

Abstract
Microdialysis coupled to an analytical system can be used to continuously monitor unbound protein analytes in any biological fluid, tissue, or organ of animals. To date, no application of microdialysis has been performed to simultaneously monitor unbound morphine and its metabolites in the placenta and fetus of pregnant rats. Our hypothesis is that morphine and its metabolite penetrate the blood–placental barrier to reach the fetus during pregnancy. To investigate this hypothesis, this study aimed to develop a microdialysis experimental animal model coupled with an analytical system to monitor morphine and morphine-3-glucuronide (M3G) in the maternal blood, placenta, fetus, and amniotic fluid of pregnant rats. To determine the analytes in dialysates, a validated ultra-high-performance liquid chromatography–tandem mass spectrometry (UHPLC-MS/MS) method was developed. The pharmacokinetic results indicated that morphine fit well to a two-compartment model and exhibited nonlinear pharmacokinetic behavior within the dosage regimen. The M3G-to-morphine metabolite ratio, determined by the area under the concentration curve (AUC) ratio (AUCM3G/AUCmorphine), was approximately 5.40 in the maternal blood. In terms of tissue distribution, the mother-to-fetus transfer ratio (AUCfetus/AUCblood) of morphine and M3G was about 0.34 and 0.18, respectively. In conclusion, the high metabolite ratio suggests that morphine has the characteristics of rapid biotransformation, and the mother-to-fetus transfer ratio indicates that morphine and M3G partially transfer the blood–placental barrier in pregnant rats. This newly developed multiple microdialysis coupled to UHPLC-MS/MS system can be applied to the studies of maternal pharmacokinetics and blood–placental transfer in pregnant rats.
Keywords: morphine, microdialysis, blood−placental barrier, transplacental transfer, pharmacokinetics
Morphine is a strong opioid analgesic that is widely used to relieve acute or chronic pain, such as that caused by myocardial infarction,1 labor,2 cancer,3 and surgery.4 In humans, morphine undergoes glucuronidation in the liver by UDP-glucuronosyltransferase-2B7 (UGT2B7).5 The predominant metabolites are morphine-3-glucuronide (M3G) and morphine-6-glucuronide (M6G). Among the metabolites, M3G is the most abundant and, compared with the others, has fewer analgesic actions,6 likely attributed to its poor affinity to opioid receptors.7 Additionally, M3G is partly related to tolerance and hyperalgesia induced by opioids.8 By contrast, M6G has an obvious analgesic effect that is more powerful than morphine.9 However, because of the species differences in morphine glucuronidase, morphine is metabolized to M3G via UGT2B1 in rats without forming M6G.10 Due to the concern of chemical analysis and morphine metabolism in various species, the method validation for the analytes of morphine, M3G, and M6G was proposed in this study.
Despite the effective clinical use of morphine, morphine abuse is increasing worldwide because of its characteristics of tolerance, dependence, and addiction. The harmful side effects of morphine and its metabolites may cause serious and irreversible consequences to drug users, particularly pregnant women. The reason is that when morphine is used in pregnancy for either analgesia during labor or by an addicted person, it influences not only the mother11 but also the fetus directly or indirectly.12 Various animal studies have been used to explore the severe adverse effects of morphine on offspring and placental developmental processes during pregnancy. For example, morphine-exposed fetuses not only show decreased birth weight but also morphine withdrawal syndrome after birth in guinea pigs.13 Others have demonstrated that prenatal morphine administration decreases fetal weight and affects neural tube development14 and the cerebrum15 in rats. Additionally, the prenatal use of morphine leads to defects in placenta formation, which subsequently induces abnormalities in the growth of the embryo.16
In conventional plasma or tissue sampling methods for the pharmacokinetics of morphine in mothers and fetuses, blood sampling and tissue and organ homogenization methods are often used to determine the drug concentration. One of the studies showed that after administration of morphine, the concentration in fetal and placental tissues was relatively high compared to maternal plasma in rats.17 However, such conventional biological sampling methods collect the overall drug concentration including protein-bound and protein-unbound forms in the sampling site. Moreover, this requires the use of more animals for experiments. In contrast, the present study uses the microdialysis technique for in vivo sampling, which measures protein-unbound drugs or endogenous substances in the extracellular fluid of almost all tissues in the body.18 Compared with commonly used sampling methods, the microdialysis does not require sample cleanup processing before analysis. Furthermore, sampling by microdialysis can continuously monitor drug concentrations in multiple tissue fluids for a long time without excessive tissue irritation or biological fluid loss from the body.19 This technique has been currently applied to various animal experiments, such as blood sampling20 and brain sampling.21 Because of these advantages, the microdialysis has made a substantial contribution to the study of drug metabolism and pharmacokinetics.
Based on the search above, we hypothesized that morphine and M3G penetrate the blood–placental barrier to reach the fetus during pregnancy. However, the PubMed database contains less relevant research on the simultaneous application of microdialysis and morphine to the placenta and fetus of pregnant rats. To investigate this hypothesis, this study aimed to develop a novel multiple microdialysis experimental animal model. Our animal model is the first to simultaneously monitor the concentrations of morphine and M3G in the maternal blood, placenta, fetus, and amniotic fluid of pregnant rats using a microdialysis system. To determine the biotransformation and tissue distribution of morphine and M3G in several tissue fluids, a validated ultra-high-performance liquid chromatography–tandem mass spectrometry (UHPLC-MS/MS) system was developed. To elucidate the pharmacokinetics of morphine, a dose-dependent regimen of morphine (10 and 30 mg/kg, i.v.) was applied to experimental pregnant rats.
Methods
Chemicals and Reagents
Morphine hydrochloride injection was obtained from the Taiwan Food and Drug Administration (Ministry of Health and Welfare, Taiwan). M3G, M6G, urethane, heparin sodium, and formic acid were obtained from Sigma-Aldrich Chemicals (St. Louis, MO, USA). Dextrose, sodium citrate, citric acid, and dimethyl sulfoxide (DMSO) were purchased from E. Merck (Darmstadt, Germany). Methanol was obtained from Macron (Hamilton, PA, USA). Acetonitrile and ammonium hydroxide (NH4OH) were purchased from J.T. Baker (Phillipsburg, NJ, USA). Triple deionized water (Millipore, Bedford, MA, USA) was used for UHPLC-MS/MS analysis.
UHPLC-MS/MS Conditions
The UHPLC-MS/MS system comprised an LC system (Shimadzu LC-20AD) and a triple-quadrupole mass spectrometer (LCMS-8030 system; Shimadzu, Kyoto, Japan) fitted with an electrospray ionization interface. A Purospher STAR RP-18 end-capped column (100 mm × 2.1 mm; 2 μm; Merck KGaA, Darmstadt, Germany), which was maintained at 40 °C, was used for chromatographic separation. Isocratic elution was performed using a mobile phase composition of methanol/acetonitrile (1:4, v/v)–0.1% aqueous formic acid (adjusted to pH 4.0 using NH4OH) (5:95, v/v) with a flow rate of 0.3 mL/min. The compounds were measured by multiple reaction monitoring (MRM) analysis operating in the positive ionization mode with an injection volume of 5 μL. The following apparatus conditions were used in the analysis process: interface voltage, 3.5 kV; nebulizing gas flow, 3.0 L/min; drying gas flow, 15.0 L/min; desolvation line temperature, 250 °C; heat temperature, 400 °C; collision-induced dissociation, 230 kPa.
Method Validation
Based on the current US Food and Drug Administration (FDA) bioanalytical method validation guidelines, method validation tests for morphine, M3G, and M6G quantification were executed and included calibration curves, precision, accuracy, and stability.22 Standard stock solutions of morphine, M3G, and M6G were prepared in methanol at concentrations of 10, 10, and 50 μg/mL. Before the analysis procedure, the solutions were frozen at −20 °C.
The calibration curves were generated by spiking blank rat maternal blood, placenta, amniotic fluid, and fetal dialysates with stock solutions of morphine and M3G at a final concentration range of 1–500 ng/mL and M6G at a range of 5–1000 ng/mL. All the calibration curves had a correlation coefficient (r2) of at least 0.995.
The precision and accuracy were estimated by analyzing five replicates at the lower limit of quantification (LLOQ): low, medium, and high concentrations on the same day (intraday); and concentrations over five consecutive days (interday). The accuracy (% bias) was defined as the difference between the nominal concentration (Cnom) and observed concentration (Cobs) and was calculated as follows: accuracy (% bias) = [(Cobs – Cnom)/Cnom] × 100%. The precision, calculated as the relative standard deviation (RSD), referred to the average difference between individual measurements and was obtained using the following formula (% RSD) = [standard deviation (SD)/Cobs] × 100%. The acceptable criteria for both precision and accuracy were within ±15%, except that the LLOQ was within ±20%.
The stability of morphine, M3G, and M6G at low and high concentrations was determined under four conditions: autosampler storage, short-term storage, long-term storage, and three freeze–thaw cycles. The autosampler stability was determined by storing the samples in an autosampler for 4 h at 10 °C. Short-term stability was assessed by placing the samples at room temperature for 4 h, while long-term stability was assessed by storing samples at −20 °C for 4 weeks. For the freeze–thaw cycle stability assay, the samples were frozen at −20 °C for 12 h and then thawed at room temperature for 12 h, repeating the cycle three times. The stability (%) was assessed by comparing the concentration of processed samples with that of freshly prepared samples, which was the following equation: stability (%) = (Cobs/Cnom) × 100%; and the value at each level was required to be within ±15%.
In vitro recovery was evaluated by measuring the increase in compound in the perfusate after passing through the microdialysis probe. The probe, perfused with anticoagulant citrate dextrose solution (ACD solution) without drug, was immersed into a sample solution containing different predetermined drug concentrations (Cs) of morphine (100, 500, and 1000 ng/mL) and M3G (100, 500, and 1000 ng/mL). The concentrations of morphine and M3G in the dialysates (Cout) were analyzed by UHPLC-MS/MS. The in vitro recovery by dialysis (Rdial) was calculated based on the formula Rdial (%) = (Cout/Cs) × 100%.
Experimental Animals
Female Sprague–Dawley rats (320 ± 20 g), on the 16th day of gestation, were obtained from the Laboratory Animal Center at National Yang Ming Chiao Tung University (Taipei, Taiwan). All the animal experimental procedures were approved by the Institutional Animal Care and Use Committee of National Yang Ming Chiao Tung University (IACUC no. 1090111) and were conducted under the guidance of the National Research Council. The animals were housed in a 12 h light/dark cycle-controlled quarter, and they were free to have food (Laboratory rodent diet 5001, PMI Feeds, Richmond, IN, USA) and water.
Microdialysis Experiments
The microdialysis equipment included a syringe pump (CMA 400; Solna, Sweden), a microfraction collector (CMA 142), and a microdialysis probe. The probe was made in the laboratory19 and comprised a concentric-shaped silica capillary covered with an 11 or 6 mm dialysis membrane at the tip for blood and transplacental dialysis sampling, respectively.
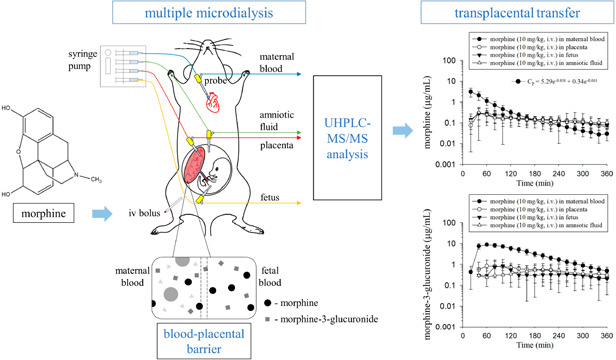
The pregnant rats were anesthetized with urethane (1 g/kg, i.p.) throughout the experiment. A blood microdialysis probe was inserted into the right jugular vein toward the heart for maternal blood dialysis sampling, according to our previous study.19 Multiple microdialysis probes were catheterized in the placenta, fetus, and amniotic fluid for transplacental dialysis sampling. The relative positions of the placenta, fetus, and amniotic fluid in the embryo were mainly based on two studies.23,24 The placenta was located on one side of the embryo and appeared dark red, while the fetus was on the other side and had a slight rat fetal shape. The amniotic fluid was transparent fluid around the fetus. When the probes were implanted into the placenta and fetus, a small amount of blood flowed out. Additionally, moving the probe in the fetus slightly moved the fetus in the direction of the probe. When the probe was implanted into the amniotic fluid, clear liquid leaked out. Polyethylene tubing (PE50) was catheterized in the left femoral vein for morphine administration. The animal model of blood and transplacental dialysis sampling is shown in Figure 1. After the surgical procedure, the probes were perfused with ACD solution, containing 13.6 mM dextrose, 7.5 mM sodium citrate, and 3.5 mM citric acid, by a syringe pump with a flow rate of 2 μL/min. Following stabilization of the dialysate levels for 1 h, morphine (dissolved in 5% DMSO and normal saline) was administered by intravenous injection through the femoral vein cannula at doses of 10 and 30 mg/kg (n = 6 for each group). Aliquots of 40 μL of blood, placenta, amniotic fluid, and fetal dialysates were collected every 20 min for a total of 6 h, and 5 μL of dialysates were analyzed by UHPLC-MS/MS without sample preparation. Before the analysis procedure, the dialysates were frozen at −20 °C.
Figure 1.
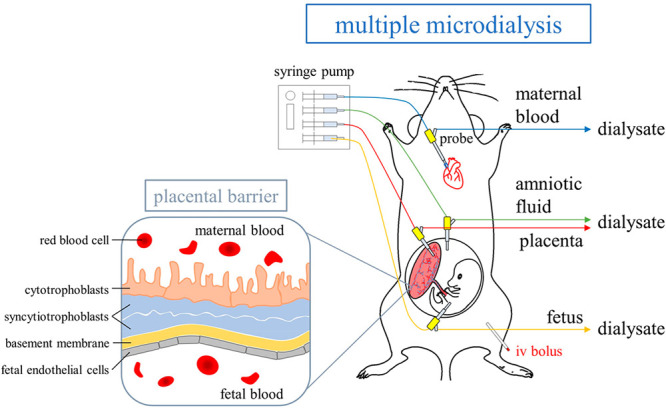
Diagram of the animal model for blood and transplacental microdialysis sampling.
Pharmacokinetic Analysis and Statistics
WinNonlin Standard Edition (ver. 5.3; Pharsight Corp., Mountain View, CA, USA) was adopted to calculate the pharmacokinetic parameters. The Akaike information criterion (AIC) value was used to determine the degree of agreement between the pharmacokinetic data and different compartment models; thus, the appropriate model could be selected for proper data presentation.25 The equation is AIC = N ln Re + 2p, where N is the number of experimental data, p is the number of parameters in an estimated model, and Re is the residual sum of squares. When the AIC value of the model is smaller, it means that the data fit the model better, which is the more suitable pharmacokinetic model. Based on the AIC value, a two-compartment model was selected to determine the pharmacokinetic parameters of morphine in maternal blood using the following equation: CP = A e–αt + B e–βt, where CP is the drug concentration at time t, A and B are zero-time plasma concentration intercepts of the biphasic disposition curve, e is the base of the natural logarithm, and α and β are rate constants related to distribution and elimination phases, respectively. Another way to analyze pharmacokinetic data is to use the noncompartment model. This simple and fast model evaluates the degree of exposure of a drug because it requires fewer assumptions about body compartments. It could easily determine and calculate the main pharmacokinetic parameters directly from the experimental data of a concentration versus time plot, such as the maximum concentration (Cmax), time of maximum concentration (Tmax), area under the concentration–time curve (AUC), clearance (CL), elimination half-life (t1/2), and mean residence time (MRT). Here, the mean residence time (MRT) is the average time a molecule stays in the body. When the drug enters the body, it will be distributed to various tissues and organs, but the residence time in different parts is not the same. Therefore, MRT is used to describe the average time that all drug molecules stay in the body, which is calculated by the equation MRT = AUMC/AUC, where AUMC is the area under the first moment curve. A noncompartment model was used to describe the pharmacokinetic parameters of the blood–placental transfer of morphine and M3G in this study. The concentration–time curves were processed using SigmaPlot (ver. 10.0; Systat Software, London, UK). The statistics were calculated by SPSS Statistics (ver. 22.0; IBM Corp., Armonk, NY, USA). The data between blood, placenta, fetus, and amniotic fluid were compared by one-way ANOVA followed by posthoc Tukey test and the data between the two dose groups at the same position were compared by the Mann–Whitney U test, with a p value <0.05 regarded as statistically significant. All data are expressed as the mean ± standard deviation (SD).
Results
Optimization of UHPLC-MS/MS Conditions
A UHPLC-MS/MS method to detect morphine, M3G, and M6G in dialysates was established. Regarding the analytical conditions, the measurements were conducted in the positive ionization mode, which provided higher sensitivity than the negative ionization mode. After optimization and modification, the monitored ion transition was m/z 286.2 to 165.0, and the collision energy was −44 V for morphine, while the monitored ion transition was m/z 462.1 to 286.1, and the collision energy was −30 V for M3G and M6G. The mass spectra are illustrated in Figure 2.
Figure 2.

Representative product ion mass spectra of (A) morphine, (B) M3G, and (C) M6G.
To optimize the separation of analytes, an RP-18 end-capped column coupled to the mobile phase of methanol/acetonitrile (1:4, v/v)-0.1% aqueous formic acid adjusted to pH 4.0 (5:95, v/v) was developed. When using only one organic solvent, acetonitrile produced a higher intensity than methanol. Additionally, the retention time of each compound was shorter in the mobile phase of acetonitrile and 0.1% formic acid; however, peak tailing of M3G was observed. After adding methanol to acetonitrile, the peak became symmetric. Additionally, by using NH4OH to adjust the pH value of aqueous formic acid to 4.0, improved peak shapes for the three analytes were obtained. Under these optimized chromatographic conditions, the retention times of M3G, M6G, and morphine were 1.73, 3.51, and 5.04 min, respectively, revealing good sensitivity, selectivity, symmetry, and peak intensity. Representative chromatograms of the maternal blood, placenta, fetus, and amniotic fluid dialysates are shown in Supporting Information Figure S1 and S2.
Analytical Method Validation
All the analytes within the calibration curve range presented good linearity, and the correlation coefficients (r2) were greater than 0.995. The lower limit of quantification (LLOQ) and limit of detection (LOD) were defined as the analyte concentrations detected in a signal-to-noise ratio (S/N ratio) of 10 and 3, respectively. The LLOQ and LOD of morphine and M3G were 1 and 0.5 ng/mL, and those of M6G were 5 and 1 ng/mL, respectively, in all the biological dialysates.
The precision (% RSD) and accuracy (% bias) were estimated by intra- and interday analyses. To conduct the assays, blank maternal blood, placenta, fetus, or amniotic fluid dialysates were spiked with varying concentrations of morphine, M3G, and M6G. The LLOQ and low, medium, and high concentrations of morphine and M3G were 1, 5, 100, and 500 ng/mL, and those of M6G were 5, 10, 500, and 1000 ng/mL, respectively. The data are presented in Supporting Information Tables S1–S3, and the RSD and bias values were all within acceptable limits of ±15% (±20% for LLOQ). The intra- and interday precision and accuracy values of the three compounds at different concentrations were within the scope of the guidelines, revealing that the experimental method was considered reproducible and reliable.
The stabilities of the compounds were examined at low and high concentrations after exposure to various conditions. The low and high concentrations of morphine and M3G were 5 and 500 ng/mL, and those of M6G were 10 and 1000 ng/mL, respectively. The data are summarized in Supporting Information Tables S4–S6. For M3G, no significant degradation was observed under the four different conditions. For morphine and M6G, no significant degradation was found in the autosampler and freeze–thaw cycle conditions, but degradation occurred in both short- and long-term conditions. Storage at room temperature for a short period or at −20 °C for a long time should be avoided when preparing samples of morphine and M6G.
Morphine and M3G at three different concentrations were used to assess the in vitro recovery of the blood and placental probes. The values are presented in Supporting Information Table S7. The mean in vitro recovery percentages of morphine and M3G were 25.36 ± 0.48% and 7.16 ± 0.25%, respectively, with the blood probe and 14.31 ± 0.50% and 4.07 ± 0.17%, respectively, with the placental probe. No significant differences were found in the recovery percentages of the blood and placental probes at the three investigated concentrations for each compound. The results of the in vitro recovery experiments demonstrated that the homemade blood and placental probes were stable and that the recovery was independent of the analyte concentration.
Blood Pharmacokinetics of Morphine and M3G
Based on the above validated UHPLC-MS/MS system, all the biological dialysate samples collected from the maternal blood, placenta, amniotic fluids, and fetus of the pregnant rats were detected (Supporting Information Figures S1 and S2). The pharmacokinetics and transplacental transfers of morphine and M3G were assessed following two doses of morphine (10 and 30 mg/kg, i.v.).
The concentration–time profiles of morphine and M3G in the rat maternal blood, placenta, fetus, and amniotic fluid dialysates are illustrated in Figures 3 and 4. Following the administration of morphine, it quickly distributed to the placenta, amniotic fluid, and fetus. The concentration of morphine in maternal blood was the highest among the four sampling tissues at first and decreased gradually. After 180 min, the morphine concentration in maternal blood became lower than that in the placenta, fetus, and amniotic fluid, and this trend remained unchanged within 360 min (Figure 3). On the other hand, after the administration of morphine, M3G appeared within 20 min in maternal blood, demonstrating a rapid conversion from morphine to M3G after dosing. M3G was also found in the placenta, amniotic fluid, and fetus at the second sampling point (40 min). Additionally, compared with those of morphine, the blood, placenta, amniotic fluid, and fetal concentrations of M3G were all higher. Furthermore, similar to the trend of the morphine concentration, the M3G concentration in maternal blood was the highest and declined gradually, while the M3G concentration in the placenta, fetus, and amniotic fluid increased steadily and then declined slowly during the sampling time (Figure 4).
Figure 3.

Concentration–time curves of unbound morphine in the rat maternal blood, placenta, fetus, and amniotic fluid after morphine administration at doses of (A) 10 mg/kg, i.v. and (B) 30 mg/kg, i.v. The data are expressed as the means ± SD (n = 6).
Figure 4.

Concentration–time curves of unbound M3G in the rat maternal blood, placenta, fetus, and amniotic fluid after morphine administration at doses of (A) 10 mg/kg, i.v. and (B) 30 mg/kg, i.v. The data are expressed as the means ± SD (n = 6).
Based on the concentrations at several sampling time points, the pharmacokinetic parameters are presented in Tables 1 and 2. Before the analysis, the one- or two-compartment pharmacokinetic model was compared to select the fittest model. The comparison was performed according to the Akaike information criterion (AIC) values. Smaller AIC model values correlate with more appropriate pharmacokinetic parameters being presented, thus best representing the concentration–time curve.25 The mean AIC values of the one-compartment model at two doses were 205.4 and 239.8, respectively, and those of the two-compartment model were 195.6 and 223.7, respectively. Therefore, a two-compartment model was more adequate than a one-compartment model to describe the pharmacokinetics of morphine in maternal blood dialysate, and the equations were CP = 5.29 e–0.03t + 0.34 e–0.01t and CP = 27.58 e–0.03t + 5.49 e–0.01t for the dosage regimens of morphine at 10 and 30 mg/kg, i.v., respectively.
Table 1. Pharmacokinetic Parameters of Morphine in the Rat Maternal Blood, Placenta, Fetus, and Amniotic Fluid after Morphine Administration (10 and 30 mg/kg, i.v.)a.
| 10 mg/kg, i.v. |
30 mg/kg,
i.v. |
|||||||
|---|---|---|---|---|---|---|---|---|
| parameter | maternal blood | placenta | fetus | amniotic fluid | maternal blood | placenta | fetus | amniotic fluid |
| AIC of two-compartment | 195.6 ± 26.85 | 223.7 ± 31.72 | ||||||
| Model | Two-compartment | Noncompartment | Two-compartment | Noncompartment | ||||
| CP = 5.29 e–0.03t + 0.34 e–0.01t | CP = 27.58 e–0.03t + 5.49 e–0.01t | |||||||
| AUC (min μg/mL) | 239.4 ± 93.08 | 62.50 ± 32.99b | 81.73 ± 17.78b | 88.02 ± 43.88b | 1231 ± 171.5c | 341.2 ± 275.6bc | 229.6 ± 103.8bc | 260.1 ± 146.6b |
| Cmax (μg/mL) | 5.63 ± 2.74 | 0.38 ± 0.25b | 0.34 ± 0.24b | 0.36 ± 0.12b | 33.07 ± 12.91c | 0.71 ± 0.99b | 0.87 ± 0.90b | 1.17 ± 0.81bc |
| t1/2, α (min) | 25 ± 9 | - | - | - | 26 ± 13 | - | - | - |
| t1/2, β (min) | 505 ± 638 | - | - | - | 278 ± 311 | - | - | - |
| Tmax(min) | - | 93 ± 94 | 87 ± 50 | 40 ± 13 | 160 ± 85 | 177 ± 69c | 97 ± 39c | |
| t1/2(min) | - | 115 ± 90 | 271 ± 299 | 213 ± 134 | - | 661 ± 1052 | 269 ± 274 | 168 ± 141 |
| CL (mL/min/kg) | 46.30 ± 14.65 | 191.7 ± 77.83b | 127.5 ± 28.89 | 138.6 ± 64.54b | 24.74 ± 3.22c | 163.7 ± 129.9 | 156.1 ± 71.65 | 193.9 ± 184.8 |
| MRT (min) | 134 ± 112 | 224 ± 122 | 433 ± 443 | 334 ± 208 | 86 ± 64 | 1038 ± 1559 | 477 ± 410 | 292 ± 201 |
| AUCtissue/AUCblood | - | 0.26 ± 0.14 | 0.34 ± 0.07 | 0.37 ± 0.18 | - | 0.28 ± 0.22 | 0.19 ± 0.08c | 0.21 ± 0.12 |
Data are expressed as means ± SD (n = 6). AUCtissue/AUCblood represents the maternal blood-to-tissue transfer ratio.
p < 0.05 compared with maternal blood within group by ANOVA with posthoc Tukey HSD test.
p < 0.05 compared with the same site in the morphine (10 mg/kg, i.v.) group by Mann–Whitney U test.
Table 2. Pharmacokinetic Parameters of M3G in the Rat Maternal Blood, Placenta, Fetus, and Amniotic Fluid after Morphine Administration (10 and 30 mg/kg, i.v.)a.
| 10 mg/kg, i.v. |
30 mg/kg,
i.v. |
|||||||
|---|---|---|---|---|---|---|---|---|
| parameter | maternal blood | placenta | fetus | amniotic fluid | maternal blood | placenta | fetus | amniotic fluid |
| model | noncompartment | noncompartment | ||||||
| AUC (min μg/mL) | 1292 ± 191.2 | 230.1 ± 59.79b | 236.3 ± 137.7b | 211.8 ± 96.51b | 6996 ± 1963d | 923.9 ± 547.6bd | 533.1 ± 150.1bd | 662.5 ± 564.5b |
| Cmax(μg/mL) | 9.30 ± 1.31 | 1.15 ± 0.56b | 1.12 ± 1.23b | 0.69 ± 0.49b | 39.38 ± 11.17d | 2.84 ± 1.82b | 1.57 ± 1.03b | 1.68 ± 1.57b |
| Tmax(min) | 63 ± 15 | 93 ± 39 | 147 ± 83 | 197 ± 77bc | 83 ± 23 | 197 ± 74bd | 220 ± 59b | 190 ± 86b |
| t1/2(min) | 62 ± 11 | 134 ± 98 | 226 ± 176 | 202 ± 169 | 91 ± 40 | 184 ± 86 | 202 ± 83 | 177 ± 103 |
| MRT (min) | 135 ± 15 | 236 ± 95 | 366 ± 254 | 397 ± 207 | 185 ± 62 | 383 ± 139b | 428 ± 111b | 384 ± 155b |
| AUCtissue/AUCblood | - | 0.18 ± 0.05 | 0.18 ± 0.11 | 0.16 ± 0.07 | - | 0.13 ± 0.08 | 0.08 ± 0.02 | 0.09 ± 0.08 |
| AUCM3G/AUCmorphine | 5.40 ± 0.80 | 3.68 ± 0.96 | 2.89 ± 1.69b | 2.41 ± 1.02b | 5.68 ± 1.59 | 2.71 ± 1.60b | 2.32 ± 0.65b | 2.55 ± 2.17b |
Data are expressed as means ± SD (n = 6). AUCtissue/AUCblood represents the maternal blood-to-tissue transfer ratio.
p < 0.05 compared with maternal blood within group by ANOVA with posthoc Tukey HSD test.
p < 0.05 compared with placenta within group by ANOVA with posthoc Tukey HSD test.
p < 0.05 compared with the same site in the morphine (10 mg/kg, i.v.) group by Mann–Whitney U test.
The morphine concentration in maternal blood reached maximum concentrations (Cmax) of 5.63 ± 2.74 and 33.0 ± 12.9 μg/mL in the two dose groups, respectively, and the ratio was 1:6. The area under the curve (AUC) of morphine in blood at a dose of 30 mg/kg was five times higher than that of 10 mg/kg, with values of 1231 ± 171 and 239 ± 93.1 min μg/mL, respectively. The clearance (CL) decreased from 46.3 ± 14.7 mL/min/kg in the low-dose group to 24.7 ± 3.22 mL/min/kg in the high-dose group. Significant differences were found in the Cmax, AUC, and CL of morphine in blood between the two dose groups. Furthermore, the Cmax and AUC values were not proportional to the increasing doses from 10 to 30 mg/kg, reflecting a nonlinear pharmacokinetic relationship for the morphine concentration within the dosage regimen (Table 1).
An extremely rapid metabolism of morphine to M3G proceeded within 20 min after dosing. The blood concentration of M3G then exceeded that of morphine after 40 min and remained higher than that of morphine at the sampling time point, suggesting an easy biotransformation of morphine to M3G at the maternal site. The Cmax for M3G was 9.30 ± 1.31 μg/mL in maternal blood in the low-dose group, and the time required to reach Cmax (Tmax) was approximately 1 h. At a dosage of 30 mg/kg, the Cmax for M3G was 39.4 ± 11.2 μg/mL at a Tmax of approximately 1 h in blood. The AUC of M3G in blood was 6996 ± 1963 min μg/mL at 30 mg/kg, which was 5-fold higher than that at 10 mg/kg (1292 ± 191 min μg/mL). The CL was 7.89 ± 1.23 and 4.58 ± 1.29 mL/min/kg in the low-dose and high-dose groups, respectively. Similar to morphine in blood, the Cmax and AUC of M3G were all significantly different between the two dose groups. The ratio of AUCM3G/AUCmorphine (defined as the metabolite-to-parent drug ratio) in maternal blood was approximately 5.5. The relationship between morphine and M3G remained similar to increasing doses from 10 to 30 mg/kg (Table 2).
Blood–Placental Barrier Transfers of Morphine and M3G
A noncompartment model was used to calculate the pharmacokinetic parameters of morphine in the placenta, amniotic fluid, and fetal dialysates and M3G in all the dialysates. The pharmacokinetic parameters are presented in Tables 1 and 2. With morphine at 10 mg/kg, the Cmax value for morphine in the placenta, fetus, and amniotic fluid was approximately 0.36 μg/mL, and the AUC was approximately 75 min μg/mL, values that were lower than those in the blood. Additionally, compared with that of morphine in the maternal blood, the mean residence time (MRT) values were increased in the placenta, fetus, and amniotic fluid, indicating that morphine lasted longer in these three distributed tissues than in maternal blood. When the dose increased to 30 mg/kg, the Cmax values became nearly 0.90 μg/mL in the placenta, fetus, and amniotic fluid, and the AUCs were higher than those in the low-dose group. The MRT in the placenta, amniotic fluid, and fetus was also higher than that in the blood in the high-dose group. Additionally, the Tmax of morphine increased with the dose.
Regarding M3G, the concentrations in the placenta, fetus, and amniotic fluid all exceeded those of morphine, with Cmax values of 0.7–1.2 μg/mL in the low-dose group and 1.6–2.8 μg/mL in the high-dose group. Additionally, the AUCs in the placenta, fetus, and amniotic fluid were significantly lower than those in the blood. The time to reach the maximum concentration (Tmax), elimination half-life (t1/2), and MRT of M3G were all longer in the placenta, amniotic fluid, and fetus than in the blood at both doses, demonstrating a longer persistence of M3G in these three distributed tissues than in maternal blood.
The mother-to-fetus transfer ratio, AUCfetus/AUCblood, of morphine was 0.34 ± 0.07 and 0.19 ± 0.08 at two doses, while the ratios for M3G were 0.18 ± 0.11 and 0.08 ± 0.02, respectively. The results revealed that morphine and M3G could pass through the placenta to the fetus (Tables 1 and 2).
Discussion
In the present study, a microdialysis system was developed and combined with a validated UHPLC-MS/MS method to simultaneously monitor morphine and M3G at multiple sites in pregnant rats. In vitro recovery was used to estimate the drug concentration in the body. Some studies have pointed out no significant difference in the recoveries obtained by the two assays in vivo and in vitro for some compounds.26,27 Additionally, in a microdialysis study of morphine, no significant difference was found in the recovery measured in vivo and in vitro.28 Furthermore, the in vitro recoveries of morphine and M3G in our study were quite low. We expected that the deviation between in vivo and in vitro recovery would not be too great. Based on the above points, we used in vitro recovery to evaluate the recovery of the probe and anticipated that the in vitro recovery of morphine and metabolites could be used to assess the in vivo drug concentration.
Since M3G is a metabolite of morphine, and considering the limited distribution by the barrier, M3G presented a biphasic concentration, which slowly appeared in the placenta, fetus, and amniotic fluid in the initial stage of administration. In addition, due to poor clearance in embryos,29 the concentration of M3G decreased at a very slow rate during the sampling time. When detecting the concentration of morphine and M3G in pregnant rats, the relationship between morphine and M3G remained similar to increasing doses (Table 2), a finding that also matched Säwe’s study findings.30 The reason may be that the metabolic pathway of morphine was not subject to saturation or autoinduction even after increasing doses in this dose range, and it may have a high glucuronidation capacity in rats.
The mother-to-fetus transfer ratios suggested that morphine and M3G partially penetrated the blood–placental barrier and reached the fetus (Tables 1 and 2). However, concerning the blood–placental barrier between the mother and fetus, higher concentrations of morphine and M3G are found in maternal blood than in fetal blood, indicating an incomplete transfer.31 The main maternal–fetal exchange of drugs occurs in an uninterrupted syncytiotrophoblast layer of the placenta. These cells mediate the transport role of the barrier. Four potential mechanisms of drug transfer can occur across the placenta: passive diffusion, active transport, facilitated diffusion, and pinocytosis.32 The first two mechanisms represent the primary method that drugs pass through the placenta; thus, they have received more research attention. According to previous studies, most drugs penetrate the placental barrier through passive diffusion with concentration gradients.33 Various factors, such as the pharmacological characteristics of the compound and physical characteristics of the barrier, contribute to different rates and extents of diffusion of compound into the placenta. In general, molecules with a low molecular weight, a low degree of ionization, poor protein binding, and high lipophilicity readily spread through the placenta.34 Among opioids, morphine is relatively insoluble in lipids (pKa = 7.9). However, because of its low molecular weight (285 Da) and low extent of protein binding (35%), morphine crosses the placenta with relative ease. Additionally, a decrease in plasma protein-bound drugs in the gestation period, resulting from the reduction in albumin,35 could lead to higher concentrations of the free-form drug and facilitate more distribution to tissues. Furthermore, the transfer of morphine is affected by the difference in pH between maternal and fetal circulations due to its weak base property. When transferred to a relatively low pH environment, morphine becomes ionized in fetal circulation and cannot return to the maternal circulation, leading to the accumulation of drugs in the fetus, termed ion trapping.34 In our study, the slowly decreased concentration and long MRT of morphine in the fetus might have been affected by this phenomenon.
Although simple diffusion is a main pathway for the transplacental transfer of drugs, several active transporters in the placenta are involved in drug translocation, including the ATP-binding cassette transporters (ABC transporters) of P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and multidrug resistance proteins (MRPs).36 These efflux transporters are abundantly present in the syncytiotrophoblast cells of the placenta and fetus during gestation.37 Several studies have investigated morphine as well as M3G and active transporters. Morphine is considered the substrate of P-gp, and various morphine blood-brain barrier studies have been conducted in vitro(38) and in vivo.39 However, morphine is not a substrate of mouse mrp3.40 Less is known about the association between morphine and BCRP. In terms of morphine metabolite, mrp2 and mrp3, a family that effluxes glucuronides,36 have affinity toward glucuronides in mice,40,41 and BCRP participates in the extrusion of other glucuronides.42 However, no evidence to date supports whether active transport participates in the transport of morphine and M3G across the placenta.
Additionally, our data revealed that the transfer ratios of M3G from the mother to the placenta, fetus, and amniotic fluid were relatively lower than those of morphine (Tables 1 and 2). We speculate that the discrepancy in the transfer ratios of morphine and M3G is attributed to the different lipid solubilities of the two compounds. As mentioned previously, highly lipophilic molecules have a greater ability to pass through the placental barrier. Because M3G is a highly polar metabolite of morphine, it has difficulty crossing lipid membranes. Additionally, another reason may account for the low maternal–fetal transfer ratio of M3G—the low activity of glucuronidation in the placenta and fetus. In rats, the mRNA expression level of UGT2B1, which is the main enzyme catalyzing glucuronidation of morphine in rats,10 in fetal tissues is much lower than that in maternal liver.43 In humans, the liver microsomes of fetuses have a low content of UGT2B7 transcripts,44 leading to a decrease in the glucuronidation activity of the fetus. However, whether most of the M3G in the fetus is attributed to its passage from the mother or formation in the fetus remains unclear.
For decades, the maternal and fetal pharmacokinetics of various drugs have been investigated. Those studies used traditional biological sampling methods to conduct animal experiments. In our study, instead of conventional sampling methods, we used microdialysis as a sampling technique in animal experiments to monitor free-form drugs in the extracellular fluid of tissues in the body. Traditional sampling methods have difficulty distinguishing the unbound portion of a drug; however, this challenge can be overcome by microdialysis.18 Additionally, minimal loss of biological fluid, minimal invasion, and long-term continuous sampling are also strengths of microdialysis.19 Furthermore, microdialysis can measure the drug concentration in multiple sites of a rat at a time and simultaneously discuss the pharmacokinetics of the drug in only one animal, providing the use of a relatively small number of animals. Considering the above advantages, the application of microdialysis makes it ideal to study the transplacental transfer and pharmacokinetics of drugs in pregnant rats. However, the limitation of the microdialysis technique is that the recovery of the compound should be considered when using it. If the drug does not easily penetrate the membrane of the probe, the recovery will be too low to detect the analyte. Therefore, a highly sensitive analytical instrument is required for accurate analysis, such as UHPLC-MS/MS.
In conclusion, a novel transplacental microdialysis animal model was successfully developed for the simultaneous collection of dialysates from multiple sites. This is the first report on the application of microdialysis in morphine, placental sampling, and fetal sampling in pregnant rats. Additionally, a validated UHPLC-MS/MS analytical method was established to analyze compounds in several tissue fluids and was applied to evaluate the pharmacokinetics and transplacental transfers of morphine and M3G. The results revealed that morphine was rapidly metabolized to M3G in pregnant rats. Furthermore, morphine and M3G quickly transferred through the blood–placental barrier and remained in the placenta, amniotic fluid, and fetus. The development of the animal model in present study opens up a new dimension to the simultaneous detection of compounds in the mother and fetus. This model can be further applied to the drug development commonly used in the gestation period, and it will bring great potential for the study of maternal–fetal pharmacokinetics and toxicology.
Acknowledgments
This study was supported in part by research grants from the Ministry of Science and Technology of Taiwan (MOST 110-2918-I-239-001; MOST 110-2113-M-A49A-503; MOST 109-2113-M-010-007) and the graduated student scholarship of the School of Medicine, National Yang Ming Chiao Tung University, Taipei, Taiwan.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsptsci.1c00142.
Analytical method validation of morphine, M3G, and M6G; Chromatograms of blank dialysates, dialysates spiked with compounds, and biological dialysates (PDF)
Author Contributions
I.H. Lin, L. Yang, and T.Y. Hsueh performed the experiments, analyzed the data, and prepared the manuscript. T.H. Tsai designed the experiments, edited the paper, and secured funding.
The authors declare no competing financial interest.
Notes
Data Availability: The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
Supplementary Material
References
- Ibanez B.; James S.; Agewall S.; Antunes M. J.; Bucciarelli-Ducci C.; Bueno H.; Caforio A. L. P.; Crea F.; Goudevenos J. A.; Halvorsen S.; Hindricks G.; Kastrati A.; Lenzen M. J.; Prescott E.; Roffi M.; Valgimigli M.; Varenhorst C.; Vranckx P.; Widimsky P.; et al. 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur. Heart J. 2018, 39 (2), 119–177. 10.1093/eurheartj/ehx393. [DOI] [PubMed] [Google Scholar]
- Sharpe E. E.; Molitor R. J.; Arendt K. W.; Torbenson V. E.; Olsen D. A.; Johnson R. L.; Schroeder D. R.; Jacob A. K.; Niesen A. D.; Sviggum H. P. Intrathecal Morphine versus Intrathecal Hydromorphone for Analgesia after Cesarean Delivery: A Randomized Clinical Trial. Anesthesiology 2020, 132 (6), 1382–1391. 10.1097/ALN.0000000000003283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosek K.; Leppert W.; Nosek H.; Wordliczek J.; Onichimowski D. A comparison of oral controlled-release morphine and oxycodone with transdermal formulations of buprenorphine and fentanyl in the treatment of severe pain in cancer patients. Drug Des., Dev. Ther. 2017, 11, 2409–2419. 10.2147/DDDT.S141007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal A. K.; Mishra S.; Bhatnagar S.; Singh R. Epidural morphine analgesia compared with intravenous morphine for oral cancer surgery with pectoralis major myocutaneous flap reconstruction. Acta Anaesthesiol. Scand. 2006, 50 (2), 234–238. 10.1111/j.1399-6576.2006.00924.x. [DOI] [PubMed] [Google Scholar]
- Coffman B. L.; Rios G. R.; King C. D.; Tephly T. R. Human UGT2B7 catalyzes morphine glucuronidation. Drug Metab. Dispos. 1997, 25 (1), 1–4. [PubMed] [Google Scholar]
- Shimomura K.; Kamata O.; Ueki S.; Ida S.; Oguri K.; Yoshimura H.; Tsukamoto H. Analgesic effect of morphine glucuronides. Tohoku J. Exp. Med. 1971, 105 (1), 45–52. 10.1620/tjem.105.45. [DOI] [PubMed] [Google Scholar]
- Ulens C.; Baker L.; Ratka A.; Waumans D.; Tytgat J. Morphine-6beta-glucuronide and morphine-3-glucuronide, opioid receptor agonists with different potencies. Biochem. Pharmacol. 2001, 62 (9), 1273–1282. 10.1016/S0006-2952(01)00761-4. [DOI] [PubMed] [Google Scholar]
- Blomqvist K. J.; Viisanen H.; Ahlstrom F. H. G.; Jokinen V.; Sidorova Y. A.; Suleymanova I.; Rauhala P. V.; Kalso E. A.; Lilius T. O. Morphine-3-glucuronide causes antinociceptive cross-tolerance to morphine and increases spinal substance P expression. Eur. J. Pharmacol. 2020, 875, 173021. 10.1016/j.ejphar.2020.173021. [DOI] [PubMed] [Google Scholar]
- Klimas R.; Mikus G. Morphine-6-glucuronide is responsible for the analgesic effect after morphine administration: a quantitative review of morphine, morphine-6-glucuronide, and morphine-3-glucuronide. Br. J. Anaesth. 2014, 113 (6), 935–944. 10.1093/bja/aeu186. [DOI] [PubMed] [Google Scholar]
- King C. D.; Rios G. R.; Green M. D.; MacKenzie P. I.; Tephly T. R. Comparison of stably expressed rat UGT1.1 and UGT2B1 in the glucuronidation of opioid compounds. Drug Metab. Dispos. 1997, 25 (2), 251–255. [PubMed] [Google Scholar]
- Carvalho F. A.; Tenório S. B. Comparative study between doses of intrathecal morphine for analgesia after caesarean. Braz J. Anesthesiol. 2013, 63 (6), 492–499. 10.1016/j.bjane.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Zanardo V.; Simbi A.; Parotto M.; Severino L.; Carta R.; Guerrini P.; Straface G. Morphine-induced supraventricular tachycardia in near-term fetus. Ital J. Pediatr. 2018, 44 (1), 111. 10.1186/s13052-018-0570-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettleton R. T.; Wallisch M.; Olsen G. D. Respiratory effects of chronic in utero methadone or morphine exposure in the neonatal guinea pig. Neurotoxicol. Teratol. 2008, 30 (5), 448–454. 10.1016/j.ntt.2008.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasiraei-Moghadam S.; Sahraei H.; Bahadoran H.; Sadooghi M.; Salimi S. H.; Kaka G. R.; Imani H.; Mahdavi-Nasab H.; Dashtnavard H. Effects of maternal oral morphine consumption on neural tube development in Wistar rats. Dev. Brain Res. 2005, 159 (1), 12–17. 10.1016/j.devbrainres.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Sadraie S. H.; Kaka G. R.; Sahraei H.; Dashtnavard H.; Bahadoran H.; Mofid M.; Nasab H. M.; Jafari F. Effects of maternal oral administration of morphine sulfate on developing rat fetal cerebrum: a morphometrical evaluation. Brain Res. 2008, 1245, 36–40. 10.1016/j.brainres.2008.09.052. [DOI] [PubMed] [Google Scholar]
- Dehghani L.; Sahraei H.; Meamar R.; Kazemi M. Time-dependent effect of oral morphine consumption on the development of cytotrophoblast and syncytiotrophoblast cells of the placental layers during the three different periods of pregnancy in Wistar rats. Clin. Dev. Immunol. 2013, 2013, 974205. 10.1155/2013/974205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVane C. L.; Simpkins J. W.; Boulton D. W.; Laizure S. C.; Miller R. L. Disposition of morphine in tissues of the pregnant rat and foetus following single and continuous intraperitoneal administration to the mother. J. Pharm. Pharmacol. 2010, 51 (11), 1283–1287. 10.1211/0022357991776859. [DOI] [PubMed] [Google Scholar]
- Azeredo F. J.; Dalla Costa T.; Derendorf H. Role of microdialysis in pharmacokinetics and pharmacodynamics: current status and future directions. Clin. Pharmacokinet. 2014, 53 (3), 205–212. 10.1007/s40262-014-0131-8. [DOI] [PubMed] [Google Scholar]
- Tsai T. H. Assaying protein unbound drugs using microdialysis techniques. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2003, 797 (1–2), 161–173. 10.1016/j.jchromb.2003.08.036. [DOI] [PubMed] [Google Scholar]
- Sadiq M. W.; Bostrom E.; Keizer R.; Bjorkman S.; Hammarlund-Udenaes M. Oxymorphone active uptake at the blood-brain barrier and population modeling of its pharmacokinetic-pharmacodynamic relationship. J. Pharm. Sci. 2013, 102 (9), 3320–3331. 10.1002/jps.23492. [DOI] [PubMed] [Google Scholar]
- Gottas A.; Oiestad E. L.; Boix F.; Vindenes V.; Ripel A.; Thaulow C. H.; Morland J. Levels of heroin and its metabolites in blood and brain extracellular fluid after i.v. heroin administration to freely moving rats. Br. J. Pharmacol. 2013, 170 (3), 546–556. 10.1111/bph.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- USFDA (2018) Bioanalytical Method Validation Guidance for Industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry.
- Charest P. L.; Vrolyk V.; Herst P.; Lessard M.; Sloboda D. M.; Dalvai M.; Haruna J.; Bailey J. L.; Benoit-Biancamano M. O. Histomorphologic Analysis of the Late-term Rat Fetus and Placenta. Toxicol. Pathol. 2018, 46 (2), 158–168. 10.1177/0192623318755135. [DOI] [PubMed] [Google Scholar]
- Furukawa S.; Tsuji N.; Sugiyama A. Morphology and physiology of rat placenta for toxicological evaluation. J. Toxicol. Pathol. 2019, 32 (1), 1–17. 10.1293/tox.2018-0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka K.; Nakagawa T.; Uno T. Application of Akaike’s information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J. Pharmacokinet. Biopharm. 1978, 6 (2), 165–175. 10.1007/BF01117450. [DOI] [PubMed] [Google Scholar]
- Evrard P. A.; Deridder G.; Verbeeck R. K. Intravenous microdialysis in the mouse and the rat: development and pharmacokinetic application of a new probe. Pharm. Res. 1996, 13 (1), 12–17. 10.1023/A:1016056628685. [DOI] [PubMed] [Google Scholar]
- Sasongko L.; Williams K. M.; Ramzan I.; McLachlan A. J. Assessment of in vitro and in vivo recovery of gallamine using microdialysis. J. Pharmacol. Toxicol. Methods 2000, 44 (3), 519–525. 10.1016/S1056-8719(00)00117-9. [DOI] [PubMed] [Google Scholar]
- Groenendaal D.; Freijer J.; de Mik D.; Bouw M. R.; Danhof M.; de Lange E. C. Population pharmacokinetic modelling of non-linear brain distribution of morphine: influence of active saturable influx and P-glycoprotein mediated efflux. Br. J. Pharmacol. 2007, 151 (5), 701–712. 10.1038/sj.bjp.0707257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland M.; Abildskov K. M.; Kiu T. W.; Daniel S. S.; Weldy P.; Stark R. I. Placental transfer and fetal elimination of morphine-3-beta-glucuronide in the pregnant baboon. Drug Metab. Dispos. 2008, 36 (9), 1859–68. 10.1124/dmd.108.021352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Säwe J.; Svensson J. O.; Rane A. Morphine metabolism in cancer patients on increasing oral doses-no evidence for autoinduction or dose-dependence. Br. J. Clin. Pharmacol. 1983, 16 (1), 85–93. 10.1111/j.1365-2125.1983.tb02148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacifici G. M. Placental transfer of antibiotics administered to the mother: A review. Int. J. Clin. Pharmacol. Ther. 2006, 44 (2), 57–63. 10.5414/CPP44057. [DOI] [PubMed] [Google Scholar]
- Syme M. R.; Paxton J. W.; Keelan J. A. Drug Transfer and Metabolism by the Human Placenta. Clin. Pharmacokinet. 2004, 43 (8), 487–514. 10.2165/00003088-200443080-00001. [DOI] [PubMed] [Google Scholar]
- Feghali M.; Venkataramanan R.; Caritis S. Pharmacokinetics of drugs in pregnancy. Semin Perinatol. 2015, 39 (7), 512–519. 10.1053/j.semperi.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren G.; Ornoy A. The role of the placenta in drug transport and fetal drug exposure. Expert Rev. Clin. Pharmacol. 2018, 11 (4), 373–385. 10.1080/17512433.2018.1425615. [DOI] [PubMed] [Google Scholar]
- Murphy M. M.; Scott J. M.; McPartlin J. M.; Fernandez-Ballart J. D. The pregnancy-related decrease in fasting plasma homocysteine is not explained by folic acid supplementation, hemodilution, or a decrease in albumin in a longitudinal study. Am. J. Clin. Nutr. 2002, 76 (3), 614–619. 10.1093/ajcn/76.3.614. [DOI] [PubMed] [Google Scholar]
- Liu L.; Liu X. Contributions of Drug Transporters to Blood-Placental Barrier. Adv. Exp. Med. Biol. 2019, 1141, 505–548. 10.1007/978-981-13-7647-4_11. [DOI] [PubMed] [Google Scholar]
- Han L. W.; Gao C.; Mao Q. An update on expression and function of P-gp/ABCB1 and BCRP/ABCG2 in the placenta and fetus. Expert Opin. Drug Metab. Toxicol. 2018, 14 (8), 817–829. 10.1080/17425255.2018.1499726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamabe W.; Maeda T.; Fukazawa Y.; Kumamoto K.; Shang L. Q.; Yamamoto A.; Yamamoto C.; Tokuyama S.; Kishioka S. P-glycoprotein ATPase activating effect of opioid analgesics and their P-glycoprotein-dependent antinociception in mice. Pharmacol., Biochem. Behav. 2006, 85 (3), 629–636. 10.1016/j.pbb.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Sanchez-Covarrubias L.; Slosky L. M.; Thompson B. J.; Zhang Y.; Laracuente M. L.; DeMarco K. M.; Ronaldson P. T.; Davis T. P. P-glycoprotein modulates morphine uptake into the CNS: a role for the non-steroidal anti-inflammatory drug diclofenac. PLoS One 2014, 9 (2), e88516 10.1371/journal.pone.0088516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N.; van de Wetering K.; Hillebrand M.; Sarton E.; Kuil A.; Wielinga P. R.; Tephly T.; Dahan A.; Beijnen J. H.; Borst P. Mice lacking multidrug resistance protein 3 show altered morphine pharmacokinetics and morphine-6-glucuronide antinociception. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (20), 7274–7279. 10.1073/pnas.0502530102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering K.; Zelcer N.; Kuil A.; Feddema W.; Hillebrand M.; Vlaming M. L.; Schinkel A. H.; Beijnen J. H.; Borst P. Multidrug resistance proteins 2 and 3 provide alternative routes for hepatic excretion of morphine-glucuronides. Mol. Pharmacol. 2007, 72 (2), 387–394. 10.1124/mol.107.035592. [DOI] [PubMed] [Google Scholar]
- Wei Y.; Wu B.; Jiang W.; Yin T.; Jia X.; Basu S.; Yang G.; Hu M. Revolving door action of breast cancer resistance protein (BCRP) facilitates or controls the efflux of flavone glucuronides from UGT1A9-overexpressing HeLa cells. Mol. Pharmaceutics 2013, 10 (5), 1736–1750. 10.1021/mp300562q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa M.; Iwano H.; Yanagisawa R.; Koike N.; Inoue H.; Yokota H. Placental transfer of conjugated bisphenol A and subsequent reactivation in the rat fetus. Environ. Health Perspect. 2010, 118 (9), 1196–1203. 10.1289/ehp.0901575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekstrom L.; Johansson M.; Rane A. Tissue distribution and relative gene expression of UDP-glucuronosyltransferases (2B7, 2B15, 2B17) in the human fetus. Drug Metab. Dispos. 2013, 41 (2), 291–295. 10.1124/dmd.112.049197. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.