

Abstract
The bromodomain and extra-terminal (BET) domain family of proteins, which include its prototypical member Brd4, is implicated in a variety of cancers and viral infections due to their interaction with cellular and viral proteins. BET proteins contain two bromodomains, a common protein motif that selectively binds acetylated lysine on histones. However, they are structurally distinct from other bromodomain-containing proteins because they encode a unique C-terminal extra-terminal (ET) domain that is important for the protein–protein interactions including jumonji C-domain-containing protein 6 (JMJD6) and histone–lysine N-methyltransferase NSD3 (NSD3). Brd4 functions primarily during transcription as a passive scaffold linking cellular and viral proteins to chromatin. The rapid development of clinical inhibitors targeting Brd4 highlights the importance of this protein as an anticancer target. Current therapeutic approaches focus on the development of small molecule acetylated lysine mimics of histone marks that block the ability of the bromodomains to bind their chromatin marks. Thus far, bromodomain-targeted agents have shown dose-limiting toxicities due to off-target effects on other bromodomain-containing proteins. Here, we exploited a viral–host protein interaction interface to design peptides for the disruption of BET protein function. A murine leukemia virus (MLV) integrase-derived peptide (ET binding motif, EBM) and its shorter minimal binding motif (pentapeptide LKIRL) were sufficient to directly bind the Brd4 ET domain and reduce cellular proliferation of an acute myeloid leukemia cell line. Using computational and biochemical approaches, we identified the minimal essential contacts between EBM and LKIRL peptides and the Brd4 ET domain. Our findings provide a structural foundation for inhibiting BET/Brd4-mediated cancers by targeting the ET domain with small peptide-based inhibitors.
Keywords: acute myeloid leukemia, murine leukemia virus, Brd4, BET proteins, extra-terminal domain, viral−host interactions
The bromodomain and extra-terminal (BET) domain family of proteins (Brd2, -3, -4, and -T) as well as its extended family (Brd1, -7, -8, and -9) are characterized by two N-terminal bromodomains, which bind acetylated lysine on H3 and H4 histone tails on chromatin.1,2 A C-terminal extra-terminal (ET) domain and SEED domain differentiates BET proteins from its extended family members. Brd4 represents the extensively studied, prototypical BET protein. Brd4 has pleiotropic functions in the cell, focused around transcriptional gene regulation. The best understood roles of BET proteins is to serve as a scaffold that associates with a variety of cellular proteins including chromatin-modifying factors, transcription factors, histone modification enzymes, and a number of viral proteins with chromatin.3 These include jumonji C-domain-containing protein 6 (JMJD6), histone–lysine N-methyltransferase NSD3 (NSD3), glioma tumor suppressor candidate region gene 1 protein (GLTSCR1), ATPase family AAA domain-containing protein 5 (ATAD5), and chromodomain helicase DNA-binding protein 4 (CHD4) as well as viral γ-2 herpesvirus latency-associated nuclear antigen and viral integrase (IN) from murine leukemia virus (MLV).4−6
Because of their well-established functions in cell cycle regulation, epigenetic sensing, hematopoietic stem cell development, and a range of disease states, there is high interest in the therapeutic targeting of BET proteins with small molecule inhibitors.7−10 For cancer therapies, interventions target BET proteins composed of acetylated lysine mimics that block the BET protein bromodomains from binding the acetylated lysine of histones in chromatin.11 In multiple clinical trials, the therapeutic potential of targeting BET/Brd4 proteins has been evaluated for a variety of disease states and cancer malignancies.12−15 For example, archetypical I-BET and JQ-1, which target the two N-terminal bromodomains, were shown to disrupt the BET/BRd4 superenhancer function in acute myeloid leukemia (AML) and mixed myeloid leukemia (MLL).16,17 Second generation pan-BET inhibitors, such as OTX015 and TEN-101, have exhibited limited therapeutic potential in clinical trials due to their dose-limiting toxicities. An alternative approach is to develop molecules that are selective for a single bromodomain. ABBV-744, a BET domain II selective inhibitor, has showed improved tolerability in a preclinical animal model.18 Currently, ABBV-744 is in phase I clinical trials. Therefore, it is important to develop selective bromodomain inhibitors with minimal toxicity19 as well as novel inhibitors that bind to other distinct sites in BET proteins.11,20
The ET domain of Brd4, which is identical to the ET domains of Brd2 and Brd3, is important for interactions with a number of cellular factors implicated in human cancers.16,17 Interactions between JMJD6 and the Brd4 ET domain have been implicated in multiple cancers including neuroblastoma, oral, breast, lung, and colon.21,22 Interactions of NSD3 with the Brd4 ET domain are required in aggressive midline carcinoma and are essential for AML maintenance.4,23−25 The role of γ-retroviral MLV vectors in human cancers was first uncovered in hematopoietic stem cell (HSC) gene-therapy studies, which showed that insertional activation of LMO-2 and CCND2 proto-oncogenes by MLV vectors can result in the development of leukemia in a subset of patients.26−29 The insertion of MLV vectors near proto-oncogenes is due to the enrichment of BET proteins near the promoters and enhancers of proto-oncogenes. Recent studies showing the selective high affinity binding of MLV IN to the BET ET domain provide a mechanistic basis for insertional activation.5 This discovery has led to functional and structural studies of MLV IN–Brd4 ET interactions to examine the protein–protein binding interface and mapping of the minimal essential interactions.6,30 Structural approaches have solved an NMR-derived solution structure of a 17 amino acid region (389′–405′) at the end of the C-terminal domain (CTD) of MLV IN, termed the ET binding motif (EBM), essential for the interaction with Brd4 ET. Because of its high binding affinity (∼160 nM), the EBM effectively outcompetes the transcription factor NSD3 for binding to full-length Brd4.30 Subsequent mutational analysis confirmed the essential nature of Brd4 E653R/D655R and V634S/I652S resides for the interaction with MLV IN and NSD3.
The central basis for this study is that MLV IN binds the ET domain of Brd4 and can outcompete other cellular binding partners of the ET domain.30 The NMR-derived solution structure of EBM bound to the BRD4 ET domain revealed a critical electrostatic and hydrophobic interaction at the protein–protein interface.30 Here, we demonstrate that the stable expression of the MLV IN-derived 17-amino acid peptide, EBM, reduces cellular viability and proliferation of MOLM-13, a BET/Brd4-mediated AML cell line. Since a 17-amino acid peptide, such as the EBM, is typically too large to be an ideal clinical drug candidate due to metabolic instability and potential low bioavailability, we focused on deriving more amenable shorter peptides. Using molecular dynamic simulations, we designed a pentapeptide (LKIRL) predicted to maintain the essential interactions of the EBM-ET domain. Structure-based computational approaches and in vitro biochemical studies demonstrate that the LKIRL peptide retains the overall binding mode and affinity for the ET domain. The LKIRL peptide also reduces cellular viability and proliferation of MOLM-13. In summary, we report the development of MLV IN-derived peptides as candidates that bind the Brd4 ET domain and reduce viability and proliferation of acute myeloid leukemia in cell culture. As there are no current therapies that specifically target the ET domain of BET proteins, our peptide narrow-down approach paves the way for the future development of peptidomimetics and small molecules using MLV IN-derived peptides as biochemical scaffolds.
Results and Discussion
Structure-Guided Design of MLV IN EBM-Derived Peptides That Bind the Brd4 ET Domain
The previously solved NMR structure of the 17 amino acid EBM of MLV IN bound to the ET domain of Brd4 (Figure 1A,B) has facilitated the identification of residues critical for interactions at the EBM-ET domain interface. The MLV IN EBM binds the Brd4 ET domain by folding into a β-sheet in the pocket between α1 and β1 of the ET domain. The negative charged acidic residues of ET β1 (E651, E653, and D655) form salt-bridges with positively charged basic residues of EBM β7′ (K400′ and R402′), and backbones of β1 and β7′ form hydrogen bonds with each other. Inside the ET pocket, the hydrophobic residues including L618, I622 (at α1), V633, V634, L630 (at α2), and F656, I654, I652 (at β1) provide accommodation for I401′, L403′, V392′, W390′, and L399′ of the EBM. Because of the electrostatic interactions and hydrophobic contacts at the interaction interface, the 17 amino acid EBM β-sheet acts as a strong binder of the ET domain. However, not all residues of EBM are involved in direct interaction with ET. Although most residues at β7′ make extensive contact with ET, the other residues such as T389′, R391′, Q393′, R394′, S395′, Q396′, N397′, and P398′ at β6′ (Figure 1B,C) do not form effective contacts with the ET pocket, but instead, they are “flipped upward” toward the outside.
Figure 1.

Graphical illustration of amino acid residues at the MLV IN EBM-Brd4 ET domain interaction interface. (A) Schematic representation of MLV integrase (IN) and Brd4 (1–1342). Key domains and structural features are indicated for both: MLV IN C-terminal domain (CTD), catalytic core domain (CCD), and N-terminal domain (NTD). The ET-binding motif (EBM) is indicated in orange, and the location of LKIRL is highlighted in red. Brd4 bromodomains (BD1 and BD2), DNA binding motifs A and B, SEED domain, and extra-terminal domain (ET). (B) Representation of amino acids residue in the β6′ strand of EBM (pink) and their interaction with the Brd4 ET domain (blue). Two perspectives of view indicate that the highlighted residues are not extensively in contact with the ET domain. (C) Annotation of the secondary structure of the EBM β-sheet and ET domain (PDB Code: 2N3K). (D) MM/GBSA per-residue energy decomposition profile of the EBM-ET complex indicating that β7′ of EBM and β1 of ET predominantly contribute to the protein–protein interaction. (E) Binding mode of LKIRL (399′–403′) with the ET domain showing that the LKIRL sequence interacts extensively with β1 of ET.
To gain detailed insights into the contributions of residues and the protein flexibility at the EBM-ET interface, 100 ns of a molecular dynamics (MD) simulation of a previously reported NMR structure (PDB Code: 2N3K) was performed, and the MM/GBSA per-residue energy decomposition was calculated when the system was converged. A higher fluctuation of RVQRSQNP (391′–398′) than LKIRLTR (399′–405′) was observed by the trajectory ensemble, indicating that the RVQRSQNP sequence was not well-stabilized by the ET domain. The energy decomposition profile suggested that the major contributions for the binding interaction belong to β7′ (Figure 1D).
Peptides and peptidomimetics have been successfully developed as biological probes and therapeutic options in the clinic. Thus far, these options have not been explored for targeting the Brd4 ET domain interface for cancer therapeutics. The development of full-length EBM into a clinical drug candidate is likely to be challenging due to the long sequence length. An attempt to shorten the β-sheet serves as an ideal alternative. Therefore, the identification of minimal critical residues can facilitate the development of a truncated version of EBM, rendering it feasible to explore the possibility of interfering with BET protein function independent of bromodomain inhibition. To this end, our calculated MM/GBSA per-residue energy decomposition suggests that W390′, V392′, P398′, L399′, K400′, I401′, R402′, L403′, T404′, and R405′ contribute the majority of the binding interactions at the EBM-ET interaction interface (Figure 1D,E). A similar binding pattern can also be found for NSD3 and JMJD6 binding to the ET domain.21,24 As shown in Figure 2B, NSD3 (amino acids 148–184) folds into a β-sheet and interacts with acidic residues at the ET interface with two positively charged side chains and three hydrophobic residues. The overall pattern of JMJD6 remains similar; however, there are slight differences when compared to MLV-IN and NSD3 binding modes, mainly because it folds into a helical coil (Figure 2C).
Figure 2.

Schematic representation of the ET domain (gray) bound with protein partners. (A) Brd4-ET domain structure bound with MLV IN-EBM (shown in pink; PDB Code: 2N3K). Side chains of K400 and R402 extend to approach acidic residues, while L399, I401, and L403 lie inside the hydrophobic groove. (B) Brd3-ET domain structure bound with NSD3 peptide with the sequence of 148–184 (shown in blue; PDB Code: 7JYN). K156 of NSD3 is slightly different from R402 of MLV-IN, but the overall binding mode is very conserved. (C) Brd4-ET domain structure bound with JMJD6 peptide (shown in brownish yellow; PDB Code: 6BNH). Although the JMJD6 binding motif folded into a helical coil upon binding with BRD4-ET, the relative pattern remains similar to MLV-IN, in that a positively charged K208 side chain extends upward and hydrophobic residues (W202, L207, and Y211) lie within the groove.
To design a minimal peptide sequence that maintains the minimal essential contact, W390′ and V392′ were eliminated as the amide backbones of these two residues are not adjacent to each other. To optimize ligand efficiency, P398′ was also eliminated since proline usually serves as a structural residue for protein and peptide folding instead of acting as an effective hydrophobic anchor. On the basis of these design principles, LKIRLTR was identified as the minimal peptide predicted to bind the ET domain, and we further hypothesized that this truncated version of EBM would bind the ET domain with a conformation similar to the EBM β7′ strand. However, our peptide docking analysis showed that LKIRLTR is likely to bend in the ET pocket and not form hydrogen bonds with the ET domain (Figure 3).
Figure 3.

Identification of minimal MLV IN EBM-derived peptide that binds the Brd4 ET domain. (A) The predicted binding mode of LKIRLTR (399′–405′) peptide. The Brd4 ET domain is shown in light blue. The peptide backbone is shown in dark green; side chain residues are shown in pink. Hydrogen bonds are shown by yellow dashed lines, and salt bridges are shown by pink dashed lines. (B) Global docking pose clustering of the LKIRL peptide. Most of the poses are concentrated at the EBM-ET binding interface. (C). Predicted binding mode of the LKIRL (399′–403′) peptide. (D) Higher dynamic perturbation in the EBM β-sheet turn and the β6′ strand (red) than the β7′ strand (gray) observed using molecular dynamic simulation. (E) Model showing overlap of the LKIRL peptide (green) and EBM (orange). The LKIRL peptide adopts a similar binding pose as the native sequence of β7′ of EBM.
Due to the high intrinsic flexibility, the linear LKIRLTR peptide bears a wide range of conformational distribution and cannot extend itself without an ordered EBM β-sheet secondary structure, resulting in a different binding pose than the EBM β7′ strand. Thus, we further shortened the peptide to restrict its degree of freedom. Given that D655 and E653 in the ET β1 strand are critical for the interaction, we eliminated T404′ and R405′ from the LKIRLTR peptide, yielding the pentapeptide LKIRL (399′–403′) as the prototype ET binder. Both local and global peptide docking indicated that LKIRL binds at the EBM-ET protein interface and shows a nearly identical binding pose as the native sequence of EBM (Figure 3E). Given the biochemical nature of the pentapeptide, the ionic interactions between the charged residue side chains and hydrophobic nature of leucine and isoleucine are likely the major reasons for the consistency of the binding pose.
Characterization of LKIRL Peptide Binding to the Brd4 ET Domain
To evaluate the detailed binding mechanism of the LKIRL peptide, we first performed in silico alanine scanning of the peptide to generate substituted peptides. Next, we compared the binding of wild-type and mutant peptides using molecular dynamics simulations. Finally, we determined the binding affinities of wild-type and mutant peptides using affinity precipitation assays. A virtual library of five mutated peptides was generated (Figure 4, bottom right) with each amino acid of LKIRL systemically mutated to alanine. Protein–peptide complex models were generated by molecular docking using the Glide program, and 100 ns of molecular dynamic simulation in the TIP3P water solvent model was performed for each complex using the GROMACS 2020.2 simulation package. As shown in Figure 4, representative snapshots of the trajectory for each system were captured after clustering analysis, and MM/GBSA per-residue energy decompositions were calculated.
Figure 4.

Contribution of individual residues of the LKIRL pentapeptide for binding the Brd4 ET domain. In silico alanine scanning of LKIRL pentapeptide and molecular dynamics simulation of mutant peptides bound to the Brd4 ET domain. Predicted binding mode of the indicated mutant peptide with the mutated alanine residue highlighted (red). The Brd4 ET domain is shown in light blue. The peptide backbone is shown in dark green; side chain residues are shown in pink. Hydrogen bonds are shown by yellow dashed lines, and salt bridges are shown by pink dashed lines. MM/GBSA per-residue energy decomposition results of each mutant peptide are shown on the left of the predicted binding mode.
When the first leucine of LKIRL was mutated into alanine, the hydrophobic interactions of mutant AKIRL with ET F656 decreased significantly, and the peptide backbone “flipped up” to form a hydrogen bond with ET E653, resulting in an unstable conformation. The lysine side chain was shifted to the left and formed a salt-bridge with ET D655 (Figure 4, top left). Although arginine and isoleucine still maintained a similar pose as in the original LKIRL peptide, the overall stability was weakened due to the backbone perturbation and lysine shifting. Compared to LKIRL, AKIRL showed weaker residue interaction energy with D650, E651, and I652 ET residues. Affinity precipitation experiments also indicated that mutant AKIRL can only maintain about 40% binding affinity compared to that of wild-type LKIRL (Figure 5). We observed that interactions with E651, E563, and I652 ET residues were also weakened when the last leucine of the LKIRL peptide was mutated to alanine (Figure 4, bottom left). Since the EIE (651–653) region of the ET β1 strand is more flexible than DFE (655–657) of the ET domain, the loss of mutant LKIRA peptide stability was not as significant as that observed for the mutant AKIRL peptide, and no lysine shifting occurred during the simulation. Although not comparable to the wild-type LKIRL peptide, LKIRA maintained slightly better binding affinity than AKIRL in the affinity precipitation experiments (Figure 5).
Figure 5.

Binding of the LKIRL pentapeptide and its alanine mutants to the Brd4 ET domain. (A, B) Affinity precipitation of the His-tagged Brd4 ET domain with FITC-peptides. Representative SDS-PAGE gels of affinity precipitation of the His-Brd4 ET domain with the indicated FITC-peptide. FITC-labeled bands were detected at 488 nm fluorescence, and the His-Brd4 ET domain was visualized with Coomassie staining. The last lane (“Blank”) indicates the control precipitation of 3 μM FITC-LKIRL without the His-Brd4 ET domain. Graphical and tabular presentation of relative binding. Relative binding was calculated by quantifying the fluorescence intensities from the SDS-PAGE gels of the indicated mutant FITC-peptide relative to the wild-type FITC-peptide bound to the His-Brd4 ET domain. (C) Surface plasmon resonance (SPR) of the His-tagged Brd4 ET domain with FITC-peptides. Peptide concentrations of 1000, 500, 200, and 100 μM were run on a BiaCore3000 with a prebound His-tagged Brd4 ET domain on NTA chips. The graphical presentation of representative SPR curves for LKIAL (Langmuir fitted curves for each concentration averaged for the final KD) is shown with the calculated binding affinities for all peptides. (D) Relative binding readout from affinity precipitation pulldown and calculated binding KD from the SPR experiment. Each data point represents the average of three independent experiments. Error bars represent standard deviations.
When the third isoleucine was mutated to alanine, the interaction of mutant LKARL peptide with the ET domain changed dramatically (Figure 4, middle left) with ∼17% observed binding to the ET domain in the affinity precipitation experiments (Figure 4). In the case of the mutant LKARL peptide, the contacts with the E651, I652, E653, I654, and F656 ET residues were largely reduced. Moreover, the hydrophobic residues within the ET domain such as I622, L630, and V633 also showed weaker interactions with the peptide. Molecular dynamics simulation trajectory revealed a major perturbation of the LKARL backbone suggesting that the entire peptide was “flipped up” and moved away from the ET binding pocket. Even though the lysine and arginine can maintain the ionic interactions with D650 and E653 ET residues, the overall binding pose was no longer stabilized. Given that the I654 of the ET domain was important in our prior mutational analysis30 and on the basis of our current analysis (Figures 4 and 5), we infer that the hydrophobic anchoring effect of the central isoleucine side chain is critical and the substitution severely affects the peptide binding conformation.
We next examined the importance of the charged residues by analyzing two mutant peptides, LAIRL (Figure 4, top right) and LKIAL (Figure 4, middle right). When lysine was mutated, the N-terminal of the mutant LAIRL peptide “flipped up”, and due to the intrinsic flexibility of the EIEIDFE (651–657) region of the ET domain, both E657 and D655 ET residues approach the protonated free amine to form salt bridges and hydrogen bonds, resulting in a major backbone perturbation. Although LAIRL showed a more favorable interaction with D655 and E657 ET residues, the overall backbone shifted into an unfavorable state since hydrophobic residues are moved away from the original binding state, and the contacts of the leucine side chain with F656 of the ET domain were largely decreased. The mutant LKIAL peptide also exhibited poor binding as the main anchor guanidine moiety was eliminated and the interactions with E651 and E653 ET residues were significantly reduced. As a reference, MM/GBSA energetical components are listed in Table 1.
Table 1. MM/GBSA Energy Calculation Results of LKIRL and Related Mutantsa.
| LKIRL | AKIRL | LAIRL | LKARL | LKIAL | LKIRA | |
|---|---|---|---|---|---|---|
| ΔEvdw | –37.8 ± 4.0 | –29.9 ± 4.2 | –28.4 ± 4.1 | –8.4 ± 6.0 | –19.7 ± 4.0 | –27.8 ± 4.9 |
| ΔEelec | –248.8 ± 21.1 | –329.6 ± 50.7 | –251.8 ± 42.3 | –452.8 ± 95.9 | –261.9 ± 32.3 | –392.1 ± 36.0 |
| ΔGGB | 251.9 ± 19.4 | 327.3 ± 44.0 | 248.3 ± 35.7 | 435.9 ± 83.3 | 267.0 ± 29.8 | 378.7 ± 33.0 |
| ΔGSA | –7.1 ± 0.4 | –5.8 ± 0.4 | –5.6 ± 0.4 | –3.5 ± 0.7 | –3.8 ± 0.4 | –6.4 ± 0.5 |
| ΔGbind | –41.8 ± 4.8 | –38.1 ± 6.8 | –37.5 ± 6.4 | –28.7 ± 11.7 | –18.4 ± 4.2 | –47.5 ± 5.1 |
Total energy contribution (ΔGbind) was divided into van der Waals energy (ΔEvdw), electrostatic energy (ΔEelec), polar solvation energy (ΔGGB), and non-polar solvation energy (ΔGSA).
Lastly, affinity precipitation and surface plasmon resonance (SPR) assays have allowed us to empirically determine the binding potency of the LKIRL peptide and its alanine mutants (Figure 5). For these binding assays, the recombinant purified ET domain was used in affinity precipitation assays with FITC-labeled peptides. Mutation of the charged residues showed that both LAIRL and LKIAL only maintain a 30% binding affinity (Figure 5B). Additionally, the central isoleucine of the LKIRL peptide was found to be critical for binding to the ET domain as its mutation to alanine resulted in an ∼6-fold reduction in binding. Next, we also performed SPR binding analysis of these peptides with the ET domain (Figure 5B). SPR analysis revealed that the binding affinity of the ET domain for LKIRL (KD of 145 nM) is comparable to its reported binding for EBM (KD of 160 nM).30 This validates our molecular modeling that LKIRL encompasses the essential contacts necessary for interaction. Second, the mutation of either of the charged residues reduced the binding affinity by >10-fold, corroborating the results of the affinity precipitation assay. Interestingly, mutation of the central isoleucine only modestly affected binding (∼2-fold reduction) in the SPR assay. It is possible that the mutation of isoleucine dramatically affects the peptide solubility of LKARL. Thus, in the SPR approach, the percentage of DMSO was increased, which may affect the binding affinity. These direct binding experiments corroborate our computational modeling findings to demonstrate that both hydrophobic and charged residues are necessary for the ET-LKIRL peptide interaction and binding pose stabilization.
MLV IN EBM and LKIRL Peptide Suppress the Proliferation of Acute Myeloid Leukemia Cells
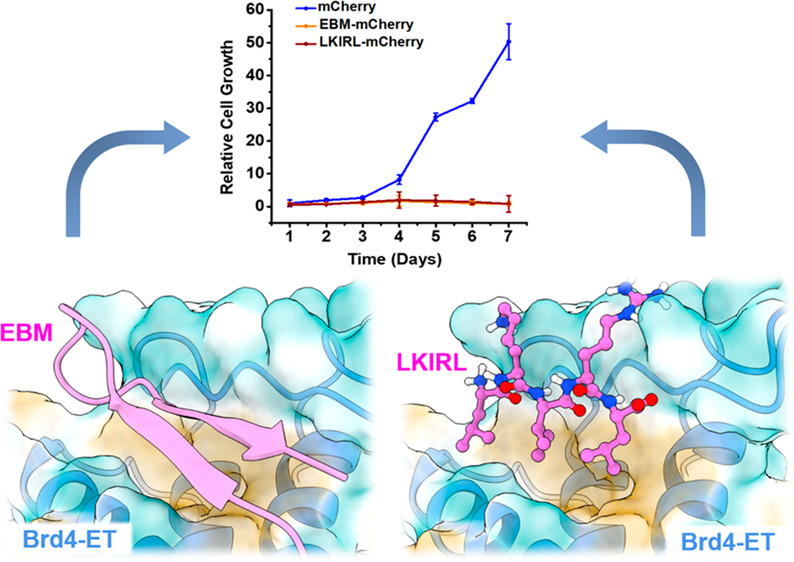
Brd4 has been implicated in multiple cancers including neuroblastoma, oral, breast, lung, and colon21,22 and is perhaps best studied for its role in AML maintenance.4,23−25 Thus, we chose to utilize AML derived cell lines as a proof of concept in the effectiveness of targeting the ET domain in Brd4 modulated cancer. Our small native peptide constructs will inform our drug development efforts due to their ability to be expressed in relevant cancer cell lines. Thus, we evaluated the ability of MLV IN EBM and LKIRL peptides to suppress the proliferation of MOLM-13, which is a Brd4-dependent acute myeloid leukemia (AML) cell line.31 MOLM-13 cells were transduced with lentiviral virus-like particles to stably express either an empty vector control, MLV IN EBM, or the short LKIRL peptide fused in frame with a fluorescent mCherry protein marker to facilitate sorting of transduced cells. Expression of peptide-mCherry fusion constructs were confirmed by immunoblotting (Figure 6A). Co-immunoprecipitation (IP) assays validated that FLAG-Brd4 interacts with both eGFP-EBM and eGFP-LKIRL fusion peptides in the cellular extracts (Figure 6B). Cellular proliferation was monitored over 7 days, and cell viability was measured at day three and at the experimental end point. The expression of EBM-mCherry resulted in a 2-fold reduction in cell viability by day three and ≥20-fold reduction at day seven (Figure 6C). The pentapeptide LKIRL-mCherry reduced the cellular proliferation and viability of MOLM-13 cells to levels similar to those observed with EBM. MOLM-13 cells expressing EBM-mCherry or LKIRL-mCherry showed an ∼50-fold reduction in cellular proliferation when compared to the empty vector control cells (Figure 6D). Thus, the LKIRL peptide, which binds at the EBM-ET protein interface and shows a nearly identical binding pose as the native sequence of EBM, is sufficient for the inhibition of AML proliferation. These findings confirm the inhibitory potential of EBM and pentapeptide LKIRL (399′–403′) for suppressing cellular proliferation of the Brd4-dependent AML cell line.
Figure 6.

MLV IN EBM and LKIRL pentapeptide suppress the proliferation of MOLM-13 cells. (A) Immunoblot of MOLM-13 cells stably expressing either an empty vector control, MLV IN EBM, or the short LKIRL peptide fused in frame with a fluorescent mCherry protein. (B) Co-immunoprecipitation (IP) of cells expressing FLAG-tagged Brd4 and eGFP alone, fused to EBM, or fused to LKIRL. Agarose containing eGFP nanobodies were used for affinity precipitation of proteins with immunoblotting with anti-FLAG and eGFP antibodies. (C) Viability of the indicated MOLM-13 stable cells at day 3 and day 7. Graphs indicate the percentage of viable cells normalized to the empty vector control. Bars represent the average of three independent experiments. Error bars represent standard deviations. Viability was compared to the empty vector control using one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test. ****: p < 0.0001. (D) Cell growth of the indicated MOLM-13 stable cells. Graph indicates cell growth measured as cellular proliferation over 7 days normalized to total cell numbers at the beginning of the experiment. Each data point represents the average of three independent experiments. Error bars represent standard deviations. For each time point, cell growth was compared to the empty vector control using one-way analysis of variance (ANOVA) followed by Dunnett’s multiple comparisons test.
Brd4 has been implicated in multiple cancers, including neuroblastoma, oral, breast, lung, and colon malignancies.21,22 For example, Brd4 interactions with NSD3 are important for aggressive midline carcinoma and essential for AML maintenance.4,23−25 The functions of Brd4 in cancer progression are mediated, in part, through its interactions with other proteins via its C-terminal ET domain.16,17 Thus far, the inhibition of the Brd4 function has focused on the dual bromodomains with limited success and no clinical approvals. For example, I-BET 151 was terminated in Phase 1 for reported cardiotoxicity whereas INCB057643 and INCB054328 were terminated in Phase 1/2 for reported thrombocytopenia.12,32−34 Approaches to develop selective inhibitors targeting bromodomain II are promising, but results from early stage clinical trials are yet-to-be known.18,19 The dose limiting toxicities or other pharmacological issues observed in 12 clinical trials could be because 10 protein families contain bromodomains. In this study, we exploited the MLV IN-ET protein interaction interface to design peptides for the disruption of BET protein function. The ET domain potentially presents a more attractive therapeutic target as it is limited to one family of proteins (the BET family) with 4 identified proteins including Brd4.33 We demonstrate that these peptides bind the ET domain, and their stable expression reduces cellular proliferation of MOLM-13, a BET/Brd4-mediated AML cell line. However, more work is needed to validate if targeting the ET domain will avoid dose limiting toxicities seen for bromodomain inhibitors in clinical trials. Thus, our approach provides a foundation for inhibiting BET/Brd4-mediated cancers by targeting the ET domain with small peptide-based inhibitors. In summary, this work paves the way for the development of peptidomimetics and small molecules that target the ET domain of BET proteins using the MLV IN-derived peptides as scaffolds.
Methods
Peptide Docking and Molecular Dynamic Simulation
The protein structure of the human Brd4 ET domain in complex with the MLV IN C-terminus was downloaded from the Protein Data Bank (PDB Code: 2N3K),30 and the first structure of the NMR ensemble was extracted (with the EBM β-sheet eliminated) for protein preparation using the Prepwizard module35 of the Schrödinger molecular modeling suite.36 Six peptide structures were generated using ChemDraw and further prepared using the LigPrep module at pH = 7. The docking grid was defined by the protein interface residues, and all six peptide ligands were docked on the basis of the Glide XP and Glide SP-Peptide protocols.37,38 Representative docking poses were picked for molecular dynamic simulations. The GROMACS 2020.2 package39−41 was applied in this work, and the CHARMM36 force field42,43 was used to parametrize the protein; the CGenFF44 web server was used to generate ligand topologies. A dodecameric space was constructed by the editconf module and was solvated in a 6.842 nm × 6.842 nm × 6.842 nm TIP3P water box. All atoms within the system were optimized by the steepest descent minimization, followed by 1000 ps of equilibration of the NVT ensemble to heat the system up to 300 K and another 1000 ps equilibration of the NPT ensemble to raise the pressure to 1 atm. Then, a 100 ns molecular dynamic simulation was produced, and the final trajectory was fit to the backbone before analysis. The MM/GBSA energy calculation was performed by the gmx_MMPBSA45 program, which implements the GB/PB and other calculations in GROMACS based on MMPBSA.py(46) (version 16.0) and AmberTools20.47,48
Peptide Design and Recombinant Proteins
MLV IN-derived peptides were designed using the following published structures: Brd4 ET bound to MLV EBM (PDB Code: 2N3K) and Brd4 ET (PDB Code: 2JNS). Once designed, synthetic peptides were purchased from Biomatik (95% purity with TFA removed) with an N-terminal FITC label (FITC-peptide). 1× phosphate buffered saline (PBS) supplemented with 10% aqueous NH3 was used as the diluent to reconstitute peptides. Recombinant His-tagged Brd4 ET was expressed and purified as previously described.6,30
Affinity Precipitation Assay
Affinity precipitation assays using Ni-NTA beads (GE Healthcare) were performed with the His-Brd4 ET domain and the indicated FITC-peptides using previously described methods.6,49 Ni-NTA beads were equilibrated in binding buffer (50 mM Tris, pH 7.5, 250 mM NaCl, 50 mM imidazole, 0.4% aqueous NH3, 0.1% NP-40, and 2 mM β-mercaptoethanol). Binding reactions were set up by incubating equilibrated Ni-NTA beads with His-Brd4 ET (1 μM) and FITC-peptide (3 μM) in the binding buffer and incubated for 1 h at 4 °C. In parallel, control reactions with 3 μM FITC-LKIRL without the His-Brd4 ET domain were performed to rule out nonspecific binding to the Ni-NTA beads. Reactions were spun and washed three times in the binding buffer to remove unbound proteins/peptides. The resulting protein–peptide complexes bound to the beads were extracted using NuPAGE lithium dodecyl sulfate sample buffer (Invitrogen), subjected to SDS-PAGE analysis, and visualized by Coomassie staining or fluorescence detection at 488 nm. Resulting FITC-labeled bands were quantified using ImageJ software.
Surface Plasmon Resonance (SPR) Assay
SPR assays using a Sensor Chip NTA (GE Healthcare) were performed with the His-Brd4 ET domain and indicated FITC-peptides on a Biacore 3000 (GE Healthcare). The chip was conditioned with 0.5 mM NiCl2 at a flow rate of 10 μL/min for 1 min followed by a 1 min wash of 3 mM EDTA at a flow rate of 10 μL/min. It was then equilibrated in binding buffer (1× phosphate buffered saline (PBS) in 7.5% DMSO) prior to the ligand capture of the His-Brd4 ET domain. Indicated peptide concentrations (1000, 500, 200, and 100 μM) in the same binding buffer were flowed over the cell for 60 s at a flow rate of 30 μL/min followed by a 4 min dissociation. The chip was regenerated with 500 mM imidazole between runs. KD values were calculated using Biacore 3000 software (GE Healthcare).
Co-immunoprecipitation (IP) Assay
Affinity precipitation assays using GFP-TRAP agarose (Chromotek) were performed with HEK293T (ATCC CRL-3216) cells expressing various constructs. Human FLAG-Brd4 (1–1362) and eGFP vector (pcDNA3.1 + N-eGFP) fused to empty vector, EBM, or LKIRL peptides were transiently expressed for 48 h. Cell pellets were lysed, and affinity precipitation was done according to the manufacturer’s recommendations (Chromoteck). Immunoblotting was performed with anti-eGFP or FLAG (AbCam ab184601 and ab18230). In parallel, control reactions without GFP-TRAP agarose were performed to rule out nonspecific binding to the tubes. The resulting protein–protein complexes bound to the agarose were extracted using NuPAGE lithium dodecyl sulfate sample buffer (Invitrogen), subjected to SDS-PAGE analysis, and visualized by immunoblotting.
Cell, Plasmid, and Lentiviral Virus-Like Particle Production
HEK293T (ATCC CRL-3216) cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco) supplemented with 10% fetal bovine serum (FBS, Gibco) at 37 °C with 5% CO2. Human acute myeloid leukemia MOLM-13 cells50 were cultured in RPMI-1640 medium (Gibco) supplemented with 10% FBS at 37 °C with 5% CO2.31
Lentiviral plasmid encoding mCherry (pLV-mCherry), plasmid encoding HIV-1 Gag-Pol (psPAX2), and plasmid encoding vesicular stomatitis virus-G protein (pMD2.G) were purchased from Addgene. The EBM and LKIRL peptide expression constructs were synthesized as DNA fragments (Genescript), digested, and ligated into pLV-mCherry using XbaI and BamHI to generate pLV-peptide-mCherry plasmids. Plasmids generated in this study were confirmed by Sanger sequencing.
Lentiviral virus-like particles (VLPs) were generated in HEK293T cells by cotransfecting pLV-peptide-mCherry, psPAX2, and pMD2.G at a ratio of 1:1:0.5 using Fugene 6 transfection reagent (Roche) following the manufacturer’s protocol. Forty-eight hours post-transfection, VLP-containing supernatants were harvested, filtered through a 0.45 μm sterile filter, and concentrated using Amicon Ultra-15 centrifugal filters (Millipore). Aliquots of VLPs were stored at −80 °C.
Generation of MOLM-13 Stable Cell Lines
MOLM-13 cells stably expressing MLV IN EBM or LKIRL peptide were generated using methods described previously.51 Briefly, MOLM-13 cells (106 cells/mL in a 6-well plate) were transduced with equivalent amounts of the indicated VLPs (mCherry vector only, EBM-mCherry, and LKIRL-mCherry) in the presence of 1 μg/mL polybrene. All transductions were carried out in duplicate. After the addition of the VLPs, the cells underwent two spinoculations at 6 h intervals. The spinoculation procedure consisted of centrifuging the cells at 1800 rpm for 45 min at 30 °C. After spinoculations, cells were washed twice, resuspended in fresh complete media, and monitored for viability every 24 h. The transduced cells were sorted to select for cell population with >85% mCherry expression. Aliquots of the cells were used to determine mCherry and peptide-mCherry expression levels using standard immunoblotting procedures with anti-mCherry antibody (AbCam ab213511). The antiproliferative effects of stably expressed EBM and LKIRL peptides were measured over a three-day time course. Cell proliferation was monitored using the commercial CellTiter 96 Aqueous One Solution Cell Proliferation (MTS) Assay (Promega) according to the manufacturer’s recommendations. Cell viability at the experimental end point was monitored using the trypan blue exclusion method and a commercial ATP assay (Cell Titer-Glo 2.0 from Promega).
Acknowledgments
This research was supported by The Ohio State University’s institutional start-up funds to R.C.L. We thank Dr. Mark Foster, Antonia Duran, and Josh Omlor for helpful discussions and technical assistance. We also thank the Ohio Supercomputer Center for the available licenses for Glide, LigPrep, and Prepwizard modules of the Schrödinger molecular modelling suite.
Author Contributions
Conceptualization: R.C.L.; research design: E.X., X.C., A.S., P.-K.L., and R.C.L.; investigation: E.X., N.S., X.K., N.C., J.D.G., and R.C.L.; writing, original draft: E.X., A.S., P.-K.L., and R.C.L.; writing, review and editing: all authors.
The authors declare no competing financial interest.
Special Issue
Published as part of the ACS Pharmacology & Translational Science special issue “Epigenetics 2022”.
References
- Moriniere J.; Rousseaux S.; Steuerwald U.; Soler-Lopez M.; Curtet S.; Vitte A. L.; Govin J.; Gaucher J.; Sadoul K.; Hart D. J.; Krijgsveld J.; Khochbin S.; Muller C. W.; Petosa C. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009, 461, 664–668. 10.1038/nature08397. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Qi J.; Picaud S.; Shen Y.; Smith W. B.; Fedorov O.; Morse E. M.; Keates T.; Hickman T. T.; Felletar I.; Philpott M.; Munro S.; McKeown M. R.; Wang Y.; Christie A. L.; West N.; Cameron M. J.; Schwartz B.; Heightman T. D.; La Thangue N.; French C. A.; Wiest O.; Kung A. L.; Knapp S.; Bradner J. E. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belkina A. C.; Denis G. V. BET domain co-regulators in obesity, inflammation and cancer. Nat. Rev. Cancer 2012, 12, 465–477. 10.1038/nrc3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman S.; Sowa M. E.; Ottinger M.; Smith J. A.; Shi Y.; Harper J. W.; Howley P. M. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol. Cell. Biol. 2011, 31, 2641–2652. 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A.; Larue R. C.; Plumb M. R.; Malani N.; Male F.; Slaughter A.; Kessl J. J.; Shkriabai N.; Coward E.; Aiyer S. S.; Green P. L.; Wu L.; Roth M. J.; Bushman F. D.; Kvaratskhelia M. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 12036–12041. 10.1073/pnas.1307157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue R. C.; Plumb M. R.; Crowe B. L.; Shkriabai N.; Sharma A.; DiFiore J.; Malani N.; Aiyer S. S.; Roth M. J.; Bushman F. D.; Foster M. P.; Kvaratskhelia M. Bimodal high-affinity association of Brd4 with murine leukemia virus integrase and mononucleosomes. Nucleic Acids Res. 2014, 42, 4868–4881. 10.1093/nar/gku135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand M.; Measures A. R.; Wilson B. G.; Cortopassi W. A.; Alexander R.; Hoss M.; Hewings D. S.; Rooney T. P.; Paton R. S.; Conway S. J. Small molecule inhibitors of bromodomain-acetyl-lysine interactions. ACS Chem. Biol. 2015, 10, 22–39. 10.1021/cb500996u. [DOI] [PubMed] [Google Scholar]
- Hewings D. S.; Rooney T. P.; Jennings L. E.; Hay D. A.; Schofield C. J.; Brennan P. E.; Knapp S.; Conway S. J. Progress in the development and application of small molecule inhibitors of bromodomain-acetyl-lysine interactions. J. Med. Chem. 2012, 55, 9393–9413. 10.1021/jm300915b. [DOI] [PubMed] [Google Scholar]
- Filippakopoulos P.; Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat. Rev. Drug Discovery 2014, 13, 337–356. 10.1038/nrd4286. [DOI] [PubMed] [Google Scholar]
- Dey A.; Yang W.; Gegonne A.; Nishiyama A.; Pan R.; Yagi R.; Grinberg A.; Finkelman F. D.; Pfeifer K.; Zhu J.; Singer D.; Zhu J.; Ozato K. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. EMBO J. 2019, 38, e100293. 10.15252/embj.2018100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perner F.; Armstrong S. A. Targeting Chromatin Complexes in Myeloid Malignancies and Beyond: From Basic Mechanisms to Clinical Innovation. Cells 2020, 9, 2721. 10.3390/cells9122721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T.; Lu W.; Luo C. A patent review of BRD4 inhibitors (2013–2019). Expert Opin. Ther. Pat. 2020, 30, 57–81. 10.1080/13543776.2020.1702645. [DOI] [PubMed] [Google Scholar]
- Zaware N.; Zhou M. M. Bromodomain biology and drug discovery. Nat. Struct. Mol. Biol. 2019, 26, 870–879. 10.1038/s41594-019-0309-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochran A. G.; Conery A. R.; Sims R. J. 3rd. Bromodomains: a new target class for drug development. Nat. Rev. Drug Discovery 2019, 18, 609–628. 10.1038/s41573-019-0030-7. [DOI] [PubMed] [Google Scholar]
- Alqahtani A.; Choucair K.; Ashraf M.; Hammouda D. M.; Alloghbi A.; Khan T.; Senzer N.; Nemunaitis J. Bromodomain and extra-terminal motif inhibitors: a review of preclinical and clinical advances in cancer therapy. Future science OA 2019, 5, FSO372. 10.4155/fsoa-2018-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J.; Hoke H. A.; Lin C. Y.; Lau A.; Orlando D. A.; Vakoc C. R.; Bradner J. E.; Lee T. I.; Young R. A. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 2013, 153, 320–334. 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J.; Shi J.; Wang E.; Rappaport A. R.; Herrmann H.; Sison E. A.; Magoon D.; Qi J.; Blatt K.; Wunderlich M.; Taylor M. J.; Johns C.; Chicas A.; Mulloy J. C.; Kogan S. C.; Brown P.; Valent P.; Bradner J. E.; Lowe S. W.; Vakoc C. R. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard G. S.; Wang L.; Fidanze S. D.; Hasvold L. A.; Liu D.; Pratt J. K.; Park C. H.; Longenecker K.; Qiu W.; Torrent M.; Kovar P. J.; Bui M.; Faivre E.; Huang X.; Lin X.; Wilcox D.; Zhang L.; Shen Y.; Albert D. H.; Magoc T. J.; Rajaraman G.; Kati W. M.; McDaniel K. F. Discovery of N-Ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]- 6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET Bromodomain Inhibitor with Selectivity for the Second Bromodomain. J. Med. Chem. 2020, 63, 5585–5623. 10.1021/acs.jmedchem.0c00628. [DOI] [PubMed] [Google Scholar]
- Faivre E. J.; McDaniel K. F.; Albert D. H.; Mantena S. R.; Plotnik J. P.; Wilcox D.; Zhang L.; Bui M. H.; Sheppard G. S.; Wang L.; Sehgal V.; Lin X.; Huang X.; Lu X.; Uziel T.; Hessler P.; Lam L. T.; Bellin R. J.; Mehta G.; Fidanze S.; Pratt J. K.; Liu D.; Hasvold L. A.; Sun C.; Panchal S. C.; Nicolette J. J.; Fossey S. L.; Park C. H.; Longenecker K.; Bigelow L.; Torrent M.; Rosenberg S. H.; Kati W. M.; Shen Y. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature 2020, 578, 306–310. 10.1038/s41586-020-1930-8. [DOI] [PubMed] [Google Scholar]
- Amorim S.; Stathis A.; Gleeson M.; Iyengar S.; Magarotto V.; Leleu X.; Morschhauser F.; Karlin L.; Broussais F.; Rezai K.; Herait P.; Kahatt C.; Lokiec F.; Salles G.; Facon T.; Palumbo A.; Cunningham D.; Zucca E.; Thieblemont C. Bromodomain inhibitor OTX015 in patients with lymphoma or multiple myeloma: a dose-escalation, open-label, pharmacokinetic, phase 1 study. Lancet. Haematology 2016, 3, e196–204. 10.1016/S2352-3026(16)00021-1. [DOI] [PubMed] [Google Scholar]
- Konuma T.; Yu D.; Zhao C.; Ju Y.; Sharma R.; Ren C.; Zhang Q.; Zhou M. M.; Zeng L. Structural Mechanism of the Oxygenase JMJD6 Recognition by the Extraterminal (ET) Domain of BRD4. Sci. Rep. 2017, 7, 16272. 10.1038/s41598-017-16588-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong M.; Sun Y.; Xi Z.; Milazzo G.; Poulos R. C.; Bartenhagen C.; Bell J. L.; Mayoh C.; Ho N.; Tee A. E.; Chen X.; Li Y.; Ciaccio R.; Liu P. Y.; Jiang C. C.; Lan Q.; Jayatilleke N.; Cheung B. B.; Haber M.; Norris M. D.; Zhang X. D.; Marshall G. M.; Wang J. Y.; Huttelmaier S.; Fischer M.; Wong J. W. H.; Xu H.; Perini G.; Dong Q.; George R. E.; Liu T. JMJD6 is a tumorigenic factor and therapeutic target in neuroblastoma. Nat. Commun. 2019, 10, 3319. 10.1038/s41467-019-11132-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French C. A.; Rahman S.; Walsh E. M.; Kuhnle S.; Grayson A. R.; Lemieux M. E.; Grunfeld N.; Rubin B. P.; Antonescu C. R.; Zhang S.; Venkatramani R.; Dal Cin P.; Howley P. M. NSD3-NUT fusion oncoprotein in NUT midline carcinoma: implications for a novel oncogenic mechanism. Cancer Discovery 2014, 4, 928–941. 10.1158/2159-8290.CD-14-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q.; Zeng L.; Shen C.; Ju Y.; Konuma T.; Zhao C.; Vakoc C. R.; Zhou M. M. Structural Mechanism of Transcriptional Regulator NSD3 Recognition by the ET Domain of BRD4. Structure 2016, 24, 1201–1208. 10.1016/j.str.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen C.; Ipsaro J. J.; Shi J.; Milazzo J. P.; Wang E.; Roe J. S.; Suzuki Y.; Pappin D. J.; Joshua-Tor L.; Vakoc C. R. NSD3-Short Is an Adaptor Protein that Couples BRD4 to the CHD8 Chromatin Remodeler. Mol. Cell 2015, 60, 847–859. 10.1016/j.molcel.2015.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo M.; Hacein-Bey S.; de Saint Basile G.; Gross F.; Yvon E.; Nusbaum P.; Selz F.; Hue C.; Certain S.; Casanova J. L.; Bousso P.; Deist F. L.; Fischer A. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science 2000, 288, 669–672. 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S.; Von Kalle C.; Schmidt M.; McCormack M. P.; Wulffraat N.; Leboulch P.; Lim A.; Osborne C. S.; Pawliuk R.; Morillon E.; Sorensen R.; Forster A.; Fraser P.; Cohen J. I.; de Saint Basile G.; Alexander I.; Wintergerst U.; Frebourg T.; Aurias A.; Stoppa-Lyonnet D.; Romana S.; Radford-Weiss I.; Gross F.; Valensi F.; Delabesse E.; Macintyre E.; Sigaux F.; Soulier J.; Leiva L. E.; Wissler M.; Prinz C.; Rabbitts T. H.; Le Deist F.; Fischer A.; Cavazzana-Calvo M. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science 2003, 302, 415–419. 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S.; Garrigue A.; Wang G. P.; Soulier J.; Lim A.; Morillon E.; Clappier E.; Caccavelli L.; Delabesse E.; Beldjord K.; Asnafi V.; MacIntyre E.; Dal Cortivo L.; Radford I.; Brousse N.; Sigaux F.; Moshous D.; Hauer J.; Borkhardt A.; Belohradsky B. H.; Wintergerst U.; Velez M. C.; Leiva L.; Sorensen R.; Wulffraat N.; Blanche S.; Bushman F. D.; Fischer A.; Cavazzana-Calvo M. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 2008, 118, 3132–3142. 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Dunbar C. E. Stem cell gene therapy: the risks of insertional mutagenesis and approaches to minimize genotoxicity. Frontiers of medicine 2011, 5, 356–371. 10.1007/s11684-011-0159-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowe B. L.; Larue R. C.; Yuan C.; Hess S.; Kvaratskhelia M.; Foster M. P. Structure of the Brd4 ET domain bound to a C-terminal motif from gamma-retroviral integrases reveals a conserved mechanism of interaction. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 2086–2091. 10.1073/pnas.1516813113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoli D.; Yang Y. C.; Clark S. C.; Kreider B. L.; Caracciolo D.; Rovera G. Synergistic and antagonistic effects of recombinant human interleukin (IL) 3, IL-1 alpha, granulocyte and macrophage colony-stimulating factors (G-CSF and M-CSF) on the growth of GM-CSF-dependent leukemic cell lines. J. Immunol. 1987, 139, 3348–3354. [PubMed] [Google Scholar]
- Morgado-Pascual J. L.; Rayego-Mateos S.; Tejedor L.; Suarez-Alvarez B.; Ruiz-Ortega M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharmacol. 2019, 10, 1315. 10.3389/fphar.2019.01315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. 10.3390/ijms17111849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali I.; Choi G.; Lee K. BET Inhibitors as Anticancer Agents: A Patent Review. Recent Pat. Anti-Cancer Drug Discovery 2017, 12, 340–364. 10.2174/1574892812666170808121228. [DOI] [PubMed] [Google Scholar]
- Madhavi Sastry G.; Adzhigirey M.; Day T.; Annabhimoju R.; Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J. Comput.-Aided Mol. Des. 2013, 27, 221–234. 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- Schrödinger. Schrödinger Release 2020-1; Schrödinger, 2020.
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J.; van der Spoel D.; van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. 10.1016/0010-4655(95)00042-E. [DOI] [Google Scholar]
- Abraham M. J.; Murtola T.; Schulz R.; Páll S.; Smith J. C.; Hess B.; Lindahl E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. 10.1016/j.softx.2015.06.001. [DOI] [Google Scholar]
- Huang J.; MacKerell A. D. Jr CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R.; Brooks C. L. III; Mackerell A. D. Jr; Nilsson L.; Petrella R. J.; Roux B.; Won Y.; Archontis G.; Bartels C.; Boresch S. CHARMM: the biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdés-Tresanco M. S.; Valdés-Tresanco M. E.; Valiente P. A.; Frías E. M.. gmx_MMPBSA (Version v1.4.2); http://doi.org/10.5281/zenodo.4569307.
- Miller B. R. III; McGee T. D. Jr; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA. py: an efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Salomon-Ferrer R.; Case D. A.; Walker R. C. An overview of the Amber biomolecular simulation package. Wiley Interdisciplinary Reviews: Computational Molecular Science 2013, 3, 198–210. 10.1002/wcms.1121. [DOI] [Google Scholar]
- Case D. A.; Cheatham T. E. III; Darden T.; Gohlke H.; Luo R.; Merz K. M. Jr; Onufriev A.; Simmerling C.; Wang B.; Woods R. J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue R. C.; Xing E.; Kenney A. D.; Zhang Y.; Tuazon J. A.; Li J.; Yount J. S.; Li P. K.; Sharma A. Rationally Designed ACE2-Derived Peptides Inhibit SARS-CoV-2. Bioconjugate Chem. 2021, 32, 215–223. 10.1021/acs.bioconjchem.0c00664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo Y.; MacLeod R. A.; Uphoff C. C.; Drexler H. G.; Nishizaki C.; Katayama Y.; Kimura G.; Fujii N.; Omoto E.; Harada M.; Orita K. Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23). Leukemia 1997, 11, 1469–1477. 10.1038/sj.leu.2400768. [DOI] [PubMed] [Google Scholar]
- Biagi E.; Bambacioni F.; Gaipa G.; Casati C.; Golay J.; Biondi A.; Introna M. Efficient lentiviral transduction of primary human acute myelogenous and lymphoblastic leukemia cells. Haematologica 2001, 86, 13–16. [PubMed] [Google Scholar]