1-(2-aminoethyl)pyrrolidine (0.04 ml, 0.32 mmol) following the general procedure A as white solid (0.01 g, 12%). 1H NMR (300 MHz, CDCl3) δ 8.67 (br. s., 0H), 6.99 – 7.33 (m, 4H), 5.96 (br. s., 1H), 3.32 (q, J = 5.2 Hz, 2H), 2.63 – 2.72 (m, 2H), 2.49 – 2.63 (m, 4H), 1.72 – 1.89 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 157.0 (1C), 138.3 (1C), 128.8 (2C), 127.5 (1C), 120.8 (2C), 56.7 (1C), 54.1 (2C), 39.7 (1C), 23.6 (2C). HRMS (ESI) m/z [M+H]+ calcd: 268.1211; found: 268.1213.
N‐(2‐{[(4‐Chlorophenyl)carbamoyl]amino}ethyl)acetamide (31) was prepared from N-acetylethylenediamine (0.03 ml, 0.32 mmol) following the general procedure A as white solid (0.02 g, 24%). 1H NMR (300 MHz, DMSO-d6) δ 8.69 (s, 1H), 7.93 (br. s., 1H), 7.42 (d, J = 8.9 Hz, 2H), 7.25 (d, J = 8.9 Hz, 2H), 6.20 (br. s., 1H), 3.03 – 3.22 (m, 4H), 1.81 (s, 3H). 13C NMR (75 MHz, DMSO-d6) δ 169.4 (1C), 155.1 (1C), 139.5 (1C), 128.4 (2C), 124.4 (1C), 119.1 (2C), 50.2 (1C), 30.6 (1C), 22.6 (1C). HRMS (ESI) m/z [M+H]+ calcd: 256.0847; found: 256.0844.
2-Methyl-2-phenylpropan-1-amine (32).
To a solution of 2-methyl-2-phenylpropanenitrile (0.15 ml, 1 mmol) in THF (4 ml) was added 2M BH3.Me2S in THF solution (0.5 ml) in an ice-water bath. The reaction was raised to room temperature and stirred for 16 h. The reaction mixture was then carefully quenched with methanol and solvent was removed in vacuo. The residue was dissolved in diethyl ether and treated with 2M HCl in diethyl ether. The precipitate was filtered and washed repeatedly with diethyl ether to give the product as white solid (0.19 g, quant.). 1H NMR (300 MHz, CD3OD) δ 7.37 – 7.48 (m, 4H), 7.26 – 7.32 (m, 1H), 3.18 (s, 2H), 1.44 (s, 6H). MS (ESI) m/z [M+H]+ calcd: 150.1; found: 150.2.
2,2-Dimethyl-3-phenylpropanoate (33).
To a solution of 2M LDA in THF (12 ml, 12 mmol) at −78 °C was added methyl isobutyrate (1.1 ml, 10 mmol) dropwise. After 1 h at −78 °C, a solution of benzyl bromide (1.2 ml, 12 mmol) in THF (5 ml) was added carefully. The reaction was continued for 1.5 h and slowly warmed to room temperature. The reaction mixture was quenched carefully with a saturated ammonium chloride solution and extracted with ethyl acetate three times. The combined organic layers were dried over anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography to yield the product as yellow liquid (1.78 g, quant.). 1H NMR (300 MHz, CDCl3) δ 7.31 – 7.35 (m, 1H), 7.20 – 7.25 (m, 2H), 7.07 – 7.13 (m, 2H), 3.66 (s, 3H), 2.85 (s, 2H), 1.18 (s, 6H).
2,2-Dimethyl-3-phenylpropanoic acid (34).
To a solution of methyl 2,2-dimethyl-3-phenylpropanoate (1.78 g, 10 mmol) (33) in methanol (20 ml) was added an aqueous solution of lithium hydroxide (1.20 g, 50 mmol) in water. The reaction mixture was stirred at room temperature for 3 h. Methanol was removed in vacuo and the reaction mixture was acidified to pH 1 and extracted with ethyl acetate three times. The combined organic layers were dried with anhydrous MgSO4, filtered, and concentrated in vacuo to yield the product as white solid (1.75 g, quant.). 1H NMR (300 MHz, CDCl3) δ 7.23 – 7.29 (m, 3H), 7.14 – 7.19 (m, 2H), 2.89 (s, 2H), 1.21 (s, 6H). MS (ESI) m/z [M-H]‒ calcd: 177.1; found: 177.2.
2-Methyl-1-phenylpropan-2-amine (35).
To a solution of 34 (0.21 g, 1.17 mmol) and triethylamine (0.17 ml, 2.25 mmol) in anhydrous acetonitrile (2 ml) was added diphenyl phosphoryl azide (0.27 ml, 1.25 ml). The resulting mixture was heated at 50 °C for 2 h. After cooling to room temperature, a 1N HCl aqueous solution was added and the reaction mixture was refluxed overnight. The acetonitrile was evaporated under reduced pressure and the aqueous portion was extracted with ethyl acetate. The aqueous layer was basified to pH 14 with saturated sodium hydroxide solution followed by extraction with ethyl acetate three times. The combined organic layer was dried over anhydrous MgSO4, filtered, and concentrated in vacuo to yield the product as colorless liquid (0.15 g, 88%). 1H NMR (300 MHz, CDCl3) δ 7.26 – 7.33 (m, 2H), 7.22 – 7.26 (m, 1H), 7.15 – 7.21 (m, 2H), 2.66 (s, 2H), 1.48 (br. s., 2H), 1.12 (s, 6H). MS (ESI) m/z [M+H]+ calcd: 150.1; found: 150.0.
2,2-Difluoro-2-phenylethanamine (36).
To a solution of 2,2-difluoro-2-phenylacetamide (0.17 g, 1 mmol) in THF (4 ml) was added 1M BH3.Me2S in THF solution (4.5 ml) in an ice-water bath. The reaction was raised to room temperature and then refluxed for 16 h. Upon cooling to room temperature, the reaction mixture was then carefully quenched with methanol and solvent was removed in vacuo. The residue was treated with methanol and then concentrated again. This process was repeated twice. The residue was then dissolved in diethyl ether and treated with 2M HCl in diethyl ether. The precipitate was filtered and washed repeatedly with diethyl ether to give the product as white solid (0.11 g, 58%). 1H NMR (300 MHz, CD3OD) δ 7.22 – 7.80 (m, 5H), 3.64 – 3.81 (m, 2H). MS (ESI) m/z [M+H]+ calcd: 158.1; found: 158.2.
trans 2-Phenylcyclopropanecarboxamide (37).
To a solution of trans 2-phenylcyclopropanecarboxylic acid (0.16 g, 1 mmol) in dichloromethane (5 ml) was added 2–3 drops of DMF and oxalyl chloride (0.1 ml, 1.21 ml). The reaction mixture was stirred at room temperature for 3 h. The reaction mixture was then cooled down to 0 °C and a solution of 28% v/v aqueous ammonium hydroxide (0.5 ml) in acetonitrile (5 ml) was added. The reaction mixture was then stirred at room temperature for 16 h. The reaction mixture was diluted with ethyl acetate (30 ml) and washed sequentially with water and brine. The organic layer was dried with anhydrous MgSO4, filtered and concentrated in vacuo to yield the product as white solid (0.16 g, quant.). 1H NMR (300 MHz, CDCl3) δ 7.27 – 7.33 (m, 2H), 7.19 – 7.24 (m, 1H), 7.08 – 7.13 (m, 2H), 5.29 – 5.69 (m, 2H), 2.48 – 2.56 (m, 1H), 1.60 – 1.71 (m, 2H), 1.27 – 1.36 (m, 1H). MS (ESI) m/z [M+H]+ calcd: 162.1; found: 162.2.
trans-1-(2-Phenylcyclopropyl)methanamine (38).
To a solution of 37 (0.16 g, 1 mmol) in THF (4 ml) was added 1M BH3.Me2S in THF solution (4.5 ml) in an ice-water bath. The reaction was raised to room temperature and then refluxed for 16 h. Upon cooling to room temperature, the reaction mixture was then carefully quenched with methanol and solvent was removed in vacuo. The residue was treated with methanol and then concentrated again. This process was repeated twice. The residue was then dissolved in diethyl ether and treated with 2 M HCl in diethyl ether. The precipitate was filtered and washed repeatedly with diethyl ether to give the product as white solid (0.14 g, 76%). 1H NMR (300 MHz, CD3OD) δ 7.06 – 7.44 (m, 4H), 3.54 – 3.62 (m, 2H), 2.95 – 3.05 (m, 1H), 1.55 – 1.64 (m, 2H), 1.03 – 1.14 (m, 1H). MS (ESI) m/z [M+H]+ calcd: 148.1; found: 148.2.
3-[2-Nitroethenyl]thiophene (39).
A solution of thiophene-3-carboxadehyde (0.17 ml, 2 mmol) in nitromethane (3 ml) was added ammonium acetate (0.15 g, 2 mmol) and refluxed for 2 h. The reaction mixture was then diluted with ethyl acetate (50 ml) and washed with water (2 × 20 ml), and brine (20 ml). The organic layer was dried with anhydrous MgSO4, filtered, and concentrated in vacuo. The residue was purified by column chromatography (SiO2, ethyl acetate/hexanes) to provide the product as yellow solid (0.30 g, 95%). 1H NMR (300 MHz, CDCl3) δ 8.01 (d, J = 13.6 Hz, 1H), 7.73 (d, J = 1.9 Hz, 1H), 7.49 (d, J = 13.6 Hz, 1H), 7.44 (dd, J = 2.9, 5.2 Hz, 1H), 7.28 (dd, J = 0.9, 5.3 Hz, 1H). MS (ESI) m/z [M+Na]+ calcd: 178.0; found: 178.0.
2-[2-Nitroethenyl]thiophene (40) was prepared from thiophene-2-carboxadehyde (0.37 ml, 4 mmol) using the same procedure as 39 to yield the product as yellow solid (0.53 g, 84%). 1H NMR (300 MHz, CDCl3) δ 8.16 (d, J = 13.4 Hz, 1H), 7.57 (d, J = 5.1 Hz, 1H), 7.44 – 7.52 (m, 2H), 7.15 (dd, J = 3.8, 5.1 Hz, 1H). MS (ESI) m/z [M+Na]+ calcd: 178.0; found: 178.0.
2-(Thiophen-3-yl)ethanamine (41).
To a solution of 39 (0.10 g, 0.64 mmol) in THF at 0 °C was added LiAlH4. The reaction mixture was raised to room temperature and stirred for 3 h. The reaction mixture was quenched with 1 N NaOH solution and extracted with ethyl acetate (3 × 20 ml). The combined organic layers were dried with anhydrous MgSO4, filtered and concentrated in vacuo to yield the crude product as dark red liquid (0.07 g, 84%) which was used for the next step without purification. MS (ESI) m/z [M+H]+ calcd: 128.1; found: 128.0.
2-(Thiophen-2-yl)ethanamine (42) was prepared from 40 (0.06 g, 0.3 mmol) using the same procedure as 41 as dark red liquid (0.04 g, 80%). MS (ESI) m/z [M+H]+ calcd: 128.1; found: 128.0.
4.2. Pharmacological characterization
4.2.1. Calcium mobilization assay.
CHO-RD-HGA16 cells (Molecular Devices, CA) stably expressing the human CB1 receptor were plated into 96-well black-walled assay plates at 25,000 cells/well in 100 μL of Ham’s F12 (supplemented with 10% fetal bovine serum, 100 units of penicillin/streptomycin, and 100 μg/mL Normocin) and incubated overnight at 37 °C, 5% CO2. Calcium 5 dye (Molecular Devices, CA) was reconstituted according to the manufacturer’s instructions. The reconstituted dye was diluted 1:40 in prewarmed (37 °C) assay buffer (1x HBSS, 20 mM HEPES, 2.5 mM probenecid, pH 7.4 at 37 °C). Growth medium was removed, and the cells were gently washed with 100 μL of prewarmed (37 °C) assay buffer. The cells were incubated for 45 min at 37 °C, 5% CO2 in 200 μL of the diluted Calcium 5 dye solution. For antagonist assays to determine IC50 values, the EC80 concentration of CP55,940 was prepared at 10x the desired final concentration in 0.25% BSA/0.5% DMSO/0.5% EtOH/assay buffer, aliquoted into 96-well polypropylene plates, and warmed to 37 °C. Serial dilutions of the test compounds were prepared at 10x the desired final concentration in 2.25% BSA/4.5% DMSO/4.5% EtOH/assay buffer. After the dye loading incubation period, the cells were pretreated with 25 μL of the test compound serial dilutions and incubated for 15 min at 37 °C. After the pretreatment incubation period, the plate was read with a FLIPR Tetra (Molecular Devices, CA). Calcium-mediated changes in fluorescence were monitored every 1 s over a 90 s time period, with the Tetra adding 25 μL of the CP55,940 EC80 concentration at the 10s time point (excitation/emission: 485/525 nm). Relative fluorescence units (RFU) were plotted against the log of compound concentrations. Data were fit to a three-parameter logistic curve to generate IC50 values (GraphPad Prism 6.0, CA). For the modulation experiments, the above procedure was followed except that cells were pretreated with a single concentration of test compound (prepared at 10x the desired concentration in 2.25% BSA/4.5% DMSO/4.5% EtOH/assay buffer) and the Tetra added serial dilutions of CP55,940 (prepared at 10x the desired concentration in 0.25% BSA/0.5% DMSO/0.5% EtOH/assay buffer). For agonist screens, the above procedure was followed except that cells were pretreated with 2.25% BSA/4.5% DMSO/4.5% EtOH/assay buffer and the Tetra added single concentration dilutions of the test compounds prepared at 10x the desired final concentration in 0.25% BSA/0.5% DMSO/0.5% EtOH/assay buffer. Test compound RFUs were compared to the CP55,940 Emax RFUs to generate % Emax values. For the CB2 agonist screens and IC50 determinations, the same procedures were followed except that stable human CB2-CHO-RD-HGA16 cells were used. CB2 antagonist screens were conducted similar to the IC50 experiments except that a single concentration of test compound was used instead of serial dilutions and test compound RFUs were compared to the CP55,940 EC80 RFUs to generate % inhibition values.
4.2.1. cAMP Assay.
Forskolin (FSK)-stimulated cyclic adenosine monophosphate (cAMP) production was measured in real-time using a transfected bioluminescence resonance energy transfer (BRET) cAMP sensor, CAMYEL.58The plasmid encodes a cAMP binding domain (Epac1) flanked by yellow fluorescent protein (YFP) and Renilla Luciferase (RLuc) assay, the latter of which can oxidize coelenterazine H and produce a photon as a byproduct. When cAMP is bound to the Epac1 domain, it separates RLuc and YFP so only Rluc emits a photon at a wavelength of 460 nm. When cAMP is not bound, closer proximity means that RLuc can resonate energy to excite YFP, which then emits light at wavelength 535 nm. A plate reader measures both wavelengths and their ratio, 460/535, is calculated to quantify cAMP levels where increases in the ratio indicate increases in cAMP. Human Embryonic Kidney 293 (HEK293) cells stably transfected with human CB1 N-terminally tagged with three haemagglutinins (3HA-hCB1)54 were maintained at 37°C at 5% CO2, and seeded in 100 mM dishes for transfection. The next day, cells were given fresh growth media and transfected with 5 µg of pcDNA3L-His-CAMYEL using linear polyethyleneimine (PEI, 25 kDa, Polysciences, Warrington, PA) in a 1:6 DNA:PEI ratio. The next day, cells were lifted with 0.05% trypsin/EDTA, counted and plated on poly-D-lysine (Sigma Aldrich, St. Louis, MO) coated white 96-well CulturPlates (Perkin Elmer, Waltham, MA) at 60,000 cells per well. The following day, working with wells for one assay run at a time, culture medium was removed by aspirating, cells were rinsed with PBS then serum starved for 30 min in assay medium, comprised of phenol-free DMEM supplemented with 1 mg/ml BSA (MP Biomedicals, Auckland, New Zealand) and 10 mM HEPES pH 7.4 (Gibco, ThermoFisher Scientific). Coelenterazine H (Prolume, Pinetop, AZ) constituted in absolute ethanol was prepared in assay medium at 10x concentration and dispensed 5 minutes prior to drug addition (final concentration in-well was 5 µM). Drug dilutions were prepared in assay medium (vehicle-controlled) at 10x concentration and pre-mixed in a V-well dispensing plate, then added simultaneously to the assay plate with a multichannel pipette. Drugs conditions included forskolin (in DMSO, to a final concentration of 5 µM), CP55,940 (in absolute ethanol, to a final concentration of 1 µM) and allosteric modulators (in DMSO, and serial dilution in DMSO-controlled assay medium). BRET signal was detected over approximately 20 minutes in a pre-warmed 37°C LUMIStar Omega plate reader (BMG Labtech, Ortenberg, Germany) using simultaneous BRET1 filters (535–30 and 475–30 nm). BRET data was exported into Excel, then inverse BRET ratios (460/535) were calculated for each time point and plotted across time in GraphPad Prism (v8, GraphPad Software, San Diego, CA). Area under the curve analysis was performed for each well, and then normalized to the mean “forskolin alone” (100%) and “vehicle only” (0%) conditions. Concentration-response curves were fit in Prism by 3-parameter non-linear regression. Three independent experiments (biological replicates – each on a different assay day) were performed, and then the pIC50 values reported in Tables 2 and 3 were calculated by taking the mean and SEM of each independent experiment’s pIC50.
Table 3.
Allosteric Modulating Activities of Representative CB1 Allosteric Modulators in the human CB1 Calcium Mobilization, [35S]GTPγS Binding, and cAMP Assays.
| Compound | hCB1 Calcium pIC50a |
hCB1 [35S]GTPγS pIC50a |
hCB1 cAMP pIC50a |
|---|---|---|---|
| Org27569 | 6.1 ± 0.07 | 6.0 ± 0.05 | 6.8 ± 0.0654 |
| PSNCBAM-1 | 7.5 ± 0.1 | 7.0 ± 0.1 | 6.4 ± 0.1 |
| 8 | 6.7 ± 0.09 | 5.2 ± 0.05 | 5.5 ± 0.03 |
| 9 | 6.1 ± 0.05 | 5.9 ± 0.3 | N.D. b |
| 10 | 6.1 ± 0.05 | 4.4 ± 0.3 | N.D. b |
| 12 | 6.6 ± 0.1 | 4.8 ± 0.2 | 5.6 ± 0.07 |
| 24 | 5.8 ± 0.03 | 5.1 ± 0.2 | N.D. b |
| 25 | 6.1 ± 0.01 | 5.1 ± 0.09 | N.D. b |
| 26 | 6.6 ± 0.06 | 4.9 ± 0.1 | 5.4 ± 0.05 |
Compounds tested against EC80 (100 nM) of CP55,940. Values are the mean ± SEM of at least three independent experiments in duplicate.
N.D.: Not determined.
Membrane preparation. Cerebella from adult male CD-1 mice were dissected on ice, snap frozen, and stored at ‒80°C until the day of the experiment. Cerebella were homogenized by polytron in membrane buffer (50 mM Tris, 3 mM MgCl2, 0.2 mM EGTA, 100 mM NaCl, pH 7.4) on ice, centrifuged for 10 min at 40,000xg at 4°C. The supernatant was discarded, and the pellet was suspended in membrane buffer, homogenized, and centrifuged again for 10 min at 40,000xg. The pellet was resuspended in membrane buffer and protein quantified by Bradford method.
4.2.3. [35S]GTP-γ-S binding assay.
Cerebellar or hCB1-expressing HEK cells membranes were preincubated in assay buffer (membrane buffer containing 1 mg/ml bovine serum albumin; BSA) for 10 min with 3 units/ml adenosine deaminase then incubated for 60 min at 30°C with 30 μM GDP and 0.1 nM [35S]GTP-γ-S or 90 min with 1 nM [3H]CP55,940. Serial dilutions of test compounds were done in 100% DMSO with final assay DMSO concentration of 0.1%. Non-specific binding was determined by adding 30 μM unlabeled GTP-γ-S or 1 μM unlabeled CP55,940. Reactions were terminated by vacuum filtration through GF/C filter plates (Perkin Elmer). GTP-γ-S inhibition curves for test compounds were normalized to CP55,940 (100 nM) stimulation in the absence of test compound (i.e. vehicle = 100%). Curvefits were accomplished using GraphPad Prism 6.0 and where GTP-γ-S data were fit to 3 parameter non-linear regression, with bottom and top constrained to >0 and =100 respectively for IC50 calculation. Values were considered significantly different when 95% confidence intervals did not overlap.
Equation 1. Operational model of allosterism. Y, observed effect; Em, maximal possible system response; n, slope factor of transducer function that links occupancy to response; τA and τB, ability of probe and modulator to promote receptor activation; [A] and [B], concentration of probe and modulator; KA and KB, affinity of probe and modulator, respectively; α, binding cooperativity between probe and modulator; β, describes magnitude at which modulator modifies probe efficacy.
4.3. Computational Methods.
4.3.1. Model Preparation from CB1 Crystal Structure Complexes.
We retained the native sequence of the reported CB1-PGSCM construct and the 5 thermostabilizing mutations for the initial crystal structure-based model used for the initial docking and induced fit studies.34 We then employed Sali’s MODELLER59 to complete the structure for disordered residues and chose the structure with the best zDOPE score (0.98) for initial model docking into the fusion model using both Autodock VINA60 and Schrodinger GLIDE61 after initial model refinement (protonation state assignment) and energy minimization using Schrodinger protein preparation preprocessing tools. Prior to docking we removed the cofactors including oleic acid (OLA), (2R)-2,3-dihydroxypropyl (9Z)-octadec-9-enoate (OLC), and polyethylene glycol (PEG) present in the crystal structure. The cofactors were part of the inverted cubic liquid crystal phase preparation facilitating crystallization and most of them were present in the extrahelical region, distant from the docking site. While we used the 6KQI fusion forms for the initial docking and MMGBSA rescoring studies to make initial assessments as close as possible to the crystallized form, we have, in selected cases, transformed the docked poses into a full N- and C-terminal model without the thermostabilizing mutations and equilibrated the docked ligand CB1 poses in a DOPC/KCl/TIPS3P water model as described in detailed simulation protocols below. All models in docking/induced-fit and dynamics studies retained the agonist CP55,940 in the orthosteric binding site in the initial configuration found in the 6KQI crystal structure.
4.3.2. Docking and Induced Fit.
Two parallel and independent approaches with different scoring functions, Schrodinger XP/Induced Fit62 and Autodock VINA with a AMBER1863 multi-trajectory (MD) molecular mechanics generalized Born surface area MMGBSA rescoring, were used to provide predictions for ligand configurations allowing for both ligand and receptor binding site flexibility. Both methods included desolvation components in the scoring function. Following initial GLIDE-XP docking we probed the importance of surface/allosteric binding site flexibility within a 5 Å window of any atom in the best docking poses using Schrodinger’s Induced Fit.62 The docking box employed for VINA was ca 15×15×15 Å3 in spatial extent, while the default box size employed in computing the GLIDE-XP docking grid was based on one of the largest ligand sizes which included residues R148, W241, G157, S158 and F237. In the case of blind docking with VINA, we employed a 40×20×40 Å3 in spatial extent to allow for docking sites across all helices in the intracellular region. Autodock VINA’s scoring function scores based on hydrophobic contacts, hydrogen bonds, lack of steric clash. This informatics-based scoring function serves primarily as a means of selecting the most plausible collection of poses from a large number of sampled docked configurations which we rescore using AMBER-MMGBSA. For VINA we collected up to 20 poses per ligand with a high “exhaustiveness” setting of 80. Similarly, the GLIDE-XP workflow and scoring function was used to obtain up to 20 poses per ligand for post-docking minimization but retaining the best Emodel scored 5 poses in the final analysis. The GLIDE-XP scoring function contains terms such as desolvation, lipophilic/hydrophobic contact and cavity costs, in addition to physics based coulombic and van der Waals and ligand strain terms.
For the initial docking/induced-fit and MMGBSA-rescoring phase we chose to use the model completed 6KQI structure, including the PGS insertion and the 5 stabilizing mutations. We then developed a AMBER18 all-hydrogen model employing the AMBER14SB forcefield. The structure prepared with TLEAP was then energy minimized for 8000 steps of conjugate gradient following a 400 steepest descents minimization to remove initial inferior contacts. The analogue of the 6KQI structure was used in order to initially explore the SAR poses in the context of the crystallographic coordinates before reverse modeling to the native human sequence with full N- and C- termini and equilibrated loop structures.
MMGBSA-Min and MMGBSA-SA. The docked poses above were then re-scored using an MMGBSA/MMPBSA method. This was performed because the top ranked docked poses are often not the crystallographically observed pose as observed by us and others in PDB-Bind assessments,64 whereas frequently the lowest MMGBSA/MMPBSA scored pose has lowest RMSD to the known crystallographic solution and provides the best affinity correlations.40, 65, 66 In order to provide a common platform for MMGBSA/MMPBSA re-scoring of all docking approaches, we have developed our own C++ and bash script base in conjunction with AMBER1863, 67 to provide an automated workflow, selecting the docked poses for all ligands (in mol2 format), performing ligand formal charge perception (C++ plus OpenBabel open-source), computation of charges based on either AM1-BCC or other ab-initio QM approaches, energy minimizing all poses in complex with the receptor/protein and optionally employing a GPU/CPU hybrid MD-simulated annealing (300K MD followed by energy minimization) procedure prior to computing the MMGBSA/MMPBSA score. Given our flexible ligand and rigid receptor docking as the initial step, we chose to allow at most short time MD i.e. 20–80 picoseconds MMGBSA/MMPBSA scoring (with optional re-minimization of the last MD-snapshot) following an initial 80ps equilibration. We collected 3–10 independent trajectories, computed a multi-trajectory MMGBSA average and obtained the standard deviation and standard error in the mean for those quantities. This multi-trajectory approach reduces the statistical uncertainty in MMGBSA/MMPBSA values with short time local sampling of the environmental fluctuations in response to the bound ligand. This approach allowed initially exploration of minor relaxation and response of the protein to the flexible ligand docked pose rather than an extensive MD exploration, thus exploring local energy wells not involving large amplitude ligand or protein motion away from the original docked pose (which requires longer trajectories). In cases where we wish to inspect the impact of larger receptor deformations we employ 300ns-2us MD starting with the poses obtained from docking/induced fit or our multi-trajectory MMGBSA exploration.
Several approaches were used to perform MMGBSA rescoring employing igb=1 and mbondi Generalized Born radii as well as igb=8/mbondi3 radii with no significant differences in conclusions for this work. The MMGBSA/MMPBSA-simulated annealing protocol we employed was based on 2000 step minimization of the complex, 20 picoseconds of MD, followed by 2000 steps of re-minimization. We collected 5 samples (every 4 ps) during the MD phase and these snapshots were re-energy minimized and used to compute and MMGBSA/MMPBSA average score for the pose. We included re-energy minimization because randomly selected dynamics snapshots may have distortions that are not long-lived and energy minimization with 1000-steps of conjugate gradient relaxes the internal structure to local minima for examination of interactions. MMPBSA calculations with alternative outer dielectrics simulating water (e=80) or membrane-aliphatic chains (e=3–12) gave numerically distinct but similar trends in which poses and ligand positions were most stable. It is unknown precisely the dielectric response at the membrane embedded allosteric site.
Quantum chemical DFT examination of a Phenyl/Phenyl versus a Phenyl-chlorophenyl π-stacking interaction. We performed a single simple model calculation to examine the difference in the π-stacking interaction with and without a chloro on the aromatic ring. Phenyl/phenyl versus a Phenyl-chlorophenyl models were constructed using the builder model in Schrodinger Small Molecule Tools. The models were geometry optimized using a B3LYP-D3 functional and a 6–31G** basis set, standard convergence criteria, and an Accurate grid setting. The energies of the final geometries of the density functional optimization were then counterpoise corrected using the Boys method to obtain estimated binding energy differences for a model mimicking the differential chlorophenyl versus phenyl π-stacking interactions observed in particular GLIDE-XP/Induced fit and VINA/MMGBSA top scoring poses.
The solvation energetics of selected cases were computed by a PBR(Poisson-Boltzmann Reaction field) approach at B3LYP-D3//6–31G** optimized geometries.
4.3.3. Molecular Dynamics.
MD was performed to equilibrate the initial modeled loops and water accessible domain with production dynamics to examine the effects of agonist and allosteric modulator biasing the receptor ensemble to examine transient salt-bridge fluctuations/breaking and other activation microswitch changes. In selected cases, we employed GLIDE XP or VINA/MMGBSA rescored poses of NAMs co-bound to the CP55940-bound receptor employing a DOPC/K+/Cl‒/TIPS3P water environment in our simulation models. For this purpose, the docked pose was employed in a full N-terminal/C-terminal representation of CB1 without the thermostabilizing mutations in 6KQI or the PGS fusion,68 using an equilibrated loop model as the start of the simulations including the Cys257/264 disulfide bond. An initial 200×200×120 Å3 DOPC membrane rectilinear box was constructed including 574 DOPC lipids on top and bottom membrane leaflets prior to deletion based on atomic overlaps along with 290 K+/Cl‒ ion pairs and 97280 water molecules. The initial DOPC/KCl/water box was obtained using the CHARMM lipid model construction server (http://www.charmm-gui.org/?doc=input/membrane.bilayer). The Sali MODELER369 and SCRWL70 rotamer modified initial model of the docked complex was equilibrated in the DOPC membrane/K+/Cl‒ water environment deleting all DOPC/K+/Cl−/water overlaps with any atom in the CB1 receptor complex within 3.5 Å. The Walker group LIPID1471 model was used for the DOPC representation and the AMBER ff14SB forcefield for the protein residues. The same GAFF/AM1-BCC representation of the ligand was employed in the membrane/complex simulations as employed in the MMGBSA/MMPBSA rescoring treatment. A cysteine-cysteine disulfide bridge was constructed between Cys257 and Cys264 in ECL2. The initial configurations of the CB1-CP55,940 complex with bound allosteric modulators immersed in the membrane/water/ion system was energy minimized using 400 steps of steepest descents minimization followed by conjugate gradients 8000 steps to remove any bad contacts. The complex model immersed in the DOPC/K+/Cl−/water model was then slowly heated to 300 K with decreasing harmonic constraints on all atoms over 200 ps followed by early equilibration equilibrated for 200 ns and then production dynamics of 300–800 ns equilibrating the loop/N-/C-termini and exploring impact of the allosteric ligands on activation microswitches in the presence of the CP55940 agonist bound in the orthosteric site. It should be noted that we observed some solvent molecules percolating into the region of the orthosteric ligands near the intracellular membrane surface and even the interface proximate to the allosteric ligands. We have examined a simulation of CB1 bound with a number of antagonists/inverse agonists such as AM6538 bound 5TGZ crystal structure,41 and analyzed the D3386.30/R2143.50 salt bridge status and the Y2003.36/W3566.48/W2795.43 aromatic toggle region. These structural features stabilized CB1 antagonist bound structures as opposed to CB1 agonists, supporting the use of MMGBSA–docking rescored poses and short simulations to illustrate the nature of the SAR environment in a membrane/ion/water environment.
Supplementary Material
Acknowledgments
This work was supported by National Institute on Drug Abuse, National Institutes of Health, U.S. A. (Grants DA040693 to Y.Z. and DA045752 to T.F.G.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data associated with this article (computational graphs, HPLC analysis, 1H and 13C of all target compounds) can be found, in the online version at doi.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Devane WA; Dysarz FAI; Johnson MR; Melvin LS; Howlett AC, Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol 1988, 34, 605–613. [PubMed] [Google Scholar]
- 2.Mechoulam R; Ben-Shabat S; Hanus L; Ligumsky M; Kaminski NE; Schatz AR; Gopher A; Almog S; Martin BR; Compton DR; et al. , Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem. Pharmacol 1995, 50 (1), 83–90. [DOI] [PubMed] [Google Scholar]
- 3.De Petrocellis L; Cascio MG; Di Marzo V, The endocannabinoid system: a general view and latest additions. Br. J. Pharmacol 2004, 141 (5), 765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pacher P; Batkai S; Kunos G, The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev 2006, 58 (3), 389–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuda LA; Lolait SJ; Brownstein MJ; Young AC; Bonner TI, Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature 1990, 346, 561–564. [DOI] [PubMed] [Google Scholar]
- 6.Munro S; Thomas KL; Abu-Shaar M, Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [DOI] [PubMed] [Google Scholar]
- 7.Pertwee RG, Targeting the endocannabinoid system with cannabinoid receptor agonists: pharmacological strategies and therapeutic possibilities. Philos. Trans. R. Soc. B Biol. Sci 2012, 367, 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Porter AC; Felder CC, The endocannabinoid nervous system: unique opportunities for therapeutic intervention. Pharmacol. Ther 2001, 90, 45–60. [DOI] [PubMed] [Google Scholar]
- 9.Harkany T; Guzmán M; Galve-Roperh I; Berghuis P; Devi LA; Mackie K, The emerging functions of endocannabinoid signaling during CNS development. Trends Pharmacol. Sci 2007, 28, 83–92. [DOI] [PubMed] [Google Scholar]
- 10.Carai MA; Colombo G; Gessa GL, Rimonabant: the first therapeutically relevant cannabinoid antagonist. Life Sci 2005, 77 (19), 2339–50. [DOI] [PubMed] [Google Scholar]
- 11.De Vries TJ; Shaham Y; Homberg JR; Crombag H; Schuurman K; Dieben J; Vanderschuren LJ; Schoffelmeer AN, A cannabinoid mechanism in relapse to cocaine seeking. Nat. Med 2001, 7 (10), 1151–4. [DOI] [PubMed] [Google Scholar]
- 12.Fattore L; Spano S; Cossu G; Deiana S; Fadda P; Fratta W, Cannabinoid CB(1) antagonist SR 141716A attenuates reinstatement of heroin self-administration in heroin-abstinent rats. Neuropharmacology 2005, 48 (8), 1097–104. [DOI] [PubMed] [Google Scholar]
- 13.FDA_Advisory_Committee, NDA 21–888 Zimulti (rimonabant) tablets 20 mg Sanofi Aventis FDA Briefing Document: 2007. [Google Scholar]
- 14.Bergman J; Delatte MS; Paronis CA; Vemuri K; Thakur GA; Makriyannis A, Some effects of CB1 antagonists with inverse agonist and neutral biochemical properties. Physiol. Behav 2008, 93 (4–5), 666–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Greasley PJ; Clapham JC, Inverse agonism or neutral antagonism at G-protein coupled receptors: A medicinal chemistry challenge worth pursuing? Eur. J. Pharmacol 2006, 553, 1–9. [DOI] [PubMed] [Google Scholar]
- 16.Chorvat RJ, Peripherally restricted CB1 receptor blockers. Bioorg. Med. Chem. Lett 2013, 23 (17), 4751–60. [DOI] [PubMed] [Google Scholar]
- 17.Nguyen T; Li JX; Thomas BF; Wiley JL; Kenakin TP; Zhang Y, Allosteric Modulation: An Alternate Approach Targeting the Cannabinoid CB1 Receptor. Med. Res. Rev 2017, 37 (3), 441–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dopart R; Lu D; Lichtman AH; Kendall DA, Allosteric modulators of cannabinoid receptor 1: developing compounds for improved specificity. Drug Metab. Rev 2018, 50 (1), 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nguyen T; Thomas BF; Zhang Y, Overcoming the psychiatric side effects of the cannabinoid CB1 receptor antagonists: Current approaches for therapeutics development. Curr. Top. Med. Chem 2019, 19, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Conn PJ; Christopoulos A; Lindsley CW, Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat. Rev. Drug. Discov 2009, 8 (1), 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Amici M; Dallanoce C; Holzgrabe U; Trankle C; Mohr K, Allosteric ligands for G protein-coupled receptors: a novel strategy with attractive therapeutic opportunities. Med. Res. Rev 2010, 30 (3), 463–549. [DOI] [PubMed] [Google Scholar]
- 22.Khurana L; Mackie K; Piomelli D; Kendall DA, Modulation of CB1 cannabinoid receptor by allosteric ligands: Pharmacology and therapeutic opportunities. Neuropharmacology 2017, 124, 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gado F; Meini S; Bertini S; Digiacomo M; Macchia M; Manera C, Allosteric modulators targeting cannabinoid cb1 and cb2 receptors: implications for drug discovery. Future Med Chem 2019, 11 (15), 2019–2037. [DOI] [PubMed] [Google Scholar]
- 24.Jing L; Qiu Y; Zhang Y; Li JX, Effects of the cannabinoid CB(1) receptor allosteric modulator ORG 27569 on reinstatement of cocaine- and methamphetamine-seeking behavior in rats. Drug Alcohol Depend 2014, 143, 251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nguyen T; German N; Decker AM; Langston TL; Gamage TF; Farquhar CE; Li JX; Wiley JL; Thomas BF; Zhang Y, Novel Diarylurea Based Allosteric Modulators of the Cannabinoid CB1 Receptor: Evaluation of Importance of 6-Pyrrolidinylpyridinyl Substitution. J. Med. Chem 2017, 60 (17), 7410–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cawston EE; Connor M; Di Marzo V; Silvestri R; Glass M, Distinct Temporal Fingerprint for Cyclic Adenosine Monophosphate (cAMP) Signaling of Indole-2-carboxamides as Allosteric Modulators of the Cannabinoid Receptors. J. Med. Chem 2015, 58 (15), 5979–5988. [DOI] [PubMed] [Google Scholar]
- 27.Mahmoud MMA, Hamed I; Ahn Kwang H.; Damaraju Aparna; Samala Sushma; Pulipati Venkata; Kolluru Srikanth; Kendall Debra A.; Lu Dai, Structure-Activity Relationship Study of Indole-2-carboxamides Identifies a Potent Allosteric Modulator for the Cannabinoid Receptor 1 (CB1). J. Med. Chem 2013, 56 (20), 7965–7975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahn KH; Mahmoud MM; Samala S; Lu D; Kendall DA, Profiling two indole-2-carboxamides for allosteric modulation of the CB1 receptor. J. Neurochem 2013, 124 (5), 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen TG, Nadezhda; Decker Ann M.; Li Jun-Xu; Wiley Jenny L.; Thomas Brian F.; Kenakin Terry P.; Zhang Yanan, Structure-activity relationships of substituted 1H-indole-2-carboxamides as CB1 receptor allosteric modulators. Bioorg. Med. Chem 2015, 23 (9), 2195–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.German ND, Ann M; Gilmour Brian P.; Gay Elaine A.; Wiley Jenny L.; Thomas Brian F.; Zhang Yanan, Diarylureas as Allosteric Modulators of the Cannabinoid CB1 Receptor: Structure-Activity Relationship Studies on 1-(4-Chlorophenyl)-3-{3-[6-(pyrrolidin-1-yl)pyridin-2-yl]phenyl}urea (PSNCBAM-1). J. Med. Chem 2014, 57 (18), 7758–7769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khurana L; Fu BQ; Duddupudi AL; Liao YH; Immadi SS; Kendall DA; Lu D, Pyrimidinyl Biphenylureas: Identification of New Lead Compounds as Allosteric Modulators of the Cannabinoid Receptor CB1. J. Med. Chem 2017, 60 (3), 1089–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen T; Gamage TF; Decker AM; Barrus D; Langston TL; Li JX; Thomas BF; Zhang Y, Synthesis and Pharmacological Evaluation of 1-Phenyl-3-Thiophenylurea Derivatives as Cannabinoid Type-1 Receptor Allosteric Modulators. J. Med. Chem 2019, 62 (21), 9806–9823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen T; Gamage TF; Decker AM; German N; Langston TL; Farquhar CE; Kenakin TP; Wiley JL; Thomas BF; Zhang Y, Diarylureas Containing 5-Membered Heterocycles as CB1 Receptor Allosteric Modulators: Design, Synthesis, and Pharmacological Evaluation. ACS Chem. Neurosci 2019, 10 (1), 518–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shao Z; Yan W; Chapman K; Ramesh K; Ferrell AJ; Yin J; Wang X; Xu Q; Rosenbaum DM, Structure of an allosteric modulator bound to the CB1 cannabinoid receptor. Nat Chem Biol 2019, 15 (12), 1199–1205. [DOI] [PubMed] [Google Scholar]
- 35.Meyers J; Carter M; Mok NY; Brown N, On the origins of three-dimensionality in drug-like molecules. Future Med Chem 2016, 8 (14), 1753–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ishikawa M; Hashimoto Y, Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem 2011, 54 (6), 1539–54. [DOI] [PubMed] [Google Scholar]
- 37.Hua T; Vemuri K; Nikas SP; Laprairie RB; Wu Y; Qu L; Pu M; Korde A; Jiang S; Ho JH; Han GW; Ding K; Li X; Liu H; Hanson MA; Zhao S; Bohn LM; Makriyannis A; Stevens RC; Liu ZJ, Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547 (7664), 468–471. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Stornaiuolo M; Bruno A; Botta L; La Regina G; Cosconati S; Silvestri R; Marinelli L; Novellino E, Endogenous vs Exogenous Allosteric Modulators in GPCRs: A dispute for shuttling CB1 among different membrane microenvironments. Sci Rep 2015, 5, 15453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ballesteros JA; Weinstein H, Integrated methods for the construction of three dimensional models and computational probing of structure-function relations in G-protein coupled receptors. Methods Neurosci 1995, 25, 366–428. [Google Scholar]
- 40.Zhang X; Wong SE; Lightstone FC, Toward fully automated high performance computing drug discovery: a massively parallel virtual screening pipeline for docking and molecular mechanics/generalized born surface area rescoring to improve enrichment. J Chem Inf Model 2014, 54 (1), 324–37. [DOI] [PubMed] [Google Scholar]
- 41.Amato GS; Manke A; Harris DL; Wiethe RW; Vasukuttan V; Snyder RW; Lefever TW; Cortes R; Zhang Y; Wang S; Runyon SP; Maitra R, Blocking Alcoholic Steatosis in Mice with a Peripherally Restricted Purine Antagonist of the Type 1 Cannabinoid Receptor. J Med Chem 2018, 61 (10), 4370–4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milton ME; Minrovic BM; Harris DL; Kang B; Jung D; Lewis CP; Thompson RJ; Melander RJ; Zeng D; Melander C; Cavanagh J, Re-sensitizing Multidrug Resistant Bacteria to Antibiotics by Targeting Bacterial Response Regulators: Characterization and Comparison of Interactions between 2-Aminoimidazoles and the Response Regulators BfmR from Acinetobacter baumannii and QseB from Francisella spp. Front Mol Biosci 2018, 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun H; Li Y; Tian S; Xu L; Hou T, Assessing the performance of MM/PBSA and MM/GBSA methods. 4. Accuracies of MM/PBSA and MM/GBSA methodologies evaluated by various simulation protocols using PDBbind data set. Phys Chem Chem Phys 2014, 16 (31), 16719–29. [DOI] [PubMed] [Google Scholar]
- 44.Price MR; Baillie GL; Thomas A; Stevenson LA; Easson M; Goodwin R; McLean A; McIntosh L; Goodwin G; Walker G; Westwood P; Marrs J; Thomson F; Cowley P; Christopoulos A; Pertwee RG; Ross RA, Allosteric Modulation of the Cannabinoid CB1 Receptor. Mol. Pharmacol 2005, 68 (5), 1484–1495. [DOI] [PubMed] [Google Scholar]
- 45.Horswill J; Bali U; Shaaban S; Keily J; Jeevaratnam P; Babbs A; Reynet C; In PWK, PSNCBAM-1, a novel allosteric antagonist at cannabinoid CB1 receptors with hypophagic effects in rats. Br. J. Pharmacol 2007, 152, 805–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hua T; Vemuri K; Nikas SP; Laprairie RB; Wu Y; Qu L; Pu M; Korde A; Jiang S; Ho JH; Han GW; Ding K; Li X; Liu H; Hanson MA; Zhao S; Bohn LM; Makriyannis A; Stevens RC; Liu ZJ, Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 47.Hua T; Vemuri K; Pu M; Qu L; Han GW; Wu Y; Zhao S; Shui W; Li S; Korde A; Laprairie RB; Stahl EL; Ho JH; Zvonok N; Zhou H; Kufareva I; Wu B; Zhao Q; Hanson MA; Bohn LM; Makriyannis A; Stevens RC; Liu ZJ, Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167 (3), 750–762 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shao Z; Yin J; Chapman K; Grzemska M; Clark L; Wang J; Rosenbaum DM, High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Janovick JA; Conn PM, Salt bridge integrates GPCR activation with protein trafficking. Proc. Natl. Acad. Sci. U. S. A 2010, 107 (9), 4454–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perrin MH; Grace CR; Digruccio MR; Fischer WH; Maji SK; Cantle JP; Smith S; Manning G; Vale WW; Riek R, Distinct structural and functional roles of conserved residues in the first extracellular domain of receptors for corticotropin-releasing factor and related G-protein-coupled receptors. J. Biol. Chem 2007, 282 (52), 37529–36. [DOI] [PubMed] [Google Scholar]
- 51.Fuxe K; Marcellino D; Leo G; Agnati LF, Molecular integration via allosteric interactions in receptor heteromers. A working hypothesis. Curr Opin Pharmacol 2010, 10 (1), 14–22. [DOI] [PubMed] [Google Scholar]
- 52.Ahn KH; Scott CE; Abrol R; Goddard WA 3rd; Kendall DA, Computationally-predicted CB1 cannabinoid receptor mutants show distinct patterns of salt-bridges that correlate with their level of constitutive activity reflected in G protein coupling levels, thermal stability, and ligand binding. Proteins 2013, 81 (8), 1304–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kulkarni AR; Garai S; Thakur GA, Scalable, One-Pot, Microwave-Accelerated Tandem Synthesis of Unsymmetrical Urea Derivatives. J. Org. Chem 2017, 82 (2), 992–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cawston EER, William J; Breen Courtney M.; Grimsey Natasha L.; Connor Mark; Glass Michelle, Real-time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br. J. Pharmacol 2013, 170 (4), 893–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nguyen T; Decker AM; Langston TL; Mathews KM; Siemian JN; Li JX; Harris DL; Runyon SP; Zhang Y, Discovery of Novel Proline-Based Neuropeptide FF Receptor Antagonists. ACS Chem. Neurosci 2017, 8 (10), 2290–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Browning C; Martin E; Loch C; Wurtz JM; Moras D; Stote RH; Dejaegere AP; Billas IM, Critical role of desolvation in the binding of 20-hydroxyecdysone to the ecdysone receptor. J Biol Chem 2007, 282 (45), 32924–34. [DOI] [PubMed] [Google Scholar]
- 57.Kenakin T; Strachan RT, PAM-Antagonists: A Better Way to Block Pathological Receptor Signaling? Trends Pharmacol. Sci 2018, 39 (8), 748–765. [DOI] [PubMed] [Google Scholar]
- 58.Jiang LI; Collins J; Davis R; Lin KM; DeCamp D; Roach T; Hsueh R; Rebres RA; Ross EM; Taussig R; Fraser I; Sternweis PC, Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1-phosphate/G13 pathway. J. Biol. Chem 2007, 282 (14), 10576–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Webb B; Sali A, Protein Structure Modeling with MODELLER. Methods Mol Biol 2017, 1654, 39–54. [DOI] [PubMed] [Google Scholar]
- 60.Trott O; Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry 2010, 31 (2), 455–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 2006, 49 (21), 6177–96. [DOI] [PubMed] [Google Scholar]
- 62.Sherman W; Day T; Jacobson MP; Friesner RA; Farid R, Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem 2006, 49 (2), 534–53. [DOI] [PubMed] [Google Scholar]
- 63.Lee TS; Cerutti DS; Mermelstein D; Lin C; LeGrand S; Giese TJ; Roitberg A; Case DA; Walker RC; York DM, GPU-Accelerated Molecular Dynamics and Free Energy Methods in Amber18: Performance Enhancements and New Features. J Chem Inf Model 2018, 58 (10), 2043–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Greenidge PA; Kramer C; Mozziconacci JC; Sherman W, Improving docking results via reranking of ensembles of ligand poses in multiple X-ray protein conformations with MM-GBSA. J Chem Inf Model 2014, 54 (10), 2697–717. [DOI] [PubMed] [Google Scholar]
- 65.Greenidge PA; Kramer C; Mozziconacci JC; Wolf RM, MM/GBSA binding energy prediction on the PDBbind data set: successes, failures, and directions for further improvement. J Chem Inf Model 2013, 53 (1), 201–9. [DOI] [PubMed] [Google Scholar]
- 66.Hou T; Wang J; Li Y; Wang W, Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. The accuracy of ranking poses generated from docking. Journal of computational chemistry 2011, 32 (5), 866–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Case DA, R. M. B., Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi R, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L and Kollman PA (2016), AMBER 2016, University of California, San Francisco. AMBER 2016, UCSF, San Francisco, 2016. [Google Scholar]
- 68.Bochevarov AD; Harder E; Hughes TF; Greenwood JR; Braden DA; Philipp DM; Rinaldo D; Halls MD; Zhang J; Friesner RA, Jaguar: A High-Performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem 2013, 113, 2110–2142. [Google Scholar]
- 69.Sali A; Blundell TL, Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol 1993, 234 (3), 779–815. [DOI] [PubMed] [Google Scholar]
- 70.Wang Q; Canutescu AA; Dunbrack RL Jr., SCWRL and MolIDE: computer programs for side-chain conformation prediction and homology modeling. Nat Protoc 2008, 3 (12), 1832–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dickson CJ; Madej BD; Skjevik AA; Betz RM; Teigen K; Gould IR; Walker RC, Lipid14: The Amber Lipid Force Field. J Chem Theory Comput 2014, 10 (2), 865–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.