

Abstract
On November 28, 2018, the Food and Drug Administration approved gilteritinib (Xospata®; Astellas, Northbrook, IL), a small molecule FMS-like tyrosine kinase 3 (FLT3) inhibitor, for treatment of relapsed or refractory (R/R) acute myeloid leukemia (AML) with a FLT3 mutation as detected by an FDA-approved test. In the ADMIRAL Study, patients were randomized 2:1 to receive gilteritinib or standard chemotherapy and stratified by response to first-line treatment and intensity of prespecified chemotherapy. Efficacy was established on interim analysis based on complete remission (CR)+CR with partial hematologic recovery (CRh) rate, duration of CR+CRh, and conversion from transfusion dependence (TD) to transfusion independence (TI) in 138 patients in the gilteritinib arm. With median follow-up of 4.6 months (95% CI 2.8–15.8 months) at interim analysis, the CR+CRh rate was 21% (95% confidence interval [CI], 15%−29%), median duration of CR+CRh was 4.6 (range, 0.1–15.8+) months, and conversion from TD to TI was 31%. Revised labeling approved May 29, 2019 included the results of the final analysis, showing an improvement in overall survival (OS) with gilteritinib compared to chemotherapy (hazard ratio [HR] 0.64 [95% CI 0.49–0.83], 1-sided p=0.0004); median OS 9.3 months vs 5.6 months). The OS benefit was observed in both high and low chemotherapy intensity subgroups. Labeling includes a boxed warning for differentiation syndrome (DS) and warnings for posterior reversible encephalopathy syndrome, QT prolongation, pancreatitis, and embryo-fetal toxicity. Safe use requires frequent monitoring of electrocardiograms and blood chemistries. Assessments of long-term safety are pending.
Introduction
Fms related tyrosine kinase 3 (FLT3) is a cytokine receptor expressed on the surface of hematopoietic cells in the early stages of hematopoiesis (1). The exact role of FLT3 in normal hematopoiesis is not entirely clarified, but it appears to be involved with early cell growth and differentiation (2). The FLT3 gene is mutated in about 25–30% of cases of acute myeloid leukemia (AML) (3). Mutations may include internal tandem duplication (ITD), resulting in the insertion of duplicated sequences of amino acids in the juxtamembrane region of the FLT3 protein, or point mutations leading to changes in the activation loop of the tyrosine kinase domain (TKD). In either situation, mutation results in constitutive activation of FLT3 kinase. FLT3-ITD is associated with a worse prognosis and more aggressive disease, whereas the effect of FLT3-TKD on prognosis is debatable (4, 5). Treatment outcomes for patients with AML who have relapsed after or are refractory to initial therapy is poor, especially for patients with early relapse (<6 months duration of remission) (6). The GOELAMS study showed that for patients with refractory or first relapse of AML, the three strongest independent adverse prognostic factors for overall survival (OS) were relapse < 12 months (including refractory patients), FLT3-ITD positive status, and high-risk cytogenetics (7).
In the past, treatment of R/R FLT3-mutated AML was largely limited to cytotoxic chemotherapy, with or without allogeneic hematopoietic stem cell transplantation. Several FLT3 inhibitors were investigated as monotherapy in patients with R/R FLT3-mutated AML, but complete remissions (CRs) with full count recovery were limited (8–17). In terms of combination therapy, a prior randomized trial with the first generation FLT3 inhibitor lestaurtinib showed no benefit to adding lestaurtinib to intensive salvage chemotherapy in patients with first relapse of FLT3-mutated AML (18). The CR rate on the chemotherapy alone arm was only 12%. In the front-line setting, however, addition of the FLT3 inhibitor midostaurin to standard intensive induction chemotherapy improved clinical outcomes in patients with FLT3 positive AML (19). In 2017, the FDA approved midostaurin in combination with standard cytarabine and daunorubicin induction and cytarabine consolidation for the treatment of newly-diagnosed FLT3-mutated AML, but due to its lack of activity as monotherapy, midostaurin is not indicated as a single-agent induction therapy (20). Thus, prior to the approval of gilteritinib, no FLT3 targeting agents were approved for use as monotherapy or in patients with FLT3-mutated relapsed or refractory (R/R) AML. Thus, there is a clear need for new treatment options for patients with R/R FLT3-mutated AML.
Herein, we provide a summary of FDA’s review of the marketing application that led to approval of gilteritinib for treatment of adult patients who have R/R AML with a FLT3 mutation as detected by an FDA-approved test.
Mechanism of action
Gilteritinib inhibits multiple receptor tyrosine kinases. In an in vitro pharmacology assay, gilteritinib demonstrated in vitro activity (> 50% inhibition at 1 nanomolar [nM]) against FLT3, nucleophosmin 1-anaplastic lymphoma kinase (NPM1-ALK), leukocyte tyrosine kinase (LTK), anaplastic lymphoma kinase (ALK), and AXL tyrosine kinase. At 5 nM, gilteritinib also produced greater than half maximal inhibition of tropomyosin receptor kinase A and proto-oncogene tyrosine-protein kinases (TRKA, ROS, RET, and MER). At nanomolar concentrations (0 control, 0.1,1, and 10 nM), gilteritinib inhibited FLT3 receptor signaling with dose response, e.g., decreasing phosphorylation of signal transducer and activator of transcription 5 (STAT5) to 0%, protein kinase B (AKT) to 9%, and extracellular signal-regulated kinase (ERK) to 1% compared to control in human AML cells expressing FLT3-internal tandem duplication (FLT3-ITD). Gilteritinib decreased proliferation in cells exogenously expressing FLT3 mutants, including FLT3-ITD and TKD mutations FLT3-D835H, FLT3-D835Y and FLT3-ITD-D835Y with concentration with 50% inhibition (IC50) of 0.92–2.1 nM, and it induced apoptosis in leukemia cells expressing FLT3-ITD. Furthermore, gilteritinib demonstrated an equivalent inhibitory effect on cells expressing several less common FLT3-TKD mutations compared with the effect on the cells expressing the FLT3-ITD mutation (21). This included testing of the I836 mutation, other D835 mutations, and variety of other mutations reported to occur in AML. However, it should be noted that it was not active against all TKD mutations, in particular the F691 TKD mutation (21).
Clinical pharmacology
Dose selection:
In a Phase 1 dose‐escalation study (2215-CL-0101), gilteritinib doses ranging from 20 to 450 mg administered orally once daily were investigated in patients with R/R AML. The maximum tolerated dose (MTD) of gilteritinib was determined to be 300 mg once daily. Gilteritinib dose of 120 mg once daily was selected as the recommended phase 2 dose (RP2D) based on the observed safety profile as well as clinical responses in study 2215-CL-0101. In this study, ex vivo FLT3 plasma inhibitory assay results showed that by day 8 of cycle 1 of gilteritinib dosing, greater than 90% inhibition of FLT3 phosphorylation at doses of ≥ 80 mg was achieved. An exposure-efficacy analysis suggested a potential threshold of gilteritinib concentration at 100 ng/mL with 22%, 48%, and 50% composite complete remission (CRc) (defined as CR + CR with incomplete hematologic recovery [CRi] + CR with incomplete platelet recovery [CRp]) rates at steady-state Ctrough values of <100, 100 to 500, and >500 ng/mL, respectively. In line with this finding, 97% (37/38) of the FLT3-mutated R/R AML patients, who achieved a best overall response of CR+CR with partial hematologic recovery (CRh) in study 2215-CL-0101 had gilteritinib plasma Ctrough ≥100 ng/mL. A Monte Carlo simulation estimated that approximately 99.4 % of patients will have a steady-state Ctrough above 100 ng/mL at the 120 mg dose. In study 2215-CL-0101, CR+CRh rates for the FLT3-mutated patients were 25.0%, 23.2%, 19.1% and 30% for the 80, 120, 200 and 300 mg dose levels, respectively. However, preliminary safety analysis of this study showed that 46% of the FLT3-mutated R/R AML patients at dose 200 mg required dose interruption compared to 19% at dose 120 mg.
The effect of dose escalation to 200 mg QD in FLT3-mutated R/R AML patients who did not achieve CRc response after 4 weeks of treatment was evaluated in the subsequent Phase 3 study 2215-CL-0301 (ADMIRAL). The CR+CRh rate in patients who had dose increase to 200 mg (6.4% with 95% CI of 2.1%−14.3%) was lower than that in patients who remained on 120 mg (25.5% with 95% CI of 17.6%−34.6%) and in patients on the chemotherapy control arm (12.9% with 95% CI of 7.6%−20.1%). Safety analysis showed higher risk of both all grades and grade 3 or higher hematological (neutropenia, anemia, and thrombocytopenia) and non-hematological (aspartate aminotransferase [AST], alanine aminotransferase [ALT], and creatine kinase elevations, and cardiac failure) treatment-emergent adverse events (AEs) in patients who had dose increased to 200 mg compared to patients who remained on dose 120 mg. Collectively, data obtained from phase 1 and phase 3 studies supported gilteritinib 120 mg QD as the dose that provided the optimal benefit risk profile in patients with FLT3-mutated R/R AML and did not support a subsequent increase to 200 mg QD.
Clinical Pharmacokinetics:
Following oral administration of gilteritinib tablet, peak concentrations were observed at a median Tmax of ~4 to 6 hours. Gilteritinib exhibited dose-proportional pharmacokinetics at doses ranging from 20 to 450 mg administered once daily with an estimated half-life (T1/2) of 113 hours. Steady-state gilteritinib concentrations were achieved by day 15 after once daily dosing with an accumulation index (Rac) range of 3.23 – 9.64. A high fat meal had no clinically significant impact on gilteritinib absorption. Based on results from a dedicated hepatic impairment study, a mass balance study, and population PK analyses, no dose adjustment is recommended for R/R AML patients with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment and mild (CrCL 50–80 mL/min) or moderate (CrCL 30–50 mL/min) renal impairment. The effect of severe hepatic (Child-Pugh Class C) and severe renal impairment (CLCr ≤ 29 mL/min) on gilteritinib pharmacokinetics has not been evaluated.
Drug interactions:
Gilteritinib is primarily metabolized by CYP3A4. A safety analysis from Study 2215-CL-0101 showed that concomitant use of strong or moderate CYP3A inhibitors was not associated with clinically significant safety issues. Co-administration of rifampicin (a strong CYP3A inducer), decreased gilteritinib exposure by approximately 70%; therefore, the concomitant use of strong CYP3A inducers with gilteritinib should be avoided.
Assessment of efficacy
Clinical trial description
ADMIRAL (NCT03182244) was an open-label, multicenter, phase 3 trial in adult patients with R/R AML having a FLT3-ITD, -D835, or -I836 mutation by the LeukoStrat CDx FLT3 Mutation Assay. Patients were randomized 2:1 to either gilteritinib or one of four prespecified salvage chemotherapy regimens. The starting dose for gilteritinib was 120 mg orally once daily in continuous 28-day cycles until unacceptable toxicity or lack of clinical benefit. Dose escalation to 200 mg orally once daily was permitted if the patient did not achieve CR, CRi or CRp after cycle 1 based on investigator assessed response. The control arm contained high-intensity (mitoxantrone, etoposide, and intermediate-dose cytarabine [MEC] or fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin [FLAG-IDA]) and low-intensity (low-dose cytarabine [LDAC] or azacitidine) regimens. Chemotherapy that would be used for patients in the control arm was chosen by the investigator prior to randomization. Randomization was stratified by response to first-line AML therapy (relapse ≤ 6 months after allogeneic hematopoietic stem cell transplantation [HSCT], relapse > 6 months after allogeneic HSCT, primary refractory without HSCT, relapse ≤ 6 months after CRc and no HSCT, and relapse after 6 months after CRc and no HSCT) and prespecified chemotherapy (high- versus low-intensity).
The co-primary efficacy endpoints were CR+CRh rate and OS. At the first interim analysis, only the coprimary endpoint of CR+CRh rate was evaluated for patients on the gilteritinib arm. The objective was to test the hypothesis that the rate of CR+CRh was > 12%. With a planned sample size of 141 patients and a true response rate of 21%, the study had 80% power to exclude a 12% response rate. The first interim analysis was to be performed when subjects were randomized into the gilteritinib arm and were at least 112 days (4 treatment cycles) post first dose or randomization (for subjects who received no study drug). The final analysis tested the co-primary endpoint of OS. The study was designed assuming a median OS for the control arm of 5 months. A sample size of 369 patients (258 OS events) was calculated to demonstrate a hazard ratio of 0.65 with 90% power at a 1-sided alpha of 0.0245. CR rate and event-free survival (EFS) were key secondary efficacy endpoints.
Demographics and disposition
The demographics of the efficacy population of the ADMIRAL trial are shown in Table 1. The arms were well balanced in terms of demographics and disease characteristics at baseline. At the time of the first interim analysis, the median follow-up on the gilteritinib arm was 4.6 (95% CI 2.8–15.8) months. At the time of the final analysis, the median follow-up was 17.8 months (95% CI 14.9–19.1 months).
Table 1:
Demographic and disease characteristics of patients included in the primary efficacy analysis and the primary safety population
| ADMIRAL Study: Randomized Population | Gilteritinib-treated Safety population (N=319) |
||
|---|---|---|---|
| Demographic parameter | Gilteritinib (N=247) |
Chemotherapy (N=124) |
|
| Sex | |||
| Male | 116 (47%) | 54 (44%) | 149 (47%) |
| Female | 131 (53%) | 70 (57%) | 170 (53%) |
| Age | |||
| Median (years) | 62 | 62 | 61 |
| Min, max (years) | 20, 84 | 19, 85 | 20, 90 |
| Age group | |||
| <65 | 141 (57%) | 75 (60%) | 183 (58%) |
| ≥65 | 106 (43%) | 49 (40%) | 136 (42%) |
| Race | |||
| White | 145 (59%) | 75 (60%) | 203 (64%) |
| Asian | 69 (28%) | 33 (27%) | 74 (23%) |
| Black/African American | 14 (6%) | 7 (6%) | 17 (5%) |
| Other1 | 19 (8%) | 9 (7%) | 25 (8%) |
| Ethnicity | |||
| Hispanic or Latino | 12 (5%) | 2 (2%) | 16 (5%) |
| Not Hispanic or Latino2 | 235 (95%) | 122 (98%) | 303 (95%) |
| Region | |||
| North America | 114 (46%) | 52 (42%) | 178 (56%) |
| Europe3 | 68 (28%) | 43 (35%) | 72 (23%) |
| Asia | 65 (26%) | 29 (23%) | 69 (22%) |
| Disease status | |||
| Untreated relapse | 151 (61%) | 74 (60%) | NA |
| Primary refractory | 96 (39%) | 49 (40%) | NA |
| Refractory relapse | 0 | 1 (1%) | NA |
| Prior relapses | |||
| 0 | 96 (39%) | 49 (40%) | NA |
| 1 | 149 (60%) | 74 (60%) | NA |
| 2+ | 2 (1%) | 1 (1%) | NA |
| FLT3 mutation | |||
| ITD | 215 (87%) | 113 (91%) | 262 (82%) |
| TKD | 21 (9%) | 10 (8%) | 27 (8%) |
| Both | 7 (3%) | 0 | 10 (3%) |
| Other4 | 4 (2%) | 1 (1%) | 20 (6%) |
Includes Native Hawaiian or other Pacific Islander, Native American/Alaskan Native, “other,” unknown race, and no race listed
Includes patients listed as not Hispanic or Latino, unknown, or with no ethnicity listed
Includes Turkey and Israel
Missing, unknown, or negative by central review
NA, not available
Median time on therapy was 4.1 months (range 0.1–29.1 months) for patients on the gilteritinib arm and 0.9 months (range 0.2–7.1 months) on the chemotherapy arm. Within the chemotherapy arm, the median duration of exposure was similar for both high and low intensity regimens. A total of 63/247 (26%) patients proceeded to HSCT after treatment with gilteritinib compared to 12/124 (15%) on the chemotherapy arm.
Efficacy Evaluation
The first interim analysis evaluated the endpoint of CR+CRh in the gilteritinib arm only. The rate of CR+CRh was 22% of the primary analysis population of 142 patients with R/R AML, with a lower 95% CI boundary of 15%. Thus, the primary objective of excluding a CR+CRh rate of 12% was met. Limiting the analysis population to only the subgroup found to be FLT3 mutation positive by the companion diagnostic test (N=138), the CR+CRh rate was 21% (Table 2). The median time to first CR or CRh was 3.6 months (range 0.9–9.6 months). Results were generally consistent across subgroups. However, among 12 patients with a FLT3-TKD mutation alone (i.e. without concomitant FLT3-ITD mutation), there were no CR or CRh.
Table 2:
Response rates (95% confidence intervals) and median (range) duration of response in months.
| Interim analysisa | Final analysis | ||
|---|---|---|---|
| Group | Gilteritinib (N=138) | Gilteritinib (N=247) | Chemotherapy (124) |
| CR/CRh | 29 (21%) (15%, 29%) |
57 (23%) (18%, 29%) |
N/Ab |
| DOCR+CRh | 4.6 (0.1, 15.8+) |
7.4 (<0.1+, 23.1+) |
N/Ab |
| CR | 16 (12%) (7%, 18%) |
35 (14%) (10%, 19%) |
13 (11%) (6%, 17%) |
| DOCR | 8.6 (1.0, 13.8) |
14.8 (0.6, 23.1+) |
1.8 (<0.1+, 1.8) |
| CRh | 13 (9%) (5%, 16%) |
22 (9%) (6%, 13%) |
N/Ab |
| DOCRh | 2.9 (0.1, 15.8+) |
2.9 (<0.1+, 9.5+) |
N/Ab |
CR, complete remission; CRh, complete remission with incomplete hematologic recovery; DOCR+CRh, duration of CR plus CRh responses; DOCR, duration of CR; DOCRh, duration of CRh; NA, not applicable; NE=not estimable
This analysis was used for the approval. The chemotherapy arm was not assessed in the interim analysis
CRh was not assessed by FDA on the chemotherapy arm given that CRh is not considered an appropriate efficacy endpoint for regimens causing drug-induced cytopenias.
Transfusion independence was a secondary endpoint in the interim analysis. Among the 106 patients who were dependent on transfusion of red blood cells (RBCs) or platelets at baseline, 33 (31%, 95%CI [22.5%, 40.9%]) became independent of RBC and platelet transfusions during any 56-day post-baseline period. Of the 32 patients who were independent of RBC and platelet transfusions at baseline, 17 (53%) remained transfusion independent during any 56-day post-baseline period.
At the time of the final analysis, a total of 371 patients were randomized, 247 to gilteritinib and 124 to chemotherapy. There was a survival benefit for the gilteritinib arm (HR, 0.64; 95% CI, 0.49–0.83; 1-sided p = 0.0004), with median survival 9.3 months for gilteritinib versus 5.6 months for chemotherapy (Figure 1). Subgroup analysis showed a consistent survival benefit for all patient groups, with the exception of patients with adverse cytogenetic risk status (see Supplementary Fig. S1). The survival benefit was similar in the high intensity and low intensity subgroups. Furthermore, the HR for OS in the TKD only subgroup (n=31; 21 gilteritinib arm, 10 control arm) was 0.69 (HR 0.29–1.64).
Figure 1:
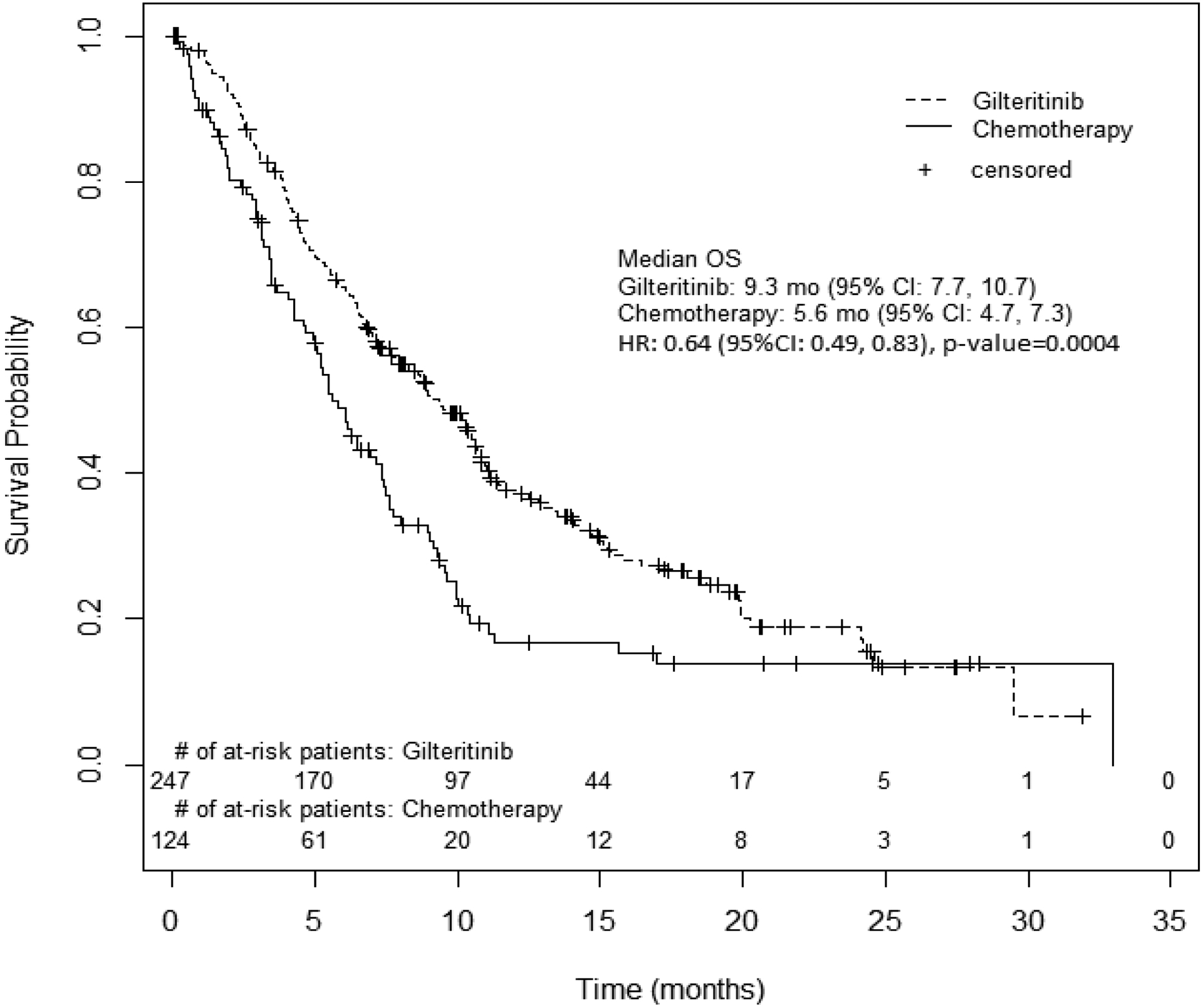
OS. Kaplan-Meier plot of OS for patients with relapsed or refractory AML on the ADMIRAL trial.
At the final analysis, the CR rate for patients on the gilteritinib arm was 14% versus 11% on the chemotherapy arm. The CR rate for patients preselected to receive high intensity chemotherapy was 15% for gilteritinib and 16% for chemotherapy, whereas the CR rate for patients preselected for low intensity chemotherapy was 12% for gilteritinib and 2% for chemotherapy (Supplementary Fig. S2). In the FLT3-TKD only subgroup (n=31), the CR rate on the gilteritinib arm was 10% (2/21) and 20% (n= 2/10) on the chemotherapy arm. The other secondary endpoint of EFS did not meet the prespecified criteria for statistical significance (hazard ratio of 0.79, 95% CI 0.58–1.09, 1-sided p-value 0.042).
Assessment of Safety
Nonclinical Toxicology
FDA identified important safety risks based on analysis of nonclinical pharmacology and toxicology data. In the 13-week oral repeated dose toxicity studies in rats and dogs, target organs of toxicity included the eye and the kidney. Eye toxicities in rats included inflammatory cell infiltration in the choroid, ciliary body, iris, and/or conjunctiva, and in dogs, included vacuolation in the rod-cone layer/outer nuclear layer of the retina. Kidney toxicities in rats included vacuolation of the renal medulla, increased mesangial matrix, tubular basophilia, hyaline droplets in the renal tubule, hyaline casts, and edematous change in the renal papilla, and in dogs, included renal tubular regeneration and focal congestion in the renal medulla. In addition, toxicities were observed in the liver in rats and dogs, and male reproductive organs in dogs only. Gilteritinib had low in vitro potency at blocking hERG currents based on IC50 was 16 μmol/L (8.84 μg/mL).
Safety events
The safety analysis results were based primarily on the results of the ADMIRAL study and the Phase 1 Studies 2215-CL-0101 (NCT02014558) and 2215-CL-0102 (NCT02181660). The primary safety population (n=319) included patients with R/R AML who received 120 mg gilteritinib, regardless of FLT3 status (Table 1). The median duration of exposure to gilteritinib in the safety population was 3.6 (range, 0.1 – 43.4) months.
Five deaths (2%) were concluded to have potentially resulted from a direct toxicity of gilteritinib. Proximate causes of death on the gilteritinib arm included cardiac arrest (n=3) and one case each of differentiation syndrome (DS) and pancreatitis. Gilteritinib was interrupted in 29% of patients, dose reduced in 6%, and discontinued treatment permanently in 7% due to an adverse reaction (AR). The most common ARs leading to dose interruption were AST increased (6%), ALT increased (6%), and fever (4%). The most common ARs resulting in discontinuation were AST increased (2%) and ALT increased (2%).
Common (≥ 25%) nonhematological ARs in the primary safety population are listed in Supplementary Table 1, which includes the incidence of toxicities over the duration of gilteritinib therapy (grouped terms as described in Supplementary Table 2 were used in the analysis). However, we focused our analysis of common ARs compared to standard chemotherapy on the Phase 3, ADMIRAL trial. On this trial, a total of 355 patients received treatment with gilteritinib (n = 246) or chemotherapy (n = 109). Median time on therapy was roughly one month on the control arm, and therefore, the comparative safety analyses were limited to the first 30 days of therapy. Analyses were performed according to whether patients were preselected for high or low intensity chemotherapy, as the comparative safety profile differed; common ARs in both groups are listed in Tables 3 and 4. Examination of ARs occurring after the first 30 days of treatment did not reveal any new toxicities, although ARs, including severe and serious ARs, continued to occur later in the course of treatment.
Table 3:
Common (≥ 10% any-grade, ≥ 5% Grade 3–5) Treatment-emergent Adverse Reactions in the Preselected High Intensity Chemotherapy Subgroup in the First 30 Days of the ADMIRAL trial Safety Populationa
| Gilteritinib (N=149) | Chemotherapy (N=68) | |||
|---|---|---|---|---|
| Adverse reactionb | Any grade | Grade ≥ 3 | Any grade | Grade ≥ 3 |
| Myalgia/arthralgia | 38% | 1% | 29% | 4% |
| Transaminase increased | 31% | 10% | 16% | 7% |
| Fatigue/malaise | 24% | 1% | 13% | 3% |
| Constipation | 20% | 0 | 15% | 0 |
| Febrile neutropenia | 17% | 17% | 44% | 44% |
| Fever | 17% | 1% | 31% | 6% |
| Nausea | 15% | 0 | 38% | 0 |
| Rash | 15% | 1% | 31% | 3% |
| Dyspnea | 13% | 1% | 13% | 9% |
| Edema | 13% | 0 | 19% | 0 |
| Neuropathy | 13% | 0 | 0 | 0 |
| Cough | 12% | 1% | 7% | 0 |
| Mucositis | 12% | 0 | 44% | 7% |
| Abdominal pain | 11% | 0 | 24% | 0 |
| Dizziness | 11% | 0 | 3% | 0 |
| Headache | 11% | 0 | 18% | 0 |
N = 217 adults with R/R AML treated with gilteritinib or high intensity chemotherapy.
Includes grouped terms. See Supplementary Table S2 for further information.
Table 4:
Common (≥ 10% any-grade, ≥ 5% Grade 3–5) Treatment-emergent Adverse Reactions in the Preselected Low Intensity Chemotherapy Subgroup in the First 30 Days of the ADMIRAL trial Safety Populationa
| Gilteritinib (N=97) | Chemotherapy (N=41) | |||
|---|---|---|---|---|
| Adverse reactionb | Any grade | Grade ≥ 3 | Any grade | Grade ≥ 3 |
| Transaminase increased | 36% | 9% | 15% | 2% |
| Febrile neutropenia | 27% | 26% | 12% | 12% |
| Myalgia/arthralgia | 22% | 2% | 17% | 0 |
| Fatigue/malaise | 21% | 4% | 22% | 2% |
| Edema | 20% | 1% | 12% | 0 |
| Mucositis | 20% | 1% | 17% | 2% |
| Constipation | 13% | 1% | 12% | 0 |
| Diarrhea | 12% | 0 | 5% | 0 |
| Dyspnea | 11% | 3% | 5% | 5% |
| Fever | 11% | 0 | 17% | 0 |
| Nausea | 10% | 0 | 17% | 0 |
| Rash | 10% | 2% | 5% | 0 |
N = 138 adults with R/R AML treated with gilteritinib or low intensity chemotherapy.
Includes grouped terms. See Supplementary Table S2 for further information.
Common laboratory abnormalities more frequent on the gilteritinib arm in the first 30 days also differed according to whether patients were preselected for high or low intensity therapy, and thus, were analyzed separately (see Supplementary Tables 3 and 4). The most common nonhematologic Grade ≥ 3 laboratory abnormalities on the gilteritinib arm (across both low and high intensity comparison groups) in the first 30 days were increased ALT (6%), increased AST (5%), and decreased sodium (5%).
Adverse events of special interest
After a sudden death was observed in study 2215-CL-0101 and suspected to be related to QTc prolongation and torsade de pointes, extra electrocardiogram (ECG) monitoring was added to the protocol and the subsequent ADMIRAL trial. Patients on study 2215-CL-0101 who received a dose of 200 mg were more likely to have QTc prolongation to >500 msec. To mitigate the risk, the ADMIRAL trial stipulated consideration for dose reduction if cycle 1 day 8 ECG revealed QTc increased > 30 msec. On ADMIRAL, <1% of patients on the gilteritinib arm had maximum post-baseline QTcF of > 500 msec, but 25% had a maximum post-baseline increase in QTcF of > 30 to ≤ 60 msec and 5% had post-baseline increase of > 60 msec. Of the 9/246 (4%) patients on the gilteritinib arm who experienced postbaseline QTcF > 480 msec, the median time to postbaseline QTcF > 480 msec was 43 days (range 8 to 168 days).
DS is a potentially fatal clinical syndrome characterized by dyspnea, unexplained fever, weight gain, unexplained hypotension, acute kidney injury, and pulmonary infiltrates or pleuropericardial effusion initially recognized with the use of all-trans retinoic acid therapy in acute promyelocytic leukemia (22). The protocol did not provide guidelines for the identification and management of DS, but investigators reported DS as an adverse event in 1% of patients in the integrated safety database. FDA performed a comprehensive analysis of AEs, laboratory, and vital sign data to further characterize the risk and determined that 3% of patients had DS. Cases occurred as early as 2 days and up to 75 days after initiation of therapy. Some cases had concomitant rash or acute febrile neutrophilic dermatosis (“Sweet’s syndrome”), which has been described with other FLT3 inhibitors (23–25). Of the 11 patients who experienced DS, most were treated with steroids, and 9 (82%) recovered after treatment or after dose interruption of gilteritinib. Of the 11 DS cases, 6 (55%) were grade 1–2, 4 (36%) were grade 3–4, and 1 (9%) was fatal.
Posterior reversible encephalopathy syndrome (PRES) occurred in two patients (1%) treated with gilteritinib out of 319 patients in the integrated safety population. Events occurred on day 67 and 276 of therapy. Symptoms included seizure and altered mental status which resolved after discontinuation of gilteritinib. Additionally, a total of 4% experienced pancreatitis, including one fatal event.
Regulatory Insights
The ADMIRAL trial provided substantial evidence of efficacy and safety of gilteritinib to support its approval for treatment of adults with R/R AML with a FLT3 mutation as detected by an FDA-approved test. Based on the durable CR+CRh rate of 21% and data on conversion to TI at the time of the first interim analysis, along with its tolerable safety profile, regular approval was granted based on the palliative benefit to patients, including sustained transfusion independence while on therapy. CR+CRh with durability has been used previously to represent direct clinical benefit as temporary control of AML and relief from burdens of the disease during treatment with relatively nontoxic and nonmyelosuppressive drugs (26, 27). At the time of the final analysis, further benefit of gilteritinib was demonstrated, based on improved OS compared to standard chemotherapy regimens (HR 0.64; p=0.0004).
The OS benefit with gilteritinib was consistent in the majority of subgroups tested, which importantly included the high and low intensity therapy subgroups (high-intensity HR 0.66 [95% CI 0.47–0.93] and low-intensity HR 0.56 [95% CI 0.38–0.84]). The only subgroup with HR for OS > 1 was patients with unfavorable risk cytogenetics (HR 1.63 [95% CI 0.69–3.85]). The CR rate in this subgroup were also lower on the gilteritinib arm, with 1/26 (4%) patients achieving CR, compared to 3/11 (27%) on the control arm. While it is biologically plausible that patients with unfavorable-risk cytogenetics may not achieve a complete remission to a single-agent targeted therapy, the significance of these findings is unclear, as the number of patients in the subgroup was small.
Another subgroup worth mentioning was patients with a FLT3-TKD mutation alone (i.e. without concomitant FLT3-ITD mutation). In accordance with FDA’s guidance on targeted therapies in low-frequency molecular subsets, which contains information on how to handle rare molecular subsets that may not have been represented on the pivotal trial (28), the approval indication was broad with respect to the mechanism of action of this FLT3 inhibitor. Given initial concern for lack of CR+CRh in the TKD only subgroup at the time of the interim analysis, a postmarketing requirement (PMR) was issued to provide data to establish that risks of gilteritinib were outweighed by the potential benefit for patients with a FLT3-TKD mutation. The PMR was ultimately fulfilled when the final analyses of the ADMIRAL trial were supportive of a positive benefit/risk balance in this subset of patients. However, the sample size was too small to allow firm conclusions concerning the relative benefit in patients with FLT3 TKD versus ITD mutations. A postmarketing commitment (PMC) was also issued with the initial approval to clarify the sensitivity of various FLT3 mutations other than the ITDs and the D835Y TKD mutation by in vitro testing. The PMC was satisfied based on a report of in vitro testing showing inhibition of FLT3 I836 mutations, other D835 mutations, and other mutations reported to occur in AML. The safety profile of gilteritinib is generally consistent with that of tyrosine kinase inhibitors as a class, but additional unique toxicities were noted. DS was observed, including a fatal event, which led to a boxed warning and Medication Guide for patients. DS has been observed with the use of targeted IDH inhibitors (29) and other FLT3 inhibitors (23–25). The finding of DS with the use of different targeted inhibitors of driver mutations across drug classes in AML suggests that DS should be assessed in future clinical trials of targeted therapy for AML early in clinical development.
QT interval prolongation was a potentially life-threatening toxicity identified by FDA and is included in Warnings and Precautions in the prescribing information (PI). QT prolongation was observed early in the development of gilteritinib. Serious events related to QT prolongation, including one sudden death and multiple events of QTc prolongation to >500 msec were observed on study 2215-CL-0101, leading to increased monitoring, restrictions on enrollment, and guidance on dose holding and decrease for changes in QT interval in the ADMIRAL study. Serious events such as torsades can largely be prevented by ECG monitoring with ECGs on days 8 and 15 of cycle 1, prior to the start of cycles 2 and 3, and as clinically indicated thereafter. Dose modifications for patients who experience QT prolongation are included in the label. of dose modifications for QT prolongation on ECGs on days 8 and 15 of cycle 1, and prior to the start of cycles 2 and 3.
Pancreatitis and PRES were other potentially life threatening ARs identified by the FDA resulting in warnings and precautions in the label. However, despite these serious risks, the drug was generally well-tolerated, with only 6% requiring a dose reduction and 7% requiring discontinuation due to an AR. This manageable safety profile was critical when assessing benefit/risk of gilteritinib in patients with FLT3-mutated R/R AML. However, at the time of the initial approval, the median follow-up was only 4.6 months (17.8 months at the time of the final analysis). Therefore, a PMR was issued to demonstrate the safety of long-term treatment with gilteritinib across the three trials in the primary safety population.
In conclusion, use of gilteritinib at 120 mg once daily in patients with FLT3-mutated R/R AML led to durable CR+CRh and a transfusion benefit, indicating a palliative benefit to patients receiving this medication. Furthermore, improved OS was observed with gilteritinib with a HR of 0.64 (95% CI 0.49–0.83) compared to patients who received standard chemotherapy regimens, with results consistent compared to both high- (HR 0.66 [95% CI 0.47–0.93]) and low-intensity (HR 0.56 [95% CI 0.38–0.84]) regimens. Serious and life-threatening toxicities were rare but included DS, QT prolongation, pancreatitis, and PRES. Long-term safety follow-up will be provided via a PMR.
Supplementary Material
Footnotes
This is a U.S. Government work. There are no restrictions on its use.
Disclosure of Potential Conflicts of Interest: The authors report no financial interests or relationships with the commercial sponsors of any products discussed in this report.
References
- 1.Matthews W, Jordan CT, Wiegand GW, Pardoll D, Lemischka IR. A receptor tyrosine kinase specific to hematopoietic stem and progenitor cell-enriched populations. Cell. 1991;65(7):1143–52. [DOI] [PubMed] [Google Scholar]
- 2.Sitnicka E, Buza-Vidas N, Larsson S, Nygren JM, Liuba K, Jacobsen SE. Human CD34+ hematopoietic stem cells capable of multilineage engrafting NOD/SCID mice express flt3: distinct flt3 and c-kit expression and response patterns on mouse and candidate human hematopoietic stem cells. Blood. 2003;102(3):881–6. [DOI] [PubMed] [Google Scholar]
- 3.Tsapogas P, Mooney CJ, Brown G, Rolink A. The Cytokine Flt3-Ligand in Normal and Malignant Hematopoiesis. Int J Mol Sci. 2017;18(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3(9):650–65. [DOI] [PubMed] [Google Scholar]
- 5.Mead AJ, Linch DC, Hills RK, Wheatley K, Burnett AK, Gale RE. FLT3 tyrosine kinase domain mutations are biologically distinct from and have a significantly more favorable prognosis than FLT3 internal tandem duplications in patients with acute myeloid leukemia. Blood. 2007;110(4):1262–70. [DOI] [PubMed] [Google Scholar]
- 6.Breems DA, Van Putten WL, Huijgens PC, Ossenkoppele GJ, Verhoef GE, Verdonck LF, et al. Prognostic index for adult patients with acute myeloid leukemia in first relapse. J Clin Oncol. 2005;23(9):1969–78. [DOI] [PubMed] [Google Scholar]
- 7.Chevallier P, Labopin M, Turlure P, Prebet T, Pigneux A, Hunault M, et al. A new Leukemia Prognostic Scoring System for refractory/relapsed adult acute myelogeneous leukaemia patients: a GOELAMS study. Leukemia. 2011;25(6):939–44. [DOI] [PubMed] [Google Scholar]
- 8.Smith BD, Levis M, Beran M, Giles F, Kantarjian H, Berg K, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103(10):3669–76. [DOI] [PubMed] [Google Scholar]
- 9.Stone RM, DeAngelo DJ, Klimek V, Galinsky I, Estey E, Nimer SD, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105(1):54–60. [DOI] [PubMed] [Google Scholar]
- 10.Fischer T, Stone RM, Deangelo DJ, Galinsky I, Estey E, Lanza C, et al. Phase IIB trial of oral Midostaurin (PKC412), the FMS-like tyrosine kinase 3 receptor (FLT3) and multi-targeted kinase inhibitor, in patients with acute myeloid leukemia and high-risk myelodysplastic syndrome with either wild-type or mutated FLT3. J Clin Oncol. 2010;28(28):4339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeAngelo DJ, Stone RM, Heaney ML, Nimer SD, Paquette RL, Klisovic RB, et al. Phase 1 clinical results with tandutinib (MLN518), a novel FLT3 antagonist, in patients with acute myelogenous leukemia or high-risk myelodysplastic syndrome: safety, pharmacokinetics, and pharmacodynamics. Blood. 2006;108(12):3674–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fiedler W, Serve H, Dohner H, Schwittay M, Ottmann OG, O’Farrell AM, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105(3):986–93. [DOI] [PubMed] [Google Scholar]
- 13.Zhang W, Konopleva M, Shi YX, McQueen T, Harris D, Ling X, et al. Mutant FLT3: a direct target of sorafenib in acute myelogenous leukemia. Journal of the National Cancer Institute. 2008;100(3):184–98. [DOI] [PubMed] [Google Scholar]
- 14.Cortes J, Roboz GJ, Kantarjian HM, Feldman EJ, Karp JE, Pratz KW, et al. A Phase I Dose Escalation Study of KW-2449, An Oral Multi-Kinase Inhibitor against FLT3, Abl, FGFR1 and Aurora in Patients with Relapsed/Refractory AML, ALL and MDS or Resistant/Intolerant CML. Blood. 2008;112(11):2967.-. [Google Scholar]
- 15.Cortes JE, Kantarjian H, Foran JM, Ghirdaladze D, Zodelava M, Borthakur G, et al. Phase I study of quizartinib administered daily to patients with relapsed or refractory acute myeloid leukemia irrespective of FMS-like tyrosine kinase 3-internal tandem duplication status. J Clin Oncol. 2013;31(29):3681–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinelli G, Perl AE, Dombret H, Kayser S, Steffen B, Rousselot PH, et al. , editors. Effect of quizartinib (AC220) on response rates and long-term survival in elderly patients with FLT3-ITD positive or negative relapsed/refractory acute myeloid leukemia. ASCO Annual Meeting; 2013. [Google Scholar]
- 17.Cortes JE, Kantarjian HM, Kadia TM, Borthakur G, Konopleva M, Garcia-Manero G, et al. , editors. Crenolanib besylate, a type I pan-FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3-ITD and D835 AML. ASCO Annual Meeting; 2016. [Google Scholar]
- 18.Levis M, Ravandi F, Wang ES, Baer MR, Perl A, Coutre S, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117(12):3294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stone RM, Mandrekar SJ, Sanford BL, Laumann K, Geyer S, Bloomfield CD, et al. Midostaurin plus Chemotherapy for Acute Myeloid Leukemia with a FLT3 Mutation. N Engl J Med. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rydapt Product Information https://www.ema.europa.eu/en/documents/product-information/rydapt-epar-product-information_en.pdf2017 [Available from: https://www.ema.europa.eu/en/documents/product-information/rydapt-epar-product-information_en.pdf.
- 21.Tarver TC, Hill JE, Rahmat L, Perl AE, Bahceci E, Mori K, et al. Gilteritinib is a clinically active FLT3 inhibitor with broad activity against FLT3 kinase domain mutations. Blood Adv. 2020;4(3):514–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frankel SR, Eardley A, Lauwers G, Weiss M, Warrell RP Jr. The “retinoic acid syndrome” in acute promyelocytic leukemia. Annals of internal medicine. 1992;117(4):292–6. [DOI] [PubMed] [Google Scholar]
- 23.Fathi AT, Le L, Hasserjian RP, Sadrzadeh H, Levis M, Chen YB. FLT3 inhibitor-induced neutrophilic dermatosis. Blood. 2013;122(2):239–42. [DOI] [PubMed] [Google Scholar]
- 24.Varadarajan N, Boni A, Elder DE, Bagg A, Micheletti R, Perl AE, et al. FLT3 Inhibitor-Associated Neutrophilic Dermatoses. JAMA Dermatol. 2016;152(4):480–2. [DOI] [PubMed] [Google Scholar]
- 25.Sexauer A, Perl A, Yang X, Borowitz M, Gocke C, Rajkhowa T, et al. Terminal myeloid differentiation in vivo is induced by FLT3 inhibition in FLT3/ITD AML. Blood. 2012;120(20):4205–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Norsworthy KJ, Luo L, Hsu V, Gudi R, Dorff SE, Przepiorka D, et al. FDA Approval Summary: Ivosidenib for Relapsed or Refractory Acute Myeloid Leukemia with an Isocitrate Dehydrogenase-1 Mutation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2019. [DOI] [PubMed] [Google Scholar]
- 27.Enasidenib Multi-Discipline Review/Summary, Clinical, Non-Clinical 2017. [Available from: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2017/209606Orig1s000MultidisciplineR.pdf.
- 28.Guidance for Industry: Developing Targeted Therapies in Low-Frequency Molecular Subsets of a Disease 2018. [Available from: https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM588884.pdf. [DOI] [PMC free article] [PubMed]
- 29.Norsworthy KJ, Mulkey F, Scott EC, Ward AF, Przepiorka D, Charlab R, et al. Differentiation syndrome with ivosidenib and enasidenib treatment in patients with relapsed or refractory IDH-mutated AML: a U.S. Food and Drug Administration systematic analysis. Clinical cancer research : an official journal of the American Association for Cancer Research. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.