

Key Points
Question
Among symptomatic but clinically stable outpatients with COVID-19, does adding antithrombotic therapy, compared with placebo, reduce major cardiopulmonary adverse outcomes over a 45-day treatment period?
Findings
This randomized trial of 657 symptomatic outpatients with COVID-19 conducted in the US was stopped early because of an unanticipated low event rate. Among randomized participants who initiated trial treatment with aspirin (81 mg once daily), apixaban (2.5 mg twice daily), apixaban (5.0 mg twice daily), or placebo, the rates of an adjudicated composite outcome (all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary cause) after 45 days were 0.0%, 0.7%, 1.4%, and 0.0%, respectively; there were no significant differences between the active groups and the placebo group.
Meaning
These data do not support the use of aspirin or apixaban in the outpatient setting to reduce the major adverse cardiovascular or pulmonary consequences associated with symptomatic but clinically stable SARS-CoV-2 infection.
Abstract
Importance
Acutely ill inpatients with COVID-19 typically receive antithrombotic therapy, although the risks and benefits of this intervention among outpatients with COVID-19 have not been established.
Objective
To assess whether anticoagulant or antiplatelet therapy can safely reduce major adverse cardiopulmonary outcomes among symptomatic but clinically stable outpatients with COVID-19.
Design, Setting, and Participants
The ACTIV-4B Outpatient Thrombosis Prevention Trial was designed as a minimal-contact, adaptive, randomized, double-blind, placebo-controlled trial to compare anticoagulant and antiplatelet therapy among 7000 symptomatic but clinically stable outpatients with COVID-19. The trial was conducted at 52 US sites between September 2020 and June 2021; final follow-up was August 5, 2021. Prior to initiating treatment, participants were required to have platelet count greater than 100 000/mm3 and estimated glomerular filtration rate greater than 30 mL/min/1.73 m2.
Interventions
Random allocation in a 1:1:1:1 ratio to aspirin (81 mg orally once daily; n = 164), prophylactic-dose apixaban (2.5 mg orally twice daily; n = 165), therapeutic-dose apixaban (5 mg orally twice daily; n = 164), or placebo (n = 164) for 45 days.
Main Outcomes and Measures
The primary end point was a composite of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary cause. The primary analyses for efficacy and bleeding events were limited to participants who took at least 1 dose of trial medication.
Results
On June 18, 2021, the trial data and safety monitoring board recommended early termination because of lower than anticipated event rates; at that time, 657 symptomatic outpatients with COVID-19 had been randomized (median age, 54 years [IQR, 46-59]; 59% women). The median times from diagnosis to randomization and from randomization to initiation of study treatment were 7 days and 3 days, respectively. Twenty-two randomized participants (3.3%) were hospitalized for COVID-19 prior to initiating treatment. Among the 558 patients who initiated treatment, the adjudicated primary composite end point occurred in 1 patient (0.7%) in the aspirin group, 1 patient (0.7%) in the 2.5-mg apixaban group, 2 patients (1.4%) in the 5-mg apixaban group, and 1 patient (0.7%) in the placebo group. The risk differences compared with placebo for the primary end point were 0.0% (95% CI not calculable) in the aspirin group, 0.7% (95% CI, –2.1% to 4.1%) in the 2.5-mg apixaban group, and 1.4% (95% CI, –1.5% to 5.0%) in the 5-mg apixaban group. Risk differences compared with placebo for bleeding events were 2.0% (95% CI, –2.7% to 6.8%), 4.5% (95% CI, –0.7% to 10.2%), and 6.9% (95% CI, 1.4% to 12.9%) among participants who initiated therapy in the aspirin, prophylactic apixaban, and therapeutic apixaban groups, respectively, although none were major. Findings inclusive of all randomized patients were similar.
Conclusions and Relevance
Among symptomatic clinically stable outpatients with COVID-19, treatment with aspirin or apixaban compared with placebo did not reduce the rate of a composite clinical outcome. However, the study was terminated after enrollment of 9% of participants because of an event rate lower than anticipated.
Trial Registration
ClinicalTrials.gov Identifier: NCT04498273
This randomized clinical trial assesses whether aspirin, prophylactic-dose apixaban, or therapeutic-dose apixaban, compared with placebo, can safely reduce major adverse cardiopulmonary outcomes among symptomatic but clinically stable US outpatients with COVID-19.
Introduction
Acutely ill patients who experience SARS-CoV-2 infection have increased risks of arterial and venous thrombosis, which often manifest as pulmonary microvascular thrombosis that can severely compromise care and complicate COVID-19 pneumonia.1,2,3 To date, a series of randomized clinical trials have evaluated the utility of anticoagulant and antiplatelet interventions among hospitalized patients with COVID-19, including those requiring intensive care and mechanical ventilation.4,5,6,7 However, whether to prescribe antithrombotic agents for clinically stable outpatients with COVID-19 is controversial, as no randomized trials in this setting have been reported. This issue is of considerable public health importance, as the majority of individuals infected with SARS-CoV-2 do not require hospitalization and are treated as outpatients.
The ACTIV-4B COVID-19 Outpatient Thrombosis Prevention Trial, funded by Operation Warp Speed and conducted as part of the National Heart, Lung, and Blood Institute (NHLBI) ACTIV platform of randomized clinical trials, addressed whether outpatients infected with COVID-19 who were symptomatic but who did not require hospitalization at the time of diagnosis would benefit from intervention with antiplatelet or anticoagulant therapies. This randomized, double-blind, minimal-contact, placebo-controlled trial was designed to address whether aspirin, prophylactic-dose apixaban, or therapeutic-dose apixaban could prevent macrovascular and microvascular thrombosis and thus slow progression to more severe disease among symptomatic but clinically stable patients with COVID-19 who were not considered by their clinicians to initially require hospitalization.
Methods
Study Organization and Oversight
This was an adaptive, randomized, double-blind, placebo-controlled trial of antiplatelet and anticoagulant agents with blinded adjudication of outcomes. The trial protocol was developed by the trial chair, principal investigator, and a protocol development committee including representatives from the NHLBI. The protocol and statistical analysis plan were approved at each of the 52 centers in the US that participated in the trial and are provided in Supplement 1 and Supplement 2.
Trial functions were coordinated by the University of Pittsburgh using a multipurpose central electronic data-capture system for data management (e-SOCDAT, SOCAR Research), and an electronic informed consent process was structured using REDCap that was augmented by standardized video presentations of the trial hypothesis, trial procedures, potential risks, and potential benefits to obtain the required informed consent from all participants.
To try to ensure consistency across sites in the setting of an outpatient COVID-19 trial, all postrandomization participant contact was conducted by weekly electronic links to REDCap surveys or by the Research Communication Center (RCC) at the University of Illinois Chicago with live telephone calls by call center agents and research pharmacists in Chicago and Pittsburgh. Direct shipment of study drug to participants homes, end point and safety adjudication, and 24-hour emergency and unblinding services were provided by investigators at the Brigham and Women’s Hospital in Boston. An independent data and safety monitoring board was assigned by the NHLBI to monitor the trial for efficacy and safety.
Study Population
Ambulatory patients between the ages of 40 and 80 years with newly diagnosed symptomatic SARS-CoV-2 infection with positive polymerase chain reaction or antigen test results were eligible. Creatinine clearance was required to be greater than 30 mL/min/1.73 m2 and platelet count greater than 100 000/mm3. Samples for analysis of D-dimer and high-sensitivity C-reactive protein levels were drawn, but no thresholds were required for enrollment. Patients were excluded who had been previously hospitalized for COVID-19, had acute leukemia, recent major bleeding, a contraindication to or other indication for anticoagulation, need for single or dual antiplatelet therapy, or who were pregnant or lactating (Supplement 1). Participants were required to have the ability to be contacted by telephone and, optionally, other electronic methods of communication. Participants self-reported race and ethnicity using fixed categories to ensure appropriate racial and ethnic representation in the trial.
Study Procedures
Given the constraints of conducting outpatient research during the COVID-19 pandemic, the trial was designed with minimal face-to-face interactions, allowing participants to quarantine in place and minimize exposure risks to study staff. Sites identified potentially eligible patients using a variety of strategies including in-person visits, daily review of institutional or regional COVID-19 test reports, and in partnership with commercial outpatient testing centers. Patients who met preliminary eligibility criteria could be randomized and qualifying laboratory measurement of creatinine clearance and platelet count, as well as D-dimer and high-sensitivity C-reactive protein levels, could be obtained either on-site prior to randomization or after randomization using home health visits conducted in partnership with commercial home health and laboratory services. Initiation and continuation of study drug was allowed only for randomized participants with demonstrated creatinine clearance greater than 30 mL/min/1.73 m2 and platelet count greater than 100 000/mm3.
Randomization and Interventions
Participants were randomized centrally in a 1:1:1:1 ratio to receive aspirin (81 mg once daily) with matching placebo, prophylactic-dose apixaban (2.5 mg twice daily), apixaban at therapeutic dose (5 mg twice daily), or placebo twice daily for 45 days, with a 30-day safety follow-up evaluation (Figure 1). Randomization code lists were computer generated using permuted blocks with block size equal to 4 during the process of drug labeling and then implemented electronically through the central electronic data-capture system.
Figure 1. Participant Flow in the ACTIV-4B Trial.
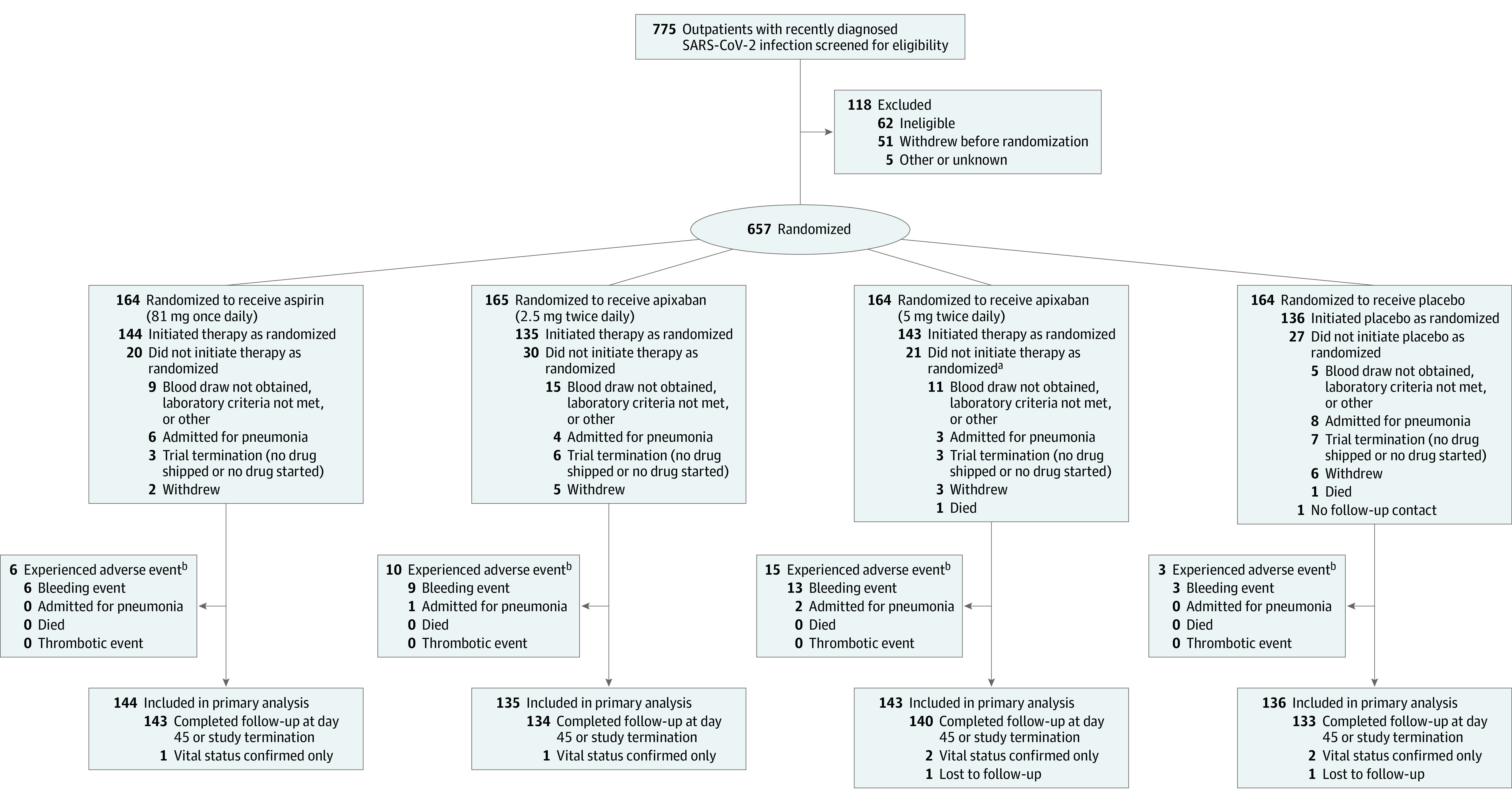
aOne additional participant in the therapeutic-dose apixaban (5 mg twice daily) group was admitted for pneumonia prior to initiating drug therapy; this event is counted in the analysis of all randomized participants. The participant subsequently initiated trial therapy in the therapeutic-dose apixaban group and then had a second cardiovascular pulmonary hospital admission (not adjudicated as pneumonia); this second event is counted in the analysis of participants who initiated therapy. Data are shown for adjudicated efficacy outcome events and for any suspected bleeding events.
bNo patients died or experienced thrombotic events to 45 days.
Surveillance and Follow-up
After randomization, drug was shipped directly to the participant’s home with subsequent follow-up conducted by the RCC central study staff. Using a combination of telephone, email, and electronic links to REDCap surveys, participants were contacted within 24 to 72 hours of drug shipment to confirm receipt and to determine start date of trial medications. Using similar methods, participants were contacted weekly using text links to REDCap survey or by the RCC staff telephone calls during the 45-day randomized treatment period to capture relevant clinical outcomes as well as bleeding events and again after a subsequent 30-day safety follow-up. Participant-reported events triggered a telephone call from a research pharmacist to confirm the event, determine type and severity, and if medically attended, obtain the contact information for the treating clinician. Medical records were obtained by the trial monitors for all events that were considered possible components of the primary efficacy or safety outcome during the 45-day treatment period or that raised any safety concerns over the additional 30-day posttreatment follow-up. Medical records were sent to the Clinical Endpoints Committee, who adjudicated events using standardized criteria. Both the medical monitors and the end points committee were unaware of randomized drug assignment.
Study End Points
The primary outcome was the composite of symptomatic deep venous thrombosis, pulmonary embolism, arterial thromboembolism, myocardial infarction, ischemic stroke, hospitalization for cardiovascular or pulmonary events, and all-cause mortality for up to 45 days after treatment initiation.
Prespecified secondary outcomes included the individual components of the primary study end point as well as mortality without antecedent hospitalization. We also conducted post hoc analyses evaluating any acute medical event (all emergency department visits, all acute outpatient clinic visits, and all hospitalizations, regardless of etiology) during the 45-day follow-up period. The principal safety outcomes were major bleeding and clinically relevant nonmajor bleeding (CRNMB) as defined by International Society on Thrombosis and Hemostasis (ISTH) criteria,8 as well as any events of disseminated intravascular coagulation. Reported bleeding events that did not meet ISTH major or CRNMB criteria were considered minor. Safety end points were analyzed during the 45-day period of randomized trial treatment and after an additional 30-day safety follow-up.
Sample Size Estimation
At the beginning of the pandemic, an analysis of past population-based data, case series, clinical trials and emerging clinical experience suggested that individuals recently infected with COVID-19 who were symptomatic but who did not require urgent hospitalization would have a rate of thromboembolic events and cardiopulmonary admissions ranging between 4% and 8%. Across this anticipated event rate range, we estimated that a sample size of 7000 (1750 participants assigned to each of the 4 treatment groups) would provide 80% to 90% power to detect relative risk reductions between 33% and 50% in the primary outcome between each active drug and placebo, with α = .025 (1-sided). Previous trials of anticoagulants for prevention of thrombotic events in ambulatory patients have noted similar event rates and relative risk reductions, and the study leadership team assumed that outpatient treatment would be adopted only if it had a large effect on thrombotic outcomes.9,10,11 Details regarding initial calculations for sample size determination are presented in Supplement 2.
Statistical Analysis
The primary sample of interest for both efficacy and safety treatment comparisons was defined on an a priori basis and consisted of all randomized participants who initiated trial therapy and had at least 1 follow-up assessment during the 45-day treatment period. All treatment comparisons were based on the assigned treatment, and end point events were counted from the time when the trial therapy was initiated through 45 days after treatment initiation. Secondary efficacy and safety analyses were conducted among all randomized participants according to assigned treatment, for which end point events were counted for 45 days beginning at the time of randomization.
Baseline characteristics of the randomized cohort and those who initiated treatment were compared across treatment groups; frequencies (percentages) are presented for categorical variables and medians (interquartile ranges, first and third quartiles) for continuous variables. The occurrence of the primary composite efficacy end point during the designated 45-day period was estimated in each treatment group as a simple proportion, regardless of whether the follow-up was incomplete. Since events were rare among patients who initiated treatment, a logistic regression analysis could not be conducted for the primary end point as originally planned. Consequently, the risk differences for the primary end point (adjudicated and suspected), any acute medical event, and any reported bleeding event were estimated for each active treatment group as compared with the placebo group. All available data through 45 days were used to identify outcomes, and patients with incomplete follow-up were assumed to have no event after the time of last contact for these analyses. Wilson (score) confidence intervals are presented for risk, and Newcombe confidence intervals for risk differences. Kaplan-Meier estimates and log rank statistics were used to estimate and compare the cumulative incidence of the adjudicated primary end point and of any acute medical event across the 4 treatment groups, and follow-up was censored at the time of last contact. Components of the primary composite efficacy end point and bleeding events, which were classified as secondary end points and safety events, are reported as estimated proportions. Because of the potential for type 1 error due to multiple comparisons, findings for analyses of secondary end points should be interpreted cautiously.
Analyses were conducted with SAS version 9.4 (SAS Institute Inc) and RStudio version 4.0.2 (RStudio). Details of the prespecified analysis plan are provided in the trial protocol (Supplement 1).
Results
Study Population
Between September 1, 2020, through June 17, 2021, 775 potential participants provided informed consent and were screened, of whom 657 met preliminary eligibility criteria and were randomized (Figure 1). On June 18, 2021, the NHLBI accepted a recommendation from the independent data and safety monitoring board to terminate the trial early because of lower than anticipated event rates (Supplement 3); for this reason, despite the adaptive clinical trial design, no additional agents underwent evaluation. At the time of early trial termination, 558 of the 657 randomized symptomatic outpatients with COVID-19 had initiated trial treatment. The median time from diagnosis to randomization was 7 days (IQR, 3-10 days), and the median time from randomization to initiation of study treatment was 3 days (IQR, 2-5 days); final follow-up was August 5, 2021.
The median age of randomized participants was 54 years (IQR, 46-59), 59.1% were women, and 12.7% self-identified as Black and 28.1% as Hispanic (Table 1). Among those randomized, the median body mass index was 30.1 (calculated as weight in kilograms divided by height in meters squared), 18.3% had diabetes, 19.9% had a history of smoking, and 35.3% reported a history of hypertension. Baseline characteristics were balanced among treatment groups in all randomized participants and among those who initiated treatment (Table 1; eTable 1 in Supplement 3).
Table 1. Baseline Characteristics of All Randomized Participants, Stratified by Assigned Treatment.
| Characteristic | No. (%) | |||
|---|---|---|---|---|
| Aspirin (81 mg once daily) (n = 164) | Apixaban (2.5 mg twice daily) (n = 165) | Apixaban (5 mg twice daily) (n = 164) | Placebo (n = 164) | |
| Age, median (IQR), y | 54.0 (46.0-59.0) | 55.0 (46.0-61.0) | 52.0 (47.0-58.0) | 54.0 (45.0-59.0) |
| Sex | ||||
| Women | 95 (57.9) | 95 (57.6) | 102 (62.2) | 96 (58.5) |
| Men | 69 (42.1) | 70 (42.4) | 62 (37.8) | 68 (41.5) |
| Race group, No. | 149 | 158 | 152 | 156 |
| American Indian or Alaska Native | 0 | 0 | 2 (1.3) | 1 (0.6) |
| Asian | 3 (2.0) | 2 (1.3) | 2 (1.3) | 2 (1.3) |
| Black or African American | 20 (13.4) | 22 (13.9) | 20 (13.2) | 16 (10.3) |
| Native Hawaiian or Other Pacific Islander | 0 | 2 (1.3) | 2 (1.3) | 0 |
| White | 119 (79.9) | 124 (78.5) | 120 (79.0) | 131 (84.0) |
| Other | 7 (4.7) | 8 (5.1) | 6 (4.0) | 6 (8.9) |
| Hispanic ethnicity, No. | 158 | 162 | 156 | 157 |
| Yes | 50 (31.7) | 48 (29.6) | 37 (23.7) | 43 (27.4) |
| No | 108 (68.4) | 114 (70.4) | 119 (76.3) | 114 (72.6) |
| Body mass index, median (IQR)a | 29.6 (26.3-34.3) | 29.9 (26.2-34.8) | 31.1 (26.2-35.4) | 30.3 (26.5-34.7) |
| Hypertensionb | 55 (33.5) | 66 (40.0) | 57 (34.8) | 54 (32.9) |
| Diabetesb | 29 (17.7) | 36 (21.8) | 31 (18.9) | 24 (14.6) |
| History of smoking | 39 (23.8) | 29 (17.6) | 32 (19.5) | 31 (18.9) |
| Platelet count,c median (IQR), /mm3 | 246.0 (195.0-316.0) | 250.0 (187.0-295.0) | 248.0 (198.0-302.0) | 238.0 (189.0-319.0) |
| Creatinine clearance,d median (IQR), mg/mL/1.73 m2 | 113.4 (90.2-138.4) | 110.2 (89.9-144.5) | 117.2 (94.2-147.4) | 115.8 (91.3-653.0) |
| D-dimer, No. (%)e | 146 | 152 | 150 | 153 |
| ≤1× ULN | 93 (63.7) | 99 (65.1) | 94 (62.7) | 101 (66.0) |
| >1 -≤2× ULN | 40 (27.4) | 33 (21.7) | 35 (23.3) | 38 (24.8) |
| >2× ULN | 13 (8.9) | 20 (13.2) | 21 (14.0) | 14 (9.2) |
| hsCRP,f median (IQR), mg/L | 3.1 (1.0-11.8) | 4.4 (1.9-14.4) | 4.0 (1.7-12.0) | 4.3 (2.0-12.1) |
Abbreviations: hsCRP, high-sensitivity C-reactive protein; ULN, upper limit of normal.
Calculated as weight in kilograms divided by height in meters squared.
Patient self-reported.
Reference range, 150-400/mm3.
Reference, greater than 90 mg/mL/1.73 m2.
D-dimer assays varied from site to site. Upper limit of normal was captured for each site with individual participant results compared with local values to determine if within the reference range or elevated above the reference range.
Reference, less than 1 mg/L.
Primary Outcome
During the period that transpired between the time of randomization and initiation of study drug (a period that often included a clinic or home-health visit to obtain and assay a blood sample to ensure laboratory eligibility criteria were met), 22 randomized participants (3.3%) became acutely unstable and were hospitalized for worsening symptoms of pneumonia prior to initiating study treatment. In this group, there were 2 pneumonia-related deaths during the 45-day observation period (one of which was attributable to pulmonary embolism) and 1 case of nonfatal deep vein thrombosis. There was an additional death in this group secondary to respiratory failure that occurred after the 45-day observation period but during the 30-day subsequent safety period.
Among the 558 participants who initiated randomized trial treatment, 556 (99.6%) had complete follow-up at 45 days after drug initiation or at the time of trial termination, whichever came earlier, and 544 (97.5%) were followed up through day 45. Five suspected primary end points and no deaths accrued during the trial treatment period (Table 2).
Table 2. Suspected and Adjudicated Efficacy Outcomes and Hemorrhagic Events Within 45 Days of Drug Initiation Among Those Who Initiated Trial Therapy, Stratified by Assigned Treatment.
| No. (%) | ||||
|---|---|---|---|---|
| Aspirin (81 mg once daily) (n = 144) | Apixaban (2.5 mg twice daily) (n = 135) | Apixaban (5 mg twice daily) (n = 143)a | Placebo (n = 136)a | |
| Suspected outcomes | ||||
| Composite primary end pointb | 1 (0.7) | 1 (0.7) | 2 (1.4) | 1 (0.7) |
| Risk difference (in percentage points) vs placebo (95% CI) | 0.0 (–3.4 to 3.2) | 0.0 (–3.4 to 3.4) | 0.7 (–2.8 to 4.3) | |
| Components of primary end point | ||||
| Cardiopulmonary hospitalizations | 0 | 1 (0.7) | 2 (1.4) | 1 (0.7) |
| Deep vein thrombosis or pulmonary embolism | 1 (0.7) | 0 | 0 | 0 |
| Myocardial infarction, stroke or other arterial embolism | 0 | 0 | 0 | 0 |
| Death | 0 | 0 | 0 | 0 |
| Any acute medical eventc | 6 (4.2) | 8 (5.9) | 13 (9.1) | 7 (5.2) |
| Risk difference (in percentage points) vs placebo (95% CI) | –1.0 (–6.6 to 4.3) | 0.8 (–5.1 to 6.7) | 4.0 (–2.4 to 10.4) | |
| Adjudicated outcomesd | ||||
| Composite primary end point | 0 | 1 (0.7) | 2 (1.4) | 0 |
| Risk difference (in percentage points) vs placebo (95% CI) | 0 | 0.7 (–2.1 to 4.1) | 1.4 (–1.5 to 5.0) | |
| Components of primary end point | ||||
| Cardiopulmonary hospitalizations | 0 | 1 (0.7) | 2 (1.4) | 0 |
| Deep vein thrombosis or pulmonary embolism | 0 | 0 | 0 | 0 |
| Myocardial infarction, stroke or other arterial embolism | 0 | 0 | 0 | 0 |
| Death | 0 | 0 | 0 | 0 |
| Suspected hemorrhagic events | ||||
| Any bleeding evente | 6 (4.2) | 9 (6.7) | 13 (9.1) | 3 (2.2) |
| Risk difference (in percentage points) vs placebo (95% CI) | 2.0 (–2.7 to 6.8) | 4.5 (–0.7 to 10.2) | 6.9 (1.4 to 12.9) | |
| Type of bleeding event | ||||
| Major bleeding | 0 | 0 | 0 | 0 |
| Clinically relevant nonmajor bleeding | 2 (1.4) | 4 (3.0) | 2 (1.4) | 0 |
| Minor bleeding | 4 (2.8) | 5 (3.7) | 11 (7.7) | 3 (2.2) |
| Adjudicated hemorrhagic eventsf | ||||
| Major bleeding | 0 | 0 | 0 | 0 |
| Clinically relevant nonmajor bleeding | 0 | 1 (0.7) | 1 (0.7) | 0 |
Two patients lost to follow-up, 1 in the apixaban 5 mg group and 1 in the placebo group, are included in the analysis through the time at which they were lost to follow-up.
The primary end point is defined as the composite of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary cause.
Any acute medical event includes all emergency department visits, all acute outpatient clinic visits, and all hospitalizations, regardless of etiology during the 45-day follow-up period.
Medical records were collected for all suspected primary end point events reported by the medical monitor, and these events were adjudicated by the Clinical Events Committee.
Suspected major and clinically relevant nonmajor bleeding events were reported by the trial medical monitor, and minor bleeding events were identified through follow-up with the research pharmacists.
All suspected major and clinically relevant nonmajor bleeding events were adjudicated by the Clinical Events Committee; minor bleeding events were not adjudicated.
In total, there were 3 adjudicated primary trial events among the 558 participants taking trial medications. The risk of the adjudicated primary composite end point was 0.0% (95% CI, 0.0% to 2.6%) in the aspirin group, 0.7% (95% CI, 0.1% to 4.1%) in the prophylactic apixaban group, 1.4% (95% CI, 0.4% to 5.0%) in the therapeutic apixaban group, and 0.0% (95% CI, 0.0% to 2.8%) in the placebo group, leading to risk differences of 0.0% (95% CI not calculable), 0.7% (95% CI, –2.1% to 4.1%), and 1.4% (95% CI, –1.5% to 5.0%) in the aspirin, prophylactic apixaban, and therapeutic apixaban groups as compared with the placebo group (Table 2).
The estimated cumulative incidence of the adjudicated primary composite end point at 45 days was 0.0% (95% CI not calculable) in the aspirin group, 0.7% (95% CI, 0.0% to 2.2%) in the prophylactic apixaban group, 1.4% (95% CI, 0.0% to 3.3%) in the therapeutic apixaban group, and 0.0% in the placebo group and did not significantly differ by assigned treatment among those who initiated treatment (log-rank P = .31) (Figure 2A). Two positively adjudicated cardiopulmonary hospitalizations occurred in the therapeutic apixaban group (an admission during which suspected myocardial infarction was ruled out and a nonfatal admission for COVID-19–associated pneumonia) and 1 positively adjudicated cardiopulmonary hospitalization occurred in the prophylactic apixaban group (a nonfatal admission for COVID-19–associated pneumonia). One positively adjudicated nonfatal myocardial infarction occurred in the aspirin group after day 45 but before the end of the 30-day safety follow-up, as did a case of suspected deep-vein thrombosis that was not positively adjudicated as a deep vein thrombosis but rather was classified as a superficial thrombophlebitis.
Figure 2. Cumulative Incidence of the Adjudicated Primary Trial End Point and the Cumulative Incidence for Any Acute Medical Event Among Randomized Trial Participants Who Initiated Trial Therapy, Stratified by Assigned Treatment.
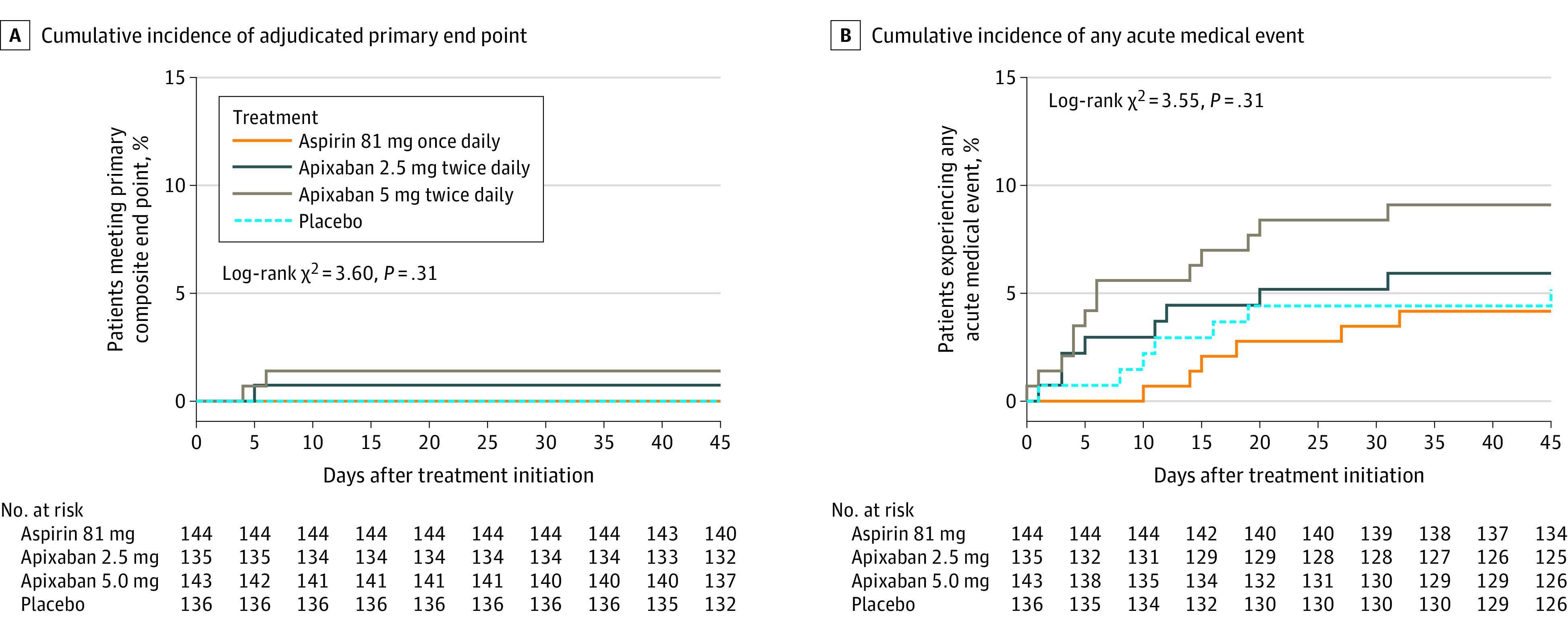
The primary end point is defined as the composite of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary cause. The end point of any acute medical event is inclusive of all emergency department visits, all acute outpatient clinic visits, and all hospitalizations during the 45-day follow-up period, regardless of etiology.
In analyses inclusive of all randomized trial participants irrespective of whether study drug was initiated, risk differences for the adjudicated primary end point were 1.2% (95% CI, –6.1% to 3.5%) in the aspirin group, –1.9% (95% CI, –6.6% to 2.7%) in the prophylactic apixaban group, and –1.8% (95% CI, –6.6% to 2.7%) in the therapeutic apixaban group, as compared with placebo (eTable 2 in Supplement 3). The cumulative incidence of the adjudicated primary trial outcomes did not significantly differ across the 4 treatment groups (log-rank P = .78) (Figure 3A).
Figure 3. Cumulative Incidence of the Adjudicated Primary Trial End Point and the Cumulative Incidence for Any Acute Medical Event Among All Randomized Trial Participants, Stratified by Assigned Treatment.
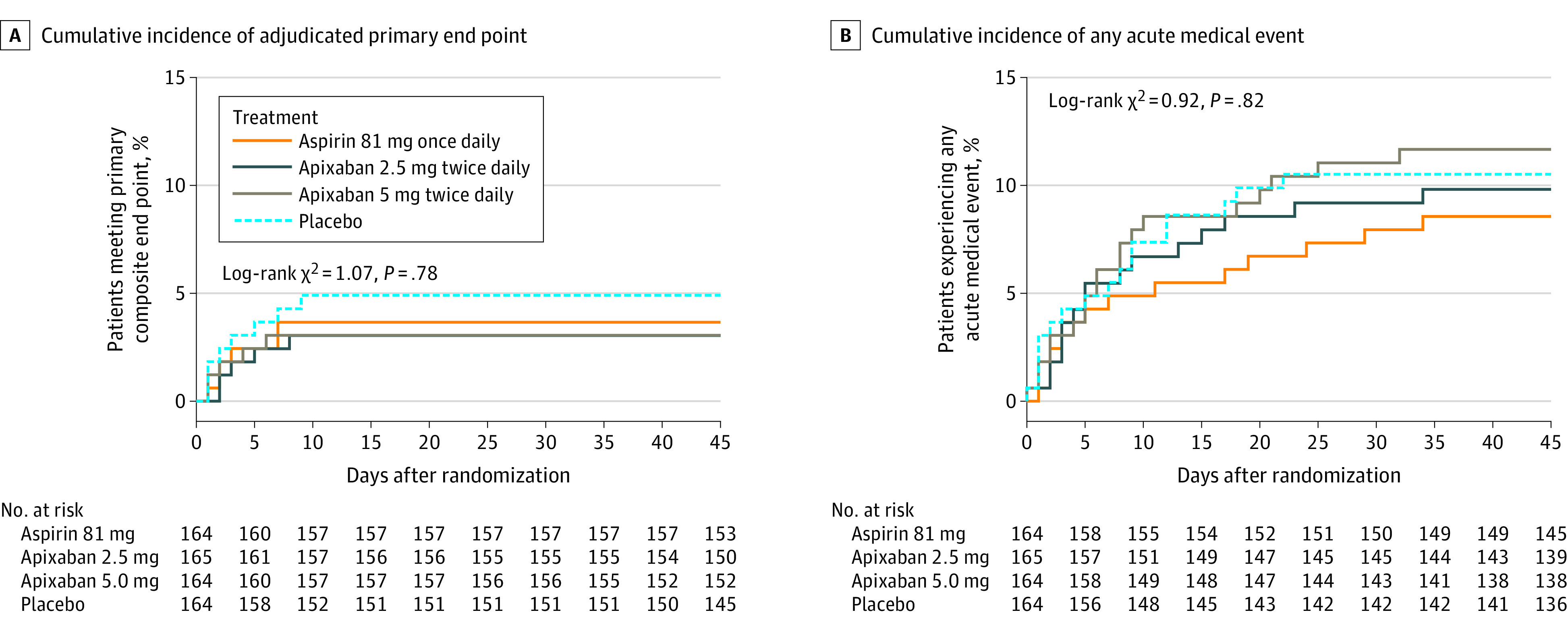
The primary end point is defined as the composite of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary cause. The end point of any acute medical event is inclusive of all emergency department visits, all acute outpatient clinic visits, and all hospitalizations during the 45-day follow-up period, regardless of etiology.
Secondary Outcomes
Table 2 provides tabular data for the individual components of the primary study end point for those who initiated trial treatment, and eTable 2 in Supplement 3 provides comparable data for all randomized participants. No episodes of death without antecedent hospitalization were reported.
Post Hoc Outcomes
Additional analyses were conducted to evaluate any acute medical event (regardless of etiology) intended to include all acute interactions with the health care system during the 45-day follow-up period. For this end point, risk differences, as compared with placebo, were –1.0% (95% CI, –6.6% to 4.3%) for the aspirin group, 0.8% (95% CI, –5.1% to 6.7%) for the prophylactic apixaban group, and 4.0% (95% CI, –2.4% to 10.4%) for the therapeutic apixaban group among participants who initiated trial treatment, and the cumulative incidence did not differ significantly according to randomized treatment assignment (log-rank P = .31) (Figure 2B). Similar results were observed for all randomized participants (log-rank P = .82) (Figure 3B; eTable 2 in Supplement 3).
Adverse Events
No major bleeding events were reported during the trial. Among patients who initiated therapy, there was an absolute excess, compared with placebo, of 2 suspected clinically relevant nonmajor bleeds in the aspirin group, 4 in the prophylactic apixaban group, and 2 in the therapeutic apixaban group (Table 2). Patients who were randomly allocated to apixaban and took at least 1 dose prior to trial termination had an overall rate of clinically relevant nonmajor bleeds of 2.2% (6/278). Among those who initiated trial treatment, any bleeding event occurred among 6 participants in the aspirin group, 9 in the prophylactic apixaban group, 13 in the therapeutic apixaban group, and 3 in the placebo group, resulting in risk differences relative to placebo of 2.0% (95% CI, –2.7% to 6.8%) in the aspirin group, 4.5% (95% CI, –0.7% to 10.2%) in the prophylactic apixaban group, and 6.9% (95% CI, 1.4% to 12.9%) in the therapeutic apixaban group. Any bleeding event occurred in 22 (7.9%) of the 278 individuals allocated to apixaban.
Among all randomized participants, there was an absolute excess, compared with placebo, of 4 suspected clinically relevant nonmajor bleeds in the aspirin group, 6 in the prophylactic apixaban group, and 4 in the therapeutic apixaban group (eTable 2 in Supplement 3).
No episodes of disseminated intravascular coagulation were reported.
Discussion
In this clinical trial of symptomatic outpatients infected with SARS-CoV-2, random allocation to aspirin or apixaban did not reduce rates of cardiopulmonary hospitalization, compared with placebo. However, the study was terminated because of a control event rate lower than anticipated.
A combination of 2 demographic shifts over time may have led to lower than anticipated event rates in the trial. First, the threshold for hospital admission has markedly declined since the beginning of the pandemic, such that hospitalization is no longer limited almost exclusively to those with severe pulmonary distress likely to require mechanical ventilation. As a result, the severity of illness among individuals with COVID-19 and destined for outpatient care has declined. Second, at least within the US where the trial was conducted, individuals currently being infected with SARS-CoV-2 tend to be younger and have fewer comorbidities when compared with individuals with incident infection at the onset of the pandemic. In addition, COVID-19 testing was quite limited early in the pandemic, and it is possible that the anticipated event rates based on data from registries available at that time were overestimated because the denominator (ie, the number of infected individuals overall) was essentially unknown.
To date, no randomized trial data addressing antithrombotic therapy have been available for outpatients with COVID-19, who comprise the majority of infected individuals. The low event rate and neutral findings in this trial should not be construed to suggest that outpatients with COVID-19 do not require close medical attention. As reported, 3.3% of those randomized became clinically unstable and required hospitalization during the median 3-day period before study drug could be initiated, 2 of whom died of COVID-related respiratory failure during the 45-day observation period and another who died during the subsequent 30-day safety follow-up. Because these hospitalized individuals did not initiate trial therapy, these findings do not address whether earlier initiation of prophylactic antithrombotic therapy would have benefited this small subset of patients.
Although the reliance on self-reported events in this study could have resulted in a reduced overall event rate despite careful adjudication processes, this seems unlikely. First, the trial design incorporated weekly systematic contact with all participants and used specific targeted questions to identify both efficacy and safety events, with a second follow-up call from trained research staff to directly obtain clinical details and hospital records. Second, for any situation in which data were uncertain, clinical sites were queried at the time of trial closeout as to whether an individual participant had been admitted locally or to an affiliated center at any point in the trial. Third, in the trial closeout process, participants were again contacted by telephone to review clinical status and evaluate any intercurrent events. Fourth, rates of bleeding complications within the apixaban groups of the trial were very similar to those previously reported from trials of treatment and secondary prevention of venous thromboembolism with similar populations,10,12 suggesting that the study captured most clinically relevant events.
Limitations
This study has several limitations. First, given the time frame of the trial, it is unlikely that many participants were infected with COVID-19 variants, such as the delta variant, that may confer greater clinical severity.13 Second, very few trial participants had been vaccinated prior to randomization, an issue that could limit generalizability. However, vaccination markedly reduces not only the likelihood of infection but also the likelihood of hospitalization.14,15,16 Third, use of outpatient interventions such as monoclonal antibody treatment was low in this trial; however, such measures would, if anything, be expected to further reduce the primary trial event rate.17,18,19 Fourth, the median time from diagnosis to randomization was 7 days, so the study findings cannot address the potential efficacy of more immediate intervention.
Conclusions
Among symptomatic clinically stable outpatients with COVID-19, treatment with aspirin or apixaban compared with placebo did not reduce the rate of a composite clinical outcome. However, the study was terminated after enrollment of 9% of participants because of a primary event rate lower than anticipated.
Study Protocol
Statistical Analysis Plan
eTable 1. Baseline Characteristics of All Randomized Participants Stratified by Assigned Treatment
eTable 2. Suspected and Adjudicated Efficacy Outcomes and Hemorrhagic Events Within 45 Days of Drug Initiation Among Those Who Initiated Trial Therapy Stratified by Assigned Treatment
Protocol Development Committee
University of Pittsburgh
University of Illinois, Chicago
Brigham and Women’s Hospital Operations Team
Brigham and Women’s Hospital Emergency Call/Unblinding Team
SOCAR
Clinical Endpoints Committee Members
DSMB
ACTIVE-4B Investigators (City, State) Enrolling at Least One Participant
NIH/NHLBI
CONNECTS Steering and Executive Committees:
CVS
Covance
Coram
Quest/ExamOne
Parexel
RTI
Clinical Endpoints Committee Charter
DSMB Termination Memo
Nonauthor Collaborators
Data Sharing Statement
References
- 1.Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in COVID-19. N Engl J Med. 2020;383(2):120-128. doi: 10.1056/NEJMoa2015432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Connors JM, Levy JH. COVID-19 and its implications for thrombosis and anticoagulation. Blood. 2020;135(23):2033-2040. doi: 10.1182/blood.2020006000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nopp S, Moik F, Jilma B, Pabinger I, Ay C. Risk of venous thromboembolism in patients with COVID-19: a systematic review and meta-analysis. Res Pract Thromb Haemost. 2020;4:1178-1191. doi: 10.1002/rth2.12439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadeghipour P, Talasaz AH, Rashidi F, et al. ; INSPIRATION Investigators . Effect of intermediate-dose vs standard-dose prophylactic anticoagulation on thrombotic events, extracorporeal membrane oxygenation treatment, or mortality among patients with COVID-19 admitted to the intensive care unit: the INSPIRATION randomized clinical trial. JAMA. 2021;325(16):1620-1630. doi: 10.1001/jama.2021.4152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopes RD, de Barros E Silva PGM, Furtado RHM, et al. ; ACTION Coalition COVID-19 Brazil IV Investigators . Therapeutic versus prophylactic anticoagulation for patients admitted to hospital with COVID-19 and elevated D-dimer concentration (ACTION): an open-label, multicentre, randomised, controlled trial. Lancet. 2021;397(10291):2253-2263. doi: 10.1016/S0140-6736(21)01203-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goligher EC, Bradbury CA, McVerry BJ, et al. ; REMAP-CAP Investigators; ACTIV-4a Investigators; ATTACC Investigators . Therapeutic anticoagulation with heparin in critically ill patients with Covid-19. N Engl J Med. 2021;385(9):777-789. doi: 10.1056/NEJMoa2103417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lawler PR, Goligher EC, Berger JS, et al. ; ATTACC Investigators; ACTIV-4a Investigators; REMAP-CAP Investigators . Therapeutic anticoagulation with heparin in noncritically ill patients with Covid-19. N Engl J Med. 2021;385(9):790-802. doi: 10.1056/NEJMoa2105911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaatz S, Ahmad D, Spyropoulos AC, Schulman S; Subcommittee on Control of Anticoagulation . Definition of clinically relevant non-major bleeding in studies of anticoagulants in atrial fibrillation and venous thromboembolic disease in non-surgical patients: communication from the SSC of the ISTH. J Thromb Haemost. 2015;13(11):2119-2126. doi: 10.1111/jth.13140 [DOI] [PubMed] [Google Scholar]
- 9.Cohen AT, Spiro TE, Büller HR, et al. ; MAGELLAN Investigators . Rivaroxaban for thromboprophylaxis in acutely ill medical patients. N Engl J Med 2013;368(6):513-523. doi: 10.1056/NEJMoa1111096 [DOI] [PubMed] [Google Scholar]
- 10.Agnelli G, Buller HR, Cohen A, et al. ; AMPLIFY-EXT Investigators . Apixaban for extended treatment of venous thromboembolism. N Engl J Med. 2013;368(8):699-708. doi: 10.1056/NEJMoa1207541 [DOI] [PubMed] [Google Scholar]
- 11.Spyropoulos AC, Ageno W, Albers GW, et al. Post-discharge prophylaxis with rivaroxaban reduces fatal and major thromboembolic events in medically ill patients. J Am Coll Cardiol. 2020;75(25):3140-3147. doi: 10.1016/j.jacc.2020.04.071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agnelli G, Buller HR, Cohen A, et al. ; AMPLIFY Investigators . Oral apixaban for the treatment of acute venous thromboembolism. N Engl J Med. 2013;369(9):799-808. doi: 10.1056/NEJMoa1302507 [DOI] [PubMed] [Google Scholar]
- 13.WHO coronavirus (COVID-19) dashboard. World Health Organization. Accessed September 29, 2021. https://covid19.who.int/
- 14.Polack FP, Thomas SJ, Kitchin N, et al. ; C4591001 Clinical Trial Group . Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N Engl J Med. 2020;383(27):2603-2615. doi: 10.1056/NEJMoa2034577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baden LR, El Sahly HM, Essink B, et al. ; COVE Study Group . Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N Engl J Med. 2021;384(5):403-416. doi: 10.1056/NEJMoa2035389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dagan N, Barda N, Kepten E, et al. BNT162b2 mRNA Covid-19 vaccine in a nationwide mass vaccination setting. N Engl J Med. 2021;384(15):1412-1423. doi: 10.1056/NEJMoa2101765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen P, Nirula A, Heller B, et al. ; BLAZE-1 Investigators . SARS-CoV-2 neutralizing antibody LY-CoV555 in outpatients with Covid-19. N Engl J Med. 2021;384(3):229-237. doi: 10.1056/NEJMoa2029849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weinreich DM, Sivapalasingam S, Norton T, et al. ; Trial Investigators . REGN-COV2, a neutralizing antibody cocktail, in outpatients with COVID-19. N Engl J Med. 2021;384(3):238-251. doi: 10.1056/NEJMoa2035002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gottlieb RL, Nirula A, Chen P, et al. Effect of bamlanivimab as monotherapy or in combination with etesevimab on viral load in patients with mild to moderate COVID-19: a randomized clinical trial. JAMA. 2021;325(7):632-644. doi: 10.1001/jama.2021.0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Study Protocol
Statistical Analysis Plan
eTable 1. Baseline Characteristics of All Randomized Participants Stratified by Assigned Treatment
eTable 2. Suspected and Adjudicated Efficacy Outcomes and Hemorrhagic Events Within 45 Days of Drug Initiation Among Those Who Initiated Trial Therapy Stratified by Assigned Treatment
Protocol Development Committee
University of Pittsburgh
University of Illinois, Chicago
Brigham and Women’s Hospital Operations Team
Brigham and Women’s Hospital Emergency Call/Unblinding Team
SOCAR
Clinical Endpoints Committee Members
DSMB
ACTIVE-4B Investigators (City, State) Enrolling at Least One Participant
NIH/NHLBI
CONNECTS Steering and Executive Committees:
CVS
Covance
Coram
Quest/ExamOne
Parexel
RTI
Clinical Endpoints Committee Charter
DSMB Termination Memo
Nonauthor Collaborators
Data Sharing Statement